

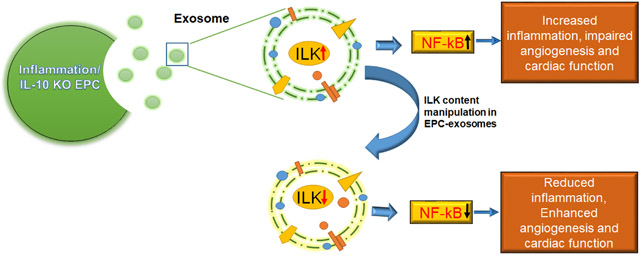
Abstract
Rationale:
Systemic inflammation compromises the reparative properties of Endothelial Progenitor Cell (EPC) and their exosomes on myocardial repair, while the underlying mechanism of loss of function of exosomes from inflamed EPCs is still obscure.
Objective:
To determine the mechanisms of Interleukin-10 (IL-10) deficient-EPC-derived exosome dysfunction in myocardial repair and to investigate if modification of specific exosome cargo can rescue reparative activity.
Methods and Results:
Using interleukin-10 (IL-10) knockout (KO) mice mimicking systemic inflammation condition, we compared therapeutic effect and protein cargo of exosomes isolated from wild type EPC and IL-10KO-EPC. In a mouse model of myocardial infarction (MI) WT-EPC-Exo treatment significantly improved left ventricle cardiac function, inhibited cell apoptosis, reduced MI scar size and promoted post-MI neovascularization, while IL-10KO-EPC-Exo treatment showed diminished and opposite effects. Mass spectrometry analysis revealed WT-EPC-Exo and IL-10KO-EPC-Exo contain different protein expression pattern. Amongst differentially expressed proteins, Integrin Linked Kinase (ILK) was highly enriched in both IL-10KO-EPC-Exo as well as TNFα treated Mouse Cardiac Endothelial Cell derived exosomes (MCEC+TNFα-Exo). ILK enriched exosomes activated NFκB pathway and NFkB-dependent gene transcription in recipient endothelial cells and this effect was partly attenuated though ILK knockdown in exosomes. Intriguingly, ILK knockdown in IL-10KO-EPC-Exo significantly rescued their reparative dysfunction in myocardial repair, improved left ventricle cardiac function, reduced MI scar size and enhanced post-MI neovascularization in MI mouse model.
Conclusions:
IL-10 deficiency/inflammation alters EPC derived exosome function, content and therapeutic effect on myocardial repair by upregulating ILK enrichment in exosomes and ILK-mediated activation of NFκB pathway in recipient cells while ILK knockdown in exosomes attenuates NFκB activation and reduces inflammatory response. Our study provides new understanding of how inflammation may alters stem cell-exosome mediated cardiac repair and identifies ILK as a target kinase for improving progenitor cell exosome-based cardiac therapies.
Keywords: Stem cell, exosome, inflammation, myocardial repair, interleukin-10, integrin-linked kinase, progenitor cell, myocardial infarction, interleukin
Subject Terms: Animal Models of Human Disease, Angiogenesis, Basic Science Research, Cell Signaling/Signal Transduction, Inflammation
Graphical Abstract
INTRODUCTION
Cardiovascular disease is the leading cause of morbidity and mortality worldwide and stem cell therapy holds great promise for ischemic cardiac repair and regeneration1, 2. Bone marrow derived EPC progressed into clinical trials and showed promising yet modest results3-5. Besides poor survival and low integration of EPCs/stem cells in the myocardium, prolonged and unresolved inflammation post ischemic heart disease also significantly compromises EPC phenotype and may compromise their reparative function in myocardial repair6, 7. Evidence suggests that almost one fifth of patients after MI are affected by complications with severe systemic inflammation as well as higher risk of death8-10. Systemic inflammation in patients is characterized with higher level of pro-inflammatory cytokine TNFα and lower level of anti-inflammatory cytokine IL-10 in the circulation. EPCs from these patients are dysfunctional with reduced cell survival, migratory and angiogenic activities11, 12. Understanding how inflammatory stimulus modulates EPC behavior is important for appropriate and timely containment and resolution of inflammation thereby enhancing EPC/progenitor cell based myocardial repair and regeneration.
Recent studies suggest that stem cell mediated myocardial repair is largely associated with paracrine effect13, 14. Exosomes have recently been identified as major paracrine functional unit of stem cells. Exosomes are secreted from all types of cells and contain cell specific small RNAs and proteins, cargo that largely determines exosome function15. Stem cell-derived exosomes show cardiac repair and regeneration properties akin to their parent cell themselves15-17. However, all exosomes, even when secreted by same cells, are not created equal; different pathophysiological conditions and stimuli of parental cells is known to alter exosome content and may modulate exosome function. Our previous studies using IL-10 knockout mice mimicking systemic inflammation conditions, indicated that IL-10-deficient EPCs secrete exosomes that are deficient in promoting cell survival, migration and angiogenesis in vitro and their cargo shows differential protein and RNA expression pattern18. However, how systemic inflammation leads to EPC exosome dysfunction is not yet clear. Understanding the molecular mechanism of exosome function/dysfunction is urgent for exosome manipulation as well as application of this cell-free therapy in cardiovascular disease.
NF-κB plays a key role in regulating inflammatory response. As a master transcription factor, after activation and nuclear translocation, it binds to DNA response elements and upregulates expression of many genes including inflammatory cytokines, chemokines and adhesion molecules19. Integrin-linked kinase (ILK) is an intracellular serine/threonine protein kinase and plays essential role in NF-κB activation20, 21. ILK can be activated via either interaction between extracellular matrix with integrins or inflammatory stimulus (LPS, TNFα, etc.) through the PI3K pathway22, 23. Activated ILK phosphorylates the p65 subunit of NFκB, triggering NFκB nuclear translocation and downstream gene expression. With the dissociation of p65, inhibitor of κBα (IκBα) is phosphorylated by IκB kinase (IKK), then rapidly ubiquitinated and degraded24, 25. It has also been shown that ILK is not required for the inflammatory stimuli-induced classical NF-κB signaling pathway involving IκB-α phosphorylation and degradation and nuclear translocation of NFkBp65. Rather, ILK utilizes alternate mechanisms for NF-κB activation and transcription activity through direct p65 phosphorylation at Ser536 and is required for LPS-induced transactivation of p65 through Ser53623.
ILK is widely distributed in mammalian tissues, but its highest expression in heart suggests an essential role in cardiac function regulation26. As upstream molecule of the Akt and GSK3β pathway, it acts in cell survival and proliferation27, but in cardiomyocytes, it appears to be pro-hypertrophic28, pro-fibrosis29, pro-inflammation30 and it is induced during cardiac aging31. In the present study, we found ILK was highly enriched in both IL-10KO-EPC-derived exosomes and TNFα-treated mouse cardiac endothelial cell-derived exosomes, suggesting its important role in exosome-mediated inflammatory response. Our study explored the role of exosome delivered ILK in NFκB activation in recipient cell and how ILK-enriched exosomes involved in inflammatory response and whether ILK knockdown in exosomes may rescue myocardial repair ability of inflamed EPC-derived exosomes.
