

Abstract
Successful development depends on the precise tissue‐specific regulation of genes by enhancers, genetic elements that act as switches to control when and where genes are expressed. Because enhancers are critical for development, and the majority of disease‐associated mutations reside within enhancers, it is essential to understand which sequences within enhancers are important for function. Advances in sequencing technology have enabled the rapid generation of genomic data that predict putative active enhancers, but functionally validating these sequences at scale remains a fundamental challenge. Herein, we discuss the power of genome‐wide strategies used to identify candidate enhancers, and also highlight limitations and misconceptions that have arisen from these data. We discuss the use of massively parallel reporter assays to test enhancers for function at scale. We also review recent advances in our ability to study gene regulation during development, including CRISPR‐based tools to manipulate genomes and single‐cell transcriptomics to finely map gene expression. Finally, we look ahead to a synthesis of complementary genomic approaches that will advance our understanding of enhancer function during development.
This article is categorized under:
Physiology > Mammalian Physiology in Health and Disease
Developmental Biology > Developmental Processes in Health and Disease
Laboratory Methods and Technologies > Genetic/Genomic Methods
Keywords: development, enhancers, genomics
The correct spatiotemporal regulation of genes by enhancers is critical for successful development from a single fertilized egg to a complex multicellular organism. Herein, we discuss current genome‐wide approaches to identify enhancers and to dissect enhancer function. Figure created with BioRender.
1. INTRODUCTION
Animal development is the dynamic process through which a single fertilized egg ultimately gives rise to a multitude of distinctive cell types. Cells maintain unique identities and are able to carry out specialized functions by expressing different sets of genes. Enhancers are cis‐regulatory elements that direct precise, tissue‐ and cell type‐specific patterns of gene expression. The correct spatiotemporal regulation of genes by enhancers is critical for successful development (Levine, 2010). Enhancers were first recognized over 30 years ago as being short DNA sequences that positively regulate expression of a gene regardless of their position or orientation relative to the transcription start site (Banerji, Rusconi, & Schaffner, 1981; Moreau et al., 1981; Shlyueva, Stampfel, & Stark, 2014). Although decades of research have focused on identifying and validating enhancers, we still do not know how many enhancers exist in any genome and currently lack a broad understanding of which sequences within an enhancer are important for function. Enhancers contain hundreds of nucleotides and several transcription factor (TF) binding sites, so deciphering how enhancer sequences encode function is a complex problem. Because enhancers can act over distances on the order of a megabase to regulate target gene expression (Amano et al., 2009), identifying which gene or genes a given enhancer regulates can be far from straightforward. Furthermore, enhancer function can be conserved without discernible sequence conservation (Blow et al., 2010; Meireles‐Filho & Stark, 2009), making it difficult to distill general principles of enhancer activity using comparative genomics in spite of significant efforts.
Despite these challenges in studying enhancer function, substantial progress has been made in recent years due to advances in sequencing technology and high‐throughput methodologies to identify and validate candidate enhancers. On the order of one million candidate enhancers have been identified in the human genome, based on genome‐wide interrogation of TF binding sites and chromatin signatures associated with active enhancers (Dunham et al., 2012; Humbert et al., 2012). Furthermore, there is an increasing appreciation that common variants associated with disease disproportionately reside within putative enhancer sequences (Corradin & Scacheri, 2014; Ernst et al., 2011; Wu & Pan, 2018). Thus, understanding how enhancers function has become an area of significant interest, not only for their roles in normal development, but also because linking enhancer sequence directly to function could provide unprecedented insight into the genetic basis of disease and evolutionary adaptation. With significant advances in sequencing and genome editing technologies, we have never been in a better position to identify and functionally validate candidate enhancers, but there remain significant bottlenecks in our ability to comprehensively understand enhancer function. In this review, we discuss current methodologies used to identify putative enhancers and the techniques that have been developed to study their function at scale. We also review two recent waves of literature that (a) use genome editing tools to interrogate candidate enhancer sequences and (b) utilize single‐cell RNA sequencing to construct developmental atlases of whole embryos. Finally, we look ahead toward a synthesis of these complementary approaches, which will broadly expand our knowledge of how enhancer sequences encode precise control of gene expression during development.
2. GENOME‐WIDE METHODOLOGIES TO IDENTIFY PUTATIVE ENHANCERS
Over the last decade, next‐generation sequencing technologies have revolutionized our ability to identify putative enhancers throughout whole genomes. Pairing classic molecular genetic approaches with high‐throughput sequencing has enabled the rapid construction of genome‐wide maps of TF binding sites, histone modifications, nucleosome positions, and long‐range genomic interactions. These efforts were spearheaded by the Encyclopedia of DNA Elements (ENCODE) Consortium (Dunham et al., 2012), and led to the identification of chromatin signatures that are strongly associated with active enhancers. Furthermore, these data demonstrated that single nucleotide polymorphisms associated with disease reside disproportionately within candidate enhancers (Corradin & Scacheri, 2014; Ernst et al., 2011; Wu & Pan, 2018), highlighting the importance of elucidating how enhancer sequences contribute to development, homeostasis, and disease.
2.1. Genome‐wide identification of TF binding sites by ChIP‐seq
Chromatin immunoprecipitation followed by deep sequencing (ChIP‐seq; Johnson, Mortazavi, Myers, & Wold, 2007; Robertson et al., 2007) has been used extensively to identify genome‐wide binding of TFs, both in vitro and in vivo (Figure 1a). In ChIP‐seq, chromatin is crosslinked using formaldehyde, which covalently links TFs to their native binding sites. The chromatin is then sheared and immunoprecipitated using antibodies specific to a TF of interest. Following immunoprecipitation, the crosslink is reversed and DNA is purified, sequenced, and computationally analyzed to identify TF binding sites. For a given TF, ChIP‐seq will typically identify thousands of regions bound by TFs, which are predominantly found in promoters, introns, and intergenic regions (X. Y. Li et al., 2008; Medina et al., 2008). ChIP‐seq is also able to identify changes in TF occupancy of binding sites throughout development (Gentleman et al., 2010; Hagman et al., 2010; Pilon et al., 2011), reflecting that TF binding is dynamic and highly specific to developmental stage.
Figure 1.
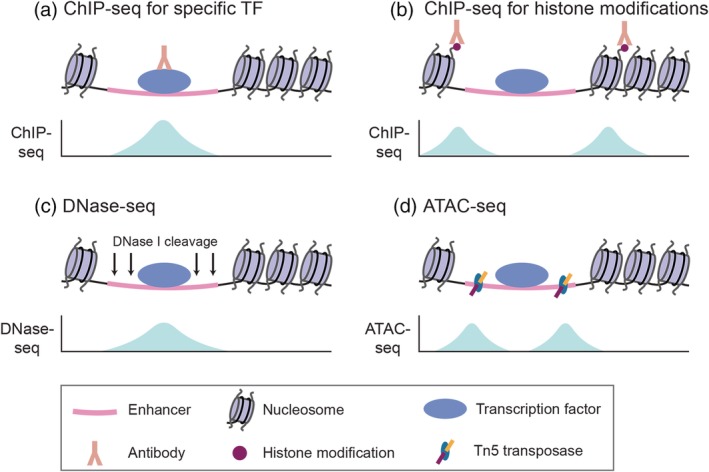
Genome‐wide methods to identify putative enhancers. The basic principle of each experimental approach and the corresponding data readout is shown for each method. (a) Chromatin immunoprecipitation followed by high‐throughput sequencing (ChIP‐seq) uses antibodies targeting a specific TF to determine the location of its binding sites genome‐wide. The broad peaks generated by ChIP‐seq cannot determine precise footprints of TF binding, but this can be achieved by adding a 5′ to 3′ exonuclease digestion step to the protocol (ChIP‐exo). (b) Nucleosomes flanking active enhancers often carry stereotypical histone modifications (e.g., H3K4me1 and H3K27ac) that can be detected with specific antibodies by ChIP‐seq. (c) Active enhancers and other cis‐regulatory elements are found within open chromatin that is depleted of nucleosomes. Accessible chromatin can be detected by DNase I digestion followed by high‐throughput sequencing (DNase‐seq). (d) Accessible chromatin can also be detected by assay for transposase‐accessible chromatin using sequencing (ATAC‐seq), where Tn5 transposase simultaneously fragments and tags accessible DNA prior to sequencing
ChIP‐seq has been extremely useful for identifying the genomic regions bound by a given TF, including putative enhancer sequences. However, several studies have demonstrated that TF binding identified by ChIP‐seq does not necessarily correspond to active cis‐regulatory elements (Kvon, Stampfel, Omar Yáññez‐Cuna, Dickson, & Stark, 2012; X. Y. Li et al., 2008; MacArthur et al., 2012), suggesting that TFs can bind DNA without having a functional role in gene expression. There are several possible explanations for this observation. First, because TFs typically bind regulatory elements in a combinatorial manner (Halfon et al., 2000; Sandmann et al., 2007; Yuh, Ransick, Martinez, Britten, & Davidson, 1994), the binding of any one TF may not be sufficient to activate transcription. Because TFs often bind in complexes, a TF of interest may be immunoprecipitated in ChIP without being directly bound to DNA. TFs also have a general affinity for DNA (Hammar et al., 2012), and may bind transiently to accessible DNA outside of functional contexts and regardless of whether a specific binding motif is present. ChIP‐seq relies on formaldehyde crosslinking and thus transient DNA–protein interactions could be captured by this technique. Indeed, even proteins that do not bind DNA are enriched at highly transcribed genes by ChIP‐seq (Teytelman, Thurtle, Rine, & van Oudenaarden, 2013). Recently, alternatives to ChIP‐seq that eliminate the need for crosslinking have been developed, including Cleavage Under Targets and Release Using Nuclease (CUT&RUN; Skene & Henikoff, 2017) and Cleavage Under Targets and TAGmentation (CUT&TAG; Kaya‐okur et al., 2019). Like ChIP‐seq, these approaches both employ a TF‐specific antibody to identify protein–DNA interactions. However, they also utilize a DNA‐cutting enzyme fused to Protein A, which binds specifically to Immunoglobin G and thus targets antibody‐bound TFs. In CUT&RUN, a Protein A‐MNase fusion protein cleaves DNA on either side of a TF, releasing TF‐DNA complexes into solution for downstream purification and library preparation steps. CUT&TAG improves upon this methodology by employing a Protein A‐Tn5 transposase fusion protein loaded with sequencing adaptors, generating DNA fragments that can be directly amplified and sequenced. Both methods are performed in situ, allow for low starting cell numbers, and produce substantially decreased background compared to ChIP‐seq.
In ChIP‐seq datasets, false positives can also arise due to experimental factors including chromatin fragment size (Rye, Sætrom, & Drabløs, 2011), read depth (Jung et al., 2014), and signal cutoff thresholds (Gomes et al., 2014). Choice of signal cutoff thresholds can be particularly important when analyzing ChIP‐seq data. If set too low, the false‐positive rate will increase but if set too high, it may not be possible to capture functional interactions that are highly dynamic or low‐affinity (Gomes et al., 2014; Landt et al., 2012). Although ChIP‐seq datasets contain both high‐ and low‐affinity TF binding sites, there is a tendency to focus on high‐affinity sites and to ignore or exclude lower affinity sequences during analysis (Nettling, Treutler, Cerquides, & Grosse, 2016). Models of TF binding specificity, such as position–weight matrices, are commonly used to predict functional TF binding sites within regions identified by ChIP‐seq. Sequences with higher affinity scores are presumed to have a greater likelihood of being functional binding sites, and many algorithms use seemingly arbitrary default cutoff thresholds, for example, the 0.8 relative log‐likelihood threshold recommended by JASPAR, an open‐source database of TF binding site profiles (Sandelin, 2004). While it is important to prioritize ChIP‐seq peaks, focusing on high affinity sites may be shortsighted, particularly because developmental enhancers are highly dynamic and often contain low‐affinity or otherwise suboptimal binding sites (Crocker et al., 2015; Farley et al., 2015; Farley, Olson, & Levine, 2016; Hosokawa et al., 2018). Many studies have recently demonstrated that suboptimal TF binding sites within enhancers can be a requirement for precise gene expression and biological function during development (Crocker et al., 2015; Crocker, Preger‐Ben Noon, & Stern, 2016; Farley et al., 2015; Farley, Levine, Olson, Zhang, & Rokhsar, 2016; Ramos & Barolo, 2013; Rowan et al., 2010). Because TFs bind degenerate DNA sequences, it remains a significant challenge to discriminate functional TF binding sites from background sequences in ChIP‐seq data. This issue can be overcome by generating position–weight matrices from datasets depleted of high‐affinity binding sites, which is more accurate at predicting low‐affinity binding sites, and surprisingly even high‐affinity binding sites, compared to conventional methods (Zandvakili, Campbell, Gutzwiller, Weirauch, & Gebelein, 2018). While ChIP strategies have identified novel enhancers (Heintzman et al., 2007), it will be important to improve our ability to identify low‐affinity TF binding and to differentiate between functional and non‐functional binding, particularly when interrogating the genome for developmental enhancers.
The utility of ChIP‐seq for the study of enhancer sequences is also limited by its resolution. In standard ChIP‐seq protocols, chromatin is randomly sheared by sonication to produce fragments within the range of 200–500 basepairs (Mahony & Pugh, 2015). The resolution of ChIP‐seq is similar to fragment size, on the order of hundreds of basepairs, while TF binding motifs are typically 6–20 basepairs (Afek, Schipper, Horton, Gordân, & Lukatsky, 2014). Thus, ChIP‐seq is unable to resolve individual TF binding events within a putative enhancer sequence. Furthermore, a TF binding motif can be found many times within a region identified by ChIP‐seq, and in these instances it is not known which motifs may be functional. ChIP‐exo is a method that provides improved resolution by adding a 5′ to 3′ exonuclease digestion step to conventional ChIP‐seq protocols, allowing the region bound by the TF to be isolated (Rhee & Pugh, 2011). Exonuclease activity is impeded by crosslinked proteins, creating a 5′ border on either side of bound TFs that can be used to precisely map TF binding sites (Rhee & Pugh, 2011). In contrast to ChIP‐seq, ChIP‐exo enables single nucleotide resolution of TF binding and reveals exact footprints of occupied TF binding sites. Because the exonuclease used in ChIP‐exo digests unbound DNA that can contaminate immunoprecipitates, this method also has an improved signal to noise ratio compared to ChIP‐seq (Rhee & Pugh, 2011). Despite these advantages of ChIP‐exo, this method is not yet widely used and thus most genome‐wide TF binding data has been obtained by ChIP‐seq, which captures relatively broad windows of TF binding. While ChIP‐seq methods to identify TF binding have certainly advanced our ability to locate putative cis‐regulatory elements in the genome, standard protocols lack the resolution to identify precise binding sites within an enhancer. Furthermore, it still remains unclear which binding events regulate transcription and are functional.
2.2. ChIP‐seq to identify transcriptional cofactor localization genome‐wide
ChIP‐seq does not rely on direct protein–DNA interactions, and thus can be used to identify cofactor proteins associated with active enhancers. Cofactors typically do not bind DNA directly, but are recruited to cis‐regulatory elements by TFs, where they can act to repress or activate transcription. The transcriptional coactivator protein p300 has been particularly useful for identifying active enhancers. p300 acts as an adaptor for protein–protein interactions and also has histone acetyltransferase activity (Visel et al., 2009), which relaxes the chromatin making it accessible to TFs and the basal transcriptional machinery. Several studies in mice and humans have demonstrated that p300 binding can be used to predict >80% of tissue‐specific enhancer activity (Blow et al., 2010; May et al., 2012; Visel et al., 2009, 2013). While p300 provides an excellent indicator of active enhancers, binding of a coactivator is not sufficient to understand why a particular sequence activates transcription, or which mutations may impact enhancer function. To further improve identification of active enhancers, the colocalization of p300 with other chromatin features, including histone modifications, is often used to locate putative enhancers.
2.3. ChIP‐seq for histone modifications associated with active enhancers
Genome‐wide mapping of histone modifications using ChIP‐seq has revealed patterns that are stereotypically found at active enhancers (Figure 1b; Bonn et al., 2012; Heintzman et al., 2007, 2009; Rada‐Iglesias et al., 2011; Roh, Cuddapah, & Zhao, 2005). While enhancers are often associated with H3K4me1 and promoters are often associated with H3K4me3, both enhancers and promoters tend to be marked by H3K27ac when active and H3K27me3 when repressed (Arnold et al., 2013; Bonn et al., 2012; Peters et al., 2002; Rada‐Iglesias et al., 2011). Together, H3K4me1 and H3K27ac are widely used to predict active enhancers (Bonn et al., 2012; Kharchenko et al., 2011; Rada‐Iglesias et al., 2011; Shen et al., 2012; Wamstad et al., 2012). Interestingly, some enhancers are associated with bivalent chromatin (Bernstein et al., 2006; Vastenhouw et al., 2010), that is, the presence of both H3K4me1 and H3K27me3. This combination of activating and repressive marks is characteristic of poised enhancers (Rada‐Iglesias et al., 2011), which are common in development (Creyghton et al., 2010). H3K79me3, which is most commonly found within actively transcribed gene bodies, has also been found at active enhancers (Barski et al., 2007; Bonn et al., 2012). In addition to epigenetically modified histones, the histone variants H2A.Z and H3.3 have been associated with putative enhancers, particularly when found together within a histone octamer (C. Jin et al., 2009).
Despite the associations between these histone modifications and enhancers, there is currently no consensus regarding which combination of histone modifications should be used to identify active enhancers. Furthermore, the histone modifications found at a given enhancer may be context‐specific. For example, latent enhancers are characterized by the absence, and then acquisition, of H3K4me1 and H3K27ac following stimulation of cell signaling pathways (Ostuni et al., 2013). None of the known histone modifications associated with enhancers correlate perfectly with enhancer activity, and even combinations of histone marks are unable to accurately predict all active enhancers (Arnold et al., 2013; Bonn et al., 2012). Indeed, many active enhancers lack characteristic histone modifications. For example, >40% of mesodermal enhancers in Drosophila melanogaster embryos could not be predicted based on the presence of H3K27ac (Bonn et al., 2012). It also remains unclear whether histone modifications have a direct role in regulating transcription, or whether they are simply useful indicators of where enhancers may reside within the genome.
2.4. Genome‐wide assays for chromatin accessibility
Active enhancers are depleted of nucleosomes, and thus assays for nucleosome positioning and chromatin accessibility are also widely used to identify putative enhancer sequences. Classic molecular biological techniques in which accessible DNA is cleaved by DNase I or micrococcal nuclease (MNase) have been paired with next‐generation sequencing to generate genome‐wide maps of chromatin accessibility. DNase‐seq is based on DNase footprinting, in which DNase I hypersensitive sites are digested and DNA bound by TFs and other proteins is protected (Figure 1c). While DNase footprinting traditionally analyzes digestion products by Southern Blot, in DNase‐seq a linker sequence is added to the protected DNA following DNase digestion to enable subsequent sequencing (Boyle et al., 2008; Humbert et al., 2012). This method generates basepair resolution of DNase I digestion, and thus footprints of TF binding. MNase‐seq utilizes micrococcal nuclease digestion, which cuts the linker DNA between adjacent nucleosomes (Schones et al., 2008; Valouev et al., 2011). Nucleosomal DNA is protected from digestion, and is subsequently purified and sequenced to reveal nucleosome positions genome‐wide. More recently, accessible chromatin has been studied by assay for transposase‐accessible chromatin using sequencing (ATAC‐seq), in which sequencing adaptors are directly transposed into native chromatin (Figure 1d; Buenrostro, Giresi, Zaba, Chang, & Greenleaf, 2013). ATAC‐seq is carried out as a simple two‐step protocol that involves insertion of a hyperactive Tn5 transposase (Adey et al., 2010; Goryshin & Reznikoff, 1998) carrying sequencing adaptors, which simultaneously fragments and tags DNA, followed by polymerase chain reaction (PCR). This method gives basepair resolution of nucleosome‐depleted genomic regions, and can be carried out rapidly because it does not depend on separate enzymatic digestion and adaptor ligation steps (Buenrostro et al., 2013).
