

Abstract
Schizophrenia is characterized by increased behavioral and neurochemical responses to dopamine-releasing drugs. This prompted the hypothesis of psychosis as a state of “endogenous” sensitization of the dopamine system although the exact basis of dopaminergic disturbances and the possible role of prefrontal cortical regulation have remained uncertain. To show that patients with first-episode psychosis release more dopamine upon amphetamine-stimulation than healthy volunteers, and to reveal for the first time that prospective sensitization induced by repeated amphetamine exposure increases dopamine-release in stimulant-naïve healthy volunteers to levels observed in patients, we collected data on amphetamine-induced dopamine release using the dopamine D2/3 receptor agonist radioligand [11C]-(+)-PHNO and positron emission tomography. Healthy volunteers (n = 28, 14 female) underwent a baseline and then a post-amphetamine scan before and after a mildly sensitizing regimen of repeated oral amphetamine. Unmedicated patients with first-episode psychosis (n = 21; 6 female) underwent a single pair of baseline and then post-amphetamine scans. Furthermore, T1 weighted magnetic resonance imaging of the prefrontal cortex was performed. Patients with first-episode psychosis showed larger release of dopamine compared to healthy volunteers. After sensitization of healthy volunteers their dopamine release was significantly amplified and no longer different from that seen in patients. Healthy volunteers showed a negative correlation between prefrontal cortical volume and dopamine release. There was no such relationship after sensitization or in patients. Our data in patients with untreated first-episode psychosis confirm the “endogenous sensitization” hypothesis and support the notion of impaired prefrontal control of the dopamine system in schizophrenia.
Subject terms: Molecular neuroscience, Schizophrenia
Introduction
Several lines of evidence demonstrate increased subcortical dopamine (DA) transmission in psychotic patients with schizophrenia (SCZ). Positron emission tomography (PET) studies show increased dopamine synthesis capacity and heightened behavioral and neurochemical responses towards DA-releasing compounds1–6. The common mechanism of action of all antipsychotic drugs—reducing DA transmission at postsynaptic D2/3 receptors—confirms the key role of DA signaling in psychosis7. While the pathophysiological basis of DA dysfunction in SCZ remains unknown, current versions of the DA theory of SCZ8 posit that upstream pathogenic factors converge on subcortical DA pathways to mediate the expression and intensity of psychotic symptoms9–11.
Sensitization denotes a process by which repeated exposure to a stimulus induces a progressive increase in responses to the very same stimulus12,13. When repeatedly administered, d-amphetamine (AMPH) induces behavioral sensitization and a progressive amplification in AMPH-induced DA release14–16. Since psychotic patients show elevated responses to AMPH without any prior drug exposure1, psychosis has been conceptualized as a state of “endogenous sensitization”10,17,18. The prefrontal cortex (PFC), origin of reciprocal regulatory connections to subcortical DA neurons19, has often been found to be structurally and functionally impaired in SCZ20,21. Earlier studies have shown particularly strong relationships between subcortical DA metabolism and the left-hemispheric dorso-lateral PFC (DLPFC) and inferior frontal gyrus (IFG) in SCZ and subjects at-risk mental state for SCZ22,23. In order to collect experimental evidence supporting this concept, we used PET and the DA D2/3 receptor agonist radioligand (+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin-9-ol ([11C]-(+)-PHNO)24 for measuring AMPH-induced changes in D2/3 receptor binding, semi-quantitative index of DA release, in drug-naïve patients with first-episode psychosis (FEP). Healthy volunteers (HV) were studied before and after exposure to a mildly sensitizing regime of repeated AMPH administration. In order to identify upstream pathogenic mechanisms of psychotic hyperdopaminergia, we analyzed volumetric parameters in the PFC for their relationship to indices of subcortical DA release.
Materials and methods
All procedures of this study (Clinical Trial Registry: EUDRACT 2010-019586-29) were approved by the ethics committee of the Medical University of Vienna and pertinent federal regulatory authorities. After a test-retest phase ensuring reliability of local [11C]-(+)-PHNO PET imaging procedures (six male HV), 42 HV and 29 antipsychotic-naïve (or minimally exposed) patients with FEP capable of providing informed consent were recruited between 2013 and 2017; HV were required to be of good health based on physical examination, history, ECG, and laboratory results. Exclusion criteria comprised any intake of drugs of abuse except nicotine, caffeine, and alcohol (occasional use only), five or more stimulant exposures lifetime, psychiatric disorders (evaluated with the DSM-IV based M.I.N.I. questionnaire25), having a first-degree relative with schizophrenia or bipolar disorder or having any contraindications against receiving a PET, MRI or d-amphetamine; in addition, FEP patients were required to have a minimum Positive and Negative Symptom Scale (PANSS26,27) score of 55 with >3 on at least two PANSS psychosis items or >4 on one psychosis item; no or minimal lifetime exposure to antipsychotics; no lifetime exposure to antipsychotic depot preparations; no antipsychotics within two weeks prior to scanning (for details see Supplemental Material). Diagnoses of FEP in SCZ according to DSM-IV were independently made by at least two experienced psychiatrists (N.M.; N.P-R., S.K., M.W.). Three patients were previously exposed to olanzapine, aripiprazole, or quetiapine. Antipsychotics had been discontinued at least two months prior to inclusion without attaining a predefined threshold of two treatment weeks or lifetime exposure up to 50 mg haloperidol-equivalent. Three patients had a history of antidepressant treatment discontinued at least two months prior to inclusion. During the study, eleven patients required symptomatic treatment for psychomotor agitation or insomnia (lorazepam 1–10 mg or zolpidem 10 mg per day). Data sets (complete or partial) of 28 + 6 HV and 21 FEP patients entered final analysis (see Supplemental Material and Supplementary Table 1 for full details).
Study setup and prospective sensitization
First, drug-naïve HV (HVUNSENS) underwent an AMPH-free [11C]-(+)-PHNO PET scan (baseline1, PET1). On a separate day less than five 5 days apart, a second scan (PET2) was performed 90–120 min after oral administration of 0.4 mg/kg body weight AMPH (Attentin®, MEDICE Arzneimittel GmbH, Iserlohn, D), a time point at which subjective effects are peaking and blood levels are still rising (blood levels are highest after 3–4 h28). The AMPH dose was chosen according to earlier studies showing that dosages between 0.3 and 0.5 mg/kg body weight induce reliable reductions in [11C]-(+)-PHNO BPND values29,30. The dose corresponds to a low to medium dose used for treating attention deficit-hyperactivity disorder in children. Administration of AMPH at this same dose was repeated two more times at intervals of two days. Two to four weeks thereafter, the now sensitized HV (HVSENS) underwent another AMPH-free scan (baseline2, PET3), and within five days, a fourth scan (PET4) preceded by AMPH dose four (Fig. 1, lower panel).