Data from our studies identify an essential role of ILK in EPC-exosome-mediated inflammatory response via the activation of NFκB pathway. We further report that knock down of ILK in inflamed exosomes attenuates the ILK-enriched-exosome-mediated inflammatory response, inhibits the NFκB pathway activation and enhances the EPC-derived exosomes reparative activity in ischemic heart.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Expanded methods are included in Online supplement.
Vertebrate animals and reagents.
All experiments conform to the protocols approved by Temple University Institutional Animal Care and Use Committee. Eight-to-ten-week-old wild-type (C57BL/6J, WT) and IL-10 knockout (B6.129P2-Il10tm1Cgn/J, IL-10KO) male mice were procured from Jackson Research Laboratory (Bar Harbor, ME) for EPC isolation. Ten-to-twelve-week-old wild type (C57BL/6J, WT) male mice were procured from Jackson Research Laboratory (Bar Harbor, ME) for myocardial infarction surgery. Studies were only performed in male mice to eliminate the possible confounding effects of reproductive hormones. Lipopolysaccharide (LPS) was obtained from Sigma Aldrich Inc (St. Louis, MO). Recombinant Mouse TNF-alpha (aa 80-235) protein was obtained from R&D Systems Inc.
BM-EPC isolation, culture and supernatant collection.
EPC isolation, ex vivo expansion and culture of EPCs were performed as previously described32. For exosome studies, all Fetal Bovine Serum (FBS) used in cell culture was Exosome depleted FBS (ThermoFisher Scientific A2720801). Supernatant of WT-EPC and IL-10KO-EPC group was collected once daily from day 7 to day 10 for exosome isolation; for ILK-KD-EPC-Exo group, siRNA transduction was administrated onetime on day 7 IL-10KO-EPCs and supernatant was collected onetime daily from day 8 to day 10 for exosome isolation.
MCEC cell culture, treatment and supernatant collection.
Immortalized mouse cardiac endothelial cell (MCEC) line was purchased from CEDARLANE (CLU510) and maintained according to the manufacturer’s instructions. Cells were cultured in DMEM with 10 mmol/L penicillin/streptomycin (Gibco), 10 mmol/L HEPES (Sigma) and 5% exosome-depleted-FBS (ThermoFisher Scientific) and plated on cell culture dishes coated with 0.2% Gelatin (Sigma). For MCEC-Exo group, supernatant was collected onetime daily; for MCEC+TNFα-Exo group, MCECs were treated with TNFα 100ng/ml for 24h, washed with PBS, replenished with fresh medium, and supernatant collected after 24h; for ILKKD-Exo group, MCECs were first treated with 100ng/ml TNFα for 24h, washed with PBS, replenished with fresh medium, then treated with siRNA for another 24h, and supernatant was collected after 24h, 48h and 72h for further studies. For experiments involving MCECs with exosome treatment, exosome number was 100-fold of cell number.
Exosome isolation and quantification.
Exosome isolation, purification, storage and characterization were performed as previously described33. Exosome protein level was quantified with the Pierce BCA Protein Assay Kit (Thermo Fisher) as per manufacturer’s instructions. Exosome particle number was quantified by NanoSight using Nanoparticle Tracking Analysis (NTA) software (Malvern Panalytical).
Exosome mass spectrometry analysis.
WT-EPC and IL-10KO-EPC-derived exosomes (50 μg protein) in PBS (duplicates in each group) were sent to Quantitative Proteomics Resource Core (QPRC) at University of Pennsylvania for mass spectrometry analysis as previously described18.
siRNA knockdown experiment.
Small interfering RNA (siRNA) against mouse ILK (Santa Cruz, sc-35667) or a negative control (siRNA-NC) were used for siRNA knockdown experiments. 24h after plating, cells were transfected with 100 nmol/L siRNA and Lipofectamine RNAiMax (Invitrogen) in OptiMEM (Invitrogen) media overnight. Cell and exosomes samples of 24h, 48h and 72h after siRNA transfection were collected for ILK knockdown efficiency tests.
Preparation of cell lysates, exosome lysates and western blotting.
Cells and exosomes lysates preparation and western blotting were conducted as previously described18, 34. As for antibody usage, beta-actin (sc-47778) was used for cell loading control while flotillin-1 (sc-74566) and CD63 (sc-15363) were used for exosome loading control. Other involved antibodies include ILK (ab52480, Abcam), NFκB p65 (# 6956S, CST), NFκB-Phospho-p65 (#3033, CST), IκBα (#9242, CST), Phospho-IκBα (#9246, CST). All antibody information can be found in Online Table III.
Tube formation assay.
4 × 104 MCECs were plated on 130 μL Matrigel (Corning) per well in a 48-well plate and simultaneously treated with MCEC-Exo (4×106), MCEC+TNFα-Exo (4×106), ILKKD-Exo (4×106) or PBS. After incubation at 37°C in an atmosphere of 5% CO2 for 6h, gels were observed by using a phase contrast microscope. The total branch points were counted in each well. Results are represented as mean ± SEM for three independent experiments.
Induction of acute myocardial infarction (AMI).
Mice underwent surgery to ligate the left anterior descending coronary artery as reported previously35 followed by administration of exosomes (WT-EPC-Exo, IL-10KO-EPC-Exo, ILK-KD-EPC-Exo all 2×109 particles per group) or PBS intramyocardially into the left ventricular wall (border zone) at three different locations immediately after left anterior descending ligation. The sham group underwent the same open chest procedure without ligation (10-13 mice/group). Heart tissue was harvested on day 5 or around a month (day 30 or day 31) after AMI for histological analysis. Whenever possible experiments were performed in a blinded fashion. For example, experiments were blinded for the surgeon performing MI or sham surgeries. Animals were excluded only under circumstances that there was surgery associated mortality (<24h post-surgery) or mice with no-MI, otherwise there were no exclusions in this study. Heart tissue was harvested on day 5 or around a month (day 30 or day 31) after AMI for histological analysis, MI area percentage in whole heart data was collected for Trichrome analysis, border zone data was collected for vessel density and TUNEL analysis, data from all slides were used for calculation and representative slides were shown in figures. Animal number calculations followed our published results of prior studies from our laboratory, which measured recovery from MI in these model systems. At a two-sided significance of .05 and a power of .90, we estimated that 8-10 mice in each treatment group at each time point will be required.
Echocardiography.
Transthoracic two-dimensional M-mode echocardiography was performed using the Vevo2100 (VisualSonics, Toronto, ON, Canada) equipped with a 30-MHz transducer as described previously35.
Tissue preparation, TUNEL assay, CD31 staining and Masson’s trichrome staining.
