

Abstract
We describe a new electrophotocatalytic strategy that harnesses the power of light and electricity to generate an excited radical anion with a reducing potential of −3.2 V vs SCE, which can be used to activate substrates with very high reduction potentials (Ered ≈ −1.9 to −2.9 V). The resultant aryl radicals can be engaged in various synthetically useful transformations to furnish arylboronate, arylstannane, and biaryl products.
Due to the unique reactivity of open-shell intermediates, the development of catalytic transformations driven by single-electron transfer (SET) has been an area of intense research in organic chemistry.1 In particular, the employment of unconventional means of activation, including photoredox catalysis2 and electrocatalysis,3 has provided unique entry to single-electron reactivities and led to new solutions to challenging synthetic problems that are not readily addressed using existing tools. In the realm of redox organic reactions, both photoredox catalysis and electrochemistry have been successfully applied to a diverse suite of oxidative transformations. However, reductive transformations, particularly those that require highly reducing potentials, remain an underdeveloped area for both of these reaction strategies.2b,3a In principle, electrochemistry can grant access to extreme reducing potentials, and significant contributions in this regard have recently been made.4 Nevertheless, the constant application of such highly biased potentials can lead to uncontrolled reactions due to accumulation of reactive intermediates near the electrode surface as well as compromised chemoselectivity in complex reaction systems. Thus, the development of catalytic strategies that are both highly reducing and also chemoselective remains a major challenge.
To this end, we envisioned a new electrophotocatalytic strategy that merged the power of electrical and photochemical energy5 to realize extremely high reduction potentials under catalyst control. The Lambert group recently described an oxidative electrophotocatalytic system that achieved an extremely high oxidation potential (3.33 V vs SCE).6 This high oxidizing power resulted from the photoexcitation of an open-shell, radical cation intermediate that was generated by electrochemical means. We reasoned that a similar potency should be achievable for reductive chemistry through the generation and photoexcitation of a radical anion intermediate. Indeed, the strongly reducing character of radical anions has long been known.7 Recently, König described systems that undergo sequential photoinduced reduction to form radical anions followed by a second photoexcitation of the resulting radical anions.8 Such a strategy, however, requires that the same catalyst must serve as both a photooxidant and, once reduced, a photoreductant. In addition, the photooxidation step can be problematic in that it necessitates the formation of oxidative byproducts (e.g., Et3N•+), which limits the effective reducing power of the system due to back electron transfer or competing side reactions.2b
We propose a new electrophotocatalytic strategy to access extremely reducing intermediates via tandem cathodic reduction and photoexcitation. The use of electrochemistry circumvents the photooxidation step in the consecutive dual photoredox approach, because the catalyst radical anion can be generated from the neutral precursor without the need for a chemical reductant. For example, cathodic reduction of dicyanoanthracene (DCA; E1/2 = −0.82 V) results in the corresponding radical anion DCA•−, which absorbs visible light and exhibits a strong fluorescence emission (excitation energy E0,0 = 2.38 eV; see Scheme 1).9 The photoexcited DCA (DCA•−*) is estimated to display an exceptionally high reducing potential of −3.2 V (vs SCE),10 which TD-DFT calculations suggest arises from a SOMO-HOMO level inversion featuring a very unstable electronic structure with a half-filled bonding orbital (ψ1) and a filled antibonding orbital (ψ2) (see Figure S3 in Supporting Information). This value is on par with some of the most reducing elemental metals such as Li (E = −3.3 V). Moreover, DCA•−* has been estimated to have a lifetime of 13.5 ns,9 sufficiently long to engage in reductive activation of suitable substrates. Importantly, the tandem electrochemical activation and photoexcitation allows the formation of this highly reducing catalyst in a very low concentration in a controllable fashion, thus providing an avenue to circumvent the chemoselectivity issues frequently associated with strongly reducing conditions using chemical reductants or direct electrolysis. In this Communication, we apply this electrophotocatalytic strategy to the reductive functionalization of a wide range of aryl halides with reduction potentials (Ered) as low as −2.94 V.