Chromatin accessibility assays are advantageous because they can predict cis‐regulatory regions independent of any specific TF, cofactor, or histone modification. However, these methods identify promoters and insulators as well as enhancers. Furthermore, open chromatin does not necessarily mean that a given DNA sequence is active or functional. Enhancers located in open chromatin can be held in an inactive state by repressive TFs, which are common in development (Gray & Levine, 1996). For example, enhancers with cell type‐specific regulatory activity have been found to be open and accessible in diverse cell types in which the enhancer is not active (Arnold et al., 2013). Repressed enhancers found within accessible chromatin may also be primed for activity at a later developmental timepoint (Zaret & Carroll, 2011). While genome‐wide profiling of chromatin accessibility cannot detect specific cis‐regulatory functions, these techniques are particularly powerful when combined with ChIP‐seq data. The overlay of transcriptional coactivators, histone modifications associated with enhancers, and chromatin accessibility at a given genomic locus can provide strong correlative evidence of active enhancers.
2.5. Genome‐wide assays for chromosomal interactions
Enhancers can act over long distances, and are thought to be brought within close spatial proximity to the promoters they regulate. Thus, methods to study chromosomal interactions have also been used to identify putative enhancer–promoter interactions. The majority of these methods are based on chromosome conformation capture (3C) (Dekker, Rippe, Dekker, & Kleckner, 2002), in which chromatin is crosslinked and sheared, followed by proximity ligation reactions that capture physical chromosomal interactions (Figure 2a). The resulting DNA molecules contain regions that are not nearby in linear DNA sequence, but represent long‐range interactions. In 3C, specific interactions are assayed using PCR, but derivatives of this technique, including 4C and 5C, are higher throughput. The highest throughput of these methods is Hi‐C, in which proximity ligation is followed by massively parallel sequencing in order to capture all genomic interactions (Lieberman‐Aiden et al., 2009). Hi‐C was originally developed to investigate the spatial organization of the genome, which revealed the presence of topologically associating domains (TADs), regions of the genome that are highly self‐interacting (Dixon et al., 2012; Nora et al., 2012; Van Steensel & Dekker, 2010). Hi‐C has since been used to map genome‐wide interactions in numerous different organisms, tissues, and cell types, and has led to the identification of hundreds of thousands of putative enhancer–promoter contacts (Ron, Globerson, Moran, & Kaplan, 2017). The resolution of Hi‐C has improved from megabase resolution (Lieberman‐Aiden et al., 2009) of interactions to a few kilobases (kb) (Rao et al., 2014). However, this technique has not yet achieved sufficient resolution to detect enhancer–promoter interactions that take place on the hundred basepair scale.
Figure 2.
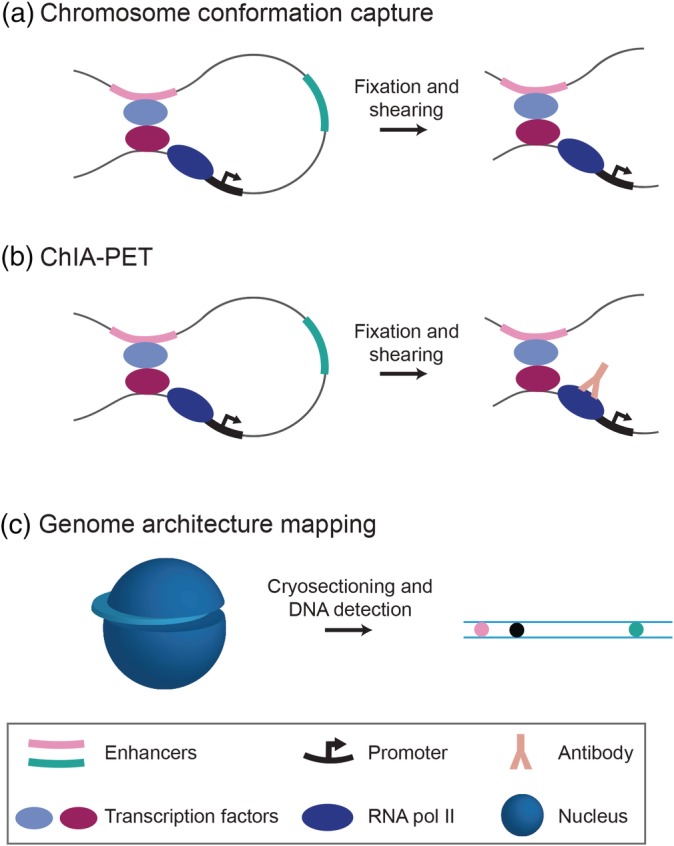
Genome‐wide methods to identify putative enhancer–promoter interactions. Distal enhancers (shown in pink) may be brought within close proximity to the promoters they regulate (shown in black) through the formation of chromatin loops. Looping is facilitated by protein–protein interactions of transcription factors that assemble at promoters and enhancers. The basic principles of methods to detect chromatin interactions are shown. Both (a) chromatin conformation capture (3C)‐based methods, including Hi‐C, and (b) chromatin interaction analysis with paired‐end tag sequencing (ChIA‐PET) preserve and detect chromatin interactions through crosslinking, fragmentation, and proximity ligation followed by high‐throughput sequencing. ChIA‐PET includes a chromatin immunoprecipitation step, often using antibodies targeting RNA pol II (shown), to enrich for complexes containing promoters. (c) Genome architecture mapping captures the distance between genomic loci by cryosectioning and laser microdissection (which allow spatial information to be preserved) followed by DNA sequencing
A related strategy to identify genome‐wide interactions is chromatin interaction analysis with paired‐end tag sequencing (ChIA‐PET) (Figure 2b; Fullwood et al., 2009). This method pairs chromatin proximity ligation with ChIP to predict genomic interactions that include a protein of interest. ChIA‐PET typically utilizes antibodies against RNA polymerase II or TFs, which may enrich for functional interactions (Fullwood et al., 2009; G. Li et al., 2012). Advantageously, ChIA‐PET can generate genome‐wide chromatin interaction maps with a resolution of ~100 bp (Z. Tang et al., 2015). ChIA‐PET can also be used to probe putative enhancer–promoter interactions by pulling down proteins involved in these interactions, including cohesin and the mediator complex (Kagey et al., 2010; Schmidt et al., 2010; Whyte et al., 2013). Most recently, a droplet‐based method termed ChIA‐Drop has been developed to study complex chromatin interactions involving multiple loci (Zheng et al., 2019). This technique relies on microfluidics and barcode‐linked sequencing to map genomic interactions from crosslinked chromatin samples. ChIA‐Drop does not employ pairwise proximity ligations, and thus can capture multiplexed chromosomal contacts that may be useful for the study of enhancer–promoter interactions. However, this method is currently limited by its resolution of 5 to 10 kb, which is not sufficient to identify individual enhancer–promoter interactions (Zheng et al., 2019). Another method to study complex genomic interactions is genome architecture mapping (GAM) (Beagrie et al., 2017), which utilizes ultrathin cryosectioning and laser microdissection followed by DNA sequencing (Figure 2c). Within randomly oriented nuclear sections, loci in close physical proximity are found together more often than distant loci, allowing three‐dimensional maps of chromatin structure to be constructed. Unlike 3C‐based methods, GAM also captures chromatin compaction, radial distributions of chromosomes, and chromatin associations with the nuclear periphery (Beagrie et al., 2017). However, its current resolution of ~30 kb presently limits its utility in studying enhancer–promoter interactions.
2.6. Biological context matters in genome‐wide assays
Enhancers direct gene expression in specific cell types, at specific times, and within specific developmental contexts, and thus it is important to use appropriate and homogeneous samples when performing genome‐wide techniques to identify putative enhancers. For example, TFs and other proteins can be expressed in a highly tissue‐specific manner, which could affect ChIP‐seq experiments as they rely on a specific protein of interest being present. Furthermore, the chromatin landscape assayed by ATAC‐seq or Hi‐C can depend on the gene regulatory programs active in a given cell type. Indeed, chromatin interactions are highly dynamic and can vary substantially between different cell types (Schmitt et al., 2016). Heterogeneity in gene expression has even been observed in single cells from seemingly homogeneous starting material (Munsky, Neuert, & Van Oudenaarden, 2012; Shalek et al., 2013). Thus, when applied to tissues composed of diverse cell types, these methods can suffer from a low signal to noise ratio. This issue can potentially be overcome by single‐cell approaches, which have been developed for many of the genome‐wide methods described above, including ChIP‐seq (Rotem et al., 2015), CUT&RUN (Hainer, Bošković, McCannell, Rando, & Fazzio, 2019), CUT&TAG (Kaya‐okur et al., 2019), DNase‐seq (W. Jin et al., 2015), MNase‐seq (Lai et al., 2018), ATAC‐seq (Buenrostro et al., 2015; DeWitt et al., 2018), and Hi‐C (Nagano et al., 2013). Although highly scalable, at present the single‐cell versions of these methods yield sparse, low‐coverage data that are difficult to analyze using conventional approaches (Ji, Zhou, & Ji, 2017; Schep, Wu, Buenrostro, & Greenleaf, 2017). While sparsity of data is inherent to current single cell methods, advances in experimental and computational approaches may increase the resolution with which we can interrogate the genome for putative enhancers. Furthermore, improved single‐cell assays could be performed in each cell of a developing embryo to give unique insight into gene regulatory dynamics during development.
3. CORRELATIVE GENOME‐WIDE DATA CAN LEAD TO MISCONCEPTIONS
Genome‐wide assays have greatly advanced our ability to predict enhancer sequences, but we cannot accurately identify active enhancers without functional validation. Genome‐wide predictions of active enhancers are based on correlative data, and the presence of a given trans‐acting factor or chromatin feature does not mean that a given DNA element truly functions as an enhancer. Furthermore, these predictions of enhancer activity cannot determine which genes might be regulated by putative enhancers and do not assess any quantitative effects on gene expression. Without functional validation, findings from genome‐wide predictions of enhancer activity could lead to misconceptions. Currently, the most stringent validation of a predicted enhancer is to test its function in its native context in animal models. This approach is inherently low‐throughput, which significantly limits its practicality, since millions of candidate enhancers have been identified. However, several recent studies in animal models have demonstrated that there are indeed misconceptions in our understanding of how chromatin features relate to enhancer function, demonstrating the value of thorough functional validation to advance our understanding of enhancer function.
3.1. Misconceptions about super‐enhancer function
Genome‐wide mapping of features associated with active enhancers led to the concept of super‐enhancers, clusters of regulatory elements that span several kilobases (Hnisz et al., 2013; Lovén et al., 2013; Whyte et al., 2013). Super‐enhancers were first identified from ENCODE data as stretch enhancers or highly occupied target regions that are cell type‐specific and associated with increased expression of genes involved in cell type‐specific processes (Kurum, Benayoun, Malhotra, George, & Ucar, 2016; Kvon et al., 2012; Parker et al., 2013). Super‐enhancers also coincide with defined locus control regions (LCRs), genomic regions that encompass a set of regulatory elements and regulate the expression of one or more genes (Grosveld, van Assendelft, Greaves, & Kollias, 1987; Q. Li, Peterson, Fang, & Stamatoyannopoulos, 2002; Pott & Lieb, 2015). Super‐enhancers are further characterized by binding of the mediator complex and H3K27ac, and are frequently flanked by insulator elements (Ing‐Simmons et al., 2015; Whyte et al., 2013). It was also observed that super‐enhancer‐associated genes are expressed at higher levels than genes associated with typical enhancers, and that super‐enhancers are highly sensitive to perturbation (Hnisz et al., 2013; Lovén et al., 2013; Whyte et al., 2013). These findings led to the idea that super‐enhancers may be more than the sum of their parts and represent a new paradigm in gene regulation. However, super‐enhancers as a class were defined based on these features without being functionally validated. Thus, it remains unclear whether they represent a simple clustering of conventional enhancers, or whether the constituent elements of a super‐enhancer may act together to produce novel and synergistic functional properties.
A cluster of regulatory elements constituting a super‐enhancer controls expression of the α‐globin gene in mouse erythroid cells (Hay et al., 2016). Each of the five individual elements in this super‐enhancer scored as an erythroid enhancer based on chromatin signatures and transient reporter assays. However, three of these elements failed to drive expression in a classic mouse transgenic assay, in which a construct containing an enhancer, minimal promoter, and lacZ reporter is stably inserted into the mouse genome (Hay et al., 2016). The authors of this study next dissected the function of this proposed super‐enhancer by generating several mouse models in which each constituent enhancer was deleted, individually or in pairs (Hay et al., 2016). Mice with homozygous deletion of any one of these enhancers were viable, had normal steady‐state levels of α‐globin mRNA, and did not exhibit any overt phenotypes (Hay et al., 2016). Two of the five enhancers were characterized by high levels of erythroid TF binding in ChIP‐seq assays, exhibited the strongest enhancer activity, and were embryonic lethal when deleted in combination. Deletion of either of these two strong enhancers caused stress erythropoiesis, which ensures normal hemoglobin production, suggesting that multiple enhancers at this locus ensure robust gene expression.
Altogether, these experiments demonstrated that components of the α‐globin super‐enhancer contribute to gene expression in an additive rather than synergistic manner, challenging the notion that super‐enhancers are a distinct class of regulatory element. However, another study found non‐additive effects within a mammary gland‐specific super‐enhancer of Wap (Shin et al., 2016). At present, few super‐enhancers have been rigorously tested and the functional differences between super‐enhancers and clusters of enhancers, if any, remain unclear (Pott & Lieb, 2015). Nevertheless, it has been known for decades that clusters of enhancers commonly regulate developmental genes, and thus it will be crucial to continue to functionally validate these clusters of enhancers identified from genome‐wide data. Indeed, the concept of super‐enhancers remains an important area of investigation, and continues to be used to describe highly active, cell type‐specific loci in development and disease.
3.2. Misconceptions about the importance of histone modifications associated with enhancers
While histone modifications are useful for identifying regions within the genome that may contain enhancers, these epigenetic marks are not necessarily required for enhancer function. H3K4me1 is a highly conserved chromatin feature associated with active enhancers, and is catalyzed by the complex of proteins associated with Set1 (COMPASS)‐like methyltransferase family (Creyghton et al., 2010; Heintzman et al., 2007). In Drosophila, this family includes the methyltransferase Trr, which is essential for successful development (Herz et al., 2012; Shilatifard, 2012). However, the requirement of enzymatically active Trr for successful development was more recently tested. Intriguingly, it was found that Drosophila embryos expressing catalytically inactive Trr, and thus lacking H3K4me1 marks, exhibit only mild phenotypes and develop to productive adulthood (Dorsett et al., 2017). Similarly, flies expressing a hyperactive Trr allele that changes the enzyme product specificity were viable and exhibited only subtle phenotypic effects, despite H3K4me1 being converted to H3K4me2 or H3K4me3 (Dorsett et al., 2017). Gene expression was largely unchanged in these mutant Drosophila lines, although diminished H3K4me1 at enhancers was associated with decreased expression of the nearest gene. Nonetheless, this study demonstrated that global loss of a conserved chromatin feature associated with active enhancers is compatible with life, challenging the idea that H3K4me1 is required for successful development. Thus, H3K4me1 appears to be only correlative with, rather than causative of, enhancer function. Moving forward, it will be important to investigate whether other epigenetic marks associated with enhancers have functional roles in gene regulation and development.
3.3. Misconceptions about topologically associated domains
The appearance of TADs revealed by Hi‐C and related methods promised to give insight into the relationship between genome architecture and gene regulation. A handful of studies found that changes in chromosome structure that affect TAD boundaries were associated with aberrant enhancer activity and gene expression (Blinka, Reimer, Pulakanti, & Rao, 2016; Franke et al., 2016; Lupiáñez et al., 2015). Developmental genes and their enhancers are commonly found within the same TAD (Williamson, Lettice, Hill, & Bickmore, 2016), and TAD boundaries tend to be demarcated with binding sites for the CCCTC‐binding factor (CTCF; Dixon et al., 2012; Phillips‐Cremins et al., 2013), which can exhibit insulator activity (Ghirlando & Felsenfeld, 2016; Merkenschlager & Nora, 2016). Together, these observations led to a model in which TAD boundaries act as insulators that prevent enhancers from regulating genes outside of their topological domains (Doyle, Fudenberg, Imakaev, & Mirny, 2014; Symmons et al., 2014). However, functional investigation of TADs has called into question the role of TADs in gene regulation. For example, global disruption of TADs by deletion of a cohesin‐loading factor has no effect on transcriptional activity (Schwarzer et al., 2017). Furthermore, several recent studies have reported that depletion of CTCF at TAD boundaries does not impact gene expression (Despang et al., 2019; Gambetta & Furlong, 2018; Kubo et al., 2017; Soshnikova, Montavon, Leleu, Galjart, & Duboule, 2010) histone modifications (Kubo et al., 2017), or even TAD structure (Kubo et al., 2017; Zuin et al., 2014).
The roles of CTCF and TAD boundaries in developmental gene regulation were recently studied further using the sonic hedgehog (Shh) locus as a model system (Williamson et al., 2019). Shh is located within a TAD that contains all of its enhancers, including the ZRS enhancer that drives restricted expression in the developing limb bud (Lettice et al., 2003). In mouse, changing the TAD structure by deleting CTCF sites or creating large inversions or deletions remarkably had no effect on Shh patterning or phenotype, despite increased distance between the Shh promoter and the ZRS by fluorescent in situ hybridization (FISH) (Williamson et al., 2019). A recent study in Drosophila utilized flies carrying highly rearranged balancer chromosomes to measure allele‐specific changes in chromatin topology and gene expression (Ghavi‐Helm et al., 2019). Rearrangement or fusing of TADs, or disruption of long‐range chromatin loops generally did not correlate with changes in gene expression, despite covering 75% of the Drosophila genome. However, gene expression at a few distinct loci did appear to be sensitive to TAD perturbations (Ghavi‐Helm et al., 2019). Indeed, another recent study found that the human endogenous retrovirus subfamily H (HERV‐H) has a role in establishing TAD boundaries in human pluripotent stem cells, and that deletion of HERV‐H elements disrupts their corresponding TADs and reduces the transcription of nearby genes (Zhang et al., 2019). While still controversial, there is a growing consensus that TADs do not globally mark regulatory boundaries in the genome. Instead, recent evidence suggests that organization of the genome into TADs may play a role in DNA replication (Jodkowska et al., 2019; Pope et al., 2014).
3.4. Misconceptions about chromatin looping
Another recent study of the Shh locus highlighted that we still do not understand precisely how enhancers interact with and activate transcription from promoters. Indeed, there are likely several different mechanisms through which enhancers and promoters interact to regulate gene expression. The physical distance between Shh and a distal brain enhancer increases rather than decreases upon enhancer activation, challenging the model that distal enhancers must interact with promoters through chromatin looping (Benabdallah et al., 2017). Instead, it appears that activation of this brain enhancer induces large‐scale chromatin decompaction, suggesting a new mechanism of long‐range gene regulation (Benabdallah et al., 2017).