Heart tissue preparation, TUNEL assay (Roche), CD31 staining (R&D, AF3628) and Masson’s trichrome staining (Sigma) were performed as previously described35, 36.
Statistics.
Results are expressed as the mean +/−standard error of the mean (SEM), computed from separate experiments. Comparison of two or more groups was performed by 1-way or 2-way ANOVA with Tukey’s multiple comparisons test. Significance for Figure 2D was calculated using unpaired Student’s t test using non-parametric measures and Mann-whitney normality test. P < 0.05 is considered statistically significant. Error bars represent ±SEM. Statistical analysis was performed using Graph Pad Prism v 7.0 software”. The images presented in the manuscript are randomly selected representative image of original data from multiple experiments and used as a visual representation of cumulative data.
Figure 2. ILK enrichment in both IL-10KO-EPC-Exo and MCEC+TNFα-Exo:

A, scatter plot of mass spectrometry analysis showed different protein expression patterns in WT-EPC-Exo and IL-10KO-EPC-Exo. B, gene ontology analysis on proteins overexpressed in IL-10KO-EPC-Exo for biological process identification. C and D, Western blotting analysis verified ILK enrichment in both IL-10KO-EPC-Exo and MCEC+TNFα-Exo (*vs MCEC-Exo, N=5, *p=0.0368, #vs WT-EPC-Exo, N=3, ##p=0.0087).
RESULTS
Exosomes from IL10KO EPCs are functionally inert.
To assess EPC-derived exosome dysfunction under IL-10 deficiency, WT-EPC-Exo, IL-10KO-EPC-Exo or PBS were intramyocardially administered in AMI mouse model. Exosome size and yield was similar in WT and IL1-0 KO EPCs suggesting loss of IL10 gene does not alter exosome secretion (Online Figure 1). Left ventricle cardiac function was measured with echocardiography serially up to a month. Subgroups of mice were sacrificed on day 5 post-MI for cardiac apoptosis analysis. Masson’s trichrome staining and CD31 staining on day 31 post-MI heart samples were used for cardiac remodeling and angiogenesis analysis. Our data revealed that compared to saline, WT-EPC-Exo treatment significantly improved left ventricle cardiac function as evident from increased percentage ejection fraction (EF%) and fractional shortening (FS%) on day 31 post-MI (Figure 1A and 1B) and significant reduction in cardiomyocyte apoptosis as shown on day 5 post-MI heart samples (Figure 1C and 1D). Histological analysis on day 31 post-MI heart samples indicated that WT-EPC-Exo treatment significantly decreased MI scar size and promoted neovascularization (Figure 1E to 1H and Online Figure III). However, no cardiac beneficial effect was observed in IL-10KO-EPC-Exo treated groups. Conversely, IL-10KO-EPC-Exo treatment enhanced apoptosis and inhibited post-MI angiogenesis. Taken together these findings demonstrated that compared to WT-EPCs exosomes which showed reparative activities, exosomes from IL-10 deficient EPCs lose their myocardial repair, angiogenic and cell survival activities.
Figure 1. IL-10KO-EPC-Exo do not improve post-MI cardiac repair:

: A and B, Echo analysis at various time points after MI. WT-EPC-Exo treated mice showed significant improvement in %EF and %FS. IL10KO-EPC-Exo treated mice did not show any improvements in cardiac function (N=10-13/group; *p=0.0103,####p-0.0003 in A; *p=0.0105, ####p=0.0004 in B) . C and D, TUNEL analysis on day 5 heart samples for post-MI cell apoptosis measurement showed enhanced apoptotic cells in IL-10KO-EPC-Exo treated mice (scale bar: 100μm, N=6, *p=0.0367, ####p<0.0001). E and F, Masson’s trichrome staining on day 31 heart samples for post-MI scar size measurement showed larger infarct size in IL10-KO-EPC-Exo group (scale bar: 2000 μm, N=6, ##p=0.0046, ***p=0.0005). G and H, CD31 staining on day 31 heart for post-MI angiogenesis measurement (scale bar: 100 μm, N=6, *p=0.0281, ##p=0.0053; *vs PBS group, #vs IL-10KO-EPC-Exo group).
IL-10 deficiency altered EPC exosome protein contents with enrichment of ILK.
To understand if differential protein cargo between exosomes might be involved in apparently opposing biological activities, mass spectrometry was conducted to evaluate the protein content of WT-EPC-Exo and IL-10KO-EPC-Exo. Protein expression pattern in two exosomes was significantly different with alteration in proteins related to multiple cellular functions/pathways (Online Table I). Proteins that were highly over-expressed in IL-10KO-EPC-Exo were categorized to biological process using gene ontology, and as expected, we found enrichment of inflammation related proteins in IL-10KO-EPC-Exo compared to WT-EPC-Exo. We performed western blotting to further validate protein expression from proteomic analysis. In order to establish that enriched proteins in IL10KO EPC exosomes are induced due to inflammation and not by loss of IL10 itself, we used exosomes from MCEC treated with TNFα as a surrogate for inflammatory model in vitro. Our results suggested IL-10 deficiency altered EPC-derived exosome protein content (Figure 2A). A number of protein kinase showed differential expression (Online Figure II). We particularly focused on ILK as its role is well documented in inflammation and it was highly enriched in IL10-KO-EPC exosomes (Figure 2B and Online Figure II). Western blotting confirmed that there was about 2 to 4-fold increase in ILK expression in both IL-10KO-EPC-Exo and MCEC+TNFα-Exo, without much difference in total cell protein content (Figure 2C and 2D). These data indicated that inflammatory stimulus changed cell-derived exosome protein content, and ILK was generally enriched in inflamed exosomes, suggesting its potential role in mediating inflammatory response.
ILK enriched exosomes activate the NFκB pathway in recipient cells.
To elucidate the role of ILK-enriched exosomes in mediating the inflammatory response, MCECs were treated with MCEC-Exo, MCEC+TNFα-Exo, PBS or TNFα. In both short-time (15min, 30min, 1h) and long-time (24h) treatment, MCEC+TNFα-Exo activated nuclear translocation of NFκB (Figure 3A and 3B) and induced IκBα degradation (Figure 4A and 4B); 24h treatment triggered NFκB downstream gene expression (Figure 4C). Similar results were observed in TNFα treated group which was designed as positive control. No significant difference was observed between MCEC-Exo and PBS group. Additionally, TNF-treated MCEC-Exo did not alter IκBα phosphorylation suggesting an alternate mechanism of ILK-mediated NFκB activation. ILK has been shown to act via alternate NFκB activation by directly phosphorylating serine 536 residue of p65 subunit of NFκB and facilitating its nuclear translocation. Indeed, MCEC=TNF-Exo treatment led to efficient serine536 phosphorylation and nuclear translocation of p65 (Online Figure IV). Collectively, these results indicated that ILK enriched exosomes activate NFκB pathway in recipient cells and enhanced inflammatory response by enhancing inflammatory gene expression.
