

Abstract
Background:
Myxomatous valve degeneration (MVD) involves the progressive thickening and degeneration of the heart valves, leading to valve prolapse, regurgitant blood flow, and impaired cardiac function. Leukocytes comprised primarily of macrophages have recently been detected in myxomatous valves, but the timing of the presence and the contributions of these cells in MVD progression are not known.
Methods:
We examined MVD progression, macrophages, and the valve micro-environment in the context of Marfan syndrome (MFS) using mitral valves from MFS mice (Fbn1C1039G/+), gene-edited MFS pigs (FBN1Glu433AsnfsX98/+), and MFS patients. Additional histologic and transcriptomic evaluation was performed using non-syndromic human and canine myxomatous valves, respectively. Macrophage ontogeny was determined using MFS mice transplanted with mTomato+ bone marrow or MFS mice harboring RFP-tagged C-C chemokine receptor type 2 (CCR2) monocytes. Mice deficient in recruited macrophages (Fbn1C1039G/+;Ccr2RFP/RFP) were generated to determine the requirements of recruited macrophages to MVD progression.
Results:
MFS mice recapitulated histopathologic features of myxomatous valve disease by 2 months-of-age, including mitral valve thickening, increased leaflet cellularity, and extracellular matrix abnormalities characterized by proteoglycan accumulation and collagen fragmentation. Concurrently, diseased mitral valves of MFS mice exhibited a marked increase of infiltrating (MHCII+, CCR2+) and resident macrophages (CD206+, CCR2-), along with increased chemokine activity and inflammatory extracellular matrix modification. Likewise, mitral valve specimens obtained from gene-edited MFS pigs and human MFS patients exhibited increased monocytes and macrophages (CD14+, CD64+, CD68+, CD163+) detected by immunofluorescence. Additionally, comparative transcriptomic evaluation of both genetic (MFS mice) and acquired forms of MVD (humans and dogs) unveiled a shared up-regulated inflammatory response in diseased valves. Remarkably, deficiency of monocytes was protective against MVD progression, resulting in a significant reduction of MHCII macrophages, minimal leaflet thickening, and preserved mitral valve integrity.
Conclusions:
Altogether, our results suggest sterile inflammation as a novel paradigm to disease progression, and we identify, for the first time, monocytes as a viable candidate for targeted therapy in MVD.
Keywords: Mitral Valve, Myxomatous Valve Degeneration, Marfan Syndrome, Macrophages, Monocytes, Extracellular Matrix, Sterile Inflammation
INTRODUCTION
Mitral valve insufficiency is a major source of morbidity and mortality worldwide and a common cause of heart failure, ventricular dysfunction, arrhythmias, and sudden cardiac death1. This loss of unidirectional blood flow is often due to myxomatous valve degeneration (MVD), a progressive deterioration of the heart valves characterized by leaflet thickening, excessive accumulation of glycosaminoglycans, and disruption of elastin and collagen fibers. MVD affects up to 2–3% of the general population, and more than 10% of individuals over 75 years of age have mitral valve insufficiency2. While MVD can be acquired with advanced age, increased incidence and earlier onset is often associated with inherited connective tissue disorders, such as Marfan syndrome (MFS), Osteogenesis imperfecta, and Ehlers-Danlos syndrome3. However, insight into early disease progression is lacking because valves are generally examined once patients are symptomatic and undergo surgery or autopsy, thereby representing advanced disease. As a result, the molecular and cellular mechanisms underlying MVD pathogenesis and disease progression are elusive and no pharmacological therapies exist to prevent or reverse this condition. Instead, surgical valve repair or replacement remains the current standard of care, despite the need for re-operation as a result of limited durability and growth potential of the transplanted valve, particularly for pediatric patients4.
Considerable insight into the molecular and cellular events of heart valve development and MVD has come from ongoing study of the two main cell populations of the heart valves: a continuous outer lining of valvular endothelial cells and fibroblast-like valvular interstitial cells within the valve leaflets. Recent discovery of immune cells in mouse and human heart valves has opened up new lines of investigation, especially pertaining to their origins and whether they play key roles in valve development and disease. We, and others, have shown that this immune cell population is comprised mostly of macrophages after birth in mice5–7. Additionally, a brief period of increased immune activity was detected by RNAseq in embryonic human aortic valves8 corroborating recent evidence that a subset of embryonically-derived macrophages are required for proper valve development5. Intriguingly, macrophages are increased in mitral valves of mouse models and human patients with MVD, although their origin and potential roles in disease are not known6, 9, 10.
In this study, we investigated the origin, timing, and requirements of macrophages during the pathogenesis of MVD using a mouse model of Marfan syndrome (Fbn1C1039G/+) that recapitulates aortic aneurysm and valve degeneration11–13. To determine whether macrophages are integral to mitral valve pathology, we performed histologic analysis using valve specimens from gene-edited MFS pigs14 and human MFS patients, in addition to a comparative transcriptomic evaluation of valves from both genetic (MFS mice) and acquired forms of MVD (humans and dogs). Here, we show that myxomatous valves exhibit a pro-inflammatory micro-environment involving the recruitment of macrophages that is evolutionarily conserved across species exhibiting MVD. To determine the requirements of sterile inflammation in the progression of MVD, we generated MFS mice lacking CCR2 monocytes (Fbn1C1039G/+;Ccr2RFP/RFP). Remarkably, deficiency of circulating CCR2+ monocytes was protective against MVD progression, resulting in valves showing minimal leaflet thickening and preserved matrix integrity. Altogether, our results suggest sterile inflammation as a novel paradigm in disease progression and we identify, for the first time, monocytes as a viable candidate for targeted therapy in MVD.
MATERIALS AND METHODS:
The authors declare that all supporting data within the article and its online supplementary files are available from the corresponding author upon reasonable request. Gene expression data are available from the GEO repository (GSE137681).
Mice:
The following mice were purchased from The Jackson Laboratory: Fbn1C1039G/+ (Stock #012885), Ccr2RFP/RFP (Stock #017586), and RosamTmG (Stock #007676). All mice were maintained on a C57BL/6 background and both males and virgin females were used in all analyses. All mouse experiments conform to NIH guidelines (Guide for the Care and Use of Laboratory Animals) and were performed with protocols approved by the Institutional Animal Care and Use Committee at the Cincinnati Children’s Research Foundation (IACUC2017–0076).
Pigs:
Gene-edited Marfan syndrome pigs (White/Landrace x Duroc crossbred strain) were generated, as previously described14. Male and female Marfan pigs were used in all analyses. All animal care and procedures were performed in accordance with the Japan Act on Welfare and Management of Animals and Regulations and approved by the Institutional Animal Care and Use Committee at Meiji University (IACUC12–0008, 15–0001). Control hearts were obtained from 2-to-4 year-old female White Yorkshire farm pigs at a local slaughterhouse. Upon removal, fresh hearts were rinsed in 1x PBS and excess myocardium and left atrial tissue were removed prior to fixation in 10% Formalin.
Human valve and aortic tissue specimens:
Healthy control valve tissue (n=10 total) was acquired postmortem from the National Disease Research Interchange (NDRI), Restore Life USA (Elizabeton, TN), and Liege University Hospital. Tissue specimens came from either healthy donors whose hearts were rejected for transplant or individuals hospitalized after motor vehicular accidents, and both cohorts had no history of cardiovascular disease. Myxomatous mitral valve tissue (n=9 total) from patients undergoing elective valvuloplasty surgery to correct mitral regurgitation (n=6), in addition to those with a confirmed diagnosis of Marfan syndrome (n=3), were obtained from Liege University Hospital and the Cardiovascular Tissue Registry at the University of British Columbia. Additionally, Marfan aortic tissue (n=6) and normal healthy controls (n=3) were also obtained from the Cardiovascular Tissue Registry. Human subject information is available in Supplemental Table 1. Cincinnati Children’s Research Foundation IRB approval was obtained (Study ID: 2017–2203; #00002988) prior to receipt of valve tissue and the study was classified as exempt due to the de-identified nature of the valve samples.
Tissue Collection and Histology
Neonatal (P0–7) and adult (1–12mo) mice were sacrificed using isoflurane, cervical dislocation, and thoracotomy. The heart was quickly removed, its apex then cut (exposing both left and right ventricles), and washed in ice-cold PBS to facilitate drainage of blood from the chambers. Hearts were then fixed in 4% paraformaldehyde (PFA) overnight at 4° C, dehydrated through a graded ethanol series, cleared in xylene, embedded in paraffin, and sectioned at 5μm thickness, as previously described15. Movat’s pentachrome staining was performed using manufacturer’s protocols (American MasterTech). For Picrosirius Red staining (American Mastertech; Cat. # KTPSRPT), a modified protocol that excluded Weigert’s Hematoxylin staining was used. Verhoeff’s staining solution was used to detect elastin (American MasterTech; Cat. # STVGIGAL). Alizarin Red (Sigma; Cat. # A5533) was used to determine the presence of calcification. Brightfield images were captured using the Olympus BX51 microscope retro-fitted with the Nikon DS-Ri1 camera and DS-U3 controller, along with the NIS-Elements BR (version 3.2) software.
