

Abstract
Clostridioides difficile (formerly Clostridium difficile; C difficile), the leading cause of nosocomial antibiotic‐associated colitis and diarrhea in the industrialized world, triggers colonic disease through the release two toxins, toxin A (TcdA) and toxin B (TcdB), glucosyltransferases that modulate monomeric G‐protein function and alter cytoskeletal function. The initial degree of the host immune response to C difficile and its pathogenic toxins is a common indicator of disease severity and infection recurrence. Thus, targeting the intestinal inflammatory response during infection could significantly decrease disease morbidity and mortality. In the current study, we sought to interrogate the influence of the pregnane X receptor (PXR), a modulator of xenobiotic and detoxification responses, which can sense and respond to microbial metabolites and modulates inflammatory activity, during exposure to TcdA and TcdB. Following intrarectal exposure to TcdA/B, PXR‐deficient mice (Nr1i2−/−) exhibited reduced survival, an effect that was associated with increased levels of innate immune cell influx. This exacerbated response was associated with a twofold increase in the expression of Tlr4. Furthermore, while broad‐spectrum antibiotic treatment (to deplete the intestinal microbiota) did not alter the responses in Nr1i2−/− mice, blocking TLR4 signaling significantly reduced TcdA/B‐induced disease severity and immune responses in these mice. Lastly, to assess the therapeutic potential of targeting the PXR, we activated the PXR with pregnenolone 16α‐carbonitrile (PCN) in wild‐type mice, which greatly reduced the severity of TcdA/B‐induced damage and intestinal inflammation. Taken together, these data suggest that the PXR plays a role in the host's response to TcdA/B and may provide a novel target to dampen the inflammatory tissue damage in C difficile infections.
Keywords: Clostridioides difficile, eosinophils, neutrophils, Nr1i2, pregnane X receptor, toxin
Abbreviations
- C difficile
Clostridioides difficile
- CDI
C difficile infection
- GI
gastrointestinal
- IEC
intestinal epithelial cells
- MyD88
myeloid differentiation primary responses 88
- NFκB
nuclear factor κ‐light‐chain‐enhancer of B cells
- PCN
pregnenolone 16α‐carbonitrile
- PXR
pregnane X receptor
- TcdA
toxin A
- TcdB
toxin B
- TLR4
Toll‐like receptor 4
1. INTRODUCTION
Clostridioides difficile (formerly Clostridium difficile; C difficile) is a gram‐positive, spore‐forming bacillus that colonizes in the gastrointestinal (GI) tract, and is recognized as the leading cause of nosocomial antibiotic‐associated colitis and diarrhea in the industrialized world.1, 2 Over the past decade, the incidence of C difficile infection (CDI) has doubled in hospitalized patients, while new cases are emerging in low‐risk populations.3, 4, 5 Progress has been made toward treatment strategies focused on reconstituting the normal microbiota (including fecal microbiota transplantation) to impede colonization and growth of C difficile to prevent persistent infections. However, when examining patient populations with CDI, the severity of disease is difficult to evaluate based on current laboratory approaches.6 Instead, markers of intestinal inflammation appear to better gauge disease severity7, 8 and predict progression and treatment success.9 Furthermore, initial disease severity and the host immune response are the best predictors of CDI recurrence.10, 11 Thus, there remains a need to target the robust intestinal inflammation and tissue damage caused by established CDI to prevent fulminant illness, morbidity, and disease recurrence.
The tissue damage and inflammation observed during CDI are incited by the release of the C difficile virulence factors, toxin A (TcdA), and toxin B (TcdB).12, 13, 14 Upon their secretion in the colon, TcdA and TcdB are taken up by intestinal epithelial cells (IEC) through receptor‐mediated endocytosis, where they subsequently catalyze the addition of a glucose to monomeric G proteins (eg, Rho, Rac, Cdc42), inhibiting their activity, disrupting the actin cytoskeleton, and triggering apoptosis, ultimately leading to the loss of epithelial barrier integrity.15, 16 Disrupted barrier integrity induced by toxin challenge allows the translocation of commensal bacteria from the intestinal lumen into the lamina propria (LP), causing the release of pro‐inflammatory cytokines and chemokines from IECs and resident immune cells, propagating a large influx of immune cells and fluid accumulation.17, 18 Combined, these events manifest as the clinical symptoms of diarrhea, pseudomembranous colitis, toxic megacolon, and in severe cases, death.15, 16
Neutrophils are one of the rapid and prominent responding innate immune cell types during CDI that help sterilize mucosal sites and control infection.17, 18, 19 The recruitment of neutrophils during CDI involves the chemokines CXCL1 and CXCL2 and is also influenced by signaling through Toll‐like receptor 4 (TLR4) and the adaptor protein myeloid differentiation primary responses 88 (MyD88).18 Monocytes and eosinophils also play important roles in modulating responses in the intestinal mucosa during CDI.18, 20, 21 but the signaling dynamics leading to the mobilization of these cells during CDI are less well characterized. The TLR4 pathway has been implicated in driving monocyte influx during CDI,18, 22 and this may be the converging pathway also influencing eosinophil influx.23, 24 Together, these innate immune cells robustly respond to C difficile and its pathogenic toxins TcdA/B to control and eliminate C difficile. However, the fulminant nature of the innate immune cell response leads to bystander tissue damage and pseudomembranous colitis that can be life threatening.
Initially recognized for its role in xenobiotic detoxification and metabolism, the pregnane X receptor (PXR; gene: Nr1i2) also significantly regulates inflammation and immune responses. The PXR is highly expressed in the liver and intestine where it can regulate nuclear factor κ‐light‐chain‐enhancer of B cells (NFκB) activation and the expression of a number of inflammatory mediators.25, 26 Importantly, PXR signaling in the intestine can dampen TLR4 signaling to promote intestinal barrier function and prevent inflammation in the GI tract.25, 27 The influence of the PXR in the modulation of enteric infections has also emerged with evidence for the involvement of a PXR‐TLR4–driven mechanism.26, 28 Interestingly, rifaximin, a luminal antibiotic and human PXR agonist, has been reported to protect Caco‐2 intestinal epithelial cells from the damaging effects of TcdA, in a PXR‐dependent TLR4/MyD88/NFκB pathway.29
These observations led us to hypothesize that signaling through the PXR plays an important role in mediating C difficile toxin‐induced tissue damage and inflammation and could be a viable target to limit the inflammatory response during CDI. Utilizing a model of intrarectal C difficile toxin challenge, we examined the innate immune responses in the colonic mucosa toward the disease‐causing effectors produced by C difficile and how these responses are mediated by the PXR. Given the strong link between PXR and TLR4 signaling in intestinal inflammation, we also explored this mechanism in underpinning the PXR in regulating damage and inflammation during CDI. Finally, we examined the ability of targeting the PXR to treat the inflammation and damage associated with C difficile toxin‐induced tissue damage and inflammation.
2. MATERIALS AND METHODS
2.1. Mice
Nr1i2+/+ (wild‐type; WT) and Nr1i2−/− mice (on the C57Bl/6 background) between 8 and 10 weeks of age were used in our studies. Mice were bred and housed at the University of Calgary. All mice were housed groups of 4 in individually ventilated cages (Tecniplast) with a standard 12‐hour light‐dark cycle in a room maintained at 21°C with free access to water and chow.
2.2. Ethics
All mouse experiments were approved by the Health Sciences Animal Care Committee of the University of Calgary (AC15‐0181) and conformed to the guidelines set forth by the Canadian Council for Animal Care.
