

Abstract
The ovarian hormones estrogen and progesterone orchestrate the transcriptional programs required to direct functions of the uterus for initiation and maintenance of pregnancy. Estrogen, acting via estrogen receptor alpha, regulates gene expression by activating and repressing distinct genes involved in signaling pathways that regulate cellular and physiological responses including cell division, water influx, and immune cell recruitment. Historically, these transcriptional responses have been postulated to reflect a biphasic physiological response. In this study, we explored the transcriptional responses of the ovariectomized mouse uterus to 17β-estradiol (E2) by RNA-seq to obtain global expression profiles of protein-coding transcripts (mRNAs) and long noncoding RNAs (lncRNAs) following 0.5, 1, 2, and 6 hours of treatment. The E2-regulated mRNA and lncRNA expression profiles in the mouse uterus indicate an association between lncRNAs and mRNAs that regulate E2-driven pathways and reproductive phenotypes in the mouse. The transient E2-regulated transcriptome is reflected in the time-dependent shifting of biological processes regulated in the uterus in response to E2. Moreover, high expression of some conserved lncRNAs that are E2 regulated in the mouse uterus are predictive of low overall survival in endometrial carcinoma patients (e.g., H19, KCNQ1OT1, MIR17HG, and FTX). Collectively, this study (1) describes a genomic approach for identifying E2-regulated lncRNAs that may serve critical function in the uterus and (2) provides new insights into our understanding of the regulation of hormone-regulated transcriptional responses with implications in pregnancy and endometrial pathologies.
Keywords: estradiol/estradiol receptor, genomics, gene expression, transcriptional regulation, uterus
Summary sentence: Estrogen regulates protein-coding genes and long noncoding RNAs with expression kinetics that reflect the shifting biological programs and functions of the uterus.
Introduction
Estrogen plays a critical role in the development and function of the female reproductive tract including the ovary, uterus, cervix, and vagina [1]. The uterus is a highly estrogen-responsive organ that undergoes cyclical waves of proliferation and differentiation through the menstrual and estrous cycles in preparation for embryo implantation and subsequently to provide a nurturing and protective environment for fetal development during pregnancy [2]. In the uterus, estrogen acts primarily through estrogen receptor alpha (ERα), a transcription factor that regulates the expression of genes required for uterine function [3]. ERα is expressed in all compartments of the uterus and exhibits spatiotemporal roles through pregnancy [4–6]. ERα acts by binding to thousands of sites across the genome to promote the coordinated recruitment of coregulatory proteins and chromatin-modifying enzymes to regulate target gene expression via long range enhancer–promoter interactions [7, 8]. Estrogen via ERα regulates a diverse and tissue-specific repertoire of transcripts, collectively called the “estrogen-regulated transcriptome” [9]. Microarray studies have begun defining the estrogen-regulated transcriptome in the murine uterus and determined that these responses occur in biphasic waves whereby direct targets activate secondary signaling cascades that mediate secondary transcriptional responses [10]. These studies have undoubtedly been informative, but they have primarily focused on the identification and functional characterization of estrogen-regulated protein-coding genes and therefore only provide a partial understanding of the estrogen-regulated transcriptome.
Long noncoding RNAs (lncRNAs) are an emerging class of long non-protein-coding RNAs exceeding 200 nucleotides in length [11, 12]. At the molecular level, lncRNAs act through a variety of mechanisms to regulate numerous cellular functions in normal physiology and disease states [13]. LncRNAs are known to have functions in cis by affecting the expression of neighboring genes and in trans by affecting genes located on different chromosomes [12]. In some cases, the act of lncRNA transcription is sufficient to positively or negatively affect the expression of nearby genes [14]. Interestingly, some lncRNAs act as effectors in functions that were previously reserved for proteins such as regulating the activity or localization of other signaling molecules. We have previously demonstrated that the predominant naturally occurring estrogen 17β-estradiol (E2) rapidly and robustly induces the expression of lncRNAs in a breast cancer model with potential implications in clinical outcomes [15]. These results uncovered unexpected roles and mechanism of action for lncRNAs in estrogen-regulated functions. Moreover, these results highlight potential implications in normal physiological states, particularly the largely unexplored role of lncRNAs in the female reproductive system. Some efforts have been made to characterize lncRNAs involved in the regulation of endometrial receptivity for embryo implantation in the mouse model and endometrial disease in clinical samples [16–18]. However, there is still a significant gap in our understanding of estrogen-regulated lncRNAs in the uterus, specifically, their interaction with mRNAs and the potential implications in uterine function and disease.
Collectively, this evidence prompts for the careful characterization of the estrogen-regulated transcriptome and the identification of lncRNAs with putative functions in the uterus. We hypothesized that lncRNAs are important effectors and modulators of estrogen-regulated functions in the uterus. In order to begin exploring this gap in knowledge, we established a genome-wide strategy to identify and categorize estrogen-regulated lncRNAs using the ovariectomized mouse model following a short time course of E2 treatment. The goals of this study are to (1) identify E2-regulated mRNAs and lncRNAs in the uterus, (2) identify putative cis roles of lncRNAs on the E2-regulated transcriptome, (3) characterize molecular features of lncRNA transcripts including exon content and size, and (4) identify their prognostic value in reproductive malignancies.
Materials and methods
Animal use and sample collection
The Institutional Animal Use and Care Committee of the UT Southwestern Medical Center approved the animal use protocol. Adult female mice (C57BL/6 J) were purchased from the UT Southwestern Breeding Core and housed in the UT Southwestern Animal Resource Center under standard light/dark cycles. Females were maintained on 16% protein rodent diet (Envigo, Huntingdon, United Kingdom). Ovariectomies were performed on adult female mice aged 6–8 weeks. Mice were rested for 2 weeks to deplete endogenous hormones. All of the mice in our studies were weighed on the day of the experiments prior to receiving the injections of E2. The average weight of the animals was 19.68 g ±0.39. Ovariectomized mice received a single subcutaneous injection of 100 ng of 17β-estradiol (E2) in corn oil (Sigma, St. Louis, MO, USA). The fixed dose of 100 ng of E2 per animal delivered an average 5.09 ng ± .01 ng of E2 per gram of body weight. Mice were sacrificed under 2,2,2-tribromoethanol (Sigma, St. Louis, MO, USA) anesthesia at 0.5, 1, 2, and 6 hours post-injection for expression analysis. Ethanol was delivered in corn oil or 0.9% saline as vehicle control for 1 hour. Cervix and vascular tissue were removed from dissected whole uteri. Uterine horns were flash frozen and stored at −80 °C for subsequent molecular analysis.
