

Abstract
The use of peptidomimetic scaffolds is a promising strategy for the inhibition of protein–protein interactions (PPIs). Herein, we demonstrate that sulfono-γ-AApeptides can be rationally designed to mimic the p53 α-helix and inhibit p53–MDM2 PPIs. The best inhibitor, with Kd and IC50 values of 26 nM and 0.891 μM toward MDM2, respectively, is among the most potent unnatural peptidomimetic inhibitors disrupting the p53–MDM2/MDMX interaction. Using fluorescence polarization assays, circular dichroism, nuclear magnetic resonance spectroscopy, and computational simulations, we demonstrate that sulfono-γ-AApeptides adopt helical structures resembling p53 and competitively inhibit the p53–MDM2 interaction by binding to the hydrophobic cleft of MDM2. Intriguingly, the stapled sulfono-γ-AApeptides showed promising cellular activity by enhancing p53 transcriptional activity and inducing expression of MDM2 and p21. Moreover, sulfono-γ-AApeptides exhibited remarkable resistance to proteolysis, augmenting their biological potential. Our results suggest that sulfono-γ-AApeptides are a new class of unnatural helical foldamers that disrupt PPIs.
Introduction
Foldamers,1−5 the synthetic unnatural oligomers that fold with high stability, have raised new prospects for mimicking the 3D structure and function of bioactive molecules with enhanced resistance to proteolytic degradation and sequence diversity compared to canonical peptides.6−15 In the past 2 decades, several important foldamer systems have been developed to target proteins16−18 and membranes.6,15,19−23 Prominent examples include β-peptides,24,25 peptoids,26,27 β-peptoids,28 oligoureas,29 azapeptides,30,31 α-aminoisobutyric acid foldamers,7 oligoproline,32 aromatic amide foldamers,33−35 and others. In particular, helical foldamers have been explored extensively for inhibition of protein–protein interactions (PPIs).36−39 For instance, the inhibition of interaction between the tumor suppressor protein 53 and human double minute 2 (p53–MDM2) using designed unnatural helical peptidomimetics has been considerably investigated because p53–MDM2 PPI plays a critical role in cancer development and progression.17,40−51 More importantly, the interaction of the p53 helical domain with MDM2 is well characterized (Figure 1A), and it has been used as a testing ground to prove the ability of peptidomimetics for the mimicry of α-helix.52
Figure 1.

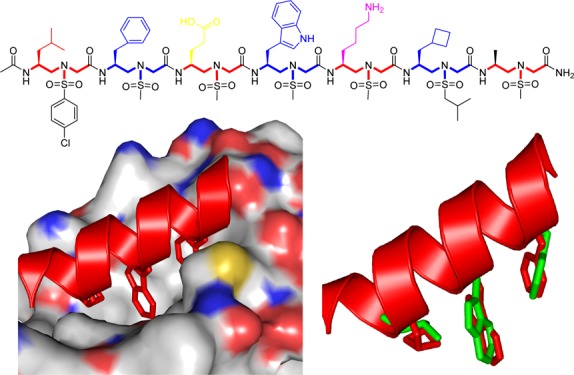
(A) Interaction of p53 with the crystal structure of MDM2 (PDB: 1YCR). p53 is shown as the cartoon, whereas MDM2 is shown as the surface representation. (B) Chemical structure of sulfono-γ-AApeptides. a and b denote the chiral side chain and the sulfonamido side chain from the building block, respectively. (C) Crystal structure of a sulfono-γ-AApeptide (CCDC: 1841094).66 (D) Top view of (C). (E,F) Schematic representation of distribution of side chains from sulfono-γ-AApeptides. (E) Side view; (F) top view, helical wheel.
MDM2 is an E3 ubiquitin ligase overexpressed or activated in many cancers.53 It specifically binds to p53 to induce its proteasomal degradation. As MDM2 negatively regulates p53 tumor suppressor activity, disrupting p53–MDM2 PPI is a promising strategy for the development of anticancer therapeutics.54 The crystal structure of p53–MDM2 (PDB: 1YCR, Figure 1A) reveals that the helical domain of p53 binds to the hydrophobic cleft of MDM2. The three critical residues, Phe19, Trp23, and Leu26, which are on the same face of the p53 helix, deeply insert into the MDM2 hydrophobic pocket and contribute bulk of the binding energy.52 Therefore, molecules that mimic the α-helix of p53 and project functional groups analogous to those three critical residues in p53 could bind to MDM2 and disrupt the interaction.52 Meanwhile, MDMX (also known as MDM4) is a structural analogue of MDM2.55−57 It also binds to p53 and blocks transactivation through N-terminal region, just like MDM2. The difference between them is the overall mechanism of regulation. P53 also induces MDMX mRNA transcription by binding to its promoter in certain cell types.58 MDMX inhibits p53 DNA binding and transcription activation function but is not a significant regulator of its degradation. The expression of MDMX is not regulated by p53 and it is not shown to target p53 to degrade; however, it does inhibit p53 transcriptional activity55,57 Because of the different physiological roles of MDMX compared to MDM2, efforts to develop new inhibitors to selectively target p53–MDM2 or p53/MDMX or simultaneously target both p53/MDM2 and p53/MDMX binding pocket are of medicinal significance.
Sulfono-γ-AApeptides are the subclass of γ-AApeptides recently developed in our lab.59,60 Similar to γ-AApeptides, half of the side chains in sulfono-γ-AApeptides are introduced by sulfonyl chlorides, providing enormous chemical diversity (Figure 1B). In comparison to α-helical peptides, sulfono-γ-AApeptides are highly resistant to proteolytic degradation.61,62 Notably, sulfono-γ-AApeptides have excellent folding stability and fold into helical structures with a well-defined hydrogen bonding pattern.63−65 We recently determined the X-ray crystal structures of a series of homogeneous l-sulfono-γ-AA foldamers.66 The oligomers fold into an unprecedented and surprisingly left-handed 414 helices (Figure 1C,D). Nonetheless, the helical parameters of sulfono-γ-AApeptide foldamers are highly consistent irrespective of side chains, suggesting that the helical propensity is intrinsic and dominated by the molecular scaffold of sulfono-γ-AApeptides. As sulfono-γ-AApeptide foldamers bear a helical pitch of 5.1 Å, which is similar to that of the α-helix (5.4 Å) and display precise arrangement of functional groups in three dimensions, it is envisioned that this class of helical foldamer could be used to mimic α-helices involved in protein–protein interactions (PPIs). Indeed, sulfono-γ-AA foldamers have exactly four side chains per turn, with all side chains aligning perfectly on top of each other in four directions (Figure 1C), resulting in a rectangular helical scaffold. The side chain arrangement of sulfono-γ-AApeptides is highly similar to the α-helix, making it straightforward to design α-helical mimetics projecting critical residues on one face of the helix. Based on this concept, we envision that sulfono-γ-AApeptides could be used to develop a new class of α-helix-like inhibitors for PPIs. We recently illustrated this α-helix-mimicking strategy by developing helical sulfono-γ-AApeptides that structurally and functionally mimic the α-helical domain of BCL9 and disrupt BCL9/β-catenin PPI.67
Results and Discussion
Design of Sulfono-γ-AApeptides and Their Biological Activity
On the basis of the structure of helical sulfono-γ-AApeptides, we set out to design sequences that mimic the α-helix of p53. Although we previously reported homogeneous γ-AApeptides for inhibition of p53–MDM2 interaction,68 because of their inability in helical structure formation, the design was highly speculative and experiential, leading to weak inhibitors (IC50 ≥ 38 μM). With the availability of helical sulfono-γ-AApeptides, the design became very straightforward.
As shown in Figure 1E,F, the first sulfono-γ-AApeptide sequence we designed (PS1) had chiral side chains at positions 2a, 4a, and 6a, which are same as the side chains of Phe19, Trp23, and Leu 26 in p53 that are critical for binding of MDM2 (Table 1 and Figure 1E,F). The binding affinity of PS1 was determined by fluorescence polarization assays (Table 1 and Figure S3).47,69 Both p53 (16–29) and Nutlin were also included in the study as the comparison.55 As shown in Table 1, the Kd of the p53 peptide is 208 nM, which is in good agreement with the literature.52,70 The fluorescence polarization (FP) competition study using a fluorescein isothiocyanate (FITC)-labeled p53 peptide led to an IC50 of 4.61 μM, again consistent with previous results. Interestingly, PS1 bound to MDM2 with Kd value of 98 nM, which is twofold more potent than the p53 peptide. The PS2 sequence was created by deleting one sulfono-γ-AA building block (two side chains) at the C-terminus of PS1. PS2 exhibited a virtually identical Kd (Table 1, PS2) to PS1 with a slightly reduced inhibitory activity (IC50 = 10.3 μM). To further understand the relationship between the binding affinity of sulfono-γ-AApeptides and side chains, we first fixed the three critical side chains (2a, 4a, and 6a) and changed the side chains at different positions relative to the sulfono-γ-AApeptide PS1 (Table 1, PS3-7). As expected, sequences PS3 and PS5-7 did not dramatically change the binding affinity, further suggesting that the binding activity was mainly governed by those three critical side chains at position 2a, 4a, and 6a. Interestingly, mutation of the methyl group to the 4-chloro phenyl group in PS4 did improve the binding affinity with Kd and IC50 values of 57 nM (∼fourfold improvement) and 2.8 μM (∼1.5-fold improvement), respectively (Table 1 and Figure S3), indicating that side chains could affect the binding activity even if they were not involved in direct contact with the MDM2 p53 binding pocket. The vital importance of critical side chains was manifested by PS8. It lacks a single key side chain at position 4a that mimics the residue Trp23 in p53 and completely lost its ability to bind MDM2. It is known that replacement of Leu26 with other bulkier residues could improve the binding affinity of p53 peptide derivatives because of their enhanced hydrophobic interactions with MDM2.71 We therefore designed the sequence PS9, in which the side chain 6a was replaced with the cyclohexylmethyl group. To our surprise, the modification slightly diminished the binding affinity, whereas its ability to inhibit p53–MDM2 interaction was better than p53(16–29). It is likely that the side chain of the cyclohexylmethyl group is too long, which prevented the close contact of PS9 with MDM2. Thus, PS10 was synthesized with the modification of 6a with the cyclobutylmethyl group, which led to the significant improvement in binding potency to MDM2 with Kd and IC50 values of 26 nM and 0.891 μM, respectively (Table 1 and Figure 2). The inhibitory activity is comparable to that of Nutlin (IC50 = 0.6 μM), which is a benchmark small molecule MDM2 inhibitor.55 To the best of our knowledge, PS10 is among the most potent peptidomimetic foldamers that antagonize p53–MDM2 interaction in the literature.52,56
Table 1. Structures of Sulfono-γ-AApeptides Investigated for the Disruption p53–MDM2 Interactiona.