2.3. Toxin preparation/administration and treatments
TcdA and TcdB were produced as described previously.30, 31 Briefly, C difficile strains (ATCC 43255—designation VPI 10463; 630—Ribotype 012) were grown in brain‐heart infusion media under anaerobic conditions. Overnight cultures were inoculated into dialysis tubing (containing phosphate‐buffered saline) suspended in 750 mL of media. The culture contents of the tubing were harvested at day 5 post‐inoculation. After centrifugation (10 000 g, 60 minutes), the toxin‐containing supernatant was removed, passed through a 0.22‐μm filter to remove spores, then through a 100‐kDa centrifugal spin filter (Chemicon, Millipore), and used as a source of TcdA/TcdB. This preparation was verified to be lipopolysaccharide‐free via HPLC, and the purity was assessed by SDS‐PAGE and Western blotting as published previously.30, 31 The isolated mixture of TcdA and TcdB (further denoted as TcdA/B) was used at concentrations of 25 μg/animal. Mice were fasted for 8 hours prior to the instillation of C difficile toxins (TcdA/B), which was performed as previously described.32 Unless noted, mice were sacrificed 4 hours after toxin exposure. The welfare of each mouse was assessed at 30‐minutes intervals following intrarectal toxin instillation. Given that death is not an acceptable experimental endpoint, based on the Canadian Council of Animal Care guidelines, mice were removed from the study and euthanized based on the following criteria: 1, animals that exhibited a hunched posture; 2, animals that remained isolated in their cages (ie, failed to socialize with cage mates); 3, animals that failed to exhibit any grooming behavior; 4, animals that did not interact with environmental enrichment; and 5, animals that failed to respond to external environmental cues (ie, did not enter their nesting following a gentle finger poke to the abdomen or flank). Mice exhibiting two or more of the behaviors indicated in criteria 1 to 4 or exhibiting the behavior described in criterion 5 alone were deemed to be in distress and removed from the study based on humane grounds.32 PCN (25mg/kg; Sigma) was dissolved in sterile corn oil and injected i.p. 48, 24, and 0.5 hours prior to intrarectal instillation of TcdA/B in mice. To block TLR4 signaling, 40 μg of anti‐mouse TLR4/MD2 complex antibody (clone MTS510; BioLegend) or isotype control (IgG2a) was injected i.p. 24 and 0.5 hours before intrarectal instillation of TcdA/B in mice. In microbiota depletion experiments, a broad‐spectrum antibiotic cocktail of ampicillin (1g/L; Sigma), metronidazole (500 mg/L; Sigma), neomycin (1 g/L; Sigma), and vancomycin (500 mg/L; Sigma) was administered ad libitum in the drinking water for two weeks, as previously described by Rakoff‐Nahoum et al,33 an approach that we have shown significantly reduces the abundance of the intestinal microbiota.34
2.4. Cell culture
Human intestinal epithelial cells (Caco‐2; adult human colorectal adenocarcinoma cells; ATCC) were grown in Dulbecco's modified Eagle Medium (DMEM; Sigma‐Aldrich) supplemented with 10% fetal bovine serum (Sigma‐Aldrich), L‐glutamine, penicillin‐streptomycin antibiotic, sodium pyruvate, and non‐essential amino acids. Cells were cultured in tissue culture flasks in a humidified incubator at 37°C with 5% CO2. Cells were split using a 1:10 dilution upon reaching 70%‐90% confluence. All experiments were performed on cells between passages 15 and 35. Cells were propagated in 24‐well plates for Western blot. All experiments were carried out with antibiotic‐free, 5% FBS reduced‐serum medium (Opti‐MEM; Life Technologies).
2.5. Western blotting
To isolate cell lysates, culture medium was aspirated, and cells were washed with ice‐cold PBS. Cell lysis buffer (150 mM NaCl, 20 mM Tris (pH 7.5), 1mM EDTA, 1mM EGTA, 1% Triton X‐100, protease inhibitor cocktail: Complete Minitab, and phosphatase inhibitor cocktail: Complete PhoStop (Roche, Laval, Canada)) was then added to cells, and they were frozen at −20°C. Total protein was quantified using the Precision Red Protein Assay (Cytoskeleton, Denver, CO), and protein concentration was normalized between samples. Western blots were performed on normalized protein extracts from Caco‐2 intestinal epithelial cells. Membranes were probed with the appropriate primary antibody (anti‐RAC1, BD Bioscience #610651; anti‐β‐actin, Abcam #mAbcam‐8226) and corresponding horseradish peroxidase (HRP)‐conjugated secondary antibody. Membranes were imaged using the MicroChemi Bio‐Imaging system.
2.6. Disease severity assessment and measurement of inflammatory mediators
Cytokine levels were assessed in colonic homogenates and serum using the Mouse Cytokine/Chemokine 31‐Plex Discovery Assay (Eve Technologies). Histological scoring was performed on blinded hematoxylin‐and‐eosin (H&E)–stained colon sections using a method as we have done previously,32 assessing the degree of inflammation, architectural changes, and epithelial damage.
2.7. Isolation of intestinal LP cells
LP cells were isolated as previously described by Denning et al, with modifications.35 Briefly, the large intestine was opened longitudinally, washed of fecal contents, and cut into pieces 0.5 cm in length. To remove epithelial cells, colon tissue was subjected to two sequential 20‐minute incubations in 30 mL RPMI with 5% FBS and 2 mM EDTA at 37°C with agitation (250 rpm). After each incubation, media containing epithelial cells and debris was discarded. The remaining colon tissue was minced and incubated for 20 minutes in RPMI with 5% FBS, 1 mg/mL collagenase IV (Sigma‐Aldrich), and 40 U/mL DNase I (Roche) at 37°C with agitation (200 rpm). Cell suspensions were collected and passed through a 100‐μm strainer and pelleted by centrifugation at 300 g.
2.8. Flow cytometry and cell sorting
The following antibodies were purchased from Thermo Fisher Scientific/eBioscience (San Diego, CA): CD11b (M1/70), CD45 (30‐F11), Ly6G (1A8), Ly6C (HK1.4), and MHCII (M5/114.15.2). Siglec‐F antibody (E50‐2440) was purchased from BD Biosciences. Fc receptors were blocked with the antibody anti‐FcγRIII/II (2.4G2). FACS was performed on a BD FACSCanto. Cells were sorted into TRIzol using the BD FACSAria to a purity >98%.
2.9. Real‐time quantitative PCR
Six‐cm‐long sections of distal colonic tissue were isolated and stored in RNAlater (Thermo Fisher Scientific, Mississauga, Canada) at −20°C. Tissue was homogenized by Bullet Blender (Next Advance, NY), with each sample containing a 5‐mm stainless steel bead (Qiagen #69989). RNA was extracted using the RNeasy Mini Kit (Qiagen #74106) as per the manufacturer's instructions. Total RNA was reverse transcribed using the QuantiTect Reverse Transcription Kit (Qiagen). Real‐time quantitative PCR was conducted on reactions containing PerfeCTa SYBR Green FastMix (Quanta Bio), cDNA, and validated RT2 primers obtained through Qiagen. In all samples, β‐actin (Actb) was used as the endogenous control. Threshold cycle (Ct) values were obtained from the amplification plots and used to calculate fold change using the ΔΔCt method.
2.10. Statistics
All statistical analyses were performed with GraphPad Prism software, version 7 (GraphPad Software). One‐way ANOVA and Tukey's multiple comparison test or Student's t test were used to determine significance. P < .05 was considered significant. *P < .05, **P < .01, ***P < .001.
3. RESULTS
3.1. C difficile TcdA/B trigger rapid innate immune cell influx into the colon
Intrarectal instillation of TcdA/B, the two main pathogenic toxins produced by C difficile, models the direct epithelial damage and inflammation in the colon of mice observed during CDI.32 To further characterize the cellular dynamics of the innate immune response following instillation of TcdA/B (25 μg/mouse), we utilized the toxin exposure model to examine the influx of immune cells into the colonic LP triggered during CDI. Colonic exposure to TcdA/B for 4 hours led to significant increases in the recruitment of myeloid cells (CD45+) into the colon, which could be further fractionated by expression of major histocompatibility complex II (MHCII) on the cell surface (CD45+MHCII+ and CD45+MHCII− cells; Figure 1A). Within the population of MHCII‐, non–antigen‐presenting myeloid cells (CD45+MHCII−), there was a large number of infiltrating cells that expressed CD11b and varying levels of Ly6C. This population was composed of three distinct innate cell populations: (1) monocytes (CD11b+Ly6Chi; green box), (2) neutrophils (CD11b+Ly6CintLy6G+; blue box), and (3) eosinophils (CD11b+Ly6CloSiglec−F+; red box). Colonic TcdA/B exposure for 4 hours led to a robust increase in the number of Ly6Chi monocytes (~50‐fold increase in cell count vs WT) and neutrophils (>100‐fold increase in cell count vs WT) and only a modest increase in eosinophils (~2‐fold increase in cell count vs WT) into the LP.
Figure 1.
