

Abstract
Background:
Obesity-related hypertension is a common disorder, and attempts to combat the underlying obesity are often unsuccessful. We previously revealed that mice globally deficient in the inhibitory IgG receptor FcγRIIB are protected from obesity-induced hypertension. However, how FcγRIIB participates is unknown. Studies were designed to determine if alterations in IgG contribute to the pathogenesis of obesity-induced hypertension.
Methods:
Involvement of IgG was studied using IgG μ heavy chain-null mice deficient in mature B cells, and by IgG transfer. Participation of FcγRIIB was interrogated in mice with global or endothelial cell-specific deletion of the receptor. Obesity was induced by high fat diet (HFD) and BP was measured by radiotelemetry or tail cuff. The relative sialylation of the Fc glycan on mouse IgG, which influences IgG activation of Fc receptors, was evaluated by SNA-lectin blotting. Effects of IgG on endothelial NO synthase (eNOS) were assessed in human aortic endothelial cells. IgG Fc glycan sialylation was interrogated in 3442 human subjects by mass spectrometry, and the relationship between sialylation and BP was evaluated. Effects of normalizing IgG sialylation were determined in HFD-fed mice administered the sialic acid precursor N-acetyl-D-mannosamine (ManNAc).
Results:
Mice deficient in B cells were protected from obesity-induced hypertension. Compared to IgG from control chow-fed mice, IgG from HFD-fed mice was hyposialylated, and it raised BP when transferred to recipients lacking IgG; the hypertensive response was absent if recipients were FcγRIIB-deficient. Neuraminidase (NA)-treated IgG lacking the Fc glycan terminal sialic acid also raised BP. In cultured endothelial cells, via FcγRIIB, IgG from HFD-fed mice and NA-treated IgG inhibited VEGF activation of eNOS by altering eNOS phosphorylation. In humans obesity was associated with lower IgG sialylation, and systolic BP was inversely related to IgG sialylation. Mice deficient in FcγRIIB in endothelium were protected from obesity-induced hypertension. Furthermore, in HFD-fed mice, ManNAc normalized IgG sialylation and prevented obesity-induced hypertension.
Conclusions:
Hyposialylated IgG and FcγRIIB in endothelium are critically involved in obesity-induced hypertension in mice, and supportive evidence was obtained in humans. Interventions targeting these mechanisms, such as ManNAc supplementation, may provide novel means to break the link between obesity and hypertension.
Keywords: endothelium, Fc receptor, hypertension, immunoglobulin, nitric oxide synthase, obesity, sialic acid
Introduction
It is well-recognized that obesity is a major risk factor for hypertension, with obesity underlying up to 75% of cases of primary, or essential, hypertension(1). Worldwide there are more than 2.5 billion adults who are obese (body mass index or BMI > 30 kg/m2) or overweight (BMI > 25 kg/m2), and in the United States 65% of adults are overweight and 36% are obese(2). Regrettably attempts to combat the underlying obesity, which focus on exercise and diet, are often insufficiently successful in the long-term(3). Furthermore, obesity often results in hypertension that is not responsive to single agent therapy(4). Most importantly, hypertension is the number one underlying cause of stroke, and it greatly increases the risk of coronary heart disease and chronic kidney disease(5). Therefore, there is a dire need for new strategies to prevent or treat obesity-related hypertension.
The basis for obesity-induced hypertension has been intensely studied, and the primary underlying processes which are currently favored relate to increases in renal tubular sodium resorption, in renin-angiotensin system activation, and in sympathetic nervous system activity(1; 6). We previously revealed that mice globally deficient in the inhibitory IgG receptor FcγRIIB are protected from the blood pressure (BP) elevation that accompanies high fat diet-induced obesity(7). However, how FcγRIIB participates in obesity-induced hypertension is unknown.