Total RNA isolation, RNA-seq library preparation, and sequencing
For RNA-seq analyses, two biological replicates were sequenced for each of the five time points [vehicle (0 hour), 0.5, 1, 2, and 6 hours]. Total RNA was isolated according to manufacturer’s instructions (RNeasy Kit; Qiagen, Hilden, Germany). Briefly, individual uterine horns were homogenized in RLT buffer with a cooled blade homogenizer (Power Gen25; Fisher, Hampton, NH, USA). Samples were centrifuged to remove debris and passed through a gDNA eliminator column. Flow through was mixed with ethanol at 1:1 ratio and passed through the RNA binding column. The column was washed once with RW1 buffer and twice with RPE buffer. RNA was eluted with RNAse-free H20 and assayed for quality by electrophoresis (Agilent Technologies, Santa Clara, CA, USA). Only samples with RINe values of greater than 9 were included in subsequent analysis. Biological replicates were generated by pooling 3 μg of total RNA from three independent animals for a total of 9 μg of total RNA per replicate. A total of six animals were used for each time point. Polyadenylated RNA (including mRNA and lncRNAs) was isolated using Oligo (dT)25 Dynabeads (Invitrogen, Carslbad, CA, USA) and reverse transcribed using SuperScript III Reverse Transcriptase (Invitrogen, Carslbad, CA, USA). Second strand synthesis was performed using DNA polymerase I. Complementary DNA ends were repaired, tailed with dATP, and ligated with barcode adapters for Illumina sequencing platform. Ligated libraries were selected for average size 250 bp, PCR amplified for 10 cycles, and sequenced using the Next-seq platform in the McDermott Center Next Generation Sequencing Core at UT Southwestern Medical Center.
Quality control, assembly of transcriptome data, and differential gene expression
The raw data were subjected to quality control analyses using the FastQC tool. The reads were then mapped to mouse genome (mm10) using the spliced reader aligner TopHat version.2.0.13 [19]. Transcriptome assembly was performed using cufflinks v.2.2.1 with default parameters [20]. The transcripts were merged into two distinct, non-overlapping sets using Cuffmerge, followed by Cuffdiff to call the differentially regulated transcripts. The significantly (q < 0.05) regulated genes upon E2 treatment at 0.5, 1, 2, and 6 hours were compared to 0 hour to find the commonly regulated gene set. The differentially expressed genes extracted from the above analysis were then used in downstream analyses. Venn diagrams were generated using jvenn for the differentially expressed genes in different conditions [21]. Sequencing data sets have been submitted to Gene Expression Omnibus accession number GSE133158.
Identification of candidate lncRNAs
After alignment, the transcripts were assembled using mouse lncRNAs GENCODE database v19. Differential expression analysis was performed with assembled lncRNAs using Cuffdiff as described above for protein-coding genes.
Molecular features and classification of transcripts
Molecular features of transcripts including biotypes, distribution of transcript lengths, and total number of exons in lncRNAs and protein-coding genes were carried out using the latest Ensembl Release 96 (April 2019) for mouse assembly (GRCm38.p6/mm10) queried via Ensembl REST API Endpoints (https://rest.ensembl.org) and compiled using custom Perl, R, and Bash scripts. Graphs were made using the R-package (ggplot2) with the function geom_bar and using standard settings with only modifications to bar colors and axis scale.
Integration of RNA-seq data with ChIP-seq data
Establishing fold change cutoffs for E2 regulation
From the differential gene expression analysis of the RNA-seq data using Cuffdiff, described above, we categorized the significantly regulated genes (q < 0.05) as E2 regulated (up- or down-regulated) or nonregulated based on the following fold change (FC) cutoffs (E2 treated versus 0 hour): up-regulated, FC > 2.0; down-regulated, FC < 0.5; unregulated, FC between 0.8 and 1.2. The mRNA and lncRNA genes falling into these categories were used in analyses integrating our RNA-seq data with RNA polymerase II (Pol II) ChIP-seq and ERα ChIP-seq data from uteri collected from vehicle- or E2-treated (1 hour) ovariectomized mice, previously published by Hewitt et al. [7].
Analysis of ChIP-seq data
The raw Pol II and ERα ChIP-seq reads from Hewitt et al. [7] were aligned to the mouse reference genome (mm10) using default parameters in Bowtie (v1.0.0) [22]. The aligned reads were subsequently filtered for quality and uniquely mappable reads using Samtools (v.0.1.19) [23] and Picard (v1.127) (https://broadinstitute.github.io/picard/). Library complexity was measured using BEDTools (v 2.17.0) [24] and met minimum ENCODE data quality standards [25]. Relaxed peaks were called using MACS (v2.1.0) [26] and a P value = 1 × 10−2 for each replicate, pooled replicates reads, and pseudoreplicates. Called peaks and peak summits were used for further analysis.
Enrichment box plots
The aligned ChIP-seq data were used to calculate enrichment of RNA Pol II (vehicle and 1 hour E2) around the transcription start sites (TSSs) of the promoters of regulated and nonregulated mRNA and lncRNA genes. The read distributions from the TSS to +5 kb were calculated and plotted using the box plot function in R.
Nearest neighboring gene analyses and Venn diagrams
The universe of significantly regulated genes at each time point was determined from the RNA-seq data using q < 0.05 (no FC cutoffs were applied for these analyses). The sets of nearest neighboring mRNA and lncRNA genes were determined using the ERα-binding sites (peak summits called from the ERα ChIP-seq in the E2-treated condition) within 20 and 50 kb of the TSSs of the regulated gene. The data were plotted in Venn diagrams to show the overlap of the E2-regulated genes with genes that have ERα-binding sites located within the defined window.
Enrichment metagenes
Metagenes (average enrichment plots) were used to illustrate the enrichment of ERα ChIP-seq reads in vehicle- and E2-treated conditions. The reads in a ± 5 kb window around the TSSs of the E2-regulated and nonregulated lncRNA genes were collected and used to generate metagene plots using the metagene function in groHMM [27], as we have described previously [8, 28]. All of the metagenes were scaled to a library size of 12 million reads to minimize differences caused by variability in sequencing depth among samples.
Gene ontology analyses
Gene ontology (GO) analyses were performed using fgsea, an R-package for fast pre-ranked gene set enrichment analysis (GSEA) (https://github.com/ctlab/fgsea). The genes included in this analysis were those that were differentially expressed at 0.5, 1, 2, and 6 hours of E2 treatment compared to 0 hour. Heat maps were generated using Java TreeView for visualizing the universe of GO terms represented in each time point with their corresponding normalized enrichment scores (NES) [29]. Cluster summaries were determined using REViGO, a clustering algorithm that relies on semantic similarity measures to assist in interpretation [30].
Prediction of cis lncRNA targets
To explore the function of lncRNAs, we first predicted putative cis-target genes of lncRNAs using Genomic Regions Enrichment of Annotations Tool (GREAT) [31]. GREAT calculates statistics by associating the query lncRNA genomic regions with nearby genes. The association rule was defined by establishing a basal gene regulatory domain extending 5 kb upstream and 1 kb downstream, plus distal up to 1000 kb of transcriptional initiation site of genes. GREAT reports significance by performing a binominal test over input genomic regions. We report the −log10 transformed binominal false discovery rate q value for gene ontologies, mouse phenotypes, human diseases, and pathways corresponding to the lncRNA–gene associations.