Statistics
Mann-Whitney U tests were used to determine the significance of observed differences using PRISM8 software package (GraphPad; La Jolla, CA). Multiple means were compared using one-way analysis of variance with the post-hoc Tukey test. Data are reported as mean +/− standard error (SEM). A p-value <0.05 (two-sided) was considered statistically significant and is indicated as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
RESULTS
Mitral valves undergo abnormal post-natal maturation of the extracellular matrix in Marfan syndrome mice
Mitral valve disease progression was examined in a murine model of Marfan syndrome13, Fbn1C1039G/+ from birth (postnatal day 0; P0) until 12 months of age. Starting at P30, MFS mice exhibited mitral valves with structural extracellular matrix (ECM) abnormalities compared to wildtype controls as determined by Movat’s pentachrome staining (Figure 1). Overall leaflet thickening was observed in MFS mitral valves by P30 and onwards with increased glycosaminoglycans (GAGs) (blue staining, arrows; Figure 1C’–E’, J) compared to that of wildtype mice (Figure 1C–E, J). More specifically, MFS mitral valves exhibited an increased deposition of hyaluronic acid binding protein (HABP) (green; Figure 1F–G’, L), indicative of overall increased proteoglycan16 and versican (VCAN) (green; Figure 1I–I’, M), the major chondroitin sulfate proteoglycan in heart valves17, as determined by immunofluorescence. MFS mitral valves also exhibited abnormal collagen deposition within the proteoglycan-rich layer (arrow) and fiber disruption (arrowheads) by 2 months of age (Figure 1H–H’). Interestingly, thickening and myxomatous alterations were not diffuse throughout the valves but rather localized to the distal tip and occasionally midway between the distal tip and the hinge region (Figure 1C’, D’). Aortic valves were also affected in MFS mice, and showed thickening, proteoglycan accumulation, and collagen deposition in the distal tips by 2 months of age (Supplemental Figure 1A–F). Hypercellularity, a feature often noted in human myxomatous mitral valves11, was also observed in the mitral valves of MFS mice by nuclear DAPI staining starting at P7 (Figure 1K). Notably, these phenotypic changes occur prior to previously reported functional changes (i.e., mitral regurgitation) detectable by echocardiography beginning at 6–8 months of age in MFS mice11, 18.
FIGURE 1: Mitral valves undergo abnormal post-natal maturation in Marfan syndrome mice.
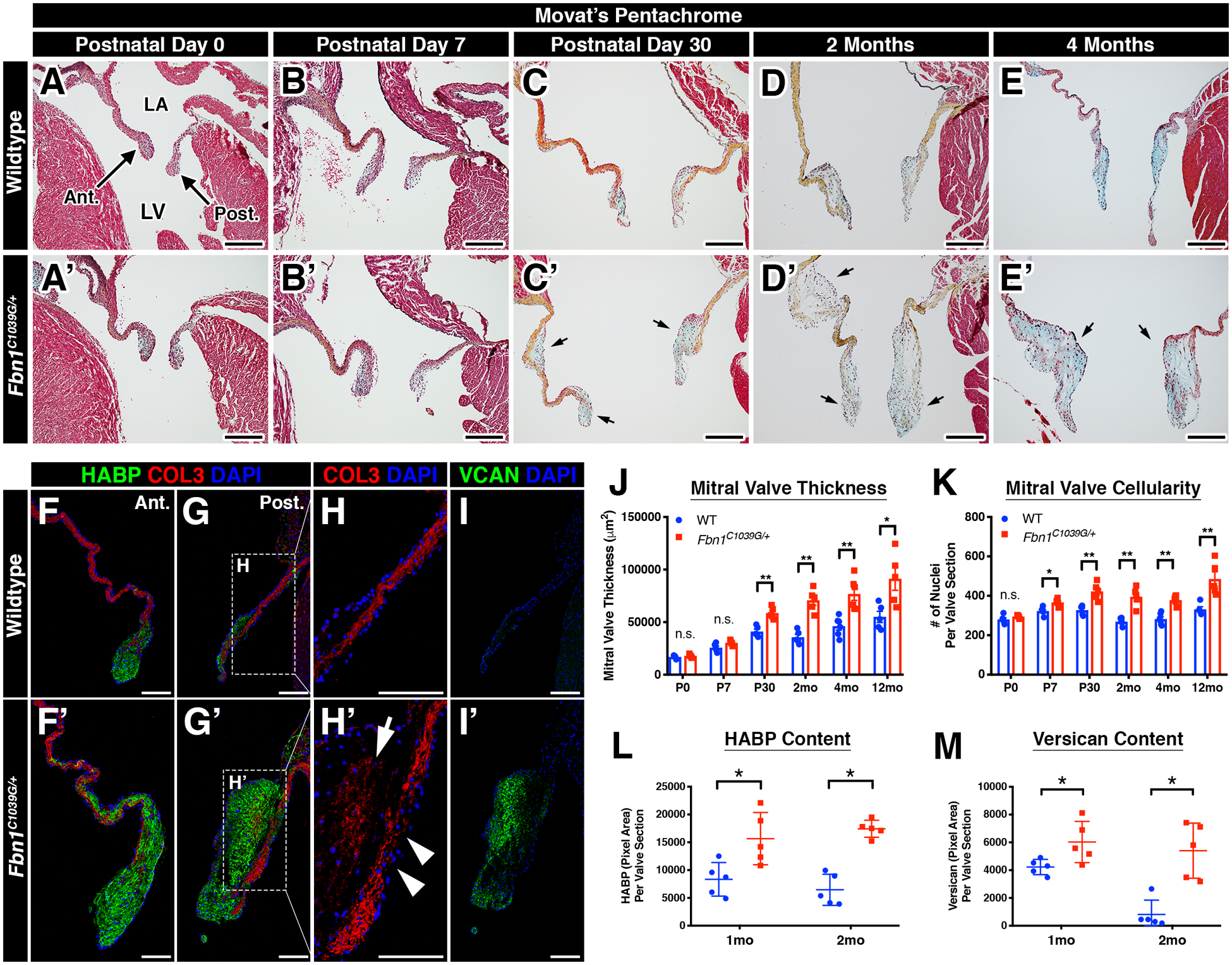
A-E’: Representative histologic images of the mitral valve at multiple timepoints in wildtype (A-E) and Marfan syndrome (A’-E’) mice using Movat’s pentachrome staining. Note that mitral valve leaflet thickening is detected (arrows) beginning at P30 in Marfan syndrome mice (C’) versus control (C). LA = left atrium; LV = left ventricle. F-I’: Diseased mitral valves exhibit ECM abnormalities, including increased HABP (green; F’, G’), collagen fragmentation (arrowheads; H’), collagen deposition (arrow; H’), and increased versican (green; I’) versus controls (F- I). The posterior leaflet is depicted. J-M: Quantification of mitral valve thickness (J), overall cellularity (K), HABP (L), and versican (M). Data are represented as mean ± SEM. *p<0.05; **p<0.01. n.s. = not significant. n=6–8 mice/genotype/timepoint. Scale Bars = 200μm (A-E’) and 100μm (F-I’). Abbreviations: Ant.=Anterior leaflet; Post.=Posterior leaflet; LA=Left Atrium; LV=Left Ventricle; HABP=Hyaluronan Binding Protein; COL=Collagen; VCAN=Versican; mo=month.
Myxomatous mitral valves exhibit a sterile pro-inflammatory micro-environment
While the heart valves are comprised mainly of valvular endothelial cells (VECs) and valvular interstitial cells (VICs), leukocytes have also been detected in both normal and diseased valves in mice and humans5–7, 9, 10, 15, 19–21. As indicated by the pan-hematopoietic cell marker, CD45, MFS mice exhibited a significant increase in CD45+ cells by 2 months of age, particularly near regions of collagen breakdown and proteoglycan expansion (Figure 2A–C’, F). Since the increase in CD45+ cells occurs after initial thickening and hypercellularity, it is likely that the immune cells contribute to progression, rather than initiation, of mitral valve disease. The majority of CD45+ cells in the wildtype and MFS valves are comprised primarily of two macrophage populations that differentially express CD206 and MHCII6. Notably, these two populations reside in two distinct regions of the mitral valve, with the CD206 population on the flow side and the MHCII population in the distal tips of the valve (Figure 2E–F’). In diseased MFS mitral valves, differential localization was lost and MHCII+ cells localized to regions of ECM breakdown and in the distal tips (Figure 2E’–F’’’).
FIGURE 2: Myxomatous mitral valves in Marfan syndrome mice exhibit a pro-inflammatory micro-environment.
