

Summary
Specialized regulatory T (Treg) cells accumulate and perform homeostatic and regenerative functions in nonlymphoid tissues. Whether common precursors for nonlymphoid-tissue Treg cells exist and how they differentiate remain elusive. Using transcription factor nuclear factor, interleukin 3 regulated (Nfil3) reporter mice and single-cell RNA-sequencing (scRNA-seq), we identified two precursor stages of interleukin 33 (IL-33) receptor ST2-expressing nonlymphoid tissue Treg cells, which resided in the spleen and lymph nodes. Global chromatin profiling of nonlymphoid tissue Treg cells and the two precursor stages revealed a stepwise acquisition of chromatin accessibility and reprogramming toward the nonlymphoid-tissue Treg cell phenotype. Mechanistically, we identified and validated the transcription factor Batf as the driver of the molecular tissue program in the precursors. Understanding this tissue development program will help to harness regenerative properties of tissue Treg cells for therapy.
Keywords: tissue Treg, precursor, Nfil3, Gata3, Batf, Areg, Foxp3, scRNA-seq, scTCR-seq, ATAC-seq
Graphical Abstract
Highlights
-
•
Two precursor stages of ST2+ nonlymphoid-tissue Tregs are evident in the spleen and LNs
-
•
Precursor stages are defined by differential expression of Nfil3, PD1, and Klrg1
-
•
Chromatin accessibility and scRNA-seq suggests a stepwise precursor reprogramming
-
•
Batf drives the molecular tissue program in the precursors
Whether a common precursor exists for nonlymphoid-tissue Treg cells is unclear. Delacher et al. identify two precursor stages for tissue-resident ST2+ Treg cells. These precursors undergo a stepwise reprogramming in the lymphoid organs toward the nonlymphoid-tissue Treg cell phenotype. Chromatin accessibility profiling identified Batf as a key driver of the tissue program in the progenitor cells.
Introduction
Foxp3-expressing regulatory T cells (Treg) are immune cells critical to restraining self-reactivity and excessive inflammation. The fatal multiorgan autoinflammatory destruction found in both immune dysregulation, polyendocrinopathy, enteropathy, and X-linked syndrome (IPEX) patients and scurfy mice is caused by mutations in the transcriptional regulator Foxp3 (Brunkow et al., 2001, Wildin et al., 2001). Treg cells modulate the functions of a variety of immune cells and thereby affect a broad range of conditions, including cancer, autoimmunity, allergy, and infectious diseases. In recent years, it became evident that Treg cells perform important tissue homeostasis and regenerative functions in nonlymphoid tissues (Panduro et al., 2016). Examples include the visceral adipose tissue (VAT) (Cipolletta et al., 2012, Feuerer et al., 2009, Vasanthakumar et al., 2015), muscle and lung (Arpaia et al., 2015, Burzyn et al., 2013), skin (Ali et al., 2017), and the central nervous system (CNS) (Dombrowski et al., 2017, Ito et al., 2019, Korn et al., 2007, Liesz et al., 2009). Amphireguling (Areg) has been suggested to contribute to these regenerative processes (Arpaia et al., 2015, Burzyn et al., 2013, Ito et al., 2019).
Recently, DNA-methylation and transcriptional data from tissue-derived Treg cells characterized a Treg cell population present in virtually all nonlymphoid tissues and identifiable by the expression of the interleukin 33 (IL-33) receptor (ST2) and killer cell lectin-like receptor subfamily G1 (Klrg1), named “tisTregST2” (Delacher et al., 2017, Delacher et al., 2019, Schmidl et al., 2018). This ST2-positive tissue Treg population readily expresses not only tissue-regenerative factors such as Areg, but also T helper 2 (Th2)-associated factors, including high levels of Gata3, Maf, and IL-10 (Delacher et al., 2017). Important tissue homeostasis and regenerative functions of ST2-positive Treg cells have been characterized in different nonlymphoid tissues including VAT, CNS, muscle, and colon (Burzyn et al., 2013, Ito et al., 2019, Schiering et al., 2014, Vasanthakumar et al., 2015). Although isolated from different tissues, Treg cells from VAT and skin showed a high degree of overlap in their epigenetic reprogramming, indicating a common developmental path preceding tissue-restricted specialization (Delacher et al., 2017). A multistep model of tissue Treg differentiation has been proposed (Li et al., 2018, Miragaia et al., 2019). Common precursors and molecular programs that drive precursor differentiation and their reprogramming toward a nonlymphoid-tissue Treg cell phenotype are not well understood. To address these questions, we generated a reporter mouse that helped to identify two precursor stages of the ST2-positive nonlymphoid-tissue Treg population in lymphoid tissues. Molecular profiling of these precursors revealed their sequence of differentiation.
Results
Chromatin Accessibility Profiling of Treg Cells from Different Tissues Reveals Shared and Distinct Gene-Regulatory Programs
Under homeostatic conditions, tisTregST2 cells comprise the majority of Treg cells in visceral adipose tissue (VAT) and skin and represent considerable fractions of Treg cells in other nonlymphoid tissues such as colon (∼30% of all Treg cells) and lung tissue (10%–20%) (Figure 1A). To study the underlying gene-regulatory programs of this ST2-positive population of tissue Treg cells, we used the assay for transposase accessible chromatin using sequencing (ATAC-seq) (Buenrostro et al., 2013) on sorted Klrg1+ST2+ tisTregST2 cells from VAT, skin, colon, and lung, and compared them with Foxp3− conventional T (Tconv) cells from the same tissue origin. In addition, CD44+ memory or activated, and CD44− naive Treg cells from lymph node (LN) were sorted and subjected to ATAC-seq for comparison. In total, we identified a robust set of 78,461 distinct peaks over all samples. Principal component analysis (PCA) of the chromatin accessibility landscapes based on the combined peak set separated tisTregST2 populations from Tconv cells and LN-derived memory or naive Treg cells (Figure 1B). Tissue Tconv cells clustered closely with LN-derived CD44+ Treg cells, indicating that the tissue Tconv chromatin profiles may be more related to an “activation or memory” program. In contrast to this, tisTregST2 cells from the given tissues separated clearly from LN-derived Treg cells and tissue Tconv cells, indicating a major impact of a tissue signature in Treg cells. For the various tissues, we found between 16,262 and 18,555 significantly different ATAC-seq peaks between tisTregST2 and naive Treg cells (Figure 1C). The comparison of tisTregST2 populations from different tissues revealed no more than 3,190 peaks as being differential between individual tissues. Next, we sought to use our chromatin accessibility data to identify a shared tisTregST2 program. Therefore, we defined differential accessible chromatin regions between the combined tisTregST2 samples and LN-derived memory Treg cells. We observed that tissue Tconv cells showed a limited “tissue signature” as they were separating from LN populations along principal component 2, similar to tisTregST2 cells (Figure 1B). Because of this, we excluded the tissue Tconv signature to retain a true tisTregST2 “core” set of open chromatin regions comprising 2,267 peaks (Figure 1D; see STAR Methods for details). This signature was not present in lung-derived Klrg1−ST2− Treg cells (Figure S1A).
Figure 1.

ATAC-Seq of Tissue-Derived T Cells Identifies a Common Gene-Regulatory Program
(A) Presence of Klrg1+ST2+ tisTregST2 among Treg cells of lung, colon, skin, and VAT (representative example).
(B) Principal component analysis (PCA) based on a consensus set of 78,461 peaks derived from ATAC-seq data of FACS-sorted lymph node (LN)-derived CD25+Foxp3(GFP)+CD44− naive Treg (turquoise), LN-derived CD25+Foxp3(GFP)+CD44+ memory Treg (blue-green) as well as colon-, lung-, VAT-, and skin-derived CD25+Foxp3(GFP)+CD44+Klrg1+ST2+ tisTregST2 or CD25−Foxp3(GFP)−CD44+ memory Tconv (pink, brown, orange, and dark blue) (n = 3–4).
(C) Pairwise comparisons of ATAC-seq data between all cell types shown in (B) except Tconv, numbers indicate differential peak sets (Benjamini-Hochberg corrected adjusted p value <0.01, log2 fold change >2, normalized mean accessibility ≥10), color code indicates number of differential peaks (n = 3–4).
(D) Unsupervised hierarchical clustering of the 2,267 “core” tisTregST2 ATAC-seq peaks with accessibility values of all cell types shown in (B), tissue Tconv signature subtracted (n = 3–4).
(E) De novo transcription factor (TF) motifs identified in the “core” tisTregST2-signature (n = 3–4).
(F) Normalized ATAC-seq signal from different cell types at “core” ATAC-seq peaks carrying a bZIP or GATA binding motif, respectively (n = 3–4).
(G) ATAC-seq data for the Ctsh and Pparg loci with all cell types shown in (B). All datasets group-normalized to maximum peak height indicated in brackets.
(H) Unsupervised hierarchical clustering of 1,345 ATAC peaks from pairwise comparisons of tisTregST2 populations from VAT, lung, skin, and colon (n = 3–4).
(I) Pathway enrichment of genes near differential peaks for tisTregST2 from different tissues (database: WikiPathways 2016).
(J) ATAC-seq data for the Liph and Tff1 loci as in (G) (n = 3–4). Data representative of independent experiments or cell sorts.
De novo motif discovery identified DNA consensus binding motifs of several transcription factor families including bZIP (containing AP-1 factors), ETS, nuclear factor κB (NF-κB), NRL and GATA in the “core” tisTregST2 cell-specific ATAC-seq peaks (Figure 1E). The expected strong ATAC-seq signals in tisTregST2 populations at respective transcription factor consensus motifs are displayed exemplarily for bZIP and GATA motifs (Figure 1F). Using gene expression data from RNA sequencing (RNA-seq) of tisTregST2 populations, Gata3 as a GATA family member and Batf (Batf1) as a bZIP family member were identified as being specifically upregulated in tisTregST2 cells and therefore likely contributing to the “core” tisTregST2 gene-regulatory program (Figures S1B and S1C). Further examples of this “core” program with tisTregST2-specific peaks include the Pparg, Ctsh, as well as Klrg1 and Mreg loci (Figures 1G and S1D).
After specifying the shared “core” tisTregST2 chromatin accessibility signature, we used the ATAC-seq data to identify tisTregST2 chromatin regions that are specific for each individual tissue (Figure 1H). Pathway analysis on the genes that were in the vicinity of these differential chromatin sites identified signaling pathways related to tissue-specific differences, such as IL-5 signaling for VAT tisTregST2 and Delta-Notch signaling for lung tisTregST2 (Figure 1I). As examples for tissue-restricted differential peaks, lipase member H (Liph) and trefoil factor 1 (Tff1) (Figure 1J) were identified as present in colon but not in VAT, while VAT tisTregST2 showed specific peaks at a potential enhancer of dual specificity phosphatase 26 (Dusp26) (Figure S1D). Taken together, ATAC-seq of tisTregST2 from different tissues revealed a prominent “core” tissue signature as well as tissue-specific flavors.
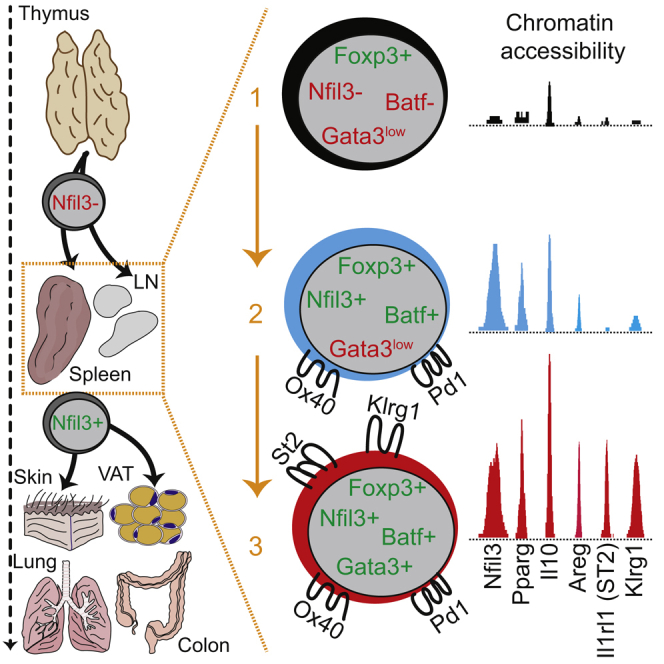
Nfil3 Identifies Putative tisTregST2 Precursor Cells in Lymphoid Organs
The finding that Pparg was among the “core” tissue signature loci (Figures 1G and S1D) was not expected because expression of Pparg has been described to be specific for Treg cells in VAT (Cipolletta et al., 2012). Our data now showed that the chromatin locus of Pparg is accessible independent of tissue location, suggesting a common step of chromatin remodeling in tisTregST2 cells outside of their resident tissues. Such a chromatin remodeling step could occur in a committed common tisTregST2 precursor. To address this possibility, we explored the gene Nfil3 (nuclear factor, interleukin 3 regulated), which showed a clear pattern of epigenetic remodeling in Treg cells from different nonlymphoid tissues (Figure 2A, top). Nfil3 was also part of our tisTregST2 “core” tissue signature with differential ATAC-seq peaks, and tisTregST2 cells showed high Nfil3 gene expression (Figures 2A and 2B). Based on this finding, we speculated that Nfil3 might help to identify early events that trigger nonlymphoid tissue Treg cell differentiation. Therefore, we generated a Nfil3-enhancedGFP (Nfil3(GFP)) reporter mouse line via a BAC-transgene and analyzed Treg cells from LN, spleen, and skin by flow cytometry (Figures 2C and S2A–S2E). In line with our RNA-seq data, the vast majority of Treg cells from the skin expressed Nfil3(GFP) in comparison to Treg cells isolated from LN and spleen (Figure 2C). We confirmed that Klrg1+ST2+ tisTregST2 from various other tissues expressed Nfil3-GFP (Figure S2C), while CD4+CD25− Tconv cells, CD4−CD8+ cytotoxic T cells and CD19+ B cells showed no or very low reporter gene expression in lymphoid or peripheral tissues (Figure S2D). Expression of Nfil3-GFP in Treg cells was accompanied by loss of CD62L expression and upregulation of CD44, indicating previous Treg cell activation (Figure S2E). In LN and spleen, we identified two small subpopulations of Nfil3(GFP)-reporter-positive Treg cells (Figures 2C, S2C, and S2D). Based on Nfil3(GFP) and Klrg1 expression, we subdivided the Treg compartment into three populations: Klrg1−Nfil3(GFP) − Treg cells, majority in spleen and LN, and almost absent in skin; Klrg1−Nfil3(GFP)+ Treg cells, constituting ∼10% in spleen and LNs; and Klrg1+Nfil3(GFP)+ Treg cells, with low frequency in spleen (∼4%) and LN (1%–2%), but dominating the Treg pool in skin or VAT (∼80%).
Figure 2.

Identification and Characterization of a Spleen tisTregST2 Precursor
(A) CG-methylation of the Nfil3 gene based on whole-genome bisulfite methylation data derived from a previous publication (Delacher et al., 2017) for LN Treg (turquoise), VAT Treg (orange), and skin Treg (blue). Ticks represent CpG sites. Below, ATAC-seq for LN-derived naive CD44−Klrg1−ST2− Treg (light blue) and lung-, VAT-, skin-, and colon-derived CD44+Klrg1+ST2+ tisTregST2 (dark brown, orange, dark blue, purple) (n = 3–4).
(B) Nfil3 gene expression in bone marrow (BM) and spleen-derived Klrg1−ST2− Treg (black) as well as lung-, liver-, VAT-, and skin-derived Klrg1+ST2+ tisTregST2 (brown, green, orange, and dark blue). Statistics based on Deseq2. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.01 and nsp > 0.05. Error bars indicate mean + SD (n = 4).
(C) Expression of GFP in Nfil3(GFP) reporter mice. Shown at the top is staining of inguinal LN Treg (CD4+TCRβ+CD25+) from control B6 animals or Nfil3(GFP) reporter mice. Show at the bottom is staining of spleen (middle) and skin (bottom) Treg. Percentage of Klrg1+Nfil3(GFP)+ of CD25+ below (unpaired t test, n = 8–9).