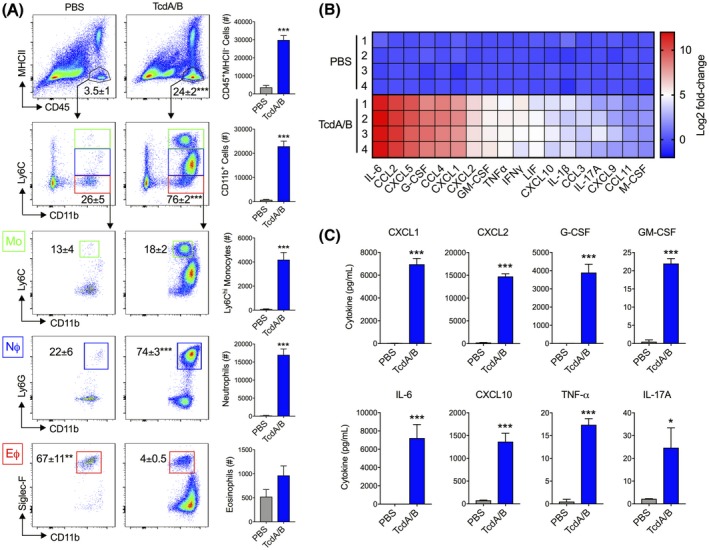
Intrarectal instillation of C difficile toxins TcdA and TcdB (TcdA/B) leads to the robust influx of innate immune cells into the colonic lamina propria and increased colonic production of inflammatory mediators. TcdA/B were intrarectally administered for 4 h (25 μg in PBS; PBS alone in control group), after which cells from the colonic lamina propria were isolated and stained for cell surface markers. A, Stained cells were fractioned based on the expression of CD45 and MHCII. CD45+MHCII− cells were further fractioned into CD11b+ cells with varying levels of Ly6C expression. These CD11b+ cells were composed of Ly6Chi inflammatory monocytes (green box, MΦ), Ly6CintLy6G+ neutrophils (blue box, NΦ), and Ly6CloSiglec‐F+ eosinophils (red box, MΦ). Cell percentage of each population is shown in the corresponding representative flow cytometry plot, and total cell count is depicted in the accompanying bar plot. B, Colonic tissue homogenates from control (PBS‐treated)‐ and TcdA/B‐treated mice were assessed for cytokine production using the Luminex discovery assay. The heatmap in panel B shows the top colonic cytokines that are differentially produced between PBS‐ and TcdA/B‐treated mice (Log2 fold change in TcdA/B vs PBS). C, Protein levels of the top cytokines upregulated in the colon of TcdA/B‐treated mice and play a role in the pathogenesis of C difficile infections (n = 4 per group). All data are expressed as SEM. Student's t test, *P < .05, ***P < .005
In parallel with increased innate immune cell infiltration was a significant upregulation in the production of a number of pro‐inflammatory cytokines and chemokines in the colon of mice following TcdA/B exposure (Figure 1B). These included the chemokines involved in neutrophil recruitment CXCL1 and CXCL2, as well as granulocyte–colony‐stimulating factor (G‐CSF), and granulocyte macrophage–colony‐stimulating factor (GM‐CSF), two cytokines/growth factors involved in the maintenance, activation, and survival of granulocytes. Importantly, one of the most highly expressed cytokines in the colon of mice exposed to TcdA/B when compared to PBS‐treated mice was the pro‐inflammatory, acute phase cytokine IL‐6, which was increased following TcdA/B exposure by >4000‐fold (Figure 1C). Other upregulated cytokines included CXCL10, TNF‐α, and IL‐17A.
3.2. Nr1i2−/− mice are hypersensitive to C difficile toxin exposure
To assess the role of the PXR in C difficile toxin‐induced injury, we utilized Nr1i2−/− (PXR‐deficient) mice to assess disease severity and immune cell influx following colonic TcdA/B exposure. While wild‐type (WT/Nr1i2+/+) mice did not show substantial morbidity after 4 hours (Figure 2A), to our surprise, Nr1i2−/− mice were extremely susceptible to toxin exposure, reaching our defined humane endpoints (see Methods for scoring scale) after 2 hours, failing to reach the pre‐determined 4‐hour experimental endpoint. Histological assessment of H&E‐stained colon sections taken from mice 2 hours after TcdA/B exposure revealed higher total damage scores in the colon of Nr1i2−/− mice compared with WT mice (Figure 2B). The total damage score accounts for epithelial damage, the inflammatory response, and architecture damage. As indicated by the black arrows in Figure 2B, when compared to control/WT mice, the colonic sections from Nr1i2−/− mice exposed to TcdA/B for 2 hours showed increased damage to the surface epithelium at the luminal interface, prominent patches of increased inflammatory cell infiltration in the mucosa, and increased edema underneath the muscularis mucosa that was also clearly populated with infiltrating cells.
Figure 2.
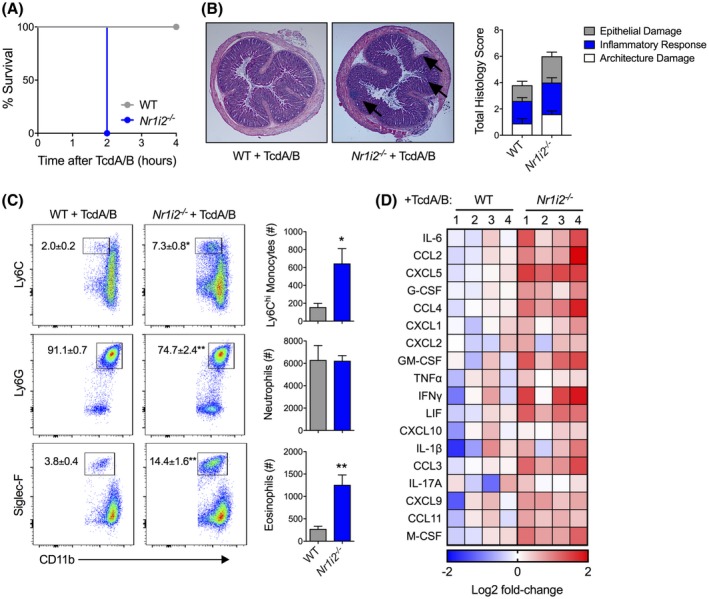
Deletion of Nr1i2 (pregnane X receptor; PXR) leads to increased susceptibility, and exacerbated tissue damage and inflammation following colonic exposure to TcdA/B. A, Survival curve of wild‐type (WT) and Nr1i2‐deficient (Nr1i2−/−) mice following intrarectal exposure to TcdA/B (n = 4 per group). B, Representative histological images and total damage scores of the colon from WT and Nr1i2−/− mice following 2 h of TcdA/B exposure. C, Representative flow cytometry plots and associated cell counts of infiltrating Ly6Chi monocytes (CD11b+Ly6C+), neutrophils (CD11b+Ly6G+), and eosinophils (CD11b+Siglec‐F+) in the colon following 2 h of TcdA/B exposure (n = 5 per group). The colon was also assessed for cytokine production, and the heatmap in panel D shows the top colonic cytokines that are differentially produced between WT and Nr1i2−/− mice following exposure to TcdA/B (Log2 fold change in Nr1i2−/− mice compared to WT mice). All data are expressed as SEM. One‐way ANOVA with Tukey's post hoc test, *P < .05, **P < .01
3.3. Nr1i2−/−mice display an abnormal immune response to TcdA/B exposure
Examination of the colonic LP of WT and Nr1i2−/− mice 2 hours after toxin exposure revealed significantly greater levels of myeloid cell infiltrate as displayed by increased CD45+MHCII− cell infiltration into the colon (Supplemental Figure 1). We next sought to characterize the cell infiltrate in TcdA/B‐treated mice. Whereas in naïve WT and Nr1i2−/− mice, the CD45 population consisted of similar levels of CD11b expression inflammatory monocytes and neutrophils, there was moderate increase (~2‐fold) in the number of eosinophils in the latter group (Supplementary Figure 2). Following TcdA/B exposure, there were significantly greater numbers of monocytes and eosinophils in the colon of Nr1i2−/− mice, while neutrophil numbers were the same between WT and Nr1i2−/− mice (Figure 2C). Colonic production of a number of cytokines and chemokines was also significantly higher in Nr1i2−/− compared with WT mice (Figure 2D). Increased levels of granulocytic infiltration in the colon of Nr1i2−/‐ mice coincided with increased cytokine and chemokine production in the colon compared with WT mice. The most differentially produced cytokine between WT and Nr1i2−/− mice was IL‐6, but also included (in order of magnitude) G‐CSF, CXCL1, CXCL2, and GM‐CSF.
