

SUMMARY
The physiological effects of the many germline mutations of TP53, encoding the tumor suppressor protein p53, are poorly understood. Here we report generating a p53 R178C knockin mouse modeling the human TP53 R181C mutation, which is notable for its prevalence and prior molecular characterization. Consistent with its weak cancer penetrance in humans, homozygous p53178C/C mice show a modest increase in tumorigenesis but, surprisingly, are lean with decreased body fat content. They display evidence of increased lipolysis and upregulation of fatty acid metabolism in their inguinal white adipose tissue (iWAT). Gene expression and chromatin immunoprecipitation sequencing (ChIP-seq) analyses show that the mutant p53 bound and trans-activated Beta-3-Adrenergic Receptor (ADRB3), a gene that is known to promote lipolysis and is associated with obesity. This study reveals that a germline mutation of p53 can affect fat metabolism, which has been implicated in cancer development.
Graphical Abstract
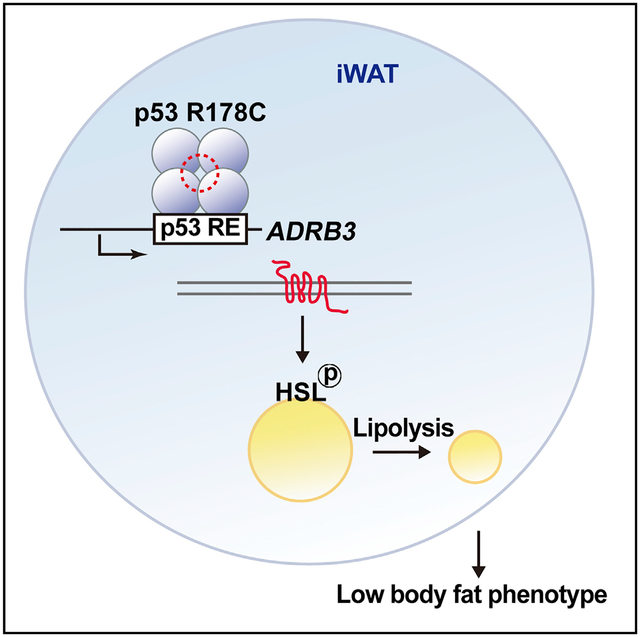
In Brief
Knockin of the mouse homolog of a human TP53 germline mutation known to cause Li-Fraumeni syndrome, a cancer predisposition disorder, results in a mouse model characterized by lower body fat content. Kang et al. show that enhancing transactivation of the lipolytic gene ADRB3 by mutant p53 contributes to this phenotype.
INTRODUCTION
Emerging studies have established p53 as a mediator of various metabolic activities, some of which are sufficient to restrain tumorigenesis even in the absence of its prototypical tumor suppressor functions, such as DNA repair, cell cycle arrest, and apoptosis (Bieging et al., 2014; Kung and Murphy, 2016; Li et al., 2012). p53 can also regulate energy homeostasis through both mitochondrial and non-mitochondrial pathways, including disposition of fatty acids, a major energy substrate and biosynthetic precursor required for cell proliferation (Berkers et al., 2013). Although wild-type p53 inhibits both fatty acid synthesis and lipid accumulation, mutant p53 has been shown to enhance fatty acid synthesis by inhibiting AMP-activated protein kinase (AMPK) (Parrales and Iwakuma, 2016; Zhou et al., 2014). Furthermore, mutant p53 cooperates with sterol regulatory element-binding proteins (SREBPs) to upregulate the mevalonate pathway to promote cancer formation (Freed-Pastor et al., 2012). Thus, alterations in lipid signaling molecules, membrane biosynthesis precursors, and substrates for fatty acid oxidation caused by mutations in p53 have the potential to support tumorigenesis (Currie et al., 2013). Whether these disparate effects of wild-type and mutant p53 on fatty acid metabolism are generalizable to other germline TP53 mutations is currently unclear.
Although many different germline mutations of TP53 have been reported, only a subset of mostly missense mutations located in the DNA binding domain have been associated with Li-Fraumeni syndrome (LFS), an autosomal dominant early-onset cancer disorder (Schneider and Garber, 2010). In a pilot study comprised of subjects with 10 different mutations, including the “hotspot” R273H and weak cancer penetrance R181C amino acid substitutions, LFS patients displayed evidence of increased oxidative metabolism that, upon inhibition, delayed tumorigenesis in a LFS mouse model (Wang et al., 2017, 2013). Given the critical role of p53 in tumor suppression, lessons from examining its metabolic activities at the organismal level in both wild-type and mutant states may provide insights to further understand their physiological and tumorigenic activities.
TP53 R181C was one of the earliest mutations described in association with breast cancer at a young age, and it currently stands out as one of the top reported germline mutations (Bouaoun et al., 2016; Sidransky et al., 1992). The incidence of this mutation was so high in a specific population studied for inherited breast cancers that it was likened to the TP53 R337H founder mutation prevalent in southern Brazil (Achatz and Zambetti, 2016; Lolas Hamameh et al., 2017). Amino acid R181 resides in the DNA binding domain of p53 and plays an important structural role by forming intermolecular salt bridges between p53 monomers for cooperative DNA binding (Schlereth et al., 2010). Because p53 R181C promoted mitochondrial biogenesis in human myoblasts (Wang et al., 2013), we investigated whether this specific mutant could have other metabolic effects in vivo. Here we report that, in addition to developing a cancer phenotype, mice with knockin of the TP53 R181C mouse homolog revealed a role for p53 in lipolysis and adipose tissue metabolism under physiologic conditions.
RESULTS
A Mouse Homolog of a LFS Mutation Reveals a Metabolic Phenotype
To examine the effect of human p53 R181C on metabolism, we generated a knockin mouse with an arginine (CGC)-to-cysteine (TGC) mutation at the corresponding amino acid residue 178 of mouse p53 using conventional embryonic stem cell (ESC)-mediated homologous recombination and the Cre-loxP strategy (Figures 1A and S1). Correctly targeted ESC clones containing a p53 C>T (c.541) missense mutation in exon 5 were identified by genomic DNA PCR, and the resulting mouse genotypes were confirmed by p53 cDNA sequencing, mouse embryonic fibroblast Southern blotting, and tail DNA PCR (Figures 1A and 1B). The homozygous mutant (p53178C/C) mice displayed a modest but significant decrease in median survival time by meeting the animal study endpoint and had a higher incidence of cancer, although the spectrum of cancer types was similar to that of wild-type mice (Figures 1C–1E). Some conventional approaches to induce p53 in p53178C/C mice and examine expression of its target genes and associated cellular activities showed partial retention of wild-type activity, with apoptosis being significantly reduced. This is consistent with previous reports of greater loss of apoptosis activity compared with cell cycle regulation in cooperative binding mutants (Figure S2; Schlereth et al., 2010; Timofeev et al., 2013). These observations in p53 R178C mice were also consistent with the relatively low cancer penetrance of the homologous TP53 R181C mutation in humans.
Figure 1. p53 R178C Knockin Mice Reveal Increased Cancer Incidence and a Metabolic Phenotype.
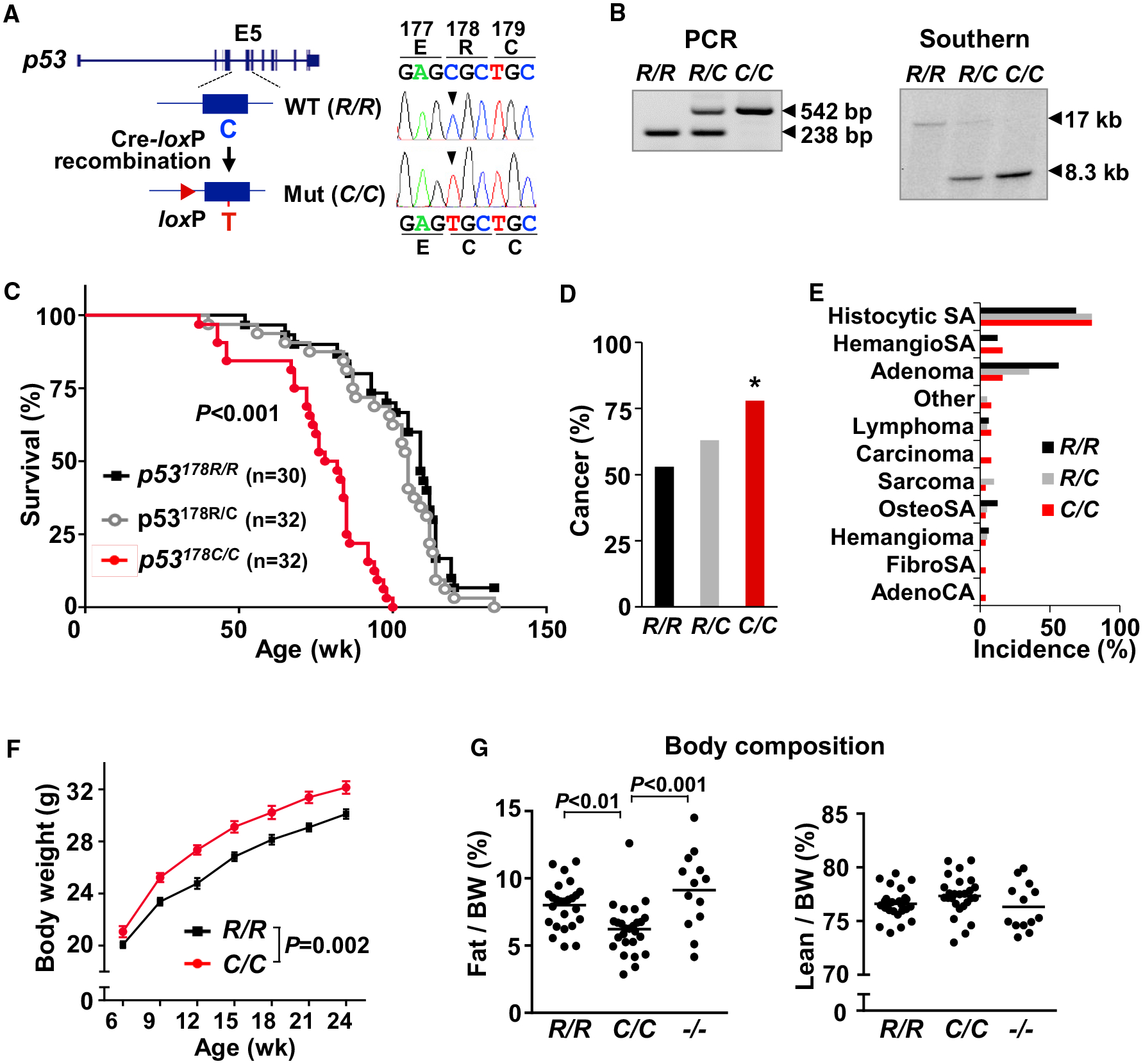
(A) Cre-loxP-mediated strategy for knockin of the amino acid R178C mutation into exon 5 (E5) of the mouse p53 gene. Mouse embryonic fibroblast (MEF) cDNA sequencing confirmed single-nucleotide substitution from C to T (right panel).