Figure 3. MCEC+TNFα-Exo activated NFκB nuclear translocation:

A and B, NFκB staining analysis showed both MCEC+TNFα-Exo and TNFα treatment triggered NFκB nuclear translocation in recipient MCEC cells. *vs PBS group (N=3, **p=0.0050, ***p=0.0004, ****p<0.0001); #vs MCEC-Exo group (N=3, #p=0.0380, ##p=0.0066, ###p=0.0004) (scale bar:10 μm).
Figure 4. MCEC+TNFα-Exo activated NFκB signaling pathway:

A and B, Western blotting showed both MCEC+TNFα-Exo and TNFα activated NFκB signaling pathway, decreased IκBα expression in recipient MCEC cells. (N=3, *p=0.0399, **p=0.0066, #p=0.0308, ##p=0.0018) C, qRT-PCR analysis showed 24h of MCEC+TNFα-Exo treatment triggered NFκB downstream gene expression in recipient MCEC cells. (N=4, *p=0.0250, ***p=0.0009); *vs PBS group #vs MCEC-Exo group (##p=0.0047).
ILK knockdown exosomes showed attenuated NFκB activation effect.
To confirm the role of ILK enriched exosomes in activating inflammatory response in recipient cells, siRNA strategy was used for ILK knockdown in cells before the isolation of exosomes. SiRNA-mediated ILK knockdown efficiency was approximately 70% to 90% in both EPCs and MCECs and their exosomes, as confirmed by western blots (Online Figure V). To rule out the possibility that siRNA may transfer to exosomes and directly alter ILK expression, MCECs were treated with ILKKD exosomes for 24 h. No changes in ILK mRNA was detected (Online Figure VI). Repeat experiments with ILK KD exosomes indicated that short time exposure (15min, 30min, 1h) significantly attenuated NFκB nuclear translocation although some degree of NFkB translocation was evident with 24 hour treatment although at levels lower than TNF α-treated cell exosomes (Figure 5A and 5B). ILKKD-Exo short-time (15min, 30min, 1h) treatment did not induce IκBα degradation (Figure 6A and 6B) and 24h treatment showed lower level of NFκB-induced downstream gene expression (Figure 6C). Taken together these observations suggest ILK enriched exosomes activate inflammatory response in recipient cells through NFκB activation. Blocking ILK in exosomes successfully inhibited NFκB activation and inflammatory response in recipient cells.
Figure 5. ILK knockdown exosomes inhibit NFκB nuclear translocation:

A and B, NFκB staining analysis indicated nuclear NFκB translocation was not activated with ILKKD-Exo short time treatment but activated with long time treatment. (N=3, **p=0.0087, ****p<0.0001; ##p=0.0015, ###p=0.0005, ####p<0.0001; Δ p=0.0121, ΔΔ p=0.0016, ΔΔΔ p=0.0001, ΔΔΔΔ p<0.0001; *vs PBS group #vs MCEC-Exo group Δ vs ILKKD-Exo group; scale bar:10 μm).
Figure 6. ILK knockdown exosomes attenuate NFκB signaling pathway activation:

A and B, Western blotting showed short time ILKKD-Exo treatment inhibited IκBα degradation. (N=3, *p=0.212, **p=0.0076, #p=0.0235) C, qRT-PCR analysis indicated 24h of ILKKD-Exo treatment showed lower level of NFκB downstream gene expression. (N=3, *p=0.0371, **p=0.0016, ****p<0.0001; ##p=0.0067, ####p<0.0001; ΔΔΔ p<0.0001; *vs PBS group, #vs MCEC-Exo group, Δ vs ILKKD-Exo group).
ILK knockdown in MCEC+TNFα-Exo rescued their angiogenic activity, in vitro.
We have previously shown that IL-10KO-EPC-Exo inhibited tube formation ability of endothelial cells in vitro18. Similar inhibition in EC tubulogenesis was observed with the MCEC+TNFα-Exo treatment. On the contrary, ILKKD-Exo treatment significantly enhanced tube formation and angiogenic capabilities of endothelial cells (Figure 7A and 7B). Therefore, the inhibiting effect on angiogenesis induced by inflamed exosomes was attenuated through ILK knockdown.
Figure 7. ILK knockdown in MCEC+TNFα-Exo rescued angiogenesis dysfunction:

A and B, MCEC+TNFα-Exo inhibited tube formation while ILK knockdown in exosomes attenuated the inhibiting effect. (N=5, **p=0.016, #p=0.0304, Δp=0.0172; *vs PBS group; #vs MCEC-Exo group; Δvs ILKKD-Exo group).
ILK knockdown in IL-10KO-EPC-Exo rescued their reparative activity, in vivo.
To further investigate ILK knockdown exosomes function in ischemic cardiac repair, we conducted same siRNA strategy on IL-10KO-EPC (Online Figure V) and collected ILK-KD-EPC-Exo for in vivo treatment in mouse AMI model and directly compared the reparative properties of IL10-KO-EPC-exo with ILK-KD exo from IL-10KO EPCs. Echocardiography, Masson’s trichome staining and CD31 staining were performed as previously described. We observed significantly increased %EF and %FS in both WT-EPC-Exo and ILK-KD-EPC-Exo treated groups on day 30 post-MI whereas IL-10EPC-exo remained nonfunctional as shown earlier in Figure 1. (Figure 8A and 8B), suggesting improved left ventricle cardiac function. Histological analysis on day 30 post-MI heart samples showed significantly reduced scar size and increased capillary density in both WT-EPC-Exo and ILK-KD-EPC-Exo groups (Figure 8C to 8F), indicating ameliorated post-MI scar formation and enhanced neovascularization. Taken together, these data suggested ILK knockdown successfully rescued IL-10 deficiency/inflammation-induced EPC exosomes dysfunction in myocardial repair.
Figure 8. ILK knockdown in IL-10KO-EPC-Exo rescued cardiac repair and regeneration dysfunction:

A and B, Echo analysis followed for a month, EF and FS were used for left ventricle cardiac function measurement (N=10, in A, *p=0.0222, ***p=0.0002, ****p<0.0001, ###p=0.0002, ###p=0.0007, ####p<0.0001, in B, *p=0.0231, **p=0.0065, #p=0.0163, ###p=0.0001, ####p<0.0001, $p=0.0103). C and D, Masson’s trichrome staining analysis on day 30 post-MI heart for scar size measurement (scale bar: 2000 μm, N=15, ###p=0.0001, ####p<0.0001, ****p<0.0001). E and F, CD31 staining on day 30 post-MI heart for angiogenesis measurement (scale bar: 100 μm, N=9, ***p=0.0005, ****p<0.0001, ####p<0.0001). (*vs PBS group; #vs IL-10KO-EPC-Exo group; $vs ILK-KD-EPC-Exo group).