The side chains mimicking Phe19, Trp23, and Leu26 in p53 are shown in blue.
Figure 2.

Kd and IC50 data of pure p53 (A) and sulfono-γ-AApeptide PS10 (B) to MDM2.
Circular Dichroism Measurements
We reasoned that the inhibitory activity of sulfono-γ-AApeptides against p53–MDM2 PPI originates from their intrinsic helical propensity. It is well established that the p53 transactivation domain is disordered and has to fold in order to bind MDM2. The loss of configurational entropy associated with p53 folding reduces the binding affinity relative to more helical peptides.57,72 The circular dichroism (CD) spectroscopy for the peptide p53(16–29) and the 10 homogeneous sulfono-γ-AApeptides were next recorded in phosphate-buffered saline (PBS) buffer between 190 and 270 nm. As shown in Figure 3A, each of the sulfono-γ-AApeptides revealed a marked cotton effect with the strong positive maximum at around 208 nm, which is consistent with the homogeneous sulfono-γ-AApeptide single crystal in the literature,66 thus suggesting that sequences PS1-10 adopt a similar left-handed helical conformation. On the other hand, a minimum of less than 200 nm and the lack of 222 nm was observed for p53(16–29), signifying random coil and almost no α-helix population. As comparison, the helical stability of sequences PS1-10 was also investigated in trifluoroethanol (TFE). The CD spectra of these sequences in TFE revealed a similar left-handed helical conformation as shown in Figure 3B, which is consistent with a previous report,66 demonstrating the robust helicity of this class of peptidomimetics.
Figure 3.

(A). CD spectra of p53 and sulfono-γ-AApeptides (100 μM) measured at room temperature in PBS buffer. (B) CD spectra of p53 and sulfono-γ-AApeptides (100 μM) in TFE at room temperature.
15N–1H HSQC NMR of PS10 in Complex with MDM2
In order to gain insight into the structural basis of sulfono-γ-AApeptide binding to MDM2, the lead linear peptide PS10 was further assessed using nuclear magnetic resonance (NMR) spectroscopy. We have shown that PS10 binds to MDM2 with nanomolar affinity and binds 10 times more tightly than p53 TAD. Chemical shift mapping was performed to determine whether the PS10 binding site overlaps with the p53 TAD binding site. NMR spectroscopy was used to measure the amide proton and nitrogen chemical shift changes of a 15N/13C-labeled fragment of MDM2 containing residues 17–125 after a stoichiometric excess of PS10 was added. Figure 4A shows an overlay of 15N HSQC spectra of MDM2 before (blue resonances) and after (red resonances) the addition of PS10. Based on the behavior of most of the resonances in the free and bound spectra, we conclude that the binding between MDM2 and PS10 is in slow exchange, which is consistent with the binding affinity we observed using fluorescence anisotropy. In Figure 4B, resonances that are not observed in the bound state are labeled with a red asterisk. We speculate that these residues are in intermediate-slow exchange and are participating in binding. An orange line marks the average chemical shift change for all the assigned MDM2 residues (0.056 ppm). Figure 4C,D shows the structure of MDM2 (residues 25–109) bound to p53 TAD. The structure of MDM2 is shown as a gray surface representation and the cyan ribbon structure shows the p53 TAD peptide. MDM2 residues with chemical shift changes greater than 0.056 ppm are colored orange and the residues for resonances in intermediate-slow exchange are colored red. The pattern of chemical shift changes for PS10 binding to MDM2 is highly similar to the results in a previous study from the Daughdrill lab of p53 TAD binding to MDM2.72 In particular, some of the MDM2 residues with the largest chemical shift changes upon binding to either PS10 or p53 TAD are the same, including M50, I54, F55, L66, T67, and Y100, indicating that the binding interface between PS10 and MDM2 overlaps with p53 TAD.
Figure 4.

Chemical shift mapping of PS10 binding to MDM2. (A) Overlay of 15N HSQC spectra of MDM2 before (blue resonances) and after (red resonances) the addition of PS10. HSQC spectra were collected with a twofold and fourfold stoichiometric excess of PS10. (B) Average chemical shift changes in part per million (ppm) for the amide proton and nitrogen resonances in MDM2 p53BD residues binding to PS10. (C,D) Surface image of the MDM2 p53BD structure.
Computational Simulations
The strong binding of PS10 to MDM2 was further supported by computer modeling using PyMOL software. PS10 was overlaid with the p53 helical structure on the MDM2 binding domain (PDB: 1YCR) so that its critical side chains could align with the side chains of p53. As shown in Figure 5A, the side chains of PS10 residues 2a, 4a, and 6a insert deeply into the hydrophobic p53 binding pocket of MDM2, with the similar binding mode observed for p53 bound to MDM2. As these side chains of PS10 closely mimic those critical residues (F19, W23, and L26) of p53 (Figure 5B), because of its intrinsic folding propensity, PS10 could potently inhibit p53–MDM2 PPI.
Figure 5.
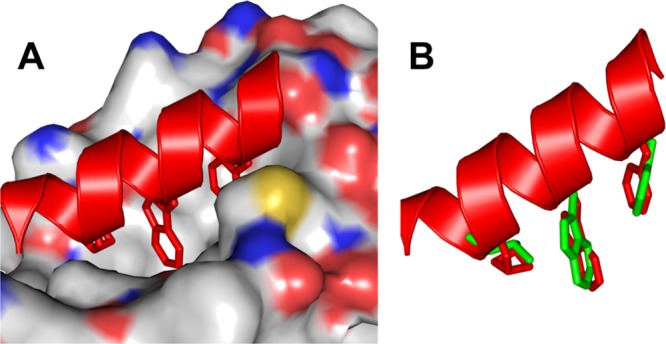
(A) Binding of PS10 to MDM2. The helical structure was built on the crystal structure of Figure 1C (CCDC: 1841094).66 (B) Overlay of side chains 2a, 4a, and 6a of PS10 with Phe19, Trp23, and Leu26 of p53 (green) using PyMOL software.
Stapled Sulfono-γ-AApeptides Induce Activation of p53 in Cells
To assess the cellular activity of sulfono-γ-AApeptides, we first attempted to investigate the activation of p53 using lead linear compounds PS4 and PS10. U2OS cells (p53 wild-type osteosarcoma) with stably integrated BP100-luc (p53-responsive luciferase reporter) and CMV-lacZ (internal control for cell mass and toxicity)70 were treated with 30 μM compounds for 18 h. Luciferase and lacZ activities were determined and the ratio of luc/lacZ indicates p53 transcriptional activity. Compared with p53 peptide, both PS4 and PS10 show marginally increased luciferase activity (Figure 6A), and none of them is active in the cellular functional assay (data not shown), suggesting that these peptides may not be cell-permeable. To circumvent this issue, the lead linear compound PS4 was chosen to prepare stapled sulfono-γ-AApeptides PS11 and PS12 (Table 2), in the hope to enhance the binding activity to protein target and cell permeability.73−75 Interestingly, although PS12 was found to have weaker binding activity compared to its linear counterpart PS4 (Table 3), which indicates that proper stapling length is needed for stapled sulfono-AApeptide to achieve optimized binding, it however displays enhanced luciferase activity (∼25%), suggesting that macrocyclic stapling indeed enhanced cell permeability. This is further evidenced by PS11, which not only displayed enhanced in vitro binding activity toward MDM2 (IC50 = 1.9 μM) compared to PS4, but also activated the luciferase reporter by 70% at 30 μM (Figure 6A). Overall, the luciferase assay suggests that stapled γ-AApeptides exhibited enhanced cell activity.
Figure 6.
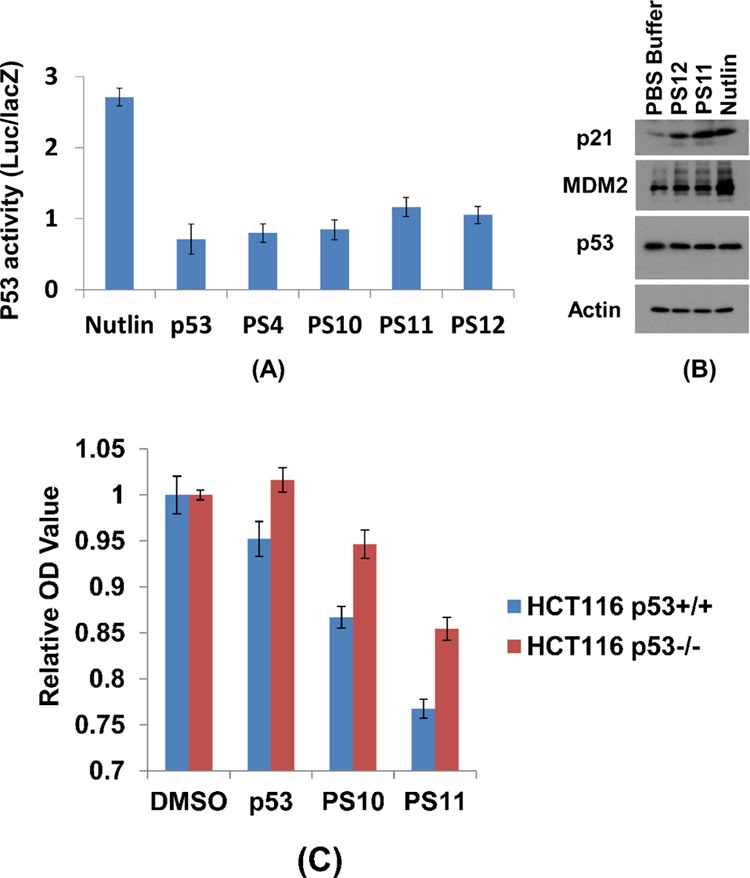
Activation of p53 by stapled sulfono-γ-AApeptides. (A). Luciferase reporter assay. The p53-dependent luciferase transcriptional activation in U2OS cells: the luciferase activities were measured at least three times and the averaged activities along with standard derivations were plotted. (B). Western blotting. Drug treatment lasted for 16 h with 30 μM peptides or 1 μM nutlin. (C) Relative optical density value after 48 h of incubation in the presence of compounds at 200 μM.