Conservation analysis
The entire list of mouse genes (3038) was queried against the latest release of Ensembl mouse genome annotation and GENCODE (GRCm38.p6/mm10). The process was done using a custom R script via the Ensembl REST API Endpoints (https://rest.ensembl.org). The resulting gene list was aligned by homology to the human genome (GRCh38/hg38) using NCBI BLAST+ program (https://blast.ncbi.nlm.nih.gov/Blast.cgi) locally on our system. Subsequently, the 434 genes were subjected to position conservation across the human genome using the UCSC Genome Browser (https://genome.ucsc.edu). Lastly, the 280 positionally conserved genes were analyzed for potential human homologs, resulting in 20 final genes with human homologs.
Tumor sample analysis
Expression of tumor samples from The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/tcga.) and normal samples from The Genotype-Tissue Expression (GTEx) were used to calculate median values for each gene of interest [32]. For this analysis, we focused on a subset of cancers arising from reproductive tissues including breast cancer, cervical squamous cell carcinoma, ovarian cancer, and uterine corpus endometrial carcinoma. Samples were scaled and converted to normalized RPKM using the recount2 project (https://jhubiostatistics.shinyapps.io/recount/). To this end, we applied the scale_counts function and used the RPKM function in R. Heat maps were made using the Plotly R package (https://help.plot.ly/citations/#step-1-citing-plotly) with standard settings excluding colors and auto scale function.
Survival analysis of endometrial carcinoma patients
To evaluate the prognostic value of the conserved E2-regulated lncRNAs, we explored their expression in 543 samples of uterine corpus endometrial carcinoma. The Kaplan-Meier Plotter tool was used to plot overall survival for these conserved E2-regulated lncRNAs [33]. Patient expression values were split into all possible upper and lower quartiles. We report the P values associated with the best performing threshold cut off for each lncRNA.
Results
Differentially expressed messenger RNAs and lncRNAs identified by RNA-seq analyses
The transcriptional response to E2 was evaluated in the ovariectomized adult mouse uterus. RNA-seq was performed to identify mRNA and lncRNAs. Functional prediction of the E2-regulated transcriptome was performed with GO and pathway analysis (Figure 1A). This analysis identified mRNAs and lncRNAs regulated at 0.5, 1, 2, and 6 hours relative to 0 hour of E2 treatment (Figure 1B and Supplementary Figure S1, respectively). The complete list of mRNAs and lncRNAs significantly regulated at each time point relative to 0 hour is found in Supplementary Tables S1 and S2, respectively.
Figure 1.
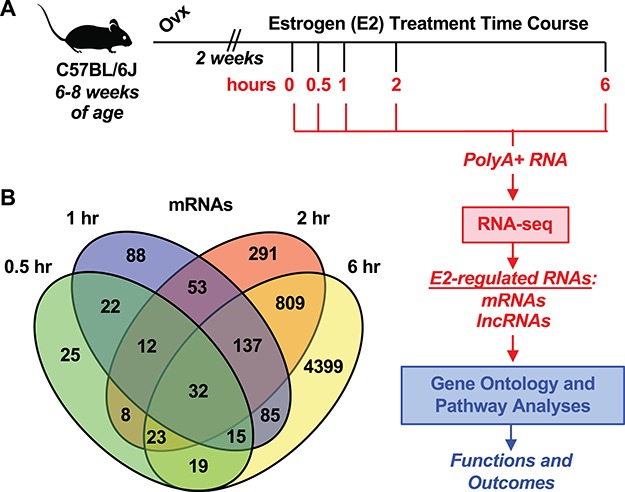
Defining the transcriptional response to E2 in the ovariectomized mouse uterus. (A) Experimental schematic for surgical preparation and treatment of female mice, genomic analysis of transcription, and downstream bioinformatics analysis. (B) Venn diagram representation of protein-coding genes (mRNAs) regulated through the time course relative to the vehicle treatment.
We compared molecular features of the E2-regulated transcripts. We observed that the majority of mRNA transcripts were greater than 4000 bp in length (Supplementary Figure S2A). In contrast, lncRNAs ranged between 300 and 4000 bp (Supplementary Figure S2B). In addition, significant differences in the distribution of exon numbers between mRNAs and lncRNAs were also observed (Supplementary Figure S2C and S2D, respectively). Notably, ~ 90% of lncRNAs contained two to four exons. The comparative analysis of the above molecular features between mRNAs and lncRNAs in this study was consistent with that of previous studies [11, 12, 15]. The universe of lncRNAs regulated by E2 (i.e., all lncRNAs significantly induced or repressed at 0.5, 1, 2, and 6 hours relative to any time point; q < 0.05) were classified according to transcript biotypes in GENCODE and Ensembl. These features are described and quantified in Supplementary Figure S2E.
Integration of RNA-seq data with RNA Pol II and ERα ChIP-seq data
Next, we integrated our RNA-seq data with published RNA Pol II and ERα ChIP-seq in uteri collected from ovariectomized mice treated with vehicle or E2 for 1 hour [7]. We defined (1) E2-regulated genes (separate mRNA and lncRNA gene sets, as well as separate up-regulated and down-regulated gene sets) based on our RNA-seq data (q value and FC cutoffs; see Materials and Methods) and (2) the relative amount of Pol II loaded at the promoters (TSS to +5 kb) of genes in the sets defined above (separate mRNA and lncRNA gene sets) based on Pol II ChIP-seq data (Supplementary Figure S3). As expected, we observed that E2-regulated mRNA genes defined by RNA-seq had up-regulated or down-regulated RNA Pol II occupancy in response to E2 treatment, as appropriate, whereas the unregulated genes showed no changes in Pol II loading in response to E2 treatment (Figure 2A). Similar results were observed for E2-regulated lncRNA genes (Supplementary Figure S4A).
Figure 2.