DISCUSSION
Over the past two decades, stem cell-based therapy has emerged as promising therapeutic approach for ischemic cardiac repair and regeneration37. Endothelial progenitor cells generated from bone marrow or peripheral blood have been applied in clinical studies. However, prolonged and unresolved inflammation after myocardial infarction appears to worsen cardiac function recovery and may alter autologous progenitor cell biology thereby compromising the therapeutic effects of EPC transplantation7. Interleukin-10 (IL-10) is a naturally occurring anti-inflammatory cytokine and has been shown to be involved in proliferation, anti-apoptotic and cell survival activities, dampens the inflammatory effects in overwhelming infections and attenuate tissue damage. Multiple studies from our and other labs have established a protective role of IL-10 in cardiac injury and repair. After MI, IL-10-deficient mice have increased infarct size and heavier myocardial necrosis with more neutrophil infiltration. IL-10 treatment in MI mouse reduces inflammation, improves left ventricle cardiac physiology, stimulates M2 macrophage polarization and fibroblast activation and improves cardiac remodeling38. However, human studies are not very consistent. Some studies find that in patients with acute MI, higher serum IL-10 within 24 h after angioplasty is associated with reduced incidence of heart failure progression whereas others show that higher IL-10 predict recurrent cardiac events or heart failure39, 40. In particular to stem cell therapy in ischemic heart disease, many studies from our lab have shown that IL-10 deficiency impairs bone marrow derived endothelial progenitor cell survival and function in ischemic myocardium and IL-10 treatment improves cardiac remodeling and function, inhibits fibroblast progenitor cell migration and trans differentiation, decreases cardiac fibrosis 35,36. Our published studies also established that EPCs were functionally impaired under inflammatory stimulus35. IL-10KO mice mimicking systemic inflammation have high level of circulating TNFα and do not express IL-10 in circulation, thus mimicking the pathological condition of systemic inflammation. Using IL-10KO mice as a model, we reported that IL-10KO-EPC-derived exosomes were functionally impaired in promoting cell survival, migration and angiogenesis, in vitro 18. In this study, we further explored the effect of IL-10 deficiency on EPC-derived exosome content and function and on mechanistic basis of inflammation induced stem cell-exosome dysfunction both in vitro and in vivo.
Recent studies revealed that functional benefits of stem cell therapy are largely mediated via paracrine effects41-43. Exosomes have emerged as a major paracrine functional units; they are involved in intercellular communication and are characterized with cell-specific cargo of proteins, small RNAs and lipids. Large number of recent studies have explored cardiac repair and regeneration benefit of exosomes from induced pluripotent stem cell(iPSCs)17, mesenchymal stem cell(MSCs)44 and bone marrow cells18 in myocardial infarction45, heart failure46, ischemia/reperfusion injury47 and many other pathologies. However, exosomes are not created equal and their cargo is actively under alteration reflecting the physiological or pathological condition of parental cells15. Cardiovascular diseases are usually accompanied with ischemia, hypoxia, intense inflammatory stress, diabetes or aging which may dramatically compromise stem cell cardiac repair ability35, 48, 49 and induce dysfunctional exosome secretion18, 49. Previous in vitro study from our lab revealed that IL-10 deficiency induced EPCs exosome dysfunction in preserving cell survival, promoting cell mobilization and angiogenesis18 and altered exosomal RNA and protein content.
To further enhance therapeutic effect of stem cell-derived exosomes, multiple studies have focused on microRNA (miR) manipulation in exosomes. Our previous studies found overexpressing miR-294 in mouse embryonic stem cell exosomes increased survival, cell cycle progression and proliferation of cardiac progenitor cells for MI treatment34. Knocking down miR-375 in IL-10KO-EPC-Exo enhanced endothelial cells ability against hypoxia stress18, 50. Despite excellent progress made in miRs, very few studies have invested efforts to investigate the functional roles for exosomal protein cargo likely due to difficulties in stem cell culture and exosome protein collection. Our data on inflammation induced augmentation of ILK in exosomes and a functional role of exosomal ILK in altering signaling and biology of recipient cells therefore opens new understanding on exosome biology and function. To rule out that observed ILK effect in IL-10KO mice are not due to loss of IL10 itself, both BM-EPC-Exo and TNF treated MCEC-derived exosomes were used for mechanistic experiments. We considered MCEC-Exo as a reasonable alternative strategy for several reasons, in context to protein content, both IL-10KO-EPC-Exo and MCEC+TNFα-Exo were enriched with ILK; functionally, we observed similar angiogenesis inhibiting effect in MCEC+TNFα-Exo. With ILK knockdown in exosomes, both IL-10KO-EPC-Exo and MCEC+TNFα-Exo showed increased angiogenic capacity. We believe that the mechanistic studies conducted with MCEC-Exo should have a relatively good reflection of IL-10KO-EPC-Exo.
The role of ILK in heart and stem cells have been investigated for years but not fully understood largely due to its cell and context specific roles. While a study suggested that ILK engineered MSCs showed enhanced cardiac therapeutic effect51 other studies indicated ILK played negative role in cardiomyocytes. ILK was reported to be highly expressed in human failing heart and in cardiomyopathy28. In myocardial hypertrophy, ILK is central mediator of oxidative stress, inflammatory cell recruitment and cardiac remodeling30 and promotes pro-fibrotic process and play critical role in cardiac fibrosis under angiotensin II stimulation52. Moderate down regulation of ILK in heart prevented the decline in cardiac performance with aging31, 52. The exact role of ILK is largely dependent on cell type and pathological condition. Specific to our study, we investigated the role of ILK enriched exosomes in NFκB activation and inflammatory response. Upon inflammatory stimulus (such as LPS, TNFα, etc.), ILK can be activated through PI3K pathway and phosphorylates p65 subunit of NFκB leading to the activation of NFκB pathway. It has also been shown that ILK is not required for the inflammatory stimuli-induced classical NF-κB signaling pathway involving IκB-α phosphorylation and degradation and nuclear translocation of NFkBp65. Rather, ILK utilizes alternate mechanisms for NF-κB activation and transcription activity through direct p65 phosphorylation at Ser536 and is required for LPS-induced transactivation of p65 through Ser53623. Our studies support that ILK contained in exosomes from inflamed cells utilizes the latter alternate mechanism of direct serine536 phosphorylation and nuclear translocation of NFκB-p65
NFκB super family is known to play an essential role in inflammatory response and cardiac cell biology19, 53. Some studies suggest NFκB is cardio-protective during acute hypoxia and reperfusion injury25, 54. However, prolonged activation of NFκB appears to be detrimental and promotes heart failure by eliciting signals that trigger chronic inflammation through enhanced elaboration of cytokines including TNFα, interleukin-1 and interleukin-6, leading to endoplasmic reticulum stress responses and cell death25, 55. In our study, TNFα treatment of ILK knockdown MCEC-derived exosomes showed attenuated NFκB activation in recipient endothelial cells. Knockdown of ILK in IL10KO-EPCs rescued the functionality of their exosomes both in vitro and in vivo. Additionally, we found ILK knockdown also decreased expression of miR-375 (Online Figure VII), which has been shown to be detrimental for EPC survival, migration, angiogenesis as well as myocardial repair. Our results indicated that ILK enriched exosomes contributed to enhanced inflammatory response through NFκB activation, ILK enrichment is at least partially responsible for compromised cardiac repair and angiogenic ability of EPCs and EPC exosomes under inflammatory stimulus, ILK manipulation significantly rescued or enhanced IL-10 deficiency induced EPC exosome dysfunction.