Scheme 1.

Reductive Electrophotocatalysis for the Functionalization of Aryl Halides
As a proof of concept, we first confirmed that DCA•− can be generated and is stable under preparative electrochemical conditions (Figure 1). Using a porous carbon cathode and a zinc plate sacrificial anode in an H-type divided cell, the application of a constant cell voltage of 3.2 V (corresponding to a cathodic potential of −1.0 V vs SCE) resulted in the formation of a deep orange solution in the cathodic compartment. The UV−vis absorption spectrum of this solution is identical to that of DCA•− reported in the literature.9
Figure 1.

Electrochemical generation of radical anion.
We next set out to investigate our proposed electrophotocatalytic system in the context of the reductive functionalization of aryl halides. In recent years, the generation of aryl radicals from aryl halides has been shown to be a productive synthetic strategy.11 However, current methods are largely limited to substrates with relatively labile carbon−halogen bonds or low reduction potentials.8,11 For example, aryl chlorides often display high reduction potentials (> −2.0 V) and strong bond dissociation energies (>97 kcal/mol),12 and thus can be difficult to engage in reductive reactions in the single-electron manifold. In addition, electron-rich aryl halides are often recalcitrant to single electron reductions. Here, we show that reductive electrophotocatalysis allows for the engagement of these challenging substrates.
We chose to study the borodechlorination of ethyl 4-chlorobenzoate (1, Ered = −2.04 V vs SCE)12 using DCA as the catalyst and bis(pinacolato)diboron (B2pin2) as the radical acceptor (Table 1). To date, only a few examples are available for the borylation of aryl chlorides under metal-free conditions.13 After optimization, we observed that the application of both blue light and current enabled efficient coupling of 1 and B2pin2 to furnish arylboronate 2 in 88% yield (entry 1). The optimal conditions employed pyridine (20 mol %) as an additive, TBAPF6 as the electrolyte, carbon foam and zinc plate as cathode and anode, respectively, in MeCN with an applied cell voltage of 3.2 V under blue LED irradiation. A set of control experiments revealed that current, light, and DCA are all necessary for reactivity (entries 2−4). Pyridine, which is known to coordinate to B2pin2 and promote the formation of a B-centered persistent radical intermediate,13a,14 was beneficial to reaction efficiency (entry 5).
Table 1.
Reaction Optimizationa
 | ||
|---|---|---|
| entry | variation from “standard conditions” | yield (%) |
| 1 | none | 90 (88) |
| 2 | no applied voltage | <5 |
| 3 | no light irradiation | <5 |
| 4 | no DCA | <5 |
| 5 | no pyridine | 58 |
| 6 | CFL (23 W) instead of blue LED | 70 |
| 7 | Al(+)/C(−) instead of Zn(+)/C(−) | 65 |
| 8 | undivided cell | 37 |
| 9 | AO as a catalyst instead of DCA | 66 |
| 10 | 3 as substrate | 57 (53) |
Cathodic chamber: 1 (0.4 mmol, 1.0 equiv), DCA (5 mol%), B2pin2 (2.0 equiv), pyridine (20 mol%), TBAPF6 (0.15 M), CH3CN (0.1 M); anodic chamber: TBAPF6 (0.15 M), CH3CN (0.1 M); cell voltage (Ucell) = 3.2 V; yields determined by 1H NMR (isolated yield in parentheses).