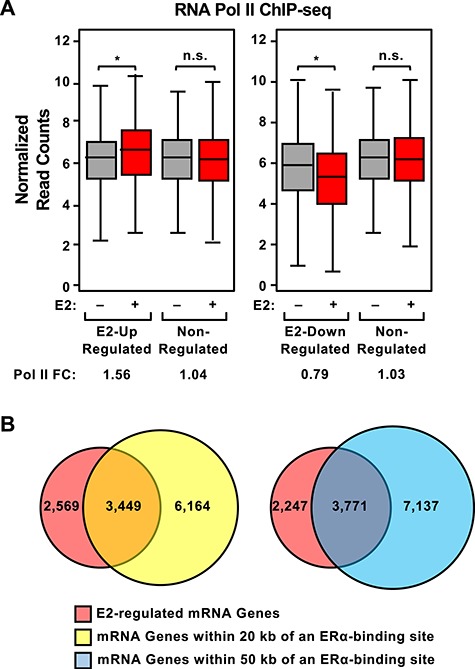
Occupancy of RNA polymerase II and ERα near E2-regulated mRNA genes. Pol II and ERα occupancy on E2-regulated and nonregulated mRNA genes in vehicle- and E2-treated ovariectomized mouse uteri, determined as shown in Supplementary Figure S3 using ChIP-seq data from Hewitt et al. [7] and RNA-seq data generated as described herein. (A) The enrichment of Pol II at the promoters (TSS to +5 kb; determined by Pol II ChIP-seq) of E2 up-regulated, E2 down-regulated, and nonregulated genes (up-regulated = FC > 2, down-regulated = FC < 0.5; determined by RNA-seq) in uteri collected from vehicle- or E2-treated mice (grey and red boxes, respectively) was determined and expressed in box plots for each condition. Significance was determined by a Wilcoxon rank sum test with P values indicated (asterisk, up-regulated P = 1.16 × 10−15, down-regulated P < 2.2 × 10−16; n.s., not significant). “Pol II FC” = the fold change in Pol II ChIP-seq normalized read counts for vehicle versus E2. (B) Comparison of mRNA genes regulated by E2 at all time points tested as determined by RNA-seq (red circle) with mRNA genes residing within 50 kb (blue circle) of an ERα-binding site determined by ChIP-seq in E2-treated mouse uteri.
We then compared the E2-regulated gene sets with nearby ERα-binding sites. To do so, we defined (1) E2-regulated genes (separate mRNA and lncRNA gene sets) based on RNA-seq (q-value only) and (2) all genes located within 20 or 50 kb of an ERα-binding site (separate mRNA and lncRNA gene sets) based on the ERα ChIP-seq data (Supplementary Figure S3). Based on these analyses, we found that 57% and 63% of the 6,018 E2-regulated mRNA genes that we identified were located within 20 and 50 kb, respectively, of an ERα-binding site identified by Hewitt et al. [7] (Figure 2B). A similar analysis with the E2-regulated lncRNA gene set revealed considerably less overlap (~12% of the 739, the E2-regulated lncRNA genes) than was observed with the E2-regulated mRNA genes (Supplementary Figure S4B). However, we did observe the E2-dependent accumulation of ERα ChIP–Pol II ChIP-seq reads at the promoters of the E2-regulated lncRNA genes (Supplementary Figure S4C and S4D, respectively). These results indicate concordance between our data and the Hewitt et al. data [7].
Kinetics of mRNA and lncRNA expression
Estrogen regulates gene expression with robust and transient kinetics [8]. We explored the kinetics of the E2-regulated uterine transcriptome by sequentially ranking gene expression magnitude of induced genes in each time point (FC relative to 0 hour). Through the time course, E2 regulates 5978 protein-coding genes. The immediate response (0.5 hour) results in the up-regulation of 75 genes, and the early response (1 and 2 hours) results in the up-regulation of 131 and 312 genes, respectively. By 6 hours, we observed 2449 up-regulated mRNAs. Throughout the time course, we observed general down-regulation of 3041 genes (Figure 3A). Through the time course, E2 regulates 736 lncRNAs. The immediate response (0.5 hour) results in the up-regulation of 257 lncRNAs, and the early response (1 and 2 hours) results in the up-regulation of 157 and 110 lncRNAs, respectively. By 6 hours, we observed 41 up-regulated lncRNAs. Throughout the time course, we observed general down-regulation of 171 lncRNAs (Figure 3B).
Figure 3.
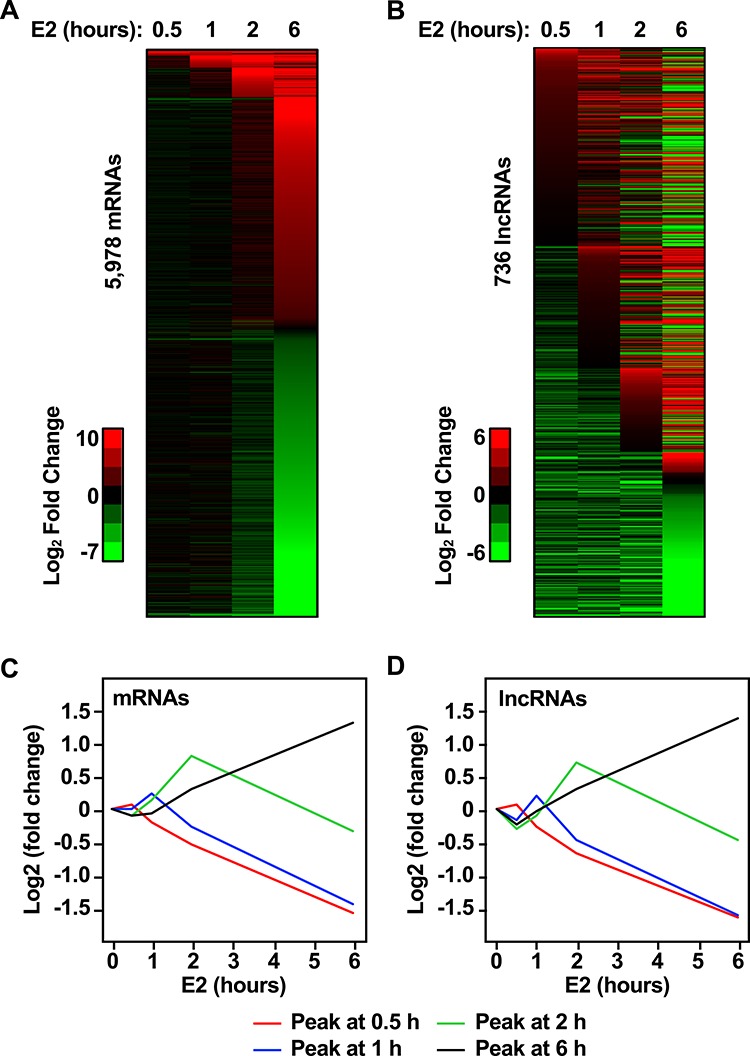
Genomic analysis of the transcriptional response to E2 in the ovariectomized mouse uterus. Heat map representation of transcriptional response to E2 for (A) protein-coding genes (mRNA) and (B) lncRNAs. Each horizontal line represents one transcript through the time course of E2 treatment. Transcripts that were increased by E2 treatment relative to the 0 hour (vehicle treatment) are indicated in red (log2 FC). Down-regulated transcripts are indicated in green. Transcripts are sequentially ranked by maximum expression through each time point. Line plot representation of average expression (log2 FC) for (C) protein-coding genes and (D) lncRNAs peaking at 0.5, 1, 2, and 6 hours.