3.4. Hypersensitivity to C difficile toxin‐induced injury in Nr1i2−/− mice is linked to increased TLR4 signaling
Previous reports have linked increased inflammatory responses in the intestine of Nr1i2−/− mice to increased TLR4 expression and potentially overactive immune signaling.27 In line with these results, we observed increased expression of Tlr4 in the colon of Nr1i2−/− mice when compared to WT mice following TcdA/B exposure (Figure 3A). To test whether this increased Tlr4 expression was linked to increased inflammation and susceptibility to TcdA/B exposure in Nr1i2−/− mice, we targeted the PXR‐TLR4 axis by administering an anti‐TLR4 antibody to mice prior to TcdA/B exposure to block TLR4 activity. Blockade of TLR4 was able to prevent the increased mortality observed in Nr1i2−/− mice, prolonging the survival of Nr1i2−/− mice to the previously defined experimental endpoint of 4 hours, as observed in WT mice. Furthermore, histological assessment of H&E‐stained colon sections taken from mice 2 hours after TcdA/B exposure (to account for increased disease severity in Nr1i2−/− mice) demonstrated that the higher total damage scores in the colon of Nr1i2−/− mice treated with isotype control antibody could be decreased with pretreatment with the anti‐TLR4 antibody and returned to levels observed in WT mice treated with isotype control antibody (Figure 3B). As indicated by the black arrows in Figure 3C, colonic sections from Nr1i2−/− mice exposed to TcdA/B for 2 hours showed increased epithelial damage, increased cellular infiltrate into the mucosa, and increased edema underneath the muscularis mucosa, observation that was absent in anti‐TLR4 pretreated Nr1i2−/− mice and WT isotype control‐treated mice.
Figure 3.
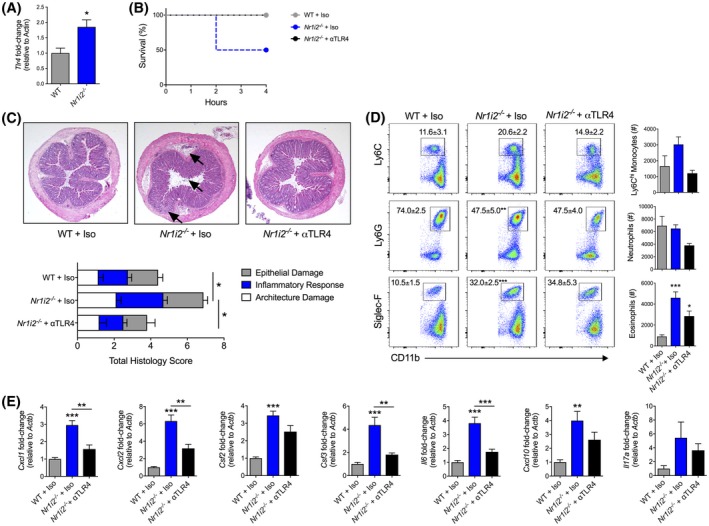
Blockade of Toll‐like receptor 4 (TLR4) alleviates the exaggerated response of Nr1i2−/− mice to intrarectal instillation of TcdA/B. A, Colonic expression of tlr4 in WT and Nr1i2−/− mice (n = 4‐5 per group). B, Survival curve of WT and Nr1i2−/− mice following pretreatment with an anti‐TLR4 antibody then intrarectal exposure to TcdA/B (n = 4 per group). C, Representative histological images and total damage scores. D, representative flow cytometry plots and associated cell counts of infiltrating Ly6Chi monocytes (CD11b+Ly6C+), neutrophils (CD11b+Ly6G+), and eosinophils (CD11b+Siglec‐F+), and (E) the expression of various colonic cytokines in the colon of WT and Nr1i2−/− mice following pretreatment with isotype or anti‐TLR4 antibody then 2 h of exposure to TcdA/B (n = 4‐10 per group). Gene expression was normalized to β‐actin (Actb). All data are expressed as SEM. Student's t test, *P < .05, **P < .01, ***P < .005
3.5. The abnormal immune response to TcdA/B in Nr1i2−/− mice is linked to TLR4 signaling
We also examined the ability of TLR4 blockade to influence the influx of immune cells into colonic LP in Nr1i2−/− mice by examining monocyte, neutrophil, and eosinophil levels in the colonic LP following TcdA/B exposure. Two hours following intrarectal instillation of TcdA/B, treatment with the anti‐TLR4 antibody was able to prevent increased influx of immune cells into the colon. Specifically, anti‐TLR4 returned the increased counts of CD45+MHCII− myeloid cells (Supplementary Figure 3), observed in Nr1i2−/− mice back to levels comparable to WT mice treated with an isotype antibody. Increased eosinophil (Siglec‐F+ cells) numbers in the colon of Nr1i2−/− mice following TcdA/B exposure were also significantly decreased following anti‐TLR4 antibody administration in Nr1i2−/− mice (Figure 3D). Increased Ly6Chi monocyte counts in Nr1i2−/− mice were also reduced toward WT levels following anti‐TLR4 treatment, and there was also a reduction in neutrophils; however, both of these changes did not reach statistical significance. Furthermore, TLR4 blockade also abrogated the increased levels of expression of Cxcl1, Cxcl2, Csf3, Il6, and Cxcl10, observed in the colon of TcdA/B‐exposed Nr1i2−/− mice, suppressing their expression levels toward WT mice (Figure 3E). Increased expression of Csf2 and Il17a in the colon of Nr1i2−/− mice following TcdA/B exposure was also suppressed with anti‐TLR4 antibody, but these changes were not statistically significant.
3.6. Innate cells from Nr1i2−/− mice display defective immune responses
To further characterize the altered immune responses and differences in cytokine expression between WT and Nr1i2−/− mice, we examined cytokine expression in neutrophils and eosinophils, the latter exhibiting an unexpected increased recruitment in Nr1i2−/− mice exposed to TcdA/B (Figure 2C; Supplementary Figure 2). Neutrophils and eosinophils in the colonic LP of TcdA/B‐exposed WT and Nr1i2−/− mice were gated into CD45+MHCII− cells and further fractioned into CD11b+ cells, then sorted into separate populations of neutrophils (Ly6G+) and eosinophils (Siglec‐F+; Figure 4A). When comparing colonic neutrophils isolated from WT and Nr1i2−/− mice following TcdA/B exposure, we found significant differences in cytokine expression including increased expression of Csf2, and reduced expression of Csf3 and Tnf‐α in Nr1i2−/− neutrophils (Figure 4B). There was no difference in the expression of Il6 between WT and Nr1i2−/− neutrophils. In contrast, there was a significant upregulation in the expression of Il6 in Nr1i2−/− eosinophils when compared to WT eosinophils (Figure 4C). The most drastic difference observed was the almost 6 times higher expression of Csf3 by Nr1i2−/− eosinophils compared with WT eosinophils. Like Nr1i2−/− neutrophils, Nr1i2−/− eosinophils also had decreased expression of Tnf‐α compared with WT counterpart cells.
Figure 4.