(B) Genomic DNA samples isolated from tail tissue and MEFs were analyzed by PCR and Southern blot, respectively. The 238-bp and 17-kb fragments correspond to the wild-type p53 allele. The 542-bp and 8.3-kb fragments correspond to the mutant p53 allele.
(C) Kaplan-Meier survival plot of the indicated p53 genotype mice. Median survival ages (weeks) were as follows: R/R, 109; R/C, 105; C/C, 80. Significance testing in comparison with wild-type p53.
(D) Cancer incidence in mice (R/R, n = 30; R/C, n = 32; C/C, n = 32).
(E) Spectrum of cancer types by p53 genotype (R/R, n = 16; R/C, n = 20; C/C, n = 25).
(F) Body weight of male mice by age (n ≥ 15).
(G) Fat and lean (muscle) body composition of 9-week-old male mice, measured by NMR analyzer (R/R, n = 25; C/C, n = 26; −/− , n = 13).
p53 R178 genotypes: wild-type p53178R/R (R/R), heterozygous mutant p53178R/C (R/C), homozygous mutant p53178C/C (C/C), and null p53−/− (−/−). Statistical difference by χ2 test in comparison with the wild-type (D), two-way ANOVA with repeated measures (F), and one-way ANOVA (G). Values are mean ± SEM. *p < 0.05. See also Figures S1–S3.
In the course of these survival studies, p53 R178C knockin mice showed a pattern of increased body weight. Therefore, we performed metabolic phenotyping focused on male homozygous mutant mice to maximize the mutant p53 effect and to control for gender dimorphism, although female mice also showed increased body weight (Figure 1F and S3A). The energy expenditure and food intake of homozygous p53178C/C mice were similar to that of wild-type mice after normalizing for body mass under both room temperature and thermoneutral conditions (Figure S3B). Notably, body composition analysis showed a significant decrease in the fraction of fat tissue in young as well as older p53178C/C mice (Figures 1G and S3C). The amount of lean tissue in p53178C/C mice accounted for their increased total body weight, but, in addition, there was a trend of increased lean mass percentage in the mutant compared with the wild-type p53 state, which may underlie the observed modest increase in glucose tolerance versus some other possible mechanism (Figures 1G, S3D, and S3E; Kung and Murphy, 2016). The decrease in fat composition of p53178C/C mice, without changes in exercise performance, as reported previously for another LFS mouse model (Figure S3F; Wang et al., 2013), appeared to be a gain of function by the p53 mutation rather than due to loss of wild-type activity because the fat composition of p53−/− mice was similar to that of wild-type controls (Figure 1G).
p53178C/C Mice Have Decreased Adipose Tissue and Increased Plasma Fatty Acids
Given the significantly lower body fat composition of p53178C/C mice, we investigated further by quantifying the amount of epididymal white adipose tissue (eWAT), inguinal white adipose tissue (iWAT), and brown adipose tissue (BAT) representing visceral, subcutaneous, and thermogenic types of fat, respectively (Figure 2A). The masses of all three adipose tissue types were lower in p53178C/C mice, with a more pronounced effect on iWAT, which showed smaller adipocytes compared with p53 wild-type or null mice (Figure 2B). Given the various contrasting reports of BAT regulation by p53 (Krstic et al., 2018), we examined BAT mitochondria and the thermogenesis phenotype of p53178C/C mice but did not observe significant differences, consistent with only a modest decrease in its mass (Figures 2A and S4A–S4C).
Figure 2. Homozygous p53178C/C Mice Display a Lean Phenotype with Increased Plasma Fatty Acids.
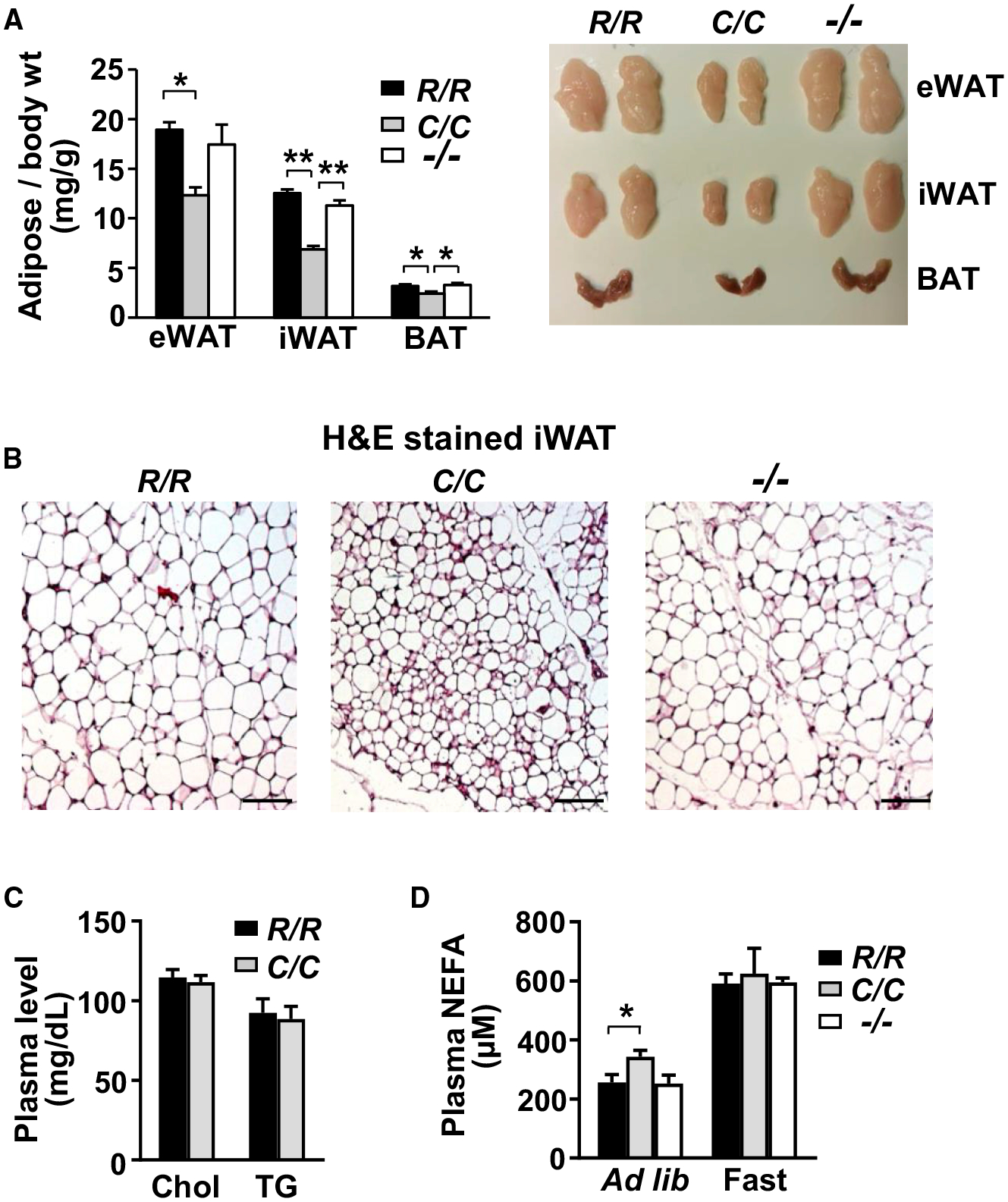
(A) Representative images of 3 different fat tissue types: inguinal white adipose tissue (iWAT), epididymal white adipose tissue (eWAT), and brown adipose tissue (BAT). The weight of fat tissue dissected from 10-week-old mice was normalized to body weight (milligrams per gram) (n = 6).
(B) H&E-stained iWAT sections. Scale bars, 100 μm.
(C) Plasma lipid levels: total cholesterol (Chol) and triglycerides (TG) (n = 10).
(D) Plasma non-esterified fatty acid (NEFA) levels measured under free feeding (ad libitum, R/R, n = 15; C/C, n = 23; −/−, n = 9) or fasting (fast for 4 h, R/R, n = 8; C/C, n = 11; −/−, n = 3) states.
p53 R178 genotypes: wild-type (R/R), homozygous mutant (C/C), and null (−/−). Statistical difference by one-way ANOVA. Values are mean ± SEM. *p < 0.05, **p < 0.01. See also Figure S4.
We next examined whether the more substantial changes in white adipose tissues were associated with alterations in blood lipid levels. Although the plasma levels of total cholesterol and triglycerides were relatively unchanged in p53178C/C mice compared with the wild-type, as also reported for p53−/− mice (Guevara et al., 1999), non-esterified fatty acid (NEFA) levels were significantly higher in the mutant p53 knockin mice under ad libitum feeding conditions (Figures 2C and 2D). This difference was attenuated by fasting, suggesting activation of lipolysis signaling by mutated p53, manifested only under basal and homeostatic conditions.
Plasma Metabolomic Analysis Reveals Increased Mobilization of Fatty Acids
To obtain a global profile of the changes in lipid metabolism, we performed metabolomic analysis of plasma obtained from 10-week-old p53178R/R, p53178C/C, and p53−/− male mice under ad libitum feeding conditions. A principal-component analysis (PCA) plot showed clustering of the metabolite levels by p53 genotype (Figure 3A). About half of the 706 identified compounds were lipid related, and ~20% were significantly changed by p53 genotype status (Figure S5A). Of the top 50 compounds whose levels were changed with the greatest statistical significance in the plasma of p53178C/C mice compared with that of p53 wild-type or null mice, 39 were lipid related with a pattern of reduced monoacylglycerol and diacylglycerol levels and increased fatty acid derivatives such as acyl carnitines and acyl glycines (Figures 3B and S5B). The increase in acyl carnitines was complemented by a decrease in free carnitine levels (Figure 3C). Interestingly, 12,13-dihydroxy-9Z-octadecenoic acid (12,13-diHOME), a lipokine reported to be induced by cold exposure and inversely correlated with body mass index in human subjects (Lynes et al., 2017), was increased in plasma of p53178C/C mice (Figure 3D). Its precursor linoleic acid and a related compound, 9,10-diHOME, were also increased, providing additional support for the specificity of the 12,13-di-HOME lipokine finding (Figure 3D). The elevation of plasma 12,13-diHOME was consistent with the low-fat phenotype of p53178C/C mice and its known promotion of fatty acid utilization in peripheral tissues (Lynes et al., 2017; Stanford et al., 2018).