Table 2. Structures of Stapled Sulfono-γ-AApeptides Investigated for the Disruption of p53–MDM2 Interaction.

Table 3. Kd and IC50 Values of the p53 and Lead Compounds to MDM2/MDMX.
| MDM2 |
MDMX |
|||
|---|---|---|---|---|
| peptide | Kd (nM) | IC50 (μM) | Kd (nM) | IC50 (μM) |
| p53 | 208 | 4.61 | 641 | 5 |
| PS1 | 98 | 3.95 | 593 | 7.1 |
| PS3 | 99 | 7.1 | 440 | 16.6 |
| PS4 | 57 | 2.8 | 370 | 6 |
| PS9 | 89.6 | 3.22 | 279 | 4.9 |
| PS10 | 26 | 0.891 | 221 | 3.6 |
| PS11 | 1.9 | 3.9 | ||
| PS12 | 3.6 | 6.6 | ||
| nutlin | 0.6 | >10 | ||
Regulation of Actin, p53, and p21 Expression by Stapled Sulfono-γ-AApeptides
To further assess the ability of the stapled sulfono-γ-AApeptides to activate p53 in cells, we next incubated exponentially growing U2OS cells with PS11 and PS12 at 30 μM for 16 h, and the levels of p21, MDM2, and p53 expression were analyzed by western blotting (Figure 6B). The MDM2 level was increased moderately after treatment, suggesting these stapled peptides disrupt p53/MDM2 PPI in cells. Moreover, the p21 level increased significantly after treatment with PS11 compared to the control (PBS buffer), confirming the significant enhancement of p53 transcriptional activity. It is intriguing that the level of p21 and MDM2 is much more elevated than p53, similar to the effect caused by nutlin. Furthermore, it seems that both PS10 and PS11 could suppress proliferation of wild-type p53 cells more efficiently than the p53 peptides (Figure 6C, MTT assay), and they also are more selective toward wild-type p53 cells than p53-deletion cells.
Binding of Sulfono-γ-AApeptides to MDMX
To confirm that sulfono-γ-AApeptides do bind to MDMX, we next investigated the binding affinity of several lead sulfono-γ-AApeptides to MDMX. Fluorescence polarization assays were used to measure the affinities of lead compounds PS1, PS3, PS4, PS9, PS10, as well as stapled sulfono-γ-AApeptides PS11 and PS12 (Table 3 and Figure S4). The ability of the sequences to disrupt the p53–MDMX interaction was also tested, in comparison to those inhibiting p53–MDM2 PPI. As shown in Table 3, the Kd and IC50 values of the p53 peptide to MDMX are 641 nM and 5 μM, respectively, which is in good agreement with the literature.55−57 Interestingly, the helical sulfono-γ-AApeptides designed to bind MDM2 also bind MDMX with good activity (Table 3). This is significant as small molecules such as nutlin (Table 3) are generally only active toward MDM2 but not MDMX, which may imply that these peptidomimetics could be an alternative strategy to small molecules for dual targeting of MDM2 and MDMX, which is needed for fully activating p53 in tumors expressing MDMX.76
Enzymatic Stability Study
One of the major reasons for the development of peptidomimetics is their enhanced stability. Although our previous results demonstrated the stability of sulfono-γ-AApeptides,77 the proteolytic stability of helical sulfono-γ-AApeptides in this study was also investigated and compared to the control peptide p53 (16–29). We incubated 0.1 mg/mL of five lead compounds (PS4, PS9, PS10, PS11, and PS12) and p53 with 0.1 mg/mL proteases in 100 mM ammonium bicarbonate buffer (pH 7.8) at 37 °C for 24 h. Both α-chymotrypsin and pronase were used as representative proteases. The stability of the examined compounds was analyzed by HPLC-MS (Figures S5–S10). The control peptide p53 was completely degraded by α-chymotrypsin and pronase and produced multiple unidentified peaks with no intact peptide remaining. Strikingly, no detectable degradation occurred for the five peptidomimetic peptides, thus demonstrating the high stability of our sulfono-γ-AApeptides against enzymatic degradation, which augments their potential for disruption of PPIs in the future.
Conclusions
In conclusion, we demonstrated that helical sulfono-γ-AApeptides could be rationally designed to mimic p53 α-helix and inhibit p53–MDM2 and p53–MDMX PPI. The lead compound PS10 bound to p53–MDM2 interaction with Kd and IC50 values of 26 nM and 0.891 μM, respectively, and, so far, is one of the most potent unnatural peptidomimetic inhibitors reported to date targeting this interaction. Analysis of the HSQC NMR provides direct evidence that the helical sulfono-γ-AApeptide inhibitor interacts with the p53-binding pocket of MDM2. Furthermore, the stapled sulfono-γ-AApeptides showed promising cellular activity by inducing p53 and MDM2 levels and enhancing p53 transcriptional activity. Enzymatic stability studies have demonstrated high resistance of this class of peptidomimetic foldamer to proteolytic degradation. Thus, this work provides a template that can be applied to explore and generate novel peptidomimetic agents to target p53–MDM2/MDMX protein–protein interactions.
Experimental Section
General Information
Fmoc-protected amino acids were purchased from Chem-impex (Wood Dale, IL). Rink Amide-MBHA resin (0.646 mmol/g) was purchased from GL Biochem (Shanghai) Ltd. 1-Hydroxybenzotriazole (HOBt) wetted with no less than 20% wt water, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, and N,N′-diisopropylcarbodiimide (DIC) was purchased from Oakwood Chemical (Estill, SC). Hoveyda–Grubbs second-generation catalyst was purchased from Sigma-Aldrich, Inc. FITC was purchased from Chemodex (Gallen, Switzerland). Thin-layer chromatography was performed on Sorbtech TLC plates (silica gel w/UV254), visualizing with UV-light 254 nm. Flash column chromatography was performed with ICN silica gel (60 Å, 230–400 mesh, 32–63 μm). Solid-phase synthesis of the peptides was conducted in the peptide synthesis vessels on a Burrell Wrist-Action shaker. All γ-AApeptides were analyzed and purified on a Waters Breeze 2 HPLC system installed with both analytic module (1 mL/min) and preparative module (16 mL/min), by employing a method using 5–100% linear gradient of solvent B [0.1% trifluoroacetic acid (TFA) in acetonitrile] in solvent A (0.1% TFA in water) for over 50 min, followed by 100% solvent B for over 15 min. The pure products were then collected and lyophilized on a Labconco lyophilizer; the purity of the compounds was determined to be >95% by analytical HPLC. Masses of γ-AApeptides were obtained on an Applied Biosystems 4700 Proteomics Analyzer. 1H NMR spectra were recorded at 400 or 500 MHz using tetramethylsilane (TMS) as the internal standard. 13C NMR spectra were recorded at 100 or 125 MHz using TMS as the internal standard. The multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), multiplet (m). Coupling constants are reported in hertz. High-resolution mass spectra were obtained on an Agilent 6220 using electrospray ionization (ESI) time-of-flight. Other chemicals and all solvents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher and used without further purification.