In recent studies of obesity-induced insulin resistance in mice, with corroborating findings in human type 2 diabetics, we made the surprising discovery that the insulin resistance is driven by an altered post-translational modification in IgG that leads to enhanced activation of FcγRIIB in endothelial cells. As a result, there is an attenuation in insulin transcytosis across endothelial cells and delivery to skeletal muscle myocytes where up to 80% of glucose disposal occurs(8). Based on these findings and our prior work implicating FcγRIIB in obesity-induced hypertension(7), in the present project we employed mice to test the hypothesis that IgG participates in obesity-induced hypertension. Additional experiments were performed to address the following questions: 1) Does the involvement of IgG entail the hyposialylation of the glycan bound to Asn297 in the IgG Fc region, which increases the affinity of Fc receptors for IgG(9); 2) Is there evidence of an inverse relationship between relative IgG Fc sialylation and BP in humans? 3) Is the hypertensive action of IgG in obesity in mice mediated by FcγRIIB, particularly FcγRIIB in endothelial cells; and 4) Does normalization of IgG sialylation in mice prevent obesity-induced hypertension?
Methods
The data and analytical methods will be made available to any researchers for purposes of reproducing the results or replicating the procedure. Information regarding materials will be made available to any interested researchers upon request.
Animal Model
All experiments were performed in male C57BL/6 mice. To study the role of B lymphocytes in obesity-induced hypertension, IgG μ heavy chain-null mice, designated B−/−, were employed. To evaluate involvement of the inhibitory IgG receptor FcγRIIB in responses to administered IgG, B−/− mice were crossed with FcγRIIB−/− mice, and the effects of IgG were compared in resulting B−/−;FcγRIIB+/+ and B−/−;FcγRIIB−/− male offspring. The participation of endothelial FcγRIIB was assessed by comparing findings in FcγRIIB floxed mice (FcγRIIBfl/fl) serving as controls and mice lacking the receptor selectively in endothelial cells, designated FcγRIIBΔEC (10). The mice received either a control diet (CON) (Research Diets, Inc. D12329, with 11 kcal% fat) or a high fat diet (HFD) (Research Diets, Inc. D12331, with 58 kcal% fat) beginning at 4–5 weeks of age. All mice were maintained in animal facilities with a 12 hour light and 12 hour dark cycle.
In experiments determining the effects of IgG, total IgG isolated from either control diet-fed or HFD-fed donor mice was injected intraperitoneally (150μg/dose) twice a week into recipient B−/− mice. To assess the effects of transfer of hyposialylated IgG, the terminal sialic acid was removed by treatment of control total IgG (500 ug) with neuraminidase (NA, 100units in 0.05M sodium citrate buffer, pH 6.0 at 37°C for 20h). Although a specific IgG subclass was determined to be hyposialylated in mice with diet-induced obesity, limitations in the abundance of separately-isolated IgG subclasses precluded their use in IgG transfer studies. The regimen for IgG transfer mirrored the approach used in prior studies(8,11). In initial studies the decline in IgG following a single dose administered to B−/− mice was evaluated (Suppl. Fig. 1). The half-life of control, HFD and NA-treated IgG was similar, with mean values ranging from 4.9 to 6.7 days. In studies of the sialic acid precursor N-acetyl-D-mannosamine (ManNAc), mice received drinking water alone or drinking water containing ManNAc (1%)(8). The care and use of mice was approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Blood Pressure (BP) Measurement by Radiotelemetry
In experiments determining the role of B cells in obesity-induced hypertension, radiotelemetry was performed as described previously(12) (7). After maintenance on control chow postweaning, instrumentation was done at 21 weeks of age, and following recovery baseline BP measurements were made 3 weeks later. One week thereafter the diet was switched to HFD, and BP measurements were repeated following 12 weeks of HFD feeding. The instrumentation entailed anesthetization with isofluorane and placement of a left carotid artery catheter and a radiotelemetry device (PA-C10, Data Sciences International, St. Paul, MN). During recording periods the BP, heart rate, and activity were recorded over 10sec duration every 15min, and values were averaged over 1h. In our prior radiotelemetry studies with comparable diets and design, hypertension was demonstrated in HFD-fed mice relative to control chow-fed mice(7).