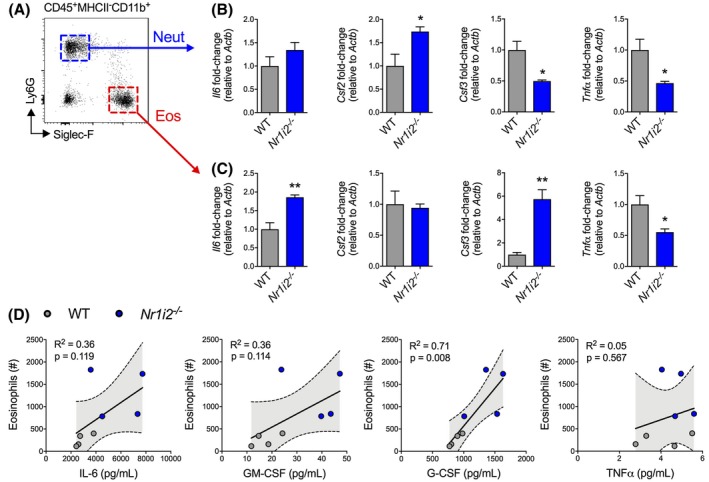
Aberrant cytokine responses in innate immune cells isolated from the colon of Nr1i2−/− mice following TcdA/B exposure. A, Two hours following colonic exposure to TcdA/B, neutrophils (blue box and arrow) and eosinophils (red box and arrow) were sorted from the colonic lamina propria of WT and Nr1i2−/− mice. B‐C, The expression of different cytokines was measured in these different responding innate immune cells via qPCR. Gene expression was normalized to β‐actin (Actb). D, Relationship between eosinophil counts and the production of different cytokines in the colon WT and Nr1i2−/− mice following TcdA/B exposure. Linear regression shown with 95% confidence bound (n = 4 per group). All data are expressed as SEM. The shaded gray area shows the SEM Student's t test, *P < .05, **P < .01
Furthermore, examination of the relationship between eosinophil influx and cytokine production in the colon of WT and Nr1i2−/− mice following TcdA/B exposure found a correlation between eosinophil influx and G‐CSF production but not IL‐6, GM‐CSF, nor TNF‐α (Figure 4D). In the case of G‐CSF, the degree to which eosinophil influx and colonic G‐CSF production are changing appears to positively correlated (r 2 = .71, P = .008). This suggests that increased eosinophil influx in Nr1i2−/− mice may be influencing increased levels of G‐CSF, with one possibility being direct production from eosinophils.36, 37
3.7. Depletion of the intestinal microbiota does not alter innate immune cell infiltration into the colon following TcdA/B exposure
Finally, the microbiota is proposed to be a driver of the inflammation associated with CDI, partly through the activation of immune pathways during breaches in the intestinal epithelium.38 Thus, we tested the ability of antibiotic depletion of the microbiota to influence innate immune cell influx into the colonic LP of Nr1i2−/− mice following TcdA/B exposure. Mice were placed on a cocktail of broad‐spectrum antibiotics in the drinking water, an approach we, and others, have used previously to dampen the impact of the intestinal microbiota in experimental models.33, 34 Mice were maintained on this cocktail (control group was drinking water only) for 2 weeks prior to intrarectal instillation of TcdA/B and during disease induction and progression. To our surprise, antibiotic administration had no effect on the infiltration of innate immune cells into the colon following TcdA/B exposure. Examination of the colonic LP 4 hours after TcdA/B exposure uncovered no differences in the number of CD45+MHCII− cells (Supplementary Figure 4), nor differences in the percent and number of inflammatory Ly6Chi monocytes, neutrophils, and eosinophils between mice given normal drinking water or antibiotic‐supplemented drinking water (Figure 5).
Figure 5.
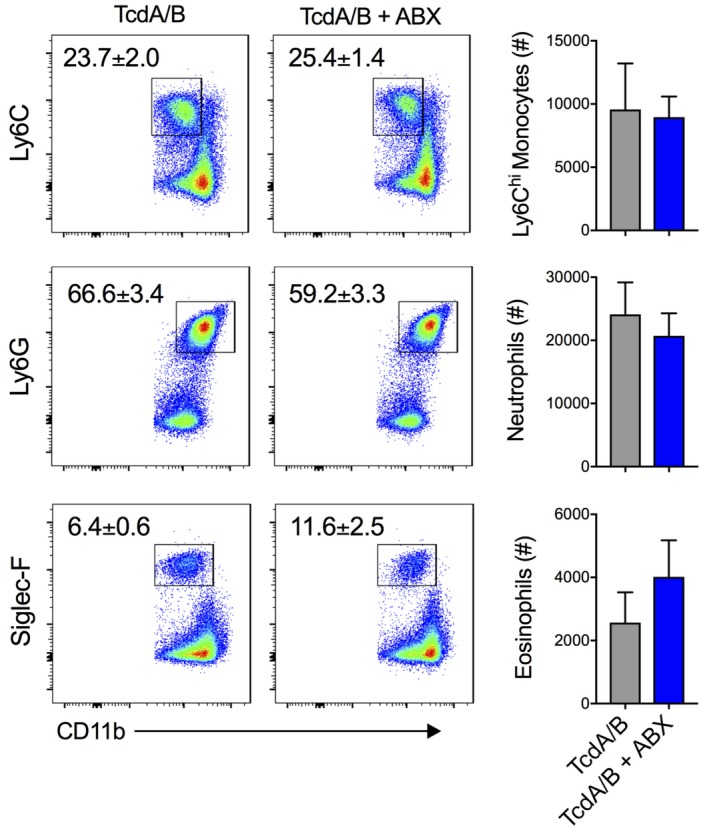
Suppression of the microbiota does not influence TcdA/B‐induced innate immune cell infiltration into the colon. The influx of innate immune cells into the colonic lamina propria was assessed 4 h after TcdA/B exposure in mice given normal water or mice given antibiotic (ABX)‐supplemented water. Representative flow cytometry plots and associated cell counts of infiltrating Ly6Chi monocytes (CD11b+Ly6C+), neutrophils (CD11b+Ly6G+), and eosinophils (CD11b+Siglec‐F+) in the colonic lamina propria of control and ABX‐treated mice following TcdA/B exposure (n = 4‐5 mice per group). All data are expressed as SEM. Student's t test
3.8. PXR activation attenuates the immune cell influx and limits colonic injury following toxin exposure
Given the role of the PXR in mediating toxin‐induced injury and inflammation, we tested the ability of a PXR agonist to mitigate colonic injury and inflammation following TcdA/B exposure. To activate the PXR in vivo, we used the selective mouse agonist PCN dissolved in sterile corn oil. To confirm activity of PCN in vivo, we examined colonic expression of the PXR target gene Cyp3a11 and found that pretreatment of mice with PCN induced an approximate 12‐fold increase in Cyp3a11 expression when compared to vehicle‐treated mice (Figure 6A). To also ensure the treatment with PCN was not inhibiting TcdA/B activity and was not the basis for protection, we used an epithelial cell line to assess the activity of TcdA/B. When internalized by epithelial cells, TcdA/B glucosylate Rac GTPases, which have a role in regulating the organization of the actin cytoskeleton. Thus, the absence of a Rac1 band on Western blot indicates that Rac1 GTPases have been glucosylated and the toxins are functional. Rac1 was not detected in colonic cells exposed to TcdA/B in the presence or absence of PCN (in sterile corn oil), indicating that PCN does not affect the uptake or activity of the TcdA/B (Supplementary Figure 5).
Figure 6.
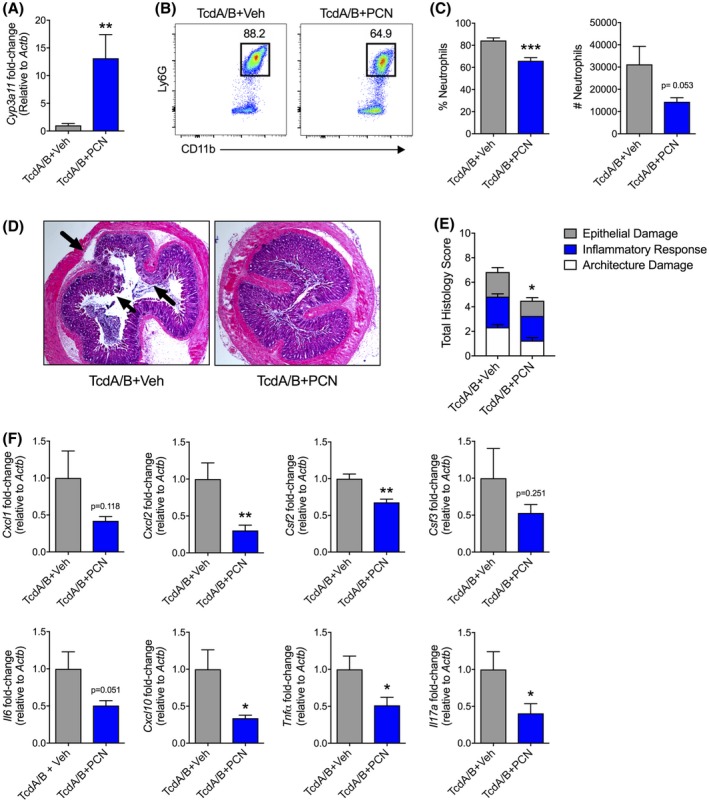
Activation of the PXR provides protection against the colonic inflammation and injury prompted by TcdA/B exposure. C57Bl/6 mice were treated with the mouse‐specific PXR agonist pregnenolone 16α‐carbonitrile (PCN; in sterile corn oil vehicle) 48, 24, and 0.5 h prior to intrarectal instillation of TcdA/B. A, Colonic expression of the PXR target gene Cyp3a11 in mice following treatment with vehicle or PCN. B, Representative FACS plots and (C) associated cell frequencies and counts of infiltrating Ly6G + neutrophils in the colon. D, representative histological images and total damage scores, and (E) the expression of various cytokines in colon 4 h after vehicle‐ or PCN‐treated mice were exposed to TcdA/B. Gene expression was normalized to β‐actin (Actb). All data are expressed as SEM (n = 4‐6 per group). Student's t test, *P < .05, ***P < .005
We next examined the effect of PCN treatment on the inflammatory response and resulting damage following TcdA/B exposure. We found that PCN treatment reduced the percentage and number of infiltrating neutrophils into the colonic LP when compared to vehicle‐treated mice (Figure 6B,C). Further histological assessment of the colon from TcdA/B‐exposed animals showed reduced damage to the colon with only minor epithelial damage and minimal immune cell influx into the mucosa, with limited edema in the layer underlying the muscularis mucosae in mice treated with PCN, which was in contrast to histological sections from vehicle‐treated mice where all parameters were clearly present (Figure 6D). These changes were reflected in the decreased total damage score in PCN‐treated mice exposed to TcdA/B when compared to vehicle‐treated, TcdA/B‐exposed mice (Figure 6E). Finally, when compared to vehicle‐treated mice, PCN significantly reduced colonic expression of Cxcl2, Csf2, Cxcl10, Tnf‐α, and Il‐17a following TcdA/B treatment (Cxcl1 and Il6 expression were also down but did not reach statistical significance; Figure 6F).