Fig. 1. Amphetamine (AMPH)-induced dopamine (DA) release and subjective AMPH effects in healthy volunteers before and after AMPH-sensitization and in drug-free patients with first-episode psychosis (FEP).

Upper panel: a AMPH-induced DA release in five subdivisions of the basal ganglia (CAU Caudate, PUT Putamen, VST Ventral Striatum, GP Globus Pallidus, SNVTA substantia nigra/ventral-tegmental area) in healthy volunteers before (HVUNSENS; sky-blue) and after prospective sensitization to AMPH (HVSENS; deep-blue), and in antipsychotic-naïve patients FEP (red). Patients show larger AMPH-induced DA release than HVUNSENS. Prospective sensitization of HVUNSENS induced by repeated AMPH administration amplifies DA release, such that the AMPH response in HVSENS no longer differs from FEP. b Statistical parametric maps displaying brain areas with the largest sensitization-induced increases in DA-release. Peak effects (up to 60 percent increase) are found in VST (MNI coordinates x = −19, y = −16, z = −10). c Subjective AMPH effects in HV undergoing prospective sensitization to AMPH. Repeated administration (four times; light to deep-blue) of AMPH at low constant dose (0.3 mg/kg body weight) successfully induced sensitization as shown by the progressive increase in the AMPH response. Patients with FEP (red; one administration of AMPH) displayed marked AMPH effects already at first contact with the drug. SSQ Subjective States Questionnaire; MNI Montreal Neurologic Institute standard space; bars represent mean ± standard error of the mean; post-hoc two-tailed t-tests: *p < 0.05, **p < 0.01). Lower panel: Study flowchart.
Patients with FEP received one baseline scan (PET1) and one AMPH-scan (PET2; protocol as above). AMPH led to temporary increases in heart rate and blood pressure (Supplementary Fig. 1). Occasionally, participants reported mild headache and insomnia the night after AMPH administration. Neither HV nor patients experienced any serious AMPH-related adverse events.
[11C]-(+)-PHNO PET and MR imaging
[11C]-(+]-PHNO was synthesized as described earlier31. Quality control was in accordance with European Pharmacopoeia. PET images were acquired on a GE Advance scanner (General Electric Medical Systems, Milwaukee, WI). Emission data were acquired over 90 min after bolus-injection of 309 [81] MBq (mean [SD]) [11C]-(+)-PHNO. Raw data were reconstructed by filtered-back projection to yield dynamic images in 15 consecutive one-minute frames followed by 15 five-minute frames. With exception of two patients who chose to terminate their participation early, all subjects underwent T1 and proton density (PD) weighted 3 T magnetic resonance (MR) imaging (see Supplemental Material).
Behavioral and hormonal measurements
Subjective AMPH effects were recorded using the drug effects questionnaire32 and the subjective states questionnaire (SSQ)33. The PANSS scale26 was administered by certified raters for measuring baseline psychopathology in FEP patients, the brief psychiatric rating scale (BPRS34) was used to measure AMPH-induced changes in psychopathology. Blood for serum AMPH levels was collected at the beginning of PET scans. Heart rate, blood pressure (systolic and diastolic were determined repeatedly before and during PET scans (see Supplemental Material).
Image analysis
Analysis in regions of interest
Frame-wise motion correction and co-registration of attenuation-corrected average PET images to T1-weighted MRIs was performed using AFNI software. PET images of two FEP patients who did not undergo MR imaging (see above) were co-registered to normalized [11C]-(+)-PHNO template images (one each for no-intervention and AMPH scans) created by averaging spatially normalized PET images (early and late low-contrast frames omitted) of 15 HV. Individually optimized bilateral regions of interest (ROIs) were obtained for the caudate nucleus (CAU), putamen (PUT), ventral striatum (VST), and cortical cerebellum (CER) using the automated image analysis software ROMI35 as described elsewhere30. Since automated algorithms provided no satisfactory ROI delineation in globus pallidus (GP) and substantia nigra/ventral tegmental area (SNVTA), GP and SN/VTA were delineated manually by a single rater on individual PD MR images (fused with PET images for aiding delineation of SNVTA). ROI delineation was evaluated independently by a second rater visually controlling for anatomical fit and assessing outliers and time drift in ROI sizes. Decay-corrected time–activity curves (TACs) were extracted from the dynamic sequence. The simplified reference tissue model (SRTM2)36,37 implemented in PMOD software (Version 3.6; PMOD Technologies Ltd, Zurich, Switzerland) was used to derive binding potential (BPND) values in each ROI. Cerebellar cortex (CER) avoiding midline structures served as reference region since it is virtually devoid of DA D2/3 receptors in humans38–40. Relative AMPH-induced change in [11C]-(+)-PHNO BPND binding was calculated as [(BPND baseline – BPND AMPH)/BPND baseline * 100] (ΔBPND; for the sake of simplicity henceforth designated “DA release”).
Parametric analysis
Voxel-wise BPND maps were calculated using the PMOD 3.6 SRTM basis function implementation41. TACs previously derived in CER (low-binding) and VST (high-binding) were used to optimize iterative model fitting procedures. Effects of AMPH and AMPH-sensitization were analyzed using the AFNI programs 3dttest++ and 3dLME.
Volumetric analyses
T1-weighted MR images were processed using Freesurfer 6.0 software (http://surfer.nmr.mgh.harvard.edu/). Cortical gray voxels were allocated to a set of regions predefined in the Destrieux atlas42 using a Bayesian algorithm. Skull-stripping, gross accuracy of delineation, and surface modeling were quality-controlled by visual inspection of processed images.
Statistical analysis
The sample size of this study was planned according to data collected with antagonist radioligands in sensitization and patients with schizophrenia3–5,15. Statistical analysis of [11C]-(+)-PHNO BPND values obtained in the ROI-based analysis of [11C]-(+)-PHNO binding was carried out using mixed linear models (MLM) as implemented in SPSS 24.0 (SPSS 24.0; IBM Corp., Armonk, NY) and R 12.0.1 (R Foundation for Statistical Computing, Vienna, Austria; https://www.r-project.org) software. Analysis of differences in DA release (ΔBPND) between HV before and after sensitization was carried out using ΔBPND as dependent variable and group (FEP vs. HVUNSENS, and FEP vs. HVSENS, respectively) as fixed variable; ROI was entered as random variable. For analyzing receptor binding, BPND was entered as dependent variable, subject status (HVUNSENS, HVSENS, FEP) as fixed between-group factor, scan condition (AMPH yes/no) as fixed repeated factor, and ROI as fixed or random repeated factor. MLM analyses testing the effects of covariates (PFC volume parameters) were carried out analogously. All main effects and all relevant interactions were entered into the respective models. The subject-identifier variable was entered as random factor where appropriate. Two-tailed t-tests (paired where appropriate) were used for post-hoc tests after assuring normal distribution. Correlations were calculated using Pearson product moment (r) or, if appropriate, Spearman rank sum (rho) correlation coefficients. Results confirming the main a-priori study hypotheses (Figs. 1, 2) were not corrected for multiple comparisons. More exploratory results (Figs. 3–5) were carried out using ROI-based and parametric methods (voxel-wise maps for PET images, vertex-wise analysis of MR images) for independent confirmation. Results of the ROI-based analysis on the relationship between PANSS items and BPND values (Fig. 3; 30 PANSS items, 5 ROIs, two hemispheres and on/off AMPH) and the relationship between regional cortical volumes as implemented in Freesurfer 6.0 software42 and ΔBPND (Fig. 4; 33 cortical regions, 5 ROIs, two hemispheres; three groups) were Bonferroni corrected, resulting in adjusted significance levels of pcorr1 = 0.00008 and pcorr2 = 0.00005, respectively.