To evaluate the transient nature of the E2-dependent gene regulation, we classified transcripts that exhibit maximum expression at 0.5, 1, 2, and 6 hours, relative to other time points. These kinetic patterns are illustrated in line plots representing the average of the log2 FC for mRNA and lncRNAs (Figure 3C and D, respectively). This classification indicated that 20% of mRNAs peak at 0.5 hour, 26% peak at 1 hour, 11% peak at 2 hours, and 43% peak at 6 hours. Similarly, 17% of lncRNAs peak at 0.5 hour, 25% peak at 1 hour, 18% peak at 2 hours, and 39% peak at 6 hours.
The uterus responds to E2 by activating and repressing canonical estrogen target genes
We performed GSEA with the protein-coding genes regulated across each time point of E2 treatment. This analysis identified common and unique pathways enriched in each time point. Analysis of the 0.5 hour time point did not result in enrichment of any specific pathways. This result was expected due to the low number of genes that are present in this data set. The genes regulated at 1 hour regulated are involved in TNFα signaling via NF-κB, ultraviolet response down-regulated, hypoxia, and estrogen response early (Supplementary Figure S5A). These pathways were also identified in the genes regulated at 2 hours. Additionally, MYC targets (V1 and V2), unfolded protein response, p53 pathway, and MTORC1 pathways were also enriched at 2 hours (Supplementary Figure S5B).
The set of genes identified at 6 hours contained many of the genes found in the earlier time points (Figure 1B). Therefore, similar pathways were represented in this analysis. In addition to the previously identified pathways, we observed the enrichment of pathways involved in oxidative phosphorylation, cholesterol homeostasis, DNA repair, and PI3K/AKT signaling (Supplementary Figure S5C). Notably, the genes regulated at 6 hours were associated with the late estrogen response and the expression of E2F targets, a transcription factor involved in DNA replication and cell cycle regulation.
Next, we performed a focused evaluation of the expression pattern of genes categorized under the “estrogen response early” and “estrogen response late” GSEA pathways. These gene sets were curated from studies primarily exploring the estrogen response in breast cancer cells and include 200 genes in each category, half of which are present in both categories. Therefore, this analysis should refine our understanding of the uterine-specific kinetics of estrogen response. We observed that a small subset of “early” genes indeed exhibit robust expression by 0.5 hour and continue to exhibit robust up-regulation through the time course. However, the majority of early induced genes are dynamically and transiently regulated between 1 and 2 hours (Supplementary Figure S6A). In contrast, “late” genes cluster closely at 6 hours (Supplementary Figure S6B).
Functional annotation of E2-regulated protein-coding genes
To relate the transcriptional changes to biological processes, we performed gene set enrichment analyses and GO analyses on E2-regulated protein-coding genes that were differentially regulated following 0.5, 1, 2, and 6 hours relative to 0 hour (Supplementary Table S3). We employed the REViGO tool to aid in the interpretation of these ontological processes and summarized each cluster with GO terms most frequently represented in each time point. The biological functions associated with the genes regulated at 0.5 hour are closely related with transcription, development, and response to hormone (cluster i). The genes regulated at 1 hour are closely related with regulation of metabolism, angiogenesis, and apoptosis (cluster ii). The genes regulated at 2 hours are associated with the response to growth factor and kinases signaling cascades (cluster iii). Finally, genes regulated at 6 hours were associated with several RNA processing events including splicing, localization, and modification (cluster iv) (Figure 4A).
Figure 4.
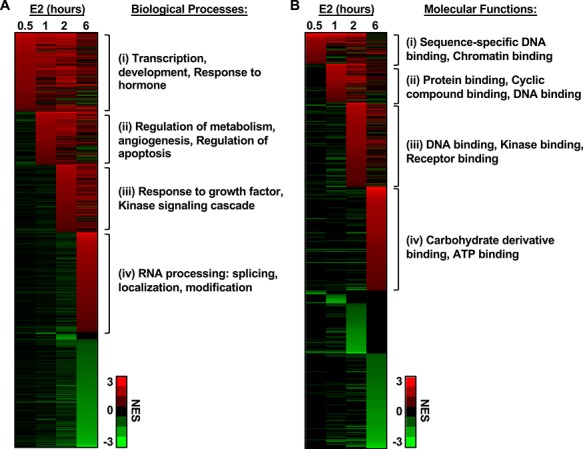
The E2-induced transcriptional response in the mouse uterus reveals a distinct and temporal regulation of biological processes. Heat map representation of NES corresponding to GO terms. (A) Biological processes and (B) molecular functions associated with the E2-regulated protein-coding transcriptome in each time point. In each representation, the x-axis (columns) is the time of E2 treatment and the y-axis is the GO terms, with those that are enriched shown in red and those that are de-enriched shown in green. Each line across represents the NES relative to 0-hour time point. The GO terms listed in each clusters (i), (ii), (iii), and (iv) indicate the most frequently represented GO terms determined by REViGO analysis.
We supplemented this analysis by evaluating the molecular functions associated with the E2-regulated protein-coding genes at each time point to identify potential mechanisms of action (Supplementary Table S4). Again, we employed the REViGO tool to aid in the interpretation of these molecular functions and summarized each cluster with GO terms most frequently represented in each time point. Interestingly, we observed that molecular features associated with DNA binding were represented in the clusters i, ii, and iii. Cluster ii was also represented by genes with protein and cyclic compound binding activities. Cluster iii was associated with kinase and receptor binding functions. In contrast, cluster iv was associated with carbohydrate derivative functions and ATP binding (Figure 4B).
Functional annotation for lncRNAs
To begin investigating the function of E2-regulated lncRNAs in the mouse uterus, we first explored the possibility that these lncRNAs could be functioning in cis, as has been previously described for other lncRNAs [14]. We predicted putative lncRNA cis-regulatory target genes and associated biological meaning using GREAT. Genomic coordinates of lncRNAs regulated by E2 at any time point were used as test regions against the mouse genome: NCBI build 38 (UCSC mm10, Dec/2011). For the association of lncRNA regions with genes, the regulatory domain of target genes was defined as “basal plus extension” (proximal 5 kb upstream, 1 kb downstream, plus distal up to 1000 kb). The lncRNA–mRNA associations are reported in Supplementary Table S5. We observed a significant enrichment of mouse phenotype associated with reproductive defects, including pale placenta, embryonic lethality early to midgestation (i.e., embryonic lethality between implantation and somite formation), abnormal uterine horn morphology, small placenta, and enlarged placenta (Figure 5A). The associated regions are also significantly linked with uterine-related diseases including uterine fibroid, neoplasm of body of uterus, and uterine corpus soft tissue neoplasm (Figure 5B). Moreover, the associated regions enrich for biological processes including RNA processing, posttranscriptional regulation of genes expression, regulation of translation, and mRNA metabolic process (Supplementary Figure S7A). The pathways enriched in these associated regions included FoxO family signaling, PDGF signaling, EGF signaling, and gene related to Wnt-mediated signal transduction (Supplementary Figure S7B).
Figure 5.