4. DISCUSSION
The host immune response is an important determinant of the severity of CDI. Indeed, fecal cytokine levels measured during CDI are a sensitive indicator of disease outcomes and risk of treatment failure.9 This highlights the need to better understand the host immune response to CDI to find ways of modulating intestinal inflammation triggered by C difficile and its pathogenic factors. In this study, we show that signaling through the xenobiotic sensing PXR is important for properly coordinating the immune response triggered by colonic exposure to the C difficile toxins TcdA/B to prevent excessive inflammation and tissue injury. This occurs, in part, through controlling TLR4 activity to prevent increased immune cell influx and cytokine production. By exploiting the role of the PXR in regulating immune responses, we also demonstrated that pharmacological activation of the PXR in mice can prevent colonic injury and inflammation following exposure to the C difficile toxins TcdA and TcdB.
Xenobiotic receptors, including the PXR, aryl hydrocarbon receptor (AhR), and constitutive androstane receptor (CAR) are recognized for their roles in sensing foreign chemicals (xenobiotics) and triggering detoxification and metabolism pathways in different host tissues. However, these receptors can also act as important regulators of inflammation and immunity, especially in the intestine.27, 39, 40, 41 The PXR, a master regulator of the human drug metabolism enzyme CYP3A4 (CYP3A11 in mice), is expressed throughout the intestine and liver where it can “sense” endogenous and exogenous substances. At these sites, activation of the PXR can also exert anti‐inflammatory and pro‐healing effects.42, 43 These protective functions occur in part through a mechanism involving the inhibition of nuclear factor κ‐light‐chain‐enhancer of B cells (NFκB), and subsequent suppression of a number of NFκB target genes including Il6, Ccl2, and Tnf‐α.42, 43, 44 In the present study, we show that the expression and production of pro‐inflammatory cytokines are induced by TcdA/B and produced in substantially higher levels in the colon of PXR‐deficient (Nr1i2−/−) mice, resulting in greater immune cell influx and tissue damage following TcdA/B exposure. Pharmacological activation of the PXR during colonic TcdA/B exposure also significantly reduced the expression of a number of cytokines including Cxcl2, Csf2, Csf3, and Tnf‐α. Together, these results further highlight the immunomodulatory role of the PXR in the intestine and uncover a role for the PXR in modulating the immune response triggered by colonic exposure to pathogenic toxins produced by C difficile.
Following uptake of the pathogenic toxins TcdA/B and the disruption of epithelial cells at the surface of the intestinal mucosa, a robust inflammatory cascade is initiated in large part by NFκB signaling in IECs and underlying immune cells.45, 46, 47, 48 An important early cytokine released during this cascade in humans is IL‐8, which acts as a robust chemoattractant for neutrophil recruitment during CDI.49, 50 In rodents, the homologs to IL‐8 are CXCL1/KC and CXCL2/MIP‐2α and are also increased in colon of mice during CDI to promote neutrophil recruitment.32, 47, 49 The role of infiltrating neutrophils during CDI is enigmatic as neutrophil function is required to help clear the C difficile bacterium, but the robust and non‐specific neutrophil response prompted by pathogenic toxin also leads to considerable bystander damage that is frequently detrimental to the host. In patients with CDI, high levels of fecal IL‐8 are strongly correlated with a worse clinical outcome.27 In mice, two separate studies have shown that blocking CXCL1/KC and the loss of CXCR1 (CXCL2/MIP‐2 receptor) can reduce inflammation and tissue damage in models of CDI,23, 24 highlighting the cytotoxic and pro‐inflammatory role neutrophils can play during CDI. In the current study, we found that pharmacologic activation of the PXR (confirmed by increased expression of Cyp3a11 in the colon) was able to significantly reduce neutrophil influx into the colonic LP following TcdA/B exposure. Decreased neutrophil influx was accompanied by a decrease in Cxcl2 expression as well as decreased Il17a expression, another cytokine shown to be upstream of CXCL1 and CXCL2 and involved in neutrophil recruitment to the intestine.51 By blocking the vital event of neutrophil recruitment, PXR activation was also able to decrease injury in the colon following TcdA/B exposure highlighting the therapeutic role the PXR could play in shaping the host immune response to mitigate bystander damage that during CDI.
The current study also highlights the dynamic responses of other innate immune cells, namely inflammatory monocytes and eosinophils, during C difficile toxin exposure. In mice, Ly6Chi monocytes are rapidly recruited into the colon during models of intestinal inflammation, even before neutrophil influx can be detected, where they acquire a pro‐inflammatory phenotype and become recognized as Ly6Chi inflammatory monocytes.52, 53 In the inflamed colonic LP, Ly6Chi inflammatory monocytes promote inflammation through TLR signaling and cytokine secretion and can potentiate inflammatory pathology and tissue damage, a process that has been described during CDI.21 In the present study, we observed an increase in Ly6Chi monocyte infiltration into the colonic LP following TcdA/B exposure, an effect that was exaggerated in mice lacking the PXR and correlated with increased levels of inflammation and tissue damage in these mice. Other studies have also shown changes in monocyte responses during enteric infections,26 suggesting the PXR may play a role somewhere in the inflammatory cascade controlling the dynamics of monocyte responses.