Fig. 2. Dopamine (DA) D2/3 receptor binding ([11C]-(+)-PHNO BPND values) in scans without (PET1 and PET3) and with prior administration of amphetamine (AMPH; PET2 and PET4).

a Panels show binding in five subcortical regions of interest (ROIs; CAU, caudate; PUT, putamen; VST, ventral striatum; GP, globus pallidus; SNVTA, substantia nigra/ventral-tegmental area) in healthy volunteers before (HVUNSENS; sky-blue) and after AMPH sensitization (HVSENS; deep-blue) and in patients with first-episode psychosis (FEP; red). X-axis break between PET2 and PET3 in HV indicates a scan-free interval (2–4 weeks) during prospective AMPH sensitization. All direct AMPH effects (PET1 vs. PET2 HVUNSENS and FEP, PET3 vs. PET4 HVsens) were significant (paired t-tests p = 0.03–2.5 × 10−5; not marked). Note that AMPH sensitization led to a significant increase in D2/3 receptor binding from PET1 to PET3 in VST. b Alternative representation of data shown in a highlighting systematic differences in D2/3 receptor binding across conditions between HV and patients with FEP. D2/3 binding in FEP is lower than in HV in D2 receptor-rich neo-striatal regions (CAU, PUT, VST), while it is elevated in D3 receptor-rich regions of the paleo-striatum (GP and SNVTA). Error bars represent 95% confidence intervals. *p < 0.05, **p < 0.01, ***p < 0.001, post-hoc two-tailed t-test.
Fig. 3. Amphetamine (AMPH)-induced changes in psychopathology and relationship between [11C]-(+)-PHNO binding and emotional withdrawal in patients with first-episode psychosis (FEP).

a Changes in brief psychiatric rating scale (BPRS) positive and b negative symptom scores at different time-points after AMPH administration in patients with FEP. Values were calculated as increase or decrease from baseline scores. Bars represent mean ± standard error of the mean. Psychopathological changes did not require medical intervention and returned to baseline levels shortly after PET scanning. c Correlation between ‘emotional withdrawal’ and [11C]-(+)-PHNO BPND values in the right putamen of patients with FEP. d Voxel-wise analysis showing the correlation between severity of the negative SCZ symptom ‘emotional withdrawal’ (PANSS item N2) and [11C]-(+)-PHNO BPND values in patients with FEP (pcorr < 0.01; t-max: 5.68 at MNI x = −21, y = 0, z = −11).
Fig. 5. Relationship between volume of the dorsolateral prefrontal cortex (in cm3) and inferior frontal gyrus (DLPFC/IFG) as measured with magnetic resonance imaging and AMPH-induced reductions in non-displaceable binding potential (BPND) values of the dopamine D2/3 receptor positron emission tomography (PET) radioligand [11C]-(+)-PHNO in healthy subjects before and after sensitization to d-amphetamine compared to patients with schizophrenia.
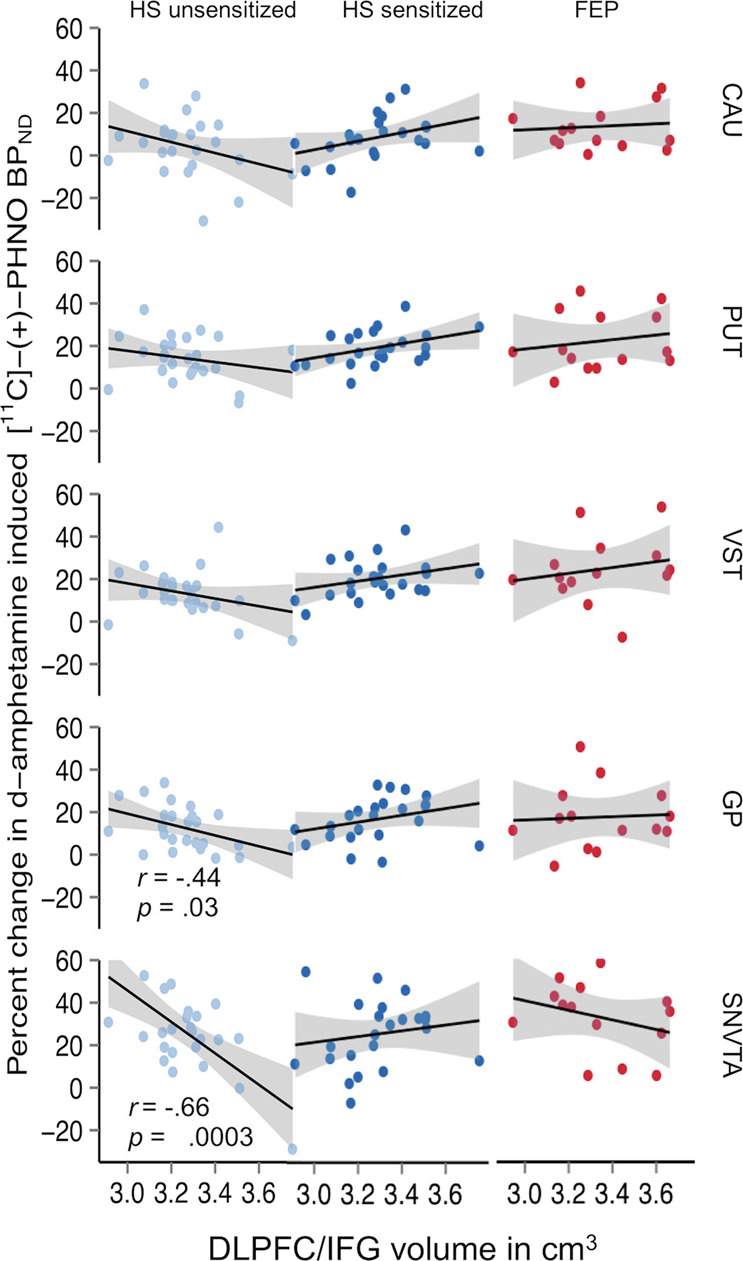
Significant interactions are found in brain regions (GP, SNVTA) where dopamine D3 (in contrast to D2) receptors are the predominant source of signal detected with [11C]-(+)-PHNO and PET. VST, ventral striatum; GP, globus pallidus; SNVTA, substantia nigra/ventral tegmental area.