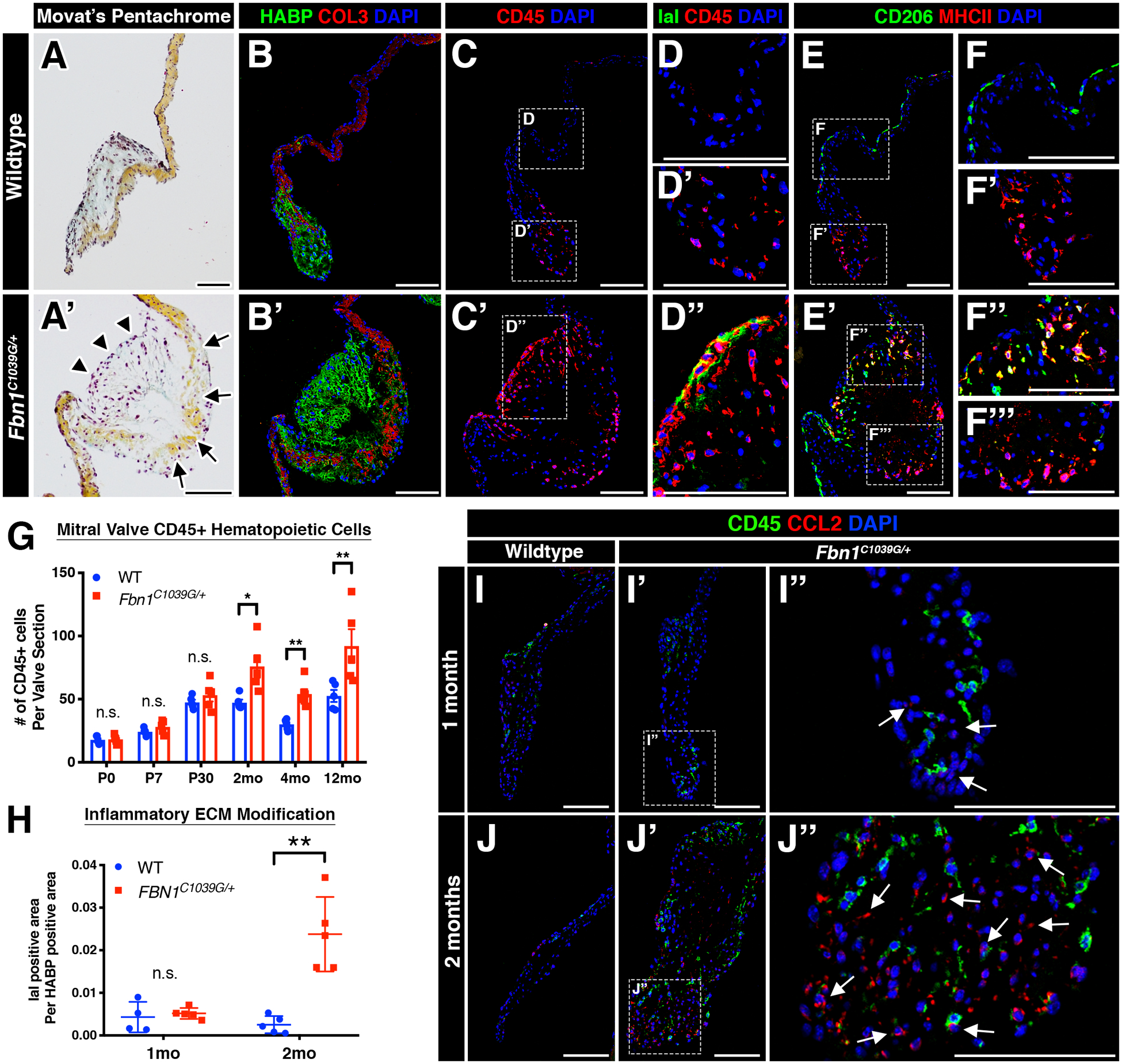
A-A’: Representative histologic images of the mitral valve at 2 months of age in wildtype (A) and Marfan syndrome (A’) mice using Movat’s pentachrome. Abnormal valvular ECM remodeling comprised of collagen fragmentation (arrows) and increased proteoglycan (arrowheads) is present in Marfan syndrome mice (A’) versus controls (A). The anterior leaflet is shown. B-B’: Increased proteoglycan deposition (green) and collagen fragmentation (red) can also be seen by confocal microscopy. Note the discontinuity of collagen (red) in mutant mitral valves (B’). C-C’: By confocal microscopy, CD45+ leukocytes are noticeably increased in MFS mitral valves (C’) versus controls (C) and are localized to regions of abnormal ECM remodeling. D-D”: MFS mitral valves exhibit immunogenic modification of the proteoglycan layer comprised of increased IaI deposition (green; D”) versus control mitral valves (D, D’). E-F’’’: CD45+ leukocytes are primarily comprised of two subsets of macrophages (CD206 and MHCII) that are spatially distinct in wildtype valves (E). Note that MHCII+ macrophages remain at the distal tips of the mitral valve (F’), whereas CD206+ macrophages run along the length of the valve leaflets (F’). In diseased valves, spatial preference of macrophages is lost, as MHCII+ and double-positive CD206+MCHII+ macrophages are located in the vicinity of CD206+ macrophages (F”, F’’’). G-H: Quantification of CD45+ leukocyte numbers (G) and IaI deposition (H). I-J”: Representative fluorescent images of CCL2 expression in WT and MFS mitral valves at one and two months of age. No significant differences in CCL2 expression was observed at one month of age (I-I”). However, CCL2 was significantly increased by two months of age in MFS mitral valves (J’-J”) versus control valves (J). CCL2 protein was predominantly expressed in CD45- populations and also deposited in the extracellular space (arrows, J”). Note that CD45+ cells are observed in the vicinity of CCL2. Data are represented as mean ± SEM. *p<0.05 and **p<0.01. n.s. = not significant. n=6–8 mice/genotype/timepoint. Scale Bar = 100μm. Abbreviations: HABP=Hyaluronan Binding Protein; COL=Collagen; IaI= Inter-alpha-trypsin inhibitor; mo=month.
Mitral valves in MFS mice exhibited immunogenic modifications of the proteoglycan layer within the valve. In addition to increased deposition of versican (Figure 1I–I’), a known inducer of sterile inflammation22, diseased valves also exhibited increased inter-alpha-trypsin inhibitor (IaI) (green; Figure 2D, D’, E), a matrix component known to attract and sequester immune cells in inflamed tissues6, 23. A major pro-inflammatory chemoattractant CCL2 was also localized predominantly in CD45-negative populations (i.e., VICs and VECs) in MFS valves at 2 months-of-age by immunofluorescence (Figure 2H–K’). Moreover, CCL2 was present in the extracellular space within regions of high GAG content24. These studies are consistent with previous demonstration of increased Ccl2 mRNA expression starting at 2 months as detected by qPCR in mouse MFS heart valves15. No significant changes in TUNEL reactivity were detected in MFS valves at 1 or 2 months-of-age, suggesting that an upregulated inflammatory response involving CD45+ cell expansion is not attributed to cell death in the valve (Supplemental Figure 2A). In addition, expression of Ly6G, indicative of neutrophils, and CD3, indicative of lymphocytes, was not detected by immunostaining of 2 month-old WT or Fbn1C1039G/+ mouse mitral valves, further supporting macrophages as the predominant immune cell population increased in MFS myxomatous valve disease (Supplemental Figure 2C). Moreover, inflammation-induced calcification was not observed upon evaluation of diseased mitral or aortic valves in aged 16-month-old mice (Supplemental Figure 3). Intriguingly, CD45+ cells were notably absent near regions of elastin fragmentation in the ascending aorta during the pathogenesis of aortic aneurysm starting from 1 to 12 months-of-age (Supplemental Figure 4), suggesting that this heightened immune activity is specific to the heart valve pathogenesis.
Comparative transcriptomic analysis of genetic and acquired forms of myxomatous valve degeneration unveils a shared up-regulated inflammatory response in disease
We further probed MVD-related gene expression changes by bulk RNA sequencing (RNAseq) of mitral valves from 5-month old MFS mice compared to age-matched WT controls. As listed in Supplemental Table 2, 494 genes were differentially expressed (335 upregulated and 159 downregulated in MFS mitral valves compared to WT controls). Gene-ontology (GO) analysis of upregulated genes from mitral valves in MFS mice versus controls showed enrichment for genes involved in the cellular response to cytokine, inflammatory response, myeloid leukocyte migration, and leukocyte chemotaxis (Supplemental Figure 5A, Supplemental Tables 3 and 4). Surprisingly, the enriched biological processes identified using ToppFun and DAVID GO analysis were nearly all related to immune cells and inflammatory response (Supplemental Tables 3 and 4). These results reinforce our histological findings as MFS mitral valves exhibited increased CD45+ cells, immunogenic remodeling of the ECM, and increased chemokine, CCL2. Additionally, our findings in 5-month old mice are evidence that the inflammatory response, first detected at 2 months-of-age in MFS mice, is chronic.
To determine whether pro-inflammatory gene expression changes in MVD are evolutionarily conserved and clinically relevant, we performed a meta-analysis of publicly available transcriptomic RNA or microarray datasets from myxomatous mitral valves obtained from human patients undergoing elective surgery for mitral valve regurgitation25 and dogs with mitral regurgitation26–28. GO analysis of genes with increased expression in human and canine MVD included gene categories related to upregulated immune responses and cytokine activity (Supplemental Figure 5B–D), including GO terms such as “inflammatory response”, “immune response”, “positive regulation of the immune system process”, among others. Altogether, these data suggest a shared up-regulated inflammatory response that is common to genetic and acquired types of myxomatous valve disease.
Increased monocyte-derived macrophages localize to regions of abnormal ECM remodeling in valves from gene-edited pigs and human patients with Marfan syndrome.
Translational applicability from mouse studies often requires validation in large animal models prior to clinical testing in human subjects. Given differences in cardiac physiology and valve ECM stratification between mice and larger mammals, including humans, we asked whether an increase in macrophages in MVD was evolutionarily conserved in a gene-edited large pig model of Marfan syndrome. These mutant pigs were generated using Zinc-finger nucleases targeting the FBN1 gene, resulting in a truncated form of the FBN1 protein. Heterozygous pigs (FBN1Glu433AsnfsX98/+) exhibit features of Marfan syndrome, including fragmentation of the aortic elastin, ocular abnormalities, and skeletal malformations14. Six mutant hearts were analyzed, five of which were obtained at two years of age. One mutant heart was obtained from a 4-year-old MFS pig that died of heart failure, which notably exhibited a significantly enlarged left atrium (Figure 3A), often indicative of chronic mitral regurgitation. Interestingly, all six mutant hearts exhibited noticeably thickened mitral valves and chordae tendineae upon gross inspection (increased leaflet opacity versus controls) and histology (arrows; Figure 3A–B). By Movat’s pentachrome, MFS mitral valves exhibited abnormal ECM remodeling comprised of an expanded proteoglycan layer and collagen disruption, characteristic of myxomatous degeneration (Figure 3B).
FIGURE 3: Myxomatous mitral valves from Fbn1Glu433AsnfsX98/+ gene-edited pigs with Marfan syndrome exhibit increased monocyte-derived macrophages localized to regions of abnormal ECM remodeling.