Figure 3. Plasma Metabolomic Profiling Reveals Altered Lipid Metabolism in p53178C/C Mice.
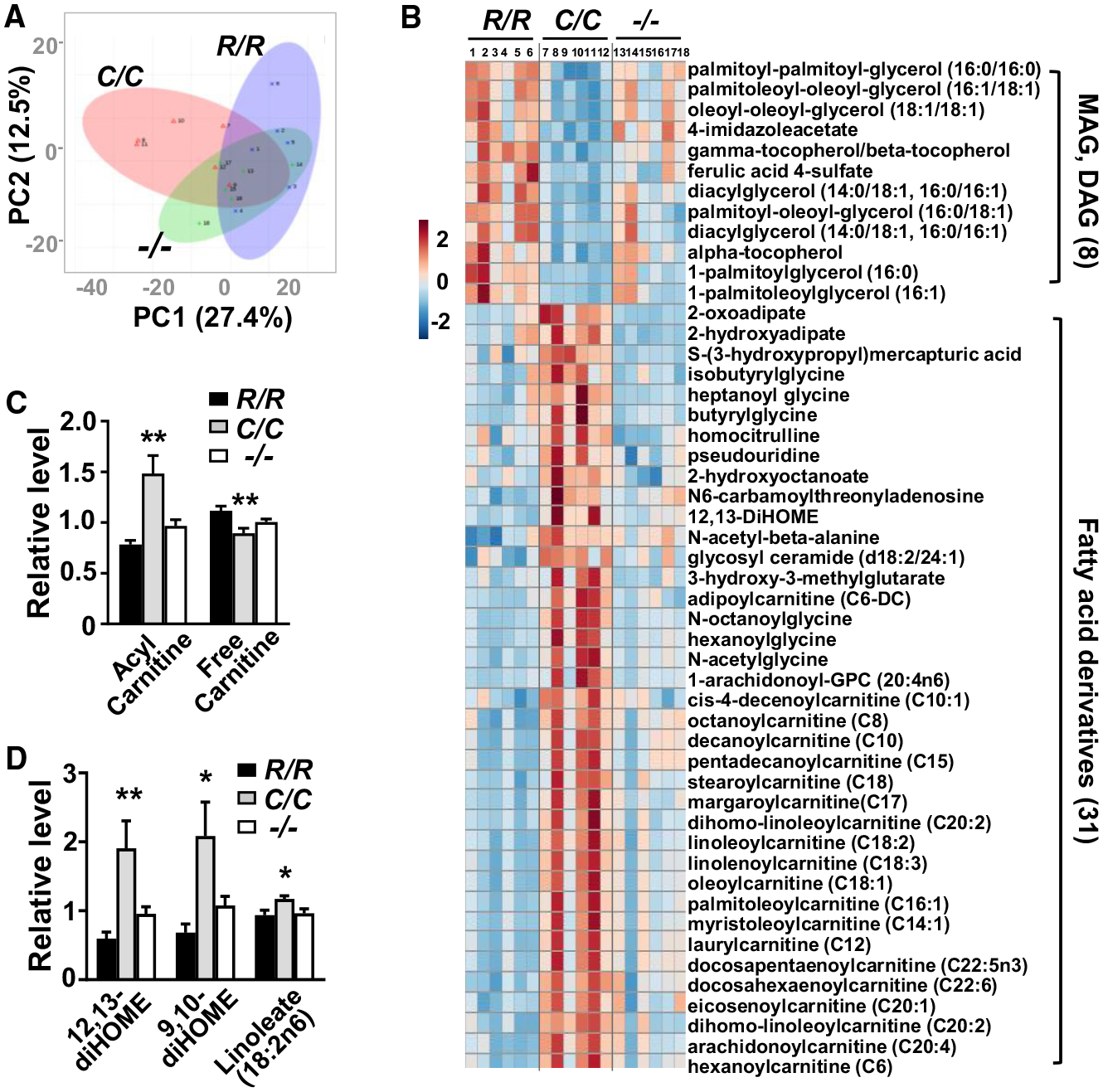
(A) Principal-component analysis (PCA) of all identified plasma metabolites of 10-week-old mice by p53 genotype (n = 6).
(B) Heatmap comparison of the top 50 most statistically different metabolites by p53 genotype shows that 39 are lipid-related compounds. Metabolite levels were normalized by median values. Relative level is color-coded in red (increased) and blue (decreased). In p53178C/C (C/C) mouse plasma, 8 of the 12 decreased metabolites are monoacylglycerols (MAGs) and diacylglycerols (DAGs), and 31 of the 38 increased metabolites are fatty acid derivatives.
(C) Relative levels of free carnitine and acyl carnitines (average of 32 different species).
(D) Relative levels of 12,13-diHOME and its isomer 9,10-diHOME and precursor linoleate.
p53 R178 genotypes: wild-type (R/R), homozygous mutant (C/C), and null (−/−). Statistical difference by one-way ANOVA. Values are mean ± SEM. *p < 0.05, **p < 0.01. See also Figure S5.
p53178C/C iWAT Gene Expression Analysis Shows Increased Expression of Lipid Metabolism Genes
The absence of the lipidomic changes in p53-null plasma raised the possibility that a metabolic activity specifically induced by mutant p53 may be conferring the low-fat phenotype of p53178C/C mice. Because mutations of p53 can affect gene expression of various biological processes, we investigated whether p53 R178C can regulate the transcription of genes related to lipolysis (Kastenhuber and Lowe, 2017; Pfister and Prives, 2017). We first examined the level of lipolytic activity associated with the p53 R178C mutation in different explanted tissues by measuring their glycerol release (Figure 4A). iWAT tissue showed a large increase in lipolytic activity associated with the p53 mutation, whereas other adipose tissue types as well as muscle and liver showed no discernable changes (Figure 4A). Therefore, we performed RNA sequencing (RNA-seq) on iWAT tissue in the basal state under which the lipolysis phenotype of p53178C/C mice was observed.
Figure 4. Increased Lipolysis and Expression of Genes Related to Fatty Acid Metabolism in iWAT of p53178C/C Mice.
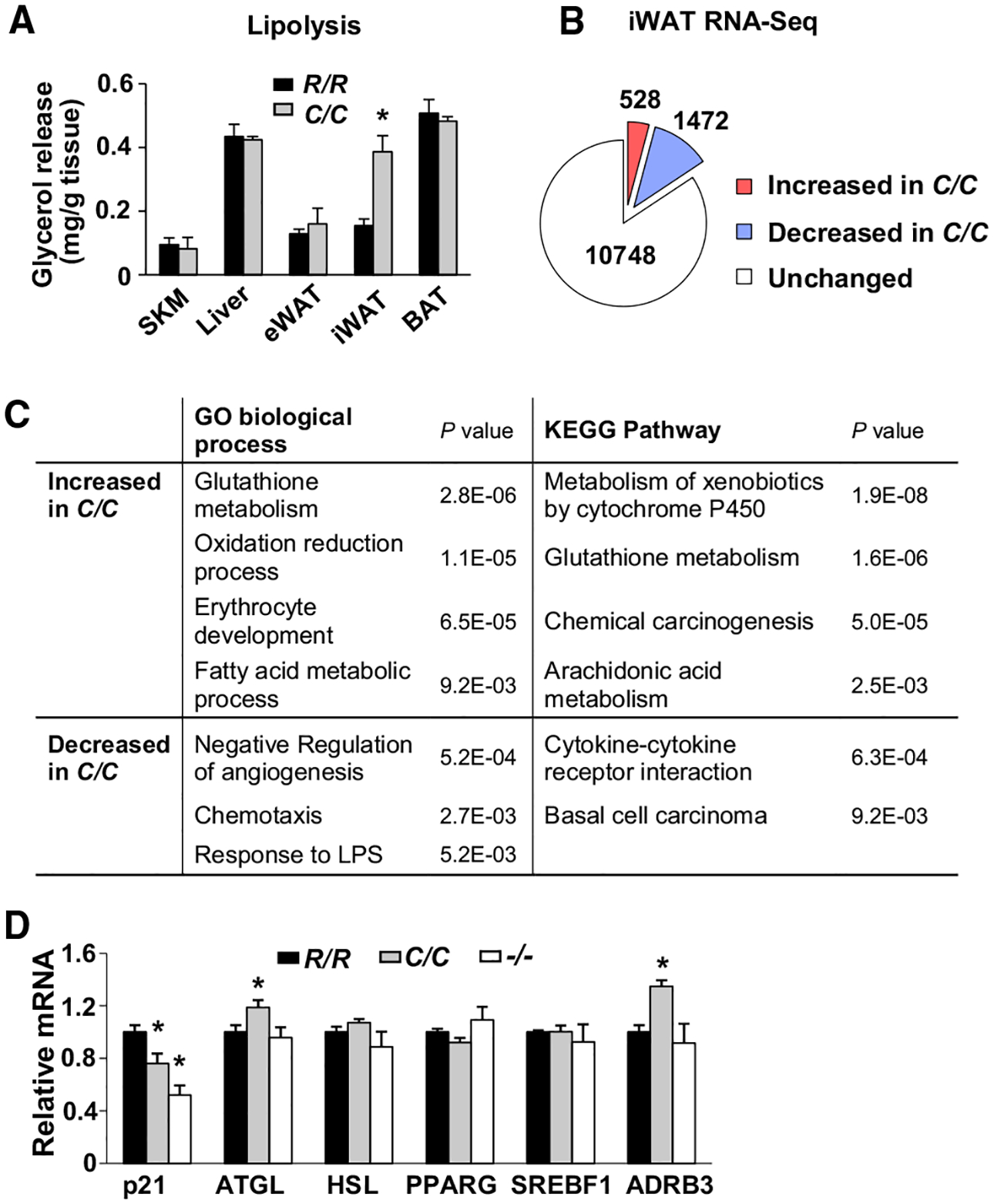
(A) Release of glycerol into culture medium as a measure of basal lipolysis in the indicated tissue explants: skeletal muscle (SKM), liver, eWAT, iWAT, and BAT (n = 3).
(B) Number of genes identified by RNA-seq with significant changes in expression (p < 0.05) in the iWAT of p53178C/C compared with p53178R/R mice (n = 4).
(C) Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of genes with significant expression changes in iWAT (>1.2-fold or <0.8-fold, p < 0.05).