Synthesis of Sulfono-γ-AApeptide Building Blocks
The sulfono-γ-AApeptide building blocks were synthesized based on a previously report63−67 and Fmoc-protected amino acids were used as the initial starting materials.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)propyl)-N-(methylsulfonyl)glycine (BB1)
1H NMR (400 MHz, DMSO-d6): δ 7.88 (d, J = 7.20 Hz, 2H), 7.68 (d, J = 6.00 Hz, 2H), 7.41 (t, J = 6.80 Hz, 2H), 7.34 (d, J = 6.80 Hz, 2H), 7.21 (d, J = 7.60 Hz, 1H), 4.27–4.34 (m, 2H), 4.21 (s, 1H), 3.99 (s, 2H), 3.76 (s, 1H), 3.13–3.20 (m, 2H), 2.94 (s, 3H), 1.04 (d, J = 5.20 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 171.3, 156.0, 144.3, 141.2, 128.0, 127.5, 125.6, 120.5, 65.7, 52.4, 49.0, 47.2, 45.8, 18.7. HRMS (ESI): ([M + H]+) calcd for C21H25N2O6S, 433.1433; found, 433.1424.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-phenylpropyl)-N-(methylsulfonyl)glycine (BB2)
1H NMR (400 MHz, DMSO-d6): δ 7.87 (d, J = 7.20 Hz, 2H), 7.61 (d, J = 6.80 Hz, 2H), 7.41 (t, J = 6.40 Hz, 2H), 7.31 (d, J = 8.40 Hz, 3H), 7.23 (s, 4H), 7.16 (s, 1H), 4.19 (d, J = 6.00 Hz, 2H), 4.13 (d, J = 6.40 Hz, 1H), 3.94–4.08 (m, 2H), 3.90 (s, 1H), 3.38 (d, J = 10.40 Hz, 1H), 3.22 (t, J = 8.00 Hz, 1H), 2.95 (s, 3H), 2.87–2.92 (m, 1H), 2.60 (t, J = 11.60 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): δ 171.3, 156.2, 144.3, 144.2, 141.1, 139.1, 129.6, 128.5, 128.0, 127.5, 126.5, 125.6, 120.5, 65.8, 51.9, 51.6, 49.0, 47.1, 37.9. HRMS (ESI): ([M + H]+) calcd for C27H29N2O6S, 509.1746; found, 509.1733.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentyl)-N-(methylsulfonyl)glycine (BB3)
1H NMR (400 MHz, DMSO-d6): δ 7.88 (d, J = 7.20 Hz, 2H), 7.68 (d, J = 6.40 Hz, 2H), 7.41 (t, J = 7.20 Hz, 2H), 7.32 (s, 2H), 7.14 (d, J = 9.20 Hz, 1H), 4.34 (s, 2H), 4.21 (d, J = 5.60 Hz, 1H), 3.97 (s, 2H), 3.71 (s, 1H), 3.24 (d, J = 11.20 Hz, 1H), 3.10 (t, J = 8.80 Hz, 1H), 2.92 (s, 3H), 1.55 (s, 1H), 1.21–1.27 (m, 2H), 0.82–0.86 (m, 6H). 13C NMR (125 MHz, DMSO-d6): δ 171.2, 156.3, 144.4, 144.3, 141.2, 128.0, 127.5, 125.6, 120.5, 65.5, 51.9, 48.9, 48.2, 47.3, 41.3, 24.7, 23.7, 22.0. HRMS (ESI): ([M + H]+) calcd for C24H31N2O6S, 475.1903; found, 475.1893.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentyl)-N-(isobutylsulfonyl)glycine (BB4)
1H NMR (500 MHz, DMSO-d6): δ 7.88 (d, J = 7.50 Hz, 2H), 7.69 (q, J = 7.00, 4.50 Hz, 2H), 7.41 (t, J = 7.50 Hz, 2H), 7.30–7.34 (m, 2H), 7.17 (d, J = 9.00 Hz, 1H), 4.35 (q, J = 10.50, 7.00 Hz, 1H), 4.28 (q, J = 10.50, 7.00 Hz, 1H), 4.20 (t, J = 7.00 Hz, 1H), 3.97 (s, 2H), 3.68–3.74 (m, 1H), 3.26 (dd, J = 14.50, 5.50 Hz, 1H), 3.12 (q, J = 14.00, 8.50 Hz, 1H), 2.92–3.01 (m, 2H), 2.03–2.13 (m, 1H), 1.51–1.59 (m, 1H), 1.19–1.31 (m, 2H), 0.97 (q, J = 6.50, 5.50 Hz, 6H), 0.84 (dd, J = 10.00, 6.50 Hz, 6H). 13C NMR (125 MHz, DMSO-d6): δ 171.2, 156.3, 144.4, 144.3, 141.2, 128.0, 127.4, 125.6, 120.5, 65.6, 59.4, 51.9, 48.6, 48.1, 47.3, 41.3, 24.7, 23.7, 22.6, 22.6, 22.1. HRMS (ESI): ([M + H]+) calcd for C27H37N2O6S, 517.2372; found, 517.2362.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentyl)-N-((4-chlorophenyl)sulfonyl)glycine (BB5)
1H NMR (500 MHz, DMSO-d6): δ 7.87 (d, J = 7.00 Hz, 2H), 7.79 (d, J = 7.50 Hz, 2H), 7.67 (d, J = 6.50 Hz, 2H), 7.60 (d, J = 8.00 Hz, 2H), 7.40 (br s, 2H), 7.31 (q, J = 14.00, 7.00 Hz, 2H), 7.09 (d, J = 9.00 Hz, 1H), 4.31 (t, J = 8.50 Hz, 1H), 4.19–4.25 (m, 2H), 4.00–4.10 (m, 2H), 3.68 (br s, 1H), 3.26 (dd, J = 14.00, 4.50 Hz, 1H), 3.15 (q, J = 13.00, 7.50 Hz, 1H), 1.52 (br s, 1H), 1.19–1.26 (m, 2H), 0.83 (d, J = 6.00 Hz, 3H), 0.78 (d, J = 5.50 Hz, 3H). 13C NMR (125 MHz, DMSO-d6): δ 170.3, 156.2, 144.4, 144.2, 141.2, 139.0, 138.0, 129.6, 129.4, 128.0, 127.4, 125.6, 120.5, 65.5, 52.5, 48.9, 48.0, 47.3, 41.2, 24.6, 23.7, 21.9. HRMS (ESI): ([M + H]+) calcd for C29H32ClN2O6S, 571.1670; found, 571.1654.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-cyclohexylpropyl)-N-(isobutylsulfonyl)glycine (BB6)
1H NMR (500 MHz, DMSO-d6): δ 7.88 (d, J = 7.50 Hz, 2H), 7.69 (d, J = 7.00 Hz, 2H), 7.41 (t, J = 7.50 Hz, 2H), 7.32 (d, J = 4.50 Hz, 2H), 7.18 (d, J = 9.00 Hz, 1H), 4.35 (t, J = 9.50 Hz, 1H), 4.21–4.27 (m, 2H), 3.98 (s, 2H), 3.74 (d, J = 4.00 Hz, 1H), 3.24–3.27 (m, 1H), 3.13 (q, J = 13.00, 8.50 Hz, 1H), 2.92–3.01 (m, 2H), 2.08 (t, J = 6.50 Hz, 1H), 1.74 (d, J = 11.50 Hz, 1H), 1.59 (d, J = 14.50 Hz, 4H), 1.27 (s, 3H), 1.16 (t, J = 10.50 Hz, 1H), 1.09 (s, 2H), 0.97 (s, 6H), 0.90 (d, J = 9.50 Hz, 1H), 0.78 (d, J = 7.00 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): δ 171.2, 156.3, 144.3, 141.2, 128.0, 127.5, 125.6, 120.6, 65.7, 59.4, 52.0, 48.6, 47.4, 47.2, 34.1, 34.0, 32.3, 26.6, 26.4, 26.1, 24.7, 22.6. HRMS (ESI): ([M + H]+) calcd for C30H41N2O6S, 557.2685; found, 557.2670.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-cyclobutylpropyl)-N-(isobutylsulfonyl)glycine (BB7)
1H NMR (500 MHz, DMSO-d6): δ 7.88 (d, J = 7.50 Hz, 2H), 7.69 (t, J = 6.50 Hz, 2H), 7.41 (t, J = 7.50 Hz, 2H), 7.30 (q, J = 12.50, 7.50 Hz, 2H), 7.10 (d, J = 9.00 Hz, 1H), 4.36 (q, J = 10.50, 7.00 Hz, 1H), 4.28 (q, J = 10.50, 7.00 Hz, 1H), 4.20 (t, J = 7.00 Hz, 1H), 3.97 (q, J = 24.00, 18.5 Hz, 2H), 3.53–3.60 (m, 1H), 3.25 (dd, J = 14.50, 5.00 Hz, 1H), 3.12 (q, J = 14.00, 8.50 Hz, 1H), 2.92–3.01 (m, 2H), 2.20–2.29 (m, 1H), 2.04–2.12 (m, 1H), 1.90–1.95 (m, 2H), 1.69–1.82 (m, 2H), 1.49–1.63 (m, 3H), 1.37–1.43 (m, 1H), 0.97 (q, J = 6.50, 4.50 Hz, 6H). 13C NMR (125 MHz, DMSO-d6): δ 171.3, 156.2, 144.4, 144.3, 141.2, 128.0, 127.4, 125.6, 120.5, 65.6, 59.4, 51.6, 48.8, 48.6, 47.3, 33.1, 28.5, 28.3, 24.7, 22.6, 22.6, 18.6. HRMS (ESI): ([M + H]+) calcd for C28H37N2O6S, 529.2372; found, 529.2377.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-((tert-butoxycarbonyl)amino)hexyl)-N-(methylsulfonyl)glycine (BB8)
1H NMR (500 MHz, DMSO-d6): δ 7.88 (d, J = 7.50 Hz, 2H), 7.69 (d, J = 6.50 Hz, 2H), 7.41 (t, J = 7.50 Hz, 2H), 7.31–7.35 (m, 2H), 7.15 (d, J = 9.00 Hz, 1H), 6.74 (t, J = 5.00 Hz, 1H), 4.33 (d, J = 6.50 Hz, 2H), 4.22 (t, J = 7.00 Hz, 1H), 3.99 (s, 2H), 3.63 (t, J = 3.50 Hz, 1H), 3.27 (dd, J = 14.50, 5.50 Hz, 1H), 3.13 (q, J = 14.50, 9.00 Hz, 1H), 2.93 (s, 3H), 2.89 (t, J = 4.50 Hz, 2H), 1.40–1.49 (m, 2H), 1.37 (s, 9H), 1.19–1.33 (m, 4H). 13C NMR (125 MHz, DMSO-d6): δ 171.3, 156.4, 156.0, 144.3, 141.2, 128.0, 127.5, 125.6, 120.5, 77.8, 65.6, 51.6, 50.1, 48.8, 47.3, 31.9, 29.8, 28.7, 23.2. HRMS (ESI): ([M + H]+) calcd for C29H40N3O8S, 590.2536; found, 590.2520.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-5-(tert-butoxy)-5-oxopentyl)-N-(methylsulfonyl)glycine (BB9)