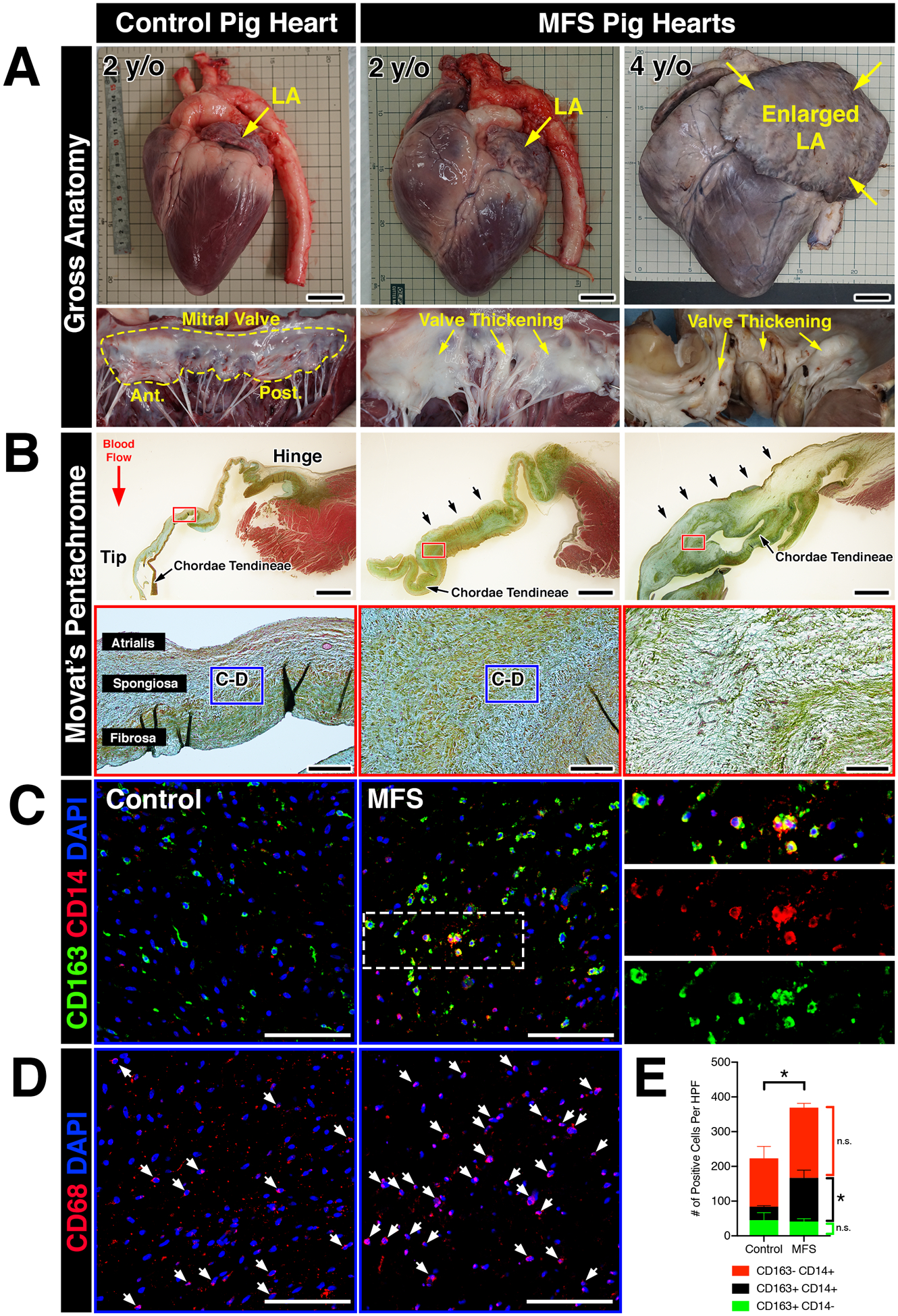
A: Whole hearts from wildtype and MFS pigs (Fbn1Glu433AsnfsX98/+) at 2- and 4-years-of-age. Note that the left atrium is enlarged in the MFS heart at 4 years-of-age (yellow arrows). Corresponding mitral valves including anterior and posterior leaflets are depicted in situ. Valve thickening is apparent by gross inspection, illustrated by increased opaqueness of the leaflets versus wildtype controls (yellow arrows). B: Histological sections of posterior leaflets of the mitral valves stained with Movat’s pentachrome are depicted and show thickening (arrows; upper panels) of the mitral valves in MFS pig hearts at 2- and 4- years-of-age versus controls. Note that the chordae tendineae are also markedly thickened in MFS hearts. At higher magnification (lower panels), MFS pig hearts exhibit loss of tri-laminar ECM organization of the atrialis (elastin), spongiosa (proteoglycan), and fibrosa (collagen) layers versus controls. C: By immunofluorescence, CD14+ monocytes and CD163 macrophages can be observed in the regions highlighted by the dotted box (B, lower panels). Overall, monocytes and macrophages were increased in Marfan syndrome pigs vs wildtype controls. Magnified view of double-positive cells is depicted in insets and separated by red and green fluorescent channels. D: CD68+ macrophages are depicted in comparable regions using sister sections. E: Quantification of macrophages in MFS pigs (n=6) versus controls (n=3). Data are represented as mean ± SEM. *p<0.05. n.s. = not significant. Scale bars = 300mm (A); 2mm (upper panels in B) and 200μm (lower panels in B); and 100μm (C, D). Abbreviations: LA=Left atrium; y/o=year-old; MFS=Marfan Syndrome; Ant.=Anterior leaflet; Post.=Posterior leaflet; HPF=High power field.
The presence of immune cells in MFS pigs was examined using well-defined monocyte and macrophage markers of the porcine immune system. Specifically, expression of CD14 and CD163 in pigs is restricted to the monocyte/macrophage lineage, with CD14 and CD163 expressed at higher levels in circulating monocytes and mature macrophages, respectively29. Confocal microscopy revealed that overall CD163+ macrophage abundance was increased in MFS mitral valves versus controls (Figure 3C, E). Interestingly, diseased valves exhibited a considerable increase in the number of double-positive CD14+CD163+ cells, suggesting that the observed increase in total numbers of CD163+ macrophages in diseased porcine valves is attributed a subpopulation derived from circulating monocytes (Figure 3E). Single-positive CD14+(CD163-) populations exhibited no significant differences between MFS pigs versus controls. CD68, a well-established pan-macrophage marker validated in mouse and human studies, exhibited similar staining patterns to that of CD163+ (CD14+ and CD14-) macrophages in comparable areas from adjacent sister sections (Figure 3D). Altogether, our results indicate that mitral valves in MFS pigs exhibit increased macrophages derived from a circulating monocyte population.
Since a pro-inflammatory micro-environment is common to both acquired and genetic forms of myxomatous valve degeneration and infiltrating monocytes were observed in MFS mice and MFS pigs, we assessed monocyte and macrophage subsets in diseased human valves (Figure 4). To test this, we analyzed mitral valve specimens from patients with a confirmed diagnosis of MFS (n=3, Figure 4A’) and those with acquired myxomatous valve degeneration (n=6), in addition to age-matched control mitral valves (n=10, Figure 4A). Using CD14, a monocytic lineage surface marker, and CD68, a pan-macrophage cytoplasmic marker, myxomatous valves exhibited a significant increase in double-positive CD14+CD68+ monocyte-derived macrophages, particularly near regions of disorganized ECM remodeling (Figure 4B, B’, D). This increase is statistically significant when comparing just the MFS mitral valves (red data points; p<0.01) or acquired MVD valves (black data points; p<0.0005) separately to control valves. Single-positive (CD14-)CD68+ macrophages, presumably a resident macrophage population, were also observed to a lesser degree, but no significant changes were observed in diseased versus control valves (Figure 4D). Additionally, as expected, CD68+ macrophages were also positive for CD64+, a surface marker highly specific to macrophages, and exhibited similar abundance to CD14+CD68+ macrophages (Figure 4C, C’). In contrast, CD45+ leukocytes are noticeably absent near regions of elastin fragmentation in the ascending aortas of MFS pigs and MFS patients (Supplemental Figure 6), similar to that of MFS mice (Supplemental Figure 4). Taken together, these results indicate that increased numbers of recruited macrophages are a common feature in the mitral valves, but not aortae, of MFS pigs, MFS patients, and patients with acquired MVD.
FIGURE 4: Myxomatous mitral valves from human patients with Marfan syndrome or acquired mitral regurgitation exhibit increased CD14+ monocyte-derived macrophage localized to regions of abnormal ECM remodeling.