Chromatin looping has been clearly illustrated between Shh and the ZRS (Symmons et al., 2016; Williamson et al., 2016), and between the beta‐globin gene and its distal LCR enhancer (Carter, Chakalova, Osborne, Dai, & Fraser, 2002; Tolhuis, Palstra, Splinter, Grosveld, & De Laat, 2002). Furthermore, in Drosophila the sex combs reduced (Scr) promoter is known to be brought into contact with its distal T1 enhancer by a promoter‐proximal tethering element (Calhoun, Stathopoulos, & Levine, 2002). It is presently unknown how many enhancer–promoter interactions are mediated by tethering elements and what other mechanisms may be involved. Indeed, only a handful of enhancer–promoter interactions have been thoroughly studied, and thus it remains unclear how pervasive chromatin looping between promoters and distal enhancers is as a gene regulatory mechanism. An emerging model of enhancer–promoter interaction posits that transcriptional control may be driven by the formation of condensates or hubs that concentrate the transcriptional machinery (Banani, Lee, Hyman, & Rosen, 2017; Boija et al., 2018; Hnisz, Shrinivas, Young, Chakraborty, & Sharp, 2017).
Condensation of TFs and cofactors is thought to be mediated by the low‐complexity, intrinsically disordered regions of their activation domains, and can result in droplets that exhibit properties of liquid–liquid phase separation (Cho et al., 2018; Chong et al., 2018; Sabari et al., 2018). Phase‐separated condensates incorporate many factors known to associate with enhancers, including the mediator complex, and may be particularly prevalent at clusters of enhancers to promote efficient activation of gene expression (Cho et al., 2018; Sabari et al., 2018). Furthermore, it has been suggested that condensates may contribute to long‐range enhancer–promoter interactions as well as higher‐order genome organization (Shrinivas et al., 2019). However, we currently lack a clear understanding of what functional properties phase separation may confer, and how phase separation should be defined and experimentally validated in biological systems (Mir, Bickmore, Furlong, & Narlikar, 2019). Indeed, several recent studies have highlighted transcriptional hubs that resemble phase‐separated condensates but appear to be driven by independent mechanisms (McSwiggen et al., 2019; Mir et al., 2018). At present, it remains unclear whether phase separation has functional roles in gene regulation, or whether the appearance of phase‐separated condensates is simply a consequence of transient protein–protein interactions (Chong et al., 2018).
While there is certainly more work to be done to work out general principles of gene regulation, genome‐wide maps of chromosomal contacts have provided a wealth of candidate enhancer–promoter interactions. When using chromatin interaction data to identify candidate enhancers, it is important to remember that physical contact between two genomic loci does not necessarily mean that a causal regulatory relationship exists. Furthermore, direct contact between a distal enhancer and promoter may not be required for transcriptional activation or successful development. For example, a recent study used circularized chromosome conformation capture (4C) to characterize the regulatory topology of Pitx1, which is required for hindlimb and mandibular arch development (Sarro et al., 2018). 4C identified a robust, hindlimb‐specific interaction between the Pitx1 promoter and a distal, putative hindlimb enhancer. Deletion of this putative enhancer in mouse completely disrupted the 4C interaction, but hindlimb expression of Pitx1 was only mildly affected; furthermore, the mice did not exhibit any developmental defects typical of Pitx1 gene deletion (Sarro et al., 2018). Although Pitx1 was still expressed in mice lacking this enhancer, 4C did not detect any compensatory interactions with the Pitx1 promoter, suggesting that gene expression may be activated by other enhancers in the absence of direct enhancer–promoter interactions. However, it is also possible that this finding may be due to limitations in the resolution of the assay (Raviram, Rocha, Bonneau, & Skok, 2014). Nevertheless, the results of this study demonstrate that tissue‐specific physical interactions involving essential developmental genes may have limited predictive power in identifying enhancers that are required for proper development and highlight the complex nature of gene regulation during development.
Like other genome‐wide methodologies used to infer active enhancer sequences, we can make better predictions when chromatin interaction data are paired with additional evidence. A recent study used this approach to identify putative causal variants for blood cell traits from genome‐wide association study (GWAS) data (Lareau et al., 2018). Fine‐mapped variants were overlaid with ATAC‐seq data to identify those that might have a role in gene regulation, and from this list ChIP‐seq datasets revealed variants that might disrupt TF binding motifs. Hi‐C data was then used to identify the putative target genes of regulatory variants. Altogether, these complementary strategies reduced the number of putative causal variants 100‐fold, providing a reasonable number of candidates that could be directly tested in functional assays. Given the vast number of putative enhancers that have been identified, combining these approaches is a viable strategy to prioritize putative regulatory elements for functional validation.
3.5. Validation is key to understanding enhancer function
Genome‐wide assays have revolutionized our ability to study the cis‐regulatory genome, but making assumptions from these data without functional validation has led to misconceptions. Such misconceptions could impeed our ability to understand the mechanisms through which enhancers regulate gene expression and prevent us from elucidating which enhancer sequences are important in development and disease processes. The studies highlighted above underscore the importance of functionally validating putative enhancers. Altogether, they provide new insight into the requirements of different chromatin features for enhancer function and successful development, and suggest that epigenetic marks are associated with enhancers rather than causative of enhancer function. Although inherently low‐throughput, transgenic animal models remain an important tool in the dissection of cis‐regulatory sequences, and highlight the need for complementary approaches to identify functional enhancers.
4. MASSIVELY PARALLEL REPORTER ASSAYS TO STUDY ENHANCER FUNCTION
Chromatin‐state mapping has identified myriad candidate enhancers across the human genome; however, these approaches are primarily descriptive and may not necessarily inform enhancer function. Furthermore, they cannot explain why a given cis‐regulatory element is active in a particular cell type, or predict the effects of specific sequence changes within an enhancer. Classic mouse transgenic approaches are a useful tool with which to test the necessity or sufficiency of individual enhancer sequences for a given biological outcome, but are highly impractical for validating putative enhancers at scale.
To overcome bottlenecks in the functional validation of candidate enhancer sequences identified by genome‐wide methods, massively parallel reporter assays (MPRAs) have been developed. These methods are based on traditional reporter assays, in which a putative regulatory element is placed upstream of a reporter coding sequence (e.g., luciferase) in a vector, transfected into cells, and assayed for reporter activity. Although the context is often heterologous, that is, using genetic elements from other organisms, these assays are a powerful tool with which to test the sufficiency of an element to regulate gene expression. MPRAs utilize advances in oligonucleotide synthesis (LeProust et al., 2010) and sequencing technology to expand upon this experimental framework. In MPRAs, libraries containing hundreds of thousands of candidate enhancers can be generated and tested for function in parallel (Figure 3a). Each enhancer is cloned upstream of a minimal promoter and a reporter gene containing a unique sequence barcode in its 3′ untranslated region. These libraries of barcoded reporter genes can then be introduced into cells or animals and expression quantified by RNA‐seq of the unique sequence barcodes. Thus, MPRAs can be used to functionally validate numerous putative enhancer sequences in a single experiment with a quantitative readout. Furthermore, because so many sequences can be tested for function at once, MPRAs enable the study of how single nucleotide changes within an enhancer affect function. This systematic dissection of enhancer sequences has given important insight into the mechanistic basis of enhancer activity and the functional constraints of enhancer sequences during development.
Figure 3.
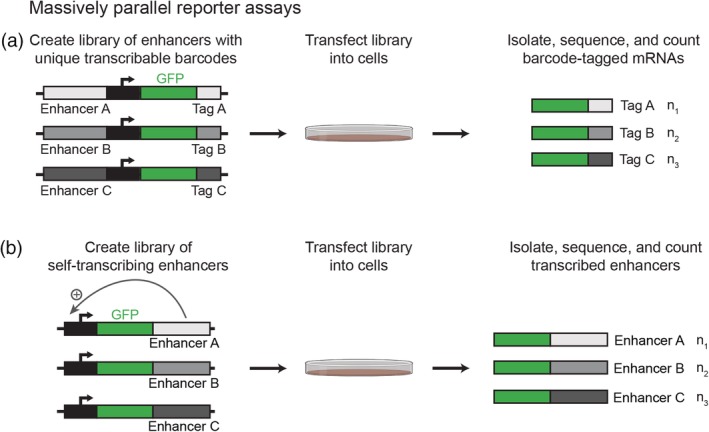
In vitro massively parallel reporter assays (MPRAs) to study enhancer function. MPRAs utilize advances in oligonucleotide synthesis and sequencing technology to test large libraries of candidate enhancer sequences for function. (a) MPRA libraries typically contain candidate enhancer sequences upstream of a minimal promoter and reporter gene. A transcribable barcode unique to each candidate enhancer is placed in the 3′ untranslated region of the reporter gene, allowing active enhancers to be identified by sequencing barcode‐tagged mRNAs. (b) Self‐transcribing active regulatory region sequencing (STARR‐seq) is a variation of the MPRA that exploits the characteristic that enhancers can function independently of their relative positions. In STARR‐seq, candidate enhancer sequences are placed downstream of a minimal promoter and reporter gene, allowing active enhancers to transcribe themselves. Figure created with BioRender
4.1. MPRA approaches to study enhancer function
MPRAs have successfully been utilized in a number of diverse biological systems to (a) interrogate the activity of candidate enhancer sequences and (b) to investigate the functional consequences of mutations within known enhancers. Several studies have used MPRAs to functionally validate putative enhancers identified from genome‐wide assays (Kheradpour et al., 2013; Kwasnieski, Fiore, Chaudhari, & Cohen, 2014). To study enhancer sequence variation, one MPRA report used the human embryonic kidney cell line HEK293T to study all possible single nucleotide variants of two inducible enhancers, a synthetic cAMP‐regulated enhancer and a virus‐induced enhancer of IFNB (Melnikov et al., 2012). The authors of this study used their data from induced versus uninduced cellular states to generate a model that could predict the activity of novel variants. This allowed for the design of synthetic enhancers that optimize different objectives, for example, minimizing basal activity while maximizing induced activity (Melnikov et al., 2012), and thus this method has important implications for synthetic biology.
In another MPRA saturation mutagenesis approach, three mammalian liver enhancers were dissected at single‐nucleotide resolution by injecting libraries containing hundreds of thousands of mutant enhancer sequences into mouse tail vein (Patwardhan et al., 2012). Following injection, livers were harvested and transcribed barcodes were sequenced. This study found that only a small fraction of enhancer mutations affected activity by more than twofold, and sequence changes with higher effects tended to fall within known liver TF binding sites (Patwardhan et al., 2012). In contrast to these modest effects, another saturation mutagenesis MPRA study found that the majority of sequence variants tested within a Rhodopsin promoter proximal enhancer had significant effects on regulatory activity in mouse retina (Mogno, Kwasnieski, Cohen, Myers, & Corbo, 2012). Not all of these effects could be explained by changes in the affinity of known TF binding sites. The different results obtained by these two studies likely reflect differences in the specific enhancers examined, study design, and biological system utilized. Indeed, an MPRA that analyzed exonic liver enhancers found different effects of enhancer mutations in liver compared to HeLa cells (Birnbaum et al., 2014), highlighting the importance of context‐specific MPRA approaches.
An alternate MPRA approach has been developed, termed self‐transcribing active regulatory region sequencing (STARR‐seq; Figure 3b; Arnold et al., 2013; Schöne et al., 2018). This method employs the characteristic that enhancers can function independently of their relative positions, and places candidate sequences downstream of a minimal promoter and open reading frame. Thus, in this system active enhancers transcribe themselves, negating the need for barcode sequences. A STARR‐seq library containing millions of randomly sheared genomic DNA fragments from Drosophila were transfected into S2 cells followed by RNA‐sequencing to identify transcribed elements (Arnold et al., 2013). This study identified thousands of genomic regions that were significantly enriched with a wide range of enhancer activity, with the strongest enhancers found next to housekeeping genes and developmental TFs. This approach also revealed that 31% of strong enhancers in S2 cells were located within closed chromatin and lacked H3K27ac, but were marked with H3K4me1, suggesting that these sequences may be silenced in their endogenous contexts. This finding highlights the utility of MPRAs as a complementary approach to genome‐wide chromatin‐state assays. Like other MPRAs, STARR‐seq is easily adaptable to the study of synthetic enhancer variants (Schöne et al., 2018).
4.2. MPRAs to study enhancer function in whole developing embryos
MPRA methodologies have also been adapted for use in whole developing embryos, providing novel insight into the function and tissue‐specificity of developmental enhancers. Enhancer‐FACS‐seq (eFS) was developed in Drosophila to identify active, tissue‐specific enhancers in whole embryos (Figure 4a; Gisselbrecht et al., 2013). This method utilizes a cross between two transgenic fly lines, one carrying a cassette containing a candidate enhancer upstream of a reporter gene and the other expressing a cell‐surface antigen in a tissue‐specific manner. The candidate enhancer‐reporter construct is integrated into the genome in a site‐specific manner to avoid position effects. A cross between these fly strains generates embryos that can be dissociated and FACS sorted, first by tissue type and then by reporter expression. Genomic DNA can then be isolated from the collected cells and sequenced to identify enhancers activated in specific tissues. eFS was first used to test hundreds of putative enhancer sequences for function in whole mesoderm, fusion‐competent myoblasts, and somatic mesoderm founder cells of developing Drosophila embryos. This identified over 100 active mesoderm enhancers that correlated well with histone marks of active enhancers and were enriched for RNA polymerase II and mesoderm‐specific TF binding by ChIP‐seq (Gisselbrecht et al., 2013).
Figure 4.
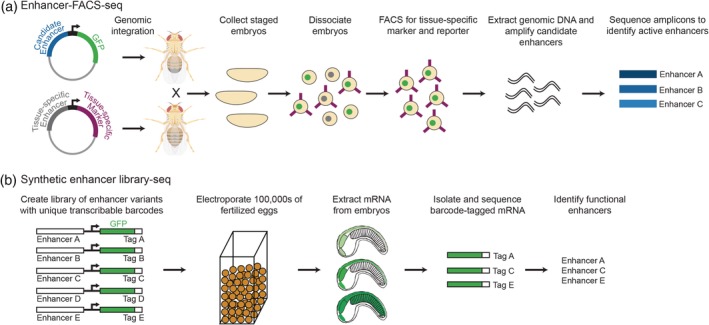
In vivo massively parallel reporter assays to study enhancer function. (a) Enhancer‐FACS‐seq identifies active, tissue‐specific enhancers in whole Drosophila embryos. Two flies are crossed, the first carrying a candidate enhancer sequence driving GFP expression, and the second expressing a cell surface marker in a tissue‐specific manner. Embryos resulting from this cross are dissociated and FACS sorted for the tissue‐specific cell surface marker and GFP. Genomic DNA can then be extracted from sorted cells to identify enhancers that are active in the specific tissue. (b) Synthetic enhancer library‐sequencing (SEL‐seq) allows millions of enhancer variants to be tested for function in whole Ciona embryos. Synthetic enhancer variants are attached to a minimal promoter, GFP coding sequence, and a unique transcribable barcode. The enhancer library is electroporated into hundreds of thousands of fertilized Ciona eggs, which are then allowed to develop until the desired developmental stage. Barcode‐tagged mRNA can then be isolated and sequenced to identify active enhancer variants, providing insight into which sequences within an enhancer are important for function. Figure created with BioRender
Because it is not feasible to test all enhancers in all tissues at all developmental timepoints, identifying which sequences within an enhancer are important for function would allow us to develop a set of rules that define how enhancers drive tissue‐specific expression. These rules could then be used to better identify enhancers in the genome prior to functional validation. To understand how enhancer sequences encode function, whole‐embryo MPRAs were developed in the tunicate Ciona intestinalis, which belongs to the sister group of all vertebrates (Delsuc, Brinkmann, Chourrout, & Philippe, 2006). Developmental programs are conserved between Ciona and vertebrates, and thus findings in Ciona are expected to be highly relevant to the study of enhancer function during vertebrate development. The synthetic enhancer library‐sequencing (SEL‐seq) method is able to test millions of enhancer variants for function in developing Ciona embryos (Figure 4b). Millions of Ciona embryos can be electroporated with reporter constructs in a single experiment, making it highly amenable to testing enhancers at scale.
SEL‐seq was first used to study a minimal, 69‐basepair Orthodenticle homeobox (Otx) enhancer that contains GATA and ETS binding sites and directs specific expression in the neural plate (Bertrand, Hudson, Caillol, Popovici, & Lemaire, 2003; Farley et al., 2015; Rothbacher, Bertrand, Lamy, & Lemaire, 2007). Millions of synthetic enhancers were created in which core GATA and ETS binding motifs remained constant and the remaining sequence was randomized (Farley et al., 2015). Each synthetic enhancer is attached to a minimal promoter, GFP coding sequence, and unique barcode sequence. The Otx enhancer library was electroporated into fertilized Ciona eggs, which developed to the stage at which the wildtype Otx enhancer becomes active and were then harvested for RNA. RNA‐seq of transcribed barcodes allowed active enhancers to be identified. Computational analysis of ~20,000 highly active enhancer variants revealed that dinucleotide motifs flanking core ETS and GATA binding sites are key determinants of enhancer function (Farley et al., 2015).
When the wildtype dinucleotide motifs were added to an otherwise inactive enhancer variant, the wildtype expression pattern was recapitulated. When these dinucleotide motifs were optimized to match the highest affinity position–weight matrices identified by in vitro binding assays, the previously inactive synthetic enhancer drove robust, ectopic expression. The strength and specificity of enhancer sequences could be further modulated by changing the spacing and orientation of TF binding sites (Farley et al., 2015; Farley, Levine, et al., 2016). The SEL‐seq method revealed that suboptimal affinity and spacing of GATA and ETS motifs within the Otx enhancer are important for tissue‐specific expression, and suggested a tradeoff between TF binding site affinity and syntax within developmental enhancers. Furthermore, analysis of ectopically expressed enhancer variants led to the identification of a novel notochord enhancer and demonstrated enhancer grammar, the interplay between TF binding site affinity and organization (Farley, Levine, et al., 2016). Defining the grammatical constraints for notochord enhancers enabled the identification of notochord‐specific enhancers within the genome, demonstrating the value of understanding grammatical constraints on enhancer function. Altogether, high‐throughput reporter assays in Ciona have made substantial progress toward elucidating the regulatory principles governing enhancer function.
4.3. Current limitations of MPRAs
MPRAs have greatly increased our ability to test the sufficiency of candidate enhancer sequences for expression at scale, and have proven to be a powerful method with which to study how variation within enhancer sequences impacts function. This has allowed us to gain significant insight into which parts of an endogenous enhancer sequence are important for function. However, there are several important considerations to be made when designing MPRAs and interpreting their results. For example, it is critical to use a minimal promoter that does not drive expression in the absence of enhancer sequences, as basal promoter activity could confound results. Furthermore, enhancer activity can vary substantially in different promoter contexts (Zabidi et al., 2015). Current limitations on the length of synthesized oligos makes it difficult to comprehensively study variation within enhancer sequences greater than 200 basepairs (Kwasnieski et al., 2014; Tewhey et al., 2016). Additionally, most MPRAs thus far have been done in cell lines which may not recapitulate the in vivo function of enhancers and limits their insight into development (Erceg et al., 2014). This limitation has been addressed by recent MPRA approaches in whole embryos, which have begun to reveal how enhancer sequences encode tissue‐specific expression during development (Farley et al., 2015; Gisselbrecht et al., 2013). Finally, MPRAs typically utilize heterologous reporter constructs that test enhancer sequences for function outside of their native genomic contexts. This potential limitation of MPRAs has recently been addressed by genome editing technology. However, manipulating enhancer sequences within the genome may produce modest phenotypes due to enhancer redundancy, or conversely, interfere with development, making it difficult to observe normal expression patterns. It is thus important to employ both MPRA and genome editing approaches to gain a comprehensive understanding of enhancer function. Indeed, MPRAs remain an essential and complementary tool because they allow enhancer sequences to be directly and systematically tested for function.