Eosinophilia in the colon was also a dominant response accompanying increased Ly6Chi monocyte influx in Nr1i2−/− mice following TcdA/B exposure. Like neutrophils, the role of eosinophils during CDI is elaborate and some reports have highlighted a protective role of eosinophils, although the mechanism through which this occurs is not completely understood but may be linked to containment of the C difficile bacteria.20 Alternatively, elevated levels of circulating eotaxin, a chemokine involved in eosinophil recruitment, are associated with more severe CDI in humans highlighting the detrimental effect of host‐driven immune responses involving eosinophils.54 Indeed, increased eosinophil recruitment and activity can trigger and exacerbate intestinal tissue damage and inflammation and the depletion of eosinophils or their recruitment can ameliorate models of intestinal inflammation.55, 56, 57 Eosinophils can produce a number of factors to drive intestinal pathology including enzymes like eosinophil peroxidase, and a number of different cytokines including IL‐6, G‐CSF, and GM‐CSF.36, 37 We observed increased expression of these cytokines in eosinophils isolated from the colon of Nr1i2−/− mice which correlates with increased eosinophil counts and increased G‐CSF, and to a lesser extent, IL‐6 production in the colon of Nr1i2−/− mice when compared to WT mice. G‐CSF.58, 59 and GM‐CSF are important in shaping the function eosinophils (and neutrophils) in tissue by promoting survival and directly increasing the cytotoxic activity of both cell types.60 G‐CSF has been reported to be elevated in the stool of patients with CDI and has been proposed as a biomarker of disease.58 IL‐6 may also be important given that it is one of the most robustly upregulated cytokines during TcdA/B exposure and, like eotaxin, increased levels of IL‐6 are associated with more severe CDI in human patients.54
Signaling through TLR4 and its associated adaptor protein MyD88 serves as an important pathway for immune activation during CDI.22, 61 For example, in mice, cellular release and new synthesis of CXCL1 and CXCL2 to recruit neutrophils can be triggered by activation of TLR4.62, 63 Engagement of the TLR4‐MyD88 signaling axis also triggers the expression of other inflammatory cytokines, in part through NFκB, to direct innate and adaptive immune responses against C difficile and it pathogenic toxins.22 These TLR4‐driven responses at the intestinal barrier are, in part, regulated by the PXR.25, 27 Indeed, Esposito et al29 reported that activation of the PXR could protect Caco‐2 intestinal epithelial cells from TcdA exposure, in part through modulation of TLR4/NFκB signaling.29 This TLR4‐PXR signaling axis is not only important in maintaining the integrity of the intestinal epithelium, but can modulate immune responses during enteric infections,26, 61 and thus drove the rationale behind examining the role of the PXR and targeting the TLR4‐PXR axis during C difficile toxin‐induced inflammation and tissue damage.26, 27, 28 This assertation was supported by our finding of increased expression of Tlr4 in the colon of Nr1i2−/− mice following TcdA/B exposure. Furthermore, blocking TLR4 was able to return the exacerbated immune cell responses and cytokine expression (including G‐CSF and IL‐6) in Nr1i2−/− mice to levels similar to WT mice following TcdA/B exposure. This highlights the role of the PXR in suppressing TcdA/B‐induced inflammation and damage, in part, through TLR4, but also suggests that activation of the TLR4 pathway may be an important event linking the mobilization and recruitment of the different innate immune cell subsets, that could be abrogated by targeting the PXR‐TLR4 signaling axis.
Finally, strategies that target the microbiota are quickly evolving as an important way to treat recurring CDI. The principles behind this involve re‐establishing a healthy microbiota after antibiotic‐driven depletion opens a niche for C difficile to germinate and colonize the colon. Furthermore, suppressing the microbiota could decrease the amount of bacterial products that cross the damaged epithelial barrier to activate underlying immune cells and recruit innate cells including monocytes, neutrophils, and eosinophils. Surprisingly, in the current study, we found that depletion of the microbiota using a broad‐spectrum antibiotic cocktail had no effect on the influx of immune cells triggered by TcdA/B exposure. Thus, the translocation of commensals following epithelial damage may not play a large contributory role in TcdA/B‐induced intestinal inflammation.
In conclusion, the data presented in the current manuscript suggest that the PXR plays a key role in the host's response to TcdA/B and may provide a novel target to dampen the inflammatory tissue damage in C difficile infections. Given the impact of the inflammatory responses on the clinical outcomes in CDI, targeting the PXR, in concert with the use of strategies to directly eradicate C difficile, may provide useful approach to reduce disease burden and enhance patient survival.
CONFLICT OF INTEREST
The authors have nothing to disclose, nor do they have any conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
S. Erickson, K. Flannigan, T. Chang, S. Mani, and S. Hirota designed the experiments and generated overall hypotheses; S. Erickson, L. Alston, K. Nieves, and K. Flannigan performed experiments and analyzed data; S. Erickson, K. Flannigan, and S. Hirota wrote the paper; all authors contributed to the editing and revising the paper.
Supporting information
ACKNOWLEDGMENTS
This work was supported by the International Microbiome Centre (IMC) and the Nicole Perkins Microbial Communities Core of the Snyder Institute, University of Calgary. The IMC is supported by the Cumming School of Medicine, Western Economic Diversification, and Alberta Economic Development and Trade.
Erickson SL, Alston L, Nieves K, et al. The xenobiotic sensing pregnane X receptor regulates tissue damage and inflammation triggered by C difficile toxins. The FASEB Journal. 2020;34:2198–2212. 10.1096/fj.201902083RR
Kyle L. Flannigan and Simon A. Hirota co‐senior authors.
Contributor Information
Kyle L. Flannigan, Email: kyle.flannigan1@ucalgary.ca.
Simon A. Hirota, Email: shirota@ucalgary.ca.
REFERENCES
- 1. Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7:526‐536. [DOI] [PubMed] [Google Scholar]
- 2. Leffler DA, Lamont JT. Clostridium difficile infection. N Engl J Med. 2015;372:1539‐1548. [DOI] [PubMed] [Google Scholar]
- 3. Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:825‐834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ghose C. Clostridium difficile infection in the twenty‐first century. Emerg Microbes Infect. 2013;2:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gerding DN, Lessa FC. The epidemiology of Clostridium difficile infection inside and outside health care institutions. Infect Dis Clin North Am. 2015;29:37‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crobach MJ, Dekkers OM, Wilcox MH, Kuijper EJ. European society of clinical microbiology and infectious diseases (ESCMID): data review and recommendations for diagnosing Clostridium difficile‐infection (CDI). Clin Microbiol Infect. 2009;15:1053‐1066. [DOI] [PubMed] [Google Scholar]
- 7. Kim J, Kim H, Oh HJ, et al. Fecal calprotectin level reflects the severity of clostridium difficile infection. Ann Lab Med. 2017;37:53‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peretz A, Tkhawkho L, Pastukh N, Brodsky D, Halevi CN, Nitzan O. Correlation between fecal calprotectin levels, disease severity and the hypervirulent ribotype 027 strain in patients with Clostridium difficile infection. BMC Infect Dis. 2016;16:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. El Feghaly RE, Stauber JL, Deych E, Gonzalez C, Tarr PI, Haslam DB. Markers of intestinal inflammation, not bacterial burden, correlate with clinical outcomes in Clostridium difficile infection. Clin Infect Dis. 2013;56:1713‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eyre, DW , Walker, AS , Wyllie, D , et al. Predictors of first recurrence of Clostridium difficile infection: implications for initial management. Clin Infect Dis. 2012;55(Suppl 2):S77‐S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Beurden YH, Nezami S, Mulder CJJ, Vandenbroucke‐Grauls C. Host factors are more important in predicting recurrent Clostridium difficile infection than ribotype and use of antibiotics. Clin Microbiol Infect. 2018;24:85 e81‐85 e84. [DOI] [PubMed] [Google Scholar]