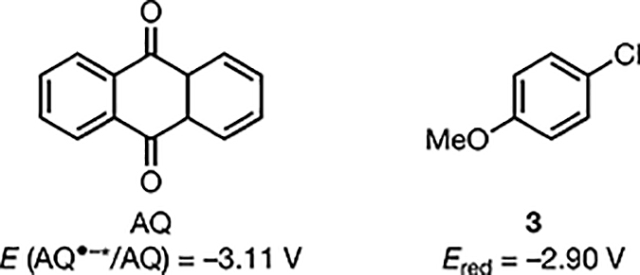
We found that the use of white CFL was also productive (entry 6), albeit with slightly reduced efficiency relative to the blue LEDs. Changing the sacrificial anode from Zn to Al did not significantly affect the reaction yield (entry 7). The reaction, in principle, can be conducted in an undivided cell, which indeed provided the desired product in 37% yield. However, the reaction was prematurely terminated due to the formation of a zinc bridge between the cathode and anode that short-circuited the electrochemical setup (entry 8). Anthraquinone (AQ), which exhibits similar light-induced redox reactivities,9 was also an effective catalyst, albeit with diminished yield (entry 9). Importantly, this reaction was also applicable to the reductive borylation of 4-chloroanisole (3), which has a highly negative reduction potential (Ered = −2.90 V vs SCE)15 (entry 10). This result highlights the extreme reducing power that this catalytic system can provide. Notably, this substrate has not been successfully engaged in high yielding reductive functionalizations using photoredox catalysis.
We have found that this reaction is capable of borylating a wide range of aryl halides. A number of functional groups potentially sensitive to strongly reducing conditions, such as ester (2 and 5), ketone (6), amide and carbamate (7 and 12), thiophene (14), and N-Boc indole16 (15) groups, were well tolerated (see Table 2). Notably, substrate 16 with an acidic C−H bond α to the ester group underwent desired transformation without epimerization of the stereogenic center. These results showcase that, although our catalytic strategy grants access to extremely reducing potentials, it does so under catalyst control with a high level of chemoselectivity that is typically uncommon in reactions promoted by potent reducing metals. In principle, the same types of products could be obtained using Pd-catalyzed borylation. Nevertheless, this metal-catalyzed method17 provided substantially lower yield for substrates bearing multiple Lewis basic coordinating groups (e.g., 16−18).18 When applied to the functionalization of indomethacin methyl ester, a pharmaceutical agent, 35% of the desired borylate was isolated (18).
Table 2.
Scope of Electrophotocatalytic Borylationa
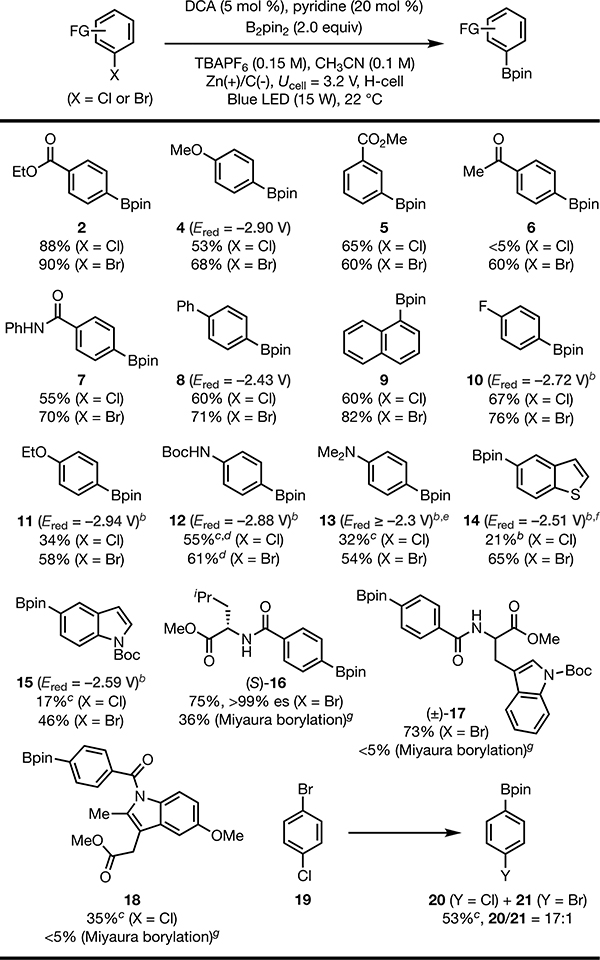 |
Isolated yields are reported. Optimal conditions from Table 1 used.
Calculated Ered values for aryl chlorides (see SI).
10 mol% DCA.
Yield determined by 1H NMR.
Ered could not be accurately determined.