Tail Cuff Blood Pressure (BP) Measurement
Tail cuff systolic BP (SBP) measurements were done using a Visitech System BP-2000 Series II (Apex, NC) BP monitor. Tail cuff SBP measurements were necessary in the IgG transfer experiments because the restraining required to administer IgG disrupted the internalized hardware in mice instrumented for radiotelemetry. Mice received tail cuff BP measurement training for 5 days following 12–14 weeks of control chow or HFD feeding, undergoing 20 measurements over a 30 min period each day. Four weeks later the mice underwent a repeat 4 day training period and on the following day (day 5) the mean of the last 10 readings was recorded as the SBP value for a given mouse. In our prior studies with comparable diets and design, an elevation in SBP in HFD-fed mice relative to control chow-fed mice was readily observed with tail cuff measurements(7).
IgG Isolation and SNA Lectin Blotting
Total IgG was isolated from control chow versus HFD-fed mice using Melon Gel IgG Purification Resin (Pierce Biotechnology). In evaluations of the terminal sialylation of the Fc glycan on mouse IgG, the three subclasses of mouse IgG that bind to FcγRIIB, IgG1, IgG2b and IgG2c, were obtained(8). Relative terminal sialylation was then assessed by lectin blotting using SNA (Vector Laboratories), which binds specifically to sialic acid attached to terminal galactose via α−2–6 linkage(8).
IgG Sialylation, FcγRIIB and NO Synthase Activity in Human Endothelial Cells
To determine how IgG impacts VEGF activation of endothelial NO synthase (eNOS), experiments were performed in primary human aortic endothelial cells (HAEC)(8). Cells were preincubated with IgG isolated from control chow versus HFD-fed mice for 20 min (10ug/ml), and VEGF (100ng/ml) activation of eNOS was then evaluated in the continued presence of the IgG by measuring 14C-L-arginine conversion to 14C-L-citrulline during a 30min incubation(13). To assess the role of IgG hyposialylation in eNOS antagonism, HAEC were incubated with control versus NA-treated total IgG from chow-fed mice; limitations in the abundance of separately-isolated IgG subclasses precluded their use. In additional experiments the impact of control, HFD and NA-treated IgG on eNOS expression was evaluated following 48h treatment by immunoblotting, and acute effects on changes in eNOS Ser1177 and Thr495 phosphorylation in response to VEGF were also assessed by immunoblotting (14). To determine the role of FcγRIIB, additional studies were done in which HAEC were transfected 48h earlier with either control siRNA (Dharmacon) or FcγRIIB-targeting siRNA (Thermo Fisher Scientific, s5074; sense: CAACAUUCUGUUUACCUUUtt; antisense: AAAGGUAAACAGAAUGUUGtt).
IgG Fc Glycan Sialylation and BP in Human Subjects
The relationship between IgG Fc glycan sialylation and BP in humans was evaluated using data from the Croatian national biobank “10,001 Dalmatians”. A total of 3442 adult volunteers 18 or more years of age were studied. They were recruited from the island of Korcula (n=951), the village of Smokvica (n=710), the island of Vis (n=794), and the city of Split (n=987). The subjects gave written informed consent, and the study was approved by the following human research committees: Multi-Centre Research Ethics Committee for Scotland (MREC/1/0/71), Ethics Committee of the Faculty of Medicine, Andrija Stampar School of Public Health, Zagreb, Croatia (reference 018057), University of Zagreb, School of Medicine, Ethics Committee (number 04-1097-2006), Lothian NHS Board (South East Scotland Research Ethics committees; REC 11/AL/0222), and the University of Split, School of Medicine, Ethics Committee (ref. 003-08/11-03/0005). BP was measured by trained clinical staff using a mercury-based sphygnomanometer while sitting with the forearm at heart level after 10 min of resting during two separate clinic visits (n= 2,491) or on one occasion (n=951). During each visit BP was measured twice and the readings were averaged, and when BP was measured on two separate clinic visits, readings were also averaged. On a weekly basis BP measurements were validated against a mannequin arm and drift analysis was performed. The N-glycan profile of the Fc region of IgG was determined using liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS)(15; 16), and findings for the N-glycome of the most abundant isoform of IgG, IgG1, were used in the analysis.
Statistical Analysis
Statistical analysis for the studies performed in mice or in cell culture was performed using Student’s t tests to compare mean values of two groups, and using ANOVA to compare means of three or more groups. Tukey’s multiple comparison test was used for post-hoc analysis. GraphPad Prism7 software was employed. Differences were considered significant at p<0.05.