(D) RNA-seq expression data of Nfil3, Gata3, Batf, and Maf in Klrg1−Nfil3(GFP)−, Klrg1−Nfil3(GFP)+, and Klrg1+Nfil3(GFP)+ spleen Treg, and lung, liver, VAT, and skin Klrg1+ST2+ Treg (Deseq2, n = 4).
(E) PCA based on 1,000 most variable genes derived from RNA-seq data of spleen Klrg1−Nfil3(GFP)− Treg (black), Klrg1−Nfil3(GFP)+ Treg (light blue), and Klrg1+Nfil3(GFP)+ Treg (red) as well as BM-, liver-, lung-, VAT-, and skin-derived tisTregST2 (gray, green, brown, orange, dark blue) (n = 4).
(F) Uniform manifold approximation and projection (UMAP) of scRNA-seq data of spleen-derived Treg cell (CD4+TCRβ+CD25+) subpopulations: Klrg1−Nfil3(GFP)− in black, Klrg1−Nfil3(GFP)+ in blue, and Klrg1+Nfil3(GFP)+ in red. Scaled expression values were clipped at a value of 4 (n = 5).
(G) UMAP plots as in (F). Color code displays gene expression of Foxp3, Klrg1, Id2, Id3, Tnfrsf4, Tnfrsf9, Icos, and Maf with gray = low and red/yellow = high expression (n = 5). Data representative of independent experiments or cell sorts.
See also Figures S2 and S3 and Tables S2 and S3.
Next, we sorted all three Treg subpopulations from the spleen, performed RNA-seq from bulk populations, and compared their gene expression profiles to those of tisTregST2 cells isolated from tissues. Both Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ Treg cells were highly positive for the tisTregST2 associated transcription factors Nfil3, Batf, and Maf, with values comparable to tisTregST2 cells from tissues, while Gata3 expression was strongly induced only in Klrg1+Nfil3(GFP)+ Treg cells (Figure 2D). To obtain a global picture, we performed principal component analysis comparing the three spleen populations with tisTregST2 cells from different organs. From these data, a sequence of differentiation from Klrg1−Nfil3(GFP) − Treg via Klrg1−Nfil3(GFP)+ to Klrg1+Nfil3(GFP)+ Treg to tisTregST2 could be inferred (Figure 2E).
Because RNA-seq from bulk populations can masks heterogeneity within a given population, we performed single-cell RNA sequencing (scRNA-seq) from all three Treg populations isolated from the spleen (16,469 single cells in total) and displayed the data by uniform manifold approximation and projection (UMAP) (Figure 2F) or t-distributed stochastic neighbor embedding analysis (t-SNE) (Figures S3A and S3B). Cells of each sorted subset formed rather homogeneous populations, with little overlap between subsets. All subpopulations expressed Foxp3, but only Klrg1+Nfil3(GFP)+ Treg cells showed Klrg1 mRNA expression (Figures 2G and S3C). Several key genes indicating activation and differentiation were identified in the transition from Klrg1−Nfil3(GFP) − Treg via Klrg1−Nfil3(GFP)+ Treg to Klrg1+Nfil3(GFP)+ cells. For example, expression of the Inhibitor of DNA-binding switched from family member 3 to 2 (Id3 to Id2), and TNF receptor superfamily members 4 (Tnfrsf4, OX40) and 9 (Tnfrsf9, 4-1BB), inducible T cell co-stimulator (Icos) and Maf were almost exclusively expressed by Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ Treg cells (Figures 2G and S3C). In summary, our data indicate that spleen Nfil3(GFP)-positive Treg cells could be precursor cells that differentiate toward Klrg1+ST2+ tisTregST2 in tissues.
Developmental Trajectories of Treg Cells from Spleen and Tissues
To investigate the differentiation pathway of tisTregST2 in an unbiased manner, we isolated CD4+CD44+CD25+Foxp3(GFP)+ memory Treg cells from spleen, inguinal LN, BM, blood, VAT, skin, lung, and liver, and performed scRNA-seq. In addition, we used our scRNA-seq data for Klrg1−Nfil3(GFP)−, Klrg1−Nfil3(GFP)+, and Klrg1+Nfil3(GFP)+ Treg cells from spleens and computed UMAP-based analysis (Figure 3A) and single-cell trajectory analysis (Figures S3D–S3E). In the UMAP plot, almost no overlap was observed between tissue-isolated memory Treg cells from lung, VAT, and skin, and the two spleen-derived Nfil3(GFP)+ putative tissue Treg precursor populations. Only a few cells from precursors were located at the different tissue positions, indicating a very minor fraction of potentially re-circulating tissue Treg cells. Trajectory analysis in the monocle-based comparison of dimension 1 versus 3 suggested a precursor-to-product relationship between Klrg1−Nfil3(GFP)+, Klrg1+Nfil3(GFP)+ Treg and the tissue Treg cells, while comparison of dimension 1 versus 2 identified precursor states (Figures S3D and S3E).
Figure 3.

Single-Cell RNA-Seq and TCR-Seq of Tissue Memory Treg Cells
(A) UMAP from scRNA-seq data of spleen Klrg1−Nfil3(GFP)− Treg, spleen Klrg1−Nfil3(GFP)+ Treg, and spleen Klrg1+Nfil3(GFP)+ Treg as well as VAT, skin, lung, and liver memory Treg (CD4+TCRβ+CD44+CD25+Foxp3(GFP)+). Number in brackets displays total number of single cells per group. Scaled expression values were clipped at a value of 4 (n = 4–11).
(B) Data derived from single-cell T cell receptor sequencing (scTCR-seq) of spleen Klrg1−Nfil3(GFP)− Treg, spleen Klrg1−Nfil3(GFP)+ Treg, and spleen Klrg1+Nfil3(GFP)+ Treg as well as colon-, skin-, and VAT-derived Klrg1+Nfil3(GFP)+ tisTregST2 from two individual mice in separate experiments. Individual clones are shown in separate colors with percentages indicating frequency. Numbers indicate total numbers of cells with successfully paired TCR α+β chains (n = 2).
(C) Inverse Simpson index of scTCR-seq data from (B) (n = 2).
(D) Tracking of skin TCR clones in spleen Klrg1−Nfil3(GFP)− Treg and spleen Klrg1−Nfil3(GFP)+ Treg as well as spleen Klrg1+Nfil3(GFP)+ Treg from two mice based on AA sequence of the TCR, plotted are skin clones (n = 2).
(E) Data derived from scTCR-seq of skin Nfil3(GFP)+ Treg, skin-draining LN Nfil3(GFP)+ Treg, colon Nfil3(GFP)+ Treg, as well as colon-draining LN Nfil3(GFP)+ Treg as in (B) (n = 2).
(F) Tracking of colon Nfil3(GFP)+ Treg TCR clones in skin-draining inguinal LN or colon-draining mesenteric LN Nfil3(GFP)+ Treg based on AA sequence of the TCR as in (D), plotted are colon clones (n = 1).
(G) Tracking of skin Nfil3(GFP)+ Treg TCR clones in inguinal or mesenteric LN Nfil3(GFP)+ Treg based on AA sequence of the TCR as in (F), plotted are skin clones (n = 1). Data representative of independent experiments or cell sorts.
Individual T cells have a natural barcoding, their T cell receptor (TCR) nucleotide sequence. Single-cell analysis of the alpha and beta chain of the TCR enables the analysis of clonal relatedness. To understand the diversity and clonal relationship of Treg cells, we used scTCR-seq of alpha and beta TCR chains of the three spleen populations as well as tisTregST2 from skin, colon, and VAT of individual mice. We plotted the clonal distribution in pie charts (Figure 3B) and calculated the inverse Simpson index (Figure 3C) and Jaccard indices (Figure S3F). Our data demonstrated a highly polyclonal TCR repertoire in Klrg1−Nfil3(GFP) − spleen Treg cells, while Klrg1−Nfil3(GFP)+ spleen Treg cells showed a much more restricted TCR repertoire. Even further restricted was the TCR repertoire of Klrg1+Nfil3(GFP)+ Treg cells, which had an inverse Simpson index comparable to those of peripheral tissue-derived tisTregST2 cells (Figures 3B and 3C). Peripheral tissue-derived tisTregST2 cells had an oligoclonal TCR repertoire, whereas the VAT-derived Treg cells were further outstanding in that 3–4 clones covered ∼50% of all TCR sequences (Figure 3B). In general, each peripheral organ had a quite distinct TCR repertoire (Figure S3G). To analyze precursor relations, we compared TCRs from Klrg1−Nfil3(GFP)−, Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ Treg cells among each other and with peripheral Treg cells with the Jaccard index. The two putative precursor populations Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ Treg were closely related (Figure S3F). We tracked TCR clones found in the skin in scTCR-data of the three spleen Treg populations and noticed very little overlap with Klrg1−Nfil3(GFP) −, but considerable overlap with Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ spleen tisTregST2 precursor cells (Figure 3D). Because skin, VAT, and colon-derived Nfil3(GFP)+ Treg TCRs showed the highest oligoclonal expansion, indicating a role of antigen-specificity, priming of tissue Treg precursors would presumably lead to an accumulation of certain TCRs in the draining compared to non-draining lymphoid organs. To investigate this, we isolated Nfil3(GFP)+ Treg cells from skin and Nfil3(GFP)+ Treg cells from skin-draining inguinal LNs as well as colon and the colon-draining mesenteric LNs from the same animal (Figure 2E). Clone tracking revealed a higher overlap of tissue Treg TCRs in Nfil3(GFP)+ Treg cells in draining as compared to non-draining LNs, which was observed for colon as well as for skin, indicating precursor priming in the draining LNs (Figures 3F and 3G). In summary, based on single-cell trajectory and TCR analysis, our data suggest a sequential differentiation of spleen Klrg1−Nfil3(GFP)+ via Klrg1+Nfil3(GFP)+ Treg cells into tisTregST2 cells.
Adoptive Transfer and Developmental Kinetics of tisTregST2 Precursors
Our data suggest that spleen Nfil3(GFP)+ Treg cells are potential precursors of tisTregST2 cells. Indeed, when we performed RNA-velocity analysis of our scRNA-seq data, a sequential differentiation from Klrg1−Nfil3(GFP) − via Klrg1−Nfil3(GFP)+ to Klrg1+Nfil3(GFP)+ was indicated by the vectors (Figure 4A). To further validate this, we performed adoptive transfer experiments with fluorescence-activated cell sorting (FACS)-purified Klrg1−Nfil3(GFP) − and Klrg1−Nfil3(GFP)+ Treg populations injected into Treg-depleted congenic (CD45.1) Foxp3DTR host mice (Figures 4B and S4A). After 6 days, we identified the transferred Treg cells in spleen by CD45.2 staining and measured the de novo expression of Klrg1 as a marker indicating differentiation into the tisTregST2 trajectory. Significantly more Klrg1−Nfil3(GFP)+ Treg cells had differentiated and upregulated Klrg1 in comparison to transferred Klrg1−Nfil3(GFP)−- or host-derived Treg cells (Figure 4B). We analyzed VAT, colon, lung, skin, and liver Treg cells 10 days after transfer of Klrg1−Nfil3(GFP)+ Treg cells. Transferred Klrg1−Nfil3(GFP)+CD45.2+ Treg cells could readily be detected in all peripheral tissues, and, in some organs, up to 60% of transferred Klrg1−Nfil3(GFP)+ had upregulated Klrg1 as a marker for differentiation (Figures 4C and S4B). To validate that Klrg1+Nfil3(GFP)+ Treg cells do not de-differentiate and lose Klrg1 expression, we sorted Klrg1+ Treg cells from spleens or lungs and transferred them into congenic animals. Two weeks after transfer, we were able to identify the population in both lymphoid and peripheral tissues, and they retained their high Klrg1 expression (Figures 4D, S4C, and S4D), rendering it unlikely that re-circulating tissue Treg cells feed the Klrg1−Nfil3(GFP)+ Treg precursor pool.
Figure 4.

Adoptive Transfer and Development Kinetics of Nfil3(GFP) Populations
(A) T-distributed stochastic neighbor embedding (t-SNE) plot with RNA velocity of spleen-derived Klrg1−Nfil3(GFP)− Treg, Klrg1−Nfil3(GFP)+ Treg, and Klrg1+Nfil3(GFP)+ Treg. Arrows represent the averaged gene expression profiles of cells within a local neighborhood. Length of arrow represents speed of development (n = 5).
(B) Klrg1−Nfil3(GFP)− and Klrg1−Nfil3(GFP)+ Treg from CD45.2+ donor animals were sorted and transferred into diphtheria toxin (DT)-pretreated Foxp3YFP,DTR,CD45.1 recipient animals. After 6 days, transferred Treg in spleen were identified as CD45.2+ population. Plots illustrate expression of Klrg1 in CD45.1+ host Treg or CD45.2+ transferred Treg (unpaired t test, n = 4–10).
(C) Identification of transferred Treg in tissues of recipient animals 10 days after transfer of spleen-derived CD45.2+Klrg1−Nfil3(GFP)+ Treg (n = 4–7).
(D) Transfer of CD45.2+Klrg1+ Treg from spleen into DT-pretreated Foxp3YFP,DTR,CD45.1 recipient animals as in (C). After 2 weeks, transferred cells were identified and stability of Klrg1 expression was measured (mes LN, mesenteric LN; paired t test, n = 3–4).
(E) Percentage of Klrg1−Nfil3(GFP)+ (light blue) and Klrg1+Nfil3(GFP)+ Treg (red) in spleen Treg (CD4+TCRβ+CD25+) measured 5 days, 8 days, 10 days, 15 days, 20 days, 25 days, and 70+ days after birth (n = 4–19).
(F) Total number of skin, lung, colon, and liver Klrg1+Nfil3(GFP)+ Treg 5 days, 8 days, 10 days, 12 days, 15 days, 20 days, 25 days, and 70+ days after birth. For all tissues, total cell numbers were normalized to tissue weight. For skin data, Treg cell fold increase was calculated by dividing cell numbers on different time intervals (shown right). Flow cytometry data of several replicates were concatenated to increase visibility (n = 2–11). Data representative of two or more independent experiments.
To investigate sequential differentiation of precursors and tissue seeding in an unperturbed system, we monitored postnatal organ seeding. To this end, we measured the percentage of Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ Treg cells in spleen and nonlymphoid tissues 5, 8, 10, 15, 20, 25, and 70+ days after birth (Figures 4E, 4F, S4E, and S4F). While Klrg1+Nfil3(GFP)+ Treg cells were not detectable in the spleen 5 days after birth, they peaked in frequency on day 10 (∼20% of all Treg cells in spleen) followed by a steady decline to base line frequency by day 20 (∼4% of Treg cells, Figure 4E, red graph). Klrg1−Nfil3(GFP)+ Treg cells were already detectable on day 5 in the spleen and peaked also on day 10, indicating that overall tissue precursor output was highest around 10 days after birth (Figures 4E and S4E). In tissues, skin tisTregST2 cells were barely detectable on day 5 after birth. Tissue seeding started on day 10, simultaneously with the described peak in spleen tisTregST2 precursor output (Figures 4F and S4F), and led to a 1,000-fold increase of Klrg1+Nfil3(GFP)+ Treg cell numbers per gram skin from day 5 until day 20 (Figure 4F, right). After day 20, seeding was complete and cell numbers and frequency achieved equilibrium. Highest seeding rates were observed around day 10 in lung and liver, and around day 25 in colon (Figure 4F). In summary, RNA velocity, transfer experiments, and unperturbed developmental kinetics after birth all indicate a sequential differentiation of spleen Klrg1−Nfil3(GFP)+ via Klrg1+Nfil3(GFP)+ precursor cells into tissue-resident Klrg1+Nfil3(GFP)+ tisTregST2 cells.