Aryl halide (0.25 mmol, 1 equiv), Pd(dppf)Cl2 (5 mol %), KOAc (3.0 equiv), B2pin2 (1.2 equiv) in DMSO (0.85 mL) under 110 °C for 12 h.
We note that certain aryl chlorides were poorly reactive and returned the majority of the starting material, despite the fact that their reduction potentials suggest that they would be suitable substrates. We reason that in these cases, back electron transfer from intermediate [Ar−Cl•−] to DCA is sufficiently fast to outcompete scission of the C−Cl bond. This hypothesis led us to investigate the corresponding aryl bromides. As expected, the reductive borylation activity was restored. An intramolecular competition experiment revealed that the mesolytic cleavage of a C−Br bond is indeed substantially faster than that of a C−Cl bond, as 1-chloro-4-bromobenzene (19) was selectively transformed to borodebrominated product 20.
Notably, in addition to haloanisoles, several other electron-rich arenes and hetereoarenes (11−15) were found to be suitable substrates. Some of these aryl halides (e.g., 10, 11, 12, 15) display very negative reduction potentials and represent highly challenging substrates for single electron reduction,8 yet were productive under the optimal conditions.
The synthetic utility of this electrophotocatalytic protocol was further expanded to the formation of C−Sn and C−C bonds by employing different radical trapping agents (Table 3). For example, using hexamethylditin as a coupling partner, we developed metal-free stannylation of aryl halides (22−26). Using N-methylpyrrole or 1,4-difluorobenzene, C−H arylation products (27−30) were isolated in moderate to high yields.
Table 3.
Diverse Functionalization of Electrophotocatalytically Generated Aryl Radicalsa
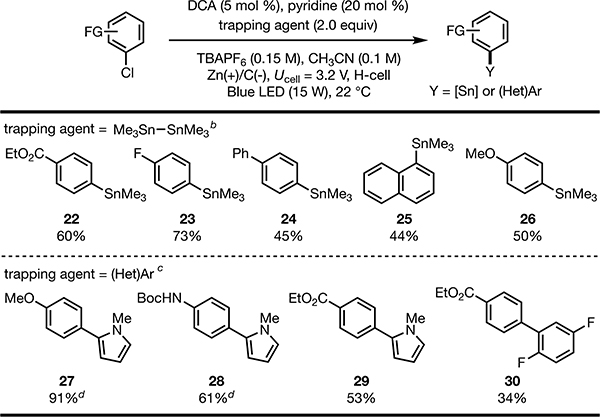 |
Isolated yields are reported unless otherwise noted. Cathodic chamber: aryl chloride (0.4 mmol, 1.0 equiv), DCA (5 mol%), Sn2Me6 (2.0 equiv), TBAPF6 (0.15 M), CH3CN (0.1 M); anodic chamber: TBAPF6 (0.15 M), CH3CN (0.1 M); cell voltage (Ucell) = 3.2 V.
Yields determined by 1H NMR.
20 equiv of (hetero)arene was used as the trapping agent.
10 mol% DCA.
A mechanistic rationale is shown in Figure 2A (pathway A). Thus, the DCA catalyst undergoes cathodic reduction to generate DCA•−, which is then photoexcited to generate DCA•−*.8e This highly reducing species can then donate an electron to the π-system of the aryl halide9 to furnish intermediate 31 and to regenerate the DCA catalyst. Aryl halide radical anions 31 are known to undergo mesolytic cleavage to form aryl radicals 32,19 which can then proceed to the functionalized products.
Figure 2.

Mechanistic experiments. In part B, red squares represent the reaction yield at a given applied cathodic potential, and the gray trace shows the cyclic voltammogram of DCA.