In the analysis of the LC-ESI-MS glycan data, to remove technical variation the data were normalized by total chromatogram area, log-transformed and batch corrected using ComBat method (R package “sva”). The percentage of sialylation was calculated as the sum of the glycan peaks with sialylated structures (after normalization and batch correction). In addition, prior to statistical modeling the percentage of sialylation was transformed to standard normal distribution by inverse transformation of ranks to normality (R package “GenABEL”). To evaluate the factors that impact IgG sialylation, a mixed model was fitted; sialylation was the dependent variable and the independent variables were sex, age, presence versus absence of antihypertensive therapy, and BMI, and the recruitment location was modelled as a random factor to account for stratified sampling. The relationship between sialylation and systolic BP (SBP) and diastolic BP (DBP) was then evaluated in subjects not on antihypertensive medication (n= 2514) using a stepwise mixed modeling method. A series of four nested models had SBP or DBP as the dependent variable, and sialylation (Model 1), sialylation and sex (Model 2), sialylation and sex and age (Model 3), and sialylation and sex and age and BMI (Model 4) as independent variables, and recruitment site as a random factors (all models).
Results
Role of IgG
To initially determine the potential role of IgG in obesity-induced hypertension, studies were performed in control B+/+ mice and B−/− mice lacking mature B cells and IgG production. The mice were instrumented for radiotelemetry, baseline BP was measured 3 weeks later, the mice were switched to HFD, and BP was remeasured after 12 weeks of HFD intake. Body weights were similar in B+/+ and B−/− mice at baseline, both groups gained weight with HFD feeding, and after 12 weeks of HFD the weights were also similar (Fig. 1A). Systolic BP (SBP) was comparable in the two genotype groups at baseline, and the expected diurnal variation was observed (Fig. 1B,C). In wild-type B+/+ mice, HFD feeding resulted in an increase in SBP during both light and dark cycles. In contrast, in B−/− mice, despite weight gain equal to that of wild-type controls, there was no increase in SBP