Gene-Regulatory Programs of Nfil3(GFP)+ tisTregST2 Precursors
To dissect the molecular programs that define tisTregST2 precursors, we sorted Klrg1−Nfil3(GFP)−, Klrg1−Nfil3(GFP)+, and Klrg1+Nfil3(GFP)+ Treg cells from spleen and performed ATAC-seq. A developmental component was visible in a heatmap representation across all 11,330 differential peaks from pairwise comparisons between samples including Klrg1−Nfil3(GFP)−, Klrg1−Nfil3(GFP)+, Klrg1+Nfil3(GFP)+ Treg cells, and a combined in silico tisTregST2 sample containing all the tisTregST2 ATAC-seq samples from all tissues (Figure 5A). In accordance with our precursor hypothesis, Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ Treg cells were already poised toward the tisTregST2 gene program, as exemplified by a stepwise increase in chromatin accessibility at tissue-specific regions as displayed for the Pparg, Mt1, and Areg loci (Figures 5B–5D), as well as Rora, Gpr55, and the TH2 control region in the Rad50 gene (Figure S5A). Contrary, Bcl-2 showed a stepwise decrease in chromatin accessibility (Figure S5A). On the other hand, genes such as Il1rl1 (ST2), Klrg1, Mreg, and Ctsh were only opened in Klrg1+Nfil3(GFP)+ Treg cells, marking these loci as indicators of more mature tisTregST2 precursors and indicating a stepwise enrichment of open chromatin regions (Figures 5E and S5A). We also investigated chromatin accessibility at genes that were expressed in tisTregST2 cells in a tissue-selective manner (VAT, lung, and skin) and found many associated loci already accessible in Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ as compared to Klrg1−Nfil3(GFP)− Treg cells (Figures S5B–S5D). The correlation between ATAC signature and RNA mean expression was 0.25 (Figure S5E).
Figure 5.

Core tisTregST2 Signature in Nfil3(GFP) Populations
(A) Heatmap across all 11,330 differential ATAC-seq peaks from pairwise comparisons of FACS-sorted spleen-derived Klrg1−Nfil3(GFP)− Treg, spleen Klrg1−Nfil3(GFP)+ Treg, spleen Klrg1+Nfil3(GFP)+ Treg, and tisTregST2 samples consisting of skin-, lung-, VAT-, or colon-derived tisTregST2 (n = 3–4).
(B) ATAC-seq data for the Pparg locus with all cell types shown in (A). All datasets group-normalized to maximum peak height indicated in brackets (n = 3–4).
(C) ATAC-seq data for the Mt1 locus with all cell types shown in (A). All datasets group-normalized to maximum peak height indicated in brackets (n = 3–4).
(D) ATAC-seq data for the Areg locus with all cell types shown in (A). All datasets group-normalized to maximum peak height indicated in brackets (n = 3–4).
(E) Overlap of core tisTregST2 peaks with ATAC-seq peaks identified in spleen Klrg1−Nfil3(GFP)− Treg (black), spleen Klrg1−Nfil3(GFP)+ Treg (light blue), and spleen Klrg1+Nfil3(GFP)+ Treg (red) (n = 4).
(F) Motif analysis with individual comparisons between spleen Klrg1−Nfil3(GFP)− Treg (black), spleen Klrg1−Nfil3(GFP)+ Treg (light blue), spleen Klrg1+Nfil3(GFP)+ Treg (red), or the core tisTregST2 signature (orange) (n = 3–4).
(G) Single-base-pair resolution ATAC-seq signal at motif-centered peaks containing the de novo discovered motifs of NF-κB, NFAT:AP1, and Gata family members from (F) in spleen Klrg1−Nfil3(GFP)− (black), Klrg1−Nfil3(GFP)+ (light blue), or spleen Klrg1+Nfil3(GFP)+ Treg (red). Transposase background signal (Tn5 background) shown in gray (n = 4).
(H) Ki67 (representative pseudocolor plots) and Gata3 (representative histograms) staining in spleen Klrg1−Nfil3(GFP)−, Klrg1−Nfil3(GFP)+, or spleen Klrg1+Nfil3(GFP)+ Treg (one-way ANOVA; Ki67 or Gata3 RNA: n = 4; Ki67 or Gata3 protein: n = 4–8). Data are representative of two or more independent experiments or cell sorts.
Next, we wanted to identify potential factors that drive differential chromatin accessibility between the precursor and tisTregST2 cells. De novo motif analysis revealed Runx, Gata, and bZIP family members to be highly enriched in the combined tissue Treg signature as compared to all the tisTregST2 precursors (Figure 5F). Forkhead, STAT and Tcf transcription factor signatures dominated the Klrg1−Nfil3(GFP)− program. In contrast to this, NF-κB, Nur, NFAT:AP1, bZIP, and BATF:JUN dominated the difference between both splenic Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ tisTregST2 precursors and Klrg1−Nfil3(GFP)− Treg cells. Single base-pair resolution mapping of the ATAC-seq data at transcription factor motifs comparing Klrg1−Nfil3(GFP)− with Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+Treg cells suggests the preferential binding of NF-κB, Nfat:AP1, and Gata transcription factors to differential regions in Nfil3(GFP)+ tisTregST2 precursors (Figure 5G). Because NF-κB and Nfat:AP1 transcription factor activity indicates T cell activation and proliferation, we measured Ki67 as a well-established marker for cell proliferation in sorted Treg cells from spleen via FACS. We detected ∼20% Ki67-positive cells in Klrg1−Nfil3(GFP)−, ∼75% in Klrg1−Nfil3(GFP)+, and ∼80% in Klrg1+Nfil3(GFP)+ Treg cells (Figure 5H). In addition, we measured Gata3 expression in the splenic populations and identified high Gata3 expression only in Klrg1+Nfil3(GFP)+ Treg cells (Figure 5H). Taken together, we identified a stepwise increase in chromatin accessibility at tisTregST2 cell-specific regions in splenic Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ tisTregST2 precursors.
Batf Is Associated with Nfil3(GFP)+ tisTregST2 Precursor Development
Because we could identify strong enrichments of binding motifs of various TF families in differential peaks between Klrg1−Nfil3(GFP)− and Nfil3(GFP)+ tisTregST2 precursor cells, we sought to identify concrete candidate TFs that could drive the tisTregST2 cell development. Therefore, we measured the overlap of differential chromatin regions between Klrg1−Nfil3(GFP)− Treg and Klrg1−Nfil3(GFP)+ or Klrg1+Nfil3(GFP)+ Treg cells with public databases, and identified, among others, Batf as a highly-enriched TF (Figure 6A). To this end, we observed a strong accumulation of ATAC-seq reads at differential peaks between precursors and Klrg1−Nfil3(GFP)− Treg cells in the vicinity of bZIP (Batf has a bZIP domain) and the composite BATF:JUN motif (Figure 6B). By re-analyzing published CD4+ and CD8+ T cell Batf chromatin immunoprecipitation followed by sequencing (ChIP-seq) data (Hasan et al., 2017, Kurachi et al., 2014), we noticed a high overlap with ATAC-seq peaks identified in our comparison between Klrg1−Nfil3(GFP)− Treg and the two Nfil3(GFP)+ precursors with experimentally validated Batf binding sites. For example, 30% of all peaks that gain accessibility in Klrg1−Nfil3(GFP)+ precursors compared to Klrg1−Nfil3(GFP)− Treg cells showed Batf binding in the Batf ChIP-seq dataset (Figure 6C), indicating that Batf may directly be responsible for considerable parts of the tisTregST2 precursor development program. Examples for Batf ChIP-seq and corresponding ATAC-seq signals are shown for the Il10 and Nfil3 loci (Figure 6D). We confirmed elevated Batf expression in both Klrg1−Nfil3(GFP)+ Treg and Klrg1+Nfil3(GFP)+ Treg cells from spleen (Figure 6E). Calculation of Batf-correlated genes in our scRNA-seq data (Figure 2F) resulted in more than 60 correlated or anti-correlated genes (Figure 6F). Correlated genes include Tnfrsf4 (CD134, Ox-40), Klrg1, and Pdcd1 (CD279, Pd1), while anti-correlated genes include Bcl2, Sell, Ccr7, Satb1, and Id3. We confirmed Pd1 and Ox40 expression in both Nfil3(GFP)+ populations (Figure 6G) and validated Pd1 as a good surrogate marker for Nfil3(GFP)+ expression by co-staining with Gata3, Ki67, and ST2 (Figure 6H). In summary, our data predict that Batf is associated with Nfil3(GFP)+ tisTregST2 precursor development, and Nfil3(GFP)+ tisTregST2 precursors can be identified by elevated expression of Pd1.
Figure 6.

Batf Is Associated in Nfil3(GFP) Precursor Populations
(A) Overlap of chromatin regions differentially opened in spleen Klrg1−Nfil3(GFP)− versus spleen Klrg1−/+Nfil3(GFP)+ Treg with public ChIP-seq datasets identified by LOLA (Sheffield and Bock, 2016) (n = 4).
(B) Single-base-pair resolution ATAC-seq signal at motif-centered peaks containing the de novo discovered motifs of BATF:JUN and bZIP family members (discovered in Figure 5F) in spleen Klrg1−Nfil3(GFP)− (black), Klrg1−Nfil3(GFP)+ (light blue), or spleen Klrg1+Nfil3(GFP)+ Treg (red). Transposase background signals (Tn5 background) shown in gray (n = 4).
(C) Chromatin regions differentially opened in spleen Klrg1−Nfil3(GFP)− versus Klrg1−Nfil3(GFP)+ (light blue) or Klrg1−Nfil3(GFP)− versus Klrg1+Nfil3(GFP)+ Treg (red) were correlated with peaks identified in public CD8 or CD4 Batf ChIP-seq datasets (Hasan et al., 2017, Kurachi et al., 2014) (n = 4).
(D) ATAC-seq data for the Il10 and Nfil3 gene and associated promoter region for spleen Klrg1−Nfil3(GFP)− (black), spleen Klrg1−Nfil3(GFP)+ (light blue), spleen Klrg1+Nfil3(GFP)+ (red), and VAT, lung, and skin tisTregST2 (orange, brown, dark blue). Top 4 lanes show public Batf ChIP-seq for CD4 or CD8 T cells with antibody control data (dark gray). All Treg ATAC-seq datasets group-normalized to maximum peak height indicated in brackets, ChIP-seq datasets group-normalized with respective control (n = 4).
(E) Batf protein and RNA expression in spleen Klrg1−Nfil3(GFP)−, Klrg1−Nfil3(GFP)+, or Klrg1+Nfil3(GFP)+ Treg (Batf-antibody [red] or isotype control antibody [black]). Histograms concatenated (one-way ANOVA, n = 4).
(F) Clustermap illustrating the Batf Pearson correlation index based on single-cell data from spleen Klrg1−Nfil3(GFP)−, Klrg1−Nfil3(GFP)+, and Klrg1+Nfil3(GFP)+ Treg cells across 30 Batf positively correlated genes (red gene names) as well as 30 Batf anti-correlated genes (blue) (n = 4).
(G) Spleen Klrg1−Nfil3(GFP)− (black), Klrg1−Nfil3(GFP)+ (light blue), or Klrg1+Nfil3(GFP)+ Treg cells (red) were pre-gated and expression of Pd1 versus Klrg1 (upper graphs) and Ox40 versus Klrg1 (lower graphs) is displayed. Representative example shown.
(H) Gata3 and Ki67 staining in Spleen Klrg1−Pd1− (black), Klrg1−Pd1+ (light blue), or Klrg1+Pd1+ Treg (red) (representative example shown). Data representative of independent experiments or cell sorts.
BATF−/− Animals Lack tisTregST2 Precursors and Cannot Initiate the Tissue Program
ATAC-seq based chromatin profiling suggests Batf as a potential key transcription factor that drives the development of tisTregST2 precursor cells in lymphatic tissues. Therefore, we investigated the presence tisTregST2 precursors and mature tisTregST2 cells in Batf-deficient animals (Schraml et al., 2009). Klrg1−Pd1+ Treg cells were reduced by ∼80%, and numbers of Klrg1+Pd1+ Treg cells were reduced by ∼95% in the spleen (Figures 7A, S6A, and S6B). This translated to a severely reduced frequency of mature tisTregST2 cells in the tissues: Batf−/− animals harbored ∼10-fold (in lung) to ∼100-fold (in VAT and colon) decreased number of Klrg1+Pd1+ tisTregST2 cells (Figure S6B). To determine whether the observed effects were Treg intrinsic, we generated mixed 1-to-1 bone-marrow chimeras with Batf+/+ and Batf−/− bone marrow. Six weeks after transfer, Batf−/−-derived Treg cells were almost completely unable to give rise to Klrg1−Pd1+ and Klrg1+Pd1+ tisTregST2 precursor cells as measured in percentage (Figure 7B) as well as total numbers (Figures S6C and S6D). This was a specific defect, because PD1−Klrg1− Treg cells in lymphoid organs were found with an equal ratio of Batf+/+ and Batf−/− Treg cells, while tissues were comprised exclusively of Batf+/+ Treg cells (Figure 7C). Finally, adoptive transfer of sorted Klrg1− spleen Treg cells, mixed in a ratio of 1:1 from congenically labeled Batf+/+ and Batf−/− animals, confirmed these results: after 14 days, only Batf+/+ Treg cells were able to give rise to Klrg1−Pd1+ and Klrg1+Pd1+ precursors cells (Figures S6E–S6G), and only Batf+/+ Treg cells were found in tissues (Figure S6G). These data demonstrate that differentiation into the tisTregST2 precursor path depends on Batf, and, in its absence, Treg cells cannot initiate the precursor program and cannot mature into tisTregST2 cells in nonlymphoid tissues.
Figure 7.

Batf Is a Lineage-Defining TF in Nfil3(GFP) Precursors
(A) Measurement of Klrg1−Pd1+ Treg (light blue) and Klrg1+Pd1+ Treg (red) in spleens of Batf+/+ versus Batf−/− animals (unpaired t test, n = 7–15).
(B) Measurement of Klrg1−Pd1+ Treg (light blue) and Klrg1+Pd1+ Treg (red) in spleens of animals reconstituted with 50% CD90.1+CD90.2−Batf+/+ and 50% CD90.1−CD90.2+Batf−/− bone marrow 6 weeks after bone marrow transfer (unpaired t test, n = 5).
(C) Analysis of Treg in mixed bone marrow chimera as in (B). Contribution of Batf+/+ (black) or Batf−/− (red) bone marrow in Treg from spleen, inguinal LN (ing LN), mesenteric LN (mes LN), colon, skin, or VAT is shown (unpaired t test, n = 5).
(D) Spleen Treg were treated for 6 days in vitro with anti-CD3/28 beads and IL-2 (Ctrl, black), IL-2 plus IFN-γ (IFNγ, blue), or IL-2 plus IL-4 and IL-33 (IL4/33, orange) followed by gene expression analysis. Graphs illustrate expression of Nfil3, Batf, Gata3, Il1rl1, or Tbx21 (Deseq2, n = 4).
(E) Spleen Treg from Nfil3(GFP) animals were treated for 6 days in vitro with anti-CD3/CD28 beads and IL-2 (Ctrl, black) or IL-2 plus IL-4 and IL-33 (IL4/33, orange) followed by flow-cytometry-based measurement of GFP (unpaired t test, n = 4).
(F) Spleen Treg from Batf+/+ or Batf−/− animals were treated for 6 days in vitro as in (D), followed by ATAC-seq. PCA based on ATAC-seq reads in all samples over a consensus set of 105,243 peaks (n = 4).
(G) Unsupervised hierarchical clustering of ATAC-seq data (28,033 unique differential peaks) with all cell types shown in (F) (n = 4).
(H) Comparison of chromatin accessibility between Batf+/+ and Batf−/− Treg treated with IL-4 and IL-33. Significant regions (log2 fold change >2, normalized mean accessibility ≥10, adjusted p value <0.01) are colored in blue (n = 4).
(I) De novo motif analysis on significant peak sets derived from (H) (n = 4).
(J) Single-base-pair resolution ATAC-seq signal at motif-centered peaks containing the de novo discovered motif highly similar to the known Batf motif (discovered in I) in IL-2-, IL-4-, and IL-33-treated Batf−/− versus Batf+/+ Treg. Transposase background shown in gray (n = 4).
(K) Pie chart illustrating the overlap of peaks that are differentially opened in Klrg1−Nfil3(GFP)− versus Klrg1+Nfil3(GFP)+ Treg (top) or Klrg1−Nfil3(GFP)− versus Klrg1−Nfil3(GFP)+ Treg (bottom) with Batf-dependent peaks identified in (H) (n = 4).
(L) ATAC-seq data for the Il1rl1 and Pparg gene and associated promoter region for in vitro-treated Batf−/− versus Batf+/+ Treg (color), spleen Klrg1−Nfil3(GFP)− (black), Klrg1−Nfil3(GFP)+ (light blue), Klrg1+Nfil3(GFP)+ (red), or VAT tisTregST2 (orange). Top 4 lanes are public Batf ChIP-seq data for CD4 or CD8 T cells including antibody control data (dark gray). All Treg ATAC-seq datasets group-normalized to maximum peak height indicated in brackets, ChIP-seq datasets group-normalized with respective control (n = 4).
(M) Pie chart illustrating the overlap of the “core” tisTregST2 peaks with Batf-dependent peaks identified in (H) (n = 4).
(N) ATAC-seq data for the Rora and IL10 loci for in vitro-treated Batf−/− versus Batf+/+ Treg as in (M) (n = 4). Data representative of independent experiments or cell sorts.
See also Figures S6 and S7 and Tables S6 and S7.
To further investigate the impact of Batf on the induction and the establishment of the tisTregST2 program, we developed an in vitro differentiation system to induce the “tissue program.” Naive (CD62L+) Treg cells were stimulated with IL-2 and anti-CD3-CD28 beads. We added the TH2-cytokine IL-4 and the ST2-ligand IL-33 to differentiate the Treg cells into the tisTregST2 program, while IL-2/interferon (IFN)-γ or IL-2-only cytokine conditions were used as controls. After 6 days, we could detect the induction of tisTregST2 signature genes such as Nfil3, Gata3, Batf, and Il1rl1 (ST2) with IL-4 and IL-33 treatment, while expression of Tbx21 was repressed (Figure 7D). Similarly, we could induce Nfil3(GFP), Batf, and Gata3 protein expression with this tissue differentiation protocol (Figures 7E and S6I). Using cytokine titration and time course experiments, we could show the cooperative effect of both IL-4 and IL-33 on the induction of Nfil3 and on the effector molecules IL-10 and Areg, both on RNA and protein level (Figures S7A–S7C). In addition, we could show that other TH2-associated cytokines such as IL-5, IL-9, or IL-13 did not show this cooperative effect with IL-4, rendering IL-33 a prototype cytokine to induce the tissue phenotype in vitro (Figure S7D).
Using this model, we treated naive Treg cells isolated from wild-type (WT) and Batf−/− animals with IL-2, IL-4, and IL-33 or IL-2-only for 6 days, followed by ATAC-seq. PCA clearly separated IL-4 and IL-33 differentiated Treg cells from WT animals from all other populations, whereas IL-4- and IL-33-treated Treg cells from Batf−/− animals grouped more closely to IL-2-only-treated Treg populations (Figure 7F). A clustered heatmap of 28,033 differential peaks across all pairwise comparisons illustrated that Batf−/− IL-4 and IL-33 differentiated Treg cells lack a large fraction of accessible sites that could be induced by these cytokines in Treg cells from WT animals (Figure 7G). This was activation-independent because loci indicating activation and proliferation (e.g., Cd69, Mki67, Stat5, or Tgfb1 were not affected) (Figure S7E). Focusing only on differential ATAC-seq signals in IL-4- and IL-33-treated cells, we identified 10,529 regions that were lost in Batf−/− cells (Figure 7H). De novo motif analysis showed highly significant enrichment of BATF, IRF:BATF, and BATF:JUN motifs in the depleted accessible regions, strongly suggesting that the observed chromatin changes are a direct effect of the transcription factor Batf (Figures 7I and 7J).
Because we propose that Batf is a driver of the tisTregST2 precursor molecular program, we measured how many chromatin regions that become accessible during the maturation of Klrg1−Nfil3−(GFP) to Klrg1−Nfil3(GFP)+ or Klrg1+Nfil3(GFP)+ were directly affected in IL-4 and IL-33 differentiated Treg cells from Batf−/− animals. Of the 2,243 and 2,169 peaks that gain accessibility from Klrg1−Nfil3−(GFP) to Klrg1−Nfil3(GFP)+ or Klrg1+Nfil3(GFP)+, respectively, 509 (22.7%) and 406 (18.7%) showed decreased accessibility in Batf−/− Treg cells compared to WT Treg cells under IL-4 and IL-33 polarizing conditions (Figure 7K). These observations are exemplified at the Pparg, Il1rl1 (ST2), and Maf loci where Batf target regions were accessible in precursors, mature VAT tisTregST2 and IL-4 and IL-33 differentiated Treg cells from WT animals but were not accessible in Batf−/− Treg cells (Figures 7L and S7F). Similar results were obtained when utilizing the “core” tisTregST2-specific peak set (Figure 7M). These observations are exemplified at the Rora and IL10 loci (Figure 7N).
In summary, our data suggests that Batf is a key transcription factor that enables the development of tisTregST2 precursors by driving a molecular precursor program, which, if not present, leads to a lack of precursor cell differentiation and consequently to the absence of mature tisTregST2 cells in nonlymphoid tissues.
Discussion
ST2-expressing tissue Treg cells perform important tissue homeostasis and regenerative functions. Using chromatin profiling, we identified tissue-specific chromatin signatures as well as substantial common accessible chromatin regions shared between all tisTregST2 cells from various tissues. This shared regulatory program indicates that tisTregST2 cells have a common precursor path. By generating a Nfil3(GFP) reporter mouse line, we were able to identify both Klrg1−Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ as lymphoid precursor stages for tisTregST2 cells. Our data did not support the notion that these lymphoid precursor stages were re-circulating tissue Treg cells. However, fate mapping studies with genetic reporter mice could further substantiate this point. Clustering of single-cell RNA-seq data, RNA-velocity analysis, adoptive transfer experiments, and the stepwise increase in chromatin accessibility of tisTregST2-specific regions suggest that Klrg1+Nfil3(GFP)+ Treg cells are a further developed precursor stage toward the tisTregST2 phenotype. The transition from the “earlier” Klrg1−Nfil3(GFP)+ to the “late” Klrg1+Nfil3(GFP)+ precursor appears to be an important one, because several tisTregST2 markers were induced only at this latter stage (Gata3, Il1rl1 [ST2], Klrg1, and a switch from Id3 to Id2). Progressive loss of Id3 correlates with differentiation of Treg cells and has recently been observed in Treg populations localized in nonlymphoid tissues (Sullivan et al., 2019). The tissue has an important contribution as the majority of tissue-specific chromatin changes were observed after tissue extravasation of progenitor cells.
Our postnatal organ seeding data of Treg cells releveled that both precursors peaked in the spleen 5–12 days after birth, followed by seeding of nonlymphoid tissues. A perinatal establishment of the Treg cell pool in nonlymphoid tissues has been described (Yang et al., 2015). TCR-repertoire, opening of certain chromatin regions, and expression of differentiation markers indicated an early activation event that takes place in Klrg1–Nfil3(GFP)+ and Klrg1+Nfil3(GFP)+ precursor cells in lymphoid organs. Indeed, we could show that TCRs of skin- or colon Treg cells were enriched in Nfil3(GFP)+ Treg cells from draining versus non-draining LNs. Our data are in line with recent publications, either based on scRNA-seq data (Miragaia et al., 2019) or on a VAT-specific TCR-transgenic mouse model (Li et al., 2018), both suggesting a priming step of nonlymphoid Treg cells in spleen or LN. It remains to be further defined which cells and external triggers contribute to the induction of the nonlymphoid Treg cell fate in lymphoid organs.
We identified the transcription factor Batf to be essential for tisTregST2 precursor cell differentiation in lymphoid organs as well as for the presence of tisTregST2 cells within nonlymphoid tissues. Loss of Treg cells in nonlymphoid tissues in Batf−/− mice has been described by us and others (Delacher et al., 2017, Hayatsu et al., 2017). Our data now show that Batf is critically required earlier than initially thought. Our findings indicate that Batf drives the differentiation of the precursor stage into the tisTregST2 phenotype by orchestrating a molecular program. Approximately 20% of all regions that gain accessibility from Klrg1−Nfil3(GFP)− to the Klrg1−Nfil3(GFP)+ precursors are likely direct Batf target loci, including regions in TH2-associated genes such as in the Il1rl1 (ST2), Il10, Maf, Pparg, and the TH2 locus control region (Rad50). It was demonstrated that Batf:Jun family protein complexes cooperate with Irf4, and that the Irf4:Jun:Batf heterotrimeric complex is critical for Irf4-mediated transcription of Il10 in TH2 cells (Li et al., 2012). Furthermore, it has been demonstrated that Batf directly modulates the TH2 locus control region (Bao et al., 2016), is required for the differentiation of TH17 cells (Glasmacher et al., 2012, Li et al., 2012, Yamazaki et al., 2017), and controls expression of the transcription factors Bcl-6 and Maf in T-follicular helper cells (Ise et al., 2011).
Will tissue Treg cells be the next frontier of immunotherapy to treat conditions like autoimmune diseases, inflammatory bowel disease, or graft-versus-host disease (Bluestone and Tang, 2018)? Understanding the ontogeny and, thereby, molecular programs that drive tissue Treg differentiation, is essential to develop future targeted therapies to leverage nonlymphoid Treg cells for tissue repair and regeneration.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified anti-mouse CD3 antibody | BD Biosciences | RRID: AB_394590 |
| APC anti-mouse CD4 antibody | Biolegend | RRID: AB_312719 |
| APC/Cy7 anti-mouse CD4 antibody | Biolegend | RRID: AB_312699 |
| Biotin anti-mouse CD4 antibody | Biolegend | RRID: AB_312711 |
| BV421 anti-mouse CD4 antibody | Biolegend | RRID: AB_11219790 |
| FITC anti-mouse CD4 antibody | Biolegend | RRID: AB_312713 |
| BV711 anti-mouse CD4 antibody | Biolegend | RRID: AB_2562099 |
| BV605 anti-mouse CD4 antibody | Biolegend | RRID: AB_2563054 |
| PE anti-mouse CD4 antibody | Biolegend | RRID: AB_312715 |
| PE/Cy7 anti-mouse CD4 antibody | Biolegend | RRID: AB_312729 |
| PerCP/Cy5.5 anti-mouse CD4 antibody | Biolegend | RRID: AB_893326 |
| PE-Dazzle 594 anti-mouse CD4 antibody | Biolegend | RRID: AB_2563684 |
| BUV395 anti-mouse CD4 antibody | BD Biosciences | RRID: AB_2738426 |
| BUV737 anti-mouse CD4 antibody | BD Biosciences | RRID: AB_2732918 |
| Biotin anti-mouse CD8a antibody | Biolegend | RRID: AB_312743 |
| BV605 anti-mouse CD8a antibody | Biolegend | RRID: AB_2562609 |
| PE/Cy7 anti-mouse CD8a antibody | Biolegend | RRID: AB_312761 |
| PerCP/Cy5.5 anti-mouse CD8a antibody | Biolegend | RRID: AB_2075238 |
| APC anti-mouse CD8a antibody | Biolegend | RRID: AB_312751 |
| FITC anti-mouse CD8a antibody | Biolegend | RRID: AB_312745 |
| BV421 anti-mouse CD8a antibody | Biolegend | RRID: AB_11204079 |
| APC/Cy7 anti-mouse CD8a antibody | Biolegend | RRID: AB_312753 |
| APC/Cy7 anti-mouse/human CD11b antibody | Biolegend | RRID: AB_830641 |
| APC/Cy7 anti-mouse CD19 antibody | Biolegend | RRID: AB_830707 |
| Pacific Blue anti-mouse CD19 antibody | Biolegend | RRID: AB_439718 |
| APC anti-mouse CD25 antibody | Biolegend | RRID: AB_312861 |
| APC anti-mouse CD25 antibody | Miltenyi | RRID: AB_2752169 |
| Biotin anti-mouse CD25 antibody | Biolegend | RRID: AB_312853 |
| Biotin anti-mouse CD25 antibody | Miltenyi | RRID: AB_2656661 |
| PE anti-mouse CD25 antibody | Biolegend | RRID: AB_312857 |
| PE anti-mouse CD25 antibody | Miltenyi | RRID: AB_2656655 |
| PE/Cy7 anti-mouse CD25 antibody | Biolegend | RRID: AB_312865 |
| BV711 anti-mouse CD25 antibody | Biolegend | RRID: AB_2564130 |
| BV421 anti-mouse CD25 antibody | Biolegend | RRID: AB_11203373 |
| AF488 anti-mouse CD25 antibody | Biolegend | RRID: AB_493333 |
| PerCP/Cy5.5 anti-mouse CD25 antibody | Biolegend | RRID: AB_893288 |
| Pacific Blue anti-mouse/human CD44 antibody | Biolegend | RRID: AB_493683 |
| BV421 anti-mouse/human CD44 antibody | Biolegend | RRID: AB_10895752 |
| BV605 anti-mouse/human CD44 antibody | Biolegend | RRID: AB_2562451 |
| APC/Cy7 anti-mouse/human CD44 antibody | Biolegend | RRID: AB_830785 |
| BV421 anti-mouse CD45 antibody | Biolegend | RRID: AB_10899570 |
| APC/Cy7 anti-mouse CD45 antibody | Biolegend | RRID: AB_312981 |
| Pacific Blue anti-mouse CD45 antibody | Biolegend | RRID: AB_493535 |
| APC anti-mouse CD45 antibody | Biolegend | RRID: AB_312977 |
| FITC anti-mouse CD45.1 antibody | Biolegend | RRID: AB_313495 |
| PerCP/Cy5.5 anti-mouse CD45.1 antibody | Biolegend | RRID: AB_893346 |
| PE/Cy7 anti-mouse CD45.1 antibody | Biolegend | RRID: AB_1134168 |
| BV605 anti-mouse CD45.1 antibody | Biolegend | RRID: AB_2562565 |
| PerCP/Cy5.5 anti-mouse CD45.2 antibody | Biolegend | RRID: AB_893350 |
| AF488 anti-mouse CD45.2 antibody | Biolegend | RRID: AB_492869 |
| AF647 anti-mouse CD45.2 antibody | Biolegend | RRID: AB_492870 |
| FITC anti-mouse CD45.2 antibody | BD Biosciences | RRID: AB_395041 |
| APC/Cy7 anti-mouse CD45.2 antibody | Biolegend | RRID: AB_830789 |
| Pacific Blue anti-mouse CD45.2 antibody | Biolegend | RRID: AB_492872 |
| APC anti-mouse CD62L antibody | Biolegend | RRID: AB_313099 |
| APC/Cy7 anti-mouse CD62L antibody | Biolegend | RRID: AB_830799 |
| PerCP/Cy5.5 anti-mouse CD62L antibody | Biolegend | RRID: AB_2285839 |
| FITC anti-mouse CD90.1 antibody | Biolegend | RRID: AB_1595653 |
| Pacific Blue anti-mouse CD90.1 antibody | Biolegend | RRID: AB_1595477 |
| APC/Cy7 anti-mouse CD90.1 antibody | Biolegend | RRID: AB_2303153 |
| PE anti-mouse CD90.1 antibody | Biolegend | RRID: AB_1595524 |
| PE/Cy7 anti-mouse CD90.1 antibody | Biolegend | RRID: AB_1659223 |
| APC anti-rat/mouse CD90.1 antibody | Biolegend | RRID: AB_1595470 |
| BV421 anti-rat /mouse CD90.1 antibody | Biolegend | RRID: AB_10899572 |
| PerCP/Cy5.5 anti-rat/mouse CD90.1 antibody | Biolegend | RRID: AB_961437 |
| APC/Cy7 anti-mouse CD90.2 antibody | Biolegend | RRID: AB_10613293 |
| APC anti-mouse CD90.2 antibody | Biolegend | RRID: AB_313183 |
| PE anti-human/mouse/rat CD278 (ICOS) antibody | Biolegend | RRID: AB_416332 |
| FITC anti-mouse/human KLRG1 antibody | Biolegend | RRID: AB_10643998 |
| PE anti-mouse/human KLRG1 antibody | Biolegend | RRID: AB_10574005 |
| PE/Dazzle 594 anti-mouse/human KLRG1 | Biolegend | RRID: AB_2564050 |
| BV711 anti-mouse/human KLRG1 antibody | Biolegend | RRID: AB_2629721 |
| BV421 anti-mouse/human KLRG1 antibody | Biolegend | RRID: AB_10918627 |
| FITC anti-mouse/human KLRG1 antibody | Biolegend | RRID: AB_10643582 |
| BV421 anti-mouse/human KLRG1 antibody | Biolegend | RRID: AB_2565613 |
| BV605 anti-mouse/human KLRG1 antibody | Biolegend | RRID: AB_2563357 |
| BV510 anti-mouse TCR-β chain antibody | Biolegend | RRID: AB_2562350 |
| APC/Cy7 anti-mouse TCR-β chain antibody | Biolegend | RRID: AB_893624 |
| PE anti-mouse TCR-β chain antibody | Biolegend | RRID: AB_313431 |
| PerCP/Cy5.5 anti-mouse TCR-β chain antibody | Biolegend | RRID: AB_1575173 |
| BV605 anti-mouse TCR-β chain antibody | Biolegend | RRID: AB_2629563 |
| BV711 anti-mouse TCR-β chain antibody | Biolegend | RRID: AB_2629564 |
| PE/Cy7 anti-mouse TCR-β chain antibody | Biolegend | RRID: AB_893625 |
| Biotin anti-mouse IL-33Ra (ST2) antibody | eBioscience | RRID: AB_2572809 |
| PE anti-mouse IL-33Ra (ST2) antibody | Biolegend | RRID: AB_2728176 |
| PerCP/Cy5.5 anti-mouse IL-33Ra (ST2) antibody | Biolegend | RRID: AB_2565636 |
| APC anti-mouse IL-33Ra (ST2) antibody | Biolegend | RRID: AB_2561917 |
| PE/Cy7 anti-mouse IL-33Ra (ST2) antibody | Biolegend | RRID: AB_2687367 |
| BV421 anti-mouse IL-33Ra (ST2) antibody | Biolegend | RRID: AB_2565634 |
| PE anti-mouse IL-33Ra (ST2) antibody | Biolegend | RRID: AB_2561915 |
| Biotin anti-mouse Amphiregulin antibody | R&D Systems | RRID: AB_2060662 |
| PE/Cy7 anti-mouse CD279 (PD-1) antibody | Biolegend | RRID: AB_10689635 |
| PE anti-mouse CD279 (PD-1) antibody | Biolegend | RRID: AB_1877231 |
| BV421 anti-mouse CD279 (PD-1) antibody | Biolegend | RRID: AB_2561447 |
| BV711 anti-mouse CD279 (PD-1) antibody | Biolegend | RRID: AB_2566158 |
| Biotin anti-mouse FoxP3 antibody | eBioscience | RRID: AB_763540 |
| PE anti-mouse FoxP3 antibody | eBioscience | RRID: AB_465936 |
| AF488 anti-mouse FoxP3 antibody | eBioscience | RRID: AB_763537 |
| APC anti-mouse Foxp3 antibody | eBioscience | RRID: AB_469457 |
| Anti-Mouse/Rat Foxp3 AF647 antibody | Biolegend | RRID: AB_763538 |
| Anti-Mouse/Rat Foxp3 Biotin antibody | Biolegend | RRID: AB_763540 |
| Anti-Mouse/Rat Foxp3 PE antibody | Biolegend | RRID: AB_465936 |
| AF647 anti-mouse Gata-3 antibody | Biolegend | RRID: AB_2563217 |
| AF647 anti-Gata-3 antibody | Biolegend | RRID: AB_2563217 |
| PE anti-Gata-3 antibody | Biolegend | RRID: AB_2562723 |
| BV421 anti-mouse/human Gata-3 antibody | Biolegend | RRID: AB_2563221 |
| PE anti-mouse/human c-MAF antibody | eBioscience | RRID: AB_2572747 |
| BV605 anti-mouse Ki-67 antibody | Biolegend | RRID: AB_2562664 |
| BV421 anti-mouse Ki-67 antibody | Biolegend | RRID: AB_2562663 |
| PE anti-mouse Ki-67 antibody | Biolegend | RRID: AB_2561525 |
| Purified anti-mouse/human BATF antibody | Cell Signaling | RRID: AB_11141425 |
| Goat Anti-Rabbit IgG (H+L) antibody, AF 647 | Thermo Fisher | RRID: AB_10562581 |
| PE anti-mouse/human BATF antibody | Cell Signaling | RRID: AB_2798938 |
| BV605 anti-mouse CD127 (IL-7Rα) ab | Biolegend | RRID: AB_2562114 |
| BV421 anti-mouse CD127 (IL-7Rα) ab | Biolegend | RRID: AB_11218800 |
| PE/Cy7 anti-mouse I-A/I-E antibody | Biolegend | RRID: AB_2290801 |
| APC/Cy7 anti-mouse I-A/I-E antibody | Biolegend | RRID: AB_1659252 |
| Pacific Blue anti-mouse I-A/I-E antibody | Biolegend | RRID: AB_493527 |
| Fixable Viability Dye eFluor 450 | eBioscience | Cat#65-0863-18 |
| Fixable Viability Dye eFluor 506 | eBioscience | Cat#65-0866-18 |
| Fixable Viability Dye eFluor 780 | eBioscience | Cat#65-0865-18 |
| AF488 Streptavidin | Biolegend | Cat#405235 |
| APC/Cy7 Streptavidin | Biolegend | Cat#405208 |
| E450 Streptavidin | eBioscience | Cat#48-4317-82 |
| FITC Streptavidin | Biolegend | Cat#405201 |
| PE Streptavidin | Biolegend | Cat#405204 |
| PE/Cy7 Streptavidin | Biolegend | Cat#405206 |
| PerCP/Cy5.5 Streptavidin | Biolegend | Cat#405214 |
| PE-Dazzle 594 Streptavidin | Biolegend | Cat#405248 |
| BUV395 Streptavidin | BD Biosciences | Cat#564176 |
| BUV737 Streptavidin | BD Biosciences | Cat#564293 |
| BV711 Streptavidin | Biolegend | Cat#405241 |
| BV605 Streptavidin | Biolegend | Cat#405229 |
| APC Streptavidin | Biolegend | Cat#405207 |
| BV421 Streptavidin | Biolegend | Cat#405225 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human IL-2 | Novartis | Proleukin S® 18 Mio U |
| Recombinant murine IL-4 | Peprotech | Cat#214-14 |
| Recombinant murine IL-5 | Peprotech | Cat#215-15 |
| Recombinant murine IL-9 | Peprotech | Cat#219-19 |
| Recombinant murine IL-13 | Peprotech | Cat#210-13 |
| Recombinant murine IL-33 | Biolegend | Cat#280504 |
| Purified anti-mouse IFN-gamma antibody | Biolegend | Cat#AB_315396 |
| Recombinant anti-mouse IFN-γ blocking mAb | Peprotech | Cat#500-P119 |
| Proteinase K | Carl Roth | Cat#7528.5 |
| HBSS | GIBCO | Cat#14025092 |
| PBS | GIBCO | Cat#10010023 |
| DMEM | GIBCO | Cat#41965 |
| Collagenase Type II | Sigma-Aldrich | Cat#C6885 |
| Collagenase Type IV | Sigma-Aldrich | Cat#C5138 |
| DNase I | Roche | Cat#11284932001 |
| Bovine Serum Albumin | Sigma | Cat#A4503 |
| Percoll | GE Healthcare | Cat#17-0891-01 |
| Power SYBR Green Master Mix | Thermo Fisher | Cat#4367659 |
| Taqman Gene Expression Master Mix | Thermo Fisher | Cat#4359016 |
| SuperScript II Reverse Transcriptase | Thermo Fisher | Cat#18064071 |
| SYBR-Green | Thermo Fisher | Cat#S7563 |
| Oligo d(T) 12-18 Primer | Thermo Fisher | Cat#18418012 |
| Compensation Beads | eBiosciences | Cat#01-1111-41 |
| Hair removal creme | Reckitt Benckiser (Veet) | N/A |
| Critical Commercial Assays | ||
| GentleMACS C tube | Miltenyi | Cat#130-096-334 |
| Lamina Propria Dissociation Kit | Miltenyi Biotec | Cat#130-097-410 |
| Anti-biotin microbeads, ultrapure | Miltenyi Biotec | Cat#130-105-637 |
| Anti-PE microbeads, ultrapure | Miltenyi Biotec | Cat#130-105-639 |
| Anti-APC microbeads | Miltenyi Biotec | Cat#130-090-855 |
| LS column | Miltenyi Biotec | Cat#130-042-401 |
| MS column | Miltenyi Biotec | Cat#130-042-201 |
| ACK lysis buffer | GIBCO | Cat#A1049201 |
| Foxp3 / Transcription Factor Buffer Set | eBiosciences | Cat#00-5523-00 |
| RNEasy Plus Mini Kit | Quiagen | Cat#74134 |
| Immuprep Analytik Jena | Analytik Jena | Cat#845-KS-2040250 |
| Dynabeads Mouse T-Activator CD3/CD28 | Thermo Fisher | Cat#11456D |
| Mouse T cell activation and expansion kit | Miltenyi Biotec | Cat#130-093-627 |
| SMARTer Ultra Low Input RNA kit | Takara | Cat#634894 |
| SMART-Seq Stranded Kit | Takara | Cat#634455 |
| NEXT CHiP-Seq Library Prep Master Mix | NEB | Cat#E6240L |
| NEBNext Multiplex Oligos | NEB | Cat#E7335L |
| NEBNext High Fidelity PCR Master mix | NEB | Cat#M0541S |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| Chromium™ Single Cell Controller & Accessory Kit | 10X | Cat#120212 |
| Chromium™ Single Cell 3′ Library and Gel Bead Kit | 10X | Cat#120237 |
| Chromium™ Single Cell 5′ Library & Gel Bead Kit | 10X | Cat#1000014 |
| Chromium™ Single Cell V(D)J Enrichment Kit, Mouse T Cell | 10X | Cat#1000071 |
| Chromium™ Single Cell A Chip Kit | 10X | Cat#120236 |
| Chromium™ Single Cell 3′/5′ Library Construction Kit | 10X | Cat#1000020 |
| Chromium™ Multiplex Kit | 10X | Cat#120262 |
| Agencourt AMPure XP beads | Beckman Coulter | Cat#A63880 |
| Illumina Nextera DNA preparation Kit | Illumina | Cat#FC1211030 |
| Qubit dsDNA HS Kit | Thermo Fisher Scientific | Cat#Q32851 |
| Qubit Protein Assay Kit | Thermo Fisher Scientific | Cat#Q33211 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat#50675584 |
| High Sensitivity D1000 Reagents | Agilent | Cat#50675585 |
| RNA ScreenTape | Agilent | Cat#50675576 |
| RNA ScreenTape Sample Buffer | Agilent | Cat#50675577 |
| Zymo clean & concentrator kit | Zymo | Cat#D4013 |
| Deposited Data | ||
| Raw and analyzed data: ATAC-seq, RNA-seq, single cell RNA-seq, single cell TCR-seq | This manuscript | GEO: GSE130884 |
| Oligonucleotides | ||
| Anti-mouse Hprt Taqman probe (FAM) | Applied Biosystems | Mm03024075_m1 |
| Anti-mouse Hprt Taqman probe (VIC) | Applied Biosystems | Mm01318746_g1 |
| Anti-mouse Gapdh Taqman probe (FAM) | Applied Biosystems | Mm99999915_g1 |
| Anti-mouse B2M Taqman probe (FAM) | Applied Biosystems | Mm00437762_m1 |
| Anti-mouse Foxp3 Taqman probe (FAM) | Applied Biosystems | Mm00475162_m1 |
| Anti-mouse Batf Taqman probe (FAM) | Applied Biosystems | Mm00479410_m1 |
| Anti-mouse Il10 Taqman probe (FAM) | Applied Biosystems | Mm01288386_m1 |
| Anti-mouse Il1rl1 Taqman probe (FAM) | Applied Biosystems | Mm00516117_m1 |
| Anti-mouse Nfil3 Taqman probe (FAM) | Applied Biosystems | Mm00600292_s1 |
| Anti-mouse Il7r Taqman probe (FAM) | Applied Biosystems | Mm00434295_m1 |
| Anti-mouse Gata3 Taqman probe (FAM) | Applied Biosystems | Mm00484683_m1 |
| Anti-mouse Areg Taqman probe (FAM) | Applied Biosystems | Mm00484683_m1 |
| Anti-mouse Klrg1 Taqman probe (FAM) | Applied Biosystems | Mm00516879_m1 |
| Anti-mouse Tbx21 Taqman probe (FAM) | Applied Biosystems | Mm00450960_m1 |
| Anti-mouse Pparg Taqman probe (FAM) | Applied Biosystems | Mm01184322_m1 |
| Software and Algorithms | ||
| Flowjo (v10.5.3) | BD Biosciences | https://www.flowjo.com/solutions/flowjo/downloads/ |
| FACS Diva (v8.0.2) | BD Biosciences | http://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| Prism (v7.0b) | GraphPad Software Inc | https://www.graphpad.com/ |
| R (v3.4.2 and v.3.5.1) | R Foundation for Statistical Computing | https://www.r-project.org/ |
| Bedtools (v2.26.0) | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io |
| Bowtie2 (v.2.2.4) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| ENRICHR | Chen et al., 2013, Kuleshov et al., 2016 | http://amp.pharm.mssm.edu/Enrichr/ |
| HOMER (v4.10.1) | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| Integrative Genomics Viewer (IGV; v2.3.97) | Robinson et al., 2011 | http://software.broadinstitute.org/software/igv/ |
| LOLA (v1.12.0) | Sheffield and Bock, 2016 | http://databio.org/lola/ |
| FASTX Toolkit (v0.0.13) | http://hannonlab.cshl.edu/fastx_toolkit/index.html | |
| STAR (v2.3) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Cell Ranger (v 2.0.1 and v.2.2.0) | 10x Genomics | https://github.com/10XGenomics/cellranger |
| sambamba (v0.6.6) | Tarasov et al., 2015 | https://github.com/biod/sambamba |
| Picard Toolkit (v1.78) | Broad Institute | https://github.com/broadinstitute/picard |
| skewer(v0.2.2) | Jiang et al., 2014 | https://github.com/relipmoc/skewer |
| DESeq2 (v1.20.0) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| UCSC genome browser Utilities | Kent et al., 2002 | https://genome.ucsc.edu/ |
| Seurat (v2.3.0 and v3.0.1) | Butler et al., 2018 | https://satijalab.org/seurat/ |
| Monocle (v2.6.3) | Trapnell et al., 2014 | http://cole-trapnell-lab.github.io/monocle-release/ |
| Velocyto (v0.17.17) | La Manno et al., 2018 | http://velocyto.org/ |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Markus Feuerer (Markus.Feuerer@ukr.de). The Nfil3 reporter mouse line generated in this study is available upon request.