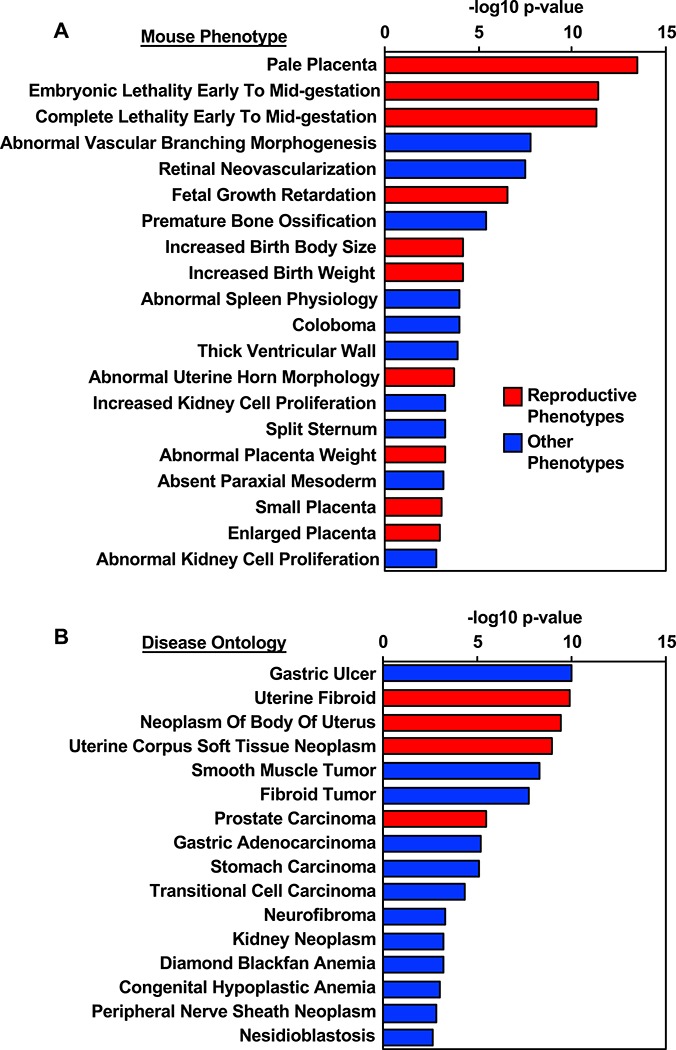
Prediction of lncRNA function based on gene neighborhood. Putative lncRNA cis-regulatory target genes and associated biological meaning were evaluated using GREAT. The genomic location of lncRNAs regulated by E2 at any given time point were associated to nearby genes through association rule defining a basal plus extension regulatory domain. (A) Genes associated with lncRNAs are associated with mouse phenotypes and morphological abnormalities. Those shown in red are associated with reproductive outcomes. (B) LncRNA–gene associations identified disease ontologies relating to uterine pathologies, shown in red.
Expression and prognostic value of conserved E2-regulated lncRNAs in endometrial cancer
The lncRNA–mRNA–ontology analysis identified a potential association between lncRNA and endometrial malignancies (Figure 5B). We followed this observation with a conservation analysis of the lncRNAs and their expression in various cancers of the reproductive system. The lncRNA filtering strategy included all lncRNA regulated by E2 in the mouse uterus at any given time point. The subsequent filtering step selected only 706 with annotation in ENSEMBL and GENCODE. Our conservation analysis was relaxed to identify 43 mouse lncRNAs with at least 78% identity to human lncRNAs and a maximum of 7% gaps. From these, we selected 28 lncRNA, which exhibit conservation in position, meaning that the neighboring genes conserved across mouse–human. We finally arrived at a group of 20 mouse lncRNA that had human homologs (Supplementary Table S6). This list included lncRNAs that were induced or repressed by E2 in the mouse uterus (Figure 6A). We explored the expression of the lncRNA human homologs in normal tissues represented in GTEx and malignant tissues represented in the TCGA cohort. Most of the lncRNAs are down-regulated in malignancies of the breast, ovary, uterus, and cervix, relative to their healthy controls (Figure 6B). To evaluate the clinical value of these lncRNAs, we explored their utility as prognostic markers of overall survival. Kaplan-Meier plots were generated from a pan-cancer resource containing 543 samples of uterine corpus endometrial carcinoma. Patients were segregated into low and high expression based on the best performing threshold cutoff. We observed that high expression of the E2-induced lncRNAs H19, KCNQ1OT1, MIR17HG, and FTX are predictive of low overall survival (Figure 7A–D, respectively).
Figure 6.
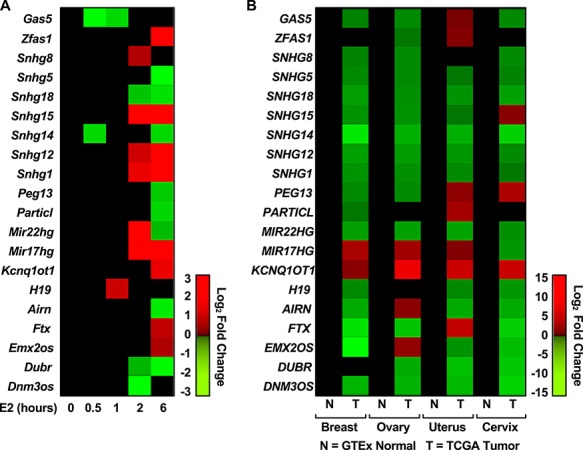
Expression of E2-regulated mouse lncRNAs with human homologs in female reproductive cancers. (A) Heat map representation of mouse uterine expression for 20 E2-regulated lncRNAs is positionally conserved in the human. (B) Conserved lncRNAs are mostly down-regulated in tumor tissues relative to healthy tissues of the breast ovary, uterus, and cervix.
Figure 7.
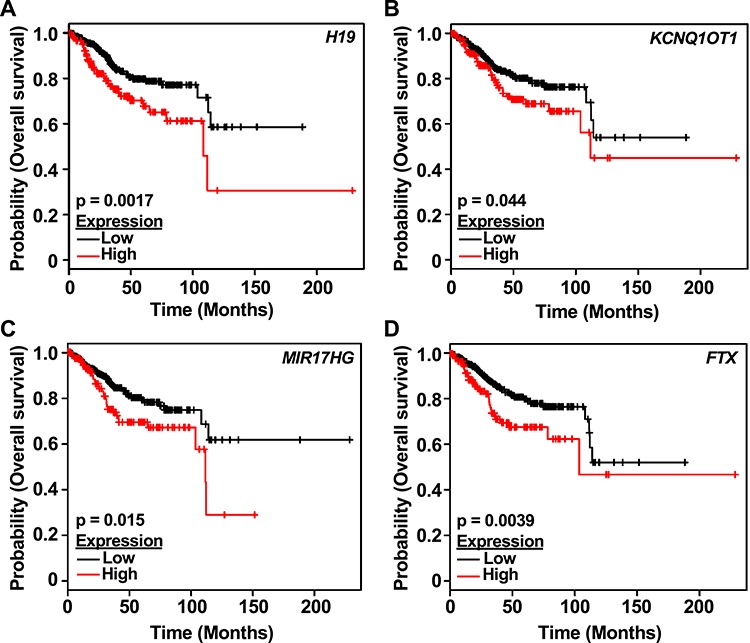
E2-regulated lncRNAs with human homologs predict overall survival to uterine cancer. Kaplan-Meier survival analyses of patients expressing high levels of E2-regulated lncRNAs human homologs (red line) exhibit a poor outcome compared to patients expressing low levels (black line) for lncRNAs (A) H19, (B) KCNQ1OT1, (C) MIR17HG, and (D) FTX. The uterine corpus endometrial carcinoma outcome-linked gene expression data were accessed and graphed using the www.kmplot.com.