1H NMR (500 MHz, DMSO-d6): δ 7.88 (d, J = 7.50 Hz, 2H), 7.69 (q, J = 7.00, 4.00 Hz, 2H), 7.41 (t, J = 7.00 Hz, 2H), 7.33 (t, J = 7.50 Hz, 2H), 7.18 (d, J = 9.00 Hz, 1H), 4.31–4.37 (m, 2H), 4.22 (t, J = 7.00 Hz, 1H), 3.98 (s, 2H), 3.65–3.68 (m, 1H), 3.28 (dd, J = 14.50, 5.50 Hz, 1H), 3.15 (q, J = 14.50, 8.50 Hz, 1H), 2.94 (s, 3H), 2.13–2.24 (m, 2H), 1.71–1.77 (m, 1H), 1.45–1.53 (m, 1H), 1.39 (s, 9H). 13C NMR (125 MHz, DMSO-d6): δ 172.3, 171.2, 156.4, 144.3, 144.3, 141.2, 128.0, 127.5, 125.6, 120.5, 80.0, 65.6, 51.2, 49.4, 48.6, 47.3, 31.8, 28.2, 27.5. HRMS (ESI): ([M + H]+) calcd for C27H35N2O8S, 547.2114; found, 547.2094.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1-(tert-butoxycarbonyl)-1H-indol-3-yl)propyl)-N-(methylsulfonyl)glycine (BB10)
1H NMR (500 MHz, DMSO-d6): δ 8.02 (d, J = 7.00 Hz, 1H), 7.85 (d, J = 7.50 Hz, 2H), 7.68 (d, J = 7.00 Hz, 1H), 7.55 (d, J = 7.50 Hz, 3H), 7.37 (d, J = 7.50 Hz, 3H), 7.31 (t, J = 7.50 Hz, 1H), 7.21–7.26 (m, 3H), 4.21 (d, J = 6.00 Hz, 2H), 4.15 (d, J = 7.00 Hz, 1H), 4.05 (s, 2H), 3.48 (dd, J = 14.00, 4.00 Hz, 1H), 3.30 (q, J = 14.00, 8.50 Hz, 1H), 2.98 (s, 3H), 2.95 (s, 1H), 2.76 (t, J = 10.00 Hz, 1H), 1.57 (s, 1H), 1.53 (s, 9H). 13C NMR (125 MHz, DMSO-d6): δ 171.3, 156.3, 144.3, 144.2, 141.1, 131.0, 128.0, 127.4, 125.6, 125.5, 124.7,122.9, 120.5, 119.8, 117.8, 115.1, 83.8, 65.9, 51.9, 50.2, 49.2, 47.1, 28.1, 27.7, 27.5. HRMS (ESI): ([M + H]+) calcd for C34H38N3O8S, 648.2380; found, 648.2358.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-5-(tert-butoxy)-5-oxopentyl)-N-((4-chlorophenyl)sulfonyl)glycine (BB11)
1H NMR (500 MHz, DMSO-d6): δ 7.88 (d, J = 7.00 Hz, 2H), 7.79 (d, J = 8.00 Hz, 2H), 7.67 (br s, 2H), 7.59 (d, J = 7.50 Hz, 2H), 7.40 (t, J = 7.00 Hz, 2H), 7.31 (br s, 2H), 7.12 (d, J = 9.00 Hz, 1H), 4.30 (d, J = 5.50 Hz, 1H), 4.22 (t, J = 6.00 Hz, 2H), 4.04 (q, J = 27.50, 19.00 Hz, 2H), 3.64 (br s, 1H), 3.31 (dd, J = 14.00, 5.00 Hz, 1H), 3.18 (q, J = 13.00, 7.50 Hz, 1H), 2.09–2.23 (m, 2H), 1.73 (d, J = 7.50 Hz, 1H), 1.49 (t, J = 8.00 Hz, 1H), 1.38 (s, 9H). 13C NMR (125 MHz, DMSO-d6): δ 172.3, 170.2, 156.2, 144.3, 144.2, 141.2, 138.9, 138.0, 129.6, 129.3, 128.0, 127.5, 125.6, 120.6, 80.0, 65.6, 51.7, 49.3, 48.6, 47.2, 31.8, 28.2, 27.5. HRMS (ESI): ([M + H]+) calcd for C32H36ClN2O8S, 643.1881; found, 643.1862.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-((tert-butoxycarbonyl)amino)hexyl)-N-(isobutylsulfonyl)glycine (BB12)
1H NMR (500 MHz, DMSO-d6): δ 7.88 (d, J = 7.50 Hz, 2H), 7.69 (dd, J = 7.50, 3.00 Hz, 2H), 7.41 (t, J = 7.50 Hz, 2H), 7.31–7.35 (m, 2H), 7.17 (d, J = 9.00 Hz, 1H), 6.74 (t, J = 5.00 Hz, 1H), 4.27–4.37 (m, 2H), 4.21 (t, J = 6.50 Hz, 1H), 3.98 (q, J = 22.50, 19.00 Hz, 2H), 3.60–3.63 (m, 1H), 3.29 (dd, J = 14.50, 5.50 Hz, 1H), 3.15 (q, J = 14.50, 9.00 Hz, 1H), 2.92–3.02 (m, 2H), 2.88 (d, J = 4.50 Hz, 2H), 2.75 (t, J = 6.00 Hz, 1H), 2.04–2.12 (m, 1H), 1.40–1.49 (m, 1H), 1.36 (s, 9H), 1.18–1.33 (m, 4H), 0.97 (q, J = 6.50, 4.50 Hz, 6H). 13C NMR (125 MHz, DMSO-d6): δ 171.3, 156.4, 156.0, 144.3, 144.3, 141.2, 128.0, 127.5, 125.6, 120.5, 77.7, 65.7, 59.4, 51.6, 50.0, 48.6, 47.3, 31.9, 29.8, 28.7, 24.7, 23.2, 22.6, 22.6. HRMS (ESI): ([M + H]+) calcd for C32H46N3O8S, 632.3006; found, 632.2988.
(R)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(tert-butoxy)propyl)-N-(methylsulfonyl)glycine (BB13)
1H NMR (500 MHz, DMSO-d6): δ 7.88 (d, J = 7.50 Hz, 2H), 7.69 (d, J = 7.50 Hz, 2H), 7.41 (t, J = 7.50 Hz, 2H), 7.30–7.34 (m, 2H), 7.17 (d, J = 8.50 Hz, 1H), 4.28–4.34 (m, 2H), 4.20–4.23 (m, 1H), 3.97–4.07 (m, 2H), 3.74–3.77 (m, 1H), 3.41 (dd, J = 15.00, 5.00 Hz, 1H), 3.19–3.32 (m, 3H), 2.96 (s, 3H), 1.11 (s, 9H). 13C NMR (125 MHz, DMSO-d6): δ 171.3, 156.3, 144.3, 141.2, 128.0, 127.5, 125.6, 120.5, 73.0, 65.8, 62.0, 51.0, 49.1, 48.9, 47.2, 27.7. HRMS (ESI): ([M + H]+) calcd for C25H33N2O7S, 505.2008; found, 505.2000.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-((tert-butoxycarbonyl)amino)hexyl)-N-(but-3-en-1-ylsulfonyl)glycine (BB14)
1H NMR (400 MHz, DMSO-d6): δ 12.83 (br s, 1H), 7.84 (d, J = 7.20 Hz, 2H), 7.65 (d, J = 4.80 Hz, 2H), 7.37 (t, J = 7.20 Hz, 2H), 7.29 (t, J = 6.80 Hz, 2H), 7.13 (d, J = 8.80 Hz, 1H), 6.70 (s, 1H), 5.68–5.78 (m, 1H), 4.99 (q, J = 39.6, 17.60 Hz, 2H), 4.23–4.34 (m, 2H), 4.17 (t, J = 6.80 Hz, 1H), 3.96 (s, 2H), 3.59 (br s, 1H), 3.26–3.30 (m, 2H), 3.10–3.16 (m, 3H), 2.84 (d, J = 3.60 Hz, 2H), 3.36 (d, J = 7.20 Hz, 2H), 1.40 (br s, 1H), 1.33 (s, 9H), 1.11–1.27 (m, 4H). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 156.4, 156.0, 144.3, 144.2, 141.2, 135.5, 128.0, 127.4, 125.6, 120.5, 116.8, 77.7, 65.6, 51.5, 50.0, 48.7, 47.2, 31.9, 29.8, 28.7, 27.6, 23.1. HRMS (ESI): ([M + H]+) calcd for C32H43N3O8S, 630.2849; found, 630.2857.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-5-(tert-butoxy)-5-oxopentyl)-N-(hex-5-en-1-ylsulfonyl)glycine (BB15)
1H NMR (400 MHz, DMSO-d6): δ 7.85 (d, J = 7.20 Hz, 2H), 7.65 (d, J = 7.20 Hz, 2H), 7.37 (t, J = 7.20 Hz, 2H), 7.29 (t, J = 7.20 Hz, 2H), 7.16 (d, J = 9.20 Hz, 1H), 5.64–5.74 (m, 1H), 4.90 (q, J = 21.20, 18.80 Hz, 2H), 4.32 (q, J = 10.00, 7.20 Hz, 1H), 4.16–4.24 (m, 2H), 3.13 (s, 2H), 3.76–3.82 (m, 1H), 3.61 (br s, 1H), 3.28 (dd, J = 14.40, 5.20 Hz, 1H), 3.13 (q, J = 14.40, 8.40 Hz, 1H), 3.04 (q, J = 8.00, 4.40 Hz, 2H), 2.46 (s, 1H), 2.07–2.20 (m, 2H), 1.94 (q, J = 13.20, 6.80 Hz, 2H), 1.67–1.69 (m, 1H), 1.55–1.62 (m, 2H), 1.40–1.50 (m, 1H), 1.35 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 172.3, 171.2, 156.4, 144.3, 141.2, 138.6, 128.0, 127.4, 125.6, 120.5, 115.5, 80.0, 65.7, 52.2, 49.2, 48.5, 47.2, 33.0, 31.7, 28.2, 27.6, 27.2, 22.8. HRMS (ESI): ([M + H]+) calcd for C32H42N2O8S, 615.2740; found, 615.2738.
(S)-N-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-((tert-butoxycarbonyl)amino)hexyl)-N-(hex-5-en-1-ylsulfonyl)glycine (BB16)
1H NMR (400 MHz, DMSO-d6): δ 7.85 (d, J = 7.20 Hz, 2H), 7.65 (d, J = 7.60 Hz, 2H), 7.37 (t, J = 7.60 Hz, 2H), 7.29 (t, J = 7.20 Hz, 2H), 7.13 (d, J = 9.20 Hz, 1H), 6.70 (br s, 1H), 5.63–5.73 (m, 1H), 4.89 (t, J = 20.00 Hz, 2H), 4.30 (q, J = 10.00, 6.80 Hz, 1H), 4.15–4.23 (m, 2H), 3.93 (s, 2H), 3.57 (br s, 5H), 3.26 (dd, J = 14.40, 4.80 Hz, 1H), 3.10 (q, J = 14.40, 8.80 Hz, 1H), 3.02 (q, J = 8.00, 4.40 Hz, 2H), 2.83 (d, J = 4.40 Hz, 2H), 2.46 (s, 1H), 1.91–1.94 (m, 2H), 1.54–1.59 (m, 2H), 1.32 (s, 9H), 1.15–1.26 (m, 3H). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 156.4, 156.0, 144.3, 141.2, 138.6, 128.0, 127.4, 125.6, 120.5, 115.5, 77.7, 65.7, 52.2, 49.9, 48.7, 47.2, 33.0, 32.0, 29.8, 28.7, 27.2, 23.1, 22.8. HRMS (ESI): ([M + H]+) calcd for C34H47N3O8S, 658.3162; found, 658.3180.
Sulfono-γ-AApeptide Preparation
The sulfono-γ-AApeptide synthesis was carried out on 100 mg of Rink Amide-MBHA resin (0.646 mmol/g) under room temperature at atmosphere pressure. The resin was swelled in dimethylformamide (DMF) for 5 min before use, followed by treatment with 20% piperidine/DMF solution (2 mL) for 15 min (×2) to remove the Fmoc-protecting group, and afterward washed three times with dichloromethane (DCM) and three times with DMF. A premixed solution of sulfono-γ-AApeptide building block (2 equiv), HOBt (4 equiv), and DIC (4 equiv) in 2 mL of DMF was added to the resin and shaken for 4 h to complete the coupling reaction. After washing with DCM and DMF, the resin was treated with 20% piperidine/DMF solution for 15 min (×2). Another sulfono-γ-AApeptide building block (2 equiv) was attached on the resin following the procedure in the first coupling step, and the Fmoc-protecting group was removed after the coupling reaction was done. The reaction cycles were repeated until the desired sulfono-γ-AApeptides were synthesized. The N-terminus of the sequence was capped with acetic anhydride (1 mL) in pyridine (2 mL) (15 min × 2), followed by treatment with TFA/DCM (6 mL, 1:1, v/v) for 3 h. The cleavage solution was collected, and the beads washed with DCM (3 mL × 2). The solution was combined and evaporated under air flow to give the crude product, which was analyzed and purified by a Water HPLC system, at the 1 and 16 mL/min flow rates for analytic and preparative HPLC, respectively. The gradient eluting method of 5–100% of solvent B (0.1% TFA in acetonitrile) in A (0.1% TFA in water) over 50 min was performed. The pure peptides were then collected and lyophilized on a Labconco lyophilizer; the purity of the compounds was determined to be >95% by analytical HPLC. All the sulfono-γ-AApeptides were obtained with moderate yield (39.39–46.72%) after prep-HPLC purification.
Fluorescence Polarization Competition Assays
The binding affinity (Kd) of the p53 and AApeptides was investigated by FP. GST-MDM2-1-150 containing human MDM2 were expressed in Escherichia coli as previously described by us. An FP experiment was carried out by incubating 50 nM FITC-labeled AApeptide with MDM2 (0.0625–2 μM) in 1× PBS. The binding affinity of the investigated AApeptides to the MDM2 protein (Kd) was obtained by incubating 50 nM FITC-labeled AApeptide in MDM2 ranging from 0.3125 to 55 μM. Dissociation constants (Kd) were determined by plotting the fluorescence anisotropy values as a function of protein concentration, and the plots were fitted to the following equation. The Lst is the concentration of the peptide and the x stands for the concentration of the protein. The experiments were performed in triplicate and repeated three times. The binding affinity of the investigated AApeptides to the MDMX protein (Kd) was obtained by using a similar procedure.
Circular Dichroism
CD spectra were measured on an Aviv 215 CD spectrometer using a 1 mm path length quartz cuvette, and compound solutions in PBS buffer (or in TFE) were prepared using dry weight of the lyophilized solid followed by dilution to give the desired concentration (100 μM) and solvent combination. Ten scans were averaged for each sample, independent experiments were conducted three times, and the spectra were averaged. The final spectra were normalized by subtracting the average blank spectra. Molar ellipticity [θ] (deg·cm2·dmol–1) was calculated using the equation
where θobs is the measured ellipticity in millidegrees, n is the number of side groups, l is the path length in centimeters (0.1 cm), and c is the concentration of the sulfono-γ-AA peptide in molar units.
Luciferase Reporter Assay
U2OS cells were stably transfected with p53-responsive luciferase reporter plasmid (BP100-luc) and cytomegalovirus (CMV)-lacZ plasmid. Cells were seeded 100,000 per well in 24-well plates 24 h before treatment. After treatment with compounds for 16 h, the cell lysate was analyzed for luciferase and β-gal expression. The ratio of luciferase/β-gal activity was used as an indicator of transcription activity.
Western Blotting
Cells were lysed in RIPA buffer (with 1 mM phenylmethylsulfonyl fluoride [PMSF], and 1× protease inhibitor mixture) and centrifuged at 4 °C for 10 min at 14,000g. The supernatant was boiled in Laemmli sample buffer for 10 min and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to Immobilon-P filters (Millipore). The filters were blocked for 1 h with PBS containing 5% nonfat dry milk and 0.1% Tween 20 and incubated with primary and secondary antibodies, and the filters were developed using SuperSignal reagent (Thermo Scientific). MDM2 was detected using monoclonal antibody 3G9. Actin antibodies were purchased from Santa Cruz Biotechnology. DO-1 for human p53 and p21 antibody were from BD Pharmingen.
MTT Assay
HCT-116 p53+/+ or p53–/– cells were seeded 4 × 104 in 24-well plates. After 2-day treatment of each compounds (p53, PS10, PS11) at a final concentration of 200 μM, cell numbers were analyzed with MTT assay (CellTiter 96 Aqueous One Solution Cell Proliferation Assay, Promega), according to the product’s instructions. Three independent experiments were conducted for statistical analysis.
Enzymatic Stability Study
Lead compounds and peptide control p53 (0.1 mg/mL) were incubated with 0.1 mg/mL proteases in 100 mM ammonium bicarbonate buffer (pH 7.8) at 37 °C for 24 h. Then, the reaction mixtures were concentrated in a speed vacuum at medium temperature to remove water and ammonium bicarbonate. The resulting residues were re-dissolved in H2O/MeCN and analyzed on a Waters analytical HPLC system with 0.8 mL/min flow rate and 5–100% linear gradient of solvent B (0.1% TFA in acetonitrile) in A (0.1% TFA in water) over the duration of 50 min. The UV detector was set to 215 nm.
15N–1H HSQC NMR of Lead Peptide PS10 in Complex with MDM2
Uniformly 15N- and 13C-labeled samples of human MDM2 residues 17–125 were expressed and purified as described in previous literature.72 Resonance assignments for free MDM2 and MDM2 bound to PS10 were made using sensitivity-enhanced 1H–15N HSQC and three-dimensional HNCA spectra using uniformly 15N- and 13C-labeled samples in 50 mM sodium phosphate buffer, 100 NaCl, 1 mM ethylenediaminetetraacetic acid, 0.02% sodium azide, and 2 mM dithiothreitol, at pH 6.8 (10% D2O). NMR spectroscopy was carried out on a Varian VNMRS 800 MHz spectrometer with a triple resonance pulse field Z-axis gradient cold probe at 30 °C. For HNCA experiments, data were acquired along 1H, 13C and 15N dimensions using 9689.9228 (t3) × 6433.1377 (t2) × 2430.4290 (t1) Hz sweep widths and 1024 (t3) × 64 (t2) × 32 (t1) complex data points. The sweep widths and complex points of the HSQC were 9689.9228 (t2) × 2430.3853 (t1) Hz and 1024 (t2) × 128 (t1), respectively. Bound spectra were collected using a stoichiometric excess of PS10. Saturation of binding was confirmed by comparing spectra from MDM2 with a twofold and fourfold stoichiometric excess of PS10. The combined average chemical shifts were calculated from the formula Δave = [((Δ1HN)2 + (Δ15N/5)2)/2]1/2. Assignments of free MDM2 were previously reported. Assignment of 81 resonances of MDM2 bound to PS10 were confirmed using data from the HNCA experiment. Assignment of 10 additional resonances in the HSQC spectrum of MDM2 bound to PS10 were assigned based on the behavior of the peaks in the free and bound spectra and assuming a minimal perturbation in the spectra. Residues 19, 61, 65, 66, 67, 72, 73, 77, 94, and 105 were assigned using these criteria. There were also several resonances that were not detected in the bound state. This includes the resonances for residues 23, 46, 58, 59, 62, 71, 74, 93, 97, and 108. Resonances that disappear in the presence of a ligand are in intermediate exchange and probably involved in binding.
All NMR spectra were processed with NMRFx and analyzed using the NMRViewJ software. Apodization was achieved in the 1H, 13C, and 15N dimensions using a squared sine bell function shifted by 70 °C. Apodization was followed by zero filling to double the number of real data points and linear prediction was used in the 15N dimension for the HNCA spectra.
Acknowledgments
This work was generously supported by NSF CAREER 1351265 (Jianfeng Cai), NIH 1R01GM112652-01A1 (Jianfeng Cai), NIH2R01CA14124406-A1 (G.D. and Jiandong Chen) and NIH1R01GM115556-01A1(G.D.).