Experimental Model and Subject Details
Mice
Wild-type C57BL/6, congenic B6.SJL-PtprcaPepcb/BoyCrl (CD45.1+), congenic B6.PL-Thy1a/CyJ (CD90.1+) mice were obtained from Charles River Breeding Laboratories (Wilmington, MA, USA) or the Jackson Laboratory (Bar Harbor, ME, USA). B6N.129(Cg)-Foxp3tm3Ayr mice (Foxp3.IRES-DTR/GFP) (Kim et al., 2007) were bred to CD45.1+ or CD90.1+ mice. Batf-deficient mice (129S-Batftm1.1Kmm/J) were obtained from Jackson Laboratories (Stock Number 013757). To generate scRNA-seq, scTCR-seq, RNA-seq and ATAC-seq data, we used adult male mice between 6 and 45 weeks of age. For all other experiments, both female and male mice between 6 and 45 weeks of age were used, unless otherwise indicated. For kinetics experiments, male and female mice between 5 days and 45 weeks of age were used. VAT was isolated exclusively from male mice.
For generation of Nfil3(GFP) BAC-transgenic mice, a fusion construct composed of the DNAs for iCre and eGFP was generated and inserted at the start codon of the Nfil3 gene in the BAC RP23-227M5 (BioScience; RPCIB731M05277Q) using Escherichia coli DH10B (Copeland et al., 2001). The final construct was linearized and injected into the pronuclei of fertilized C57BL/6N mouse eggs at the animal facility of the German Cancer Research Center. Genotyping was carried out by PCR from genomic DNA as described in the following paragraph. Details are shown in Figure S2A. Animals were housed under specific pathogen-free conditions at the DKFZ animal care facility or the Regensburg University Clinics animal care facility, and the governmental committee for animal experimentation (Regierungspräsidium Karlsruhe, Germany for DKFZ Heidelberg or Regierungspräsidium Unterfranken, Würzburg for Regensburg) approved all experiments involving animals.
Method Details
Genotyping of Batf−/− and Nfil3(GFP) animals
Tissue was digested in digest buffer (50 mM KCl, 20 mM Tris–HCl pH 8.8, 0.045% Tween20 and Igepal CA-630) with proteinase K (Carl Roth #7528.5) overnight at 56°C, followed by inactivation for 10 min at 96°C. For Batf genotyping, a PCR reaction with Taq polymerase, dNTPs, 2.5 mM MgCl2, 1X PCR buffer (50mM KCl, 20mM TRIS-HCl pH 8.8), and 0.4 μM Batf primers (TGCTGAGCATCTTTCCAGTCC, AGAATCCAGTGACTCCCTATCCC, CTGCTCAGTCTCAGTTTTCAC) was performed (94°C-10min; 94°C-30 s 61°C-30 s 72°C-60 s × 35; 74°C10min). For Nfil3 genotyping, a PCR reaction with Taq polymerase, dNTPs, 2.5 mM MgCl2, 1X PCR buffer (50mM KCl, 20mM TRIS-HCl pH 8.8), 0.4 μM Nfil3-GFP primers (AGCTGACCCTGAAGTTCATCTG, CATGATATAGACGTTGTGGCTGTT) and 0.1 μM RAG1 primers (CCCCTTTATTGATATGCACCA, AGATGTCTCAAAGTCATGGGC) was performed (95°C-5min; 95°C-30 s 58°C-30 s 72°C-30 s × 35; 72°C5min). Samples were separated on an agarose gel (2%) and gene status analyzed.
Tissue digestion for flow cytometric analysis and FACS sorting of cells
To isolate T cells from VAT tissue, gonadal fat pads of male mice were excised, cut into small pieces and digested for 45 minutes at 37°C (base medium DMEM (GIBCO #41965), 1 mg/ml collagenase type II (Sigma-Aldrich #C6885), 20 μg/ml DNase I (Roche #11284932001), 20 mg/ml bovine serum albumin (Sigma-Aldrich #A4503)) in an orbitally-shaking waterbath, followed by incubation with 2 mM EDTA-PBS for 2 minutes and centrifugation and filtration steps.
To isolate T cells from skin tissue, hair and hair follicles from the back of the animal were removed with an electric shaver and depilatory cream. Skin was separated from the dorsal surface, cut into small pieces and digested (base medium DMEM (GIBCO #41965), 4 mg/ml collagenase type IV (Sigma-Aldrich #C5138), 10 μg/ml DNase I (Roche #11284932001), 2% fetal bovine serum). Digestion was performed for 45-60 minutes in an orbitally-shaking waterbath pre-heated to 37°C or directly in a GentleMACS C tube (Miltenyi Biotec #130-096-334) and the program “37_C_Multi_H” for 90 minutes, followed by centrifugation and filtration steps.
To isolate T cells from liver or lung tissue, animals were perfused by opening the inferior vena cava and flushing the left ventricle with 10ml PBS to clear the body circulation. Liver and lung were excised, cut in small pieces and digested (base medium DMEM (GIBCO #41965), 1 mg/ml collagenase type IV (Sigma-Aldrich #C5138), 20 μg/ml DNase I (Roche #11284932001), 5 mg/ml bovine serum albumin (Sigma-Aldrich #A4503)) for 30-45 minutes in an orbitally-shaking waterbath pre-heated to 37°C, followed by centrifugation and filtration steps. Liver tissue was further separated using an 80%/40% Percoll-PBS gradient (GE Healthcare #17-0891-01), centrifugation (2000 x g, 20 min, 4°C, low acceleration, low deceleration) and isolation of the lymphocyte-containing interface. Lung samples were pre-enriched using biotinylated or fluorochrome-labeled CD25 antibody (Clone PC61, Biolegend) and anti-biotin ultrapure, anti-PE ultrapure or anti-APC microbeads (Miltenyi Biotec #130-105-637, #130-105-639, #130-090-855).
To isolate T cells from colon tissue, colons were isolated, cleared of feces and prepared according to manufacturer’s instructions with a lamina propria dissociation kit (Miltenyi #130-097-410). Samples were either pre-enriched with CD4 (Clone RM4-5, Biolegend) or CD25 (Clone REA568, Miltenyi Biotec) antibody and microbeads or measured directly. More detailed protocols about T cell isolation from murine tissues are published (Cossarizza et al., 2019).
To isolate T cells from blood, peripheral blood was collected via cardiac puncture and incubated in heparin-coated capsules. Blood samples were centrifuged and red blood cells were lysed using ACK lysis buffer (GIBCO #A1049201).
To isolate T cells from bone marrow, femurs were collected and femoral head and femoral medial and lateral epicondyle were removed. Bone shafts were flushed with PBS and bone marrow collected by centrifugation followed by red blood cell lysis. T cells were pre-enriched with CD25-biotin antibody (Clone PC61) and microbeads.
To isolate T cells from spleen and lymph nodes, tissues were harvested and mechanically disintegrated. Samples were either pre-enriched with CD4 or CD25 antibody and microbead-based enrichment or measured directly.
Preparation of samples for flow cytometry
T cells were isolated and pre-enriched as described previously. Samples were stained either in 1.5 mL Eppendorf tubes or 96-well plates in FACS buffer (1%FCS in PBS). Surface staining was performed at 4°C for 20 minutes in 50-100 μL staining volume. Antibodies were used, if not indicated otherwise, as recommended by the manufacturer. Following antibodies were used for surface staining: CD3 (Clone 145-2C11), TCR-β chain (H57-597), CD4 (RM4-5), CD8α (53-6.7), CD19 (6D5), CD25 (PC61 and REA568), CD44 (IM7), CD45 (30-F11), CD45.1 (A20), CD45.2 (104), CD90.1 (OX-7), CD90.2 (30-H12), CD120b (TR79-89), CD278 (C398.A4), CD279 (29F.1A12), I-A/I-E/MHCII (M5/114.15.2), Klrg1 (2F1), IL-33R/ST2 (DIH9 and RMST2-33). Intracellular staining was performed with the Foxp3 / Transcription Factor Buffer Set (eBiosciences) according to manufacturer’s protocol with the following adaptations: intracellular staining steps were performed for 60 minutes at room temperature. Antibodies for intracellular staining include Gata3 (16E10A23), Foxp3 (JFK-16S), KI67 (16A8), and Batf (D7C4, 1:200 dilution). For Batf staining, secondary intracellular staining was performed with AF647-coupled anti-rabbit antibody at 1:400 dilution. Dead cells were excluded with a fixable live/dead dye (eBioscience Fixable Viability Dye eFluor450, eFluor506 or eFluor780). All antibodies are listed in the Key Resources Table.
Flow cytometry of T cells from tissues
T cells were isolated, pre-enriched and stained as described previously. Afterward, they were filtered with a 40 μm filter unit and acquired on a BD™ LSRII™, LSR Fortessa™, or ARIA II™ cytometer. BD CS&T™ beads were used to validate machine functionality. Fluorescence spillover compensation was performed with either compensation beads (eBiosciences OneComp eBeads) or lymphocytes stained with CD4 (RM4-5) in the respective colors. Flow cytometry data were analyzed using FlowJo™ (BD, Version 10 for Mac). Files were concatenated using FlowJo™.
FACS sorting of T cells from tissues for bulk ATAC-seq and bulk RNA-seq
T cells were isolated and pre-enriched as described previously. Afterward, they were stained with live/dead dye and surface antibodies as described in the section “Preparation of samples for flow cytometry.” Cells were then sorted into 1.5 mL tubes on an BD™ FACSAria™ IIu, III, or FACSAria™ FUSION cell sorting system using 70 μm or 85 μm nozzle size and 4-way-purity sort mode into 1.5 mL tubes. If required, cells were sorted in a double-sort strategy with primary sort (85 μm, yield mode, into FACS buffer) and secondary sort (85 μm, 4-way-purity mode, into RLT+ buffer or FACS buffer). Sample and post-sort quality control were recorded where applicable and are displayed in the source data file linked to this manuscript. For bulk ATAC-seq, cells were sorted into FACS buffer. For bulk RNA-seq, cells were sorted into RLT+ lysis buffer (QIAGEN RNeasy Plus MiniKit).
Real time PCR
For RNA isolation, cells were resuspended in RLT+ lysis buffer and purified according to manufacturer’s protocol (RNEasy mini Kit, Quiagen). RNA was reversely transcribed into cDNA according to manufacturer’s protocol (Reverse Transcriptase II, life technologies). cDNA was used with Taqman probes and Taqman master mix in a Viia7 real-time PCR system (all ThermoFisher). Gene expression was normalized to housekeeping gene expression (Hprt) with the formula: relative gene expression = 2ˆ - (Ct (gene X) – Ct (Hprt)). Primer designations are listed in Key Resources Table.
DT-depletion, bone marrow chimera and transfer studies in-vivo
Cells for transfer were FACS-sorted as described above. Congenic Foxp3CD45.1, GFP, DTR animals were injected intraperitoneally with 1000 ng DT one day before transfer and immediately following transfer. Cells for transfer were resuspended in sterile PBS with 1% FCS and transferred via retroorbital injection. 10-14 days later, cells were isolated from various tissues and identified based on CD45.1/CD45.2/CD90.1/CD90.2 expression. To generate mixed bone-marrow chimeras, recipient animals were treated with 9 Gray of radiation, followed by transfer of 5 million red blood cell-depleted bone marrow cells. Bone marrow was mixed from 50% WT and 50% Batf-deficient bone marrow. Cells from donor (Batf+/+ or Batf−/−) and recipient were identified based on CD45.1/CD45.2/CD90.1/CD90.2 expression.
In-vitro differentiation of Treg and Tconv cells
Treg cells and Tconv cells were purified from Foxp3GFP, DTR animals by FACS-based high-purity sorting. Then, cells were stimulated with anti-mouse CD3/28 microbeads at a 4 to 1 bead to cell ratio. For Treg cell cultures, IL-2 was supplemented at 5000 U/ml. For Tconv cell cultures, IL-2 was supplemented at 50 U/ml, unless otherwise indicated. For tissue Treg induction experiments with IL-4 and IL-33, Treg cells were treated with 100 ng/ml IL-4, 100 ng/ml IL-33 as well as anti-mouse IFNγ blocking antibodies (10,000 U/ml) for 6 days at 37° in-vitro. For IFN-γ differentiation, cells were treated with 100 ng/mL IFN-γ for 6 days at 37° in-vitro. For cytokine titrations with IL-4 versus IL-33, IL-4 versus IFN-γ, IL-4 versus IL-5, IL-4 versus IL-9, and IL-4 versus IL-13, cytokines were used as indicated in the Figures (1+9 titrations). After 6 days, cells were either analyzed via flow cytometry or RNA was harvested, followed by cDNA synthesis and RT-PCR. Protein MFI or relative gene expression were normalized to untreated expanded Treg cells. For RNA induction kinetics, Treg cells were harvested on day 1, 3, 5, 7, 9, and 11 after seeding. Medium was exchanged on day 7.
Assay for transposable-accessible chromatin with sequencing (ATAC-seq)
Chromatin accessibility mapping was performed using the ATAC-seq method as previously described (Buenrostro et al., 2013, Corces et al., 2017), with minor adaptations. Briefly, in each experiment 2,000 - 15,000 sorted cells were pelleted by centrifuging for 10 min at 4°C at 500 x g. After centrifugation, the pellet was carefully lysed in 50 μl resuspension buffer supplemented with NP-40 (Sigma), Tween-20 and Digitonin (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 0.1% Tween-20, 0.01% Digitonin) and incubated for 3 minutes on ice. Then, 1 mL of ice-cold resuspension buffer supplemented with Tween-20 was added, and the sample was centrifuged at 4°C at 500 x g for 10 minutes. The supernatant was discarded, and the cell pellet was carefully resuspended in the transposition reaction (25 μl 2 x TD buffer (Illumina), 2.5 μl TDE1 (Illumina), 16.5 μl PBS, 5 μl nuclease-free water, 0.5 μl 1% Digitonin (Promega), 0.5 μl 10% Tween-20 (Sigma)) for 30 min at 37°C on a shaker at 1000 rpm. Following DNA purification with the Clean and Concentrator-5 kit (Zymo) eluting in 23 μl, 2 μl of the eluted DNA was used in a quantitative 10 μL PCR reaction (1.25 μM forward and reverse custom Nextera primers (Corces et al., 2017), 1x SYBR green final concentration) to estimate the optimum number of amplification cycles with the following program 72°C 5 min; 98°C 30 s; 25 cycles: 98°C 10 s, 63°C 30 s, 72°C 1 min; the final amplification of the library was carried out using the same PCR program and the number of cycles according to the Cq value of the qPCR. Library amplification using custom Nextera primers was followed by SPRI size selection with AmpureXP beads to exclude fragments larger than 1,200 bp. DNA concentration was measured with a Qubit fluorometer (Life Technologies). The libraries were sequenced using the Illumina HiSeq3000/4000 or NextSeq platforms.
Preprocessing and analysis of ATAC-seq data
ATAC-seq reads were trimmed using Skewer (Jiang et al., 2014) and aligned to the mm10 assembly of the murine genome using Bowtie2 (Langmead and Salzberg, 2012) with the ‘-very-sensitive’ parameter and a maximum fragment length of 2,000 bp. Duplicate and unpaired reads were removed using the sambamba (Tarasov et al., 2015) ‘markdup’ command, and reads with mapping quality > 30 and alignment to the nuclear genome were kept. All downstream analyses were performed on these filtered reads. All reads aligning to the “+” strand were offset by +4 bp, and all reads aligning to the “–“ strand were offset −5 bp to represent the center of the transposon binding event as described previously (Buenrostro et al., 2013). For visualization purposes only, coverage files from filtered bam files were produced using bedtools (Quinlan and Hall, 2010) genomeCoverageBed, each position was normalized by dividing to the total library size and multiplying by 106, followed by conversion to a bigwig using the bedGraphToBigWig command from the UCSC genome browser tools.
Bioinformatic analysis of chromatin accessibility data
Peak calling for each sample was performed using HOMER (Heinz et al., 2010) with the following approach: Two different peak sets were called, once we used the options “-style factor -fragLength 150 -size 250 -minDist 250 -L 4 -fdr 0.0001 -tbp 1” parameters, to identify highly robust small regions of open chromatin, and in a second approach we used the parameters”-region -fragLength 150 -size 150 -minDist 250 -L 4 -fdr 0.0001 -tbp 1” to identify larger regions of open chromatin. The two peak sets were then intersected using the bedtools intersect function with the “-u” option to create the peak set that was used for downstream analysis. Peaks overlapping blacklisted features as defined by the ENCODE project (ENCODE Project Consortium, 2012) were discarded. For the analysis of sample sets we always created a consensus region set by merging the called peaks from all involved samples, and we quantified the accessibility of each region in each sample by counting the number of reads from the filtered BAM file that overlapped each region.