5. CRISPR/CAS9‐BASED METHODS TO IDENTIFY CANDIDATE ENHANCERS
The development of genome editing technology using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and the CRISPR‐associated protein 9 (Cas9) (Cong et al., 2013; Mali et al., 2013) has revolutionized our ability to interrogate putative enhancer sequences for function within their native genomic context. In this system, the Cas9 nuclease is guided by a single guide RNA (sgRNA; Jinek et al., 2012) to a specific DNA target, where it induces a double strand break (DSB). Cas9‐induced DSBs are typically repaired by non‐homologous end joining (NHEJ), an error‐prone DNA repair pathway that creates random insertion or deletion mutations (indels) at the site of cleavage. CRISPR/Cas9 is advantageous over traditional genetic manipulations because it can target any region of the genome containing a protospacer‐adjacent motif (PAM) (Figure 5a) and has been shown to effectively edit the genomes of diverse organisms (Friedland et al., 2013; Nakayama et al., 2013; Shan et al., 2013; Stolfi, Gandhi, Salek, & Christiaen, 2014; Wang et al., 2013; Yang et al., 2014; Yu et al., 2013). Furthermore, editing by CRISPR/Cas9 has improved efficiency compared to traditional reverse genetics approaches (Burgio, 2018). Studying putative cis‐regulatory elements individually can be extremely informative and is currently the standard for functional validation. However, due to the large number of putative enhancer sequences identified by genome‐wide assays, a major focus has been to develop massively parallel cell‐based CRISPR/Cas9 screens to functionally test the effects of cis‐regulatory changes on gene expression and phenotype. While this cutting‐edge technology has not yet been applied to developing embryos, advances in high‐throughput CRISPR screens could provide unprecedented insight into the regulation of gene expression during development.
Figure 5.
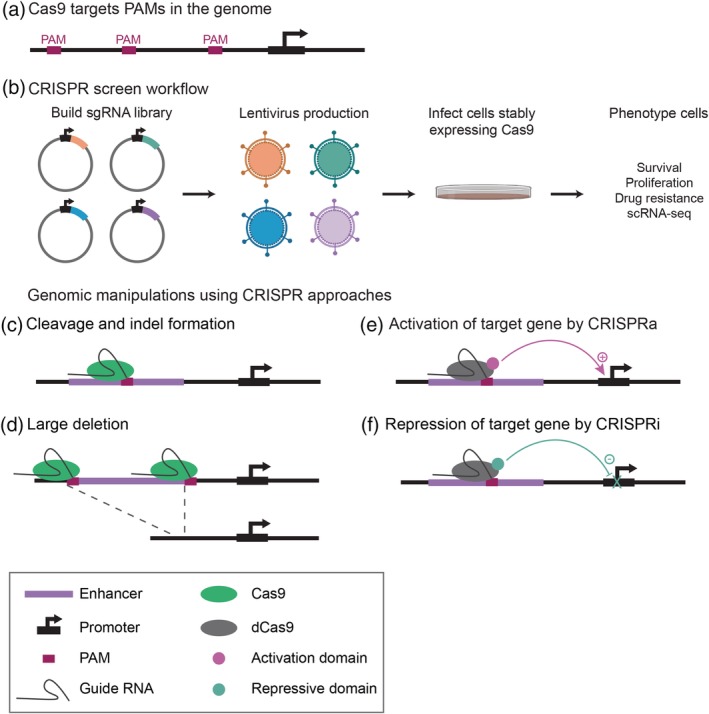
CRISPR‐based methods to identify and study candidate enhancers. (a) Any genomic region containing a protospacer‐adjacent motif (PAM) can be targeted by CRISPR/Cas9. (b) The workflow for high‐throughput CRISPR screens includes building a library of sgRNAs, cloning the library into lentiviral vectors, infecting cells that stably express Cas9 or one of its variant forms, and phenotyping cells. The sgRNA(s) carried by cells of a given phenotype can easily be identified by sequencing, and can give insight into candidate cis‐regulatory regions that affect phenotype. CRISPR screens can utilize (c) one sgRNA to mutate a particular locus, or (d) two sgRNAs to delete a genomic region. In both cases, DNA cut by Cas9 will be repaired by the error‐prone non‐homologous end joining (NHEJ) pathway, creating indels. Variant forms of Cas9 that are used in high‐throughput screens include (e) CRISPRa to activate transcription and (f) CRISPRi to repress transcription. Both CRISPRa and CRISPRi utilize catalytically dead Cas9 fused to an effector protein and impact gene expression through chromatin remodeling. Figure created with BioRender
5.1. High‐throughput CRISPR screens to identify candidate enhancers
To identify candidate enhancer sequences, high‐throughput CRISPR/Cas9 screens typically utilize tens of thousands of sgRNAs targeting the noncoding genome, which are cloned into lentiviral vectors and delivered as a pool to cells at a low multiplicity of infection (MOI) along with Cas9 (Figure 5b). The use of a low MOI ensures that, on average, cells are infected with only one sgRNA construct and thus bear only one CRISPR‐induced mutation (Figure 5c). Cells are then selected for a phenotype, such as survival, proliferation, or drug resistance, and sgRNAs that are enriched or depleted in cells give insight into putative cis‐regulatory regions that affect phenotype. Findings from these pooled screens can then be prioritized and validated in follow‐up experiments using individual sgRNAs.
High‐throughput CRISPR/Cas9 screens (Canver et al., 2015; Gasperini et al., 2017; Korkmaz et al., 2016; Sanjana et al., 2016; Shen et al., 2017) have been used to identify putative enhancer sequences by (a) mutating the sequence proximal to a specific gene or (b) mutating the binding sites of a specific TF genome‐wide. For example, a pooled CRISPR screen was used for saturating mutagenesis across an erythroid lineage‐specific enhancer associated with hemoglobin disorders, which provided a detailed map of the organization and function of this enhancer (Canver et al., 2015). A similar approach was used to identify novel enhancers that regulate genes involved in BRAF inhibitor resistance in melanoma (Sanjana et al., 2016). Another study mutated p53 binding sites genome‐wide to understand enhancers that control oncogene‐induced senescence, a cell‐cycle arrest program induced by p53 (Korkmaz et al., 2016). A less biased approach is to make tiling deletions along a genomic region by delivering two sgRNAs to each cell (Figure 5d; Gasperini et al., 2017; Shen et al., 2017), which successfully identified novel enhancers of POU5F1 using human embryonic stem cells (ESCs; Shen et al., 2017).
At present, a major limitation of CRISPR screens is that indels produced by NHEJ may not be sufficient to disrupt enhancer function, and even if function is disrupted by indels or larger deletions, this may fail to produce detectable phenotypes due to enhancer redundancy. Instead of phenotyping mutants based on discrete cellular phenotypes, some studies have utilized knock‐in GFP reporters as a quantitative and continuous readout of gene regulatory activity (Rajagopal et al., 2016; Shen et al., 2017). Furthermore, single cell RNA‐seq (scRNA‐seq) has recently been applied to pooled CRISPR screens, particularly those that cause multi‐locus perturbation, as it allows for comprehensive cell phenotyping (Dixit et al., 2016; Jaitin et al., 2016). Following large‐scale CRISPR/Cas9 screens, genome‐wide data from ChIP‐seq, ATAC‐seq, Hi‐C, and related methods are typically used to prioritize putative enhancers for targeted functional validation.
5.2. CRISPR activation and CRISPR interference to identify candidate enhancers
CRISPR screens that utilize variant forms of Cas9 also have the potential to be useful for the functional identification of putative enhancers. Catalytically dead Cas9 (dCas9) can be fused to an effector domain that either activates (CRISPRa) (Gilbert et al., 2014; Maeder et al., 2013; Perez‐Pinera et al., 2013; Figure 5e) or interferes with (CRISPRi) (Gilbert et al., 2013, 2014; Figure 5f) the expression of target genes. These Cas9 fusion proteins do not cut DNA, but instead, when paired with a specific sgRNA, recruit effector domains to specific genomic loci. CRISPRa strategies to activate gene expression commonly utilize dCas9 fused to the C‐terminal VP64 acidic transactivation domain derived from herpes simplex virus protein VP16 (Beerli, Dreier, & Barbas, 2000; Perez‐Pinera et al., 2013; Seipel, Georgiev, & Schaffner, 1992). A pooled screen successfully identified stimulus‐responsive enhancers when CRISPRa was recruited to the autoimmunity risk loci CD69 and IL2RA (Simeonov et al., 2017).
A more popular approach for studying enhancer function has been CRISPRi, in which dCas9 is fused to a Krüppel‐associated box (KRAB) domain that represses gene expression (Gilbert et al., 2014). The KRAB domain works by recruiting repressive cofactors that methylate or deacetylate histones, which ultimately results in heterochromatin spreading and subsequent impacts on gene expression (Groner et al., 2010; Reynolds et al., 2012; Schultz, Ayyanathan, Negorev, Maul, & Rauscher, 2002; Sripathy, Stevens, & Schultz, 2006). A CRISPRi screen using tiled sgRNAs in the vicinity of MYC and GATA1 loci revealed nine distal enhancers that control cellular proliferation (Fulco et al., 2016). Putative enhancers that correlated well with H3K27ac, Hi‐C data, and DNase hypersensitive sites were thoroughly validated by luciferase assays, qPCR for the predicted target gene, and loss of function experiments (Fulco et al., 2016). Other studies have used CRISPRi with multiple sgRNAs per cell to interrogate combinatorial enhancer activity, followed by scRNA‐seq to phenotype cells (Gasperini et al., 2019; Xie, Duan, Li, Zhou, & Hon, 2017). sgRNAs targeting 71 constituent enhancers from 15 super‐enhancers revealed some individual enhancer sequences that had strong effects on gene expression, while others only showed an effect when silenced in combination and thus appear to be redundant (Xie et al., 2017).
A recent CRISPRi screen delivered lentiviral sgRNA constructs at a high MOI to achieve an average of 28 random perturbations per cell (Gasperini et al., 2019). Rather than focusing on a particular gene or TF, this study silenced combinations of candidate enhancers genome‐wide and then used scRNA‐seq data to infer hundreds of putative enhancer‐gene regulatory relationships, a handful of which were validated by homozygous deletion of the enhancer and gene expression assays (Gasperini et al., 2019). As expected due to the redundancy of enhancer sequences, only 10% of candidate enhancers that were targeted by CRISPRi affected gene expression (Gasperini et al., 2019). Indeed, enhancer redundancy may significantly limit the ability of CRISPR perturbations to identify functional enhancers, demonstrating the challenge in identifying enhancers within the endogenous locus and highlighting the value of MPRAs as a complementary approach to study enhancer function.
5.3. Current limitations of high‐throughput CRISPR screens
Altogether, high‐throughput screens using CRISPR/Cas9 and its derivatives have allowed for the analysis of numerous cis‐regulatory elements in their native genomic context. Although several novel enhancers have been functionally identified, these methodologies are still in their infancy and there are several important considerations moving forward. CRISPR screens typically utilize data from ChIP‐seq, DNase‐seq, Hi‐C and related assays to prioritize hits. However, a CRISPR screen focused on ESC‐specific genes identified “unmarked” regulatory elements that lacked stereotypical epigenetic features of enhancers (Rajagopal et al., 2016). This finding highlights the value of pooled CRISPR screens as a complementary approach to identify candidate cis‐regulatory elements. Another study found that DNase I hypersensitive sites, Hi‐C interactions, and H3K27ac were able to predict enhancers from a CRISPRi screen, but ranked them in the wrong order (Fulco et al., 2016). For example, the enhancer with the most significant DNase‐seq peak had the smallest effect on gene expression. An additional consideration is that random indels produced by NHEJ may be insufficient to disrupt enhancer function. On the other hand, the heterochromatin spreading induced by dCas9‐KRAB can span across 1 to 2 kb (Gasperini et al., 2019), and thus lacks the resolution to discern which enhancer, or which parts of a putative enhancer sequence, are important for function. This could cause off‐target effects that are unrelated to sgRNA specificity. Even if a candidate enhancer sequence is successfully and specifically disrupted, perturbation of a single enhancer may not produce a detectable phenotype. Indeed, current CRISPR screens are likely unable to identify some functional enhancers because of enhancer redundancy. Finally, it remains unclear whether all enhancers are equally susceptible to perturbation by CRISPR or CRISPRi. Indeed, nucleosome positioning has been shown to play a key role in the ability of Cas9 to interact with DNA, and thus it may be difficult for Cas9 to act at enhancers located in nucleosome dense regions (Horlbeck et al., 2016).
Chromatin accessibility is also an important consideration when choosing a cell type for high‐throughput CRISPR screens, as results could be impacted by a non‐relevant chromatin context. A number of studies have employed the human immortalized myelogenous leukemia cell line K562 (Dixit et al., 2016; Fulco et al., 2016; Gasperini et al., 2019; Xie et al., 2017). These cells are advantageous because they can easily be grown in suspension and have been engineered to stably express Cas9 (Deans et al., 2016), but it is unclear whether findings in this cell line can be extrapolated to other cellular contexts. One strategy to mitigate this issue has been to do an initial CRISPR screen in K562 cells followed by a sub‐screen of the most significant hits in a more relevant cell type (Kramer et al., 2018). At present, large‐scale CRISPR screens may be particularly useful for identifying putative regulatory elements that lack the stereotypical epigenetic signatures associated with enhancers. These methodologies have just begun to be applied to the whole genome for the unbiased identification of candidate enhancer sequences (Gasperini et al., 2019), and will no doubt have a significant impact on our ability to understand enhancer function. Moving forward, it will be important to validate putative enhancer sequences found in high‐throughput CRISPR assays at scale. The sufficiency of these sequences to drive gene expression could be tested in MPRAs, while necessity could be assessed in scaled up CRISPR deletion assays. Thus, we predict that MPRAs and CRISPR screens will provide complementary insight into enhancer function and will together advance our ability to identify bona fide enhancer sequences.
5.4. CRISPR approaches in animal models
A primary advantage of CRISPR screens is the ability to identify and manipulate candidate enhancer sequences within the native chromatin context in a high‐throughput manner, but these studies currently lack any direct insight into development. Animal models remain a critical tool for validating putative enhancer sequences and understanding enhancer function within a developmental context, but there are significant challenges in scaling up these approaches. CRISPR screens have been performed in whole animals, in which exogenous, edited cancer cells are introduced into immunocompromised mice and scored for tumor growth and metastasis (S. Chen et al., 2015). However, at present the utility of this approach is likely limited to studies of hematopoiesis, blood disorders, and cancer.
Recently, a gene drive strategy was developed in mice that biases the inheritance of desired alleles and has the potential to greatly increase the efficiency with which transgenic mouse lines can be generated (Grunwald et al., 2019). This approach could be used to efficiently manipulate a handful of enhancer sequences at once, which would be particularly useful given the number of predicted redundant enhancers present in genomes. Furthermore, CRISPR strategies could be used alongside anti‐CRISPRs (Bubeck et al., 2018; Nakamura et al., 2019), phage‐derived proteins that abrogate CRISPR activity, to facilitate spatiotemporally restricted genomic perturbations during development. This approach would allow for finely tuned functional studies and could provide novel insight into enhancer function. High‐throughput CRISPR screens have rapidly provided a wealth of information regarding enhancer function and are a tantalizing method with which to study developmental enhancers, but there are still bottlenecks that must be overcome to gain direct insight into organismal development.
6. SINGLE‐CELL TRANSCRIPTOMIC APPROACHES TO CONSTRUCT ATLASES OF ANIMAL DEVELOPMENT
A major objective in developmental biology is to understand how changes in gene expression direct the progression of embryonic cell lineages from pluripotency to diverse cell fates. Because enhancers control patterns of gene expression during development, significant efforts have focused on locating enhancer sequences in the genome, elucidating the specific tissues in which they are active, and identifying the genes that they regulate. Although it remains a substantial challenge to directly link enhancer sequences to tissue‐specific gene expression at scale, single‐cell transcriptomics approaches promise to play an integral role in achieving this goal. Early scRNA‐seq was performed after FACS sorting (Meador et al., 2014) or microfluidics processing (e.g., the Fluidigm C1 platform) (Pollen et al., 2014) to isolate individual cells within separate compartments of a multiwell plate (F. Tang et al., 2009, 2010). While these methods offer some advantages, isolating individual cells can be expensive and time consuming (Kolodziejczyk, Kim, Svensson, Marioni, & Teichmann, 2015; Trapnell & Liu, 2016), limiting the number of single cells that can be analyzed in a single experiment. Recent advances in scRNA‐seq have greatly increased the number of cells that can economically be analyzed (J. Cao et al., 2017; Macosko et al., 2015), making it possible to assemble comprehensive, single‐cell transcriptional atlases of developing embryos. These developmental atlases provide a description of gene expression in each cell of a developing embryo, revealing the transcriptional blueprints for development.
To generate an atlas representing a developmental timepoint, embryos are dissociated into single cells, and then subjected to scRNA‐seq. scRNA‐seq is compatible with fixed cells, which can minimize the effects of dissociation and handling on cell state (J. Cao et al., 2017). In droplet‐based scRNA‐seq approaches, cells are introduced into a microfluidic system in which single cells are encapsulated in oil droplets containing uniquely barcoded primers on a bead (Macosko et al., 2015; Figure 6a). Primers contain two barcodes, one that is cell‐specific (i.e., represented on every primer for a given bead) and one that is transcript‐specific (i.e., unique to each primer on a given bead). mRNA is then reverse‐transcribed and amplified, after which barcoded DNA can be pooled and sequenced. An alternate method is single‐cell combinatorial indexing (sci)‐RNA‐seq (J. Cao et al., 2017), which utilizes split‐pool barcoding to uniquely label single cells (Figure 6b). Briefly, cells are distributed to multiwell plates, where a unique barcode is introduced to each well during reverse transcription (RT). Cells are then pooled and redistributed, and a second unique barcode is introduced to each well during PCR. Amplicons can then be pooled and subjected to massively parallel sequencing. Following scRNA‐seq by either method, single‐cell transcriptomic information is then deconvoluted using cell‐specific barcodes, and transcript abundance quantified using unique molecular identifiers. Computational approaches are then used for the unsupervised clustering of cells based on the similarity of their transcriptomes (Figure 6c; Kiselev, Andrews, & Hemberg, 2019). Cell clusters are typically represented as a t‐distributed stochastic neighbor embedding (t‐SNE) or uniform manifold approximation and projection plot (UMAP), the latter of which preserves data structure in such a way that inter‐cluster relationships can be elucidated (Becht et al., 2019). Because cells are dissociated, spatial information is lost, but can be reconstructed using known cell type‐specific markers and in situ hybridization data (Figure 6d).
Figure 6.