Glossary
Abbreviations
- MDM2
murine double minute-2
- MDMX
murine double minute-X
- TFE
trifluoroethanol
- DMF
dimethylformamide
- DCM
dichloromethane
- HOBt
1-hydroxybenzotriazole
- DIPEA
N,N-diisopropylethylamine
- DIC
N,N′-diisopropylcarbodiimide
- TFA
trifluoroacetic acid
- FITC
fluorescein isothiocyanate
- Kd
dissociation constant
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b00993.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 1YCR. The authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions
# P.S., Y.S., and J.L. contributed equally. Jianfeng Cai led and supervised the project. P.S., Y.S., J.L. and W.B. performed the experiments. P.S., Y.S., Q.L., G.D., Jiandong Chen, and Jianfeng Cai analyzed the data. P.S., G.D., Jiandong Chen, and Jianfeng Cai wrote the paper. All the authors discussed the results, commented on, and proofread the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Roy A.; Prabhakaran P.; Baruah P. K.; Sanjayan G. J. Diversifying the structural architecture of synthetic oligomers: the hetero foldamer approach. Chem. Commun. 2011, 47, 11593–11611. 10.1039/c1cc13313f. [DOI] [PubMed] [Google Scholar]
- Guichard G.; Huc I. Synthetic foldamers. Chem. Commun. 2011, 47, 5933–5941. 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]
- Horne W. S.; Gellman S. H. Foldamers with heterogeneous backbones. Acc. Chem. Res. 2008, 41, 1399–1408. 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman C. M.; Choi S.; Shandler S.; DeGrado W. F. Foldamers as versatile frameworks for the design and evolution of function. Nat. Chem. Biol. 2007, 3, 252–262. 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht S.; Huc I.. Foldamers: Structure, Properties, and Applications; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- De Poli M.; Zawodny W.; Quinonero O.; Lorch M.; Webb S. J.; Clayden J. Conformational photoswitching of a synthetic peptide foldamer bound within a phospholipid bilayer. Science 2016, 352, 575–580. 10.1126/science.aad8352. [DOI] [PubMed] [Google Scholar]
- Jones J. E.; Diemer V.; Adam C.; Raftery J.; Ruscoe R. E.; Sengel J. T.; Wallace M. I.; Bader A.; Cockroft S. L.; Clayden J.; Webb S. J. Length-Dependent Formation of Transmembrane Pores by 310-Helical α-Aminoisobutyric Acid Foldamers. J. Am. Chem. Soc. 2016, 138, 688–695. 10.1021/jacs.5b12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collie G. W.; Pulka-Ziach K.; Lombardo C. M.; Fremaux J.; Rosu F.; Decossas M.; Mauran L.; Lambert O.; Gabelica V.; Mackereth C. D.; Guichard G. Shaping quaternary assemblies of water-soluble non-peptide helical foldamers by sequence manipulation. Nat. Chem. 2015, 7, 871–878. 10.1038/nchem.2353. [DOI] [PubMed] [Google Scholar]
- Kwon S.; Kim B. J.; Lim H.-K.; Kang K.; Yoo S. H.; Gong J.; Yoon E.; Lee J.; Choi I. S.; Kim H.; Lee H.-S. Magnetotactic molecular architectures from self-assembly of β-peptide foldamers. Nat. Commun. 2015, 6, 8747–8753. 10.1038/ncomms9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloha R. W.; Maeda A.; Dean T.; Gardella T. J.; Gellman S. H. Backbone modification of a polypeptide drug alters duration of action in vivo. Nat. Biotechnol. 2014, 32, 653–655. 10.1038/nbt.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratto J.; Colombo C.; Stupfel M.; Dawson S. J.; Dolain C.; Langlois d’Estaintot B.; Fischer L.; Granier T.; Laguerre M.; Gallois B.; Huc I. Structure of a Complex Formed by a Protein and a Helical Aromatic Oligoamide Foldamer at 2.1 Å Resolution. Angew. Chem., Int. Ed. 2014, 53, 883–887. 10.1002/anie.201309160. [DOI] [PubMed] [Google Scholar]
- Mayer C.; Müller M. M.; Gellman S. H.; Hilvert D. Building proficient enzymes with foldamer prostheses. Angew. Chem., Int. Ed. 2014, 53, 6978–6981. 10.1002/anie.201400945. [DOI] [PubMed] [Google Scholar]
- Wang P. S. P.; Nguyen J. B.; Schepartz A. Design and High-Resolution Structure of a β3-Peptide Bundle Catalyst. J. Am. Chem. Soc. 2014, 136, 6810–6813. 10.1021/ja5013849. [DOI] [PubMed] [Google Scholar]
- Wolffs M.; Delsuc N.; Veldman D.; Anh N. V.; Williams R. M.; Meskers S. C. J.; Janssen R. A. J.; Huc I.; Schenning A. P. H. J. Helical aromatic oligoamide foldamers as organizational scaffolds for photoinduced charge transfer. J. Am. Chem. Soc. 2009, 131, 4819–4829. 10.1021/ja809367u. [DOI] [PubMed] [Google Scholar]
- Hamuro Y.; Schneider J. P.; DeGrado W. F. De Novo Design of Antibacterial β-Peptides. J. Am. Chem. Soc. 1999, 121, 12200. 10.1021/ja992728p. [DOI] [Google Scholar]
- Sadowsky J. D.; Schmitt M. A.; Lee H.-S.; Umezawa N.; Wang S.; Tomita Y.; Gellman S. H. Chimeric (α/β + α)-Peptide Ligands for the BH3-Recognition Cleft of Bcl-xL: Critical Role of the Molecular Scaffold in Protein Surface Recognition. J. Am. Chem. Soc. 2005, 127, 11966–11968. 10.1021/ja053678t. [DOI] [PubMed] [Google Scholar]
- Kritzer J. A.; Lear J. D.; Hodsdon M. E.; Schepartz A. Helical β-Peptide Inhibitors of the p53-hDM2 Interaction. J. Am. Chem. Soc. 2004, 126, 9468–9469. 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]
- Gademann K.; Ernst M.; Hoyer D.; Seebach D. Synthesis and Biological Evaluation of a Cyclo--tetrapeptide as a Somatostatin Analogue. Angew. Chem., Int. Ed. 1999, 38, 1223–1226. . [DOI] [PubMed] [Google Scholar]
- Werder M.; Hauser H.; Abele S.; Seebach D. β-Peptides as Inhibitors of Small-Intestinal Cholesterol and Fat Absorption. Helv. Chim. Acta 1999, 82, 1774–1783. . [DOI] [Google Scholar]
- Porter E. A.; Wang X.; Lee H.-S.; Weisblum B.; Gellman S. H. Non-haemolytic β-amino-acid oligomers. Nature 2000, 404, 565. 10.1038/35007145. [DOI] [PubMed] [Google Scholar]
- Seurynck S. L.; Patch J. A.; Barron A. E. Simple, Helical Peptoid Analogs of Lung Surfactant Protein B. Chem. Biol. 2005, 12, 77–88. 10.1016/j.chembiol.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Patch J. A.; Barron A. E. Helical peptoid mimics of magainin-2 amide. J. Am. Chem. Soc. 2003, 125, 12092–12093. 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- Choi S.; Clements D. J.; Pophristic V.; Ivanov I.; Vemparala S.; Bennett J. S.; Klein M. L.; Winkler J. D.; DeGrado W. F. The Design and Evaluation of Heparin-Binding Foldamers. Angew. Chem., Int. Ed. 2005, 117, 6843–6847. 10.1002/anie.200501279. [DOI] [PubMed] [Google Scholar]
- Berlicki Ł.; Pilsl L.; Wéber E.; Mándity I. M.; Cabrele C.; Martinek T. A.; Fülöp F.; Reiser O. Unique α,β- and α,α,β,β-Peptide Foldamers Based on cis-β-Aminocyclopentanecarboxylic Acid. Angew. Chem., Int. Ed. 2012, 51, 2208–2212. 10.1002/anie.201107702. [DOI] [PubMed] [Google Scholar]
- Cheng R. P.; Gellman S. H.; DeGrado W. F. β-Peptides: From Structure to Function. Chem. Rev. 2001, 101, 3219–3232. 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- Stringer J. R.; Crapster J. A.; Guzei I. A.; Blackwell H. E. Extraordinarily Robust Polyproline Type I Peptoid Helices Generatedviathe Incorporation of α-Chiral AromaticN-1-Naphthylethyl Side Chains. J. Am. Chem. Soc. 2011, 133, 15559–15567. 10.1021/ja204755p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R. J.; Kania R. S.; Zuckermann R. N.; Huebner V. D.; Jewell D. A.; Banville S.; Ng S.; Wang L.; Rosenberg S.; Marlowe C. K. Peptoids: a modular approach to drug discovery. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 9367–9371. 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen J. S.; Harris P.; Fristrup P.; Olsen C. A. Triangular prism-shaped β-peptoid helices as unique biomimetic scaffolds. Nat. Commun. 2015, 6, 7013–7022. 10.1038/ncomms8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L.; Claudon P.; Pendem N.; Miclet E.; Didierjean C.; Ennifar E.; Guichard G. The canonical helix of urea oligomers at atomic resolution: insights into folding-induced axial organization. Angew. Chem., Int. Ed. 2009, 49, 1067–1070. 10.1002/anie.200905592. [DOI] [PubMed] [Google Scholar]
- Proulx C.; Sabatino D.; Hopewell R.; Spiegel J.; García Ramos Y.; Lubell W. D. Azapeptides and their therapeutic potential. Future Med. Chem. 2011, 3, 1139–1164. 10.4155/fmc.11.74. [DOI] [PubMed] [Google Scholar]
- Malachowski W. P.; Tie C.; Wang K.; Broadrup R. L. The Synthesis of Azapeptidomimetic β-Lactam Molecules as Potential Protease Inhibitors. J. Org. Chem. 2002, 67, 8962–8969. 10.1021/jo026280d. [DOI] [PubMed] [Google Scholar]
- Wilhelm P.; Lewandowski B.; Trapp N.; Wennemers H. A crystal structure of an oligoproline PPII-helix, at last. J. Am. Chem. Soc. 2014, 136, 15829–15832. 10.1021/ja507405j. [DOI] [PubMed] [Google Scholar]
- Zhang D.-W.; Zhao X.; Hou J.-L.; Li Z.-T. Aromatic amide foldamers: structures, properties, and functions. Chem. Rev. 2012, 112, 5271–5316. 10.1021/cr300116k. [DOI] [PubMed] [Google Scholar]
- Garric J.; Léger J.-M.; Huc I. Molecular apple peels. Angew. Chem., Int. Ed. 2005, 44, 1954–1958. 10.1002/anie.200462898. [DOI] [PubMed] [Google Scholar]
- Helsel A. J.; Brown A. L.; Yamato K.; Feng W.; Yuan L.; Clements A. J.; Harding S. V.; Szabo G.; Shao Z.; Gong B. Highly conducting transmembrane pores formed by aromatic oligoamide macrocycles. J. Am. Chem. Soc. 2008, 130, 15784–15785. 10.1021/ja807078y. [DOI] [PubMed] [Google Scholar]
- Barnard A.; Long K.; Martin H. L.; Miles J. A.; Edwards T. A.; Tomlinson D. C.; Macdonald A.; Wilson A. J. Selective and potent proteomimetic inhibitors of intracellular protein-protein interactions. Angew. Chem., Int. Ed. 2015, 54, 2960–2965. 10.1002/anie.201410810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