Fig. 4. Vertex-wise analysis on the relationship between sensitization in dopamine (DA) release (increase in amphetamine-induced changes in [11C]-(+)-PHNO BPND values) and whole-cortex volumetric measures in healthy volunteers (HV).

a The analysis identified largest effects in a left-hemispheric cluster comprising the dorsolateral prefrontal cortex (BA46) and Broca’s area (BA 44 and 45; cluster thresholded at t = −4.3; peak signal at MNI coordinates x = 6, y = −13, z = −7). b Positive relationship between sensitization of DA release and volume of the inferior frontal gyrus (IFG).
Results
DA release in FEP and sensitization
The concept of “endogenous sensitization” in psychosis17 predicts that the difference in AMPH-induced DA release between FEP patients and HV should disappear or substantially diminish after HV are sensitized to AMPH. Analysis of AMPH-induced changes in [11C]-(+)-PHNO binding (ΔBPND) showed significant differences between unsensitized HV and FEP (HVUNSENS vs. FEP: F(1;184.04) = 11.6, p = .0008: Fig. 1a). After HV were sensitized, groups were no longer significantly different (HVSENS vs. FEP: F(1;175.6)) = 0.99, p = .32; Fig. 1a, Supplementary Table 2). Including alcohol and nicotine consumption and sex as covariates into the model did not relevantly alter results (HVUNSENS vs. FEP: F(1;139.9) = 11.6, p = 0.0008, see Supplemental Material for details). In good agreement with the ROI-based analysis, parametric ΔBPND maps showed the most robust effects of sensitization in the VST (Fig. 1b). Indicating behavioral sensitization in HVSENS, repeated AMPH-administration induced progressive enhancement of subjective AMPH effects up to levels observed in FEP (Fig. 1c, Supplementary Fig. 2).
Analysis of group differences and AMPH-effects at the level of D2/3 receptor binding before sensitization ([11C]-(+)-PHNO BPND values by group, condition, and ROI) showed significant two-way interactions between group and condition (HVUNSENS/FEP * no-AMPH/AMPH: F(1;305.7) = 9.9, p = .009), group and ROI (F(4;140) = 10.7, p = 1.3 × 10−7), and a significant three-way interaction between group, condition, and ROI (F(8;140) = 3.0, p = .004).
The same model, when applied to FEP and HV after sensitization, did no longer show a significant two-way interaction between group and condition (F(1;275.8) = 0.6, p = 0.42). However, the group*ROI (F(4;123) = 11.2, p = 9.4 × 10−8), and a group*condition* ROI interactions remained significant, (F(8;123.1) = 5.5, p = 5 × 10−5; Fig. 2a, b). At least in part, this is due to the fact that irrespective of AMPH pretreatment, D2/3 receptor binding was consistently lower in FEP than in HVs in neo-striatal ROIs (CAU, PUT, VST) but higher in GP (and trend-wise also in SNVTA; Fig. 2b; Supplementary Table 3). Baseline D2/3 receptor binding and sensitization showed a significant positive correlation for all regions in HV (see Supplementary Fig. 3). An analysis of functional subdivisions of the basal ganglia as previously published did not reflect our findings (Supplementary Fig. 4).
Indices of DA release were positively correlated with behavioral AMPH effects (see Supplemental Material). It is known that benzodiazepines might interact with dopamine neurotransmission43. Thus we compared DA release between patients receiving benzodiazepines and those who did not require sedation, we found no significant difference (16 available datasets for calculating DA release, 7 not receiving lorazepam, 9 receiving lorazepam; two-sided t-test p > 0.05 for all ROIs).
AMPH effects on psychopathology
With a mean [SD] PANSS score of 82.2 [6.9] (subscales for positive, negative, and general SCZ symptoms: 21.4 [6.8], 20.1 [6.1], and 40.8 [9.4]), FEP patients were suffering from acute psychosis of moderate to marked severity at the time of PET scanning. Administration of AMPH prompted a temporary increase in psychotic symptoms. Symptoms returned to baseline after 3–4 h without specific intervention. While positive symptoms of SCZ (in particular hallucinations, delusions, and thought-disorder) increased with AMPH, negative (especially blunted affect and emotional withdrawal) and affective symptoms (depressed mood) improved (Fig. 3a, b; Supplementary Table 4). In contrast to earlier studies3–6,44–46 but in line with a recent study using the D2/3 receptor agonist radioligand [11C]N-propyl-apomorphine ([11C]NPA47), we did not observe significant relationships between positive symptoms and DA release. However, there were significant relationships between [11C]-(+)-PHNO BPND values in the AMPH condition and negative symptoms. Correlations were strongest in PUT and driven mainly by the PANSS item N2 (“emotional withdrawal”; right PUT: rho = 0.93, p = 7.7 × 10−8; Fig. 3c, d; Supplementary Fig. 5). Typical behavioral effects of AMPH in healthy volunteers included increased production of speech, extraversion, alertness, and enhanced energy.
PFC volume and subcortical DA release
Dysfunctional PFC-DA interactions are prime candidates for “upstream” pathogenic mechanisms underlying subcortical hyperdopaminergia in patients with SCZ. Furthermore, brain circuits involved in cognitive functions encompassing the PFC are often found to be compromised in patients with SCZ48. Thus we derived volumetric parameters from MR images and, in a first step, analyzed the relationship between indices of DA release and overall PFC volume. Consistent with inhibition of DA release by the PFC19,22, we observed an inverse relationship between PFC volume and DA release in AMPH naïve HVUNSENS. After AMPH sensitization, the association was lost or even reversed and more resembled the pattern observed in FEP patients. An anatomically unbiased vertex-wise analysis of the relationship between sensitization of DA release in SNVTA (ΔBPND-SENS − ΔBPND-UNSENS) and whole-cortex volumetric parameters identified a left-hemispheric cluster spanning the boundaries of middle and inferior frontal gyrus as main driver of the whole-PFC signal (Fig. 4a, b). In a next step, we analyzed correlations between DA release and volumes of individual PFC sub-regions as implemented into Freesurfer 6.0 software according to anatomical and functional priors42. In perfect agreement with the vertex-wise analysis, we observed the by far strongest correlations in three anatomically adjacent left-hemispheric regions: Brodmann areas (BA) 46 (dorsolateral prefrontal cortex; DLPFC), and BA 44 and BA 45, or pars opercularis and triangularis of the left inferior frontal gyrus (IFG), together forming “Broca’s area”49.