with HFD feeding. Parallel observations were made for diastolic BP (DBP, Fig. 1D,E) and for mean arterial pressure (MAP, Fig. 1F,G). However, the increase in heartrate with diet-induced obesity occurred similarly in the two genotype groups (Fig. 1H,I). Thus, B cell-deficient mice incapable of IgG production are protected from obesity-induced hypertension but not from obesity-induced tachycardia.
Figure 1:
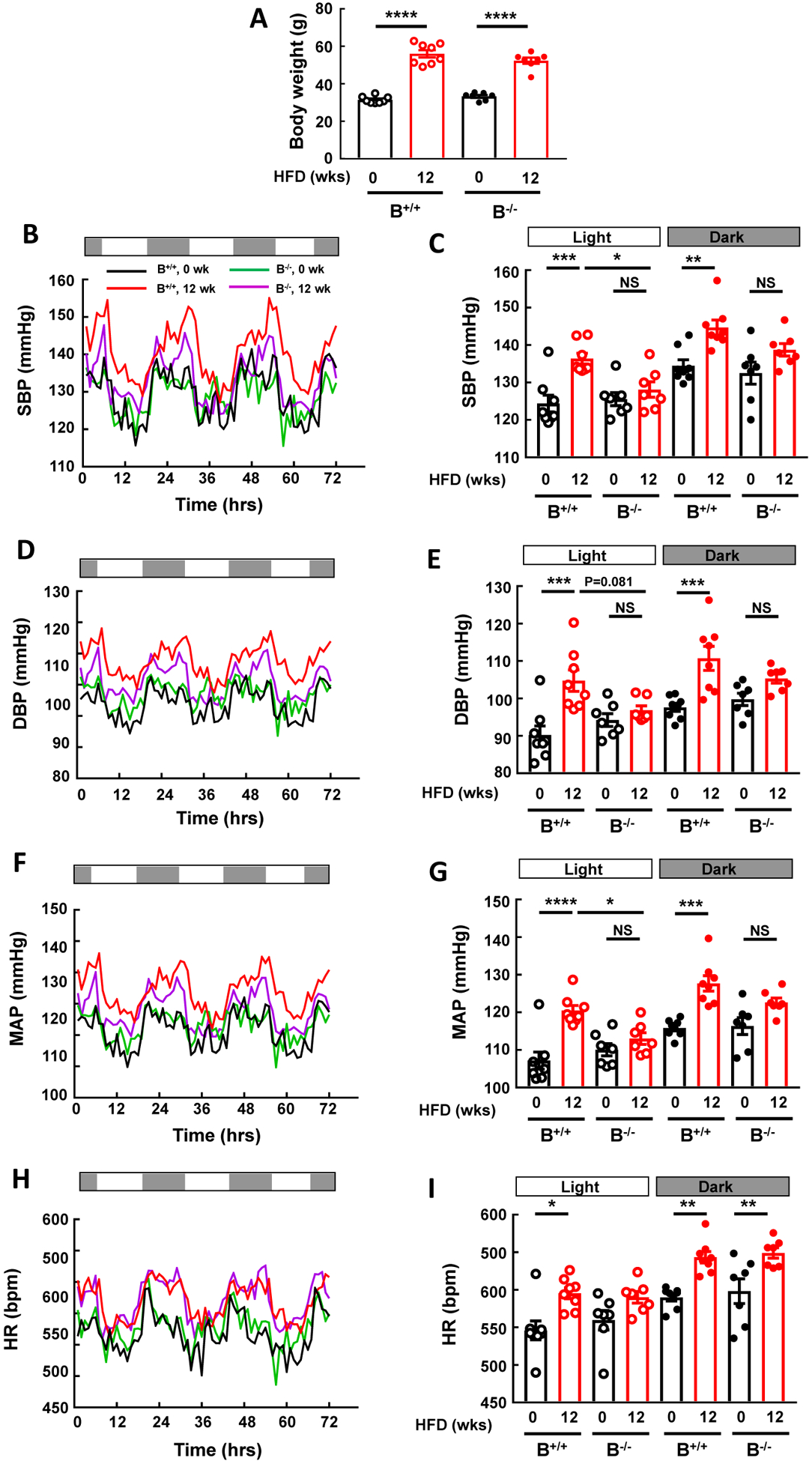
B cell deficiency provides protection from obesity-induced hypertension. Blood pressure was measured by radiotelemetry at baseline and following 12 weeks of high fat diet (HFD) feeding in wild-type mice with normal B cell abundance (B+/+) and in IgG μ heavy chain-null mice, designated B−/−, which lack mature B cells and IgG production. Body weights were similar in the two genotype groups at the start and after HFD feeding (A). B-C. Systolic BP (SBP) recordings over 72h (B) and summary data during light and dark periods (C). D-E. Diastolic BP (DBP) recordings over 72h (D) and summary data during light and dark periods (E). F-G. Mean arterial pressure (MAP) recordings over 72h (F) and summary data during light and dark periods (G). H-I. Heartrate (HR) recordings over 72h (H) and summary data during light and dark periods (I). Values in bar graphs are mean±SEM, n=7–8 per group, *p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
To further determine if IgG participates in obesity-induced hypertension, IgG transfer experiments were performed in mice that underwent tail cuff SBP measurements. B−/− mice, which are protected from obesity-induced hypertension (Fig. 1), were placed on HFD at weaning, and beginning 12–14 weeks later the mice received IP injections of IgG from control chow-fed versus HFD-fed mice twice a week. SBP was evaluated 4 weeks later. At that time body weights were similar in the control IgG and HFD IgG groups (Fig. 2A). However, SBP was greater in the mice who received HFD IgG (Fig. 2B). As such, IgG from mice with diet-induced obesity invokes an increase in BP when transferred into recipient mice incapable of IgG production. When combined with the results in Fig. 1, these findings indicate that IgG plays an important role in obesity-induced hypertension.