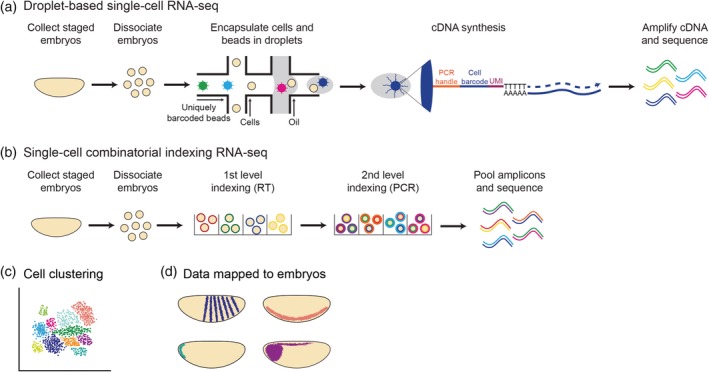
scRNA methodologies to build atlases of animal development. Single cell RNA‐sequencing (scRNA‐seq) methods used to generate atlases of animal development are illustrated using Drosophila as a model system. Staged embryos are collected and dissociated before being subjected to (a) droplet‐based scRNA‐seq or (b) single‐cell combinatorial indexing RNA‐seq (sciRNA‐seq). Droplet‐based scRNA‐seq methods utilize beads bearing primers that contain two unique barcodes, one that is cell‐specific and one that is transcript‐specific (Unique Molecular Identifier). The primers also contain a PCR handle for subsequent amplification. For sciRNA‐seq, cells are distributed to multiwell plates where they receive a well‐specific barcode during reverse transcription. Cells are then pooled and distributed to new multiwell plates where they receive a second well‐specific barcode during amplification. A third level of indexing can increase the number of cells processed in a single experiment (not shown). (c) Single‐cell transcriptomic data is represented as a t‐distributed stochastic neighbor embedding (t‐SNE) or uniform manifold approximation and projection (UMAP) plot, which cluster cells based on the similarity of their transcriptomes. (d) Cell type‐specific markers and in situ hybridization data can be used to map single‐cell transcriptomes back to a virtual embryo
6.1. Single‐cell transcriptomics can provide new insight into development
Single‐cell transcriptomic approaches were applied to the construction of a comprehensive developmental atlas for C. elegans (J. Cao et al., 2017). In L2 stage C. elegans embryos, many distinct neuronal cell types were identified, including some known to correspond to only one or two cells in the larval worm. The ability to detect extremely rare cell types highlights the detail with which this approach can map single‐cell transcriptomes from whole embryos. In Drosophila, this approach has been used to characterize stage 6 embryos, which represent the onset of gastrulation (Karaiskos et al., 2017). Single‐cell transcriptome data were correlated with high‐resolution in situ expression patterns for 84 marker genes, allowing single cells to be mapped to specific positions on a virtual embryo. This information was used to predict “virtual” in situ hybridization patterns for transcripts expressed in the stage 6 embryo, many of which were validated by in situ hybridization and found to be accurate across a wide range of expression patterns (Karaiskos et al., 2017). Interestingly, long noncoding RNAs (lncRNAs) were some of the most variable transcripts between clusters, and many exhibited patterned expression, suggesting that lncRNAs may have a role in early embryonic patterning and development (Karaiskos et al., 2017). Furthermore, the authors of this study identified patterned expression of Hippo signaling components, which are major regulators of organ size, cell cycle, and proliferation and had not previously been implicated in early embryonic development. Indeed, increased nuclear localization of Yorkie, a TF downstream of Hippo signaling, was found in cells that were undergoing mitosis, suggesting a new role of Hippo signaling in cell cycle regulation during early Drosophila development (Karaiskos et al., 2017). Thus, while scRNA‐seq of whole embryos is primarily a descriptive method, new biological insights are already being gleaned from these datasets.
Atlases of animal development have not yet been directly applied to the study of enhancers, although additional insight into developmental gene regulation could be achieved by combining scRNA‐seq with single‐cell ATAC‐seq to assay chromatin accessibility, which may reveal putative developmental enhancers. This principle was recently demonstrated in Ciona embryos by bulk RNA‐seq and ATAC‐seq performed on FACS sorted cardiac progenitor cells, which led to the identification of novel enhancers (Racioppi, Wiechecki, & Christiaen, 2019). Pairing single‐cell approaches within the context of a whole developing embryo will likely expand our ability to identify and functionally validate enhancer sequences.
6.2. Phenotyping mutants with single‐cell resolution
Beyond facilitating the construction of developmental atlases, scRNA‐seq allows for rapid, unbiased phenotyping of mutant embryos at single‐cell resolution. Several studies have directly compared the transcriptional landscapes of wildtype embryos and embryos bearing mutations in genes known to be important for development. Intriguingly, it appears thus far that single cells from mutant embryos adopt a subset of wildtype transcriptomic states, but do not exhibit markedly distinct transcriptomes compared to wildtype.
In maternal‐zygotic one‐eyed pinhead (MZoep) mutant zebrafish, which are defective in nodal signaling, no mutant‐specific transcriptomic states were identified, although wildtype states were significantly reduced or absent (Farrell et al., 2018). Similarly, disruption of the chordin locus in zebrafish, which is required for patterning the early dorsal ventral axis (Sasai et al., 1994; Schulte‐Merker, Lee, McMahon, & Hammerschmidt, 1997), did not change the overall transcriptional landscape, but dramatically altered cell state abundances (Wagner et al., 2018). In mouse, scRNA‐seq was used to study an embryonic lethal mutation in Tal1, a gene that is critical for hematopoiesis (Pijuan‐Sala et al., 2019). Challenges in studying this lethal mutation were overcome by injecting tdTomato‐positive, homozygous Tal1 mutant ESCs into wildtype blastocysts, and then dissociating and sorting cells from the resulting embryos prior to scRNA‐seq. Similar to findings in zebrafish, mutant cells did not form novel clusters. However, tdTomato‐positive, Tal1‐null cells from chimeric embryos were depleted of multiple blood cell clusters (Pijuan‐Sala et al., 2019).
Moving forward, comparing scRNA‐seq data from embryos bearing mutations within enhancers to stage‐matched wildtype transcriptomic atlases will be a valuable method for systematically assessing how enhancer sequences contribute to development on a global scale. This approach will provide unprecedented insight into the specific cell types that are affected by enhancer manipulations within the context of developing embryos.
6.3. Reconstruction of developmental trajectories
When cells from embryos representing different developmental timepoints are subjected to scRNA‐seq, trajectories of differentiation can be reconstructed over time. Single‐cell trajectories are built by a computational approach termed pseudotime ordering (Trapnell et al., 2014), which reflects developmental progress rather than absolute time. In zebrafish, many different embryonic stages have been evaluated by this method, in both wildtype and mutant animals (Farrell et al., 2018; Wagner et al., 2018). In wildtype zebrafish, trees constructed by pseudotime ordering substantially mirrored the developmental trajectories expected from classic embryological studies (Farrell et al., 2018). However, some branch points contained intermediate cells that expressed genes characteristic of many disparate cell fates (Farrell et al., 2018). One study complemented its reconstructed atlas of the zebrafish embryo with a transposon‐based lineage tracing approach to interrogate whether individual cell histories correlated with inferred developmental trajectories (Wagner et al., 2018). This revealed that some clonally distinct lineages can give rise to similar cell types, while in other cases, transcriptional changes can drive related cells toward distinct fates. This finding highlights the complexity of development and the need for complementary strategies to study developmental processes. While at present treelike hierarchies cannot fully represent the dynamics of differentiating cells in the zebrafish embryo, using single‐cell transcriptome analysis in combination with lineage tracing can help us to better understand developmental trajectories. If combined with single cell ATAC‐seq, this approach could contribute new insights into gene regulatory dynamics during development.
6.4. Methods to study developmental trajectories have broad implications
Moving forward, we predict that methods to reconstruct developmental trajectories will be broadly applicable to the study of biological processes that share similarities with development, including cancer progression and tissue regeneration. Indeed, regeneration trajectories have been described in the regenerative planarian Schmidtea mediterranea, an immortal organism that continuously regenerates its tissues and has remarkable regenerative capabilities (Plass et al., 2018). All cell types and trajectories identified in regenerating planarians were present in the intact planarian atlas, demonstrating that there are not regeneration‐specific trajectories. However, regenerating planarians showed a large increase in neoblast numbers, consistent with increased mitotic activity, and increased neural progenitor cells, reflecting active neurogenesis to replace missing head structures (Plass et al., 2018). This study also highlighted the potential of developmental atlases to rapidly accelerate our understanding of developmental processes in emergent model organisms.
The construction of developmental atlases may also be applicable to questions in evolutionary developmental biology. In a comparison of developmental trajectories from zebrafish and Xenopus, lineage topologies were found to be broadly conserved between species, while the observed abundances of different cell types were markedly different between species (Briggs et al., 2018). Furthermore, a recent study of developmental trajectories in C. intestinalis provided new insight into the origins of the vertebrate telencephalon (C. Cao et al., 2019). As more developmental atlases become available, more cross‐species comparisons can be made. These studies are undoubtedly just the first of a new wave of literature that will give significant insights into the transcriptomic dynamics of embryonic development, and will provide a foundation for future studies of enhancer function in development, regeneration, and disease.
6.5. Future directions in single‐cell studies of developing embryos
Several groups have demonstrated that these single‐cell methods can be performed in an efficient and cost‐effective manner, facilitating their application to the study of diverse developmental contexts. A current technical limitation is the inability to capture all of the different transcripts from a single cell, meaning that some transcripts will inevitably be undetected (J. Cao et al., 2017). However, developmental atlases of mid‐gestational mouse embryos revealed novel notochord markers (J. Cao et al., 2019), indicating that this lack of depth does not preclude new biological insights. Furthermore, rare cell types including germ cells have reproducibly been identified by scRNA‐seq of whole embryos (Briggs et al., 2018; J. Cao et al., 2019; Farrell et al., 2018; Pijuan‐Sala et al., 2019; Wagner et al., 2018), demonstrating that these methods can provide fine detail of single‐cell transcriptomes.
In future experiments, scRNA‐seq could be combined with single‐cell ATAC‐seq or Hi‐C to investigate the transcriptomic and chromatin landscapes of whole embryos. Although these combined approaches would not directly test enhancer function, they could identify putative developmental enhancers and provide insight into cis‐regulatory dynamics in developing embryos. Further functional insight could come from scRNA‐seq of embryos bearing mutations in enhancer sequences. Because spatial information is lost when embryos are dissociated, construction of developmental atlases currently requires human annotation based on known marker genes. While human annotation can be semi‐automated and has been useful for validating these methodologies, in the future this limitation could be overcome by methods that couple spatial and transcriptomic data. For example, multiplexed fluorescence in situ hybridization techniques allow for the detection of thousands of RNA species to be imaged in single cells in tissue sections (K. H. Chen, Boettiger, Moffitt, Wang, & Zhuang, 2015; Lubeck & Cai, 2012; Lubeck, Coskun, Zhiyentayev, Ahmad, & Cai, 2014; Moffitt et al., 2018). One of these methods, termed multiplexed error‐robust FISH (MERFISH), has been combined with scRNA‐seq to generate a comprehensive map of the hypothalamic preoptic region in mouse (Moffitt et al., 2018), an approach that could easily be extended to the study of whole embryos. Additionally, spatial transcriptomic methods have recently been developed that enable in situ barcoding and cDNA synthesis, such that high‐quality RNA‐seq data can be generated with retained spatial information (Ståhl et al., 2016). These methods will advance our ability to construct accurate and high‐resolution atlases of embryonic development. Looking ahead, we expect that advances in single‐cell transcriptomics approaches will have a substantial impact on our understanding of how enhancer sequences affect cell type‐specific gene expression in developing embryos.
7. CONCLUSION
Since the discovery of enhancers over 30 years ago, technological advancements have greatly improved our ability to identify and study enhancers. Advances in high‐throughput sequencing, oligonucleotide synthesis, and genome editing technology have enabled new methodologies that have been critical in elucidating how enhancer sequences relate to function. The development of genome‐wide assays has enabled the identification of millions of putative enhancer sequences that are associated with particular chromatin signatures. Genome‐wide assays have also led to an increased interest in understanding enhancer function because it is now clear that the majority of disease‐causing genetic variants reside within putative enhancer sequences.
Bottlenecks in our ability to functionally validate candidate enhancer sequences are starting to be overcome by MPRAs, although these are most valuable when done in a context‐specific way. Advantageously, MPRAs can be performed in whole embryos to give direct insight into tissue‐specific enhancer function during development. CRISPR screens are also useful because they allow enhancers to be identified and studied within the native genomic context, but such screens have not yet been done in developing embryos. The major challenge will be designing these in a way that is informative despite significant enhancer redundancy.
Similarly, a recent wave of developmental atlases has given unprecedented insight into the transcriptomes of whole developing organisms with single‐cell resolution, but these methods are primarily descriptive in their present state. Given the current limitations of these methods, animal models are still important for their direct relevance to development and have proven to be a valuable tool with which to correct misconceptions that have arisen from genome‐wide data. Moving forward, technological advances in high‐throughput CRISPR screens and single‐cell transcriptomic analyses of developing embryos will likely allow for the study of enhancer function in developmental contexts. Together with MPRAs, these approaches will allow us to gain a broad understanding of which enhancer sequences are functional and important for successful development.
CONFLICT OF INTEREST
The authors have declared no conflicts of interest for this article.
AUTHOR CONTRIBUTIONS
Genevieve E. Ryan: Conceptualization; writing‐original draft; writing‐review and editing. Emma K. Farley: Conceptualization; writing‐original draft; writing‐review and editing.
RELATED WIREs ARTICLE
ACKNOWLEDGMENTS
We thank members of the Farley lab for helpful discussions.
Ryan GE, Farley EK. Functional genomic approaches to elucidate the role of enhancers during development. WIREs Syst Biol Med. 2020;12:e1467 10.1002/wsbm.1467
Funding information National Heart, Lung, and Blood Institute, Grant/Award Number: T32HL007444; National Human Genome Research Institute, Grant/Award Number: DP2HG010013
REFERENCES
- Adey, A. , Morrison, H. G. , Asan, Xun, X. , Kitzman, J. O. , Turner, E. H. , … Shendure, J. (2010). Rapid, low‐input, low‐bias construction of shotgun fragment libraries by high‐density in vitro transposition. Genome Biology, 11(12), R119 10.1186/gb-2010-11-12-r119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afek, A. , Schipper, J. L. , Horton, J. , Gordân, R. , & Lukatsky, D. B. (2014). Protein−DNA binding in the absence of specific base‐pair recognition. Proceedings of the National Academy of Sciences, 111(48), 17140–17145. 10.1073/pnas.1410569111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano, T. , Sagai, T. , Tanabe, H. , Mizushina, Y. , Nakazawa, H. , & Shiroishi, T. (2009). Chromosomal dynamics at the Shh locus: Limb bud‐specific differential regulation of competence and active transcription. Developmental Cell, 16(1), 47–57. 10.1016/j.devcel.2008.11.011 [DOI] [PubMed] [Google Scholar]
- Arnold, C. D. , Gerlach, D. , Stelzer, C. , Boryń, Ł. M. , Rath, M. , & Stark, A. (2013). Genome‐wide quantitative enhancer activity maps identified by STARR‐seq. Science, 339(6123), 1074–1077. 10.1126/science.1232542 [DOI] [PubMed] [Google Scholar]
- Banani, S. F. , Lee, H. O. , Hyman, A. A. , & Rosen, M. K. (2017). Biomolecular condensates: Organizers of cellular biochemistry. Nature Reviews Molecular Cell Biology, 18(5), 285–298. 10.1038/nrm.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji, J. , Rusconi, S. , & Schaffner, W. (1981). Expression of a β‐globin gene is enhanced by remote SV40 DNA sequences. Cell, 27, 299–308. 10.1016/0092-8674(81)90413-X [DOI] [PubMed] [Google Scholar]
- Barski, A. , Cuddapah, S. , Cui, K. , Roh, T. Y. , Schones, D. E. , Wang, Z. , … Zhao, K. (2007). High‐resolution profiling of histone methylations in the human genome. Cell, 129, 823–837. 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- Beagrie, R. A. , Scialdone, A. , Schueler, M. , Kraemer, D. C. A. , Chotalia, M. , Xie, S. Q. , … Pombo, A. (2017). Complex multi‐enhancer contacts captured by genome architecture mapping. Nature, 543(7646), 519–524. 10.1038/nature21411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht, E. , McInnes, L. , Healy, J. , Dutertre, C. A. , Kwok, I. W. H. , Ng, L. G. , … Newell, E. W. (2019). Dimensionality reduction for visualizing single‐cell data using UMAP. Nature Biotechnology, 37(1), 38–47. 10.1038/nbt.4314 [DOI] [PubMed] [Google Scholar]
- Beerli, R. R. , Dreier, B. , & Barbas, C. F. (2000). Positive and negative regulation of endogenous genes by designed transcription factors. Proceedings of the National Academy of Sciences, 97(4), 1495–1500. 10.1073/pnas.040552697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benabdallah, N. S. , Williamson, I. , Illingworth, R. S. , Boyle, S. , Grimes, G. R. , Therizols, P. , & Bickmore, W. (2017). PARP mediated chromatin unfolding is coupled to long‐range enhancer activation. BioRxiv. 10.1101/155325 [DOI]
- Bernstein, B. E. , Mikkelsen, T. S. , Xie, X. , Kamal, M. , Huebert, D. J. , Cuff, J. , … Lander, E. S. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell, 125, 315–326. 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- Bertrand, V. , Hudson, C. , Caillol, D. , Popovici, C. , & Lemaire, P. (2003). Neural tissue in ascidian embryos is induced by FGF9/16/20, acting via a combination of maternal GATA and Ets transcription factors. Cell, 115(5), 615–627. 10.1016/S0092-8674(03)00928-0 [DOI] [PubMed] [Google Scholar]
- Birnbaum, R. Y. , Patwardhan, R. P. , Kim, M. J. , Findlay, G. M. , Martin, B. , Zhao, J. , … Ahituv, N. (2014). Systematic dissection of coding exons at single nucleotide resolution supports an additional role in cell‐specific transcriptional regulation. PLoS Genetics, 10(10), e1004592 10.1371/journal.pgen.1004592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinka, S. , Reimer, M. H. , Pulakanti, K. , & Rao, S. (2016). Super‐enhancers at the Nanog locus differentially regulate neighboring pluripotency‐associated genes. Cell Reports, 17, 19–28. 10.1016/j.celrep.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow, M. J. , McCulley, D. J. , Li, Z. , Zhang, T. , Akiyama, J. A. , Holt, A. , … Pennacchio, L. A. (2010). ChIP‐seq identification of weakly conserved heart enhancers. Nature Genetics, 42(9), 806–810. 10.1038/ng.650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija, A. , Klein, I. A. , Sabari, B. R. , Dall'Agnese, A. , Coffey, E. L. , Zamudio, A. V. , … Young, R. A. (2018). Transcription factors activate genes through the phase‐separation capacity of their activation domains. Cell, 175(7), 1842–1855.e16. 10.1016/j.cell.2018.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn, S. , Zinzen, R. P. , Girardot, C. , Gustafson, E. H. , Perez‐Gonzalez, A. , Delhomme, N. , … Furlong, E. E. M. (2012). Tissue‐specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nature Genetics, 44, 148–156. 10.1038/ng.1064 [DOI] [PubMed] [Google Scholar]