(D) Relative expression levels of p21 and some lipid metabolism genes in iWAT by p53 genotype.
p53 R178 genotypes: wild-type (R/R), homozygous mutant (C/C), and null (−/−). Statistical difference by two-tailed unpaired t test (A) and one-way ANOVA in comparison with the wild-type (R/R) (D). Values are mean ± SEM. *p < 0.05. See also Figure S6.
Of all identified genes expressed in iWAT, 528 (~4%) and 1,472 (~12%) were up- and downregulated, respectively, in p53178C/C compared with wild-type mice, using a significance threshold of p < 0.05 (Figure 4B). Functional pathway analyses of the RNA-seq data showed upregulation of genes involved in fatty acid metabolism as well as arachidonic acid and cytochrome P450 pathways in the iWAT of p53178C/C mice (Figure 4C). In particular, the upregulation of genes involved in biosynthesis of 12,13-diHOME was in agreement with its elevated plasma levels in p53178C/C mice (Figure S6A). Because the human equivalent of the mouse p53 R178 residue mediates DNA binding cooperativity, a mutation at this amino acid would be expected to show reduced p53 target gene transactivation, evidenced by decreased expression of the prototypical p53 target gene CDKN1A (p21) (Figure 4D; Schlereth et al., 2010). In contrast, the mRNA expression level of a key lipolysis enzyme, adipose tissue triglyceride lipase (ATGL), was significantly increased, consistent with the higher lipolytic activity of p53178C/C iWAT, whereas another important lipolysis enzyme, hormone-sensitive lipase (HSL), and the adipocyte transcription factors PPARG and SREBF were relatively unchanged (Figure 4D). Notably, the expression of b3-adrenergic receptor (ADRB3), which is associated with obesity and promotes lipolysis by activating protein kinase A and phosphorylating HSL (Clément et al., 1995; Cypess et al., 2015; Widén et al., 1995; Zechner et al., 2012), was markedly increased in p53178C/C mouse adipose tissue. These data indicate that the hydrolysis of triglycerides to free fatty acids and their subsequent modification are upregulated by p53 R178C in iWAT.
ChIP-Seq Analysis Identifies ADRB3 as a p53 Target Gene and Potential Mediator of the p53178C/C Fat Metabolism Phenotype
To further understand the mechanism underlying the gene expression changes in p53178C/C mice, we performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) to create a genomic profile of wild-type and mutant p53 binding. Because of the difficulties associated with handling low levels of endogenous p53 in tissues, we utilized a homogeneous primary culture of adipocytes differentiated from the stromal vascular fraction (SVF) of iWAT tissue. Adipocytes differentiated from the p53−/− SVF were also utilized for ChIP-seq to screen out non-specific p53 binding peaks. Compared with wild-type cells, immunoblotting of p53178C/C SVF adipocytes showed higher levels of mutant p53 but lower levels of p21, consistent with its diminished transcriptional activity and increased stabilization, possibly because of loss of its negative feedback regulation (Figure 5A). In contrast to the 6,094 high-confidence (false discovery rate [FDR] < 1E – 10) p53 binding sites identified in wild-type adipocytes by ChIP-seq analysis, only 794 such sites were observed in p53178C/C adipocytes, the majority (683 sites, 86%) of which were shared with wild-type p53 binding sites (Figure 5B). Because mutated p53 can gain function through association with other transcription factors, we performed a motif search and confirmed that the p53 motif was the most significantly enriched binding sequence for both wild-type and mutant p53 ChIP-seq data (Table S1A; Do et al., 2012; Pfister and Prives, 2017). These analyses indicate that the mutant p53 R178C retains DNA binding specificity, albeit with lower affinity, as suggested previously for other cooperativity mutants of p53 (Schlereth et al., 2010).
Figure 5. Identification of ADRB3 as a Target Gene of p53 R178C Using ChIP-seq Analysis.
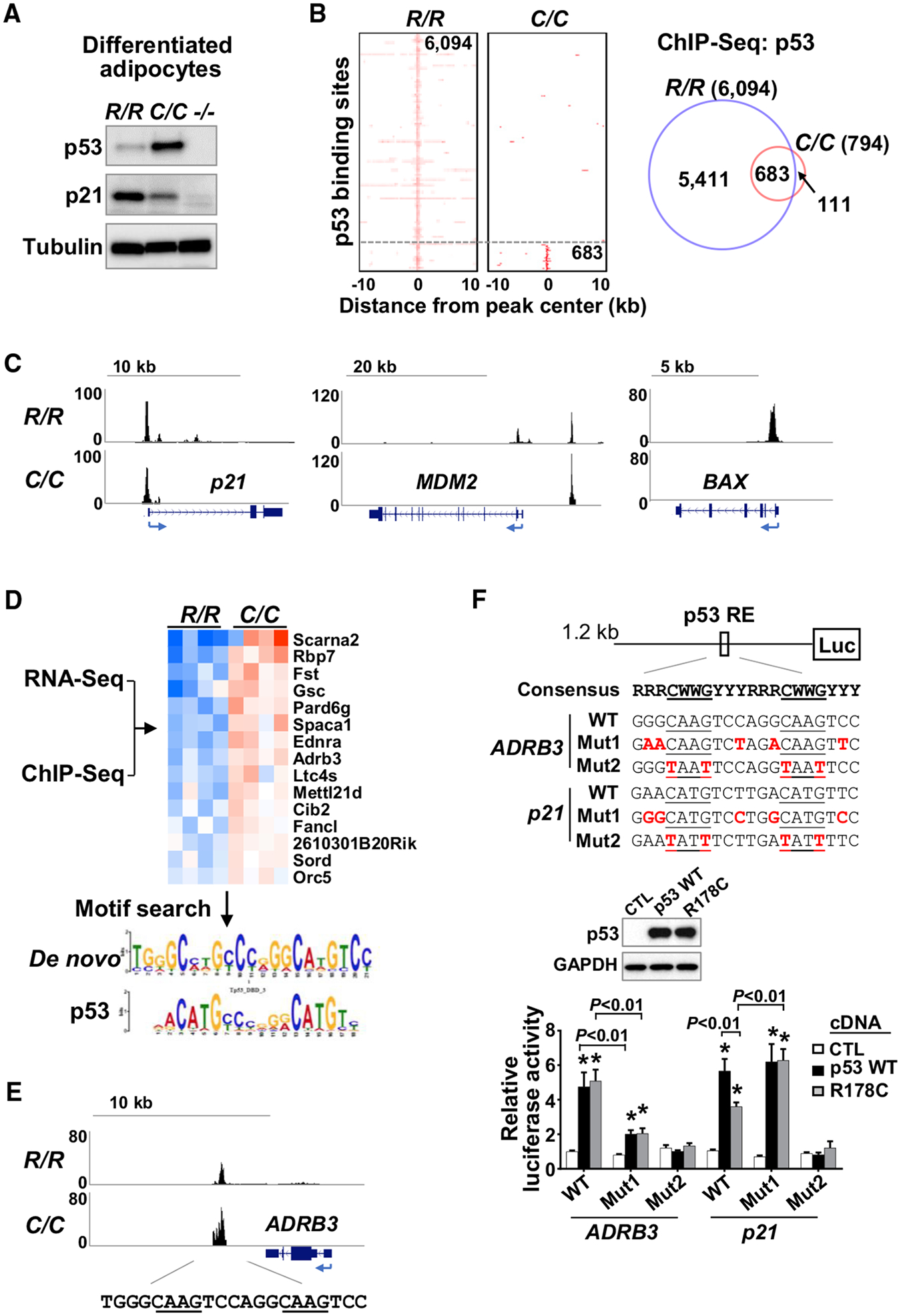
(A) Stromal vascular fractions (SVFs) isolated from iWAT of the indicated p53 genotype mice, differentiated into adipocytes and analyzed by immunoblotting.
(B) Heatmaps and Venn diagram of high-confidence (FDR < 1E 10) p53 binding sites from ChIP-seq analysis of R/R and C/C differentiated adipocytes described in (A).
(C) Genome browser view of p53 binding peaks on known wild-type p53 target genes.
(D) RNA-seq and ChIP-seq data were integrated to identify genes that are upregulated and bound by p53 R178C. p53-bound genomic sequences were subjected to a transcription factor motif search, which yielded a de novo motif (E value = 2E–44) matching the p53 binding motif (E value = 3E–5).
(E) ChIP-seq profile of the ADRB3 gene region with sequences containing the consensus p53 RE.
(F) Effect of GC-rich sequences in the p53 RE of ADRB3 on transactivation by p53, assessed by luciferase reporter assay. The GC-rich flanking sequences were mutated from GC to AT (Mut 1), and the p53 RE core sequence was also mutated as an additional negative control (Mut 2). The luciferase reporter constructs were co-transfected with wild-type p53, p53 R178C, or a control pcDNA empty vector (CTL) into p53−/− 3T3L1 cells. p53 expression levels were assessed by immunoblotting (center panel), and luciferase activity levels of the respective constructs were measured (n = 4).
p53 R178 genotypes: wild-type (R/R), homozygous mutant (C/C), and null (−/−). Statistical difference by two-tailed unpaired t test in comparison with the vector control. Values are mean ± SEM. *p < 0.05. See also Table S1.
Although hotspot mutations of p53 in its DNA binding domain can abolish recognition of p53 binding sequences, we observed a range of binding signals retained by p53 R178C (Figure 5C). Reflecting its partial transactivation of p21, p53 R178C binding to the p21 genomic region appeared to be reduced compared with that of the wild-type protein. It also bound to only one of the two genomic binding sites of MDM2 compared with wild-type p53, whereas there was complete loss of binding to BAX (Figure 5C). Gene Ontology (GO) analysis showed that p53 R178C retains binding to a subset of genomic sites that regulate diverse activities under basal conditions beyond its reported loss of apoptotic activity (Table S1B; Schlereth et al., 2010). Because the increased lipolytic activity of p53178C/C suggested a “gain of function,” we looked for genes induced by mutant p53 in the iWAT RNA-seq data (>1.2-fold, p < 0.05) and integrated them with the p53 R178C ChIP-seq data, resulting in identification of 15 upregulated genes that were bound by mutant p53 (Figure 5D). Performing a de novo motif search on these genes yielded the p53 binding sequence as the only significantly enriched transcription factor motif, confirming a direct interaction with the mutant p53 (Figure 5D). Among these upregulated genes, ADRB3 stood out as a potential mediator of the adipocyte lipolysis phenotype, possessing a conserved p53 response element (RE) proximal to the 3′ region of the gene (Figure 5E).