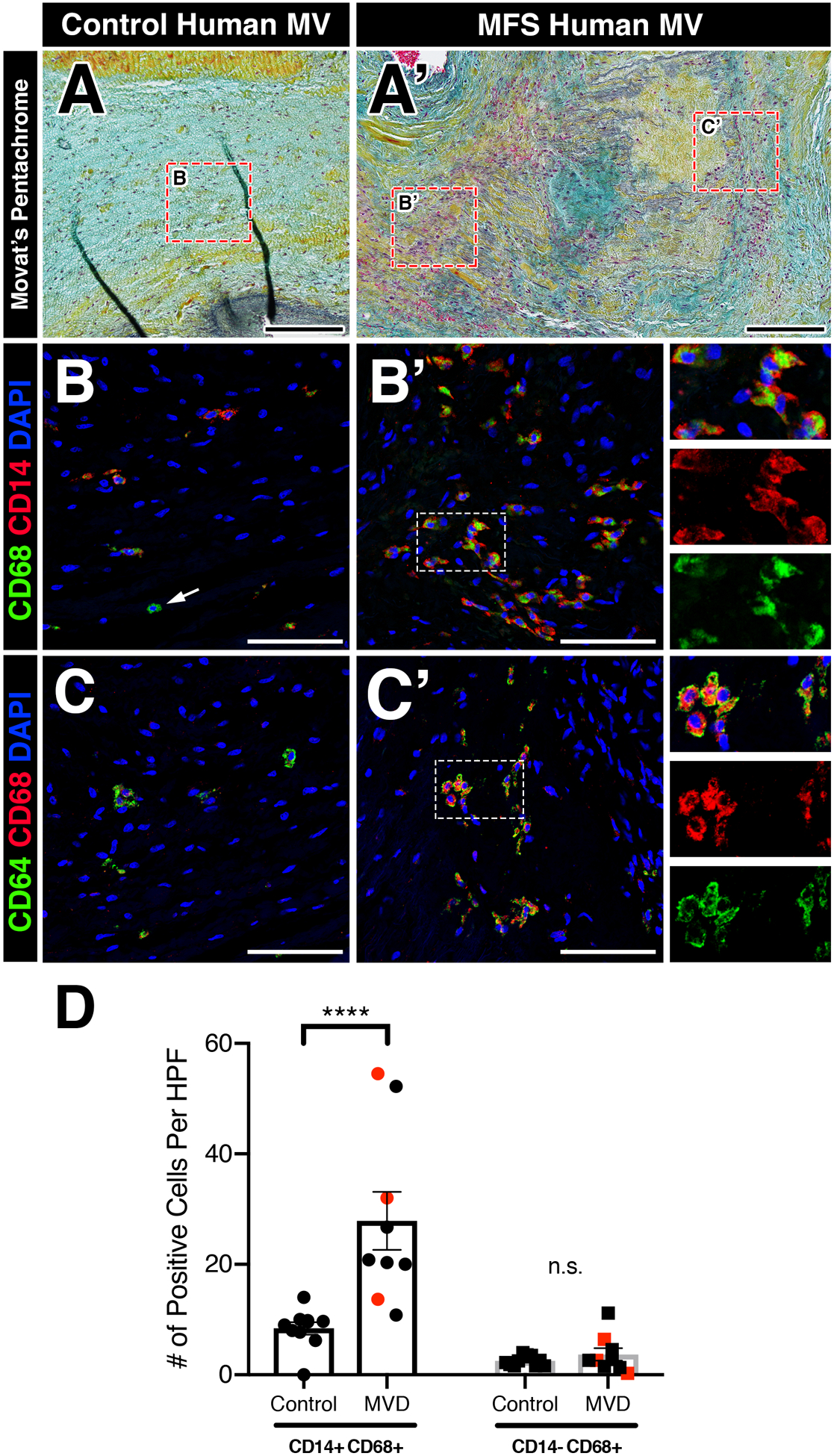
A-A’: Histological sections of healthy (A) and myxomatous (A’) human mitral valves from patients with Marfan syndrome are depicted. Diseased valves show loss of tri-laminar ECM organization of the atrialis (black), spongiosa (blue), and fibrosa (yellow) layers versus normal controls by Movat’s pentachrome staining. B-B’: Fluorescent detection of monocyte-derived macrophages and tissue-resident macrophages using CD68 (pan-macrophage cytoplasmic marker) and CD14 (circulating monocyte lineage surface marker). Myxomatous mitral valves in Marfan syndrome patients exhibit an increase in CD14+CD68+ monocyte-derived macrophages (B’) versus healthy controls (B). Note that tissue-resident CD68+ macrophages that lack CD14 are observed (arrow, B). Magnified view of double-positive cells is depicted in insets (B’) and separated by red and green fluorescent channels. C-C’: CD68+ macrophages are also positive for CD64+ (pan-macrophage surface marker). D: Quantification of macrophages in human myxomatous mitral valves (n=9) versus controls (n=10). Red data points indicate MFS valve specimens while black data points indicate acquired myxomatous mitral valves. Data are represented as mean±SEM. ****p<0.0001. n.s. = not significant. Scale bars = 200μm (A, A’); and 100μm (B-C’). Abbreviations: MV=Mitral valve; MFS=Marfan Syndrome; MVD=Mitral valve disease; HPF=High power field.
Bone marrow-derived circulating monocytes contribute to one of two distinct macrophage populations in the murine heart valve.
In order to determine the origins of increased CD45+ cells in the MFS valves, we generated bone marrow (BM) chimeras in which irradiated WT and MFS recipient mice were transplanted with congenic age-matched mTomato+ bone marrow from RosamTmG/+ donor mice. BM transplantation was performed at 6 weeks-of-age, prior to the observed increase in CD45+ cells in MFS mice, and harvested 3 weeks later (Figure 5H). Upon histological evaluation, MFS valves exhibited a significant increase in the number of BM-derived mTomato+ cells versus wildtype controls (Figure 5A, A’, F). Notably, in wildtype mice, BM-derived mTomato+ cells were observed near the distal tips (Figure 5C), whereas this spatial preference was not observed in MFS mitral valves (Figure 5C’). Interestingly, MFS valves also exhibited a modest increase in the number of mTomato-negative CD45+ cells, which could be attributed to the presence of resident pHH3+ CD45+ cells observed at 1 month-of-age in non-irradiated MFS mice (Figure 5F; Supplemental Figure 2B).
FIGURE 5: Inflammatory micro-environment in myxomatous valves recruits bone-marrow-derived monocytes that contribute to one of two distinct macrophage populations in the heart valve.
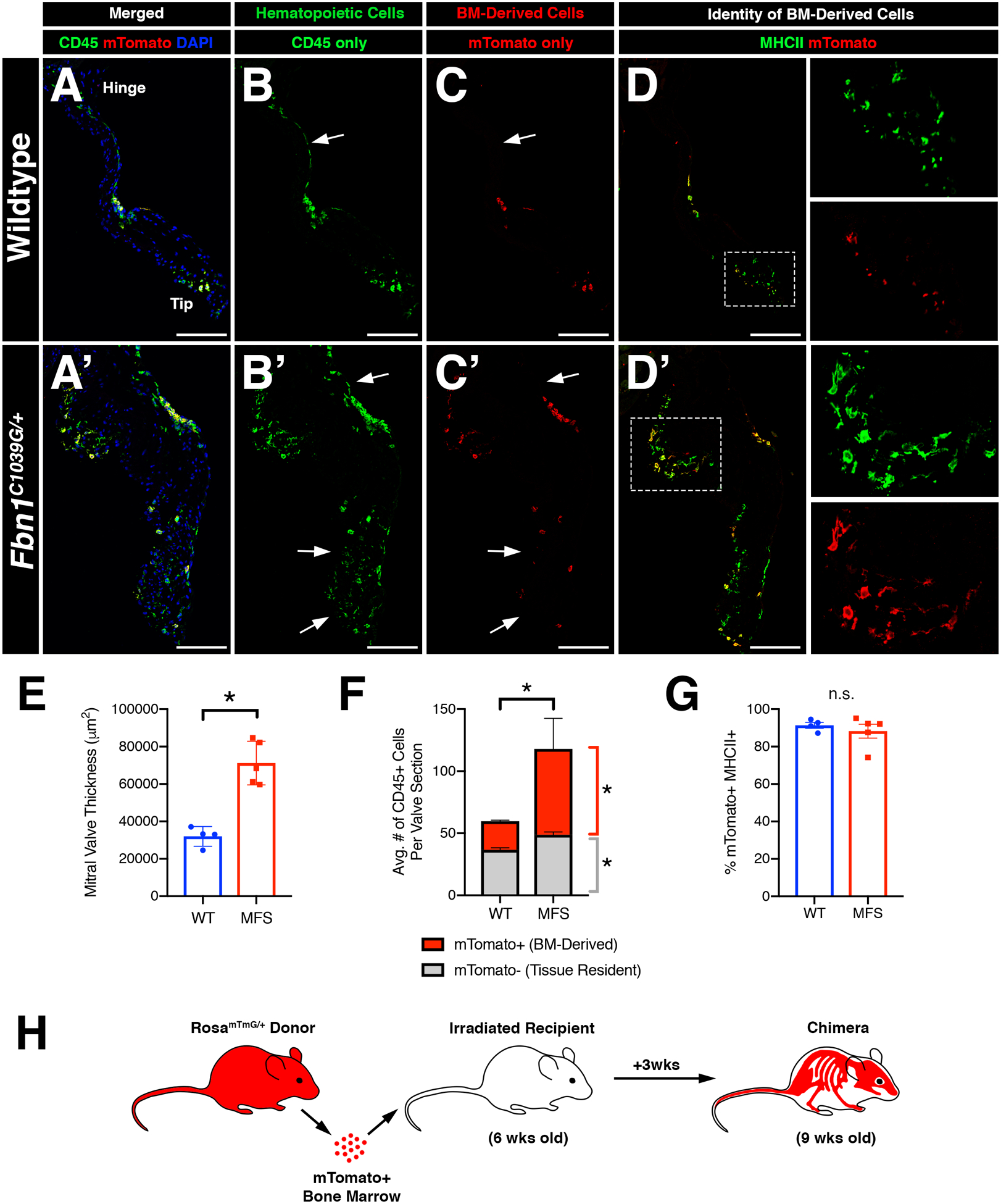
Infiltrating CD45+ cells populate the valve leaflets during homeostasis and disease. A-H: Bone marrow (BM) transplantation was performed on irradiated 6 week-old Fbn1C1039G/+ mice and age-matched controls and analyzed 3 weeks later. Immunofluorescence demonstrates the presence of mTomato+ donor cells in the mitral valve (red; C, C’) which are also CD45+ (green; B, B’; yellow; A, A’). Note the presence of host-derived CD45+ cells in merged panels (green; A’, A’). mTomato+ donor cells were predominantly MHCII+ (D, D’). E: Mitral valve leaflets remained thickened in Marfan syndrome mice versus controls after BM transplant, as expected. F: Marfan syndrome mice exhibit increased BM-derived CD45+ cells versus controls. Tissue-resident CD45+ cells also show a modest increase in number in Marfan syndrome mice. G: Quantification of MHCII+ cells among mTomato+ cells in the mitral valve indicates that BM-derived cells are predominantly MHCII+. H: Schematic of experiment. Data are represented as mean±SEM. *p<0.05. n.s. = not significant. n=4–5 mice/genotype. Scale Bar = 100μm. Abbreviations: BM=Bone marrow; MFS=Marfan Syndrome; wks=weeks.