Discussion
In this study, we explored the transcriptional response of the ovariectomized mouse uterus to E2 by RNA-seq to obtain global expression profiles of mRNAs and lncRNAs. Using GO and gene set enrichment analyses, we found an association between the expression of mRNAs and lncRNAs that regulate E2-driven pathways and reproductive phenotypes in the mouse. Comparative analyses of conserved E2-regulated lncRNAs in the mouse uterus with human homologs allowed us to infer additional biological functions, including potential roles of some E2-regulated lncRNAs in endometrial carcinoma. Collectively, this study (1) describes a genomic approach for identifying E2-regulated lncRNAs that may serve critical function in the uterus and (2) provides new insights into our understanding of the regulation of hormone-regulated transcriptional responses with implications in pregnancy and endometrial pathologies.
Kinetics of E2-regulated gene expression in the mouse uterus
Microarray studies in the mouse uterus have revealed temporal features of the transcriptional response to estrogen [10]. These responses have been described to mirror a biphasic physiological response associated with key biological outcomes including cellular proliferation, water influx, and immune cell infiltration [10]. Aspects of this gene regulatory response in the uterus are mirrored in other E2-responsive cells and tissues, such as the mammary gland [8, 9]. We have revisited these observations by employing RNA-seq to define the early transcriptional response to E2 with greater sensitivity. To this end, we identified mRNAs and lncRNAs regulated by E2 during the immediate (0.5 hour), early (1 and 2 hours), and late (6 hours) response. We identified four classes of mRNA and lncRNA expression kinetics, represented in Figure 3C and D, which show the different gene sets exhibit maximal expression at different time points. From our GO analyses (Figure 4), we can postulate that the early induced targets can act as secondary signaling molecules and effectors. Ultimately, these signaling cascades have temporal roles mediating major E2-regulated processes including proliferation, apoptosis, water influx, and immune cell traffic.
Our laboratory has previously demonstrated in breast cancer cells that E2-regulated gene expression kinetics are associated with different enrichment of ERα binding [8]. Transcripts that peak at 10or 40 minutes of E2 treatment have a greater enrichment of ERα-binding sites within 10 kb of the promoter compared to late expressed or down-regulated genes, suggesting direct regulation by liganded ERα [8]. In comparing our E2-regulated gene sets with previously published ERα ChIP-seq data from the mouse uterus, we also observe an enrichment of ERα binding within the likely regulatory regions of the genes (i.e., within 50 kb) (Figure 2B). We previously observed that the promoters of E2-regulated lncRNA genes are enriched for ERα binding [15], but this finding was less evident in our current analyses (Supplementary Figure 4B), although we did observe some accumulation of ERα ChIP-seq reads in response to E2 treatment at the lncRNA promoters (Supplementary Figure 4C).
Association of biological functions with E2-regulated gene expression in the mouse uterus
The dynamic transcriptome changes in response to E2 measured by RNA-seq were reflected in the time-dependent shifting of biological processes in the uterus (Figure 4). This observation is consistent with the notion that the early transcriptional response to estrogen results in the regulation of signaling components, which mediate secondary signaling events. The enrichment of molecular functions associated with DNA binding or kinase binding reveal that most of these signaling components are transcription factors and regulators of kinase signaling cascades. Additionally, our work provides new insights into the estrogen-dependent regulation of protein-coding genes involved in RNA processing in the late estrogen response in the uterus. Our laboratory has previously reported a similar pattern of distinct biological effects in the E2-dependent response, whereby the early induced genes (40 minutes) are involved in regulation of gene expression, and intracellular signaling, while the late response (160 minutes) is associated with ribonucleoprotein complex biogenesis, and assembly, translation, protein synthesis, and metabolism [8]. Collectively these observations highlight a conserved mechanism of action for estrogen in the uterus and malignant breast at the level of gene expression.
Discerning biological functions of E2-regulated lncRNAs
We have previously explored the role of noncoding E2-regulated transcripts and their role in the regulation of transcription in breast cancer [8, 15]. Those studies led us to explore the potential biology and association between mRNAs and lncRNAs in the healthy mouse uterus. Functional annotation and prioritization of lncRNAs is challenging due to the lack of functional ontologies associated with such type of transcript. This remains an active area of development in the field [34]. We approached this challenge by employing two strategies to interrogate the putative functions of these lncRNAs. Our first strategy employed a genomic analysis tool, GREAT [31], to identify putative cis roles for lncRNAs by exploring gene ontologies, phenotypes, and diseases associated with the gene neighborhoods. Notably, this analysis identified an association between cis lncRNA targets and RNA processing, as well as other RNA-directed regulatory processes (Supplementary Figure 7A), a connection that we noted previously in breast cancer cells [8]. In addition, we identified lncRNA–mRNA associations with potential impacts on mouse phenotypes related to reproductive defects and endometrial malignancies (Figure 5A). As a result of this observation, we employed a computational approach to explore lncRNA function in the context of human disease.