In summary, our study demonstrated that IL-10 deficiency which mimic post-MI systemic inflammation condition, altered EPC-derived exosome content and function in myocardial repair largely via enhanced ILK packaging and ILK-mediated NFkB-dependent augmentation in inflammatory gene expression. Our study identified ILK as a potential target for ischemic heart disease, broadened understanding of exosome mediated inflammatory response and raised possibility of exosome protein manipulation for cell free therapeutic strategy for ischemic heart repair.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Bone marrow-derived endothelial progenitor cells (BM-EPC) contribute to ischemic myocardial neovascularization.
Systemic inflammation such as loss of Interleukin-10 (IL-10) leads to BM-EPC dysfunction.
Exosomes derived from BM-EPCs mimic parental cell reparative properties.
What New Information Does This Article Contribute?
Exosomes from IL-10 knock out mice (systemic inflammation) lose their reparative activity.
Inflammation alters protein contents of EPC exosomes including enrichment of integrin-linked kinase (ILK).
Exosomes enriched in ILK activate NFkB signaling in recipient cells and induce inflammatory gene expression.
Small-interfering RNA (siRNA)-mediated knock down of ILK in IL-10KO EPCs rescue their cardiac repair ability
Bone marrow stem cell therapies in clinical trials showed little or modest effect. It is known that pre-existing comorbid conditions like systemic inflammation and diabetes in cardiac patients may compromise the functional activity of autologous stem/progenitor cells. Lately, exosomes or extracellular vesicles from obtained from stem cells have been shown to mimic parental cell reparative activity in models of myocardial infarction. Whether exosomes obtained from stem cells from mice with systemic inflammation retain their reparative properties and whether inflammation alters protein cargo of exosomes is not known. Using IL-10 knock out mice a model of systemic inflammation, here we report that exosomes obtained from endothelial progenitor cells (EPCs) of these mice lose their angiogenic and cardiac reparative properties. We also show specific enrichment of integrin linked kinase (ILK) in these exosomes which induces NFkB activation and NFkB-mediated inflammatory gene activation in recipient cells. Finally, we report a strategy of ILK-knockdown in exosomes to rescue inflammation-induced loss of their reparative activity.
Acknowledgments
SOURCES OF FUNDING
This work was supported, in part, by funding from the National Institute of Health grants HL091983, HL126186, and HL134608 to Raj Kishore and AHA Pre-doctoral Fellowship 17PRE33370001 to Yujia Yue.
Nonstandard Abbreviations and Acronyms:
- (AMI)
Acute Myocardial Infarction
- (BM-EPCs)
Bone Marrow-derived Endothelial Progenitor Cells
- (Echo)
Echocardiography
- (EF)
Ejection Fraction
- (Exo)
Exosomes
- (FS)
Fractional Shortening
- (ILK)
Integrin-Linked Kinase
- (IL-10)
Interleukin-10
- (IκBα)
Inhibitor of κBα
- (LPS)
Lipopolysaccharide
- (miRs)
MicroRNAs
- (MCEC)
Mouse Cardiac Endothelial Cell
- (NF-κB)
Nuclear factor kappa-light-chain-enhancer of activated B cells
- (PBS)
Phosphate-Buffered Saline
- (TNFα)
Tumor Necrosis Factor-α
- (WT-EPC-Exo)
Wild Type EPC-derived exosome
- (IL-10KO-EPC-Exo)
IL-10 knockout EPC-derived exosome
- (ILK-KD-EPC-Exo)
IL-10 knockout EPC with ILK knockdown-derived exosome
- (MCEC-Exo)
Mouse Cardiac Endothelial Cell-derived exosome
- (MCEC+TNFα-Exo)
TNFα inflamed MCEC-derived exosome
- (ILKKD-Exo)
TNFα inflamed MCEC with ILK knockdown-derived exosome
Footnotes
DISCLOSURES
Authors have nothing to disclose
REFERENCES
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS, American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019:CIR0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Muller P, Lemcke H and David R. Stem Cell Therapy in Heart Diseases - Cell Types, Mechanisms and Improvement Strategies. Cell Physiol Biochem. 2018;48:2607–2655. [DOI] [PubMed] [Google Scholar]
- 3.Naseri MH, Madani H, Ahmadi Tafti SH, Moshkani Farahani M, Kazemi Saleh D, Hosseinnejad H, Hosseini S, Hekmat S, Hossein Ahmadi Z, Dehghani M, Saadat A, Mardpour S, Hosseini SE, Esmaeilzadeh M, Sadeghian H, Bahoush G, Bassi A, Amin A, Fazeli R, Sharafi Y, Arab L, Movahhed M, Davaran S, Ramezanzadeh N, Kouhkan A, Hezavehei A, Namiri M, Kashfi F, Akhlaghi A, Sotoodehnejadnematalahi F, Vosough Dizaji A, Gourabi H, Syedi N, Shahverdi AH, Baharvand H and Aghdami N. COMPARE CPM-RMI Trial: Intramyocardial Transplantation of Autologous Bone Marrow-Derived CD133+ Cells and MNCs during CABG in Patients with Recent MI: A Phase II/III, Multicenter, Placebo-Controlled, Randomized, Double-Blind Clinical Trial. Cell J. 2018;20:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang J, Guo Y, Tan S, Li Z, Xie H, Chen P, Wang K, He Z, He P, Ke Y, Jiang X and Chen Z. Autologous Endothelial Progenitor Cells Transplantation for Acute Ischemic Stroke: A 4-Year Follow-Up Study. Stem Cells Transl Med. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J, Lin L and Zong W. Bone Marrow Mononuclear Cells Transfer for Patients after ST-Elevated Myocardial Infarction: A Meta-Analysis of Randomized Control Trials. Yonsei Med J. 2018;59:611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin CP, Lin FY, Huang PH, Chen YL, Chen WC, Chen HY, Huang YC, Liao WL, Huang HC, Liu PL and Chen YH. Endothelial progenitor cell dysfunction in cardiovascular diseases: role of reactive oxygen species and inflammation. Biomed Res Int. 2013;2013:845037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Desouza CV, Hamel FG, Bidasee K and O’Connell K. Role of inflammation and insulin resistance in endothelial progenitor cell dysfunction. Diabetes. 2011;60:1286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazzerini PE, Capecchi PL and Laghi-Pasini F. Systemic inflammation and arrhythmic risk: lessons from rheumatoid arthritis. Eur Heart J. 2017;38:1717–1727. [DOI] [PubMed] [Google Scholar]