To normalize the chromatin accessibility signal across samples, we performed quantile normalization followed by GC content normalization by regression using the cqn R package (Hansen et al., 2012). Principal component analysis (PCA) was performed using the R function prcomp with the parameter “scale=TRUE” (version 3.4.2) on the normalized chromatin accessibility values of all chromatin-accessible regions across all samples. DESeq2 (Love et al., 2014) was used on the raw count values for each sample and regulatory element to identify differential chromatin accessibility between samples. Significant regions were defined as having an FDR-corrected p value below 0.01, an absolute log2 fold change above 2, and a mean accessibility equal or greater than 10.
For the common tisTregST2 signature, differential peaks between LN memory Treg and tisTregST2 were used and filtered based on FDR adjusted p values equal or smaller than 0.01, an absolute log2 fold-change equal or greater than 2 and a base mean equal or greater than 10. For each remaining peak the fold change between tisTregST2 and Tconv was calculated. Peaks were kept if their change exceeded the median of the distribution and were clustered using the pheatmap R package with default settings.
For tisTregST2 tissue-specific programs, differential peaks of tisTregST2 samples based on pairwise comparisons were filtered for FDR adjusted p values smaller than 0.01, an absolute log2 fold-change greater than 2 and a base mean equal or greater than 10. For each tissue (skin, fat, colon, lung), the fold change between the tissue (e.g., skin) and all other tissues (e.g., fat, colon, lung) was calculated. In a tissue dependent manner, peaks were kept if their fold change exceeded the upper quartile value of the distribution. In addition to differentiate tisTregST2 to Tconv cells of the same tissue, similarly, for each tissue-specific peak the fold change between tisTregST2 and Tconv cells was calculated and peaks kept which exceeded the upper quartile value of the distribution. Kept peaks were clustered using pheatmap and R with default settings. P values for differential ATAC-seq p values were Benjamini-Hochberg corrected.
Motif enrichment analysis was done using HOMER (Heinz et al., 2010) with the function findMotifsGenome using “-size 500 -len 8,10,12 -h” parameters. Statistical significances of motif enrichment using HOMER were calculated using hypergeometric testing indicated by the “-h” parameter. Motifs from analyses were combined to a merged motif file and filtered for redundant motifs using HOMER’s compareMotifs function with the parameters “-reduceThresh 0.6 -matchThresh 10 -info 0.6 –pvalue 1e-25” and were compared to the vertebrate known motifs from the HOMER database. The enrichment of this reduced motif set was then calculated in the peaks of the respective comparisons using the annotatePeaks function. For Figure 7G, de novo motifs with a similarity value to a known motif less than 0.75 were discarded. Histograms of reads around transcription factor binding motifs were generated using HOMER by centering the peaks of interest on the investigated motifs using the annotatePeaks function, followed by counting reads from individual experiments at single base pair resolution in a radius of 1000 bp (or 150 bp) around the peak centers using the annotatePeaks function with the parameters “-hist -fragLength 1.” Tn5 background was calculated by processing publically available sequencing data of tagmented “naked” DNA fragments (ERR2213810_1, ERR2213810_2, ERR2213806_1, and ERR2213806_2) (Henriksson et al., 2019) similar to all other ATAC-seq data.
Peaks were assigned to their nearest TSS using the HOMER promoter annotation. Region set enrichment analysis of genes in the vicinity of differential peaks was performed using LOLA (Sheffield and Bock, 2016) with its core databases. Enrichment of genes associated with regulatory elements (annotated with the nearest transcription start site from Ensembl) was performed through the Enrichr API (Kuleshov et al., 2016) for the WikiPathways 2016 database.
Bioinformatic analysis of publicly available ChIP-seq datasets
Public CD8 or CD4 Batf ChIP-seq datasets including IgG or input controls (Hasan et al., 2017, Kurachi et al., 2014), GSE54191 and GSE86535, respectively, were reanalyzed and preprocessed similar to ATAC-seq data, with the difference that read positions were not shifted. Peak calling was performed using the HOMER “findPeaks” command with the standard settings and the “-style factor” option. Overlaps with other peak sets from ATAC-seq data was performed using the HOMER “mergePeaks” command.
Isolation of RNA, library preparation and sequencing
RNA was isolated with the RNEasy Plus Mini Kit (QIAGEN) according to manufacturer’s recommendations. cDNA was generated and amplified with the SMARTer Ultra Low Input RNA kit for Illumina Sequencing (Clontech Laboratories, Inc.) or the SMART-Seq Stranded Kit (Takara Bio) according to the manufacturer’s protocol. For ultra-low applications, sequencing libraries were prepared using the NEXT ChIP-Seq Library Prep Master Mix Set for Illumina (New England Biolabs) according to the manufacturer’s instructions with the following modifications: The adaptor-ligated double-stranded cDNA (10μl) was amplified using NEBNext Multiplex Oligos for Illumina (New England Biolabs, 25 μM primers), NEBNext High-Fidelity 2x PCR Master Mix (New England Biolabs) and 15 cycles of PCR. For all samples, final libraries were validated using Agilent 2100 Bioanalyzer (Agilent Technologies) and Qubit fluorometer (Invitrogen), normalized and pooled in equimolar ratios. 50bp single-read sequencing was performed on the Illumina HiSeq™ 2000 v4 or 75bp single-read sequencing on the Illumina Nextseq™ 500 according to the manufacturer’s protocol.
Mapping of RNA sequencing data, statistical evaluation and plotting
For all samples, low quality bases were removed with Fastq_quality_filter from the FASTX Toolkit 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/index.html) with 90% of the reads needing a quality phred score > 20. Homertools 4.7 (Heinz et al., 2010) were used for PolyA-tail trimming, and reads with a length < 17 were removed. PicardTools 1.78 (https://broadinstitute.github.io/picard/) were used to compute the quality metrics with CollectRNASeqMetrics. With STAR 2.3 (Dobin et al., 2013), the filtered reads were mapped against mouse genome 38 using default parameters. Count data and RPKM tables were generated by mapping filtered reads against union transcripts (derived from Mouse Ensembl 90) using a custom pipeline. Mapping was carried out with bowtie2 version 2.2.4 (Langmead and Salzberg, 2012) against union mouse genes: every gene is represented by a union of all its transcripts (exons). The count values (RPKM and raw counts) were calculated by running CoverageBed from Bedtools v2.26.0 (Quinlan and Hall, 2010) of the mapped reads together with the mouse annotation file (Ensembl 90) in gtf format and parsing the output with custom perl scripts. The input tables containing the replicates for groups to compare were created by a custom perl script. For DESeq2 (Love et al., 2014), DESeqDataSetFromMatrix was applied, followed by estimateSizeFactors, estimateDispersions, and nbinomWald testing. The result tables were annotated with gene information (gene symbol, gene type) derived from the gencode.vM16.annotation.gtf file. The results were then filtered for protein-coding genes according to the gencode.vM16.annotation.gtf file. The 1000 most variable transcripts were extracted and used for principal component analysis (PCA) using the R function prcomp with the parameter “scale=TRUE” (version 3.4.2). To measure correlation of RNA-seq with ATAC-seq data, peaks of the common signature were used if the nearest gene was no further away than 10,000 kb. In case of multiple peaks associated to the same gene, a mean was calculated. For each peak, a mean of the normalized ATAC-seq scores from fat, skin and lung was then plotted against the associated mean gene expression from the same tissues.
FACS sorting of T cells from tissues for scRNA-seq and scTCR-seq
T cells were isolated and pre-enriched as described above. Afterward, they were stained with live/dead dye and surface antibodies as described. Cells were then sorted on a BD™ FACSAria™ IIu, III, or FACSAria™ FUSION cell sorter using 70 μm or 85 μm nozzle size and 4-way-purity sort mode into 1.5 mL tubes filled with FACS buffer. Sample and post-sort quality control were recorded where applicable and are displayed in the source data file linked to this manuscript. For scRNA-seq, we sorted 15000 spleen CD25+Klrg1-Nfil3(GFP)- Treg, 15000 spleen CD25+Klrg1-Nfil3(GFP)+ Treg, 15,000 spleen CD25+Klrg1+Nfil3(GFP)+ Treg, 10000 spleen CD44+CD25+Foxp3(GFP)+ Treg, 7500 VAT CD44+CD25+Foxp3(GFP)+ Treg, 3000 skin CD44+CD25+Foxp3(GFP)+ Treg, 1500 blood CD44+CD25+Foxp3(GFP)+ Treg, 10000 inguinal lymph-node CD44+CD25+Foxp3(GFP)+ Treg, 10000 bone marrow CD44+CD25+Foxp3(GFP)+ Treg, 10000 lung CD44+CD25+Foxp3(GFP)+ Treg, and 1000 liver CD44+CD25+Foxp3(GFP)+ Treg. Samples were loaded onto the 10X Chromium Controller and processed the Single Cell 3′ Library Gel and Bead Kit v2 (10X). Samples were pooled from several Foxp3(GFP) or Foxp3(YFP) or Nfil3(GFP) reporter mice.
For scTCR-seq, we sorted 15000 (mouse 1) and 15000 (mouse 2) spleen CD25+Klrg1-Nfil3(GFP)- Treg, 15000 (mouse 1) and 15000 (mouse 2) spleen CD25+Klrg1-Nfil3(GFP)+ Treg, 8000 (mouse 1) and 15000 (mouse 2) spleen CD25+Klrg1+Nfil3(GFP)+ Treg, 3000 (mouse 1) and 15000 (mouse 2) skin CD25+Nfil3(GFP)+ Treg, 3000 (mouse 1) and 15000 (mouse 2) skin CD25+Nfil3(GFP)+ Treg, and 3000 (mouse 1) and 15000 (mouse 2) skin CD25+Nfil3(GFP)+ Treg. For mouse 3, we sorted 8200 skin CD25+Nfil3(GFP)+ Treg, 800 skin-draining inguinal LN CD25+Nfil3(GFP)+ Treg, 11000 colon CD25+Nfil3(GFP)+ Treg, and 80000 colon-draining mesenteric LN CD25+Nfil3(GFP)+ Treg. Samples were loaded onto the 10X Chromium Controller and processed the Single Cell 5′ Library Gel and Bead Kit (10X). Samples were sorted from single male Nfil3(GFP) animals.
scRNA-seq and scTCR-seq library preparation and sequencing
Samples for scRNA-seq were loaded on the Chromium Single cell Controller (10x Genomics #120212) using the Single Cell 3′ Library & Gel Bead Kit v2 (10x Genomics #120237). cDNA was amplified using 14 cycles of PCR. Library was constructed using 13 cycles of PCR. Products were purified using Ampure XP beads and quality was controlled using Agilent Tapestation. Samples were sequenced using 50bp single-read sequencing on the Illumina HiSeq™ 2000 v4. Samples for scTCR-seq were loaded on the Chromium Single cell Controller using the Single Cell 5′ Library & Gel Bead Kit (10x Genomics #1000014) and the Single Cell V(D)J Enrichment Kit, Mouse T Cell (10x Genomics #1000071). TCR samples were sequenced on a Illumina NextSeq 500 with 300 cycles paired-end sequencing.
Analysis of scTCR-seq data
Fastq files were processed using Cell Ranger (version 2.2.0) based on 10xGenomics provided mm10 reference genome (refdata-cellranger-vdj-GRCm38-alts-ensembl-2.2.0). Clones were matched by TRA and TRB sequence and clonal diversity was estimated using the inverse Simpson index. Samples from each mouse were compared in a pairwise fashion using the Jaccard index. Clonal abundance pie charts were generated using ggplot2 (version 2.2.1) and R (version 3.4.2).
Analysis of scRNA-seq data
Fastq files were processed using Cell Ranger version 2.0.1 based on 10xGenomics provided mm10 reference genome (refdata-cellranger-mm10-1.2.0). Cell Ranger was run per sample (using cellranger count). For downstream analysis the R package Seurat (Butler et al., 2018) (version 2.3.0) together with R (version 3.4.2) was used. Cells with fewer than 500 or more than 2500 transcripts were discarded as well as cells exceeding a 5% threshold of mitochondrial transcripts. The data was log normalized (using NormalizeData) and scaled (using ScaleData) which includes regressing for mitochondrial DNA content and the number of UMIs using a negative binomial distribution. Highly variable genes (HVGs) were identified (using FindVariableGenes) with default parameter settings. In the tissue Treg precursor analysis alone, as well as in the combined precursor and tisTregST2 analysis, 10 most contributing principle components (PCs) were used for downstream analysis. TSNE (using RunTSNE) was used for dimensionality reduction and visualization. For UMAP dimensionality reduction, Seurat (version 3.0.1, using RunUMAP) was used. UMAP reductions for single tissue analysis are based on the 10 most contributing PCs, whereas the 20 most contributing PCs were used for all other UMAP projections. Two-dimensional kernel densities were estimated for each of the three tissue Treg precursor populations (using kde2d) from the MASS package (version 7.3-50). The 70% contour levels were estimated with the function kde from the ks package (version 1.11.3) in R.
To reconstruct a developmental trajectory, Monocle (Trapnell et al., 2014) (version 2.6.3) was used. Steps included size factor estimation (using estimateSizeFactors), estimation of dispersions (using estimateDispersions), applying a minimal expression threshold of 0.1 (using detectGenes), HVGs were set to be used (using setOrderingFilter) and reducing dimensionality to three components (using reduceDimension(method = ‘DDRTree’, max_components = 3, …)). For visualization purposes, cells were binned in hexagons and expression was calculated as a mean based on the gene of interest.
To investigate developmental dynamics, Velocyto (La Manno et al., 2018), version 0.17.17) was used. Velocyto annotates reads as spliced, unspliced and ambiguous. The pipeline was run individually for each sample and data from resulting loom files were combined. Ambiguous counts were excluded and cells having spliced and unspliced count below 800 and 300, respectively, were removed. 3000 most variable genes were selected for downstream analysis. Counts were normalized based on the initial molecule count. For cell nearest neighbor pooling, a k-nearest neighbor graph (k = 500) was computed based on Euclidean distance utilizing the top 20 principal components. Gamma coefficients, velocity and extrapolation of gene expression profile at time t = 1 were calculated. Finally, for visualization, velocity vectors were plotted as locally average vector fields on a tSNE embedding.
Pearson correlation was calculated on HVGs of precursor tisTregST2 cells. The 30 most correlated and anti-correlated genes to Batf were clustered using pheatmap and R (version 3.4.2) with default settings.
RNA-seq and ATAC-seq integrative analysis
DESeq2 (version 1.20.0) (Love et al., 2014) was used for differential gene expression analysis in a pairwise fashion between tisTregST2 cells from fat, skin and lung. Differentially expressed genes were kept if the adjusted p value was equal or below 0.05 and if a log2 fold-change of 1 or greater was observed. A gene was defined as exclusively expressed for a tissue if it was found to be upregulated in the tissue for all pairwise comparisons. ATAC-seq peaks from precursor and tisTregST2 samples were extracted if they could be associated to exclusively expressed genes within a distance of 30kb up- or downstream. Next, the ratio of precursor ATAC-seq peaks to tissue ATAC-seq peaks was calculated and visualized.
Quantification and Statistical Analysis
Data were analyzed with Prism software or algorithm. Statistical details are indicated in the figure legend. Population size is described in the figure legend. Results of statistical tests are listed in Tables S1, S2, S3, S4, S5, S6, and S7.
Data and Code Availability
The accession numbers for the RNA-Seq, scRNA-Seq, scTCR-Seq, and ATAC-seq data reported in this paper are: Gene Expression Omnibus (GEO) GSE130884.