Interestingly, the de novo motif analysis of the 15 p53 R178C upregulated genes revealed that the mutant protein preferentially binds to the p53 RE, with its core (CWWG) flanked by GC-rich sequences (5′ PuGG and 3′ PyCC), as reported for other cooperative binding mutants of p53 (Figure 5D; Ciribilli et al., 2013; Schlereth et al., 2013). Because the p53 RE of ADRB3 also showed higher GC content compared with p21, we tested whether it was required for more effective transactivation by the mutant p53 with loss of cooperative binding (Figure 5F). Indeed, a luciferase reporter construct with the ADRB3 p53 RE flanking sequences mutated from GC to AT showed marked loss of transactivation by both wild-type and mutant p53 protein, indicating their critical importance for p53 interaction with its target gene (Figure 5F). In contrast, increasing the GC content of the p53 RE of p21 resulted in increased transactivation by p53 R178C to a level comparable with that of wild-type p53, further demonstrating that the flanking GC sequences may obviate the need for cooperative p53 binding to its target genes, as suggested previously (Figure 5F; Jordan et al., 2012).
p53 R178C Regulates ADRB3 and Lipolysis in Adipocytes
Given the direct interaction between mutant p53 and the ADRB3 gene, we set out to clarify its functional significance. The increased expression of ADRB3 in the iWAT of p53178C/C mice relative to p53178R/R and p53−/− mice correlated with the levels of basal p53 and phosphorylated HSL, known to be activated by protein kinase A (PKA) in β-adrenergic receptor-stimulated lipolysis (Figures 6A and 6B; Zechner et al., 2012). To confirm that mutant p53 regulates this pathway, SVF cells isolated from p53178C/C iWAT were stably transduced with lentiviral shRNA to knock down p53 and then differentiated into adipocytes. Both non-specific (NS) and p53 shRNA-transduced cells showed similar degrees of adipocyte differentiation, as assessed by oil red O cell staining and HSL expression levels (Figures 6C, 6D, and S6B). The depletion of mutant p53 specifically decreased ADRB3 mRNA and protein levels in association with decreased PKA activity, HSL phosphorylation and release of glycerol (Figures 6C–6E). We controlled for potential shRNA off-target effects by rescuing these changes with re-expression of p53 R178C and further demonstrating that the human homolog p53 R181C can also confer HSL phosphorylation and ADRB3 mRNA expression activities in p53−/− SVF-derived adipocytes (Figures S6C and S6D).
Figure 6. p53 R178C Promotes Lipolysis via ADRB3 Signaling.
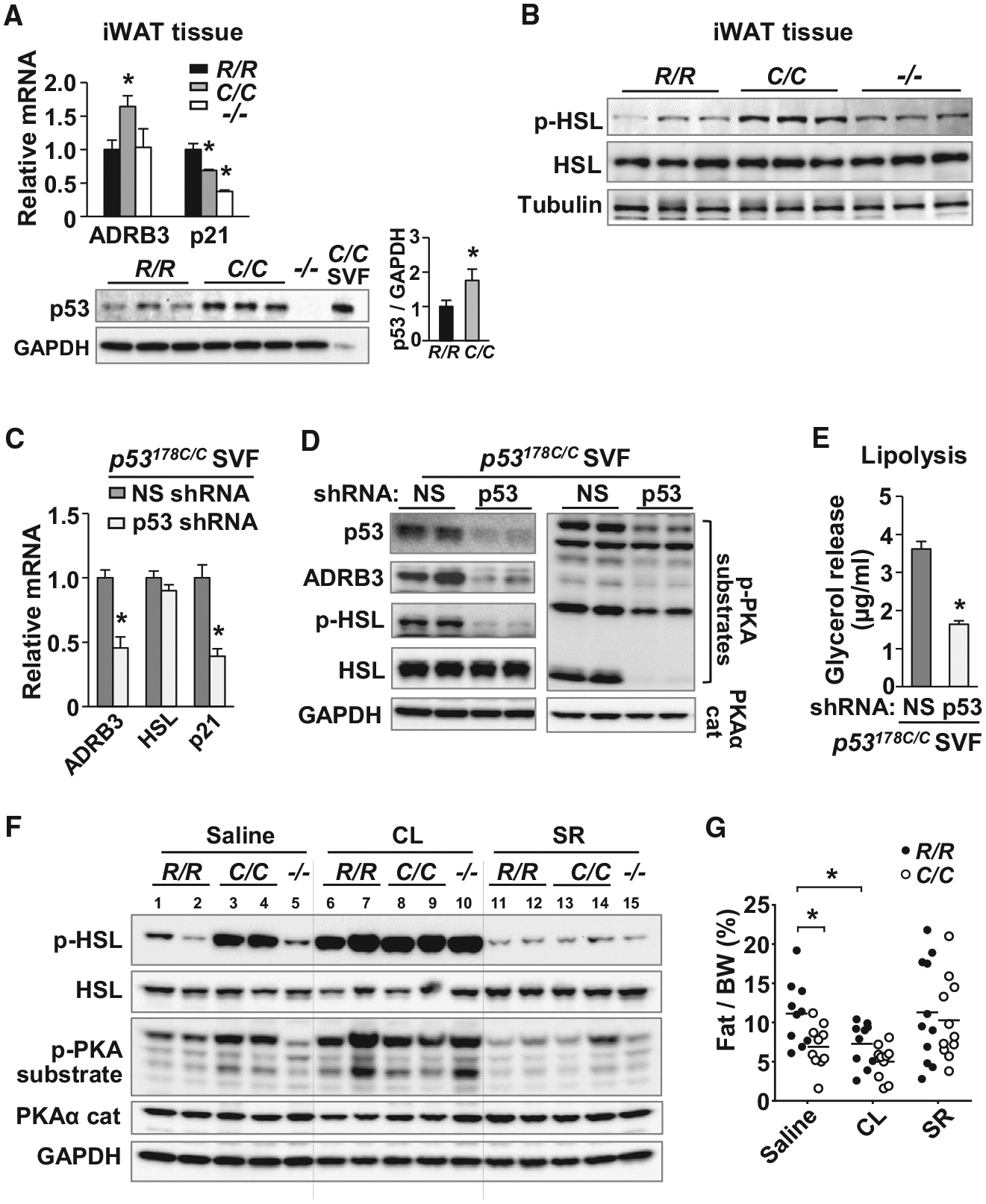
(A) ADRB3 and p21 mRNA levels measured by RT-PCR (top) and p53 protein expression evaluated by immunoblotting in iWAT of the indicated p53 genotype (n = 4). Adipocytes differentiated from p53178C/C SVF cells are shown as a positive control. p53 protein levels were quantified relative to GAPDH by densitometric scanning.
(B) Immunoblots of total and phosphorylated HSL (Ser-660, p-HSL) in iWAT (n = 3).
(C) SVFs isolated from iWAT of p53178C/C mice were transduced with a non-specific (NS) or p53 shRNA lentivirus, differentiated into adipocytes, and analyzed by RT-PCR (n = 3).
(D) The C/C adipocytes described in (C) were immunoblotted with antibodies, including those against phospho-PKA substrates (RRXS/T motif) and the PKAα catalytic subunit. Shown is one representative of 4 experiments with 3 biological replicates.
(E) Glycerol release into culture medium by C/C adipocytes transduced with shRNA as an in vitro measure of lipolysis activity (n = 4).
(F) Immunoblot of iWAT samples from the indicated p53 genotype mice after control saline, ADRB3 agonist CL316243 (CL), or ADRB3 antagonist SR59230A (SR) injection.
(G) Effect of daily injections of control saline, CL, or SR for 5 days on body fat composition of mice (~15 weeks old), measured by NMR analyzer (n = 10–11).
p53 R178 genotypes: wild-type (R/R), homozygous mutant (C/C), and null (−/−). Statistical difference by one-way ANOVA (A) and two-tailed unpaired t test (B, E, and G). Values are mean ± SEM. *p < 0.05. See also Figure S6.
To demonstrate the role of ADRB3 in mutant p53-induced lipolysis in vivo, we pharmacologically modulated ADRB3 signaling in wild-type and p53178C/C mice. Remarkably, the increased levels of PKA activity and phosphorylated HSL associated with the mutant p53 was reduced to wild-type levels by treatment with the ADRB3 antagonist SR-59230A (Figure 6F). In contrast, the low basal levels of phosphorylated HSL and PKA substrate in wild-type p53 iWAT were increased to p53178C/C levels by treatment with the ADRB3 agonist CL-316243 (Figure 6F). These molecular alterations in the mediators of lipolysis were accompanied by corresponding changes in fat tissue body composition. Daily injections of the antagonist SR-59230A for 5 days reverted the fat tissue phenotype in p53178C/C mice to that of the wild-type, whereas treatment with the agonist CL-316243 reproduced the lower fat composition of the p53 mutant state in wild-type mice (Figure 6G). Taken together, these data demonstrated that p53 R178C-regulated ADRB3 expression plays an important role in activating the lipolysis signaling pathway of white adipose tissue and in mediating the lower body fat composition of p53178C/C mice.
DISCUSSION
Because physiologic characteristics can be difficult to discern under heterozygous states in humans, we have created a p53 R178C knockin mouse to explore its role in metabolism. In our current study, we have demonstrated that this mouse homolog of the human TP53 R181C LFS mutation can confer a lipolytic activity that likely contributes to its low body fat phenotype. As observed in TP53 R181C LFS patients, p53 R178C knockin mice displayed a mild cancer phenotype, even in the homozygous mutant state, with a longer median survival time than that of hotspot LFS mouse models (Lang et al., 2004; Olive et al., 2004). Nonetheless, p53178C/C mice showed a unique metabolic phenotype of decreased white adipose tissue mass with evidence of increased fatty acid mobilization by lipolysis that has not been observed in p53−/−or other LFS models, such as p53172H/H mice (Park et al., 2009; Wang et al., 2013). Most genome-wide p53 target gene studies have been performed with cancer cell lines under stimulated conditions (Fischer, 2017). Here we examined the adipose tissue and cells relevant to the phenotype under basal conditions to demonstrate that p53 R178C retains the ability to transactivate ADRB3 and mediate lipolysis.