Plotting the correlations between volume of these regions (henceforth referred to as DLPFC/IFG) and DA release in the five subcortical ROIs (CAU, PUT, VST, GP, and SNVTA in rostro-caudal anatomical order) revealed a remarkable “herring bone”-like pattern (Fig. 5). This pattern is shaped by negative regression lines—indicating DLPFC/IFG-inhibition of DA release in HVUNSENS—and positive regression lines observed after the same subjects had undergone AMPH sensitization (HVUNSENS vs. HVSENS vs. FEP: F(2;278.9)) = 10.14, p = .00005). We did not observe any significant relationships between subcortical DA release and cortical volumes in FEP (Fig. 5). To account for possible changes in brain volume induced by AMPH sensitization, we derived volumetric parameters in an independent group of AMPH naïve, sex and age matched HVs. There were no significant differences between our group of HVSENS and the independent HV group (Supplementary Fig. 6). Although of restricted power, these results argue against gross volumetric effects arising from our sensitization procedure.
Discussion
This study is the first to directly compare AMPH-induced DA release in patients with FEP to DA release in HV before and after prospective AMPH sensitization. Our data confirm the main hypothesis of this work: subcortical DA transmission in patients with FEP is in a state of “endogenous sensitization” towards AMPH. They thus replicate and extend the results of earlier studies that have used D2/3 receptor antagonist-radioligands for studying DA release in SCZ3–6 and the effects of AMPH sensitization in HV15. In contrast to these studies, we found no relationship between DA release and change in positive symptoms of SCZ. However, we observed a significant correlation between [11C]-(+)-PHNO BPND values after AMPH in the putamen and negative symptoms, in particular emotional withdrawal. Although the interpretation of this finding is not straightforward, the finding was highly significant and is somewhat supported another [11C]-(+)-PHNO study50 that found a negative relationship between stress-induced DA release in SNVTA and negative symptoms in individuals with clinical high risk for schizophrenia (and at trend level, also patients with FEP). Together, these results corroborate the notion that reduced DA transmission is a relevant element in the pathogenesis of negative schizophrenic symptoms.
Our results are at odds with those of a recent study using the D2/3 receptor agonist radioligand [11C]NPA47. In contrast to our results and those obtained with D2/3 antagonist radioligands3–6, the study by Frankle et al. study did not find enhanced AMPH-induced DA release in schizophrenia. Besides differences in clinical variables (as for example a smaller proportion of medication-naïve patients in the Frankle et al. study), the discrepancy could also reflect different properties of the two radioligands. Since the method used in both studies does not allow for reliably distinguishing between the absolute number of receptors and receptor affinity (including changes in apparent affinity induced by competition with endogenous DA51), it remains open how these parameters exactly relate to each other in dopaminergic dysfunction of SCZ52.
Relationships between volumetric and functional data suggest that AMPH-induced DA release in HV at first exposure to the drug is under inhibitory control of the left-hemispheric DLPFC/IFG. At the same time, AMPH sensitization increased DA release to a greater extent in subjects with larger DLPFC/IFG volumes. This relationship was particularly pronounced in GP and SNVTA (Fig. 4), where the [11C]-(+)-PHNO signal is predominantly reflecting binding to DA D3 rather than D2 receptor subtypes53–55. Since it is unlikely, according to our data obtained in an independent HV sample, that AMPH sensitization had induced major volumetric changes, the “herring bone” pattern most likely reflects a true functional shift in control of subcortical DA release by the DLPFC/IFG from inhibition in HVUNSENS towards facilitation in HVSENS. This interpretation is supported by rodent data showing that expression of the sensitized state is critically dependent on the integrity of PFC neuronal tissue and its interaction with subcortical D3 receptors56.
In our study, prospective sensitization of HV led to an increase in DA release to the magnitude observed in FEP. DLPFC/IFG volumes, while showing strong correlations to DA release in HVUNSENS, did not show any significant relationship to DA release in patients with FEP (Fig. 4). While we cannot rule out that this is due to lack of power, the very same region was found to show abnormal cortical folding in SCZ patients with a mean illness duration of two decades and extensive exposure to antipsychotics57. Thus, our data support the long-standing conjecture that the hyperdopaminergic state in SCZ is directly related to a dysfunction in top-down control of subcortical DA transmission by the PFC22,58, and they are consistent with the notion that antipsychotics act by rectifying the consequences of upstream pathogenic factors, which persist despite antipsychotic treatment.
Antipsychotic-sensitive hyperlocomotion is a standard measure for sensitization in rodents and a widely accepted animal model of SCZ. Although not formally quantified in this study, the most evident behavioral effect of AMPH in our participants was an increase in quantitative speech production. Thus, while expressing on a motor level in rodents, enhanced DA transmission in human AMPH-sensitive brain circuits seems to affect primarily neuronal functions involved in the processing of language and speech, making speech being the human equivalent to rodent hyperlocomotion. Alterations in cytoarchitecture introduced into the rodent PFC by disrupting the cytoskeleton of dendritic spines increase subcortical DA levels and induce hyper-locomotion via direct projections between the PFC and SNVTA59. Similar alterations in the DLPFC/IFG of patients with SCZ may cause dysfunctions in hierarchical control within DLPFC/IFG—SNVTA–striato-thalamic loops and may be involved in mediating the autonomous (i.e., not subject to will) production of language in auditory verbal hallucinations of patients with SCZ. Simultaneously activated by certain grammatical constructs60, altered interactions between the DLPFC/IFG and the evolutionarily ancient SNVTA may also be involved in generating the fascinating and inherently grammatical symptom-complex of schizophrenic “ego disturbances”.
As prospective AMPH sensitization in HV successfully mimics the endophenotype of increased DA release observed in “psychotic sensitization”, our data prompt the question why the endophenotype associates with psychotic symptoms in SCZ but is occurring—unless re-exposed to AMPH—without any relevant behavioral effects in HVs. Symptom-provocation studies61 and ecological evidence strongly suggest this to be a matter of dose and intensity of exposure: While an escalation of the AMPH dose induces transient psychotic symptoms in psychiatrically healthy subjects61, subjects with stimulant-use disorders indeed exhibit disproportionally high rates of psychotic disorders62. Interestingly, subjects abusing methamphetamine or cocaine exhibit increased [11C]-(+)-PHNO binding in the GP63,64. What seems to differentiate AMPH-induced from “endogenous psychotic” sensitization in our data—higher [11C]-(+)-PHNO BPND values in GP—may just be a matter of dose and length of exposure to AMPH.