Figure 2:
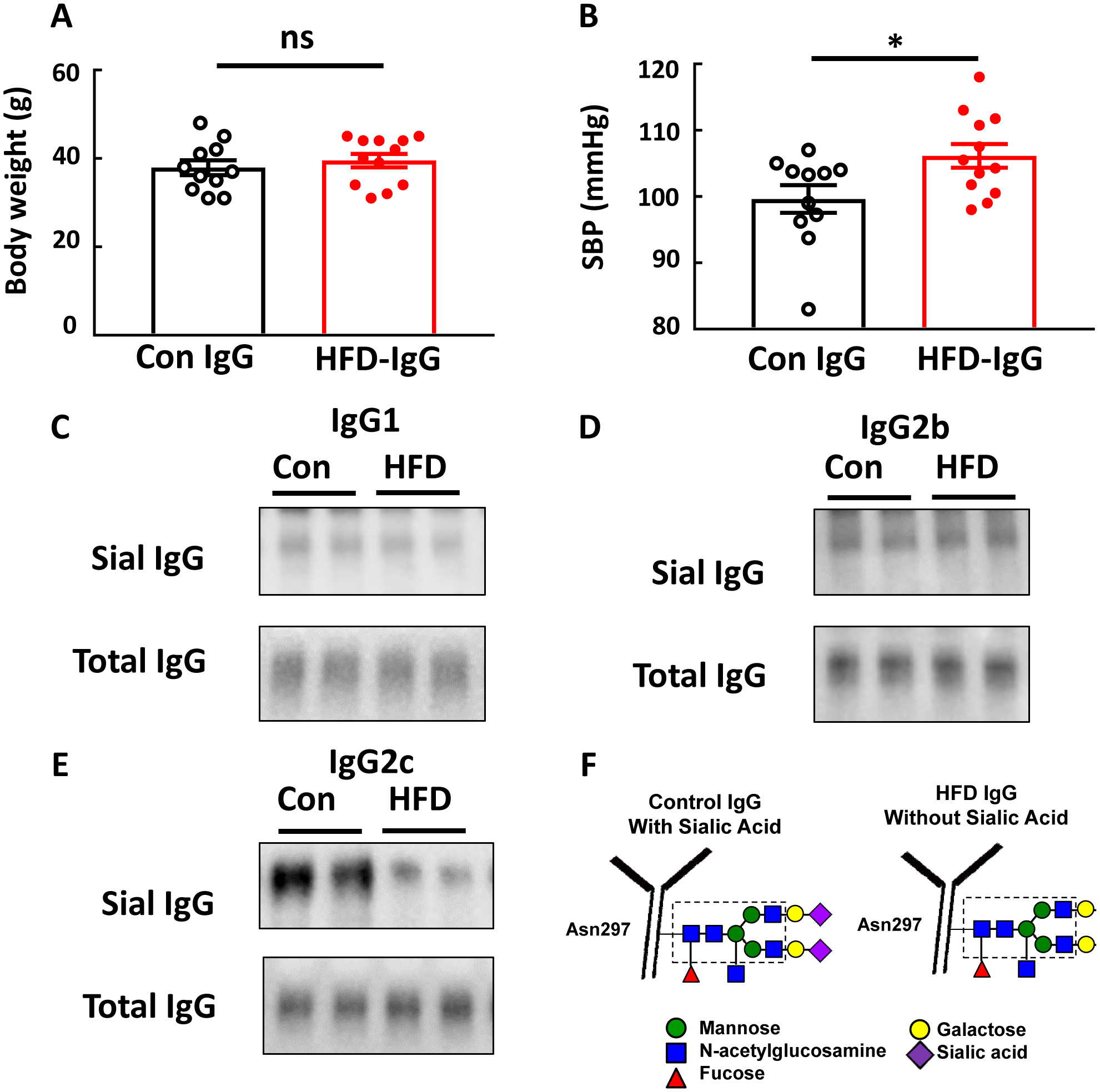
IgG isolated from high fat diet (HFD)-fed mice raises blood pressure in B−/− mice and is hyposialylated. Male B−/− mice were fed a HFD for 12–14 weeks, and over the ensuing 4 weeks while remaining on HFD, they were i.p. injected with IgG (150μg/mouse, 2 times/week) isolated from wild-type mice fed either a control diet (Con IgG) or a HFD (HFD IgG). Body weight was determined (A), and the mice underwent systolic blood pressure (SBP) measurements by tail cuff (B). In parallel studies the three subclasses of IgG which bind to FcγRIIB, IgG1, IgG2b and IgG2c were isolated from control diet- and HFD-fed mice and lectin blotting was performed using SNA (C-E, respectively). In F, the relative loss of the terminal sialic acid bound to Asn297 on the Fc glycan of IgG from HFD-fed mice is illustrated. All blots show findings for two samples per group. In A,B, values are mean±SEM, n=11–12 per group, * p<0.05.