- 9.Tan Y, Tu Y, Tian D, Li C, Zhong JK and Guo ZG. ST-elevation myocardial infarction following systemic inflammatory response syndrome. Cardiovasc J Afr. 2015;26:e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang L, Moore XL, Dart AM and Wang LM. Systemic inflammatory response following acute myocardial infarction. J Geriatr Cardiol. 2015;12:305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eming SA, Wynn TA and Martin P. Inflammation and metabolism in tissue repair and regeneration. Science. 2017;356:1026–1030. [DOI] [PubMed] [Google Scholar]
- 12.Kohsaka S, Menon V, Lowe AM, Lange M, Dzavik V, Sleeper LA, Hochman JS and Investigators S. Systemic inflammatory response syndrome after acute myocardial infarction complicated by cardiogenic shock. Arch Intern Med. 2005;165:1643–50. [DOI] [PubMed] [Google Scholar]
- 13.Tachibana A, Santoso MR, Mahmoudi M, Shukla P, Wang L, Bennett M, Goldstone AB, Wang M, Fukushi M, Ebert AD, Woo YJ, Rulifson E and Yang PC. Paracrine Effects of the Pluripotent Stem Cell-Derived Cardiac Myocytes Salvage the Injured Myocardium. Circ Res. 2017;121:e22–e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rafatian G and Davis DR. Heart-Derived Cell Therapy 2.0: Paracrine strategies to increase therapeutic repair of injured myocardium. Stem Cells. 2018. [DOI] [PubMed] [Google Scholar]
- 15.Khan M and Kishore R. Stem Cell Exosomes: Cell-FreeTherapy for Organ Repair. Methods Mol Biol. 2017;1553:315–321. [DOI] [PubMed] [Google Scholar]
- 16.Kishore R and Khan M. Cardiac cell-derived exosomes: changing face of regenerative biology. Eur Heart J. 2017;38:212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garikipati VNS and Kishore R. Induced Pluripotent Stem Cells Derived Extracellular Vesicles: A Potential Therapy for Cardiac Repair. Circ Res. 2018;122:197–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yue Y, Garikipati VNS, Verma SK, Goukassian DA and Kishore R. Interleukin-10 Deficiency Impairs Reparative Properties of Bone Marrow-Derived Endothelial Progenitor Cell Exosomes. Tissue Eng Part A. 2017;23:1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon JW, Shaw JA and Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011;108:1122–32. [DOI] [PubMed] [Google Scholar]
- 20.Ahmed AU, Yim HCH, Alorro M, Ernst M and Williams BRG. Integrin-Linked Kinase Expression in Myeloid Cells Promotes Inflammatory Signaling during Experimental Colitis. J Immunol. 2017. [DOI] [PubMed] [Google Scholar]
- 21.Hsu EC, Kulp SK, Huang HL, Tu HJ, Chao MW, Tseng YC, Yang MC, Salunke SB, Sullivan NJ, Chen WC, Zhang J, Teng CM, Fu WM, Sun D, Wicha MS, Shapiro CL and Chen CS. Integrin-linked kinase as a novel molecular switch of the IL-6-NF-kappaB signaling loop in breast cancer. Carcinogenesis. 2016;37:430–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alique M, Civantos E, Sanchez-Lopez E, Lavoz C, Rayego-Mateos S, Rodrigues-Diez R, Garcia-Redondo AB, Egido J, Ortiz A, Rodriguez-Puyol D, Rodriguez-Puyol M and Ruiz-Ortega M. Integrin-linked kinase plays a key role in the regulation of angiotensin II-induced renal inflammation. Clin Sci (Lond). 2014;127:19–31. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed AU, Sarvestani ST, Gantier MP, Williams BR and Hannigan GE. Integrin-linked kinase modulates lipopolysaccharide- and Helicobacter pylori-induced nuclear factor kappaB-activated tumor necrosis factor-alpha production via regulation of p65 serine 536 phosphorylation. J Biol Chem. 2014;289:27776–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oeckinghaus A and Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdullah M, Berthiaume JM and Willis MS. Tumor necrosis factor receptor-associated factor 6 as a nuclear factor kappa B-modulating therapeutic target in cardiovascular diseases: at the heart of it all. Transl Res. 2018;195:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hannigan GE, Coles JG and Dedhar S. Integrin-linked kinase at the heart of cardiac contractility, repair, and disease. Circ Res. 2007;100:1408–14. [DOI] [PubMed] [Google Scholar]
- 27.Cho HJ, Youn SW, Cheon SI, Kim TY, Hur J, Zhang SY, Lee SP, Park KW, Lee MM, Choi YS, Park YB and Kim HS. Regulation of endothelial cell and endothelial progenitor cell survival and vasculogenesis by integrin-linked kinase. Arterioscler Thromb Vasc Biol. 2005;25:1154–60. [DOI] [PubMed] [Google Scholar]
- 28.Lu H, Fedak PW, Dai X, Du C, Zhou YQ, Henkelman M, Mongroo PS, Lau A, Yamabi H, Hinek A, Husain M, Hannigan G and Coles JG. Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation. 2006;114:2271–9. [DOI] [PubMed] [Google Scholar]
- 29.Thakur S, Li L and Gupta S. NF-kappaB-mediated integrin-linked kinase regulation in angiotensin II-induced pro-fibrotic process in cardiac fibroblasts. Life Sci. 2014;107:68–75. [DOI] [PubMed] [Google Scholar]
- 30.Bettink SI, Werner C, Chen CH, Muller P, Schirmer SH, Walenta KL, Bohm M, Laufs U and Friedrich EB. Integrin-linked kinase is a central mediator in angiotensin II type 1- and chemokine receptor CXCR4 signaling in myocardial hypertrophy. Biochem Biophys Res Commun. 2010;397:208–13. [DOI] [PubMed] [Google Scholar]
- 31.Nishimura M, Kumsta C, Kaushik G, Diop SB, Ding Y, Bisharat-Kernizan J, Catan H, Cammarato A, Ross RS, Engler AJ, Bodmer R, Hansen M and Ocorr K. A dual role for integrin-linked kinase and beta1-integrin in modulating cardiac aging. Aging Cell. 2014;13:431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garikipati VNS and Kishore R. Endothelial Progenitor Cells: Procedure for Cell Isolation and Applications. Methods Mol Biol. 2017;1553:85–89. [DOI] [PubMed] [Google Scholar]
- 33.Greening DW, Xu R, Ji H, Tauro BJ and Simpson RJ. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol Biol. 2015;1295:179–209. [DOI] [PubMed] [Google Scholar]