- Boyle, A. P. , Davis, S. , Shulha, H. P. , Meltzer, P. , Margulies, E. H. , Weng, Z. , … Crawford, G. E. (2008). High‐resolution mapping and characterization of open chromatin across the genome. Cell, 132(2), 311–322. 10.1016/j.cell.2007.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs, J. A. , Weinreb, C. , Wagner, D. E. , Megason, S. , Peshkin, L. , Kirschner, M. W. , & Klein, A. M. (2018). The dynamics of gene expression in vertebrate embryogenesis at single‐cell resolution. Science, 360(6392), eaar5780 10.1126/science.aar5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck, F. , Hoffmann, M. D. , Harteveld, Z. , Aschenbrenner, S. , Bietz, A. , Waldhauer, M. C. , … Niopek, D. (2018). Engineered anti‐CRISPR proteins for optogenetic control of CRISPR‐Cas9. Nature Methods, 15(11), 924–927. 10.1038/s41592-018-0178-9 [DOI] [PubMed] [Google Scholar]
- Buenrostro, J. D. , Giresi, P. G. , Zaba, L. C. , Chang, H. Y. , & Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nature Methods, 10(12), 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro, J. D. , Wu, B. , Litzenburger, U. M. , Ruff, D. , Gonzales, M. L. , Snyder, M. P. , … Greenleaf, W. J. (2015). Single‐cell chromatin accessibility reveals principles of regulatory variation. Nature, 523(7561), 486–490. 10.1038/nature14590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgio, G. (2018). Redefining mouse transgenesis with CRISPR/Cas9 genome editing technology. Genome Biology, 19(1), 18–20. 10.1186/s13059-018-1409-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun, V. C. , Stathopoulos, A. , & Levine, M. (2002). Promoter‐proximal tethering elements regulate enhancer‐promoter specificity in the Drosophila Antennapedia complex. Proceedings of the National Academy of Sciences of the United States of America, 99(14), 9243–9247. 10.1073/pnas.142291299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver, M. C. , Smith, E. C. , Sher, F. , Pinello, L. , Sanjana, N. E. , Shalem, O. , … Bauer, D. E. (2015). BCL11A enhancer dissection by Cas9‐mediated in situ saturating mutagenesis. Nature, 527(7577), 192–197. 10.1038/nature15521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, C. , Lemaire, L. A. , Wang, W. , Yoon, P. H. , Choi, Y. A. , Parsons, L. R. , … Chen, K. (2019). Comprehensive single‐cell transcriptome lineages of a proto‐vertebrate. Nature, 571, 349–354. 10.1038/s41586-019-1385-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, J. , Packer, J. S. , Ramani, V. , Cusanovich, D. A. , Huynh, C. , Daza, R. , … Shendure, J. (2017). Comprehensive single‐cell transcriptional profiling of a multicellular organism. Science, 357, 661 10.1126/science.aam8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, J. , Spielmann, M. , Qiu, X. , Huang, X. , Ibrahim, D. M. , Hill, A. J. , … Shendure, J. (2019). The single‐cell transcriptional landscape of mammalian organogenesis. Nature, 566, 496–502. 10.1038/s41586-019-0969-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, D. , Chakalova, L. , Osborne, C. S. , Dai, Y. f. , & Fraser, P. (2002). Long‐range chromatin regulatory interactions in vivo. Nature Genetics, 32, 623–626. 10.1038/ng1051 [DOI] [PubMed] [Google Scholar]
- Chen, K. H. , Boettiger, A. N. , Moffitt, J. R. , Wang, S. , & Zhuang, X. (2015). Spatially resolved, highly multiplexed RNA profiling in single cells. Science, 348(6233), 1360–1363. 10.1126/science.aaa6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Sanjana, N. E. , Zheng, K. , Shalem, O. , Lee, K. , Shi, X. , … Sharp, P. A. (2015). Genome‐wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell, 160(6), 1246–1260. 10.1016/j.cell.2015.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, W. K. , Spille, J. H. , Hecht, M. , Lee, C. , Li, C. , Grube, V. , & Cisse, I. I. (2018). Mediator and RNA polymerase II clusters associate in transcription‐dependent condensates. Science, 361(6400), 412–415. 10.1126/science.aar4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, S. , Dugast‐Darzacq, C. , Liu, Z. , Dong, P. , Dailey, G. M. , Cattoglio, C. , … Tjian, R. (2018). Imaging dynamic and selective low‐complexity domain interactions that control gene transcription. Science, 361(6400), eaar2555 10.1126/science.aar2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L. , Ran, F. A. , Cox, D. , Lin, S. , Barretto, R. , Habib, N. , … Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science, 339(6121), 819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin, O. , & Scacheri, P. C. (2014). Enhancer variants: Evaluating functions in common disease. Genome Medicine, 6(10), 85 10.1186/s13073-014-0085-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton, M. P. , Jaenisch, R. , Young, R. A. , Kooistra, T. , Lodato, M. A. , Carey, B. W. , … Hanna, J. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences, 107(50), 21931–21936. 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker, J. , Abe, N. , Rinaldi, L. , McGregor, A. P. , Frankel, N. , Wang, S. , … Stern, D. L. (2015). Low affinity binding site clusters confer HOX specificity and regulatory robustness. Cell, 160, 191–203. 10.1016/j.cell.2014.11.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker, J. , Preger‐Ben Noon, E. , & Stern, D. L. (2016). The soft touch: Low‐affinity transcription factor binding sites in development and evolution. Current Topics in Developmental Biology, 117, 455–469. 10.1016/bs.ctdb.2015.11.018 [DOI] [PubMed] [Google Scholar]
- Deans, R. M. , Morgens, D. W. , Ökesli, A. , Pillay, S. , Horlbeck, M. A. , Kampmann, M. , … Bassik, M. C. (2016). Parallel shRNA and CRISPR‐Cas9 screens enable antiviral drug target identification. Nature Chemical Biology, 12(5), 361–366. 10.1038/nchembio.2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker, J. , Rippe, K. , Dekker, M. , & Kleckner, N. (2002). Capturing chromosome conformation. Science, 295(5558), 1306–1311. 10.1126/science.1067799 [DOI] [PubMed] [Google Scholar]
- Delsuc, F. , Brinkmann, H. , Chourrout, D. , & Philippe, H. (2006). Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature, 439, 965–968. 10.1038/nature04336 [DOI] [PubMed] [Google Scholar]
- Despang, A. , Schöpflin, R. , Franke, M. , Ali, S. , Jerković, I. , Paliou, C. , … Ibrahim, D. M. (2019). Functional dissection of the Sox9‐Kcnj2 locus identifies nonessential and instructive roles of TAD architecture. Nature Genetics, 51, 1263–1271. 10.1038/s41588-019-0466-z [DOI] [PubMed] [Google Scholar]
- DeWitt, W. S. , Shendure, J. , Steemers, F. J. , Regalado, S. G. , Berletch, J. B. , Trapnell, C. , … Cusanovich, D. A. (2018). A single‐cell atlas of in vivo mammalian chromatin accessibility. Cell, 174(5), 1309–1324.e18. 10.1016/j.cell.2018.06.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit, A. , Parnas, O. , Li, B. , Chen, J. , Fulco, C. P. , Jerby‐Arnon, L. , … Regev, A. (2016). Perturb‐Seq: Dissecting molecular circuits with scalable single‐cell RNA profiling of pooled genetic screens. Cell, 167(7), 1853–1866. 10.1016/j.cell.2016.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, J. R. , Selvaraj, S. , Yue, F. , Kim, A. , Li, Y. , Shen, Y. , … Ren, B. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485(7398), 376–380. 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett, D. , Gause, M. , Piunti, A. , Takahashi, Y. , Wang, L. , Shilatifard, A. , … Bartom, E. T. (2017). Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS‐like proteins at enhancers is dispensable for development and viability. Nature Genetics, 49(11), 1647–1653. 10.1038/ng.3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, B. , Fudenberg, G. , Imakaev, M. , & Mirny, L. A. (2014). Chromatin loops as allosteric modulators of enhancer‐promoter interactions. PLoS Computational Biology, 10(10), e1003867 10.1371/journal.pcbi.1003867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham, I. , Kundaje, A. , Aldred, S. F. , Collins, P. J. , Davis, C. A. , Doyle, F. , … Lochovsky, L. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erceg, J. , Saunders, T. E. , Girardot, C. , Devos, D. P. , Hufnagel, L. , & Furlong, E. E. M. (2014). Subtle changes in motif positioning cause tissue‐specific effects on robustness of an enhancer's activity. PLoS Genetics, 10(1), e1004060 10.1371/journal.pgen.1004060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst, J. , Kheradpour, P. , Mikkelsen, T. S. , Shoresh, N. , Ward, L. D. , Epstein, C. B. , … Bernstein, B. E. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature, 473(7345), 43–49. 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley, E. K. , Levine, M. S. , Olson, K. M. , Zhang, W. , & Rokhsar, D. S. (2016). Syntax compensates for poor binding sites to encode tissue specificity of developmental enhancers. Proceedings of the National Academy of Sciences, 113(23), 6508–6513. 10.1073/pnas.1605085113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley, E. K. , Olson, K. M. , & Levine, M. S. (2016). Regulatory principles governing tissue specificity of developmental enhancers. Cold Spring Harbor Symposia on Quantitative Biology, 80, 27–32. 10.1101/sqb.2015.80.027227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley, E. K. , Olson, K. M. , Zhang, W. , Brandt, A. J. , Rokhsar, D. S. , & Levine, M. S. (2015). Suboptimization of developmental enhancers. Science, 350, 325–328. 10.1126/science.aac6948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell, J. A. , Wang, Y. , Riesenfeld, S. J. , Shekhar, K. , Regev, A. , & Schier, A. F. (2018). Single‐cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science, 360(6392), eaar3131 10.1126/science.aar3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke, M. , Ibrahim, D. M. , Andrey, G. , Schwarzer, W. , Heinrich, V. , Schöpflin, R. , … Mundlos, S. (2016). Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature, 538(7624), 265–269. 10.1038/nature19800 [DOI] [PubMed] [Google Scholar]
- Friedland, A. E. , Tzur, Y. B. , Esvelt, K. M. , Colaiácovo, M. P. , Church, G. M. , & Calarco, J. A. (2013). Heritable genome editing in C. elegans via a CRISPR‐Cas9 system. Nature Methods, 10(8), 741–743. 10.1038/nmeth.2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco, C. P. , Munschauer, M. , Anyoha, R. , Munson, G. , Grossman, S. R. , Perez, E. M. , … Engreitz, J. M. (2016). Systematic mapping of functional enhancer‐promoter connections with CRISPR interference. Science, 354(6313), 769–773. 10.1126/science.aag2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood, M. J. , Liu, M. H. , Pan, Y. F. , Liu, J. , Xu, H. , Mohamed, Y. B. , … Ruan, Y. (2009). An oestrogen‐receptor‐α‐bound human chromatin interactome. Nature, 462(7269), 58–64. 10.1038/nature08497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetta, M. C. , & Furlong, E. E. M. (2018). The insulator protein CTCF is required for correct Hox gene expression, but not for embryonic development in Drosophila . Genetics, 210(1), 129–136. 10.1534/genetics.118.301350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini, M. , Findlay, G. M. , McKenna, A. , Milbank, J. H. , Lee, C. , Zhang, M. D. , … Shendure, J. (2017). CRISPR/Cas9‐mediated scanning for regulatory elements required for HPRT1 expression via thousands of large programmed genomic deletions. American Journal of Human Genetics, 101(2), 192–205. 10.1016/j.ajhg.2017.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini, M. , Hill, A. J. , McFaline‐Figueroa, J. L. , Martin, B. , Kim, S. , Zhang, M. D. , … Shendure, J. (2019). A genome‐wide framework for mapping gene regulation via cellular genetic screens. Cell, 176(1–2), 377–390.e19. 10.1016/j.cell.2018.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman, R. C. , Parker, M. H. , Sanchez, G. J. , Yao, Z. , Davison, J. , MacQuarrie, K. L. , … Sarkar, D. (2010). Genome‐wide MyoD binding in skeletal muscle cells: A potential for broad cellular reprogramming. Developmental Cell, 18(4), 662–674. 10.1016/j.devcel.2010.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavi‐Helm, Y. , Jankowski, A. , Meiers, S. , Viales, R. R. , Korbel, J. O. , & Furlong, E. E. M. (2019). Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nature Genetics, 51, 1272–1282. 10.1038/s41588-019-0462-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirlando, R. , & Felsenfeld, G. (2016). CTCF: Making the right connections. Genes and Development, 30(8), 881–891. 10.1101/gad.277863.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, L. A. , Horlbeck, M. A. , Adamson, B. , Villalta, J. E. , Chen, Y. , Whitehead, E. H. , … Weissman, J. S. (2014). Genome‐scale CRISPR‐mediated control of gene repression and activation. Cell, 159(3), 647–661. 10.1016/j.cell.2014.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, L. A. , Larson, M. H. , Morsut, L. , Liu, Z. , Brar, G. A. , Torres, S. E. , … Qi, L. S. (2013). CRISPR‐mediated modular RNA‐guided regulation of transcription in eukaryotes. Cell, 154(2), 442–451. 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisselbrecht, S. S. , Barrera, L. A. , Porsch, M. , Aboukhalil, A. , Estep, P. W. , Vedenko, A. , … Bulyk, M. L. (2013). Highly parallel assays of tissue‐specific enhancers in whole Drosophila embryos. Nature Methods, 10(8), 774–780. 10.1038/nmeth.2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes, A. L. C. , Abeel, T. , Peterson, M. , Azizi, E. , Lyubetskaya, A. , Carvalho, L. , & Galagan, J. (2014). Decoding ChIP‐seq with a double‐binding signal refines binding peaks to single‐nucleotides and predicts cooperative interaction. Genome Research, 24, 1686–1697. 10.1101/gr.161711.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryshin, I. Y. , & Reznikoff, W. S. (1998). Tn5 in vitro transposition. Journal of Biological Chemistry, 273(13), 7367–7374. 10.1074/jbc.273.13.7367 [DOI] [PubMed] [Google Scholar]
- Gray, S. , & Levine, M. (1996). Transcriptional repression in development. Current Opinion in Cell Biology, 8(3), 358–364. 10.1016/S0955-0674(96)80010-X [DOI] [PubMed] [Google Scholar]
- Groner, A. C. , Meylan, S. , Ciuffi, A. , Zangger, N. , Ambrosini, G. , Dénervaud, N. , … Trono, D. (2010). KRAB‐zinc finger proteins and KAP1 can mediate long‐range transcriptional repression through heterochromatin spreading. PLoS Genetics, 6(3), e1000869 10.1371/journal.pgen.1000869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosveld, F. , van Assendelft, G. B. , Greaves, D. R. , & Kollias, G. (1987). Position‐independent, high‐level expression of the human β‐globin gene in transgenic mice. Cell, 51(6), 975–985. 10.1016/0092-8674(87)90584-8 [DOI] [PubMed] [Google Scholar]
- Grunwald, H. A. , Gantz, V. M. , Poplawski, G. , Xu, X. S. , Bier, E. , & Cooper, K. L. (2019). Super‐Mendelian inheritance mediated by CRISPR–Cas9 in the female mouse germline. Nature, 566(7742), 105–109. 10.1038/s41586-019-0875-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagman, J. , Mansson, R. , Dutkowski, J. , Jhunjhunwala, S. , Benner, C. , Murre, C. , … Lin, Y. C. (2010). A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nature Immunology, 11(7), 635–643. 10.1038/ni.1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainer, S. J. , Bošković, A. , McCannell, K. N. , Rando, O. J. , & Fazzio, T. G. (2019). Profiling of pluripotency factors in single cells and early embryos. Cell, 177(5), 1319–1329. 10.1016/j.cell.2019.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfon, M. S. , Carmena, A. , Gisselbrecht, S. , Sackerson, C. M. , Jiménez, F. , Baylies, M. K. , & Michelson, A. M. (2000). Ras pathway specificity is determined by the integration of multiple signal‐activated and tissue‐restricted transcription factors. Cell, 103(1), 63–74. 10.1016/S0092-8674(00)00105-7 [DOI] [PubMed] [Google Scholar]
- Hammar, P. , Leroy, P. , Mahmutovic, A. , Marklund, E. G. , Berg, O. G. , & Elf, J. (2012). The lac repressor displays facilitated diffusion in living cells. Science, 336(6088), 1595–1598. 10.1126/science.1221648 [DOI] [PubMed] [Google Scholar]
- Hay, D. , Hughes, J. R. , Babbs, C. , Davies, J. O. J. , Graham, B. J. , Hanssen, L. L. P. , … Higgs, D. R. (2016). Genetic dissection of the α‐globin super‐enhancer in vivo. Nature Genetics, 48(8), 895–903. 10.1038/ng.3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman, N. D. , Hon, G. C. , Hawkins, R. D. , Kheradpour, P. , Stark, A. , Harp, L. F. , … Ren, B. (2009). Histone modifications at human enhancers reflect global cell‐type‐specific gene expression. Nature, 459(7243), 108–112. 10.1038/nature07829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman, N. D. , Stuart, R. K. , Hon, G. , Fu, Y. , Ching, C. W. , Hawkins, R. D. , … Ren, B. (2007). Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genetics, 39(3), 311–318. 10.1038/ng1966 [DOI] [PubMed] [Google Scholar]
- Herz, H. M. , Mohan, M. , Garruss, A. S. , Liang, K. , Takahashi, Y. H. , Mickey, K. , … Shilatifard, A. (2012). Enhancer‐associated H3K4 monomethylation by trithorax‐related, the Drosophila homolog of mammalian MLL3/MLL4. Genes and Development, 26(23), 2604–2620. 10.1101/gad.201327.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz, D. , Abraham, B. J. , Lee, T. I. , Lau, A. , Saint‐André, V. , Sigova, A. A. , … Young, R. A. (2013). Super‐enhancers in the control of cell identity and disease. Cell, 155(4), 934 10.1016/j.cell.2013.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz, D. , Shrinivas, K. , Young, R. A. , Chakraborty, A. K. , & Sharp, P. A. (2017). A phase separation model for transcriptional control. Cell, 169(1), 13–23. 10.1016/j.cell.2017.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck, M. A. , Witkowsky, L. B. , Guglielmi, B. , Replogle, J. M. , Gilbert, L. A. , Villalta, J. E. , … Weissman, J. S. (2016). Nucleosomes impede cas9 access to DNA in vivo and in vitro. eLife, 5, e12677 10.7554/eLife.12677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa, H. , Ungerbäck, J. , Wang, X. , Matsumoto, M. , Nakayama, K. I. , Cohen, S. M. , … Rothenberg, E. V. (2018). Transcription factor PU.1 represses and activates gene expression in early T cells by redirecting partner transcription factor binding. Immunity, 48(6), 1119–1134. 10.1016/j.immuni.2018.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert, R. , Thurman, R. E. , Giste, E. , Lee, B.‐K. , Ebersol, A. K. , Furey, T. S. , … Dekker, J. (2012). The accessible chromatin landscape of the human genome. Nature, 489(7414), 75–82. 10.1038/nature11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ing‐Simmons, E. , Seitan, V. C. , Faure, A. J. , Flicek, P. , Carroll, T. , Dekker, J. , … Merkenschlager, M. (2015). Spatial enhancer clustering and regulation of enhancer‐proximal genes by cohesin. Genome Research, 25, 504–513. 10.1101/gr.184986.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin, D. A. , Weiner, A. , Yofe, I. , Lara‐Astiaso, D. , Keren‐Shaul, H. , David, E. , … Amit, I. (2016). Dissecting immune circuits by linking CRISPR‐pooled screens with single‐cell RNA‐Seq. Cell, 167(7), 1883–1896. 10.1016/j.cell.2016.11.039 [DOI] [PubMed] [Google Scholar]
- Ji, Z. , Zhou, W. , & Ji, H. (2017). Single‐cell regulome data analysis by SCRAT. Bioinformatics, 33(18), 2930–2932. 10.1093/bioinformatics/btx315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, C. , Zang, C. , Wei, G. , Cui, K. , Peng, W. , Zhao, K. , & Felsenfeld, G. (2009). H3.3/H2A.Z double variant‐containing nucleosomes mark “nucleosome‐free regions” of active promoters and other regulatory regions. Nature Genetics, 41(8), 941–945. 10.1038/ng.409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, W. , Tang, Q. , Wan, M. , Cui, K. , Zhang, Y. , Ren, G. , … Zhao, K. (2015). Genome‐wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature, 528, 142–146. 10.1038/nature15740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek, M. , Chylinski, K. , Fonfara, I. , Hauer, M. , Doudna, J. A. , & Charpentier, E. (2012). A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096), 816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodkowska, K. , Pancaldi, V. , Almeida, R. , Rigau, M. , Pisano, D. , Valencia, A. , … Genomics, S. (2019). Three‐dimensional connectivity and chromatin environment mediate the activation efficiency of mammalian DNA replication origins. BioRxiv.