CCR2 is a surface receptor on pro-inflammatory monocyte subsets required for bone marrow exit and homing to sites of inflammation, often in a CCL2-dependent manner30. Because mitral valves exhibit increased CCL2 expression (Figure 2J–J”), we crossed MFS mice with CCR2-red fluorescent protein (RFP) knock-in reporter mice to generate MFS mice harboring fluorescently labeled CCR2 monocytes (Fbn1C1039G/+;Ccr2RFP/+). As expected, MFS mice exhibited mitral valves with increased numbers of CD45+ cells, including a significant increase in CCR2-expressing RFP+ monocytes (Supplemental Figure 7). As mentioned previously, the majority of CD45+ cells in the wildtype and MFS valves are comprised primarily of two macrophage populations that differentially express CD206 and MHCII6 with MHCII+ cells localized to regions of ECM breakdown and in the distal tips in diseased MFS mitral valves (Figure 2E’–F’’’). BM-derived cells are predominantly MHCII+ (Figure 5D, D’, G). Similarly, ~80% of all CCR2 RFP+ cells were also MHCII positive in WT and MFS mice (Supplemental Figure 7), suggesting that circulating CCR2+ monocytes contribute primarily to the MHCII+ subpopulation of valve macrophages.
Deficiency of infiltrating CCR2+ monocytes inhibits the progression of myxomatous degeneration in Marfan syndrome mice.
Because monocytic infiltration was significantly increased in myxomatous valve degeneration, we sought to determine if the loss of monocyte recruitment would be protective against valve disease. To test this, we generated MFS mice deficient in monocytes by crossing MFS heterozygous CCR2 reporter mice (Ccr2RFP/+) with homozygous CCR2 deficient mice (Ccr2RFP/RFP) and analyzed the mitral valves at 2 and 6 months-of-age. In the CCR2-heterozygous state at 2 months-of-age, MFS mice exhibited significantly thickened mitral valves compared to Fbn1+/+ controls, as expected (Figure 6A, A’, G). In addition, CCR2+ RFP monocytes are increased in Ccr2RFP/+ MFS mitral valves versus Fbn1+/+ controls (Figure 6C, C’, H). Interestingly in control Fbn1+/+ mitral valves, CCR2 deficiency had no effect on mitral valve thickness (Figure 6B, G) despite showing a significant reduction in the number of RFP+ CCR2 monocytes (Figure 6D, H). In contrast, Ccr2RFP/RFP MFS mice deficient in CCR2 exhibited mitral valves that were significantly reduced in thickness relative to Ccr2RFP/+ MFS mitral valves, and were indistinguishable from that of Fbn1+/+ controls (Figure 6B’, D’, G, H). Depleted MFS valves also exhibited a preservation of ECM integrity, indicated by a significant reduction in proteoglycan content and lack of collagen dysregulation detected by Movat’s pentachrome staining. This was accompanied by a significant decrease in the MHCII+ macrophage subpopulation (Figure 6I). Therefore, CCR2 deficiency leads to decreased numbers of MHCII macrophages along with reduced progression of valve thickening in MFS mice.
FIGURE 6: Deficiency of infiltrating CCR2+ monocytes inhibits the progression of myxomatous degeneration in Marfan syndrome mice.
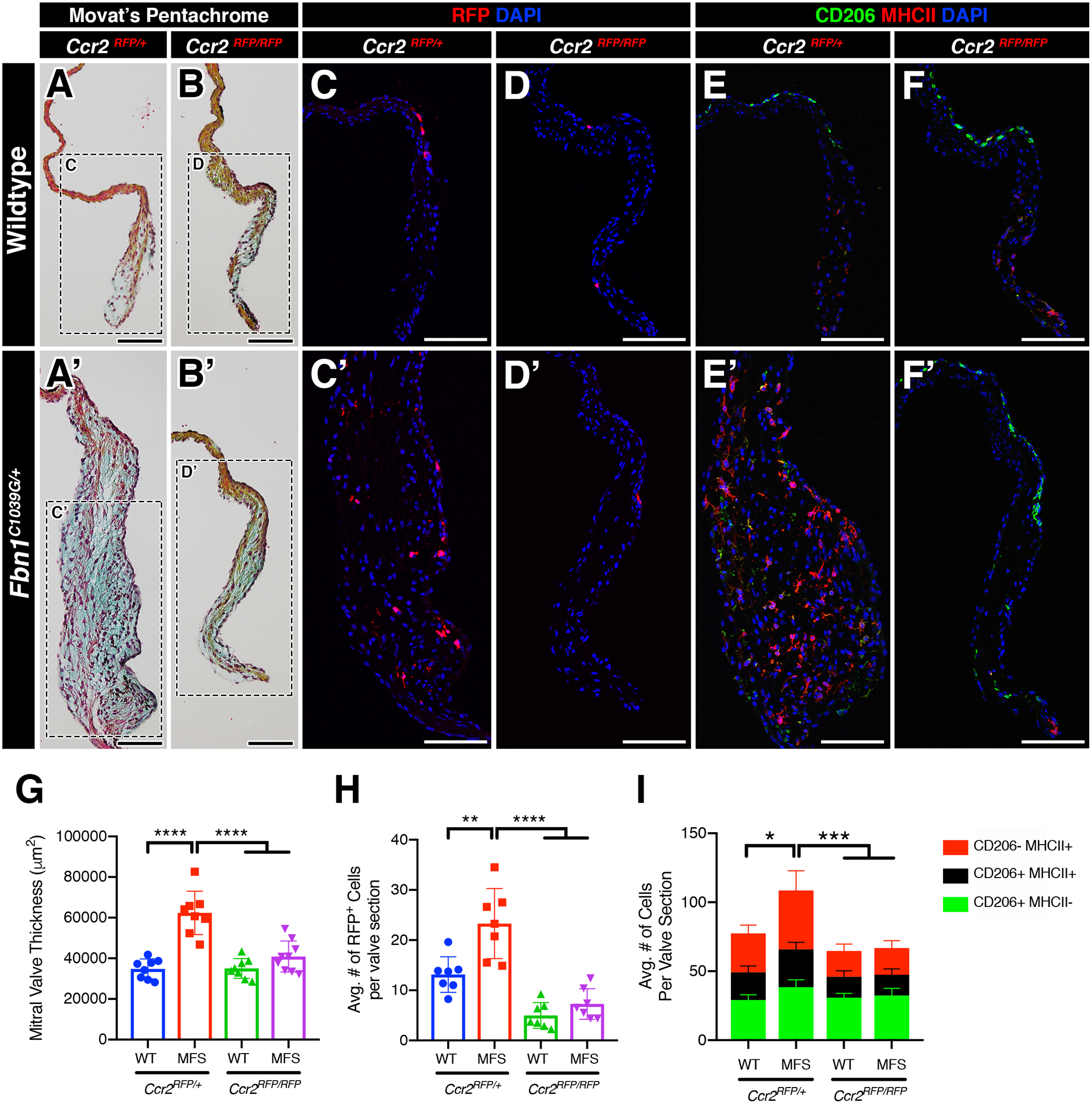
A-B’: Representative Movat’s pentachrome stained sections of the mitral valves (anterior leaflet) in 2-month-old WT and MFS mice in the normal (Ccr2RFP/+; A, A’) and CCR2-depleted (Ccr2RFP/RFP; B, B’) state. Note that the leaflet thickness is notably reduced in MFS mitral valves in the depleted (B’) vs normal (A’) state. C-D’: By immunofluorescence, CCR2+ monocytes (red) are significantly increased in MFS mitral valves (C’) versus WT (C). CCR2-depleted mice exhibit significant reduction in monocytes in both WT (D) and MFS (D’) mitral valves. E-F’: CCR2+ monocytes contribute mainly to the MHC-II macrophage subpopulation and an increase in monocyte population is correlated to the increase in the MHC-II macrophage subpopulation versus controls. Deficiency of CCR2 results in a significant reduction of the MHC-II+ population in both wildtype and MFS mice. G-I: Quantification of mitral valve thickness (G), number of CCR2+ monocytes (H), and number of macrophage subpopulations (I) in WT and MFS mice in the normal and CCR2-depleted state. Data are represented as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. n=8–9 mice/genotype. Data are represented as mean ± SEM. Scale Bar = 100μm. Abbreviations: WT=Wild Type; MFS = Marfan Syndrome; RFP=Red fluorescent protein.
In contrast to the mitral valves, elastin fragmentation is similar in aortae from Ccr2RFP/RFP deficient and Ccr2RFP/+ MFS, supporting a specific effect of CCR2 cell infiltration in mitral valve disease of MFS (Supplemental Figure 8). Similar results were obtained in 6 month-old MFS mice, in which CCR2 deficiency leads to normalization of mitral valve thickness but no improvement in elastin fragmentation in the aorta in Fbn1C1039G/+;Ccr2RFP/RFP animals (Supplemental Figure 9). Additional evidence for the specificity of macrophage increases in the mitral valve is that total monocytes (CD11b+;Ly6G-), CCR2+ monocytes (CD11b+;Ly6G-;RFP+), and neutrophils (CD11b+;Ly6G+) in circulating blood, as determined by FACS, are not different between WT and MFS mice at 6 months-of-age (Supplemental Figure 9E). As expected, the FACS analysis also confirmed loss of circulating CCR2+ monocytes in Ccr2RFP/RFP deficient mice. Taken together, these results indicate that the increase in CCR2+ macrophages is relatively higher in the MFS valves than other tissues and that deficiency of CCR2+ macrophages is sufficient to prevent MVD progression in vivo.