Acknowledgments
We thank A. Rudensky (Memorial Sloan Kettering Cancer Center) for providing mice and Günter Küblbeck (DKFZ) for help with the BAC construct. We thank the DKFZ core facilities Preclinical Research, Flow Cytometry and Genomics & Proteomics for technical support. We thank Sabine Schmitt (DKFZ), Marina Wuttke, Brigitte Ruhland, Kathrin Schambeck, Veronika Hofmann, Brigitte Wild, Manuela Kovacs-Sauter, and David Dittmar (all RCI) for technical support. We thank the RCI flow cytometry core facility, the RCI NGS core facility, and Animal Facility Regensburg University for technical support. This work was supported by grants from the European Research Council (ERC-CoG, 648145 REGiREG) to M.F.; the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (Projektnummer 324392634-TRR 221) to M.F. (Project B08) and P.H., M.E., and M.R. (Project B07); and a faculty grant from the University of Regensburg (ReForM C program) to M.F. M.D. was supported by the German-Israeli Helmholtz Research School in Cancer Biology. B.B. received funding from the German Ministry of Research and Education (BMBF) (031L0076A and 01KU1216B).
Author Contributions
M.D. and M.F. designed the study. M.D., C.S., and M.F. designed the experiments. M.D., J.-P.M., K.B., D.R., S.B., L. Sanderink, A.P., L. Schmidleithner., K.L.B., B.E., and A.F. performed the experiments. M.D., C.D.I., A.H.-W., M.S., A.F.R., V.G., P.H., M.E., C.B., M.R., B.B., C.S., and M.F. analyzed data. M.D., C.S., and M.F. wrote the manuscript.
Declaration of Interests
The authors declare no competing financial interests.
Published: January 7, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.immuni.2019.12.002.
Supplemental Information
Figure 1A (gating example for tisTregST2); Figure 1B (sort layout for tisTregST2 from ing LN, VAT, skin, lung and colon; data used to calculate PCA); Figure 1C (data for heatmap); Figures 1D and 1H (dataset for heatmaps); Figure 1 Core Signature (data underlying the tissue Treg core signature); Figure 1E (de novo motif data); Figure 1F (distance from motif spreadsheet); Figure 1I (pathway enrichment dataset); Figure S1A (data table used to calculate heatmap); Figures S1B and S1C (Rpkm table and Deseq2).
Figure 2B (Rpkm table and Deseq2); Figure 2C (flow-cytometry data table and statistics); Figure 2D (Rpkm table and Deseq2); Figure 2E (data used to calculate PCA); Figure S2A (nucleotide sequence of BAC insert); Figure S2C (flow-cytometry data and statistics); Figure S2D (flow cytometry data and statistics); Figure S2E (flow cytometry data and statistics).
Figure 3A (sort layout and post-sort QC); Figures 3B and 3D (TCR sequences); Figure 3C (inverse Simpson Index); Figures 3E–3G (TCR sequences); Figure S3B (Seurat data output); Figure S3C (Rpkm table and Deseq2).
Figure 4A (RNA velocity coordinates and vectors); Figure 4B (flow-cytometry data and statistics); Figure 4C (flow cytometry data and statistics); Figure 4D (flow-cytometry data and statistics); Figure 4E (flow-cytometry data and statistics); Figure 4F (flow-cytometry data and statistics).
Figure 5A (data used to generate heatmap); Figure 5E (number of regions open); Figure 5F (raw data and p values); Figure 5G (distance from motif spreadsheet); Figure 5H (flow-cytometry data and statistics).
Figure 6A (Dataset collection, p value (log), description of dataset, cell type used for chromatin IP, antibody used for chromatin IP, data source identifier, comparison name, direction, number of regions in public dataset, and number of overlapping regions (opening chromatin regions in progenitors with target public region set); Figure 6B (distance from motif spreadsheet); Figure 6C (percentage of overlapping regions); Figure 6E (flow-cytometry data and statistics); Figure S6B (flow-cytometry data and statistics); Figure S6C (flow-cytometry data and statistics); Figure S6H (flow-cytometry data and statistics).
Figure 7A (flow-cytometry data and statistics); Figure 7B (flow-cytometry data and statistics); Figure 7C (flow-cytometry data and statistics); Figure 7D (flow-cytometry data and statistics); Figure 7E (flow-cytometry data and statistics); Figure 7F (raw data for PCA); Figure 7G (data for heatmap); Figure 7H (fold change versus p value dataset); Figure 7I (de novo motif analysis data); Figure 7J (distance from motif spreadsheet); Figure 7K (peak opening table); Figure 7M (peak opening table); Figure S7A (gene expression data for heatmap); Figure S7B (gene expression and flow-cytometry data for heatmap); Figure S7D (gene expression and flow cytometry data for heatmap).
References
- Ali N., Zirak B., Rodriguez R.S., Pauli M.L., Truong H.A., Lai K., Ahn R., Corbin K., Lowe M.M., Scharschmidt T.C. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell. 2017;169:1119–1129. doi: 10.1016/j.cell.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N., Green J.A., Moltedo B., Arvey A., Hemmers S., Yuan S., Treuting P.M., Rudensky A.Y. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. 2015;162:1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao K., Carr T., Wu J., Barclay W., Jin J., Ciofani M., Reinhardt R.L. BATF Modulates the Th2 Locus Control Region and Regulates CD4+ T Cell Fate during Antihelminth Immunity. J. Immunol. 2016;197:4371–4381. doi: 10.4049/jimmunol.1601371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone J.A., Tang Q. Treg cells-the next frontier of cell therapy. Science. 2018;362:154–155. doi: 10.1126/science.aau2688. [DOI] [PubMed] [Google Scholar]
- Brunkow M.E., Jeffery E.W., Hjerrild K.A., Paeper B., Clark L.B., Yasayko S.A., Wilkinson J.E., Galas D., Ziegler S.F., Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D., Kuswanto W., Kolodin D., Shadrach J.L., Cerletti M., Jang Y., Sefik E., Tan T.G., Wagers A.J., Benoist C., Mathis D. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155:1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta D., Feuerer M., Li A., Kamei N., Lee J., Shoelson S.E., Benoist C., Mathis D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–553. doi: 10.1038/nature11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland N.G., Jenkins N.A., Court D.L. Recombineering: a powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001;2:769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossarizza A., Chang H.D., Radbruch A., Acs A., Adam D., Adam-Klages S., Agace W.W., Aghaeepour N., Akdis M., Allez M. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition) Eur. J. Immunol. 2019;49:1457–1973. doi: 10.1002/eji.201970107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacher M., Imbusch C.D., Weichenhan D., Breiling A., Hotz-Wagenblatt A., Träger U., Hofer A.C., Kägebein D., Wang Q., Frauhammer F. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol. 2017;18:1160–1172. doi: 10.1038/ni.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacher M., Schmidl C., Herzig Y., Breloer M., Hartmann W., Brunk F., Kägebein D., Träger U., Hofer A.C., Bittner S. Rbpj expression in regulatory T cells is critical for restraining TH2 responses. Nat. Commun. 2019;10:1621. doi: 10.1038/s41467-019-09276-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski Y., O’Hagan T., Dittmer M., Penalva R., Mayoral S.R., Bankhead P., Fleville S., Eleftheriadis G., Zhao C., Naughton M. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017;20:674–680. doi: 10.1038/nn.4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerer M., Herrero L., Cipolletta D., Naaz A., Wong J., Nayer A., Lee J., Goldfine A.B., Benoist C., Shoelson S., Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasmacher E., Agrawal S., Chang A.B., Murphy T.L., Zeng W., Vander Lugt B., Khan A.A., Ciofani M., Spooner C.J., Rutz S. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science. 2012;338:975–980. doi: 10.1126/science.1228309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K.D., Irizarry R.A., Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics. 2012;13:204–216. doi: 10.1093/biostatistics/kxr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan Z., Koizumi S.I., Sasaki D., Yamada H., Arakaki N., Fujihara Y., Okitsu S., Shirahata H., Ishikawa H. JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat. Commun. 2017;8:15628. doi: 10.1038/ncomms15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayatsu N., Miyao T., Tachibana M., Murakami R., Kimura A., Kato T., Kawakami E., Endo T.A., Setoguchi R., Watarai H. Analyses of a Mutant Foxp3 Allele Reveal BATF as a Critical Transcription Factor in the Differentiation and Accumulation of Tissue Regulatory T Cells. Immunity. 2017;47:268–283. doi: 10.1016/j.immuni.2017.07.008. [DOI] [PubMed] [Google Scholar]
- Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson J., Chen X., Gomes T., Ullah U., Meyer K.B., Miragaia R., Duddy G., Pramanik J., Yusa K., Lahesmaa R., Teichmann S.A. Genome-wide CRISPR Screens in T Helper Cells Reveal Pervasive Crosstalk between Activation and Differentiation. Cell. 2019;176:882–896. doi: 10.1016/j.cell.2018.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ise W., Kohyama M., Schraml B.U., Zhang T., Schwer B., Basu U., Alt F.W., Tang J., Oltz E.M., Murphy T.L., Murphy K.M. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat. Immunol. 2011;12:536–543. doi: 10.1038/ni.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M., Komai K., Mise-Omata S., Iizuka-Koga M., Noguchi Y., Kondo T., Sakai R., Matsuo K., Nakayama T., Yoshie O. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. 2019;565:246–250. doi: 10.1038/s41586-018-0824-5. [DOI] [PubMed] [Google Scholar]
- Jiang H., Lei R., Ding S.W., Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics. 2014;15:182. doi: 10.1186/1471-2105-15-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.M., Rasmussen J.P., Rudensky A.Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- Korn T., Reddy J., Gao W., Bettelli E., Awasthi A., Petersen T.R., Bäckström B.T., Sobel R.A., Wucherpfennig K.W., Strom T.B. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1) doi: 10.1093/nar/gkw377. W90-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi M., Barnitz R.A., Yosef N., Odorizzi P.M., DiIorio M.A., Lemieux M.E., Yates K., Godec J., Klatt M.G., Regev A. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 2014;15:373–383. doi: 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., DiSpirito J.R., Zemmour D., Spallanzani R.G., Kuswanto W., Benoist C., Mathis D. TCR Transgenic Mice Reveal Stepwise, Multi-site Acquisition of the Distinctive Fat-Treg Phenotype. Cell. 2018;174:285–299. doi: 10.1016/j.cell.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Spolski R., Liao W., Wang L., Murphy T.L., Murphy K.M., Leonard W.J. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature. 2012;490:543–546. doi: 10.1038/nature11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A., Suri-Payer E., Veltkamp C., Doerr H., Sommer C., Rivest S., Giese T., Veltkamp R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009;15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miragaia R.J., Gomes T., Chomka A., Jardine L., Riedel A., Hegazy A.N., Whibley N., Tucci A., Chen X., Lindeman I. Single-Cell Transcriptomics of Regulatory T Cells Reveals Trajectories of Tissue Adaptation. Immunity. 2019;50:493–504. doi: 10.1016/j.immuni.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panduro M., Benoist C., Mathis D. Tissue Tregs. Annu. Rev. Immunol. 2016;34:609–633. doi: 10.1146/annurev-immunol-032712-095948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiering C., Krausgruber T., Chomka A., Fröhlich A., Adelmann K., Wohlfert E.A., Pott J., Griseri T., Bollrath J., Hegazy A.N. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl C., Delacher M., Huehn J., Feuerer M. Epigenetic mechanisms regulating T-cell responses. J. Allergy Clin. Immunol. 2018;142:728–743. doi: 10.1016/j.jaci.2018.07.014. [DOI] [PubMed] [Google Scholar]
- Schraml B.U., Hildner K., Ise W., Lee W.L., Smith W.A., Solomon B., Sahota G., Sim J., Mukasa R., Cemerski S. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield N.C., Bock C. LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 2016;32:587–589. doi: 10.1093/bioinformatics/btv612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan J.M., Höllbacher B., Campbell D.J. Cutting Edge: Dynamic Expression of Id3 Defines the Stepwise Differentiation of Tissue-Resident Regulatory T Cells. J. Immunol. 2019;202:31–36. doi: 10.4049/jimmunol.1800917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A., Vilella A.J., Cuppen E., Nijman I.J., Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasanthakumar A., Moro K., Xin A., Liao Y., Gloury R., Kawamoto S., Fagarasan S., Mielke L.A., Afshar-Sterle S., Masters S.L. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol. 2015;16:276–285. doi: 10.1038/ni.3085. [DOI] [PubMed] [Google Scholar]
- Wildin R.S., Ramsdell F., Peake J., Faravelli F., Casanova J.L., Buist N., Levy-Lahad E., Mazzella M., Goulet O., Perroni L. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- Yamazaki S., Tanaka Y., Araki H., Kohda A., Sanematsu F., Arasaki T., Duan X., Miura F., Katagiri T., Shindo R. The AP-1 transcription factor JunB is required for Th17 cell differentiation. Sci. Rep. 2017;7:17402. doi: 10.1038/s41598-017-17597-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Fujikado N., Kolodin D., Benoist C., Mathis D. Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science. 2015;348:589–594. doi: 10.1126/science.aaa7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1A (gating example for tisTregST2); Figure 1B (sort layout for tisTregST2 from ing LN, VAT, skin, lung and colon; data used to calculate PCA); Figure 1C (data for heatmap); Figures 1D and 1H (dataset for heatmaps); Figure 1 Core Signature (data underlying the tissue Treg core signature); Figure 1E (de novo motif data); Figure 1F (distance from motif spreadsheet); Figure 1I (pathway enrichment dataset); Figure S1A (data table used to calculate heatmap); Figures S1B and S1C (Rpkm table and Deseq2).
Figure 2B (Rpkm table and Deseq2); Figure 2C (flow-cytometry data table and statistics); Figure 2D (Rpkm table and Deseq2); Figure 2E (data used to calculate PCA); Figure S2A (nucleotide sequence of BAC insert); Figure S2C (flow-cytometry data and statistics); Figure S2D (flow cytometry data and statistics); Figure S2E (flow cytometry data and statistics).
Figure 3A (sort layout and post-sort QC); Figures 3B and 3D (TCR sequences); Figure 3C (inverse Simpson Index); Figures 3E–3G (TCR sequences); Figure S3B (Seurat data output); Figure S3C (Rpkm table and Deseq2).
Figure 4A (RNA velocity coordinates and vectors); Figure 4B (flow-cytometry data and statistics); Figure 4C (flow cytometry data and statistics); Figure 4D (flow-cytometry data and statistics); Figure 4E (flow-cytometry data and statistics); Figure 4F (flow-cytometry data and statistics).
Figure 5A (data used to generate heatmap); Figure 5E (number of regions open); Figure 5F (raw data and p values); Figure 5G (distance from motif spreadsheet); Figure 5H (flow-cytometry data and statistics).
Figure 6A (Dataset collection, p value (log), description of dataset, cell type used for chromatin IP, antibody used for chromatin IP, data source identifier, comparison name, direction, number of regions in public dataset, and number of overlapping regions (opening chromatin regions in progenitors with target public region set); Figure 6B (distance from motif spreadsheet); Figure 6C (percentage of overlapping regions); Figure 6E (flow-cytometry data and statistics); Figure S6B (flow-cytometry data and statistics); Figure S6C (flow-cytometry data and statistics); Figure S6H (flow-cytometry data and statistics).
Figure 7A (flow-cytometry data and statistics); Figure 7B (flow-cytometry data and statistics); Figure 7C (flow-cytometry data and statistics); Figure 7D (flow-cytometry data and statistics); Figure 7E (flow-cytometry data and statistics); Figure 7F (raw data for PCA); Figure 7G (data for heatmap); Figure 7H (fold change versus p value dataset); Figure 7I (de novo motif analysis data); Figure 7J (distance from motif spreadsheet); Figure 7K (peak opening table); Figure 7M (peak opening table); Figure S7A (gene expression data for heatmap); Figure S7B (gene expression and flow-cytometry data for heatmap); Figure S7D (gene expression and flow cytometry data for heatmap).
Data Availability Statement
The accession numbers for the RNA-Seq, scRNA-Seq, scTCR-Seq, and ATAC-seq data reported in this paper are: Gene Expression Omnibus (GEO) GSE130884.