An inherent limitation of our method is that it does not discriminate the effects of extracellular DA levels from changes in maximal D2/3 receptor binding capacity. In addition, although more sensitive towards fluctuations in extracellular DA, the differences between HVs and FEP patients observed with the agonist radioligand [11C]-(+)-PHNO are by no means larger than those observed with antagonist radioligands1–6. However, this could in part also be a consequence of increased D2/3 receptor occupancy due to higher baseline extracellular DA levels in patients with FEP (Fig. 3b). Another limitation might be the fact that some patients required benzodiazepines due to agitation or anxiety. Although there was no significant difference between medicated and unmedicated patients we cannot rule out the possibility that this might be a confounding factor. Furthermore, although sex did not play a role when entered as a covariate, the imbalance between male and female patients in the FEP sample has to be mentioned as a limitation. Lastly, our design has the limitation that we did not expose, mainly for ethical reasons, our FEP patients to a sensitizing regime of repeated AMPH. However, rarely cited earlier work by Strakowski et al.65 has shown that FEP patients fail to show a progressive enhancement in the response to repeated AMPH. This suggests that FEP patients, at least at the level of behavior, are already maximally sensitized. Our data support and extend this finding to a neurochemical level. In summary, we feel confident that our work provides the first direct experimental proof for the hypothesis that the pathogenic substrate underlying psychosis in SCZ is indeed a state of “endogenous sensitization” in subcortical DA systems.
Supplementary information
Acknowledgements
This study was funded by the Austrian Science Fund FWF [Grant No. P23585-B09], the Anniversary Fund of the Austrian National Bank [Grant No. 16723], the Medical Scientific Fund of the Mayor of Vienna (Medizinisch-Wissenschaftlichen Fonds des Bürgermeisters der Bundeshauptstadt Wien) [Grant No. 15189], and the Vienna Science and Technology Fund (WWTF) [Grant No. CS15-033], all granted to M.W., and the Austrian Science Funds FWF [Grant No. F3506-B11] granted to H.H.S.
Author contributions
Drs. Weidenauer, Sauerzopf, Bauer, and Willeit had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Concept and design: Willeit, Bauer. Acquisition, analysis, or interpretation of data: All authors. Drafting of the manuscript: Weidenauer, Bauer, Sauerzopf, Praschak-Rieder, Willeit. Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: Dorffner, Weidenauer, Bauer, Sauerzopf, Rusjan, Willeit. Planning, analysis and interpretation of radiochemistry, laboratory and genetic analyses, structural MR imaging: Pichler, Pfaff, Berrotéran-Infante, Nics, Philippe, Wadsak, Mitterhauser, Sitte, Mitterhauser, Zimprich, Rusjan, Stimpfl, Pezawas, Meyer, Sezen, Rabl.
Conflict of interest
Without relevance to this work, Matthäus Willeit declares to having received speaker honoraria and consulting fees from Janssen-Cilag Pharma GmbH, Austria. Without relevance to this work, Wolfgang Wadsak declares to having received speaker honoraria from GE Healthcare, research grants from Ipsen Pharma, Eckert-Ziegler AG, Scintomics and ITG. WW is a part time eployee of CBmed Ltd (Center for Biomarker Research in Medicine, Graz, Austria). Without relevance to this work Georg Dorffner is employee and shareholder of The Siesta Group GmbH, a clinical trial service provider in the area of electrophysiological measurements such as EEG. Harald Sitte received grants/research support, consulting fees and/or honoraria from AbbVie, Aesca, Amgen, Astellas, AstraZeneca, Astropharma, Chiesi, Gebro, IMH, IIR, Janssen-Cilag Lundbeck, MSD, Mundipharma, Pfizer, Ratiopharm, Roche, Sandoz, Serumwerk Bernburg, Shire, Vertex and is a member of advisory boards of Amgen, Chieri and Sanofi-Aventis. Marcus Hacker received consulting fees and/or honoraria from Bayer Healthcare BMS, Eli Lilly, EZAG, GE Healthcare, Ipsen, ITM, Janssen, Roche, Siemens Healthineers. Rupert Lanzenberger received travel grants and/or conference speaker honoraria from Shire, AstraZeneca, Lundbeck A/S, Dr. Willmar Schwabe GmbH, Orphan Pharmaceuticals AG, Janssen-Cilag Pharma GmbH, and Roche Austria GmbH. Siegfried Kasper received grants/research support, consulting fees and/or honoraria within the last three years from Angelini, AOP Orphan Pharmaceuticals AG, Celegne GmbH, Eli Lilly, Janssen-Cilag Pharma GmbH, KRKA-Pharma, Lundbeck A/S, Mundipharma, Neuraxpharm, Pfizer, Sanofi, Schwabe, Servier, Shire, Sumitomo Dainippon Pharma Co. Ltd. and Takeda. Ana Weidenauer, Martin Bauer, Ulrich Sauerzopf, Lucie Bartova, Lukas Nics, Sarah Pfaff, Cecile Phelippe, Neydher Berroterán-Infante, Verena Pichler, Bernhard Meyer, Ulrich Rabl, Patrick Sezen, Paul Cumming, Thomas Stimpfl, Nilufar Mossaheb, Alexander Zimprich, Pablo Rusjan, Markus Mitterhauser, Lukas Pezawas, and Nicole Praschak-Rieder have no conflict of interest to declare.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Ana Weidenauer, Martin Bauer
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41398-019-0681-5).