We also recognized the possibility of a photoinduced reduction of DCA at the cathode to form DCA•− (Figure 2A, pathway B), in analogy to König’s dual photocatalytic system. However, this pathway is unlikely because DCA (λmax < 420 nm) is not efficiently excited by the blue LEDs used in our study. Furthermore, controlled potential electrolysis at different cathodic potentials from 0 to −1.2 V (Figure 2B) revealed that the onset potential for the reductive borodehalogenation is about −0.4 V (5% yield), whereas the optimal reactivity occurs above the thermodynamic reduction potential of DCA (−0.82 V). The low potential threshold for an outer sphere electron transfer event is typically ca. 500 mV below the thermodynamic potential of the reductant.20 Meanwhile, because of the highly oxidizing nature of DCA* (E ≈ 2.0 V),2b an applied potential of 0 V should have been more than sufficient to reduce DCA*. Thus, these data are fully consistent with the proposed mechanism involving direct electrochemical activation of DCA.
The second alternative pathway entails the formation of an electron-donor acceptor complex between DCA•− and the aryl halide substrate,2b followed by light-promoted inner sphere electron transfer from DCA•− to the substrate. However, mixing DCA•− with 3 produced no changes to the UV−vis spectrum (see Supporting Information for details), which argues against this proposal.2c,21
In summary, we report a new electrophotocatalytic strategy to access radical anion species with exceedingly high reducing power through tandem electrochemical activation and photoexcitation. This strategy made possible the chemoselective activation of inert substrates with very negative reduction potentials, which are challenging using existing redox modules that rely solely on photoredox activation. We anticipate that this conceptual advance will be broadly applicable in organic synthesis for reactions that require extreme redox power.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided by Cornell University and NIGMS [R35GM127135 (T.H.L.) and R01GM130928 (S.L.)]. This study made use of the NMR facility supported by the NSF (CHE-1531632). We thank Dr. Terry McCallum (Cornell University) for helpful discussions and Prof. Paul Zimmerman (University of Michigan) for providing computation resources.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b10678.
Procedures and characterization data (PDF)
Contributor Information
Hyunwoo Kim, Cornell University, Ithaca, New York.
Hyungjun Kim, Incheon National University, Incheon, Republic of Korea.
Tristan H. Lambert, Cornell University, Ithaca, New York.
Song Lin, Cornell University, Ithaca, New York.
REFERENCES
- (1) (a).Zhang N; Samanta SR; Rosen BM; Percec V Single Electron Transfer in Radical Ion and Radical-Mediated Organic, Materials and Polymer Synthesis. Chem. Rev 2014, 114, 5848–5958. [DOI] [PubMed] [Google Scholar]; (b) van der Vlugt JI Radical-Type Reactivity and Catalysis by Single-Electron Transfer to or from Redox-Active Ligands. Chem. -Eur. J 2019, 25, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Plesniak MP; Huang H-M; Procter DJ Radical Cascade Reactions Triggered by Single Electron Transfer. Nat. Rev. Chem 2017, 1, 0077. [Google Scholar]; (d) Ashby EC Single-Electron Transfer, a Major Reaction Pathway in Organic Chemistry. An Answer to Recent Criticisms. Acc. Chem. Res 1988, 21, 414–421. [Google Scholar]; (e) Zhang C; Tang C; Jiao N Recent Advances in Copper-Catalyzed Dehydrogenative Functionalization via a Single Electron Transfer (SET) Process. Chem. Soc. Rev 2012, 41, 3464–3484. [DOI] [PubMed] [Google Scholar]; (f) Broggi J; Terme T; Vanelle P Organic Electron Donors as Powerful Single-Electron Reducing Agents in Organic Synthesis. Angew. Chem., Int. Ed 2014, 53, 384–413. [DOI] [PubMed] [Google Scholar]; (g) Roth HG; Romero NA; Nicewicz DA Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- (2).(a) For representative reviews on visible-light photocatalysis, see: Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]; (c) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem 2017, 1, 0052. [Google Scholar]; (e) Narayanam JMR; Stephenson CRJ Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]; (f) Fukuzumi S; Ohkubo K Organic synthetic transformations using organic dyes as photoredox catalysts. Org. Biomol. Chem 2014, 12, 6059–6071. [DOI] [PubMed] [Google Scholar]
- (3).For representative reviews on electrochemistry, see: (a)Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sauer GS; Lin S An Electrocatalytic Approach to the Radical Difunctionalization of Alkenes. ACS Catal. 2018, 8, 5175–5187. [Google Scholar]; (c) Meyer TH; Finger LH; Gandeepan P; Ackermann L Resource Economy by Metallaelectrocatalysis: Merging Electrochemistry and C−H Activation. Trends in Chem. 2019, 1, 63–76. [Google Scholar]; (d) Moeller KD Using Physical Organic Chemistry To Shape the Course of Electrochemical Reactions. Chem. Rev 2018, 118, 4817–4833. [DOI] [PubMed] [Google Scholar]; (e) Waldvogel SR; Lips S; Selt M; Riehl B; Kampf CJ Electrochemical Arylation Reaction. Chem. Rev 2018, 118, 6706–6765. [DOI] [PubMed] [Google Scholar]; (f) Nutting JE; Rafiee M; Stahl SS Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev 2018, 118, 4834–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Tang S; Liu Y; Lei A Electrochemical Oxidative Cross-coupling with Hydrogen Evolution: A Green and Sustainable Way for Bond Formation. Chem. 2018, 4, 27–45. [Google Scholar]
- (4) (a).Peters BK; Rodriguez KX; Reisberg SH; Beil SB; Hickey DP; Kawamata Y; Collins M; Starr J; Chen L; Udyavara S; Klunder K; Gorey TJ; Anderson SL; Neurock M; Minteer SD; Baran PS Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-Ion Battery Chemistry. Science 2019, 363, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Perkins RJ; Pedro DJ; Hansen EC Electrochemical Nickel Catalysis for Sp2-Sp3 Cross-Electrophile Coupling Reactions of Unactivated Alkyl Halides. Org. Lett 2017, 19, 3755–3758. [DOI] [PubMed] [Google Scholar]; (c) Sun G; Ren S; Zhu X; Huang M; Wan Y Direct Arylation of Pyrroles via Indirect Electroreductive C−H Functionalization Using Perylene Bisimide as an Electron-Transfer Mediator. Org. Lett 2016, 18, 544–547. [DOI] [PubMed] [Google Scholar]; (d) Edinger C; Waldvogel SR Electrochemical Deoxygenation of Aromatic Amides and Sulfoxides. Eur. J. Org. Chem 2014, 2014, 5144–5148. [Google Scholar]
- (5).Recent examples of combining electrochemistry and photochemistry for organic synthesis: (a)Wang F; Stahl SS Merging Photochemistry with Electrochemistry: Functional-Group Tolerant Electrochemical Amination of C(sp3)−H Bonds. Angew. Chem., Int. Ed 2019, 58, 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang L; Liardet L; Luo J; Ren D; Gratzel M; Hu X Photoelectrocatalytic Arene C−H Amination. Nat. Catal 2019, 2, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yan H; Hou Z-W; Xu H-C Photoelectrochemical C−H Alkylation of Heteroarenes with Organo-trifluoroborates. Angew. Chem., Int. Ed 2019, 58, 4592–4595. [DOI] [PubMed] [Google Scholar]; (d) Zhang W; Carpenter K; Lin S Electrochemistry Broadens the Scope of Flavin Photocatalysis: Photoelectrocatalytic Oxidation of Unactivated Alcohols. Angew. Chem., Int. Ed 2020, 59, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Huang H; Strater ZM; Rauch M; Shee J; Sisto TJ; Nuckolls C; Lambert TH Electrophotocatalysis with a Trisaminocyclopropenium Radical Dication. Angew. Chem., Int. Ed 2019, 58, 13318–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang H; Lambert TH Electrophotocatalytic SNAr Reactions of Unactivated Aryl Fluorides at Ambient Temperature and Without Base. Angew. Chem., Int. Ed 2020, 59, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7) (a).Lund H; Carlsson HS; Nishida T; Enzell CR; Matsuno T Photochemistry of Radical Ions. Acta Chem. Scand 1978, 32, 505–509. [Google Scholar]; (b) Carlsson HS; Lund H; Heby O; von Bahr C; Glaumann H Photochemistry of Radical Ions. II. Influence of Thermal Convection and Photoexcitation on Voltammetric Curves. Acta Chem. Scand 1980, 34, 409–412. [Google Scholar]; (c) Fox MA The Photoexcited States of Organic Anions. Chem. Rev 1979, 79, 253–273. [Google Scholar]
- (8) (a).Ghosh I; Ghosh T; Bardagi JI; König B Reduction of Aryl Halides by Consecutive Visible Light-Induced Electron Transfer Processes. Science 2014, 346, 725–728. [DOI] [PubMed] [Google Scholar]; (b) Ghosh I; Marzo L; Das A; Shaikh R; König B Visible Light Mediated Photoredox Catalytic Arylation Reactions. Acc. Chem. Res 2016, 49, 1566–1577. [DOI] [PubMed] [Google Scholar]; (c) Ghosh I; König B Chromoselective Photocatalysis: Controlled Bond Activation through Light-Color Regulation of Redox Potentials. Angew. Chem., Int. Ed 2016, 55, 7676–7679. [DOI] [PubMed] [Google Scholar]; (d) Bardagi JI; Ghosh I; Schmalzbauer M; Ghosh T; König B Anthraquinones as Photoredox Catalysts for the Reductive Activation of Aryl Halides. Eur. J. Org. Chem 2018, 2018, 34–40. [Google Scholar]; (e) Neumeier M; Sampedro D; Májek M; de la Peña O’Shea VA; Jacobi von Wangelin A; Pérez-Ruiz R Dichromatic Photocatalytic Substitutions of Aryl Halides with a Small Organic Dye. Chem. - Eur. J 2018, 24, 105–108. [DOI] [PubMed] [Google Scholar]
- (9).Eriksen J; Lund H; Nyvad AI; Yamato T; Mitchell RH; Dingle TW; Williams RV; Mahedevan R Electron-Transfer Fluorescence Quenching of Radical Ions. Acta Chem. Scand 1983, 37, 459–466. [Google Scholar]
- (10).The reduction potential of radical anion photocatalyst was estimated by Rehm−Weller formalism. For a reference, see: Rehm D; Weller A Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr. J. Chem 1970, 8, 259–262. [Google Scholar]
- (11).For representative recent examples, see: (a)Nguyen JD; D’Amato EM; Narayanam JMR; Stephenson CRJ Engaging Unactivated Alkyl, Alkenyl and Aryl Iodides in Visible-Light-Mediated Free Radical Reactions. Nat. Chem 2012, 4, 854–859. [DOI] [PubMed] [Google Scholar]; (b) Devery JJ; Nguyen JD; Dai C; Stephenson CRJ Light-Mediated Reductive Debromination of Unactivated Alkyl and Aryl Bromides. ACS Catal. 2016, 6, 5962–5967. [Google Scholar]; (c) Boyington AJ; Riu M-LY; Jui NT Anti-Markovnikov Hydroarylation of Unactivated Olefins via Pyridyl Radical Intermediates. J. Am. Chem. Soc 2017, 139, 6582–6585. [DOI] [PubMed] [Google Scholar]; (d) Seath CP; Vogt DB; Xu Z; Boyington AJ; Jui NT Radical Hydroarylation of Functionalized Olefins and Mechanistic Investigation of Photocatalytic Pyridyl Radical Reactions. J. Am. Chem. Soc. 2018, 140, 15525–15534. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Aycock RA; Vogt DB; Jui NT A Practical and Scalable System for Heteroaryl Amino Acid Synthesis. Chem. Sci 2017, 8, 7998–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Qiao Y; Schelter EJ Lanthanide Photocatalysis. Acc. Chem. Res 2018, 51, 2926–2936. [DOI] [PubMed] [Google Scholar]; (g) Li L; Liu W; Zeng H; Mu X; Cosa G; Mi Z; Li C-J Photoinduced Metal-Catalyst-Free Aromatic Finkelstein Reaction. J. Am. Chem. Soc 2015, 137, 8328–8331. [DOI] [PubMed] [Google Scholar]; (h) Liu W; Yang X; Gao Y; Li C-J Simple and Efficient Generation of Aryl Radicals from Aryl Triflates: Synthesis of Aryl Boronates and Aryl Iodides at Room Temperature. J. Am. Chem. Soc 2017, 139, 8621–8627. [DOI] [PubMed] [Google Scholar]
- (12).Costentin C; Robert M; Savéant J-M Fragmentation of Aryl Halide π Anion Radicals. Bending of the Cleaving Bond and Activation vs Driving Force Relationships. J. Am. Chem. Soc 2004, 126, 16051–16057. [DOI] [PubMed] [Google Scholar]
- (13) (a).Zhang L; Jiao L Visible-Light-Induced Organocatalytic Borylation of Aryl Chlorides. J. Am. Chem. Soc 2019, 141, 9124–9128. [DOI] [PubMed] [Google Scholar]; (b) Chen K; Cheung MS; Lin Z; Li P Metal-Free Borylation of Electron-Rich Aryl (Pseudo)halides Under Continuous-Flow Photolytic Conditions. Org. Chem. Front 2016, 3, 875–879. [Google Scholar]; (c) Mfuh AM; Nguyen VT; Chhetri B; Burch JE; Doyle JD; Nesterov VN; Arman HD; Larionov OV Additive- and Metal-Free, Predictably 1,2- and 1,3-Regioselective, Photoinduced Dual C−H/C−X Borylation of Haloarenes. J. Am. Chem. Soc 2016, 138, 8408–8411. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mfuh AM; Doyle JD; Chhetri B; Arman HD; Larionov OV Scalable, Metal- and Additive-Free, Photoinduced Borylation of Haloarenes and Quaternary Arylammonium Salts. J. Am. Chem. Soc 2016, 138, 2985–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhang L; Jiao L Pyridine-Catalyzed Radical Borylation of Aryl Halides. J. Am. Chem. Soc 2017, 139, 607–610. [DOI] [PubMed] [Google Scholar]
- (15).Haimerl J; Ghosh I; König B; Vogelsang J; Lupton JM Single-Molecule Photoredox Catalysis. Chem. Sci 2019, 10, 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ragnarsson U; Grehn L; Maia HLS; Monteiro LS Enhancing Reductive Cleavage of Aromatic Carboxamides. Org. Lett 2001, 3, 2021–2023. [DOI] [PubMed] [Google Scholar]
- (17).Ishiyama T; Murata M; Miyaura N Palladium(0)-Catalyzed Cross-Coupling Reaction of Alkoxydiboron with Haloarenes: A Direct Procedure for Arylboronic Esters. J. Org. Chem 1995, 60, 7508–7510. [Google Scholar]
- (18).For example, aminoesters have been used as ligands for Pd catalysis: Lu Y; Wang D-H; Engle KM; Yu J-Q Pd(II)-Catalyzed Hydroxyl-Directed C−H Olefination Enabled by Monoprotected Amino Acid Ligands. J. Am. Chem. Soc 2010, 132, 5916–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Takeda N; Poliakov PV; Cook AR; Miller JR Faster Dissociation: Measured Rates and Computed Effects on Barriers in Aryl Halide Radical Anions. J. Am. Chem. Soc 2004, 126, 4301–4309. [DOI] [PubMed] [Google Scholar]
- (20) (a).Francke R; Little RD Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev 2014, 43, 2492–2521. [DOI] [PubMed] [Google Scholar]; (b) Steckhan E Indirect Electro-organic Syntheses—A Modern Chapter of Organic Electrochemistry [New Synthetic Methods (59)]. Angew. Chem., Int. Ed. Engl 1986, 25, 683–701. [Google Scholar]
- (21) (a).Arias-Rotondo DM; McCusker JK The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]; (b) Fukuzumi S; Kochi JK Nature of Electrophiles and Electron Acceptors. Comparison of Their Molecular Complexes with Aromatic Donors. J. Org. Chem 1981, 46, 4116–4126. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.