- Johnson, D. S. , Mortazavi, A. , Myers, R. M. , & Wold, B. (2007). Genome‐wide mapping of in vivo protein–DNA interactions. Science, 316(5830), 1497–1502. 10.1126/science.1141319 [DOI] [PubMed] [Google Scholar]
- Jung, Y. L. , Luquette, L. J. , Ho, J. W. K. , Ferrari, F. , Tolstorukov, M. , Minoda, A. , … Park, P. J. (2014). Impact of sequencing depth in ChIP‐seq experiments. Nucleic Acids Research, 42(9), e74 10.1093/nar/gku178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey, M. H. , Newman, J. J. , Bilodeau, S. , Zhan, Y. , Orlando, D. A. , Van Berkum, N. L. , … Young, R. A. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature, 467(7314), 430–435. 10.1038/nature09380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaiskos, N. , Wahle, P. , Alles, J. , Boltengagen, A. , Ayoub, S. , Kipar, C. , … Zinzen, R. P. (2017). The Drosophila embryo at single‐cell transcriptome resolution. Science, 358(6360), 194–199. 10.1126/science.aan3235 [DOI] [PubMed] [Google Scholar]
- Kaya‐okur, H. S. , Wu, S. J. , Codomo, C. A. , Pledger, E. S. , Bryson, T. D. , Henikoff, J. G. , … Henikoff, S. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communications, 10(1930), 1–10. 10.1038/s41467-019-09982-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko, P. V. , Alekseyenko, A. A. , Schwartz, Y. B. , Minoda, A. , Riddle, N. C. , Ernst, J. , … Park, P. J. (2011). Comprehensive analysis of the chromatin landscape in Drosophila melanogaster . Nature, 471(7339), 480–485. 10.1038/nature09725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour, P. , Ernst, J. , Melnikov, A. , Rogov, P. , Wang, L. , Zhang, X. , … Kellis, M. (2013). Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Research, 23, 800–811. 10.1101/gr.144899.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselev, V. Y. , Andrews, T. S. , & Hemberg, M. (2019). Challenges in unsupervised clustering of single‐cell RNA‐seq data. Nature Reviews Genetics, 20(5), 273–282. 10.1038/s41576-018-0088-9 [DOI] [PubMed] [Google Scholar]
- Kolodziejczyk, A. A. , Kim, J. K. , Svensson, V. , Marioni, J. C. , & Teichmann, S. A. (2015). The technology and biology of single‐cell RNA sequencing. Molecular Cell, 58(4), 610–620. 10.1016/j.molcel.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Korkmaz, G. , Lopes, R. , Ugalde, A. P. , Nevedomskaya, E. , Han, R. , Myacheva, K. , … Agami, R. (2016). Functional genetic screens for enhancer elements in the human genome using CRISPR‐Cas9. Nature Biotechnology, 34(2), 192–198. 10.1038/nbt.3450 [DOI] [PubMed] [Google Scholar]
- Kramer, N. J. , Haney, M. S. , Morgens, D. W. , Jovičić, A. , Couthouis, J. , Li, A. , … Gitler, A. D. (2018). CRISPR‐Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide‐repeat‐protein toxicity. Nature Genetics, 50(4), 603–612. 10.1038/s41588-018-0070-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo, N. , Ishii, H. , Gorkin, D. , Meitinger, F. , Xiong, X. , Fang, R. , … Ren, B. (2017). Preservation of chromatin organization after acute loss of CTCF in mouse embryonic stem cells. BioRxiv. 10.1101/118737 [DOI]
- Kurum, E. , Benayoun, B. A. , Malhotra, A. , George, J. , & Ucar, D. (2016). Computational inference of a genomic pluripotency signature in human and mouse stem cells. Biology Direct, 11, 47 10.1186/s13062-016-0148-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvon, E. Z. , Stampfel, G. , Omar Yáññez‐Cuna, J. , Dickson, B. J. , & Stark, A. (2012). HOT regions function as patterned developmental enhancers and have a distinct cis‐regulatory signature. Genes and Development, 26(9), 908–913. 10.1101/gad.188052.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski, J. C. , Fiore, C. , Chaudhari, H. G. , & Cohen, B. A. (2014). High‐throughput functional testing of ENCODE segmentation predictions. Genome Research, 24, 1595–1602. 10.1101/gr.173518.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, B. , Gao, W. , Cui, K. , Xie, W. , Tang, Q. , Jin, W. , … Zhao, K. (2018). Principles of nucleosome organization revealed by single‐cell micrococcal nuclease sequencing. Nature, 562, 281–285. 10.1038/s41586-018-0567-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt, S. G. , Marinov, G. K. , Kundaje, A. , Kheradpour, P. , Pauli, F. , Batzoglou, S. , … Snyder, M. (2012). ChIP‐seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research, 22(9), 1813–1831. 10.1101/gr.136184.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau, C. A. , Ulirsch, J. C. , Bao, E. L. , Ludwig, L. S. , Finucane, H. K. , Aryee, M. J. , … Sankaran, V. G. (2018). Interrogation of human hematopoiesis at single‐cell and single‐variant resolution. BioRxiv. 10.1101/255224 [DOI] [PMC free article] [PubMed]
- LeProust, E. M. , Peck, B. J. , Spirin, K. , McCuen, H. B. , Moore, B. , Namsaraev, E. , & Caruthers, M. H. (2010). Synthesis of high‐quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Research, 38(8), 2522–2540. 10.1093/nar/gkq163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettice, L. A. , Heaney, S. J. H. , Purdie, L. A. , Li, L. , de Beer, P. , Oostra, B. A. , … de Graaff, E. (2003). A long‐range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Human Molecular Genetics, 12(14), 1725–1735. 10.1093/hmg/ddg180 [DOI] [PubMed] [Google Scholar]
- Levine, M. (2010). Transcriptional enhancers in animal development and evolution. Current Biology, 20(17), R754–R763. 10.1016/j.cub.2010.06.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. , Ruan, X. , Auerbach, R. K. , Sandhu, K. S. , Zheng, M. , Wang, P. , … Ruan, Y. (2012). Extensive promoter‐centered chromatin interactions provide a topological basis for transcription regulation. Cell, 148(1), 84–98. 10.1016/j.cell.2011.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Peterson, K. R. , Fang, X. , & Stamatoyannopoulos, G. (2002). Locus control regions. Blood, 100(9), 3077–3086. 10.1182/blood-2002-04-1104.BLOOD [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. Y. , MacArthur, S. , Bourgon, R. , Nix, D. , Pollard, D. A. , Iyer, V. N. , … Biggin, M. D. (2008). Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biology, 6(7), e190 10.1371/journal.pbio.0060027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman‐Aiden, E. , Van Berkum, N. L. , Williams, L. , Imakaev, M. , Ragoczy, T. , Telling, A. , … Dekker, J. (2009). Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science, 326(5950), 289–293. 10.1126/science.1181369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén, J. , Hoke, H. A. , Lin, C. Y. , Lau, A. , Orlando, D. A. , Vakoc, C. R. , … Young, R. A. (2013). Selective inhibition of tumor oncogenes by disruption of super‐enhancers. Cell, 153(2), 320–334. 10.1016/j.cell.2013.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck, E. , & Cai, L. (2012). Single‐cell systems biology by super‐resolution imaging and combinatorial labeling. Nature Methods, 9(7), 743–748. 10.1038/nmeth.2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubeck, E. , Coskun, A. F. , Zhiyentayev, T. , Ahmad, M. , & Cai, L. (2014). Single‐cell in situ RNA profiling by sequential hybridization. Nature Methods, 11(4), 360–361. 10.1038/nmeth.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez, D. G. , Kraft, K. , Heinrich, V. , Krawitz, P. , Brancati, F. , Klopocki, E. , … Mundlos, S. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene‐enhancer interactions. Cell, 161(5), 1012–1025. 10.1016/j.cell.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur, S. , Pfeiffer, B. D. , Eisen, M. B. , Stamatoyannopoulos, J. A. , Bickel, P. J. , Li, J. J. , … Celniker, S. E. (2012). DNA regions bound at low occupancy by transcription factors do not drive patterned reporter gene expression in Drosophila . Proceedings of the National Academy of Sciences, 109(52), 21330–21335. 10.1073/pnas.1209589110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko, E. Z. , Basu, A. , Satija, R. , Nemesh, J. , Shekhar, K. , Goldman, M. , … McCarroll, S. A. (2015). Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell, 161(5), 1202–1214. 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder, M. L. , Linder, S. J. , Cascio, V. M. , Fu, Y. , Ho, Q. H. , & Joung, J. K. (2013). CRISPR RNA‐guided activation of endogenous human genes. Nature Methods, 10(10), 977–979. 10.1038/nmeth.2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony, S. , & Pugh, B. F. (2015). Protein‐DNA binding in high‐resolution. Critical Reviews in Biochemistry and Molecular Biology, 50(4), 269–283. 10.3109/10409238.2015.1051505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P. , Yang, L. , Esvelt, K. M. , Aach, J. , Guell, M. , DiCarlo, J. E. , … Church, G. M. (2013). RNA‐guided human genome engineering via Cas9. Science, 339(6121), 823–826. 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May, D. , Blow, M. J. , Kaplan, T. , McCulley, D. J. , Jensen, B. C. , Akiyama, J. A. , … Visel, A. (2012). Large‐scale discovery of enhancers from human heart tissue. Nature Genetics, 44(1), 89–93. 10.1038/ng.1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSwiggen, D. T. , Hansen, A. S. , Teves, S. S. , Marie‐Nelly, H. , Hao, Y. , Heckert, A. B. , … Darzacq, X. (2019). Evidence for DNA‐mediated nuclear compartmentalization distinct from phase separation. eLife, 8, 1–31. 10.7554/eLife.47098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meador, J. P. , Lech, J. J. , Rice, S. D. , Hose, J. E. , Short, J. W. , Rice, S. D. , … White, E. (2014). Massively parallel single‐cell RNA‐Seq for marker‐free decomposition of tissues into cell types. Science, 343, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina, C. , Valouev, A. , Sidow, A. , Batzoglou, S. , Sundquist, A. , Myers, R. M. , … Johnson, D. S. (2008). Genome‐wide analysis of transcription factor binding sites based on ChIP‐Seq data. Nature Methods, 5(9), 829–834. 10.1038/nmeth.1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meireles‐Filho, A. C. , & Stark, A. (2009). Comparative genomics of gene regulation‐conservation and divergence of cis‐regulatory information. Current Opinion in Genetics and Development, 19(6), 565–570. 10.1016/j.gde.2009.10.006 [DOI] [PubMed] [Google Scholar]
- Melnikov, A. , Murugan, A. , Zhang, X. , Tesileanu, T. , Wang, L. , Rogov, P. , … Mikkelsen, T. S. (2012). Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nature Biotechnology, 30(3), 271–277. 10.1038/nbt.2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager, M. , & Nora, E. P. (2016). CTCF and Cohesin in genome folding and transcriptional gene regulation. Annual Review of Genomics and Human Genetics, 17(1), 17–43. 10.1146/annurev-genom-083115-022339 [DOI] [PubMed] [Google Scholar]
- Mir, M. , Bickmore, W. , Furlong, E. E. M. , & Narlikar, G. (2019). Chromatin topology, condensates and gene regulation: Shifting paradigms or just a phase? Development, 146, dev182766 10.1242/dev.182766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir, M. , Stadler, M. R. , Ortiz, S. A. , Hannon, C. E. , Harrison, M. M. , Darzacq, X. , & Eisen, M. B. (2018). Dynamic multifactor hubs interact transiently with sites of active transcription in Drosophila embryos. eLife, 7, 1–27. 10.7554/eLife.40497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt, J. R. , Bambah‐Mukku, D. , Eichhorn, S. W. , Vaughn, E. , Shekhar, K. , Perez, J. D. , … Zhuang, X. (2018). Molecular, spatial, and functional single‐cell profiling of the hypothalamic preoptic region. Science, 362(6416), eaau5324 10.1126/science.aau5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogno, I. , Kwasnieski, J. C. , Cohen, B. A. , Myers, C. A. , & Corbo, J. C. (2012). Complex effects of nucleotide variants in a mammalian cis‐regulatory element. Proceedings of the National Academy of Sciences, 109(47), 19498–19503. 10.1073/pnas.1210678109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau, P. , Hen, R. , Wasylyk, B. , Everett, R. , Gaub, M. P. , & Chambon, P. (1981). The SV40 72 base repair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Research, 9(22), 6047–6068. 10.1093/nar/9.22.6047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munsky, B. , Neuert, G. , & Van Oudenaarden, A. (2012). Using gene expression noise to understand gene regulation. Science, 336, 183–187. 10.1126/science.1216379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano, T. , Lubling, Y. , Stevens, T. J. , Schoenfelder, S. , Yaffe, E. , Dean, W. , … Fraser, P. (2013). Single‐cell Hi‐C reveals cell‐to‐cell variability in chromosome structure. Nature, 502(7469), 59–64. 10.1038/nature12593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, M. , Srinivasan, P. , Chavez, M. , Carter, M. A. , Dominguez, A. A. , La Russa, M. , … Qi, L. S. (2019). Anti‐CRISPR‐mediated control of gene editing and synthetic circuits in eukaryotic cells. Nature Communications, 10(1), 194 10.1038/s41467-018-08158-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama, T. , Fish, M. B. , Fisher, M. , Oomen‐Hajagos, J. , Thomsen, G. H. , & Grainger, R. M. (2013). Simple and efficient CRISPR/Cas9‐mediated targeted mutagenesis in Xenopus tropicalis . Genesis, 51(12), 835–843. 10.1002/dvg.22720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettling, M. , Treutler, H. , Cerquides, J. , & Grosse, I. (2016). Detecting and correcting the binding‐affinity bias in ChIP‐seq data using inter‐species information. BMC Genomics, 17, 347 10.1186/s12864-016-2682-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora, E. P. , Lajoie, B. R. , Schulz, E. G. , Giorgetti, L. , Okamoto, I. , Servant, N. , … Heard, E. (2012). Spatial partitioning of the regulatory landscape of the X‐inactivation centre. Nature, 485(7398), 381–385. 10.1038/nature11049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni, R. , Piccolo, V. , Barozzi, I. , Polletti, S. , Termanini, A. , Bonifacio, S. , … Natoli, G. (2013). Latent enhancers activated by stimulation in differentiated cells. Cell, 152(1), 157–171. 10.1016/j.cell.2012.12.018 [DOI] [PubMed] [Google Scholar]
- Parker, S. C. J. , Stitzel, M. L. , Taylor, D. L. , Orozco, J. M. , Erdos, M. R. , Akiyama, J. A. , … Collins, F. S. (2013). Chromatin stretch enhancer states drive cell‐specific gene regulation and harbor human disease risk variants. Proceedings of the National Academy of Sciences of the United States of America, 110(44), 17921–17926. 10.1073/pnas.1317023110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan, R. P. , Hiatt, J. B. , Witten, D. M. , Kim, M. J. , Smith, R. P. , May, D. , … Shendure, J. (2012). Massively parallel functional dissection of mammalian enhancers in vivo. Nature Biotechnology, 30(3), 265–270. 10.1038/nbt.2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Pinera, P. , Kocak, D. D. , Vockley, C. M. , Adler, A. F. , Kabadi, A. M. , Polstein, L. R. , … Gersbach, C. A. (2013). RNA‐guided gene activation by CRISPR‐Cas9‐based transcription factors. Nature Methods, 10(10), 973–976. 10.1038/nmeth.2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, A. H. , Mermoud, J. E. , O'Carroll, D. , Pagani, M. , Schweizer, D. , Brockdorff, N. , & Jenuwein, T. (2002). Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nature Genetics, 30(1), 77–80. 10.1038/ng789 [DOI] [PubMed] [Google Scholar]
- Phillips‐Cremins, J. E. , Sauria, M. E. G. , Sanyal, A. , Gerasimova, T. I. , Lajoie, B. R. , Bell, J. S. K. , … Corces, V. G. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell, 153(6), 1281–1295. 10.1016/j.cell.2013.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijuan‐Sala, B. , Griffiths, J. A. , Guibentif, C. , Hiscock, T. W. , Jawaid, W. , Calero‐Nieto, F. J. , … Göttgens, B. (2019). A single‐cell molecular map of mouse gastrulation and early organogenesis. Nature, 566(7745), 490–495. 10.1038/s41586-019-0933-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon, A. M. , Ajay, S. S. , Kumar, S. A. , Steiner, L. A. , Cherukuri, P. F. , Wincovitch, S. , … Bodine, D. M. (2011). Genome‐wide ChIP‐Seq reveals a dramatic shift in the binding of the transcription factor erythroid Kruppel‐like factor during erythrocyte differentiation. Blood, 118(17), 139–149. 10.1182/blood-2011-05-355107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plass, M. , Solana, J. , Alexander Wolf, F. , Ayoub, S. , Misios, A. , Glažar, P. , … Rajewsky, N. (2018). Cell type atlas and lineage tree of a whole complex animal by single‐cell transcriptomics. Science, 360(6391), eaaq1723 10.1126/science.aaq1723 [DOI] [PubMed] [Google Scholar]
- Pollen, A. A. , Nowakowski, T. J. , Shuga, J. , Wang, X. , Leyrat, A. A. , Lui, J. H. , … West, J. A. A. (2014). Low‐coverage single‐cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nature Biotechnology, 32(10), 1053–1058. 10.1038/nbt.2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope, B. D. , Ryba, T. , Dileep, V. , Yue, F. , Wu, W. , Denas, O. , … Gilbert, D. M. (2014). Topologically associating domains are stable units of replication‐timing regulation. Nature, 515(7527), 402–405. 10.1038/nature13986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott, S. , & Lieb, J. D. (2015). What are super‐enhancers? Nature Genetics, 47(1), 8–12. 10.1038/ng.3167 [DOI] [PubMed] [Google Scholar]
- Racioppi, C. , Wiechecki, K. A. , & Christiaen, L. (2019). Combinatorial chromatin dynamics foster accurate cardiopharyngeal fate choices. BioRxiv. [DOI] [PMC free article] [PubMed]
- Rada‐Iglesias, A. , Bajpai, R. , Swigut, T. , Brugmann, S. A. , Flynn, R. A. , & Wysocka, J. (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature, 470(7333), 279–283. 10.1038/nature09692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal, N. , Srinivasan, S. , Kooshesh, K. , Guo, Y. , Edwards, M. D. , Banerjee, B. , … Sherwood, R. I. (2016). High‐throughput mapping of regulatory DNA. Nature Biotechnology, 34(2), 167–174. 10.1038/nbt.3468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, A. I. , & Barolo, S. (2013). Low‐affinity transcription factor binding sites shape morphogen responses and enhancer evolution. Philosophical Transactions of the Royal Society, B: Biological Sciences, 368, 20130018 10.1098/rstb.2013.0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S. S. P. , Huntley, M. H. , Durand, N. C. , Stamenova, E. K. , Bochkov, I. D. , Robinson, J. T. , … Aiden, E. L. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159(7), 1665–1680. 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviram, R. , Rocha, P. P. , Bonneau, R. , & Skok, J. A. (2014). Interpreting 4C‐Seq data: How far can we go? Epigenomics, 6(5), 455–457. 10.2217/epi.14.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, N. , Salmon‐Divon, M. , Dvinge, H. , Hynes‐Allen, A. , Balasooriya, G. , Leaford, D. , … Hendrich, B. (2012). NuRD‐mediated deacetylation of H3K27 facilitates recruitment of Polycomb repressive complex 2 to direct gene repression. The EMBO Journal, 31(3), 593–605. 10.1038/emboj.2011.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, H. S. , & Pugh, B. F. (2011). Comprehensive genome‐wide protein‐DNA interactions detected at single‐nucleotide resolution. Cell, 147(6), 1408–1419. 10.1016/j.cell.2011.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, G. , Bernier, B. , Zhao, Y. , Varhol, R. , Snyder, M. , Euskirchen, G. , … Bilenky, M. (2007). Genome‐wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature Methods, 4(8), 651–657. 10.1038/nmeth1068 [DOI] [PubMed] [Google Scholar]