Our second strategy for predicting lncRNA functions used the expression of conserved lncRNAs in a subset of reproductive tissues found in the GTEx and TCGA databases to define potential roles for the human lncRNA homologs. Many of these lncRNAs have been explored in cancer, and their expression is associated with aberrant cancer phenotypes (Supplementary Table S7). We focused on the expression of lncRNAs in endometrial cancer, one of the most common type of gynecological malignancies, given that recent studies have shown that lncRNAs are associated with carcinogenesis and disease progression [18]. We demonstrated that a subset of the E2 up-regulated lncRNAs was shown to have prognostic value in endometrial cancer. Notably, higher expression of these E2-regulated lncRNAs was associated with poor overall survival in patients with uterine corpus endometrial carcinoma (Figure 7). Among these is H19, a lncRNA expressed in epithelial cells of endometrial hyperplasia [35]. H19 is widely studied in breast cancer and implicated in the sequestration of microRNAs regulating pluripotency, proliferation, and invasion [36]. We also identified the E2-dependent regulation of Kcnq1ot1 in the mouse uterus. In some biological systems, KCNQ1OT1 works by silencing multiple genes in cis by establishing a repressive higher-order chromatin structure [37]. In breast cancer cells, high expression of KCNQ1OT1 promotes tumor growth through a mechanism involving miRNA sponging [38]. Finally, we report two E2-regulated lncRNAs with potential function and prognostic value in endometrial cancer, MIR17HG and FTX. MIR17HG is an lncRNA that carries the miR-17/92 cluster under MYC transcriptional control. Interestingly, MYC targets are represented in the early E2 response (2 hours), when we observed expression of MIR17HG. In contrast, Ftx expression peaks at 6 hours. FTX is expressed in aggressive forms of hepatocellular carcinoma (HCC), where it is implicated in promoting the Warburg effect and supporting proliferation, invasion, and migration of HCC cells [39]. It remains to be determined if these E2-regulated lncRNAs promote tumorigenesis in endometrial cancer through similar mechanisms. Our results are in general agreement with the observations that lncRNAs may have diagnostic and/or prognostic significance. The potential function of estrogen-regulated lncRNAs as prospective therapeutic or prognostic targets and the utility of the mouse model to evaluate these questions require further investigation.
Supplementary Material
Acknowledgments
We thank members of the Kraus lab and the Cecil and Ida Green Center for Reproductive Biology Sciences for their helpful comments and support.
References
- 1. Findlay JK, Liew SH, Simpson ER, Korach KS. Estrogen signaling in the regulation of female reproductive functions. Handb Exp Pharmacol 2010; 198:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Robertshaw I, Bian F, Das SK. Mechanisms of uterine estrogen signaling during early pregnancy in mice: an update. J Mol Endocrinol 2016; 56:R127–R138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walker VR, Korach KS. Estrogen receptor knockout mice as a model for endocrine research. ILAR J 2004; 45:455–461. [DOI] [PubMed] [Google Scholar]
- 4. Pawar S, Laws MJ, Bagchi IC, Bagchi MK. Uterine epithelial estrogen receptor-alpha controls decidualization via a paracrine mechanism. Mol Endocrinol 2015; 29:1362–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winuthayanon W, Lierz SL, Delarosa KC, Sampels SR, Donoghue LJ, Hewitt SC, Korach KS. Juxtacrine activity of estrogen receptor alpha in uterine stromal cells is necessary for estrogen-induced epithelial cell proliferation. Sci Rep 2017; 7:8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Winuthayanon W, Hewitt SC, Orvis GD, Behringer RR, Korach KS. Uterine epithelial estrogen receptor alpha is dispensable for proliferation but essential for complete biological and biochemical responses. Proc Natl Acad Sci U S A 2010; 107:19272–19277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hewitt SC, Li L, Grimm SA, Chen Y, Liu L, Li Y, Bushel PR, Fargo D, Korach KS. Research resource: whole-genome estrogen receptor alpha binding in mouse uterine tissue revealed by ChIP-seq. Mol Endocrinol 2012; 26:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 2011; 145:622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vasquez YM. Estrogen-regulated transcription: mammary gland and uterus. Steroids 2018; 133:82–86. [DOI] [PubMed] [Google Scholar]
- 10. Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S, Afshari CA, Korach KS. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol Endocrinol 2003; 17:2070–2083. [DOI] [PubMed] [Google Scholar]
- 11. Ransohoff JD, Wei Y, Khavari PA. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol 2018; 19:143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun M, Kraus WL. From discovery to function: the expanding roles of long noncoding RNAs in physiology and disease. Endocr Rev 2015; 36:25–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DiStefano JK. The emerging role of long noncoding RNAs in human disease. Methods Mol Biol 2018; 1706:91–110. [DOI] [PubMed] [Google Scholar]
- 14. Long Y, Wang X, Youmans DT, Cech TR. How do lncRNAs regulate transcription? Sci Adv 2017; 3:eaao2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun M, Gadad SS, Kim DS, Kraus WL. Discovery, annotation, and functional analysis of long noncoding RNAs controlling cell-cycle gene expression and proliferation in breast cancer cells. Mol Cell 2015; 59:698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Q, Wang N, Cai R, Zhao F, Xiong Y, Li X, Wang A, Lin P, Jin Y. Genome-wide analysis and functional prediction of long non-coding RNAs in mouse uterus during the implantation window. Oncotarget 2017; 8:84360–84372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vallone C, Rigon G, Gulia C, Baffa A, Votino R, Morosetti G, Zaami S, Briganti V, Catania F, Gaffi M, Nucciotti R, Costantini FM et al. Non-coding RNAs and endometrial cancer. Genes (Basel) 2018; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li BL, Wan XP. The role of lncRNAs in the development of endometrial carcinoma. Oncol Lett 2018; 16:3424–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bardou P, Mariette J, Escudie F, Djemiel C, Klopp C. Jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 2014; 15:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP. The sequence alignment/map format and SAMtools. Bioinformatics 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 2012; 22:1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc 2012; 7:1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chae M, Danko CG, Kraus WL. groHMM: a computational tool for identifying unannotated and cell type-specific transcription units from global run-on sequencing data. BMC Bioinformatics 2015; 16:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 2013; 23:1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics 2004; 20:3246–3248. [DOI] [PubMed] [Google Scholar]
- 30. Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 2011; 6:e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagy A, Lanczky A, Menyhart O, Gyorffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep 2018; 8:9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cao H, Wahlestedt C, Kapranov P. Strategies to annotate and characterize long noncoding RNAs: advantages and pitfalls. Trends Genet 2018; 34:704–721. [DOI] [PubMed] [Google Scholar]
- 35. Tanos V, Ariel I, Prus D, De-Groot N, Hochberg A. H19 and IGF2 gene expression in human normal, hyperplastic, and malignant endometrium. Int J Gynecol Cancer 2004; 14:521–525. [DOI] [PubMed] [Google Scholar]
- 36. Collette J, Le Bourhis X, Adriaenssens E. Regulation of human breast cancer by the long non-coding RNA H19. Int J Mol Sci 2017; 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kanduri C. Kcnq1ot1: a chromatin regulatory RNA. Semin Cell Dev Biol 2011; 22:343–350. [DOI] [PubMed] [Google Scholar]
- 38. Feng W, Wang C, Liang C, Yang H, Chen D, Yu X, Zhao W, Geng D, Li S, Chen Z, Sun M. The dysregulated expression of KCNQ1OT1 and its interaction with downstream factors miR-145/CCNE2 in breast cancer cells. Cell Physiol Biochem 2018; 49:432–446. [DOI] [PubMed] [Google Scholar]
- 39. Li X, Zhao Q, Qi J, Wang W, Zhang D, Li Z, Qin C. lncRNA Ftx promotes aerobic glycolysis and tumor progression through the PPARgamma pathway in hepatocellular carcinoma. Int J Oncol 2018; 53:551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.