Role of IgG Hyposialylation
We previously demonstrated that the inhibitory IgG receptor FcγRIIB participates in obesity-induced hypertension(7). The binding affinity of IgG for Fc receptors is influenced by the relative sialylation of the glycan bound to Asn297 in the Fc region of IgG(9), and in mice the IgG1, IgG2b and IgG2c subclasses bind to FcγRIIB(17). To further understand how IgG participates in obesity-induced hypertension, we assessed the degree of sialylation of these IgG subclasses isolated from total IgG from control chow- versus HFD-fed mice. Paralleling our prior observations(8), whereas SNA lectin blotting revealed that the relative sialylation of the glycan on IgG1 and on IgG2b was similar for control chow- and HFD-fed mice, compared with IgG2c from control chow-fed mice, IgG2c from HFD-fed mice was hyposialylated (Fig. 2C–F).
To determine if the hyposialylation of IgG participates in obesity-induced hypertension, IgG transfer experiments were performed administering either control-treated or neuraminidase (NA)-treated IgG to B−/− mice lacking endogenous IgG production. Fig. 3A shows an SNA-lectin blot demonstrating effective loss of sialic acid from the IgG Fc glycan with NA treatment, which is illustrated in Fig. 3B. Following the period of IgG injection, body weights were similar in the control IgG and NA IgG groups (Fig. 3C). However, SBP was greater in the mice who received NA-IgG (Fig. 3D).
Figure 3:
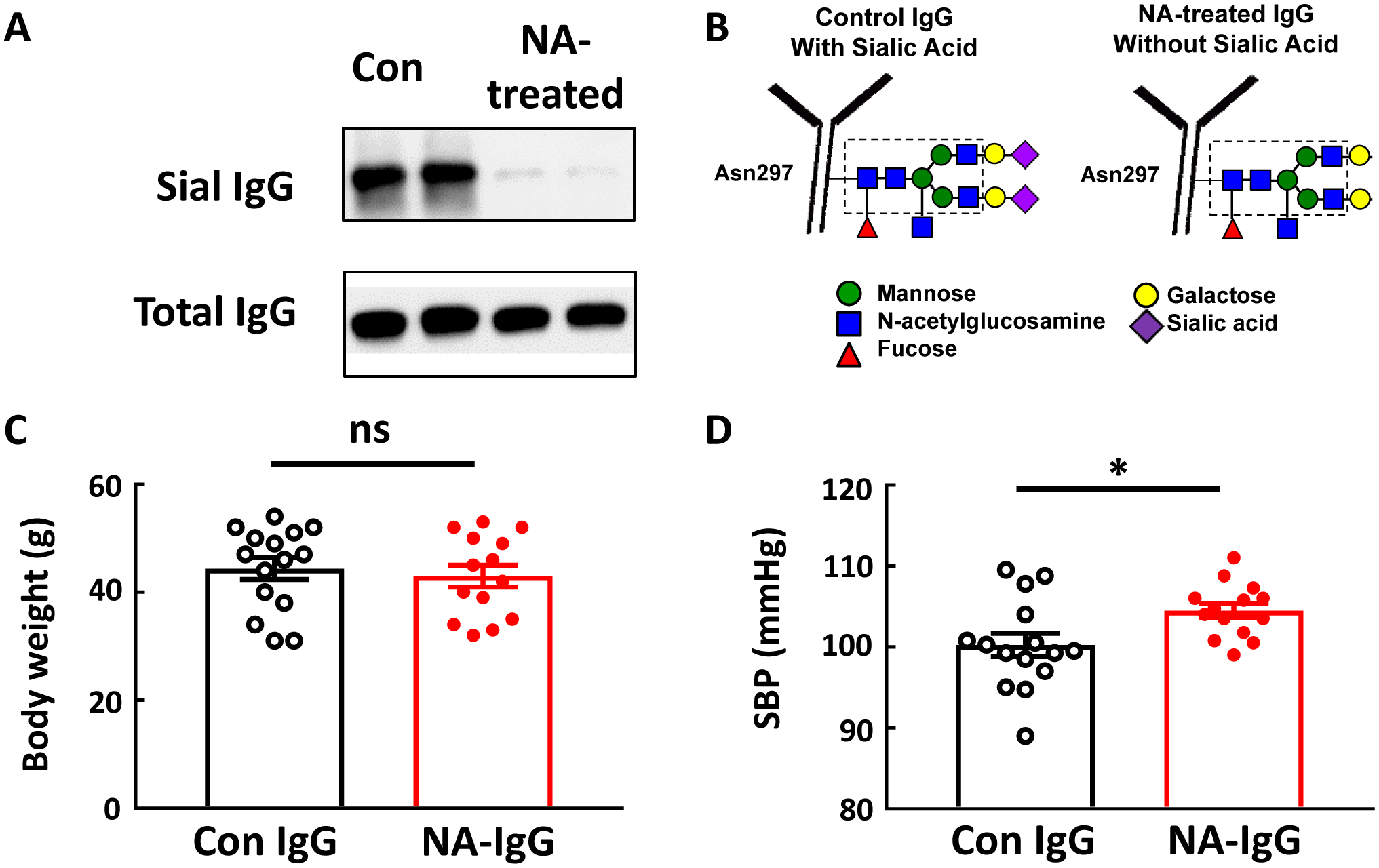
IgG hyposialylated by neuraminidase treatment raises blood pressure in B−/− mice. IgG isolated from control chow-fed mice was incubated with vehicle versus neuraminidase (NA) to remove the terminal sialic acid from the Fc glycan, and SNA-lectin blotting was performed (A). The blot shows findings for two samples per group. The NA treatment-induced loss of sialic acid