- 34.Khan M, Nickoloff E, Abramova T, Johnson J, Verma SK, Krishnamurthy P, Mackie AR, Vaughan E, Garikipati VN, Benedict C, Ramirez V, Lambers E, Ito A, Gao E, Misener S, Luongo T, Elrod J, Qin G, Houser SR, Koch WJ and Kishore R. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ Res. 2015;117:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnamurthy P, Thal M, Verma S, Hoxha E, Lambers E, Ramirez V, Qin G, Losordo D and Kishore R. Interleukin-10 deficiency impairs bone marrow-derived endothelial progenitor cell survival and function in ischemic myocardium. Circ Res. 2011;109:1280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verma SK, Garikipati VNS, Krishnamurthy P, Schumacher SM, Grisanti LA, Cimini M, Cheng Z, Khan M, Yue Y, Benedict C, Truongcao MM, Rabinowitz JE, Goukassian DA, Tilley D, Koch WJ and Kishore R. Interleukin-10 Inhibits Bone Marrow Fibroblast Progenitor Cell-Mediated Cardiac Fibrosis in Pressure-Overloaded Myocardium. Circulation. 2017;136:940–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edlinger C, Schreiber C, Wernly B, Anker A, Ruzicka K, Jung C, Hoppe UC and Lichtenauer M. Stem Cell Therapy for Myocardial Infarction 2001-2013 Revisited. Stem cell reviews. 2015;11:743–51. [DOI] [PubMed] [Google Scholar]
- 38.Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR and Lindsey ML. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. 2017;112:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Falcao RA, Christopher S, Oddi C, Reznikov L, Grizzard JD, Abouzaki NA, Varma A, Van Tassell BW, Dinarello CA and Abbate A. Interleukin-10 in patients with ST-segment elevation myocardial infarction. Int J Cardiol. 2014;172:e6–8. [DOI] [PubMed] [Google Scholar]
- 40.Welsh P, Murray HM, Ford I, Trompet S, de Craen AJ, Jukema JW, Stott DJ, McInnes IB, Packard CJ, Westendorp RG, Sattar N and Group PS. Circulating interleukin-10 and risk of cardiovascular events: a prospective study in the elderly at risk. Arterioscler Thromb Vasc Biol. 2011;31:2338–44. [DOI] [PubMed] [Google Scholar]
- 41.Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, Ito A, Liu T, Kamide C, Agrawal H, Perlman H, Qin G, Kishore R and Losordo DW. Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circ Res. 2011;109:724–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallet R, Dawkins J, Valle J, Simsolo E, de Couto G, Middleton R, Tseliou E, Luthringer D, Kreke M, Smith RR, Marban L, Ghaleh B and Marban E. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. European heart journal. 2017;38:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma S, Mishra R, Bigham GE, Wehman B, Khan MM, Xu H, Saha P, Goo YA, Datla SR, Chen L, Tulapurkar ME, Taylor BS, Yang P, Karathanasis SK, Goodlett DR and Kaushal S. A Deep Proteome Analysis Identifies the Complete Secretome as the Functional Unit of Human Cardiac Progenitor Cells. Circulation research. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun X, Shan A, Wei Z and Xu B. Intravenous mesenchymal stem cell-derived exosomes ameliorate myocardial inflammation in the dilated cardiomyopathy. Biochem Biophys Res Commun. 2018;503:2611–2618. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Ding N, Guan G, Liu G, Huo D, Li Y, Wei K, Yang J, Cheng P and Zhu C. Rapid Delivery of Hsa-miR-590-3p Using Targeted Exosomes to Treat Acute Myocardial Infarction Through Regulation of the Cell Cycle. J Biomed Nanotechnol. 2018;14:968–977. [DOI] [PubMed] [Google Scholar]
- 46.Ye W, Tang X, Yang Z, Liu C, Zhang X, Jin J and Lyu J. Plasma-derived exosomes contribute to inflammation via the TLR9-NF-kappaB pathway in chronic heart failure patients. Mol Immunol. 2017;87:114–121. [DOI] [PubMed] [Google Scholar]
- 47.Huang X, Ding J, Li Y, Liu W, Ji J, Wang H and Wang X. Exosomes derived from PEDF modified adipose-derived mesenchymal stem cells ameliorate cerebral ischemia-reperfusion injury by regulation of autophagy and apoptosis. Exp Cell Res. 2018;371:269–277. [DOI] [PubMed] [Google Scholar]
- 48.Kaur I, Rawal P, Rohilla S, Bhat MH, Sharma P, Siddiqui H and Kaur S. Endothelial progenitor cells from aged subjects display decreased expression of sirtuin 1, angiogenic functions, and increased senescence. Cell Biol Int. 2018. [DOI] [PubMed] [Google Scholar]
- 49.Hassanpour M, Cheraghi O, Brazvan B, Hiradfar A, Aghamohammadzadeh N, Rahbarghazi R and Nouri M. Chronic Exposure of Human Endothelial Progenitor Cells to Diabetic Condition Abolished the Regulated Kinetics Activity of Exosomes. Iran J Pharm Res. 2018;17:1068–1080. [PMC free article] [PubMed] [Google Scholar]
- 50.Garikipati VN, Krishnamurthy P, Verma SK, Khan M, Abramova T, Mackie AR, Qin G, Benedict C, Nickoloff E, Johnson J, Gao E, Losordo DW, Houser SR, Koch WJ and Kishore R. Negative Regulation of miR-375 by Interleukin-10 Enhances Bone Marrow-Derived Progenitor Cell-Mediated Myocardial Repair and Function After Myocardial Infarction. Stem Cells. 2015;33:3519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Song SW, Chang W, Song BW, Song H, Lim S, Kim HJ, Cha MJ, Choi E, Im SH, Chang BC, Chung N, Jang Y and Hwang KC. Integrin-linked kinase is required in hypoxic mesenchymal stem cells for strengthening cell adhesion to ischemic myocardium. Stem Cells. 2009;27:1358–65. [DOI] [PubMed] [Google Scholar]
- 52.Chen X, Li Z, Feng Z, Wang J, Ouyang C, Liu W, Fu B, Cai G, Wu C, Wei R, Wu D and Hong Q. Integrin-linked kinase induces both senescence-associated alterations and extracellular fibronectin assembly in aging cardiac fibroblasts. J Gerontol A Biol Sci Med Sci. 2006;61:1232–45. [DOI] [PubMed] [Google Scholar]
- 53.Valen G, Yan ZQ and Hansson GK. Nuclear factor kappa-B and the heart. J Am Coll Cardiol. 2001;38:307–14. [DOI] [PubMed] [Google Scholar]
- 54.Valen G Signal transduction through nuclear factor kappa B in ischemia-reperfusion and heart failure. Basic Res Cardiol. 2004;99:1–7. [DOI] [PubMed] [Google Scholar]
- 55.Kumar R, Yong QC and Thomas CM. Do multiple nuclear factor kappa B activation mechanisms explain its varied effects in the heart? Ochsner J. 2013;13:157–65. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.