References
- 1.Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology. 1987;91:415–433. doi: 10.1007/BF00216006. [DOI] [PubMed] [Google Scholar]
- 2.Fusar-Poli P, Meyer-Lindenberg A. Striatal presynaptic dopamine in schizophrenia, part II: meta-analysis of [(18)F/(11)C]-DOPA PET studies. Schizophrenia Bull. 2013;39:33–42. doi: 10.1093/schbul/sbr180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laruelle M, et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc. Natl Acad. Sci. USA. 1996;93:9235–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breier A, et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc. Natl Acad. Sci. USA. 1997;94:2569–2574. doi: 10.1073/pnas.94.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abi-Dargham A, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am. J. Psychiatry. 1998;155:761–767. doi: 10.1176/ajp.155.6.761. [DOI] [PubMed] [Google Scholar]
- 6.Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol. Psychiatry. 1999;46:56–72. doi: 10.1016/s0006-3223(99)00067-0. [DOI] [PubMed] [Google Scholar]
- 7.Howes OD, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch. Gen. Psychiatry. 2012;69:776–786. doi: 10.1001/archgenpsychiatry.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Rossum JM. The significance of dopamine-receptor blockade for the mechanism of action of neuroleptic drugs. Arch. Internationales de Pharmacodynamie et de Therapie. 1966;160:492–494. [PubMed] [Google Scholar]
- 9.Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am. J. Psychiatry. 2003;160:13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- 10.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III–the final common pathway. Schizophr. Bull. 2009;35:549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winton-Brown TT, Fusar-Poli P, Ungless MA, Howes OD. Dopaminergic basis of salience dysregulation in psychosis. Trends Neurosci. 2014;37:85–94. doi: 10.1016/j.tins.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Pinsker HM, Hening WA, Carew TJ, Kandel ER. Long-term sensitization of a defensive withdrawal reflex in Aplysia. Science. 1973;182:1039–1042. doi: 10.1126/science.182.4116.1039. [DOI] [PubMed] [Google Scholar]
- 13.Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pierce RC, Kalivas PW. Amphetamine produces sensitized increases in locomotion and extracellular dopamine preferentially in the nucleus accumbens shell of rats administered repeated cocaine. J. Pharmacol. Exp. Ther. 1995;275:1019–1029. [PubMed] [Google Scholar]
- 15.Boileau I, et al. Modeling sensitization to stimulants in humans: an [11C]raclopride/positron emission tomography study in healthy men. Arch. Gen. Psychiatry. 2006;63:1386–1395. doi: 10.1001/archpsyc.63.12.1386. [DOI] [PubMed] [Google Scholar]
- 16.Steinkellner T, et al. In vivo amphetamine action is contingent on alphaCaMKII. Neuropsychopharmacology. 2014;39:2681–2693. doi: 10.1038/npp.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laruelle M. The role of endogenous sensitization in the pathophysiology of schizophrenia: implications from recent brain imaging studies. Brain Res. Rev. 2000;31:371–384. doi: 10.1016/s0165-0173(99)00054-5. [DOI] [PubMed] [Google Scholar]
- 18.Weidenauer A, et al. Making sense of: sensitization in schizophrenia. Int. J. Neuropsychopharmacol. 2017;20:1–10. doi: 10.1093/ijnp/pyw081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pycock CJ, Kerwin RW, Carter CJ. Effect of lesion of cortical dopamine terminals on subcortical dopamine receptors in rats. Nature. 1980;286:74–76. doi: 10.1038/286074a0. [DOI] [PubMed] [Google Scholar]
- 20.Falkai P, et al. Kraepelin revisited: schizophrenia from degeneration to failed regeneration. Mol. Psychiatry. 2015;20:671–676. doi: 10.1038/mp.2015.35. [DOI] [PubMed] [Google Scholar]
- 21.Guo JY, Ragland JD, Carter CS. Memory and cognition in schizophrenia. Mol. Psychiatry. 2019;24:633–642. doi: 10.1038/s41380-018-0231-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer-Lindenberg A, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat. Neurosci. 2002;5:267–271. doi: 10.1038/nn804. [DOI] [PubMed] [Google Scholar]
- 23.Fusar-Poli P, et al. Abnormal prefrontal activation directly related to pre-synaptic striatal dopamine dysfunction in people at clinical high risk for psychosis. Mol. Psychiatry. 2011;16:67–75. doi: 10.1038/mp.2009.108. [DOI] [PubMed] [Google Scholar]
- 24.Wilson AA, et al. Radiosynthesis and evaluation of [11C]-(+)-4-propyl-3,4,4a,5,6,10b-hexahydro-2H-naphtho[1,2-b][1,4]oxazin-9-ol as a potential radiotracer for in vivo imaging of the dopamine D2 high-affinity state with positron emission tomography. J. Med. Chem. 2005;48:4153–4160. doi: 10.1021/jm050155n. [DOI] [PubMed] [Google Scholar]
- 25.Sheehan DV, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J. Clin. psychiatry. 1998;59:22–33. [PubMed] [Google Scholar]
- 26.Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr. Bull. 1987;13:261–276. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- 27.Muller MJ, Rossbach W, Davids E, Wetzel H, Benkert O. [Evaluation of standardized training for the “Positive and Negative Syndrome Scale” (PANSS)] Der Nervenarzt. 2000;71:195–204. doi: 10.1007/s001150050029. [DOI] [PubMed] [Google Scholar]
- 28.Brauer LH, de Wit H. Subjective responses to d-amphetamine alone and after pimozide pretreatment in normal, healthy volunteers. Biol.Psychiatry. 1996;39:26–32. doi: 10.1016/0006-3223(95)00110-7. [DOI] [PubMed] [Google Scholar]
- 29.Shotbolt P, et al. Within-subject comparison of the sensitivity of [11C]-(+)-PHNO and [11C]raclopride to amphetamine induced changes in endogenous dopamine in healthy human volunteers. Neuroimage. 2010;52(Supplement 1):S42–S43. [Google Scholar]
- 30.Willeit M, et al. First human evidence of d-amphetamine induced displacement of a D2/3 agonist radioligand: A [11C]-(+)-PHNO positron emission tomography study. Neuropsychopharmacology. 2008;33:279–289. doi: 10.1038/sj.npp.1301400. [DOI] [PubMed] [Google Scholar]
- 31.Rami-Mark C, et al. Reliable set-up for in-loop (1)(1)C-carboxylations using Grignard reactions for the preparation of [carbonyl-(1)(1)C]WAY-100635 and [(1)(1)C]-(+)-PHNO. Appl. Radiat. Isot. 2013;82:75–80. doi: 10.1016/j.apradiso.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White TL, Justice AJ, de Wit H. Differential subjective effects of D-amphetamine by gender, hormone levels and menstrual cycle phase. Pharm. Biochem. Behav. 2002;73:729–741. doi: 10.1016/s0091-3057(02)00818-3. [DOI] [PubMed] [Google Scholar]
- 33.Justice AJ, de Wit H. Acute effects of estradiol pretreatment on the response to d-amphetamine in women. Neuroendocrinology. 2000;71:51–59. doi: 10.1159/000054520. [DOI] [PubMed] [Google Scholar]
- 34.Mombour W, Kockott G, Fliege K. The use of the brief psychiatric rating scale (BPRS) by overall and gorham for the diagnosis of acute paranoid psychoses: evaluation of a german translation of the BPRS (author's transl) Pharmakopsychiatr. Neuropsychopharmakol. 1975;8:279–288. doi: 10.1055/s-0028-1094458. [DOI] [PubMed] [Google Scholar]
- 35.Rusjan P, et al. An automated method for the extraction of regional data from PET images. Psychiatry Res. 2006;147:79–89. doi: 10.1016/j.pscychresns.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 36.Wu Y, Carson RE. Noise reduction in the simplified reference tissue model for neuroreceptor functional imaging. J. Cereb. Blood Flow. Metab. 2002;22:1440–1452. doi: 10.1097/01.WCB.0000033967.83623.34. [DOI] [PubMed] [Google Scholar]
- 37.Ginovart N, et al. Positron emission tomography quantification of [11C]-(+)-PHNO binding in the human brain. J. Cereb. Blood Flow. Metab. 2007;27:857–871. doi: 10.1038/sj.jcbfm.9600411. [DOI] [PubMed] [Google Scholar]
- 38.Camps M, Cortes R, Gueye B, Probst A, Palacios JM. Dopamine receptors in human brain: autoradiographic distribution of D2 sites. Neuroscience. 1989;28:275–290. doi: 10.1016/0306-4522(89)90179-6. [DOI] [PubMed] [Google Scholar]
- 39.Hall H, et al. Autoradiographic localization of extrastriatal D2-dopamine receptors in the human brain using [125I]epidepride. Synapse. 1996;23:115–123. doi: 10.1002/(SICI)1098-2396(199606)23:2<115::AID-SYN7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 40.Levant B. Differential distribution of D3 dopamine receptors in the brains of several mammalian species. Brain Res. 1998;800:269–274. doi: 10.1016/s0006-8993(98)00529-0. [DOI] [PubMed] [Google Scholar]
- 41.Gunn RN, Lammertsma AA, Hume SP, Cunningham VJ. Parametric imaging of ligand-receptor binding in PET using a simplified reference region model. NeuroImage. 1997;6:279–287. doi: 10.1006/nimg.1997.0303. [DOI] [PubMed] [Google Scholar]
- 42.Destrieux C, Fischl B, Dale A, Halgren E. Automatic parcellation of human cortical gyri and sulci using standard anatomical nomenclature. NeuroImage. 2010;53:1–15. doi: 10.1016/j.neuroimage.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vilkman H, et al. The effects of lorazepam on extrastriatal dopamine D(2/3)-receptors-A double-blind randomized placebo-controlled PET study. Psychiatry Res. 2009;174:130–137. doi: 10.1016/j.pscychresns.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Farde L, Hall H, Pauli S, Halldin C. Variability in D2-dopamine receptor density and affinity: a PET study with [11C]raclopride in man. Synapse. 1995;20:200–208. doi: 10.1002/syn.890200303. [DOI] [PubMed] [Google Scholar]
- 45.Abi-Dargham A, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J. Neurosci. 2002;22:3708–3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abi-Dargham A, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc. Natl Acad. Sci. USA. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frankle WG, et al. Amphetamine-induced striatal dopamine release measured with an agonist radiotracer in schizophrenia. Biol. Psychiatry. 2018;83:707–714. doi: 10.1016/j.biopsych.2017.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Driesen NR, et al. Impairment of working memory maintenance and response in schizophrenia: functional magnetic resonance imaging evidence. Biol. Psychiatry. 2008;64:1026–1034. doi: 10.1016/j.biopsych.2008.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Broca P. Remarques sur le siège de la faculté du langage articulé, suivies d’une observation d’aphémie (perte de la parole) Bull de la Soci.été d’anatomie. 1861;6:330–357. [Google Scholar]
- 50.Tseng HH, et al. Nigral stress-induced dopamine release in clinical high risk and antipsychotic-naive schizophrenia. Schizophr. Bull. 2018;44:542–551. doi: 10.1093/schbul/sbx042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ginovart N, Wilson AA, Houle S, Kapur S. Amphetamine pretreatment induces a change in both D2-receptor density and apparent affinity: a [11C]raclopride positron emission tomography study in cats. Biol. Psychiatry. 2004;55:1188–1194. doi: 10.1016/j.biopsych.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 52.Slifstein M, Abi-Dargham A. Is it pre-synaptic or postsynaptic? Imaging striatal dopamine excess in schizophrenia. Biol. Psychiatry. 2018;83:635–637. doi: 10.1016/j.biopsych.2018.02.015. [DOI] [PubMed] [Google Scholar]
- 53.Graff-Guerrero A, et al. Blockade of [11C](+)-PHNO binding in human subjects by the dopamine D3 receptor antagonist ABT-925. Int. J. Neuropsychopharmacol. 2010;13:273–287. doi: 10.1017/S1461145709990642. [DOI] [PubMed] [Google Scholar]
- 54.Rabiner EA, et al. In vivo quantification of regional dopamine-D3 receptor binding potential of (+)-PHNO: Studies in non-human primates and transgenic mice. Synapse. 2009;63:782–793. doi: 10.1002/syn.20658. [DOI] [PubMed] [Google Scholar]
- 55.Tziortzi AC, et al. Imaging dopamine receptors in humans with [11C]-(+)-PHNO: dissection of D3 signal and anatomy. NeuroImage. 2011;54:264–277. doi: 10.1016/j.neuroimage.2010.06.044. [DOI] [PubMed] [Google Scholar]
- 56.Guillin O, et al. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature. 2001;411:86–89. doi: 10.1038/35075076. [DOI] [PubMed] [Google Scholar]
- 57.Wisco JJ, et al. Abnormal cortical folding patterns within Broca’s area in schizophrenia: evidence from structural MRI. Schizophr. Res. 2007;94:317–327. doi: 10.1016/j.schres.2007.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am. J. Psychiatry. 1991;148:1474–1486. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- 59.Kim IH, et al. Spine pruning drives antipsychotic-sensitive locomotion via circuit control of striatal dopamine. Nat. Neurosci. 2015;18:883–891. doi: 10.1038/nn.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Newman AJ, Supalla T, Hauser P, Newport EL, Bavelier D. Dissociating neural subsystems for grammar by contrasting word order and inflection. Proc. Natl Acad. Sci. USA. 2010;107:7539–7544. doi: 10.1073/pnas.1003174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Angrist BM, Gershon S. The phenomenology of experimentally induced amphetamine psychosis–preliminary observations. Biol. Psychiatry. 1970;2:95–107. [PubMed] [Google Scholar]
- 62.Yui K, Ikemoto S, Ishiguro T, Goto K. Studies of amphetamine or methamphetamine psychosis in Japan: relation of methamphetamine psychosis to schizophrenia. Ann. N. Y. Acad. Sci. 2000;914:1–12. doi: 10.1111/j.1749-6632.2000.tb05178.x. [DOI] [PubMed] [Google Scholar]
- 63.Boileau I, et al. Higher binding of the dopamine D3 receptor-preferring ligand [11C]-(+)-propyl-hexahydro-naphtho-oxazin in methamphetamine polydrug users: a positron emission tomography study. J. Neurosci. 2012;32:1353–1359. doi: 10.1523/JNEUROSCI.4371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Worhunsky PD, et al. Regional and source-based patterns of [11C]-(+)-PHNO binding potential reveal concurrent alterations in dopamine D2 and D3 receptor availability in cocaine-use disorder. NeuroImage. 2017;148:343–351. doi: 10.1016/j.neuroimage.2017.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strakowski SM, Sax KW, Setters MJ, Stanton SP, Keck PE., Jr. Lack of enhanced response to repeated d-amphetamine challenge in first-episode psychosis: implications for a sensitization model of psychosis in humans. Biol. Psychiatry. 1997;42:749–755. doi: 10.1016/s0006-3223(97)00052-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.