DISCUSSION
In this study, we show that a sterile inflammatory response, involving immunogenic ECM components and circulating monocytes, is a key driver of MVD. Using the heterozygous Fbn1C1039G/+ MFS mouse model that recapitulates aortic aneurysm and myxomatous valve degeneration11–13, we identified a marked increase of monocyte-derived macrophages, along with increased chemokine activity and inflammatory extracellular matrix modification, in diseased mitral valves. Likewise, mitral valve specimens obtained from gene-edited MFS pigs and human MFS patients exhibited a marked expansion of monocyte-derived macrophages. Additionally, comparative transcriptomic evaluation of both genetic (MFS mice) and acquired forms of MVD (humans and dogs) unveiled a shared up-regulated inflammatory response in diseased valves, suggesting that increased immune activity is an integral component of the progression of disease. Remarkably, inhibiting macrophage recruitment by CCR2 deficiency in monocytes was protective against MVD progression in MFS mice, resulting in valves showing minimal leaflet thickening and preserved matrix integrity. Altogether, our results suggest sterile inflammation as a novel paradigm underpinning disease progression, and we identify, for the first time, monocytes as a viable candidate for targeted therapy in MVD.
The mitral valve is highly plastic and can have notably distinct disease etiologies and histopathological features in the context of mitral insufficiency. Primary MVD with genetic etiologies is characterized by valve abnormalities at birth, commonly associated with mutations in genes encoding components of the ECM, as in Marfan syndrome3, cell-cell adhesion31 and primary cilia32. This is in contrast to secondary MVD, which often manifests past the fifth decade of life33 as a result of deformities of the valve apparatus (e.g., papillary muscles and ventricles) and cardiac insufficiency (as reviewed by Levine et al.1). Interestingly, CD45+ immune cells have been detected in the valve leaflets of both primary and secondary MVD6, 9, 10, 15, 34, challenging the traditional classification of MVD as a “non-inflammatory” degenerative valve disease. Unlike valvulitis classically associated with rheumatic heart disease35, 36 and endocarditis37, whereby the valve injury is extrinsic to the leaflets (i.e., autoantibodies and bacterial colonization), the initiating “injury” in primary MVD is likely intrinsic to the valve structure and function. The pathologic accumulation and remodeling of valve ECM components, including hyaluronan and versican, which contain microbial motifs known to interact with receptors of the innate immune system38, could trigger an immune response potentially related to GO enrichment of biological processes such as “defense response” and “response to other organisms” in our RNAseq analysis (Supplementary Figure 5). Athough the biological processes related to differentially expressed genes were predominantly immune-related, increased expression of a few Wnt pathway genes was detected in MFS mitral valves, consistent with our previous observation of increased Wnt signaling at early stages of valve disease in 2 month-old MFS mice34. Still, additional studies are needed to determine what initiates leaflet thickening and matrix accumulation prior to monocyte infiltration as a result of Fbn1 mutations in the context of MFS.
Currently there are limited treatment options for heart valve disease of MFS aside from surgical replacement or repair. Previous studies by the Dietz group and others have linked altered transforming growth factor beta (TGFβ) signaling with the clinical manifestations of MFS, including in the mitral valves in neonatal Fbn1C1039G/+ mice11. In addition, treatment of MFS mice with the angiotensin receptor blocker losartan prevents progression of aortic dilation, and specific TGFβ receptor blockade can inhibit mitral valve thickening. However, a recent clinical trial found no additional benefit from losartan therapy compared to standard beta-blocker treatment in managing aortic aneurysm, suggesting that other pathways might contribute to disease progression39. While mitral valves have not been as extensively evaluated in MFS patients treated with losartan, there is increasing evidence that pathogenesis of heart valves may be distinct from that of aortic dilation and aneurism. Moreover, increased macrophage recruitment was not observed in the diseased aortas of MFS mice, pigs, or human patients in this study. Therefore, treatment strategies for MVD and aortic disease in MFS may need to be considered separately.
Macrophages populate all tissues, exhibiting organ-specific functions that impact not just host immunity, but also embryonic development, homeostasis, repair, and disease40. In the heart, distinct populations of macrophages arise from embryonic and adult origins that can be differentiated by the expression of CCR241. The majority of resident macrophages in heart valves lack CCR2 expression and are located subjacent to the valve endothelial cells along the length of the mitral valve leaflets. These resident macrophages have also been detected in developing heart valves, appearing as early as E10.5 in mice, and are critical in the proper remodeling of the valve during embryogenesis5. A recent RNAseq study of human embryonic aortic valves reveals a temporary upregulation in immune activity during mid-late gestation8, possibly reflecting valve macrophage activity seen in the developing mouse valve, although histological analysis of embryonic human valves is needed to verify this. Whereas embryonically-sourced macrophages are critical for proper valve development, we identify a distinct cohort of recruited CCR2+ macrophages that promote MVD. While additional studies are needed to determine the specific functions of CCR2+ macrophages in MVD, our data are in line with evidence in other cardiovascular diseases that recruitment of CCR2+ macrophages is pathogenic and associated with negative cardiac outcomes, including myocardial infarct expansion, atherosclerotic plaque progression, and left ventricular dysfunction41.
Over the past few decades, studies in animal models have implicated inflammation in the development of a wide variety of cardiovascular diseases, including atherosclerosis and myocardial infarction42. As a result, promising new pathways and inflammatory mediators have been identified as therapeutic targets. Clinical translation of these studies to the bedside has been difficult, however, due to the complex and often overlapping roles that cellular and molecular mediators of inflammation play in homeostasis and disease43. Steroids are widely used clinically but lack the specificity to target pathologic inflammation and preserve its protective functions, such as wound healing and defense against infection44. Alternative more targeted strategies directed towards specific cytokines, such as the landmark CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcome Study) clinical trial targeting a key pro-inflammatory chemokine, interleukin-1β, have provided significant therapeutic benefits by mitigating adverse cardiovascular outcomes45. In light of this study, there has been a renewed interest in dissecting the cellular and molecular mediators of the immune system and their influence on a broad range of cardiovascular diseases. Here we show that loss of CCR2+ monocyte-derived macrophages, classically viewed as pro-inflammatory, improves MFS-related valve thickening and ECM dysregulation. Ongoing efforts are directed towards developing effective CCR2-specific inhibitors, several of which are currently in clinical trials for a wide range of diseases46–48. Additional studies are needed to determine if pharmacological CCR2+ macrophage inhibition is an effective therapeutic approach to prevent or inhibit the progression of MVD in MFS patients.
Supplementary Material
CLINICAL PERSPECTIVE.
What is New?
Myxomatous mitral valves exhibit a pro-inflammatory micro-environment involving the recruitment of monocyte-derived macrophages that is evolutionarily conserved across species (mouse, pig, dog, human) with myxomatous valve degeneration.
Deficiency in circulating CCR2+ monocytes is protective against myxomatous valve degeneration but not aortic aneurysm formation in a mouse model of Marfan syndrome.
What are the Clinical Implications?
Sterile inflammation is a novel paradigm in myxomatous valve disease progression.
CCR2+ monocytes were identified as novel candidates for targeted therapy in myxomatous valve degeneration.
Pathogenesis and immune cell involvement in Marfan Syndrome-related myxomatous mitral valve disease and aortic aneurism are distinct.
ACKNOWLEDGEMENTS:
We would like to thank Jeff Bailey (Cincinnati Children’s Hospital Medical Center) for technical assistance; Minzhe Guo (CCHMC) for assistance with bioinformatics; Kimimasa Tobita (Biogen Ltd.) for assistance with Marfan pig phenotyping; and Jordan Miller (Mayo Clinic) for providing human mitral valve gene expression data. We also thank Dr. Michael Seidman (Cardiovascular Tissue Registry) for his collaborative support and Dr. Marc Radermecker from the Department of Cardiovascular and Thoracic Surgery and Human Anatomy (CHU-Sart-Tilman, Belgium) for sharing human mitral valve samples. All flow cytometric data were acquired using equipment maintained by the Research Flow Cytometry Core in the Division of Rheumatology at CCHMC.
FUNDING SOURCES:
This study was supported by grants from the American Heart Association (16PRE30180000) to A.J.K and the National Institutes of Health (T32GM063483 and T32HL125204) to A.J.K and (R01HL143881) to K.E.Y. R.J.V. was supported by a Career Development Award from the American Heart Association (19CDA34670044). H.N. and K.U. were supported by the Leading Advanced Projects for Medical Innovation (LEAP), the Japan Agency for Medical Research and Development (AMED), and a Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Non-standard Abbreviations and Acronyms
- BM
Bone marrow
- CANTOS
Canakinumab Anti-inflammatory Thrombosis Outcome Study
- E
Embryonic day
- ECM
Extracellular matrix
- GAG
Glycosaminoglycan
- GO
Gene ontology
- HABP
Hyaluronan binding protein
- HPF
High power field
- IaI
Inter-alpha-trypsin inhibitor
- LA
Left atrium
- LV
Left ventricle
- MFS
Marfan Syndrome
- MV
Mitral valve
- MVD
Myxomatous valve disease
- P
Postnatal day
- RFP
Red fluorescent protein
- VCAN
Versican
- VEC
Valve endothelial cell
- VIC
Valve interstitial cell
- WT
Wild type
Footnotes
DISCLOSURES:
The authors have no conflicts of interest to declare.