- Roh, T. Y. , Cuddapah, S. , & Zhao, K. (2005). Active chromatin domains are defined by acetylation islands revealed by genome‐wide mapping. Genes and Development, 19(5), 542–552. 10.1101/gad.1272505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron, G. , Globerson, Y. , Moran, D. , & Kaplan, T. (2017). Promoter‐enhancer interactions identified from Hi‐C data using probabilistic models and hierarchical topological domains. Nature Communications, 8(1), 2237 10.1038/s41467-017-02386-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem, A. , Ram, O. , Shoresh, N. , Sperling, R. A. , Goren, A. , Weitz, D. A. , & Bernstein, B. E. (2015). Single‐cell ChIP‐seq reveals cell subpopulations defined by chromatin state. Nature Biotechnology, 33(11), 1165–1172. 10.1038/nbt.3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbacher, U. , Bertrand, V. , Lamy, C. , & Lemaire, P. (2007). A combinatorial code of maternal GATA, Ets and ‐catenin‐TCF transcription factors specifies and patterns the early ascidian ectoderm. Development, 134(22), 4023–4032. 10.1242/dev.010850 [DOI] [PubMed] [Google Scholar]
- Rowan, S. , Siggers, T. , Lachke, S. A. , Yue, Y. , Bulyk, M. L. , & Maas, R. L. (2010). Precise temporal control of the eye regulatory gene Pax6 via enhancer‐binding site affinity. Genes and Development, 24(10), 980–985. 10.1101/gad.1890410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye, M. B. , Sætrom, P. , & Drabløs, F. (2011). A manually curated ChIP‐seq benchmark demonstrates room for improvement in current peak‐finder programs. Nucleic Acids Research, 39, e25 10.1093/nar/gkq1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari, B. R. , Dall'Agnese, A. , Boija, A. , Klein, I. A. , Coffey, E. L. , Shrinivas, K. , … Young, R. A. (2018). Coactivator condensation at super‐enhancers links phase separation and gene control. Science, 361(6400), eaar3958 10.1126/science.aap9195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin, A. (2004). JASPAR: An open‐access database for eukaryotic transcription factor binding profiles. Nucleic Acids Research, 32, D91–D94. 10.1093/nar/gkh012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmann, T. , Girardot, C. , Brehme, M. , Tongprasit, W. , Stolc, V. , & Furlong, E. E. M. (2007). A core transcriptional network for early mesoderm development in Drosophila melanogaster . Genes and Development, 21(4), 436–449. 10.1101/gad.1509007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana, N. E. , Wright, J. , Zheng, K. , Shalem, O. , Fontanillas, P. , Joung, J. , … Zhang, F. (2016). High‐resolution interrogation of functional elements in the noncoding genome. Science, 353(6307), 1545–1549. 10.1126/science.aaf7613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarro, R. , Kocher, A. A. , Emera, D. , Uebbing, S. , Dutrow, E. V. , Weatherbee, S. D. , … Noonan, J. P. (2018). Disrupting the three‐dimensional regulatory topology of the Pitx1 locus results in overtly normal development. Development, 145(7), dev158550 10.1242/dev.158550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai, Y. , Lu, B. , Steinbeisser, H. , Geissert, D. , Gont, L. K. , & De Robertis, E. M. (1994). Xenopus chordin: A novel dorsalizing factor activated by organizer‐specific homeobox genes. Cell, 79(5), 779–790. 10.1016/0092-8674(94)90068-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep, A. N. , Wu, B. , Buenrostro, J. D. , & Greenleaf, W. J. (2017). ChromVAR: Inferring transcription‐factor‐associated accessibility from single‐cell epigenomic data. Nature Methods, 14(10), 975–978. 10.1038/nmeth.4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, D. , Schwalie, P. C. , Ross‐Innes, C. S. , Hurtado, A. , Brown, G. D. , Carroll, J. S. , … Odom, D. T. (2010). A CTCF‐independent role for cohesin in tissue‐specific transcription. Genome Research, 20(5), 578–588. 10.1101/gr.100479.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, A. D. , Hu, M. , Jung, I. , Xu, Z. , Qiu, Y. , Tan, C. L. , … Ren, B. (2016). A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Reports, 17(8), 2042–2059. 10.1016/j.celrep.2016.10.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöne, S. , Meijsing, S. H. , Thomas‐Chollier, M. , Bothe, M. , Vingron, M. , Borschiwer, M. , … Einfeldt, E. (2018). Synthetic STARR‐seq reveals how DNA shape and sequence modulate transcriptional output and noise. PLoS Genetics, 14(11), e1007793 10.1371/journal.pgen.1007793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones, D. E. , Cui, K. , Cuddapah, S. , Roh, T. Y. , Barski, A. , Wang, Z. , … Zhao, K. (2008). Dynamic regulation of nucleosome positioning in the human genome. Cell, 132(5), 887–898. 10.1016/j.cell.2008.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte‐Merker, S. , Lee, K. J. , McMahon, A. P. , & Hammerschmidt, M. (1997). The zebrafish organizer requires chordino. Nature, 387(6636), 862–863. 10.1038/43092 [DOI] [PubMed] [Google Scholar]
- Schultz, D. C. , Ayyanathan, K. , Negorev, D. , Maul, G. G. , & Rauscher, F. J. (2002). SETDB1: A novel KAP‐1‐associated histone H3, lysine 9‐specific methyltransferase that contributes to HP1‐mediated silencing of euchromatic genes by KRAB zinc‐finger proteins. Genes and Development, 16(8), 919–932. 10.1101/gad.973302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer, W. , Abdennur, N. , Goloborodko, A. , Pekowska, A. , Fudenberg, G. , Loe‐Mie, Y. , … Spitz, F. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature, 551(7678), 51–56. 10.1038/nature24281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seipel, K. , Georgiev, O. , & Schaffner, W. (1992). Different activation domains stimulate transcription from remote (‘enhancer’) and proximal (‘promoter’) positions. The EMBO Journal, 11(13), 4961–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek, A. K. , Satija, R. , Adiconis, X. , Gertner, R. S. , Gaublomme, J. T. , Raychowdhury, R. , … Regev, A. (2013). Single‐cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature, 498(7453), 236–240. 10.1038/nature12172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan, Q. , Wang, Y. , Li, J. , Zhang, Y. , Chen, K. , Liang, Z. , … Gao, C. (2013). Targeted genome modification of crop plants using a CRISPR‐Cas system. Nature Biotechnology, 31(8), 686–688. 10.1038/nbt.2623 [DOI] [PubMed] [Google Scholar]
- Shen, Y. , Diao, Y. , Li, B. , Ren, B. , Fang, R. , Lin, K. C. , … Huang, H. (2017). A tiling‐deletion‐based genetic screen for cis‐regulatory element identification in mammalian cells. Nature Methods, 14(6), 629–635. 10.1038/nmeth.4264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y. , Yue, F. , Mc Cleary, D. F. , Ye, Z. , Edsall, L. , Kuan, S. , … Lobanenkov, V. V. (2012). A map of the cis‐regulatory sequences in the mouse genome. Nature, 488(7409), 116–120. 10.1038/nature11243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard, A. (2012). The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annual Review of Biochemistry, 81, 65–95. 10.1146/annurev-biochem-051710-134100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, H. Y. , Willi, M. , Yoo, K. H. , Zeng, X. , Wang, C. , Metser, G. , & Hennighausen, L. (2016). Hierarchy within the mammary STAT5‐driven Wap super‐enhancer. Nature Genetics, 48(8), 904–911. 10.1038/ng.3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva, D. , Stampfel, G. , & Stark, A. (2014). Transcriptional enhancers: From properties to genome‐wide predictions. Nature Reviews Genetics, 15(4), 272–286. 10.1038/nrg3682 [DOI] [PubMed] [Google Scholar]
- Shrinivas, K. , Sabari, B. R. , Coffey, E. L. , Klein, I. A. , Boija, A. , Zamudio, A. V. , … Chakraborty, A. K. (2019). Enhancer features that drive formation of transcriptional condensates. Molecular Cell, 75, 549–561. 10.1016/j.molcel.2019.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov, D. R. , Ansel, K. M. , Gagnon, J. D. , Tobin, V. R. , Farh, K. K. , Schumann, K. , … Satpathy, A. T. (2017). Discovery of stimulation‐responsive immune enhancers with CRISPR activation. Nature, 549(7670), 111–115. 10.1038/nature23875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene, P. J. , & Henikoff, S. (2017). An efficient targeted nuclease strategy for high‐resolution mapping of DNA binding sites. eLife, 6, 1–35. 10.7554/eLife.21856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soshnikova, N. , Montavon, T. , Leleu, M. , Galjart, N. , & Duboule, D. (2010). Functional analysis of CTCF during mammalian limb development. Developmental Cell, 19(6), 819–830. 10.1016/j.devcel.2010.11.009 [DOI] [PubMed] [Google Scholar]
- Sripathy, S. P. , Stevens, J. , & Schultz, D. C. (2006). The KAP1 corepressor functions to coordinate the assembly of de novo HP1‐demarcated microenvironments of heterochromatin required for KRAB zinc finger protein‐mediated transcriptional repression. Molecular and Cellular Biology, 26(22), 8623–8638. 10.1128/mcb.00487-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhl, P. L. , Salmén, F. , Vickovic, S. , Lundmark, A. , Navarro, J. F. , Magnusson, J. , … Frisén, J. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science, 363(6294), 78–82. 10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- Stolfi, A. , Gandhi, S. , Salek, F. , & Christiaen, L. (2014). Tissue‐specific genome editing in ciona embryos by CRISPR/Cas9. Development, 141(21), 4115–4120. 10.1242/dev.114488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons, O. , Pan, L. , Remeseiro, S. , Aktas, T. , Klein, F. , Huber, W. , & Spitz, F. (2016). The Shh topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Developmental Cell, 39(5), 529–543. 10.1016/j.devcel.2016.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmons, O. , Uslu, V. V. , Tsujimura, T. , Ruf, S. , Nassari, S. , Schwarzer, W. , … Spitz, F. (2014). Functional and topological characteristics of mammalian regulatory domains. Genome Research, 24(3), 390–400. 10.1101/gr.163519.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, F. , Barbacioru, C. , Bao, S. , Lee, C. , Nordman, E. , Wang, X. , … Surani, M. A. (2010). Tracing the derivation of embryonic stem cells from the inner cell mass by single‐cell RNA‐seq analysis. Cell Stem Cell, 6(5), 468–478. 10.1016/j.stem.2010.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, F. , Barbacioru, C. , Wang, Y. , Nordman, E. , Lee, C. , Xu, N. , … Surani, M. A. (2009). mRNA‐Seq whole‐transcriptome analysis of a single cell. Nature Methods, 6(5), 377–382. 10.1038/nmeth.1315 [DOI] [PubMed] [Google Scholar]
- Tang, Z. , Luo, O. J. , Li, X. , Zheng, M. , Zhu, J. J. , Szalaj, P. , … Ruan, Y. (2015). CTCF‐mediated human 3D genome architecture reveals chromatin topology for transcription. Cell, 163(7), 1611–1627. 10.1016/j.cell.2015.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey, R. , Kotliar, D. , Park, D. S. , Liu, B. , Winnicki, S. , Reilly, S. K. , … Sabeti, P. C. (2016). Direct identification of hundreds of expression‐modulating variants using a multiplexed reporter assay. Cell, 165(6), 1519–1529. 10.1016/j.cell.2016.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teytelman, L. , Thurtle, D. M. , Rine, J. , & van Oudenaarden, A. (2013). Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proceedings of the National Academy of Sciences, 110(46), 18602–18607. 10.1073/pnas.1316064110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhuis, B. , Palstra, R. J. , Splinter, E. , Grosveld, F. , & De Laat, W. (2002). Looping and interaction between hypersensitive sites in the active β‐globin locus. Molecular Cell, 10(6), 1453–1465. 10.1016/S1097-2765(02)00781-5 [DOI] [PubMed] [Google Scholar]
- Trapnell, C. , Cacchiarelli, D. , Grimsby, J. , Pokharel, P. , Li, S. , Morse, M. , … Rinn, J. L. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology, 32(4), 381–386. 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , & Liu, S. (2016). Single‐cell transcriptome sequencing: Recent advances and remaining challenges. F1000Research, 5, 182 10.12688/f1000research.7223.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev, A. , Johnson, S. M. , Boyd, S. D. , Smith, C. L. , Fire, A. Z. , & Sidow, A. (2011). Determinants of nucleosome organization in primary human cells. Nature, 474(7352), 516–520. 10.1038/nature10002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Steensel, B. , & Dekker, J. (2010). Genomics tools for unraveling chromosome architecture. Nature Biotechnology, 28(10), 1089–1095. 10.1038/nbt.1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw, N. L. , Zhang, Y. , Woods, I. G. , Imam, F. , Regev, A. , Liu, X. S. , … Schier, A. F. (2010). Chromatin signature of embryonic pluripotency is established during genome activation. Nature, 464(7290), 922–926. 10.1038/nature08866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel, A. , Blow, M. J. , Li, Z. , Zhang, T. , Akiyama, J. A. , Holt, A. , … Pennacchio, L. A. (2009). ChIP‐seq accurately predicts tissue‐specific activity of enhancers. Nature, 457(7231), 854–858. 10.1038/nature07730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel, A. , Taher, L. , Girgis, H. , May, D. , Golonzhka, O. , Hoch, R. V. , … Rubenstein, J. L. R. (2013). A high‐resolution enhancer atlas of the developing telencephalon. Cell, 152(4), 895–908. 10.1016/j.cell.2012.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, D. E. , Weinreb, C. , Collins, Z. M. , Briggs, J. A. , Megason, S. G. , & Klein, A. M. (2018). Single‐cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science, 360(6392), 981–987. 10.1126/science.aar4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamstad, J. A. , Alexander, J. M. , Truty, R. M. , Shrikumar, A. , Li, F. , Eilertson, K. E. , … Bruneau, B. G. (2012). Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell, 151(1), 206–220. 10.1016/j.cell.2012.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Yang, H. , Shivalila, C. S. , Dawlaty, M. M. , Cheng, A. W. , Zhang, F. , & Jaenisch, R. (2013). One‐step generation of mice carrying mutations in multiple genes by CRISPR/cas‐mediated genome engineering. Cell, 153(4), 910–918. 10.1016/j.cell.2013.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte, W. A. , Orlando, D. A. , Hnisz, D. , Abraham, B. J. , Lin, C. Y. , Kagey, M. H. , … Young, R. A. (2013). Master transcription factors and mediator establish super‐enhancers at key cell identity genes. Cell, 153(2), 307–319. 10.1016/j.cell.2013.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, I. , Kane, L. , Devenney, P. S. , Anderson, E. , Kilanowski, F. , Hill, R. E. , … Lettice, L. A. (2019). Developmentally regulated Shh expression is robust to TAD perturbations. BioRxiv. 10.1101/609941 [DOI] [PMC free article] [PubMed]
- Williamson, I. , Lettice, L. A. , Hill, R. E. , & Bickmore, W. A. (2016). Shh and ZRS enhancer colocalisation is specific to the zone of polarising activity. Development, 143(16), 2994–3001. 10.1242/dev.139188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C. , & Pan, W. (2018). Integration of enhancer‐promoter interactions with GWAS summary results identifies novel schizophrenia‐associated genes and pathways. Genetics, 209(3), 699–709. 10.1534/genetics.118.300805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, S. , Duan, J. , Li, B. , Zhou, P. , & Hon, G. C. (2017). Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Molecular Cell, 66(2), 285–299.e5. 10.1016/j.molcel.2017.03.007 [DOI] [PubMed] [Google Scholar]
- Yang, D. , Xu, J. , Zhu, T. , Fan, J. , Lai, L. , Zhang, J. , & Chen, Y. E. (2014). Effective gene targeting in rabbits using RNA‐guided Cas9 nucleases. Journal of Molecular Cell Biology, 6(1), 97–99. 10.1093/jmcb/mjt047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Z. , Ren, M. , Wang, Z. , Zhang, B. , Rong, Y. S. , Jiao, R. , & Gao, G. (2013). Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila . Genetics, 195(1), 289–291. 10.1534/genetics.113.153825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuh, C. H. , Ransick, A. , Martinez, P. , Britten, R. J. , & Davidson, E. H. (1994). Complexity and organization of DNA–protein interactions in the 5′‐regulatory region of an endoderm‐specific marker gene in the sea urchin embryo. Mechanisms of Development, 47, 165–186. 10.1016/0925-4773(94)90088-4 [DOI] [PubMed] [Google Scholar]
- Zabidi, M. A. , Arnold, C. D. , Schernhuber, K. , Pagani, M. , Rath, M. , Frank, O. , & Stark, A. (2015). Enhancer‐core‐promoter specificity separates developmental and housekeeping gene regulation. Nature, 518, 556–559. 10.1038/nature13994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandvakili, A. , Campbell, I. , Gutzwiller, L. M. , Weirauch, M. T. , & Gebelein, B. (2018). Degenerate Pax2 and senseless binding motifs improve detection of low‐affinity sites required for enhancer specificity. PLoS Genetics, 14(4), e1007289 10.1371/journal.pgen.1007289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret, K. S. , & Carroll, J. S. (2011). Pioneer transcription factors: Establishing competence for gene expression. Genes and Development, 25(21), 2227–2241. 10.1101/gad.176826.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Li, T. , Preissl, S. , Amaral, M. L. , Grinstein, J. D. , Farah, E. N. , … Ren, B. (2019). Transcriptionally active HERV‐H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nature Genetics, 51, 1380–1388. 10.1038/s41588-019-0479-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, M. , Tian, S. Z. , Capurso, D. , Kim, M. , Maurya, R. , Lee, B. , … Ruan, Y. (2019). Multiplex chromatin interactions with single‐molecule precision. Nature, 566, 558–562. 10.1038/s41586-019-0949-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin, J. , Dixon, J. R. , van der Reijden, M. I. J. A. , Ye, Z. , Kolovos, P. , Brouwer, R. W. W. , … Wendt, K. S. (2014). Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proceedings of the National Academy of Sciences, 111(3), 996–1001. 10.1073/pnas.1317788111 [DOI] [PMC free article] [PubMed] [Google Scholar]