- 12. Abt MC, McKenney PT, Pamer EG. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol. 2016;14:609‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lyerly DM, Lockwood DE, Richardson SH, Wilkins TD. Biological activities of toxins A and B of Clostridium difficile . Infect Immun. 1982;35:1147‐1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hunt JJ, Ballard JD. Variations in virulence and molecular biology among emerging strains of Clostridium difficile . Microbiol Mol Biol Rev. 2013;77:567‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nusrat A, von Eichel‐Streiber C, Turner JR, Verkade P, Madara JL, Parkos CA. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect Immun. 2001;69:1329‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Voth DE, Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18:247‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kelly CP, Becker S, Linevsky JK, et al. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest. 1994;93:1257‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88‐mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun. 2012;80:2989‐2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jose S, Madan R. Neutrophil‐mediated inflammation in the pathogenesis of Clostridium difficile infections. Anaerobe. 2016;41:85‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buonomo EL, Cowardin CA, Wilson MG, Saleh MM, Pramoonjago P, Petri WA Jr. Microbiota‐Regulated IL‐25 increases eosinophil number to provide protection during Clostridium difficile infection. Cell Rep. 2016;16:432‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McDermott AJ, Falkowski NR, McDonald RA, et al. Role of interferon‐gamma and inflammatory monocytes in driving colonic inflammation during acute Clostridium difficile infection in mice. Immunology. 2017;150:468‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ryan A, Lynch M, Smith SM, et al. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog. 2011;7:e1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dheer R, Santaolalla R, Davies JM, et al. Intestinal epithelial toll‐like receptor 4 signaling affects epithelial function and colonic microbiota and promotes a risk for transmissible colitis. Infect Immun. 2016;84:798‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McAlees JW, Whitehead GS, Harley IT, et al. Distinct Tlr4‐expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol. 2015;8:863‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garg A, Zhao A, Erickson SL, et al. Pregnane X receptor activation attenuates inflammation‐associated intestinal epithelial barrier dysfunction by inhibiting cytokine‐induced myosin light‐chain kinase expression and c‐Jun N‐terminal kinase 1/2 activation. J Pharmacol Exp Ther. 2016;359:91‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qiu Z, Cervantes JL, Cicek BB, et al. Pregnane X receptor regulates pathogen‐induced inflammation and host defense against an intracellular bacterial infection through toll‐like receptor 4. Sci Rep. 2016;6:31936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Venkatesh M, Mukherjee S, Wang H, et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll‐like receptor 4. Immunity. 2014;41:296‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang K, Mukherjee S, DesMarais V, et al. Targeting the PXR‐TLR4 signaling pathway to reduce intestinal inflammation in an experimental model of necrotizing enterocolitis. Pediatr Res. 2018;83:1031‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Esposito G, Nobile N, Gigli S, et al. Rifaximin improves clostridium difficile toxin a‐induced toxicity in caco‐2 cells by the PXR‐dependent TLR4/MyD88/NF‐kappaB pathway. Front Pharmacol. 2016;7:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hirota SA, Fines K, Ng J, et al. Hypoxia‐inducible factor signaling provides protection in Clostridium difficile‐induced intestinal injury. Gastroenterology. 2010;139(259–269):e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ng J, Hirota SA, Gross O, et al. Clostridium difficile toxin‐induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology. 2010;139:542–552, 552 e541–543. [DOI] [PubMed] [Google Scholar]
- 32. Hirota SA, Iablokov V, Tulk SE, et al. Intrarectal instillation of Clostridium difficile toxin A triggers colonic inflammation and tissue damage: development of a novel and efficient mouse model of Clostridium difficile toxin exposure. Infect Immun. 2012;80:4474‐4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rakoff‐Nahoum S, Paglino J, Eslami‐Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll‐like receptors is required for intestinal homeostasis. Cell. 2004;118:229‐241. [DOI] [PubMed] [Google Scholar]
- 34. Miller KA, Vicentini FA, Hirota SA, Sharkey KA, Wieser ME. Antibiotic treatment affects the expression levels of copper transporters and the isotopic composition of copper in the colon of mice. Proc Natl Acad Sci U S A. 2019;116:5955‐5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17‐producing T cell responses. Nat Immunol. 2007;8:1086‐1094. [DOI] [PubMed] [Google Scholar]
- 36. Weller PF, Spencer LA. Functions of tissue‐resident eosinophils. Nat Rev Immunol. 2017;17:746‐760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. 2013;13:9‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hasegawa M, Kamada N, Jiao Y, Liu MZ, Nunez G, Inohara N. Protective role of commensals against Clostridium difficile infection via an IL‐1beta‐mediated positive‐feedback loop. J Immunol. 2012;189:3085‐3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hudson GM, Flannigan KL, Erickson SL, et al. Constitutive androstane receptor regulates the intestinal mucosal response to injury. Br J Pharmacol. 2017;174:1857‐1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ranhotra HS, Flannigan KL, Brave M, et al. Xenobiotic receptor‐mediated regulation of intestinal barrier function and innate immunity. Nucl Receptor Res. 2016;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lamas B, Natividad JM, Sokol H. Aryl hydrocarbon receptor and intestinal immunity. Mucosal Immunol. 2018;11:1024‐1038. [DOI] [PubMed] [Google Scholar]
- 42. Shah YM, Ma X, Morimura K, Kim I, Gonzalez FJ. Pregnane X receptor activation ameliorates DSS‐induced inflammatory bowel disease via inhibition of NF‐kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1114‐G1122. [DOI] [PubMed] [Google Scholar]
- 43. Terc J, Hansen A, Alston L, Hirota SA. Pregnane X receptor agonists enhance intestinal epithelial wound healing and repair of the intestinal barrier following the induction of experimental colitis. Eur J Pharm Sci. 2014;55:12‐19. [DOI] [PubMed] [Google Scholar]
- 44. Zhou C, Tabb MM, Nelson EL, et al. Mutual repression between steroid and xenobiotic receptor and NF‐kappaB signaling pathways links xenobiotic metabolism and inflammation. J Clin Invest. 2006;116:2280‐2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jefferson KK, Smith MF Jr, Bobak DA. Roles of intracellular calcium and NF‐kappa B in the Clostridium difficile toxin A‐induced up‐regulation and secretion of IL‐8 from human monocytes. J Immunol. 1999;163:5183‐5191. [PubMed] [Google Scholar]
- 46. Kim JM, Lee JY, Yoon YM, Oh YK, Youn J, Kim YJ. NF‐kappa B activation pathway is essential for the chemokine expression in intestinal epithelial cells stimulated with Clostridium difficile toxin A. Scand J Immunol. 2006;63:453‐460. [DOI] [PubMed] [Google Scholar]
- 47. Lee JY, Park HR, Oh YK, et al. Effects of transcription factor activator protein‐1 on interleukin‐8 expression and enteritis in response to Clostridium difficile toxin A. J Mol Med (Berl). 2007;85:1393‐1404. [DOI] [PubMed] [Google Scholar]
- 48. Warny M, Keates AC, Keates S, et al. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL‐8 production, and enteritis. J Clin Invest. 2000;105:1147‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Castagliuolo I, Keates AC, Wang CC, et al. Clostridium difficile toxin A stimulates macrophage‐inflammatory protein‐2 production in rat intestinal epithelial cells. J Immunol. 1998;160:6039‐6045. [PubMed] [Google Scholar]
- 50. Hammond ME, Lapointe GR, Feucht PH, et al. IL‐8 induces neutrophil chemotaxis predominantly via type I IL‐8 receptors. J Immunol. 1995;155:1428‐1433. [PubMed] [Google Scholar]
- 51. Flannigan KL, Ngo VL, Geem D, et al. IL‐17A‐mediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol. 2017;10:673‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bain CC, Scott CL, Uronen‐Hansson H, et al. Resident and pro‐inflammatory macrophages in the colon represent alternative context‐dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6:498‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zigmond E, Varol C, Farache J, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen‐presenting cells. Immunity. 2012;37:1076‐1090. [DOI] [PubMed] [Google Scholar]
- 54. Rao K, Erb‐Downward JR, Walk ST, et al. The systemic inflammatory response to Clostridium difficile infection. PLoS ONE. 2014;9:e92578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Forbes E, Murase T, Yang M, et al. Immunopathogenesis of experimental ulcerative colitis is mediated by eosinophil peroxidase. J Immunol. 2004;172:5664‐5675. [DOI] [PubMed] [Google Scholar]
- 56. Maltby S, Wohlfarth C, Gold M, Zbytnuik L, Hughes MR, McNagny KM. CD34 is required for infiltration of eosinophils into the colon and pathology associated with DSS‐induced ulcerative colitis. Am J Pathol. 2010;177:1244‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vieira AT, Fagundes CT, Alessandri AL, et al. Treatment with a novel chemokine‐binding protein or eosinophil lineage‐ablation protects mice from experimental colitis. Am J Pathol. 2009;175:2382‐2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Darkoh C, Turnwald BP, Koo HL, et al. Colonic immunopathogenesis of Clostridium difficile infections. Clin Vaccine Immunol. 2014;21:509‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pawlowski SW, Calabrese G, Kolling GL, et al. Murine model of Clostridium difficile infection with aged gnotobiotic C57BL/6 mice and a BI/NAP1 strain. J Infect Dis. 2010;202:1708‐1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Griseri T, Arnold IC, Pearson C, et al. Granulocyte macrophage colony‐stimulating factor‐activated eosinophils promote interleukin‐23 driven chronic colitis. Immunity. 2015;43:187‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hung YP, Lin HJ, Wu TC, et al. Risk factors of fecal toxigenic or non‐toxigenic Clostridium difficile colonization: impact of Toll‐like receptor polymorphisms and prior antibiotic exposure. PLoS ONE. 2013;8:e69577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. De Filippo K, Henderson RB, Laschinger M, Hogg N. Neutrophil chemokines KC and macrophage‐inflammatory protein‐2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J Immunol. 2008;180:4308‐4315. [DOI] [PubMed] [Google Scholar]
- 63. De Filippo K, Dudeck A, Hasenberg M, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood. 2013;121:4930‐4937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials