

Abstract
There is a critical need to understand mechanisms of resistance and develop combinatorial strategies to improve responses to checkpoint blockade immunotherapy (CBI). Here we uncover a novel mechanism by which the human papillomavirus (HPV) inhibits the activity of CBI in head and neck squamous cell carcinoma (HNSCC). Using orthotopic HNSCC models we show that radiation combined with anti-PD-L1 immunotherapy significantly enhanced local control, CD8+ memory T-cells, and induced preferential T-cell homing via modulation of vascular endothelial cells. However, the HPV E5 oncoprotein suppressed immune responses by downregulating expression of MHC and interfering with antigen presentation in murine models and patient tumors. Furthermore, tumors expressing HPV E5 were rendered entirely resistant to anti-PD-L1 immunotherapy and patients with high expression of HPV16 E5 had worse survival. The anti-viral E5 inhibitor rimantadine demonstrated remarkable single agent anti-tumor activity. This is the first report that describes HPV E5 as a mediator of resistance to anti-PD-1/PD-L1 immunotherapy and demonstrates the anti-tumor activity of rimantadine. These results have broad clinical relevance beyond HNSCC to other HPV-associated malignancies and reveal a powerful mechanism of HPV-mediated immunosuppression which can be exploited to improve response rates to checkpoint blockade.
Keywords: Head and neck squamous cell carcinoma, Radiotherapy, Radiation, Immunotherapy, Checkpoint blockade, PD-1/PD-L1, Resistance, HPV, HPV E5, MHC, Rimantadine
Graphical Abstract
Introduction
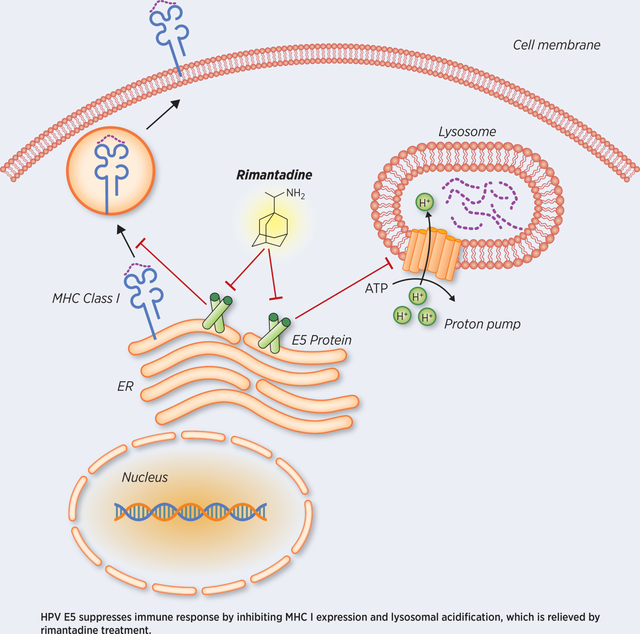
Human papillomavirus (HPV) is associated with 5% of overall cancer worldwide including cervical cancer and head and neck squamous cell carcinoma (HNSCC), and the percentage of HPV-associated malignancy has been increasing. In 2016 the FDA approved anti-PD-1 checkpoint blockade immunotherapy (CBI) for recurrent or metastatic HNSCC after platinum-based chemotherapy (1–3). However, the objective response rate to single agent CBI in these landmark studies remains low on the order of 15–20% (1,3). Thus, there is a critical need to develop combinatorial strategies to enhance response rates as well as identify mechanisms of resistance to CBI in HNSCC. Our group has focused on strategies incorporating radiation therapy combined with CBI (4–7). We previously reported that radiation therapy can synergize with anti-PD-1 immunotherapy by enhancing antigen cross-presentation and antigen specific anti-tumor immune responses in breast cancer and melanoma models (5). Preclinical studies support this combination in head and neck cancer and multiple large Phase II/Phase III clinical trials are underway testing this combination (7–14). In this study, we investigated whether radiation combined with CBI could enhance local tumor control and immune responses using novel orthotopic and HPV-associated HNSCC models. We observed that radiation augmented development of memory T-cells in HNSCC and enhanced T-cell extravasation and infiltration via modulation of tumor endothelial cell adhesion molecules and chemokines. However, radiation combined with CBI was not able to completely control tumor growth in all mice. In order to explore mechanisms of resistance to CBI, we engineered our HNSCC models to express the HPV16 E5, E6, and E7 oncogenes. The HPV16 genome has undergone intense selective pressure which has resulted in a virus which is capable of causing malignant transformation by targeting the critical tumor suppressors p53 and pRb which are effectively the “Achilles heel” of genomic stability (15,16). However, less is known about how HPV genes help HPV-infected cells evade immune responses (17). Viruses have developed powerful immunosuppressive mechanisms including targeting antigen processing and antigen presentation which is required for effective adaptive immune responses to evade the immune system (17,18). HPV E5 is a small hydrophobic protein which has been reported to have multiple functions including regulation of tumor cell differentiation and apoptosis, modulation of H+ ATPase responsible for acidification of late endosomes, and immune-modulation including direct binding and downregulation of major histocompatibility complex (MHC) class I and MHC class II (19–23). These later two functions of E5 inhibit processes which are required for proper antigen processing and presentation. Here we report that HPV E5 is a potent immuno-suppressive molecule that mediates resistance to CBI and decreases surface expression of MHC on tumors. HNSCC lines engineered to express HPV16 E5 were rendered entirely resistant to anti-PD-L1 CBI. In attempting to block the effects of HPV E5, we tested an anti-viral drug and known HPV E5 inhibitor rimantadine, which is currently FDA approved to treat influenza. Remarkably we discovered that rimantadine has broad anti-cancer activity in multiple tumor types tested including HPV E5-expressing HNSCC. This is the first report to our knowledge demonstrating that HPV E5 impairs the activity of anti-PD-L1 immunotherapy and the first report demonstrating the anti-cancer activity of the drug rimantadine. These results have broad clinical relevance to HPV-associated malignancies and elucidate a novel HPV-mediated resistance mechanism whereby HPV E5 targets antigen presentation and MHC as a potential “Achilles heel” of the immune system.
Materials and Methods
Cell lines
HPV16 E7-expressing AT-84 HNSCC cells (AT-84-E7) were kindly provided by Dr. Aldo Venuti (Regina Elena National Cancer Institute; Italy) on November 2016 (24). B16-OVA cells were a kind gift from Dr. Hyam Levitsky (Johns Hopkins University). AT-84-E7 and B16-OVA were grown in RPMI 1640 containing 10% FBS, 1% L-Glutamine, 1% Penicillin/Streptomycin, 1% Sodium Pyruvate, and 200 μg/ml G418. DC2.4 mouse dendritic cells, RAW264.7 macrophages, and HEK293T cells were kindly provided by Dr. Dong-Er Zhang [University of California, San Diego (UCSD)] on September 2017. B3Z T-cell hybridoma cells were a kind gift from Dr. Nilabh Shastri (University of California, Berkeley) on September 2013. MC38 cells were a kind gift from Dr. Mark Smyth (QIMR Berghofer Medical Research Institute, Melbourne, Australia) on April 2013. 4T1 and B16 cells were purchased from ATCC (Manassas, VA). DC2.4, RAW264.7, B3Z, 4T1, B16 and MC38 were grown in RPMI 1640 containing 10% FBS, 1% L-Glutamine, 1% Penicillin/Streptomycin, and 1% Sodium Pyruvate. HEK293T was grown in DMEM containing 10% FBS, 1% L-Glutamine, and 1% Penicillin/Streptomycin. MEER cells were a kind gift from Dr. Judith Varner (UCSD) on March 2018 and grown in the media previously described (25). MOC2, 4MOSC1, CAL-27, CAL-33, and SCC-47 murine and human head and neck squamous carcinoma cells were kindly provided by Dr. J. Silvio Gutkind (UCSD) on March 2018. MOC2 and 4MOSC1 were cultured in the media previously described (26,27). CAL-27, CAL-33, and SCC-47 were grown in DMEM containing 10% FBS, 1% L-Glutamine, and 1% Penicillin/Streptomycin. Routine monitoring for Mycoplasma contamination was performed using the MycoAlert PLUS Detection Kit (Lonza, Basel, Switzerland). All cell lines were used within ten passages after thawing.
Mouse studies
All experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the UCSD (#S15281). Animal experiments were performed in specific pathogen-free facilities at Moores Cancer Center accredited by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). Female 6-to 8-week-old mice were used for experiments. C3H/HeN mice were purchased from Charles River (Wilmington, MA). C57BL/6 and BALB/c were purchased from The Jackson Laboratory (Bar Harbor, ME). OT-1 mice were kindly provided by Dr. Dong-Er Zhang (UCSD). Mice were injected subcutaneously with 1.0 to 5.0 × 105 AT-84-E7, 1.5 × 105 B16-OVA, 5.0 × 105 4T1, or 1,0 × 105 MOC2 cells resuspended in 100 μl of PBS in the right flank. For orthotopic models, 1.0 × 105 AT-84-E7 or 1.0 × 106 4MOSC1 in 30 μl of PBS were injected into tongue. Tumor diameter was measured every 2 to 3 days with an electronic caliper and reported as volume using the formula; tumor volume (mm3) = (length × width2)/2. Once tumors become palpable, mice were treated with 200 μg of anti-PD-L1 antibody (BioXcell, West Lebanon, NH) via i.p. injection every 3 days for a total of three or four injections per mouse, or mice were treated with 10 mg/kg body weight of rimantadine (Sigma, St. Louis, MO) via i.p. injection daily for 7 days. For adoptive transfer experiments, single-cell suspension of spleen from OT-1 mice were cultured in media containing 10 ng/ml OVA SIINFEKL peptide (InvivoGen, San Diego, CA) and 2 ng/ml recombinant IL-2 (PeproTech, Rocky Hill, NJ) for 5 days, and then 4.0 × 106 cells were intravenously injected into B16-OVA-bearing mice.
Mouse Irradiation
Mice received radiation to the tumor site, chest, or abdomen using a JL Shepherd Cs-137 Irradiator (JL Shepherd and Associates, San Fernando, CA). Customized shielding is manually installed to direct focal radiation. Mice were anesthetized and placed in a custom jig to immobilize the region receiving focal radiation. Mice received radiation as a single fraction (8–12 Gy) or using a Quad-shot regimen (3.7 Gy × 4 fractions given twice daily, at least 8 hours apart, for 2 consecutive days). The dose rate was 2.53 Gy/min.
Flow cytometry
Single-cell suspensions were prepared from, lung, liver, tumor-draining lymph node, and tumor by mechanical dissociation and then filtered using a 70 μm cell strainer. AT-84-E7 and MOC2 tumors were incubated in collagenase D (Roche, Basel, Switzerland) at 37°C for 1 hour prior to mechanical dissociation. Density gradient centrifugation on 40%/80% Percoll (GE Healthcare, Chicago, IL) gradient was performed for single-cell suspension from tumors.
After obtaining single-cell suspensions, each sample was incubated with an Fc blocking reagent (anti-CD16/32 antibody; BioLegend, San Diego, CA). Following Fc blockade, cells were stained with fluorescent-labeled antibodies [BioLegend, BD Bioscience (San Jose, CA), or eBiosciences (Thermo Fisher Scientific, Waltham, MA)]. LIVE/DEAD Fixable Cell Staining Kit (Invitrogen) was used for viability staining. For intracellular staining, cells were processed with Foxp3/Transcription Factor Fixation/Permeabilization Concentrate and Diluent (Invitrogen). Cells were analyzed using a BD FACS Aria II or LSR II flow cytometer (BD). Data was analyzed on FlowJo (FlowJo, LLC, Ashland, OR).
For each antibody, the following clones were used: CD45.2 (104), CD3e (145–2C11), CD4 (RM4–5), CD8a (5H10), CD25 (3C7, PC61), CD44 (IM7), CD62L (MEL-14), IFN-γ (XMG1.2), Foxp3 (MF23), H-2Kb (AF6–88.5), H-2Kk (36–7-5), H-2Kd (SF1–1.1), H-2Kb/SIINFEKL (eBio25-D1.16), I-A/I-E (2G9), CD49b (DX5), CD11b (M1/70), FLAG (L5), CD31 (MEC13.3), NK-T/NK Cell Antigen (U5A2–13), CD102 (3C4 (MIC2/4)), CD62P (RMP-1), CD105 (MJ7/18), CD106 (429 (MVCAM.A)), and CD162 (2PH1). H-2Kb/SIINFEKL tetramer was purchased from MBL International (Woburn, MA).
Plasmid construction and HPV16 E5-expressing stable cell line
Codon-optimized HPV16 E5 (from Dr. Frank Suprynowicz, Georgetown University Medical School) were amplified. Either C-terminal or N-terminal FLAG-tagged full-length HPV16 E5 and deletion mutants were cloned into MIP (MSCV-IRES-Puro) vector (from Dr. Dong-Er Zhang, UCSD). All the constructs were confirmed by DNA sequencing. For establishing HPV16 E5-expressing cell line, HEK293T cells were co-transfected with MIP-HPV16 E5 and Ecopac (pIK6.1MCV.ecopac.UTd) using PEI reagent (Sigma). Retroviruses from the culture medium of these cells were then used to infect AT-84-E7, MEER, MOC2, and CAL-27 cells, and the infected cells were selected by puromycin.
Immunoprecipitation and immunoblotting
Cells were lysed in RIPA buffer composed of 25 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40, and protease inhibitors (Roche). The cell lysates were centrifuged (15,000 × g) at 4°C for 5 min. For immunoprecipitation, soluble fractions were precleared using a Protein G/A-Agarose Suspension (EMD Millopore, Billerica, MA) at 4 °C for 15 min. Precleared cell lysates were immunoprecipitated for 2 hours with anti-FLAG M2 antibody (Sigma). Immunocomplexes were adsorbed onto the protein G/A-Agarose Suspension and washed three times. All samples were denatured in 1x sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 2-mercaptethanol, 10% glycerol, and 1% bromophenol blue) for 5 min at 100°C. Proteins were electroblotted onto nitrocellulose membranes (GE Healthcare, Chicago, IL). HRP-conjugated secondary antibodies (Thermo Fisher Scientific, Waltham, MA) were used for detection with Western ECL Substrates (BIO-RAD, Hercules, CA).
Immunofluorescence
HPV16 E5 constructs were transiently transfected into HEK293T with PEI reagent. After 24 hours, cells were fixed with cold methanol for 10 min and then permeabilized with 0.05% Triton X-100/PBS for 15 min. Localization of FLAG-tagged E5 was determined by staining with anti-FLAG antibody (Sigma) followed by Alexa Fluor 568-conjugated secondary antibody (Invitrogen). For E5-expressing AT-84-E7 cells, FLAG-tagged E5 was stained with anti-FLAG antibody followed by Alexa Fluor 488-conjugated secondary antibody, and MHC class I (H-2Kk) was stained with Alexa Fluor 647-conjugated anti-H-2Kk antibody (BioLegend). Nuclei were labeled with DAPI. Images were taken using a BZ-X710 fluorescence microscope (Keyence, Osaka, Japan).
Cell cycle and proliferation assays
Cell cycle progression was analyzed on the basis of BrdU incorporation following cell staining with BrdU-APC and 7-AAD using BD Pharmingen BrdU Flow Kit. (BD, Franklin Lakes, NJ) according to the manufacture’s protocol. Cells were analyzed using flow cytometry.
Cell proliferation was assessed by using MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]. First, cells were seeded in 96-well plate and cultured for 2–3 days. Next, culture media was replaced with fresh media containing 0.5 mg/ml of MTT (Sigma) and the plates were incubated for 4 hours at 37°C. Then, purple formazan crystals were dissolved in lysis buffer (4 mM HCl and 0.1% NP-40 in isopropanol) and the absorbance was recorded on a TECAN infinite M200 microplate reader (Tecan, Männedorf, Switzerland) at a wavelength of 570 nm with absorbance at 650 nm as reference.
B3Z activation assay
B16-OVA cells were seeded into 96-well plate and treated with 100 μM rimantadine for 24 hours, prior to addition of B3Z cells. After 24 hours of co-culture, medium was removed and 100 μl of lysis buffer [0.155 mM chlorophenol red β-D-galactopyranoside (CPRG) (Roche), 0.125% Nonidet P-40 Alternative (EMD-Calbiochem), and 9 mM MgCl2 (Sigma) in PBS] was added. After incubation for 4 hours at 37°C, the absorbance at 570 nm was determined on a TECAN infinite M200 microplate reader.
Phagocytosis assay
DC2.4 cells were co-cultured with FLAG-tagged E5-expressing AT-84-E7 for 24 hours. Phagocytosis of E5 was analyzed by using anti-FLAG (L5) and anti-CD11b (M1/70) antibodies. 7-AAD was used for viability staining. Cells were analyzed on flow cytometer.
Reverse transcription and quantitative PCR
Total RNA were extracted using TRIzol Reagent (Invitrogen) and reverse transcribed with qScript cDNA Synthesis Kit (Quanta BioSciences, Beverly, MA) according to the manufacturer’s instructions. Quantitative PCR analysis were conducted by using KAPA SYBR FAST (KAPA Biosystems, Wilmington, MA) on the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA).
T-cell proliferation assay
T-cells from spleens and lymph nodes from C57BL/6 mice were labeled in 2 μM CFSE for 8 minutes at 37°C on shaker. CFSE-labeled cells were seeded into 96-well plate coated with anti-CD3/CD28 antibody (BioLegend) with or without rimantadine. Three days later, cells were analyzed on flow cytometer.
Clinical Patient Cohorts
HPV-positive OPSCCs from Johns Hopkins University (JHU) patient cohort were analyzed as previously described (28). Patients were recruited with written informed consent under protocol approved by the institutional review board of JHU (#NA_00–36235). The data includes normal (n = 25), E245 high (n = 25), and E67 high (n = 10). HPV status were classified based on the expression of high-risk HPV 16, 33, and 35. TMN and stage were classified based on AJCC 8th edition. mRNA expression was analyzed by RNAseq and assessed as RSEM.
NanoString mRNA profiling
Biospecimens were collected by the Moores Cancer Center Biorepository from consented patients under a University of California, San Diego Human Research Protections Program Institutional Review Board approved protocol (HRPP# 181755). Biorepository subjects provide a written consent which is maintained in the Biorepository archives. Biospecimens were processed for RNA using the RNeasy kit (QIAGEN Silicon Valley, Redwood City, CA). RNA integrity number (RIN) was determined by RNA ScreenTape Analysis (Agilent Santa Clara, CA). RNA expression values were determined for 572 endogenous genes using nCounter PanCancer Immune Profiling Panel (Nanostring Technologies, Seattle, WA). Expression values were normalized using positive controls (geometric means) to eliminate platform-related variation, negative controls (max) to eliminate background effect, and housekeeping genes (geometric mean) to remove variation due to sample input. Expression values from different runs were then quantile normalized and then combined for subsequent analyses. Linear models for microarray (limma-trend approach, using R-limma package, R v3.4.1) were built to compare HPV+ vs HPV- groups utilizing log2 expression values. The Benjamini-Hochberg procedure was applied to control the false discovery rate (FDR). If log2 fold change in expression for an HPV+ vs HPV- was greater than 0, it was classified as up-regulated in HPV+ samples; otherwise, it was classified as being down-regulated. Volcano plots and heatmaps (data standardized by row) were used to show significantly differentially expressed genes.
Statistical analysis
Statistical analysis was performed using Prism 7 (GraphPad Software, La Jolla, CA). Unpaired two-tailed t-test or Ordinary One-way ANOVA multiple comparison test with post hoc Tukey were conducted. Chi-square test and residual analysis was used for mRNA expression analysis. Spearman’s correlation coefficient was used for correlation analysis. P-value <0.05 was considered to be statistically significant; P < 0.05 (*), 0.01 (**), 0.001 (***), and 0.0001 (****).
Results
Radiation combined with CBI enhances tumor control, T-cell infiltration and activation in HPV associated HNSCC
To better understand the effects of radiation combined with PD-L1 blockade in non-HPV-associated and HPV-associated HNSCC, we utilized the syngeneic 4MOSC1 and AT-84 expressing HPV16 E7 (AT-84-E7) tumor models. Radiotherapy (XRT) combined with anti-PD-L1 immunotherapy resulted in significantly enhanced local tumor control in both flank and orthotopic tongue models (Fig 1A and 1B). When analyzing T-cell infiltrates into these models, we observed that XRT alone resulted in a significant increase in CD4+ and CD8+ T-cells which was further enhanced when combined with PD-L1 blockade (Fig 1C and 1D). Notably we did not observe significant increases in tumor-infiltrating lymphocytes (TIL) populations with PD-L1 blockade alone (Fig 1C and 1D). We also observed increased activation of CD8+ T-cells in the tumor-draining lymph node (TDLN) and TIL by intracellular IFN-γ staining in the XRT + PD-L1 blockade groups, indicating an increase in antigen specific adaptive immunity (Fig 1E and Supp Fig 1A). To study the effect of XRT and CBI on T-cell subsets including memory T-cells and regulatory T-cells (Treg) we used the markers CD62L, CD44, CD25, and Foxp3 to distinguish CD8+CD62L+CD44– naïve, CD8+CD62L+CD44+ central memory, CD8+CD62L–CD44+ effector memory, and CD4+CD25+Foxp3+ Treg subsets in the TDLN and TIL of mice 21 days after tumor inoculation. We identified that XRT and XRT + PD-L1 blockade significantly reduce the frequency of naïve T-cells in the TDLN and increase populations of central and effector memory subsets (Fig 1F), indicating that XRT is promoting antigen-experienced T-cell differentiation in the TDLN. XRT alone has been reported to increase the percentage of Treg in other tumor types and we also observed an increase in Treg in the TDLN using this HPV-associated tumor model (Fig 1G and Supp Fig 1B). Importantly, the addition of PD-L1 blockade to XRT prevented further increases in Tregs in the TDLN compared to PD-L1 blockade alone (Fig 1G and Supp Fig 1B). We additionally observed that the majority of TIL possessed an effector phenotype (Supp Fig 1C). Additionally, within the tumor we did not observe any increase in Treg cells with XRT, suggesting differential effects of radiation in the tumor microenvironment versus TDLN (Supp Fig 1D).
Figure 1:

Radiation combined with anti-PD-L1 immunotherapy increases local tumor control, T-cell infiltration, and T-cell memory formation in an orthotopic head and neck cancer model. A) Tumor volume of mice inoculated with 5 × 105 AT-84-E7 cells into the flank of C3H mice. On Day 10, mice were irradiated (1 × 12 Gy) and/or treated with anti-PD-L1 antibodies given every 3 days for a total of 3 injections (n = 5–7 per group). B) Photograph demonstrating the orthotopic HNSCC model. Mice were inoculated 1 × 106 cells 4MOSC1 into the tongue of C57BL/6 mice and irradiated (1 × 8 Gy) on Day 7 and/or treated with anti-PD-L1 antibodies for a total of 3 injections. Tumor volume on Day 21 was shown (n = 5 per group). C, D) Representative dot plots (C) and bar graphs (D) of CD4+ and CD8+ TIL from AT-84-E7-bearing mice on Day 21. E) Percentage of IFN-γ+ cells in CD8+ T-cells in the TDLN from an AT-84-E7 flank model (left) or orthotopic model (right). F) Representative dot plots demonstrating memory subsets of CD8+ T-cells in the TDLN from an AT-84-E7 flank model. G) Frequency of CD25+Foxp3+ regulatory T-cells within CD4+ T-cells in the TDLN from an AT-84-E7 flank model. Data are shown as mean ± S.E.M.
Radiation upregulates the cell adhesion molecules on vascular endothelial cells
To elucidate the mechanism by which radiation modulates TIL populations we next studied the tumor vasculature and endothelial cells. T-cell extravasation is a well-orchestrated process in which endothelial cells become activated by pro-inflammatory cytokines and chemokines and upregulate intraluminal cell adhesion molecules to permit T-cell extravasation (29). To study the effect of radiation on normal vasculature versus tumor vasculature, we irradiated mouse lung, liver, and tumor with varying radiation doses and fractionation patterns including a single fraction dose using clinically for palliation (8–10 Gy × 1 fraction) and a multi-fraction dose used clinically for palliation and tumor control in head and neck cancer (‘Quad-shot regimen’ of 3.7 Gy × 4 fractions). XRT resulted in significant upregulation of ICAM-1, ICAM-2, VCAM-1, P-Selectin, and Endoglin at 48 hours on CD31+ endothelial cells from liver and lung parenchyma (Fig 2A upper and middle panels). Interestingly XRT single fraction radiation to tumor vascular endothelium resulted in increases in ICAM-1, VCAM-1, P-Selectin, but a dramatic decrease in ICAM-2 and Endoglin (Fig 2A lower panels), demonstrating differential effects of radiation on normal parenchymal versus tumor endothelial cells. Quad-shot to tumors also had more variable effects on these cell adhesion molecules which could be related to the sequencing of radiation therapy and the induction of overlapping signaling cascades. Next, we evaluated whether a single fraction of XRT can enhance directional homing and tumor infiltration of adoptively transferred T-cells to an irradiated site. To study this, we established bilateral B16-OVA tumors and irradiated one tumor while shielding the contralateral flank. 48 hours later we injected activated OT-1 T-cells intravenously and then harvested both tumors to assess for T-cell infiltration. We observed a significant, approximate 2-fold increase in OT-1 T-cells present in TIL and TDLN of the irradiated site compared to the contralateral non-irradiated tumor site (Fig 2B). This indicates that radiation-induced upregulation of cell adhesion molecules on tumor-associated endothelial cells can result in preferential homing of T-cells to irradiated sites, and possibly contribute to local tumor control.
Figure 2:

Radiation modulates endothelial cell adhesion molecules and chemokines to enhance T-cell migration and preferential homing to irradiated sites. A) Percentage of cell adhesion molecules in CD45–CD31+ endothelial cells. (upper and middle) Naïve C57BL/6 mice received chest/abdominal irradiation (4 × 3.7 Gy or 1 × 12 Gy), and then the liver and lung were harvested 48 hours after the last irradiation. (lower) AT-84-E7 tumors in the flank were irradiated (4 × 3.7 Gy or 1 × 10 Gy) on Day 18, and tumors were harvested 48 hours after the last irradiation. Experiments repeated 3 times with similar results. B) Antigen-specific CD8+ T-cells in TDLN (upper) or TIL (lower) were detected by H-2Kb/SIINFEKL tetramer. Mice were inoculated with 1.5 × 105 B16-OVA into unilateral or bilateral flank, and tumors on one side were irradiated (12 Gy) on Day 14. After 48 hours, 4 × 106 T-cells from OT-1 mice were injected intravenously. Antigen-specific T-cells from both the irradiated tumor and contralateral tumor were analyzed 3 days after adoptive transfer. Data are shown as mean ± S.E.M. C) Differently expressed genes in 17 patient-derived HNSCC tissue samples comparing HPV-positive patients to HPV-negative patients analyzed using NanoString technology. D) Expression changes in mRNA detected by NanoString comparing an irradiated level 2 LN to a non-irradiated level 4 LN from the same patient. (Top) Heat map of the log2 value comparison between irradiated LN and non-irradiated LN mRNA. ≤5 (blue) shows low expression and ≥15 (red) showing high expression. (Bottom) Log2 fold changes of mRNA expression comparing an irradiated level 2 LN to a non-irradiated level 4 LN. mRNA Expression fold changes of ≥1.5 or ≤−1.5 are shown.
HPV-positive tumors have increased Chemokine expression and radiation therapy enhances chemokine expression
To examine whether these finding translate into human HNSCC, we performed NanoString analysis on 17 different head and neck tumors and tissues collected under an Institutional Biorepository specimen protocol (Fig 2C). This included one patient with recurrent HNSCC who received stereotactic body radiation therapy (SBRT) to a recurrent level 2 cervical lymph node metastasis and then underwent salvage neck dissection of the entire level 2–4 cervical lymph node chain (Fig 2D). It has been reported that HPV-positive tumors tend to have an increased tumor-associated immune cell infiltrate relative to HPV-negative tumors (30,31). However, the mechanism for these differences is not fully understood. We observed significantly increased expression of multiple chemokines including CXCL9, CXCL10, and CXCL11 in HPV-positive tumors compared to HPV-negative tumors and tissues sampled (Fig 2C). Furthermore, when comparing an irradiated to non-irradiated LN within the same patient, we observed significant increases in multiple chemokines, chemokine receptors, and MHC class II in the irradiated LN (Fig 2D and Supp Fig 1E). These findings taken together suggest that the increased T-cell infiltrates observed in HPV-positive tumors and after radiation therapy are likely due to increased expression of specific chemokines and cell adhesion molecules in the tumor microenvironment.
HPV E5 downregulates expression of MHC
As PD-1/PD-L1 blockade alone has limited activity and XRT + PD-1/PD-L1 blockade was unable to completely eradicate tumors in our HNSCC models, we wished to explore mechanisms of resistance to PD-1/PD-L1 blockade. Given the unique pathogenesis of HPV-associated HNSCC, we investigated inhibitory properties of HPV viral proteins. While the functions of HPV E6 and E7 have been studied extensively, the function of HPV E5 protein is far less understood (19). To study the effect of HPV E5, we generated tagged constructs and multiple stable cell lines expressing HPV16 E5 and observed that N-terminal FLAG-tagged E5 had the proper subcellular localization in the endoplasmic reticulum, Golgi apparatus, and plasma membrane (Fig 3A and Supp Fig 2A). To study the localization of HPV16 E5 and MHC, we performed immunohistochemistry using anti-FLAG and anti-MHC antibodies and observed striking co-localization of E5 and MHC (Fig 3B). We then examined MHC expression in HPV16 E5-expressing MEER and AT-84-E7 stable cell lines as well as CAL-27 human squamous cell carcinoma. We observed that tumor cells which expressed HPV16 E5 had significant decreases in cell-surface MHC (Fig 3C). The degree of downregulation was varied among the cell lines. MEER cells showed much higher level of downregulation of MHC I compared to AT-84-E7 and CAL-27 lines. Antigen presenting cells (APCs) including dendritic cells and macrophages phagocytose HPV E5-expressing tumor cells; however, the effect of E5 on APCs which have phagocytosed cell expressing HPV E5 is unknown. To test whether HPV E5 is also detectable in non-transformed host APCs, we setup MHC mismatched co-cultures of APC with tumor cells expressing FLAG-tagged E5. After co-culture, APC phagocytosed cells expressing FLAG-tagged E5, and CD11b+ APC containing FLAG-tagged E5 were detected (Fig 3D), raising the possibility that HPV E5 could also impact professional antigen processing and cross-presentation by APCs. Since MHC class I is required for antigen cross-presentation as well as the activation and effector function of classical CD8+ αβ T-cells, HPV E5-mediated downregulation of MHC is a powerful novel mechanism to inhibit the development and function of adaptive immune responses.
Figure 3:

HPV16 E5 downregulates MHC surface expression and mediates resistance to anti-PD-L1 immunotherapy. A) (left) Expression of FLAG-tagged E5 in AT-84-E7 was confirmed by western blot after immunoprecipitation with anti-FLAG antibody. (right) Localization of FLAG-tagged E5 in AT-84-E7 was determined by staining with anti-FLAG antibody followed by Alexa Fluor 568-conjugated secondary antibody (red). Nuclei were labeled with DAPI (blue). B) Immunofluorescence of AT-84-E7-expressing empty vector or N-terminal FLAG-tagged E5 (N-FLAG E5). FLAG-tagged E5 was stained with anti-FLAG antibody followed by Alexa Fluor 488-conjugated secondary antibody (green). MHC I (H-2Kk) was stained with AF647-conjugated anti-H-2Kk (purple). Nuclei were labeled with DAPI (blue). C) MHC I cell surface expression on control or E5-expressing cell line was analyzed by flow cytometry. D) DC2.4 was co-cultured with FLAG-tagged E5-expressing AT-84-E7 for 24 hours. Phagocytosis of E5 by DC2.4 was then analyzed by flow cytometry. E, F) 5 × 105 AT-84-E7 (E) or 1 × 105 MOC2 (F) cells expressing empty vector or FLAG-tagged E5 were subcutaneously injected and treated with anti-PD-L1 for a total of 4 injections; n = 6 in each group. Data are shown as mean ± S.E.M.
HPV E5 mediates resistance to anti-PD-L1 immunotherapy
Acquired loss of MHC has been reported as a mechanism of resistance to anti-PD-1/PD-L1 immunotherapy (32). In order to test whether HPV E5 confers resistance to CBI, we established tumor models in C3H/HeN and C57BL/6 mice. It is well-known that single-agent PD-1/PD-L1 blockade has modest activity against murine HNSCC models, which we also observed. However, we observed that expression of HPV16 E5 in AT-84-E7 and MOC2 lines completely abrogated the activity of anti-PD-L1 immunotherapy in these syngeneic murine models (Fig 3E and 3F). Interestingly, there are reports that HPV E5 can induce epithelial-mesenchymal transition and we observed that mice harboring tumors expressing HPV16 E5 had an increased frequency of lung metastases (Supp Fig 2B and 2C) (33). The mechanism for this increased metastatic potential deserves further investigation. When we analyzed the tumor-associated immune cell infiltrates of these tumors, we did not observe any significant differences in CD4+ or CD8+ T-cell or natural killer (NK)-cell infiltrates (Supp Fig 2D). While NK cells would classically serve to kill cells which have lost MHC expression, it has been reported that HPV can selectively downregulate certain HLA while retaining other cell surface molecules to prevent NK cell-mediated cytotoxicity (22). This data indicates that HPV E5 may confer resistance or decrease the response rate to anti-PD-1/PD-L1 immunotherapy in patients with HPV-associated HNSCC or other HPV-associated malignancies.
HPV E5 expression correlates with decreased HLA expression in HNSCC patients
In humans the major and minor MHC class I subtypes are termed HLA A-C and HLA E-G, respectively (34). In order to evaluate whether HPV E5 has an effect on HLA expression in human tumors, we analyzed a prospectively collected RNAseq database of 35 HNSCC patient tumors and 25 healthy volunteers [Johns Hopkins University (JHU) cohort] (Supp Table 1). We observed that high expression of E5 correlated with significantly decreased expression of multiple HLA genes including HLA-B, HLA-C, and HLA-F, and trended towards decreased HLA-A and HLA-E (Fig 4A). Interestingly we also observed a significant inverse correlation with E5 expression and Antigen Peptide Transporter 2 (TAP2) which is critical for antigen processing and MHC loading (Fig 4A). Furthermore, when we analyzed the expression level of HLA, we observed the lowest levels of HLA-A, HLA-B, and HLA-C in patient tumors expressing high E2E4E5 compared to normal healthy volunteers or HNSCC patients expressing high E6E7 (Fig 4B left, Supp Fig 3A, 3B, and Supp Table 2). Conversely patients expressing high E6E7 had unchanged or increased relative HLA expression (Fig 4B right, Supp Fig 3A, 3B, and Supp Table 2). These findings indicate that HPV E5 has additional regulatory functions outside of direct binding to MHC and inhibition of antigen processing and loading. Taken together, these findings demonstrate that HPV E5 colocalizes with MHC and downregulates MHC expression in murine and human tumors expressing HPV E5.
Figure 4:

Patients with high expression of HPV16 E5 have decreased HLA expression associated with worse disease-free survival. A, B) mRNA expression analyzed by RNAseq in HNSCC from normal, E245 high, and E67 high patients in Johns Hopkins University (JHU) cohort. (Normal; n = 25, E245 high: n = 26, E67 high: n = 10) (A) Correlation analysis of HPV16 E5 expression and HLA-subtype or TAP expression (n = 36, Spearman’s correlation coefficient) (B) Patients were assigned to 3 groups based on the mRNA expression level of HLA (low, average, and high). Patients with low and high expression of HLA are shown. (Chi-square test and residual analysis) C) Disease-free survival and overall survival of patients in JHU cohort. Patients were assigned to groups based on E5 and HLA expression level.
Patients with high expression of HPV E5 and low expression of HLA have worse disease-free survival
To determine whether expression of HPV E5 impacts HNSCC patient outcomes including overall survival and disease-free survival, patients in our database were stratified into high versus low HPV16 E5 expression based upon median expression level (Supp Fig 3C). HPV16 E5 expression level alone did not independently impact disease-free survival or overall survival (Fig 4C). However, when we included high versus low expression of HLA, we observed that patients with high expression of E5 and low expression of HLA had significantly worse disease-free survival and trended towards diminished overall survival (Fig 4C). These findings demonstrate that expression levels of HPV E5 and HLA may impact patient outcomes or potentially responses to therapy.
The anti-viral drug and HPV E5 inhibitor Rimantadine has novel anti-tumor activity
Due to the immunosuppressive activity of HPV E5, we screened compounds that inhibited HPV E5 to determine if they possess anti-tumor or immuno-modulatory activity. Rimantadine, FDA approved to treat influenza, was one compound which was identified though our screen and has been previously reported to inhibit the E5 protein (35,36). However, there are currently no published reports demonstrating any anti-tumor activity of rimantadine. Thus, we evaluated the anti-tumor activity of rimantadine in our AT-84-E7/E5 tumor model. Remarkably rimantadine alone had potent anti-tumor activity and significantly decreased tumor growth (Fig 5A). The anti-tumor effect of rimantadine was decreased in AT-84-E7 tumors which didn’t express E5 (Supp Fig 4A). Surprisingly we also observed that rimantadine has significant anti-tumor activity in 4MOSC1 HNSCC models as well as B16-OVA melanoma and 4T1 breast cancer models (Fig 5A). We next tested the ability of rimantadine to upregulate MHC and observed significant increases in surface expression of MHC in multiple cell lines (Fig 5B, Supp Fig 4B and 4C). Also, cell surface expression of MHC I on E5-positive AT-84-E7 was restored with rimantadine treatment (Fig 5C). To test the ability of rimantadine to enhance functional antigen presentation on tumor cells, we used B16 cells expressing OVA as a model tumor antigen and co-cultured them with B3Z cells which respond to OVA SIINFEKL peptide presented within MHC as previously described (5). Treatment of B16-OVA cells with rimantadine resulted in a significant 3-fold increase in recognition of this model tumor antigen by B3Z cells (Fig 5D). We also observed that rimantadine combined with anti-PD-L1 immunotherapy resulted in a significant improvement in survival in mice harboring B16-OVA tumors (Fig 5E). We next tested the ability for rimantadine to increase expression of MHC on APCs using the RAW264.7 cell line and observed significant increases in both MHC class I and MHC class II surface expression (Fig 5F). These findings demonstrate that rimantadine has novel anti-tumor activity in multiple pre-clinical tumor models and functions to enhance antigen presentation by upregulating MHC.
Figure 5:

Rimantadine has novel anti-tumor activity and enhances MHC expression on tumor cells and antigen presenting cells. A) Tumor growth of AT-84-E7/E5, B16-OVA, and 4T1 (flank), and 4MOSC1 (tongue); n = 6 in each group. B, C) MHC I expression and antigen-presentation (H-2Kb/SIINFEKL) were analyzed by flow cytometry after rimantadine treatment (100 μM, 48 hours). D) Antigen-specific T-cell activation was analyzed by using B3Z after rimantadine treatment (48 hours). E) Survival curve of B16-OVA-bearing mice treated with rimantadine and/or anti-PD-L1 antibody; n = 6 in each group. F) Cell surface expression of MHC I (H-2Kd) and MHC II (I-A/I-E) on rimantadine-treated RAW264.7 48 hours after treatment. Data are shown as mean ± S.E.M.
While rimantadine may function to enhance immune responses via MHC upregulation, in vitro assays indicate an additional direct cytotoxic mechanism. Rimantadine alone resulted in significant increases in G0/G1 cell cycle arrest and significant decreases in S phase in both AT-84-E7 and B16-OVA models (Fig 6A). Suppression of cell proliferation was also observed (Supp Fig 5A). Proliferation assays using CFSE-labeled T-cells in the presence or absence of rimantadine did not demonstrate a significant effect on T cells (Supp Fig 5B). We screened for changes in gene expression of cell cycle proteins using RT-qPCR and identified significant decreases in microtubule and cell cycle regulatory molecule Stathmin after rimantadine treatment (Fig 6B), and also observed decreases in microtubule associated molecule Tau (Supp Fig 5C). To confirm rimantadine has activity against human HNSCC lines, we performed BrdU incorporation assays and proliferation assays. We observed significant cell cycle arrest and decreased proliferation with rimantadine alone in human CAL-27, CAL-33, and SCC-47 HNSCC lines (Fig 6C). Finally, rimantadine induced cell cycle arrest in murine and human cell lines expressing HPV16 E5 (Fig 6D), indicating that rimantadine was able to functionally reverse effects of HPV E5. This is the first report demonstrating anti-tumor activity of the drug rimantadine and these data support clinical trials repurposing this generic drug to treat HNSCC.
Figure 6:

Rimantadine causes cell cycle arrest in HPV E5-expressing human head and neck cancer cell lines and inhibits cell cycle regulatory proteins. A) BrdU cell cycle was analyzed 24 hours following treatment with rimantadine. B) Stathmin mRNA expression was analyzed 24 hours following treatment with rimantadine. C) Effect of rimantadine on human HNSCC lines. BrdU cell cycle analysis (upper) and MTT proliferation assay (lower). Cells were treated with rimantadine for 24 or 48 hours for BrdU assay or MTT assay, respectively. D) Cell cycle and proliferation assay of E5-expressing cell lines. Cells were treated with rimantadine for 24 hours for BrdU assay and qPCR analysis, or for 48 hours for MTT assay at the concentration as indicated. Data are shown as mean ± S.E.M.
Discussion
Radiation therapy and radiation combined with concurrent chemotherapy are effective curative treatment options and part of the standard of care for management of head and neck cancers. While radiation kills cancer cells primarily through DNA damage, the role that the host immune system plays in radiation tumor control has been underappreciated. Indeed, initial studies in preclinical models of HNSCC reported that the ability for radiation to control tumors was dependent on CD4+ and CD8+ T-cells (14,37,38). There is now an established body of literature that radiation can modulate immune responses and the tumor microenvironment in both beneficial as well as potentially detrimental ways (39–45). Multiple groups have shown that radiation can have immunogenic effects of increasing CD8+ T-cell infiltration, upregulating MHC and FAS on tumor cell surface and activating antigen presenting cells and inducing antigen cross-presentation (5,39–41,46). On the other hand, radiation can have potentially detrimental or immunosuppressive effects causing upregulation of PD-L1 on tumor cells or antigen presenting cells and increasing in CD4+Foxp3+ T-regulatory cell populations in the tumor microenvironment (47–50). A better understanding of these diverse effects would facilitate strategies to enhance or harness the immunostimulatory effects of radiation while actively blocking the immunosuppressive effects to ultimately enhance the efficacy of radiation and loco-regional control rates in HNSCC. To this end we used novel orthotopic HNSCC models to better understand the effects of radiation and radiation combined with CBI. We observed significant increases in CD4+ and CD8+ T-cell infiltration after radiation therapy and significantly improved local control when combining radiation with CBI (Fig 1A–D). We also observed significantly enhanced CD8+ T-cell memory responses after RT or RT + CBI (Fig 1F). As a potentially detrimental effect we did observe an increase in CD4+CD25+Foxp3+ T-regulatory cells after radiation therapy alone; however, addition of anti-PD-L1 mitigated this increase, perhaps due to an increased influx or proliferation of cytotoxic effector CD8+ T-cells and alteration of the immunosuppressive tumor microenvironment with CBI.
The mechanism by which radiation induces robust immune cell infiltrates is not fully understood. Here we identified that radiation therapy can modulate tumor vascular endothelial cells to enhance expression of cell adhesion molecules and chemokines and induce preferential T-cell homing and infiltration to irradiated sites (Fig 2). Importantly this immuno-modulatory effect is entirely independent of any potential synergistic activity of radiation and CBI. When comparing fractionation patterns, we observed that Quad-shot regimen resulted in a more robust upregulation of ICAM-1, VCAM-1, and P-Selectin in tumors (Fig 2). In order for endogenous T-cells or adoptively transferred T-cells to be able to eradicate a tumor, they must be able to infiltrate or invade into that tumor. Thus, our findings that radiation can cause preferential homing to irradiated sites could have implications for enhancing the activity of cellular therapies for cancer. For example, a strategy of using a single fraction of radiation 48–72 hours prior to adoptive T-cell transfer could potentially help improve the activity of cellular therapy for solid tumors. Additionally, after irradiation damaged tumor cells upregulate MHC and FAS rendering them more sensitive and susceptible to killing by cytotoxic T-cells (39). One limitation of our studies is the use of OVA model and transgenic OT-1 T-cells which are artificially engineered and may not recapitulate the function of endogenous effector T-cells. These findings deserve further investigation in therapeutic adoptive transfer models and could help to guide strategies incorporating radiation in clinical trials prior to adoptive T-cell transfer.
While checkpoint blockade immunotherapy has revolutionized oncology, the objective response rate in unselected patients with HNSCC treated with single agent anti-PD-1/PD-L1 immunotherapy remains as low as 13–14% (1–3). Thus, there is a critical need to identify mechanisms of resistance and combinatorial strategies to improve response rates to CBI. The presence of foreign HPV antigens would otherwise be expected to elicit robust anti-tumor immune responses, yet it is interesting to note that response rates to CBI are not drastically different between HPV-positive and HPV-negative patients (1–3). HPV-positive tumors are known to have decreased tumor mutational burden compared to HPV-negative tumors, which may be one reason why objective responses are similar. Moreover, while prior studies have suggested that HPV may subvert or inhibit antigen presentation as a mechanism of immune evasion (51), the ability for HPV proteins to directly inhibit the activity of anti-PD-1/PD-L1 has not been clearly described. Here we identify that the HPV protein E5 is a novel and potent inhibitor of the activity of anti-PD-L1 immunotherapy and alters of antigen processing and MHC expression. Future studies could investigate whether HPV E5 blocks the activity of other CBI such as CTLA-4. Interestingly, loss of β2 microglobulin and MHC expression due to mutation has been described as a mechanism of acquired resistance to anti-PD-1 immunotherapy (32), but active MHC downregulation and inhibition of antigen loading by HPV oncogenes has not been previously described as a mechanism of resistance to CBI to our knowledge. These findings may have important implications for other HPV-associated diseases including cervical cancer, anal cancer, and other genitourinary cancers.
As HPV E6 and E7 have been studied more extensively, it has previously reported that these proteins suppress expression of inflammatory cytokines and chemokines, such as IL-8, IL-18, CCL2, and CCL20. In addition to transcriptional regulation, E6 and E7 can modulate immune responses at the post-translational level, including inhibition of type I and II interferon signaling and STING pathway. Moreover, there is evidence that E7 and E5 may work in concert to impair immune responses. Ultimately in order to integrate, replicate, and spread, HPV likely utilizes multiple complex mechanisms to evade the immune system. Much less is known about HPV E5 proteins compared to HPV E6 and E7. HPV E5 has diverse effects, including downregulation of MHC class I and inhibition of acidification of endosome, which directly impairs host immune responses. Specifically, the N-terminal transmembrane domain of E5 interacts to MHC class I heavy chain and retains them in the Golgi apparatus to prevent them from trafficking to the cell surface (21,52,53). Additionally E5 perturbs expression and stability of MHC class II, by blocking peptide loading and subsequent transport of MHC class II to the cell surface (54). By directly binding MHC, blocking trafficking of MHC to the plasma membrane, HPV E5 may severely impair antigen processing and presentation and help HPV evade the immune system. Because MHC is required for activation and engagement of classical CD8+ αβ T-cells, MHC or HLA may represent an ‘Achilles heel’ of the immune system which HPV targets similar to targeting p53 and pRb as the ‘Achilles heel’ of genomic stability. However, a more subtle modification of antigen presentation may be taking place whereby HPV E5-mediated inhibition of endosome acidification (55) may prevent proper protein degradation, peptide formation, and antigen loading into MHC complexes. Thus, even if cytotoxic T-cells are generated against HPV antigens these CD8+ T-cells would be rendered largely ineffective due to loss of MHC or lack of HPV antigens being presenting on MHC. This novel HPV E5-mediated mechanism of action may have developed to suppress development of adaptive immune responses and permit viral latency while avoiding natural killer cells, which would otherwise detect and kill cells which have completely lost expression of MHC. Quite interestingly, HPV-infected cells have been reported to leave alternative forms of HLA on the cell surface to likely prevent NK-cell mediated cell death (22). We also detected the presence of E5 within APCs which phagocytosed HPV E5-expressing tumor cells, raising the question of whether HPV E5 remains active in APCs to inhibit professional antigen presentation. Overall, the ability for viruses to block immune responses to prolong their lifecycle and proliferation is not a new concept. However, in the era of immunotherapy the immunosuppressive activity of viruses is of paramount importance as these mechanisms likely serve to impair the activity of immunotherapy and reduce patient survival.
Our RNA sequencing data demonstrated an inverse correlation of E5 and multiple HLAs. Interestingly, we were also able to see an inverse correlation between E5 and TAP, which is essential for transporting peptides from the cytoplasm into the lumen of the endoplasmic reticulum to be loaded on MHC molecules. These findings indicate that HPV E5 may modulate antigen processing and presentation via transcriptional regulation in addition to direct post-translational regulation of MHC I. It is likely that this transcriptional regulation is occurring indirectly through HPV E5-mediated modulation of transcription factors, although this mechanism requires further investigation. Additionally, Gamerio et al. reported lower expression of MHC in HPV-positive HNSCC patients, although stratification by HPV oncogenes was not performed (56). Prospective studies using technologies to further characterize the specific cell populations in the tumor microenvironment that are modulated by HPV oncogenes are warranted. In addition to these effects, E5 also has other reported functions, including upregulation of EGFR and its downstream signaling (57–60), impairment of cell differentiation (61), and inhibition of apoptosis induced by Fas ligand (FasL) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) (62), which can influence host immune responses. Within our HPV-positive cohort we did not observe that E5 expression level alone significantly impacted patient survival (Fig 4C), suggesting that E5 may work in concert with other HPV proteins including E6 and E7. However, when combined with expression level of HLA, there were significant differences in survival (Fig 4C). Clearly, further investigation is deserved into the function and molecular pathways disrupted by HPV E5 and the impact of HPV E5 on patient outcomes.
As HPV E5-expressing tumors were resistant to anti-PD-1/PD-L1 immunotherapy, we screened and tested reported HPV E5 inhibitors including rimantadine. Rimantadine has been reported to inhibit the channel forming ability of HPV E5 (35,36), although the anti-cancer activity of rimantadine has not been previously reported in tumor models. Rimantadine had single agent anti-tumor activity and functionally increased MHC expression in our HPV E5-expressing tumor models and non-transformed APCs. Mechanistically, rimantadine has pleiotropic activity as we also observed direct cytotoxic activity and inhibition of cell cycling and cell cycle proteins in multiple murine and human cell lines. This is the first report demonstrating the anti-tumor and immune-modulating activity of rimantadine and supports clinical studies repurposing it as an anti-cancer and immuno-modulatory drug. Due to the robust anti-tumor activity of radiation and PD-L1 blockade we did not observe synergistic anti-tumor activity when adding rimantadine to radiation and PD-L1 blockade in our murine models. Nevertheless, further evaluation may be needed in scenarios involving patient tumors which are resistant or refractory to radiation therapy.
In summary, we have identified that radiation therapy combined with checkpoint blockade enhances CD8+ memory T-cell formation in HNSCC and that T-cells preferentially home to irradiated sites. We have identified HPV E5 as a novel mediator of resistance to PD-L1 checkpoint blockade which downregulates MHC expression in mouse and human HNSCC tumors. Lastly, we report novel anti-tumor activity of the drug rimantadine, supporting clinical trials repurposing this drug in head and neck cancers. These findings have broad relevance to other HPV-associated malignancies and furthers our understanding of the mechanisms of viral mediated immunosuppression and mechanisms of resistance to checkpoint blockade immunotherapy which may be exploited to improve outcomes for HNSCC patients.
Supplementary Material
Significance.
This study identifies a novel mechanism of resistance to anti-PD-1/PD-L1 immunotherapy mediated by HPV E5 which can be exploited using the HPV E5 inhibitor Rimantadine to improve outcomes for head and neck cancer patients.
Acknowledgements
We thank Dr. Frank Suprynowicz (Georgetown University Medical School) for HPV16 E5 vectors, Dr. Judith Varner (UCSD) for MEER cells, and Dr. Dong-Er Zhang (UCSD) for cell lines and vectors. We also thank Biorepository and Tissue technology shared resource for Biospecimen collection.
This work was supported in part by NIH grants (1KL2TR001444, 1R01DE028563, 1U01DE028227) and the San Diego Center for Precision Immunotherapy to A.B.S. Biorepository and Tissue technology shared resource is supported by CCSG Grant P30CA23100.
Footnotes
Conflict of interest statement
The authors declare competing financial interests: J.S.G reports research funding from Kura Oncology and Mavupharma, and consultant fees from Oncoceutics Inc and Vividion Therapeutics. E.E.W.C reports research funding from Pfizer, Merck, AstraZeneca, and Bristol-Myers Squibb outside the submitted work. A.B.S. reports research funding and honoraria from Pfizer and Varian Medical Systems, consultant fees from Astrazeneca, and other fees from Raysearch and Merck. A.B.S. is the scientific founder and has an equity interest in Toragen Inc. that could potentially benefit from the research results. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies.
References
- 1.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med 2016;375(19):1856–67 doi 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol 2016;17(7):956–65 doi 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 3.Cohen EEW, Soulieres D, Le Tourneau C, Dinis J, Licitra L, Ahn MJ, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet 2019;393(10167):156–67 doi 10.1016/S0140-6736(18)31999-8. [DOI] [PubMed] [Google Scholar]
- 4.Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol 2015;16(13):e498–509 doi 10.1016/S1470-2045(15)00007-8. [DOI] [PubMed] [Google Scholar]
- 5.Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francica BJ, Velarde E, et al. Stereotactic Radiation Therapy Augments Antigen-Specific PD-1-Mediated Antitumor Immune Responses via Cross-Presentation of Tumor Antigen. Cancer Immunol Res 2015;3(4):345–55 doi 10.1158/2326-6066.CIR-14-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guram K, Nunez M, Einck J, Mell LK, Cohen E, Sanders PD, et al. Radiation Therapy Combined With Checkpoint Blockade Immunotherapy for Metastatic Undifferentiated Pleomorphic Sarcoma of the Maxillary Sinus With a Complete Response. Front Oncol 2018;8:435 doi 10.3389/fonc.2018.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyauchi S, Kim SS, Pang J, Gold KA, Gutkind JS, Califano J, et al. Immune modulation of head and neck squamous cell carcinoma and the tumor microenvironment by conventional therapeutics. Clin Cancer Res 2019. doi 10.1158/1078-0432.CCR-18-0871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu Y, Lee NY. JAVELIN Head and Neck 100: a Phase III trial of avelumab and chemoradiation for locally advanced head and neck cancer. Future Oncol 2019;15(7):687–94 doi 10.2217/fon-2018-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bahig H, Aubin F, Stagg J, Gologan O, Ballivy O, Bissada E, et al. Phase I/II trial of Durvalumab plus Tremelimumab and stereotactic body radiotherapy for metastatic head and neck carcinoma. BMC Cancer 2019;19(1):68 doi 10.1186/s12885-019-5266-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonomo P, Desideri I, Loi M, Mangoni M, Sottili M, Marrazzo L, et al. Anti PD-L1 DUrvalumab combined with Cetuximab and RadiOtherapy in locally advanced squamous cell carcinoma of the head and neck: A phase I/II study (DUCRO). Clin Transl Radiat Oncol 2018;9:42–7 doi 10.1016/j.ctro.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gillison ML, Ferris RL, Zhang Q, Colevas AD, Mell LK, Kong C, et al. A randomized phase II study of chemoradiation (CRT) +/− nivolumab (Nivo) with sequential safety evaluations of Nivo +/− lirilumab (Liri) or ipilumumab (Ipi) concomitant with (C) RT in intermediate (IR) and high-risk (HR) head and neck squamous cell carcinoma (HNSCC) (RTOG 3504, ). Journal of Clinical Oncology 2017;35(15_suppl):TPS6097–TPS doi 10.1200/JCO.2017.35.15_suppl.TPS6097. [DOI] [Google Scholar]
- 12.Siu LL, Licitra L, Tao YG, Yen C-J, Rischin D, Waldron J, et al. Abstract CT163: KEYNOTE-412: Pembrolizumab plus chemoradiation vs chemoradiation alone for locally advanced head and neck squamous cell carcinoma. Cancer Research 2018;78(13 Supplement):CT163–CT doi 10.1158/1538-7445.am2018-ct163. [DOI] [Google Scholar]
- 13.Powell SF, Gitau MM, Sumey CJ, Reynolds JT, Lohr M, McGraw S, et al. Safety of pembrolizumab with chemoradiation (CRT) in locally advanced squamous cell carcinoma of the head and neck (LA-SCCHN). Journal of Clinical Oncology 2017;35(15_suppl):6011- doi 10.1200/JCO.2017.35.15_suppl.6011. [DOI] [Google Scholar]
- 14.Oweida A, Lennon S, Calame D, Korpela S, Bhatia S, Sharma J, et al. Ionizing radiation sensitizes tumors to PD-L1 immune checkpoint blockade in orthotopic murine head and neck squamous cell carcinoma. Oncoimmunology 2017;6(10):e1356153 doi 10.1080/2162402X.2017.1356153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conway MJ, Meyers C. Replication and assembly of human papillomaviruses. J Dent Res 2009;88(4):307–17 doi 10.1177/0022034509333446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoppe-Seyler K, Bossler F, Braun JA, Herrmann AL, Hoppe-Seyler F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol 2018;26(2):158–68 doi 10.1016/j.tim.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Senba M, Mori N. Mechanisms of virus immune evasion lead to development from chronic inflammation to cancer formation associated with human papillomavirus infection. Oncol Rev 2012;6(2):e17 doi 10.4081/oncol.2012.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Brien PM, Saveria Campo M. Evasion of host immunity directed by papillomavirus-encoded proteins. Virus Res 2002;88(1–2):103–17. [DOI] [PubMed] [Google Scholar]
- 19.Venuti A, Paolini F, Nasir L, Corteggio A, Roperto S, Campo MS, et al. Papillomavirus E5: the smallest oncoprotein with many functions. Mol Cancer 2011;10:140 doi 10.1186/1476-4598-10-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campo MS, Graham SV, Cortese MS, Ashrafi GH, Araibi EH, Dornan ES, et al. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 2010;407(1):137–42 doi 10.1016/j.virol.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 21.Cortese MS, Ashrafi GH, Campo MS. All 4 di-leucine motifs in the first hydrophobic domain of the E5 oncoprotein of human papillomavirus type 16 are essential for surface MHC class I downregulation activity and E5 endomembrane localization. Int J Cancer 2010;126(7):1675–82 doi 10.1002/ijc.25004. [DOI] [PubMed] [Google Scholar]
- 22.Ashrafi GH, Haghshenas MR, Marchetti B, O’Brien PM, Campo MS. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int J Cancer 2005;113(2):276–83 doi 10.1002/ijc.20558. [DOI] [PubMed] [Google Scholar]
- 23.DiMaio D, Petti LM. The E5 proteins. Virology 2013;445(1–2):99–114 doi 10.1016/j.virol.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paolini F, Massa S, Manni I, Franconi R, Venuti A. Immunotherapy in new pre-clinical models of HPV-associated oral cancers. Hum Vaccin Immunother 2013;9(3):534–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoover AC, Spanos WC, Harris GF, Anderson ME, Klingelhutz AJ, Lee JH. The role of human papillomavirus 16 E6 in anchorage-independent and invasive growth of mouse tonsil epithelium. Arch Otolaryngol Head Neck Surg 2007;133(5):495–502 doi 10.1001/archotol.133.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Judd NP, Allen CT, Winkler AE, Uppaluri R. Comparative analysis of tumor-infiltrating lymphocytes in a syngeneic mouse model of oral cancer. Otolaryngol Head Neck Surg 2012;147(3):493–500 doi 10.1177/0194599812442037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Wu VH, Allevato MM, Gilardi M, He Y, Callejas-Valera JL, et al. Syngeneic animal model of tobacco-associated oral cancer reveals the activity of in situ anti-CTLA-4. Nat Commun 2019;in press doi 10.1038/s41467-019-13471-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo T, Gaykalova DA, Considine M, Wheelan S, Pallavajjala A, Bishop JA, et al. Characterization of functionally active gene fusions in human papillomavirus related oropharyngeal squamous cell carcinoma. Int J Cancer 2016;139(2):373–82 doi 10.1002/ijc.30081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res 2014;74(24):7168–74 doi 10.1158/0008-5472.CAN-14-2458. [DOI] [PubMed] [Google Scholar]
- 30.Lechner A, Schlosser H, Rothschild SI, Thelen M, Reuter S, Zentis P, et al. Characterization of tumor-associated T-lymphocyte subsets and immune checkpoint molecules in head and neck squamous cell carcinoma. Oncotarget 2017;8(27):44418–33 doi 10.18632/oncotarget.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Partlova S, Boucek J, Kloudova K, Lukesova E, Zabrodsky M, Grega M, et al. Distinct patterns of intratumoral immune cell infiltrates in patients with HPV-associated compared to non-virally induced head and neck squamous cell carcinoma. Oncoimmunology 2015;4(1):e965570 doi 10.4161/21624011.2014.965570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168(4):707–23 doi 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ranieri D, Belleudi F, Magenta A, Torrisi MR. HPV16 E5 expression induces switching from FGFR2b to FGFR2c and epithelial-mesenchymal transition. Int J Cancer 2015;137(1):61–72 doi 10.1002/ijc.29373. [DOI] [PubMed] [Google Scholar]
- 34.Paaso A, Jaakola A, Syrjanen S, Louvanto K. From HPV Infection to Lesion Progression: The Role of HLA Alleles and Host Immunity. Acta Cytol 2019;63(2):148–58 doi 10.1159/000494985. [DOI] [PubMed] [Google Scholar]
- 35.Wetherill LF, Wasson CW, Swinscoe G, Kealy D, Foster R, Griffin S, et al. Alkyl-imino sugars inhibit the pro-oncogenic ion channel function of human papillomavirus (HPV) E5. Antiviral Res 2018;158:113–21 doi 10.1016/j.antiviral.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wetherill LF, Holmes KK, Verow M, Muller M, Howell G, Harris M, et al. High-risk human papillomavirus E5 oncoprotein displays channel-forming activity sensitive to small-molecule inhibitors. J Virol 2012;86(9):5341–51 doi 10.1128/JVI.06243-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spanos WC, Nowicki P, Lee DW, Hoover A, Hostager B, Gupta A, et al. Immune response during therapy with cisplatin or radiation for human papillomavirus-related head and neck cancer. Arch Otolaryngol Head Neck Surg 2009;135(11):1137–46 doi 10.1001/archoto.2009.159. [DOI] [PubMed] [Google Scholar]
- 38.Williams R, Lee DW, Elzey BD, Anderson ME, Hostager BS, Lee JH. Preclinical models of HPV+ and HPV- HNSCC in mice: an immune clearance of HPV+ HNSCC. Head Neck 2009;31(7):911–8 doi 10.1002/hed.21040. [DOI] [PubMed] [Google Scholar]
- 39.Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res 2004;64(21):7985–94 doi 10.1158/0008-5472.CAN-04-1525. [DOI] [PubMed] [Google Scholar]
- 40.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol 2005;174(12):7516–23. [DOI] [PubMed] [Google Scholar]
- 41.Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med 2006;203(5):1259–71 doi 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys 2004;58(3):862–70 doi 10.1016/j.ijrobp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 43.Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med 2018;24(12):1845–51 doi 10.1038/s41591-018-0232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520(7547):373–7 doi 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young KH, Baird JR, Savage T, Cottam B, Friedman D, Bambina S, et al. Optimizing Timing of Immunotherapy Improves Control of Tumors by Hypofractionated Radiation Therapy. PLoS One 2016;11(6):e0157164 doi 10.1371/journal.pone.0157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Filatenkov A, Baker J, Mueller AM, Kenkel J, Ahn GO, Dutt S, et al. Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clin Cancer Res 2015;21(16):3727–39 doi 10.1158/1078-0432.CCR-14-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res 2014;74(19):5458–68 doi 10.1158/0008-5472.CAN-14-1258. [DOI] [PubMed] [Google Scholar]
- 48.Battaglia A, Buzzonetti A, Martinelli E, Fanelli M, Petrillo M, Ferrandina G, et al. Selective changes in the immune profile of tumor-draining lymph nodes after different neoadjuvant chemoradiation regimens for locally advanced cervical cancer. Int J Radiat Oncol Biol Phys 2010;76(5):1546–53 doi 10.1016/j.ijrobp.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 49.Wu L, Wu MO, De la Maza L, Yun Z, Yu J, Zhao Y, et al. Targeting the inhibitory receptor CTLA-4 on T cells increased abscopal effects in murine mesothelioma model. Oncotarget 2015;6(14):12468–80 doi 10.18632/oncotarget.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muroyama Y, Nirschl TR, Kochel CM, Lopez-Bujanda Z, Theodros D, Mao W, et al. Stereotactic Radiotherapy Increases Functionally Suppressive Regulatory T Cells in the Tumor Microenvironment. Cancer Immunol Res 2017;5(11):992–1004 doi 10.1158/2326-6066.CIR-17-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bashaw AA, Leggatt GR, Chandra J, Tuong ZK, Frazer IH. Modulation of antigen presenting cell functions during chronic HPV infection. Papillomavirus Res 2017;4:58–65 doi 10.1016/j.pvr.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashrafi GH, Haghshenas M, Marchetti B, Campo MS. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int J Cancer 2006;119(9):2105–12 doi 10.1002/ijc.22089. [DOI] [PubMed] [Google Scholar]
- 53.Ashrafi GH, Brown DR, Fife KH, Campo MS. Down-regulation of MHC class I is a property common to papillomavirus E5 proteins. Virus Res 2006;120(1–2):208–11 doi 10.1016/j.virusres.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 54.Zhang B, Li P, Wang E, Brahmi Z, Dunn KW, Blum JS, et al. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virology 2003;310(1):100–8. [DOI] [PubMed] [Google Scholar]
- 55.Straight SW, Herman B, McCance DJ. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J Virol 1995;69(5):3185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gameiro SF, Zhang A, Ghasemi F, Barrett JW, Nichols AC, Mymryk JS. Analysis of Class I Major Histocompatibility Complex Gene Transcription in Human Tumors Caused by Human Papillomavirus Infection. Viruses 2017;9(9) doi 10.3390/v9090252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Straight SW, Hinkle PM, Jewers RJ, McCance DJ. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J Virol 1993;67(8):4521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crusius K, Auvinen E, Steuer B, Gaissert H, Alonso A. The human papillomavirus type 16 E5-protein modulates ligand-dependent activation of the EGF receptor family in the human epithelial cell line HaCaT. Exp Cell Res 1998;241(1):76–83 doi 10.1006/excr.1998.4024. [DOI] [PubMed] [Google Scholar]
- 59.Tomakidi P, Cheng H, Kohl A, Komposch G, Alonso A. Modulation of the epidermal growth factor receptor by the human papillomavirus type 16 E5 protein in raft cultures of human keratinocytes. Eur J Cell Biol 2000;79(6):407–12 doi 10.1078/0171-9335-00060. [DOI] [PubMed] [Google Scholar]
- 60.Gu Z, Matlashewski G. Effect of human papillomavirus type 16 oncogenes on MAP kinase activity. J Virol 1995;69(12):8051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wasson CW, Morgan EL, Muller M, Ross RL, Hartley M, Roberts S, et al. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017;8(61):103581–600 doi 10.18632/oncotarget.21658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kabsch K, Alonso A. The human papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J Virol 2002;76(23):12162–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.