REFERENCES
- 1.Levine RA, Hagege AA, Judge DP, Padala M, Dal-Bianco JP, Aikawa E, Beaudoin J, Bischoff J, Bouatia-Naji N, Bruneval P, et al. Mitral valve disease--morphology and mechanisms. Nat Rev Cardiol. 2015;12:689–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG and Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368:1005–1011. [DOI] [PubMed] [Google Scholar]
- 3.Le Tourneau T, Merot J, Rimbert A, Le Scouarnec S, Probst V, Le Marec H, Levine RA and Schott JJ. Genetics of syndromic and non-syndromic mitral valve prolapse. Heart. 2018;104:978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lincoln J and Garg V. Etiology of valvular heart disease-genetic and developmental origins. Circ J. 2014;78:1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shigeta A, Huang V, Zuo J, Besada R, Nakashima Y, Lu Y, Ding Y, Pellegrini M, Kulkarni RP, Hsiai T, et al. Endocardially Derived Macrophages Are Essential for Valvular Remodeling. Dev Cell. 2019;48:617–630 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hulin A, Anstine LJ, Kim AJ, Potter SJ, DeFalco T, Lincoln J and Yutzey KE. Macrophage Transitions in Heart Valve Development and Myxomatous Valve Disease. Arterioscler Thromb Vasc Biol. 2018;38:636–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hulin A, Hortells L, Gomez-Stallons MV, O’Donnell A, Chetal K, Adam M, Lancellotti P, Oury C, Potter SS, Salomonis N and Yutzey KE. Maturation of heart valve cell populations during postnatal remodeling. Development. 2019;146:pii:dev173047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottlieb Sen D, Halu A, Razzaque A, Gorham JM, Hartnett J, Seidman JG, Aikawa E and Seidman CE. The Transcriptional Signature of Growth in Human Fetal Aortic Valve Development. Ann Thorac Surg. 2018;106:1834–1840. [DOI] [PubMed] [Google Scholar]
- 9.Geirsson A, Singh M, Ali R, Abbas H, Li W, Sanchez JA, Hashim S and Tellides G. Modulation of transforming growth factor-beta signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers. Circulation. 2012;126:S189–S197. [DOI] [PubMed] [Google Scholar]
- 10.Sauls K, Toomer K, Williams K, Johnson AJ, Markwald RR, Hajdu Z and Norris RA. Increased Infiltration of Extra-Cardiac Cells in Myxomatous Valve Disease. J Cardiovasc Dev Dis. 2015;2:200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP and Dietz HC. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY and Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Umeyama K, Watanabe K, Watanabe M, Horiuchi K, Nakano K, Kitashiro M, Matsunari H, Kimura T, Arima Y, Sampetrean O, et al. Generation of heterozygous fibrillin-1 mutant cloned pigs from genome-edited foetal fibroblasts. Sci Rep. 2016;6:24413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim AJ, Alfieri CM and Yutzey KE. Endothelial Cell Lineage Analysis Does Not Provide Evidence for EMT in Adult Valve Homeostasis and Disease. Anat Rec (Hoboken). 2019;302:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dina C, Bouatia-Naji N, Tucker N, Delling FN, Toomer K, Durst R, Perrocheau M, Fernandez-Friera L, Solis J, PROMESA Investigators, et al. Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat Genet. 2015;47:1206–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grande-Allen KJ, Griffin BP, Ratliff NB, Cosgrove DM and Vesely I. Glycosaminoglycan profiles of myxomatous mitral leaflets and chordae parallel the severity of mechanical alterations. J Am Coll Cardiol. 2003;42:271–277. [DOI] [PubMed] [Google Scholar]
- 18.Tae HJ, Petrashevskaya N, Marshall S, Krawczyk M and Talan M. Cardiac remodeling in the mouse model of Marfan syndrome develops into two distinctive phenotypes. Am J Physiol Heart Circ Physiol. 2016;310:H290–H299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Hjortnaes J, Beaudoin J, Szymanski C, Bartko PE, Seybolt MM, Handschumacher MD, et al. Myocardial Infarction Alters Adaptation of the Tethered Mitral Valve. J Am Coll Cardiol. 2016;67:275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartko PE, Dal-Bianco JP, Guerrero JL, Beaudoin J, Szymanski C, Kim DH, Seybolt MM, Handschumacher MD, Sullivan S, Garcia ML, et al. Effect of Losartan on Mitral Valve Changes After Myocardial Infarction. J Am Coll Cardiol. 2017;70:1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi JH, Do Y, Cheong C, Koh H, Boscardin SB, Oh YS, Bozzacco L, Trumpfheller C, Park CG and Steinman RM. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med. 2009;206:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wight TN, Kang I and Merrilees MJ. Versican and the control of inflammation. Matrix Biol. 2014;35:152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrey AC and de la Motte CA. Hyaluronan, a crucial regulator of inflammation. Front Immunol. 2014;5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN and Kosco-Vilbois MH. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci U S A. 2003;100:1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thalji NM, Hagler MA, Zhang H, Casaclang-Verzosa G, Nair AA, Suri RM and Miller JD. Nonbiased Molecular Screening Identifies Novel Molecular Regulators of Fibrogenic and Proliferative Signaling in Myxomatous Mitral Valve Disease. Circ Cardiovasc Genet. 2015;8:516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu CC, Liu MM, Culshaw G, Clinton M, Argyle DJ and Corcoran BM. Gene network and canonical pathway analysis in canine myxomatous mitral valve disease: a microarray study. Vet J. 2015;204:23–31. [DOI] [PubMed] [Google Scholar]
- 27.Li Q, Freeman LM, Rush JE, Huggins GS, Kennedy AD, Labuda JA, Laflamme DP and Hannah SS. Veterinary Medicine and Multi-Omics Research for Future Nutrition Targets: Metabolomics and Transcriptomics of the Common Degenerative Mitral Valve Disease in Dogs. OMICS. 2015;19:461–470. [DOI] [PubMed] [Google Scholar]
- 28.Oyama MA and Chittur SV. Genomic expression patterns of mitral valve tissues from dogs with degenerative mitral valve disease. Am J Vet Res. 2006;67:1307–1318. [DOI] [PubMed] [Google Scholar]
- 29.Fairbairn L, Kapetanovic R, Beraldi D, Sester DP, Tuggle CK, Archibald AL and Hume DA. Comparative analysis of monocyte subsets in the pig. J Immunol. 2013;190:6389–6396. [DOI] [PubMed] [Google Scholar]
- 30.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M and Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durst R, Sauls K, Peal DS, deVlaming A, Toomer K, Leyne M, Salani M, Talkowski ME, Brand H, Perrocheau M, et al. Mutations in DCHS1 cause mitral valve prolapse. Nature. 2015;525:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toomer KA, Yu M, Fulmer D, Guo L, Moore KS, Moore R, Drayton KD, Glover J, Peterson N, Ramos-Ortiz S,et al. Primary cilia defects causing mitral valve prolapse. Sci Transl Med. 2019;11:pii:eaax0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kolibash AJ Jr., Kilman JW, Bush CA, Ryan JM, Fontana ME and CF Wooley. Evidence for progression from mild to severe mitral regurgitation in mitral valve prolapse. Am J Cardiol. 1986;58:762–767. [DOI] [PubMed] [Google Scholar]
- 34.Hulin A, Moore V, James JM and Yutzey KE. Loss of Axin2 results in impaired heart valve maturation and subsequent myxomatous valve disease. Cardiovasc Res. 2017;113:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carapetis JR, Beaton A, Cunningham MW, Guilherme L, Karthikeyan G, Mayosi BM, Sable C, Steer A, Wilson N, Wyber R and Zuhlke L. Acute rheumatic fever and rheumatic heart disease. Nat Rev Dis Primers. 2016;2:15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meier LA, Auger JL, Engelson BJ, Cowan HM, Breed ER, Gonzalez-Torres MI, Boyer JD, Verma M, Marath A and Binstadt BA. CD301b/MGL2(+) Mononuclear Phagocytes Orchestrate Autoimmune Cardiac Valve Inflammation and Fibrosis. Circulation. 2018;137:2478–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thiene G and Basso C. Pathology and pathogenesis of infective endocarditis in native heart valves. Cardiovasc Pathol. 2006;15:256–263. [DOI] [PubMed] [Google Scholar]
- 38.Chen GY and Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, Pearson GD, Selamet Tierney ES, Levine JC, Atz AM, et al. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014;371:2061–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ginhoux F and Guilliams M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity. 2016;44:439–449. [DOI] [PubMed] [Google Scholar]
- 41.Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, Godwin J, Rosenthal N and Kovacic JC. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J Am Coll Cardiol. 2018;72:2213–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen H, Kreisel D and Goldstein DR. Processes of sterile inflammation. J Immunol. 2013;191:2857–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noels H, Weber C and Koenen RR. Chemokines as Therapeutic Targets in Cardiovascular Disease. Arterioscler Thromb Vasc Biol. 2019;39:583–592. [DOI] [PubMed] [Google Scholar]
- 44.Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 46.Struthers M and Pasternak A. CCR2 antagonists. Curr Top Med Chem. 2010;10:1278–1298. [DOI] [PubMed] [Google Scholar]
- 47.Zheng Y, Qin L, Zacarias NV, de Vries H, Han GW, Gustavsson M, Dabros M, Zhao C, Cherney RJ, Carter P, et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature. 2016;540:458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor BC, Lee CT and Amaro RE. Structural basis for ligand modulation of the CCR2 conformational landscape. Proc Natl Acad Sci U S A. 2019;116:8131–8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.