- Pelay-Gimeno M.; Glas A.; Koch O.; Grossmann T. N. Structure-based design of inhibitors of protein-protein interactions: mimicking peptide binding epitopes. Angew. Chem., Int. Ed. 2015, 54, 8896–8927. 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S.; Nguyen H. D.; Phan T. T. P.; Burton M. F.; Nieto L.; de Vries-van Leeuwen I. J.; Schmidt A.; Goodarzifard M.; Agten S. M.; Rose R.; Ottmann C.; Milroy L.-G.; Brunsveld L. Proline primed helix length as a modulator of the nuclear receptor-coactivator interaction. J. Am. Chem. Soc. 2013, 135, 4364–4371. 10.1021/ja311748r. [DOI] [PubMed] [Google Scholar]
- Grison C. M.; Miles J. A.; Robin S.; Wilson A. J.; Aitken D. J. An α-Helix-Mimicking 12,13-Helix: Designed α/β/γ-Foldamers as Selective Inhibitors of Protein-Protein Interactions. Angew. Chem., Int. Ed. 2016, 55, 11096–11100. 10.1002/anie.201604517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzarito V.; Miles J. A.; Fisher J.; Edwards T. A.; Warriner S. L.; Wilson A. J. Stereocontrolled protein surface recognition using chiral oligoamide proteomimetic foldamers. Chem. Sci. 2015, 6, 2434–2443. 10.1039/c4sc03559c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T.; Durell S. R.; Myers M. C.; Appella D. H. Probing the Structural Requirements of Peptoids That Inhibit HDM2–p53 Interactions. J. Am. Chem. Soc. 2006, 128, 1995–2004. 10.1021/ja056344c. [DOI] [PubMed] [Google Scholar]
- Lao B. B.; Drew K.; Guarracino D. A.; Brewer T. F.; Heindel D. W.; Bonneau R.; Arora P. S. Rational design of topographical helix mimics as potent inhibitors of protein-protein interactions. J. Am. Chem. Soc. 2014, 136, 7877–7888. 10.1021/ja502310r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista A. D.; Appelbaum J. S.; Craig C. J.; Michel J.; Schepartz A. Bridged β3-peptide inhibitors of p53-hDM2 complexation: correlation between affinity and cell permeability. J. Am. Chem. Soc. 2010, 132, 2904–2906. 10.1021/ja910715u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Lee G.-i.; Park H. S.; Payne G. A.; Rodriguez J. M.; Sebti S. M.; Hamilton A. D. Terphenyl-based helical mimetics that disrupt the p53/HDM2 interaction. Angew. Chem., Int. Ed. 2005, 44, 2704–2707. 10.1002/anie.200462316. [DOI] [PubMed] [Google Scholar]
- Shaginian A.; Whitby L. R.; Hong S.; Hwang I.; Farooqi B.; Searcey M.; Chen J.; Vogt P. K.; Boger D. L. Design, Synthesis, and Evaluation of an α-Helix Mimetic Library Targeting Protein–Protein Interactions. J. Am. Chem. Soc. 2009, 131, 5564–5572. 10.1021/ja810025g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchey L. K.; Porter J. R.; Ghosh I.; Arora P. S. High Specificity in Protein Recognition by Hydrogen-Bond-Surrogate α-Helices: Selective Inhibition of the p53/MDM2 Complex. ChemBioChem 2010, 11, 2104–2107. 10.1002/cbic.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Song T.; Feng Y.; Guo Z.; Fan Y.; Xu W.; Liu L.; Wang A.; Zhang Z. Bcl-2/MDM2 Dual Inhibitors Based on Universal Pyramid-Like α-Helical Mimetics. J. Med. Chem. 2016, 59, 3152–3162. 10.1021/acs.jmedchem.5b01913. [DOI] [PubMed] [Google Scholar]
- Patgiri A.; Joy S. T.; Arora P. S. Nucleation effects in peptide foldamers. J. Am. Chem. Soc. 2012, 134, 11495–11502. 10.1021/ja301953j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakaran P.; Barnard A.; Murphy N. S.; Kilner C. A.; Edwards T. A.; Wilson A. J. Aromatic Oligoamide Foldamers with a ″Wet Edge″ as Inhibitors of the α-Helix-Mediated p53-hDM2 Protein-Protein Interaction. Eur. J. Org. Chem. 2013, 2013, 3504–3512. 10.1002/ejoc.201300069. [DOI] [Google Scholar]
- Saraogi I.; Hamilton A. D. α-Helix mimetics as inhibitors of protein–protein interactions. Biochem. Soc. Trans. 2008, 36, 1414–1417. 10.1042/bst0361414. [DOI] [PubMed] [Google Scholar]
- Murray J. K.; Gellman S. H. Targeting protein-protein interactions: lessons from p53/MDM2. Biopol.—Pept. Sci. 2007, 88, 657–686. 10.1002/bip.20741. [DOI] [PubMed] [Google Scholar]
- Zhan C.; Zhao L.; Wei X.; Wu X.; Chen X.; Yuan W.; Lu W.-Y.; Pazgier M.; Lu W. An Ultrahigh Affinityd-Peptide Antagonist Of MDM2. J. Med. Chem. 2012, 55, 6237–6241. 10.1021/jm3005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown C. J.; Lain S.; Verma C. S.; Fersht A. R.; Lane D. P. Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- Gilkes D. M.; Chen J. Distinct roles of MDMX in the regulation of p53 response to ribosomal stress. Cell Cycle 2007, 6, 151–155. 10.4161/cc.6.2.3719. [DOI] [PubMed] [Google Scholar]
- Michel J.; Harker E. A.; Tirado-Rives J.; Jorgensen W. L.; Schepartz A. In Silico Improvement of β3-Peptide Inhibitors of p53hDM2 and p53hDMX. J. Am. Chem. Soc. 2009, 131, 6356–6357. 10.1021/ja901478e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan J.; Li Z.; Kasprzak A.; Li B.; Sebti S.; Guida W.; Schönbrunn E.; Chen J. Structure-based design of high affinity peptides inhibiting the interaction of p53 with MDM2 and MDMX. J. Biol. Chem. 2010, 285, 2174–2183. 10.1074/jbc.m109.073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Cheng Q.; Li Z.; Chen J. p53 inactivation by MDM2 and MDMX negative feedback loops in testicular germ cell tumors. Cell Cycle 2010, 9, 1411–1420. 10.4161/cc.9.7.11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; She F.; Gao W.; Prince A.; Li Y.; Wei L.; Mercer A.; Wojtas L.; Ma S.; Cai J. The synthesis of head-to-tail cyclic sulfono-γ-AApeptides. Org. Biomol. Chem. 2015, 13, 672–676. 10.1039/c4ob02232g. [DOI] [PubMed] [Google Scholar]
- Wu H.; Teng P.; Cai J. Rapid Access to Multiple Classes of Peptidomimetics from Common γ-AApeptide Building Blocks. Eur. J. Org. Chem. 2014, 2014, 1760–1765. 10.1002/ejoc.201301841. [DOI] [Google Scholar]
- Shi Y.; Teng P.; Sang P.; She F.; Wei L.; Cai J. γ-AApeptides: Design, Structure, and Applications. Acc. Chem. Res. 2016, 49, 428–441. 10.1021/acs.accounts.5b00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng P.; Shi Y.; Sang P.; Cai J. γ-AApeptides as a New Class of Peptidomimetics. Chem.—Eur. J. 2016, 22, 5458–5466. 10.1002/chem.201504936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng P.; Ma N.; Cerrato D. C.; She F.; Odom T.; Wang X.; Ming L.-J.; van der Vaart A.; Wojtas L.; Xu H.; Cai J. Right-handed helical foldamers consisting of de novo d-AApeptides. J. Am. Chem. Soc. 2017, 139, 7363–7369. 10.1021/jacs.7b03007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng P.; Niu Z.; She F.; Zhou M.; Sang P.; Gray G. M.; Verma G.; Wojtas L.; van der Vaart A.; Ma S.; Cai J. Hydrogen-bonding-driven 3D supramolecular assembly of peptidomimetic zipper. J. Am. Chem. Soc. 2018, 140, 5661–5665. 10.1021/jacs.7b11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng P.; Gray G. M.; Zheng M.; Singh S.; Li X.; Wojtas L.; van der Vaart A.; Cai J. Orthogonal Halogen-Bonding-Driven 3D Supramolecular Assembly of Right-Handed Synthetic Helical Peptides. Angew. Chem., Int. Ed. 2019, 58, 7778–7782. 10.1002/anie.201903259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She F.; Teng P.; Peguero-Tejada A.; Wang M.; Ma N.; Odom T.; Zhou M.; Gjonaj E.; Wojtas L.; van der Vaart A.; Cai J. De novo left-handed synthetic peptidomimetic foldamers. Angew. Chem., Int. Ed. 2018, 57, 9916–9920. 10.1002/anie.201805184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang P.; Zhang M.; Shi Y.; Li C.; Abdulkadir S.; Li Q.; Ji H.; Cai J. Inhibition of β-catenin/B cell lymphoma 9 protein–protein interaction using α-helix-mimicking sulfono-γ-AApeptide inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 10757–10762. 10.1073/pnas.1819663116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y.; Hu Y.; Li X.; Chen J.; Cai J. γ-AApeptides: design, synthesis and evaluation. New J. Chem. 2011, 35, 542–545. 10.1039/c0nj00943a. [DOI] [Google Scholar]
- Knight S. M. G.; Umezawa N.; Lee H.-S.; Gellman S. H.; Kay B. K. A fluorescence polarization assay for the identification of inhibitors of the p53-DM2 protein-protein interaction. Anal. Biochem. 2002, 300, 230–236. 10.1006/abio.2001.5468. [DOI] [PubMed] [Google Scholar]
- Chen L.; Yin H.; Farooqi B.; Sebti S.; Hamilton A. D.; Chen J. p53 -Helix mimetics antagonize p53/MDM2 interaction and activate p53. Mol. Cancer Ther. 2005, 4, 1019–1025. 10.1158/1535-7163.mct-04-0342. [DOI] [PubMed] [Google Scholar]
- Sawyer S. A.; Parsch J.; Zhang Z.; Hartl D. L. Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6504–6510. 10.1073/pnas.0701572104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borcherds W.; Theillet F.-X.; Katzer A.; Finzel A.; Mishall K. M.; Powell A. T.; Wu H.; Manieri W.; Dieterich C.; Selenko P.; Loewer A.; Daughdrill G. W. Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat. Chem. Biol. 2014, 10, 1000–1002. 10.1038/nchembio.1668. [DOI] [PubMed] [Google Scholar]
- Dougherty P. G.; Sahni A.; Pei D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. 10.1021/acs.chemrev.9b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdine G. L.; Hilinski G. J.. Stapled peptides for intracellular drug targets. In Methods in Enzymology; Wittrup K. D., Verdine G. L., Eds.; Academic Press, 2012; Vol. 503, pp 3–33. [DOI] [PubMed] [Google Scholar]
- Chu Q.; Moellering R. E.; Hilinski G. J.; Kim Y.-W.; Grossmann T. N.; Yeh J. T.-H.; Verdine G. L. Towards understanding cell penetration by stapled peptides. MedChemComm 2015, 6, 111–119. 10.1039/c4md00131a. [DOI] [Google Scholar]
- Gilkes D. M.; Pan Y.; Coppola D.; Yeatman T.; Reuther G. W.; Chen J. Regulation of MDMX expression by mitogenic signaling. Mol. Cell. Biol. 2008, 28, 1999–2010. 10.1128/mcb.01633-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Wu H.; Teng P.; Bai G.; Lin X.; Zuo X.; Cao C.; Cai J. Helical Antimicrobial Sulfono-γ-AApeptides. J. Med. Chem. 2015, 58, 4802–4811. 10.1021/acs.jmedchem.5b00537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.