The mobilization of fatty acid stores by lipolysis observed in p53178C/C mice is notable because another LFS mouse model with the p53 R172H knockin mutation has been shown to have increased mitochondrial β-oxidation, suggesting that concerted promotion of oxidative metabolism by p53 occurs through more than one specific gene or pathway (Wang et al., 2013). As suggested previously, germline p53 mutations could be a double-edged sword from an evolutionary perspective (Wang et al., 2012). On one hand, mutant p53 promotion of fatty acid metabolism could improve endurance running or thermogenesis, both of which are beneficial for organismal survival, but in transformed cancer cells, increased mitochondrial activity can contribute to greater malignancy and metastatic potential (LeBleu et al., 2014; Vazquez et al., 2013). Although likely challenging to detect in LFS patient-derived cells, as observed in heterozygous p53178R/C mice, it will be important to determine whether there are also alterations in lipid metabolism in humans (for example, using adipocytes derived from homozygous TP53 R181C human iPS cells), but the effects of p53 activity on in vitro cell differentiation may be difficult to control (Krstic et al., 2018).
The dual nature of p53 regulation of metabolism under normal conditions and in cancer cells is further exemplified by the genes and metabolites identified in this study. Although increased plasma levels of 12,13-diHOME in p53178C/C mice have the potential to improve metabolic health, they can also stimulate proliferation of breast cancer cells (Lynes et al., 2017; Markaverich et al., 2005; Stanford et al., 2018). The increased expression of arachidonic acid metabolism genes may also promote breast cancer growth through generation of eicosanoids (Figure S6A; Jiang et al., 2005; Mitra et al., 2011). Consistent with the elevated lipolytic activity, bile acid metabolites were the most highly increased compounds identified in p53178C/C plasma and have been reported to mediate tumor metastasis to lymph nodes (Figure S5B; Lee et al., 2019). There are limited data on the physiological effects of germline LFS mutations, but our p53178C/C mouse model reveals an activity of a relatively common LFS mutation that regulates basal metabolism and may also affect cancer development.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Paul M Hwang (hwangp@mail.nih.gov). Unique reagents generated in this study will be made available under a standard material transfer agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
All mice were maintained and handled in accordance with the NHLBI Animal Care and Use Committee protocol. p53+/− mice (Jackson Laboratory) were of the C57BL6 strain. p53 R178C knockin mice were generated using the conventional embryonic stem (ES) cell-mediated homologous recombination method (Lang et al., 2004; Park et al., 2018). Briefly, a gene-targeting construct containing the CGC (Arg) to TGC (Cys) point mutation in exon 5 of p53 and neomycin resistance gene flanked by loxP sites was electroporated into the V6.5 mouse ES cell line. G418-resistant colonies were genotyped by PCR, and two correctly targeted ES clones were injected into blastocysts collected from C57BL/6 mice for producing chimeric mice. The chimeras were then bred with wild-type C57BL/6 mice for germline transmission of the mutant allele. After crossing with CMV-Cre mice (Jackson Laboratory) to remove the neomycin resistance gene, the knockin mice were backcrossed for 8 generations into the C57BL/6 genetic background. Male mice (9–32 wk old) were used for animal experiments, except for the cancer-free survival studies in which both female (F) and male (M) mice were used (wild-type R/R, 17 F and 13 M; heterozygous R/C, 17 F and 15 M; homozygous C/C, 17 F and 15 M).
Cell culture
Adipose tissue stromal vascular fraction (SVF) was prepared and differentiated as previously described (Ma et al., 2015). Briefly, iWAT was dissected from mice, minced, digested in buffer (123 mM NaCl, 5 mM KCl, 1.3 mM CaCl2, 5 mM glucose, 100 mM HEPES, and 4% BSA) containing collagenase (1 mg/ml) at 37°C for 45 min, filtered through a 100 m nylon screen, and centrifuged at 150x g for 5 min at room temperature. Cell pellets were washed twice, resuspended, and cultured in DMEM medium containing 10% FBS and 1% penicillin/streptomycin with daily media changes. For adipocyte differentiation, the isolated SVF cells were stimulated with 0.5 mM isobutylmethylxanthine, 125 μM indomethacin, 5 μM dexamethasone, 20 nM insulin, 1 nM T3 and 1 μM rosiglitazone for 48 h and subsequently maintained with 20 nM insulin and 1 nM T3 for 3–4 d, both in complete DMEM medium. Mouse embryonic fibroblasts (MEF) were prepared from 13.5 d embryos and cultured in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin.
p53 null (p53−/−) preadipocyte 3T3-L1 cell line was generated using CRISPR-Cas9 technique (Ran et al., 2013). Cells transfected with mouse p53 CRISPR/Cas9 KO plasmid (Santa Cruz, sc-423509) were subjected to serial dilution for isolating single clones and subsequently treated with 5 μM nutlin-3 (Sigma) for selection. p53 null clones were selected by p53 western blot and confirmed with genomic DNA sequencing.
METHOD DETAILS
Mouse phenotyping studies
For the cancer-free survival study, the mice were euthanized if any external mass exceeded 2 cm in its largest dimension or when the mouse met moribund criteria. Necropsies and histopathologic diagnoses were performed by qualified veterinary pathologists (Division of Veterinary Resources, NIH). To induce p53 in vivo, mice were exposed to 5 Gy total body γ-irradiation (TBI) using a Gammacell 40 (K2K 1 × 8; MDS Nordion) (Chen et al., 2004) or injected with doxorubicin (20 mg/kg, i.p.) 6 or 18 h, respectively, prior to tissue harvest.
Metabolic phenotyping was performed in male mice to control for gender dimorphism and due to resource limitations. Body composition of non-anesthetized mice was measured using the Minispec NMR analyzer (Bruker). Energy expenditure, food intake, and activity under room temperature (22°C) or thermoneutral condition (29.5°C) were measured using the Open Circuit Calorimetry system (CLAMS, Columbus Instruments). For the glucose tolerance test (GTT) and insulin tolerance test (ITT), mice were fasted for 6 h for after which glucose (1.5 mg/g BW) or insulin (0.5 mU/g BW) was administered by i.p. injection and blood glucose levels were measured after injection using a glucometer (AlphaTrak2, Zoetis) over a 120 min period. Treadmill exercise testing was performed as described previously (Park et al., 2009). For thermogenesis testing, mice were individually caged and exposed to 4°C for 5 h during which hourly core body temperatures were measured per rectum using a mouse thermometer probe (THM-100, Indus Instruments).
For ADRB3 pharmacologic studies, iWAT tissue samples were collected from mice 20 min after injection with control saline or CL316243 (1 mg/kg BW, i.p., Sigma-Aldrich), or 1.5 h after SR59230A (3 mg/kg BW, i.p., Sigma-Aldrich). Longer term effect of these agents on body composition was measured 5 d after daily injections of saline, CL316243, or SR59230A at the same doses noted above.
Southern blotting
Mouse embryonic fibroblasts (MEF) genomic DNA was digested with EcoRI and analyzed by Southern blotting. The genomic blot was hybridized with 32P random primed probe (Exon 1 region, Figure S1; Table S2) in Ultrahyb solution (Ambion) at 42°C overnight. The blot was washed with 0.3% SSC + 0.1% SDS at 60°C for 1 h followed by 0.1% SSC + 0.1% SDS at room temperature for 1 h and exposed to X-ray film at −80°C.
Histology and tissue biochemistry
Tissues were fixed in 4% formaldehyde, paraffin-embedded, sectioned, and hematoxylin and eosin stained. Differentiated adipocytes were fixed in 4% formaldehyde, stained with Oil Red O solution (American MasterTech) for 20 min, and washed in PBS. The stain was eluted with 100% isopropanol and quantified by measuring A490 nm. iWAT tissue norepinephrine (NE) levels were measured using a noradrenaline ELISA Kit (AVIVA systems biology).
Lipid analysis and metabolomic profiling
Blood from 10 wk old male mice were collected in EDTA tubes and the resulting plasma samples were frozen in liquid nitrogen. Total cholesterol and triglycerides levels were measured by the NCI Pathology/Histology Laboratory (Frederick, MD). Plasma levels of non-esterified fatty acids (NEFA) were measured using a NEFA kit (Wako). Metabolomic profiling was performed by Metabolon (Durham, NC), and the median value-normalized data were analyzed in MetaboAnalyst 3.0.
Lipolysis assay
Freshly isolated tissue or SVF differentiated adipocytes were incubated in DMEM + 1% fatty acid-free BSA for 1 h. Lipolysis rate was determined by measuring the amount of glycerol released into the medium using Free Glycerol Reagent (Sigma) per manufacturer’s protocol. Briefly, 5 μL of culture media were mixed with 200 ul of glycerol reagent, incubated at room temperature for 15 min, and measured at 550 nm with a plate reader.
Mitochondrial respiratory complex activity assays
Mitochondria were isolated from brown adipose tissue using standard techniques as previously described (Frezza et al., 2007). Respiratory complex I and IV enzymatic activities were measured using blue-native (BN) gel and in-gel assays. The mitochondrial complexes were resolved using a Invitrogen NativePAGE 4%–16% Bis-Tris gel according to the manufacturer’s protocol, and the enzymatic activities were visualized by incubating the gel as follows: respiratory complex I–50 mM potassium phosphate (pH 7.0) buffer containing 0.2 mg/ml Nitro BlueTetrazolium (NBT, Sigma-Aldrich) and 0.1 mg/ml NADH (Sigma-Aldrich); and respiratory complex IV–50 mM sodium phosphate (pH 7.2) buffer containing 0.4 mg/ml diaminobenzidine (DAB, Sigma-Aldrich) and 1 mg/ml cytochrome c (Sigma-Aldrich).
Cell cycle and cellular metabolic assays
CD4+ T cells were isolated from mouse spleen using CD4 MicroBeads (Miltenyi) according to the manufacturer’s protocol. To measure apoptosis, T cells were stimulated with anti-CD3/anti-CD28 antibodies (eBioscience) for 3.5 d, stained with Annexin V (BD Phar-Mingen), and the apoptotic cells were analyzed by flow cytometry (BD LSR II). For the proliferation assay, splenic CD4+ T cells were stained with 5 μM CellTrace Violet reagent (Thermo Fisher Scientific) prior to stimulation with anti-CD3/anti-CD28 and successive generations of live cells were analyzed by flow cytometry. Proliferation was quantified using cell populations that underwent more than 5 divisions normalized to total live cells. Senescence in MEFs was quantified by staining for b-galactosidase activity as previously described (Senescence β-Galactosidase Staining Kit, #9860, Cell Signaling Technology) (Park et al., 2018). Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in MEFs were measured by Seahorse XF 24 Analyzer (Agilent) as previously described (Kang et al., 2014).
Antibodies and immunoblotting
Antibodies used in this study were as follows: ADRB3 and GAPDH (Thermo Fisher Scientific); α-tubulin (Sigma-Aldrich); p21 (CDKN1A, Millipore Sigma); PKAα cat (A-2, Santa Cruz Biotechnology); HSL, phospho-HSL (Ser660), phospho-PKA Substrate (RRXS*/T*) and p53 (1C12) (Cell Signaling Technology); TFAM, Total OXPHOS Rodent Antibody Cocktail and UCP1 (Abcam).
Protein samples were solubilized in cold RIPA buffer supplemented with protease/phosphatase inhibitors (Roche), resolved by Tris-glycine SDS-PAGE, and transferred to Immobilon-P membrane (Millipore) for standard ECL immunoblotting. For p53 immunoblotting in iWAT tissue, endogenous IgG in the tissue lysate was depleted with Dynabeads Protein G (Thermo Fisher Scientific) prior to gel electrophoresis as previously described (Park et al., 2018).
Lentiviruses and cell transduction
Plasmids containing the sequences of non-specific shRNA and mouse p53 shRNA (Open Biosystems, clone # TRCN0000012361) were used to prepare lentivirus according to manufacturer’s protocol (Sigma-Aldrich). The p53 R178C and R181C mutations were introduced into mouse or human p53 cDNA, respectively, using QuickChange II site-directed mutagenesis kit (Agilent technologies), and the human p53 R175H cDNA has previously been described (Wang et al., 2017). Mutations were confirmed by sequencing, and the mutant p53 cDNAs were subcloned into pLEX-MCS plasmid (OpenBiosystems) for lentivirus preparation. The lentivirus and polybrene (6 μg/ml) were added to the SVF cells, and the transduced cells were grown for 2 d followed puromycin (2.5 μg/ml) selection. For transient p53 expression, cells were transduced with the p53 cDNA containing lentivirus and grown for 2 d prior to use.
Luciferase reporter assay
The ~1.2 -kb p21 and ADRB3 genomic fragments containing the p53 binding site were cloned into the pGL4.10-luciferase vector (Promega) using the In-Fusion Cloning kit (Takara). Mutations were introduced into these constructs using the QuickChange II kit (Agilent technologies) (see Table S2 for primer sequences). Luciferase activity was measured in p53−/− 3T3 L1 cells 24 h after cotransfection with wild-type p53, p53 R178C, or empty vector pcDNA and pGL4.74 plasmid containing the TK promoter and Renilla luciferase as a transfection efficiency control (Dual -Luciferase Reporter Assay System, Promega).
Gene expression analysis
Total tissue RNA was purified using QIAzol Lysis Reagent and the RNeasy kit (QIAGEN), and mRNA was isolated using the Dynabead mRNA Purification Kit (Thermo Fisher Scientific). For RT-PCR, cDNA was synthesized using Superscript IV (Thermo Fisher Scientific), and real-time PCR was performed using SYBR green fluorescence on a HT7900 thermal cycler (ABI) as previously described (Patino et al., 2006). The average cycle threshold (Ct) of the respective gene was normalized to housekeeping gene TIF (EIF3F) (see Table S2 for primer sequences). For RNA-Seq, mRNAs purified from 2 μg of iWAT total RNA were used to synthesize double-stranded cDNAs using the SuperScript Double-Stranded cDNA Synthesis Kit (Thermo Fisher Scientific).
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed as previously described with some modifications (Lee et al., 2017). Briefly, cells were treated with 1.5% formaldehyde to cross-link at room temperature for 10 min, and their nuclei were isolated by incubating in lysis buffer (20 mM Tris pH 8.0, 85 mM KCl, 0.5% NP-40, supplemented with protease inhibitors) on ice for 10 min followed by centrifugation at 3000x g for 5 min. The nuclear pellet was resuspended in RIPA buffer (10 mM Tris-Cl, pH 7.5, 1mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, supplemented with protease inhibitors) and sonicated at 20% amplitude for 15 s, repeated 20 times. The sheared nuclear extracts were immunoprecipitated with anti-p53 (FL393, Santa Cruz #sc-6243) or control rabbit IgG (Santa Cruz #sc-2027) antibodies (pre-bound with Dynabeads Protein A, Thermo Fisher Scientific) at 4°C overnight. Antibody-bound DNA fragments were eluted in 100 μL of elution buffer (1% SDS, 0.1 M NaHCO3, supplemented with proteinase K to a final concentration of 100 μg/ml) at 65°C for 5 h and purified by QIAquick PCR Purification Kit (QIAGEN).
Sequencing and computational analysis
Sequencing libraries were constructed using NEBNext Ultra II DNA Library Prep Kit forIllumina (NEB). All ChIP-Seq and RNA-Seq samples were sequenced on the Illumina HiSeq 2500. Sequencing reads were aligned to mouse reference genome mm9 using Bowtie 2. To identify p53 binding regions, we used the ‘SICER’ method with a window and gap sizes of 50 bp. To eliminate non-specific binding of p53 antibody, we compared p53 ChIP-Seq data of p53−/− cells with that of p53178R/R and p53178C/C cells, and a FDR of < 1E 10 was used to find high-confidence p53-binding regions. Heatmaps of p53 binding sites were generated with Python 2.7 scripts and graphics package in R with 50 bp resolution. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) analyses were performed with DAVID Bioinformatics Resources (https://david.ncifcrf.gov/; Huang et al., 2009). Motif search (Table S1A) on the ChIP-Seq data was performed by SeqPos motif tool in Galaxy Cistrome as previously described (Lee et al., 2017). Motif search with a limited number of sequences (< 50, Figure 5D) was done with MEME and TOTOM (http://meme-suite.org/tools/meme).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise specified, data were analyzed using the two-tailed unpaired t test. For data containing more than 2 experimental groups, one-way ANOVA with Tukey’s posttest analysis was performed using GraphPad Prism (v7.02). Principal component analysis, ANOVA analysis, and heatmap clustering of metabolomic data were performed using MetaboAnalyst 3.0 (https://www.metaboanalyst.ca/) (Xia et al., 2015).
DATA AND CODE AVAILABILITY
The accession number for the RNA-seq and ChIP-seq datasets reported in this paper is NCBI Gene Expression Omnibus: GSE132715.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ADRB3 | Thermo Fisher Scientific | Cat# PA5–50914; RRID:AB_2636362 |
| α-Tubulin | Sigma-Aldrich | Cat# T5168; RRID:AB_477579 |
| CD3e | eBioscience | Cat# 16–0031–86; RRID:AB_468849 |
| CD28 | eBioscience | Cat# 16–0281–86; RRID:AB_468923 |
| GAPDH | Thermo Fisher Scientific | Cat# AM4300; RRID:AB_2536381 |
| HSL | Cell Signaling Technology | Cat# 4107; RRID:AB_2296900 |
| phospho-HSL (Ser660) | Cell Signaling Technology | Cat# 4126; RRID:AB_490997 |
| phospho-PKA substrate (RRXS*/T*) | Cell Signaling Technology | Cat# 9624; RRID:AB_331817 |
| PKAα cat (A-2) | Santa Cruz Biotechnology | Cat# sc-28315; RRID:AB_628136 |
| p53 (1C12) | Cell Signaling Technology | Cat# 2524; RRID:AB_331743 |
| p53 (FL393) | Santa Cruz Biotechnology | Cat# sc-6243; RRID:AB_653753 |
| p21 (CDKN1A) | Millipore Sigma | Cat# OP76 |
| TFAM | Abcam | Cat# ab131607; RRID:AB_11154693 |
| Total OXPHOS Rodent Antibody Cocktail | Abcam | Cat# ab110413; RRID:AB_2629281 |
| UCP1 | Abcam | Cat# ab10983; RRID:AB_2629281 |
| Control rabbit IgG | Santa Cruz Biotechnology | Cat# sc-2027; RRID: AB_737197 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CL316243 | Sigma-Aldrich | Cat# C5976 |
| Dexamethasone | Sigma-Aldrich | Cat# D1881 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat# 10001D |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat# 10007D |
| Indomethacin | Sigma-Aldrich | Cat# I7378 |
| Insulin | Sigma-Aldrich | Cat# I5500 |
| Isobutylmethylxanthine | Sigma-Aldrich | Cat# I7018 |
| Oil Red O stain | American MasterTech | Cat# STORO100 |
| SR59230A | Sigma-Aldrich | Cat# S8688 |
| T3 | Sigma-Aldrich | Cat# T2877 |
| Ultrahyb solution | Ambion | N/A |
| Critical Commercial Assays | ||
| Free Glycerol Reagent | Sigma | Cat# F6428 |
| NEFA kit | Wako | Cat# 995–34791 |
| RNeasy kit | Qiagen | Cat# 74104 |
| Dual -Luciferase Reporter Assay System | Promega | Cat# E1960 |
| QuikChange II Site-Directed Mutagenesis Kit | Agilent | Cat# 200523 |
| In-Fusion HD Cloning Plus | Takara | Cat# 638920 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7645 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling Technology | Cat# 9860 |
| Deposited Data | ||
| RNA-Seq | This paper | GSE132715 |
| p53 ChIP-Seq | This paper | GSE132715 |
| Experimental Models: Cell Lines | ||
| 3T3-L1 | ATCC | ATCC CL-173 |
| 3T3-L1 p53−/− | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL6/J | Jackson Laboratory | JAX 000664 |
| Mouse: B6.129S2-Trp53tm1Tyj/J | Jackson Laboratory | JAX 002101 |
| Mouse: C57BL6J < i > Trp53-R178C KI | This paper | N/A |
| Oligonucleotides | ||
| See Table S2 | This paper | N/A |
| Recombinant DNA | ||
| Mouse p53 shRNA lentiviral plamid | OpenBiosystems | TRCN0000012361 |
| Lentiviral Packaging Mix | Sigma-Aldrich | SHP001 |
| pcDNA-p53R178C | This paper | N/A |
| pLex-p53R178C | This paper | N/A |
| Mouse p53 CRISPR/Cas9 KO plasmid | Santa Cruz Biotechnology | Cat# sc-423059 |
| Software and Algorithms | ||
| GraphPad Prism 7.0 | GraphPad Software | N/A |
| MetaboAnalyst 3.0 | MetaboAnalyst | N/A |
| MEME/TOMTOM | MEME Suite | N/A |
| DAVID Bioinformatics Resources | National Institutes of Health | N/A |
| SICER | https://home.gwu.edu/~wpeng/Software.htm | N/A |
| SeqPos motif tool | Galaxy Cistrome | N/A |
| R | https://www.r-project.org/ | N/A |
| Other | ||
| Comprehensive Laboratory Animal Monitoring System (CLAMS) | Columbus Instruments | N/A |
| Minispec NMR analyzer | Bruker | N/A |
Highlights.
Mouse homolog of a TP53 germline mutation reveals a metabolic phenotype
Mutant p53 R178C differentially retains wild-type p53 activity
ChIP-seq analysis identifies adrenergic receptor ADRB3 as a p53 target gene
p53 R178C promotes lipolysis in adipocytes by increased ADRB3 signaling
ACKNOWLEDGMENTS
We thank Danielle A. Springer, Michele D. Allen (NHLBI Murine Phenotyping Core), Zu-Xi Yu (NHLBI Pathology Core), William M. Kamp, Somin Kwon, Ryoon Hwan Kim, Deborah Simon, Komudi Singh, and Haiming Cao for helpful advice and assistance during the course of this study. This work was supported by the National Heart, Lung, and Blood Institute Division of Intramural Research (HL005101-14 to P.M.H.). J.K. was supported by research funding from Chungnam National University, South Korea.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.074.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Achatz MI, and Zambetti GP (2016). The Inherited p53 Mutation in the Brazilian Population. Cold Spring Harb. Perspect. Med 6, a026195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkers CR, Maddocks OD, Cheung EC, Mor I, and Vousden KH (2013). Metabolic regulation by p53 family members. Cell Metab. 18, 617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, and Attardi LD (2014). Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, and Olivier M (2016). TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat 37, 865–876. [DOI] [PubMed] [Google Scholar]
- Chen J, Lipovsky K, Ellison FM, Calado RT, and Young NS (2004). Bystander destruction of hematopoietic progenitor and stem cells in a mouse model of infusion-induced bone marrow failure. Blood 104, 1671–1678. [DOI] [PubMed] [Google Scholar]
- Ciribilli Y, Monti P, Bisio A, Nguyen HT, Ethayathulla AS, Ramos A, Foggetti G, Menichini P, Menendez D, Resnick MA, et al. (2013). Transactivation specificity is conserved among p53 family proteins and depends on a response element sequence code. Nucleic Acids Res. 41, 8637–8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clément K, Vaisse C, Manning BS, Basdevant A, Guy-Grand B, Ruiz J, Silver KD, Shuldiner AR, Froguel P, and Strosberg AD (1995). Genetic variation in the beta 3-adrenergic receptor and an increased capacity to gain weight in patients with morbid obesity. N. Engl. J. Med 333, 352–354. [DOI] [PubMed] [Google Scholar]
- Currie E, Schulze A, Zechner R, Walther TC, and Farese RV Jr. (2013). Cellular fatty acid metabolism and cancer. Cell Metab. 18, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elía E, Kessler SH, Kahn PA, English J, Chatman K, Trauger SA, Doria A, and Kolodny GM (2015). Activation of human brown adipose tissue by a b3-adrenergic receptor agonist. Cell Metab. 21, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do PM, Varanasi L, Fan S, Li C, Kubacka I, Newman V, Chauhan K, Daniels SR, Boccetta M, Garrett MR, et al. (2012). Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 26, 830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M (2017). Census and evaluation of p53 target genes. Oncogene 36, 3943–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. (2012). Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, and Scorrano L (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc 2, 287–295. [DOI] [PubMed] [Google Scholar]
- Guevara NV, Kim HS, Antonova EI, and Chan L (1999). The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat. Med 5, 335–339. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, Ning YG, Xiao X, Zeldin DC, and Wang DW (2005). Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res. 65, 4707–4715. [DOI] [PubMed] [Google Scholar]
- Jordan JJ, Menendez D, Sharav J, Beno I, Rosenthal K, Resnick MA, and Haran TE (2012). Low-level p53 expression changes transactivation rules and reveals superactivating sequences. Proc. Natl. Acad. Sci. USA 109, 14387–14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JG, Wang PY, and Hwang PM (2014). Cell-based measurements of mitochondrial function in human subjects. Methods Enzymol. 542, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber ER, and Lowe SW (2017). Putting p53 in Context. Cell 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krstic J, Reinisch I, Schupp M, Schulz TJ, and Prokesch A (2018). p53 Functions in Adipose Tissue Metabolism and Homeostasis. Int. J. Mol. Sci 19, E2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung CP, and Murphy ME (2016). The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol 231, R61–R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. (2004). Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872. [DOI] [PubMed] [Google Scholar]
- LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, et al. (2014). PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol 16, 992–1003, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Park YK, Park S, Jang Y, Waring N, Dey A, Ozato K, Lai B, Peng W, and Ge K (2017). Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun 8, 2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Jeong SH, Jang C, Bae H, Kim YH, Park I, Kim SK, and Koh GY (2019). Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 363, 644–649. [DOI] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, and Gu W (2012). Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolas Hamameh S, Renbaum P, Kamal L, Dweik D, Salahat M, Jaraysa T, Abu Rayyan A, Casadei S, Mandell JB, Gulsuner S, et al. (2017). Genomic analysis of inherited breast cancer among Palestinian women: Genetic heterogeneity and a founder mutation in TP53. Int. J. Cancer 141, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynes MD, Leiria LO, Lundh M, Bartelt A, Shamsi F, Huang TL, Takahashi H, Hirshman MF, Schlein C, Lee A, et al. (2017). The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat. Med 23, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Xu L, Alberobello AT, Gavrilova O, Bagattin A, Skarulis M, Liu J, Finkel T, and Mueller E (2015). Celastrol Protects against Obesity and Metabolic Dysfunction through Activation of a HSF1-PGC1a Transcriptional Axis. Cell Metab. 22, 695–708. [DOI] [PubMed] [Google Scholar]
- Markaverich BM, Crowley JR, Alejandro MA, Shoulars K, Casajuna N, Mani S, Reyna A, and Sharp J (2005). Leukotoxin diols from ground corncob bedding disrupt estrous cyclicity in rats and stimulate MCF-7 breast cancer cell proliferation. Environ. Health Perspect 113, 1698–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R, Guo Z, Milani M, Mesaros C, Rodriguez M, Nguyen J, Luo X, Clarke D, Lamba J, Schuetz E, et al. (2011). CYP3A4 mediates growth of estrogen receptor-positive breast cancer cells in part by inducing nuclear translocation of phospho-Stat3 through biosynthesis of (±)-14,15-epoxyeicosatrienoic acid (EET). J. Biol. Chem 286, 17543–17559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, and Jacks T (2004). Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. [DOI] [PubMed] [Google Scholar]
- Park JY, Wang PY, Matsumoto T, Sung HJ, Ma W, Choi JW, Anderson SA, Leary SC, Balaban RS, Kang JG, et al. (2009). p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ. Res 105, 705–712, 11 p following 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Li J, Starost MF, Liu C, Zhuang J, Chen J, Achatz MI, Kang JG, Wang PY, Savage SA, and Hwang PM (2018). Mouse Homolog of the Human TP53 R337H Mutation Reveals Its Role in Tumorigenesis. Cancer Res. 78, 5375–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrales A, and Iwakuma T (2016). p53 as a Regulator of Lipid Metabolism in Cancer. Int. J. Mol. Sci 17, E2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino WD, Kang JG, Matoba S, Mian OY, Gochuico BR, and Hwang PM (2006). Atherosclerotic plaque macrophage transcriptional regulators are expressed in blood and modulated by tristetraprolin. Circ. Res 98, 1282–1289. [DOI] [PubMed] [Google Scholar]
- Pfister NT, and Prives C (2017). Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53. Cold Spring Harb. Perspect. Med 7, a026054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlereth K, Beinoraviciute-Kellner R, Zeitlinger MK, Bretz AC, Sauer M, Charles JP, Vogiatzi F, Leich E, Samans B, Eilers M, et al. (2010). DNA binding cooperativity of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 38, 356–368. [DOI] [PubMed] [Google Scholar]
- Schlereth K, Heyl C, Krampitz AM, Mernberger M, Finkernagel F, Scharfe M, Jarek M, Leich E, Rosenwald A, and Stiewe T (2013). Characterization of the p53 cistrome–DNA binding cooperativity dissects p53’s tumor suppressor functions. PLoS Genet. 9, e1003726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider K, and Garber J (2010). Li-Fraumeni Syndrome In GeneReviews, Pagon RA, et al. , (University of Washington; ). https://www.ncbi.nlm.nih.gov/books/NBK1311/. [Google Scholar]
- Sidransky D, Tokino T, Helzlsouer K, Zehnbauer B, Rausch G, Shelton B, Prestigiacomo L, Vogelstein B, and Davidson N (1992). Inherited p53 gene mutations in breast cancer. Cancer Res. 52, 2984–2986. [PubMed] [Google Scholar]
- Stanford KI, Lynes MD, Takahashi H, Baer LA, Arts PJ, May FJ, Lehnig AC, Middelbeek RJW, Richard JJ, So K, et al. (2018). 12,13-di- HOME: An Exercise-Induced Lipokine that Increases Skeletal Muscle Fatty Acid Uptake. Cell Metab 27, 1111–1120.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev O, Schlereth K, Wanzel M, Braun A, Nieswandt B, Pagenstecher A, Rosenwald A, Elsässer HP, and Stiewe T (2013). p53 DNA binding cooperativity is essential for apoptosis and tumor suppression in vivo. Cell Rep. 3, 1512–1525. [DOI] [PubMed] [Google Scholar]
- Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, and Puigserver P (2013). PGC1a expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 23, 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PY, Zhuang J, and Hwang PM (2012). p53: exercise capacity and metabolism. Curr. Opin. Oncol 24, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PY, Ma W, Park JY, Celi FS, Arena R, Choi JW, Ali QA, Tripodi DJ, Zhuang J, Lago CU, et al. (2013). Increased oxidative metabolism in the Li-Fraumeni syndrome. N. Engl. J. Med 368, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PY, Li J, Walcott FL, Kang JG, Starost MF, Talagala SL, Zhuang J, Park JH, Huffstutler RD, Bryla CM, et al. (2017). Inhibiting mitochondrial respiration prevents cancer in a mouse model of Li-Fraumeni syndrome. J. Clin. Invest 127, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widén E, Lehto M, Kanninen T, Walston J, Shuldiner AR, and Groop LC (1995). Association of a polymorphism in the beta 3-adrenergic-receptor gene with features of the insulin resistance syndrome in Finns. N. Engl. J. Med 333, 348–351. [DOI] [PubMed] [Google Scholar]
- Xia J, Sinelnikov IV, Han B, and Wishart DS (2015). MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res 43, W251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, and Madeo F (2012). FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 15, 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Wang J, Zhao M, Xie TX, Tanaka N, Sano D, Patel AA, Ward AM, Sandulache VC, Jasser SA, et al. (2014). Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 54, 960–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq and ChIP-seq datasets reported in this paper is NCBI Gene Expression Omnibus: GSE132715.