from the IgG glycan attached to Asn297 in the Fc region is illustrated in B. Male B−/− mice were fed a high fat diet (HFD) for 12–14 weeks, and over the ensuing 4 weeks while remaining on HFD, they were i.p. injected with either the control-treated IgG (Con IgG, 150μg/mouse, 2 times/week) or NA-treated IgG. Body weight was determined (C), and the mice underwent systolic blood pressure (SBP) measurements by tail cuff (D). Values are mean±SEM, n=14–15 per group, * p<0.05.
In prior work we discovered that IgG from HFD-fed mice inhibits eNOS activation by VEGF. However, the characteristic(s) of the HFD IgG responsible and the mechanisms underlying the inhibition of VEGF action remained unknown(7). We evaluated how IgG hyposialylated by NA treatment impacts VEGF stimulation of eNOS in HAEC, and we determined the role of FcγRIIB in the actions of HFD IgG and NA IgG. Whereas VEGF activation of eNOS was unchanged in cells exposed to control IgG, the activation was fully attenuated in a parallel manner in cells treated with HFD IgG or NA IgG (Fig. 4A). Similar findings were obtained in siRNA control cells expressing endogenous FcγRIIB (Fig. 4B,C). In contrast, the knockdown of FcγRIIB prevented the inhibition of eNOS activation by VEGF caused by either HFD IgG or NA IgG. The effects of HFD IgG and NA-treated IgG on eNOS did not entail changes in eNOS expression (Fig. 4D,E). Instead, they involved blunting of VEGF-induced eNOS Ser1177 phosphorylation and eNOS Thr 495 dephosphorylation (Fig. 4F–H). Thus, hyposialylated IgG antagonizes VEGF activation of eNOS via effects on enzyme phosphorylation, and the effects of purposefully hyposialylated IgG both in culture and in vivo paralleled those observed with IgG from HFD-fed mice. Collectively these findings reveal that IgG hyposialylation contributes to obesity-induced hypertension.
Figure 4:
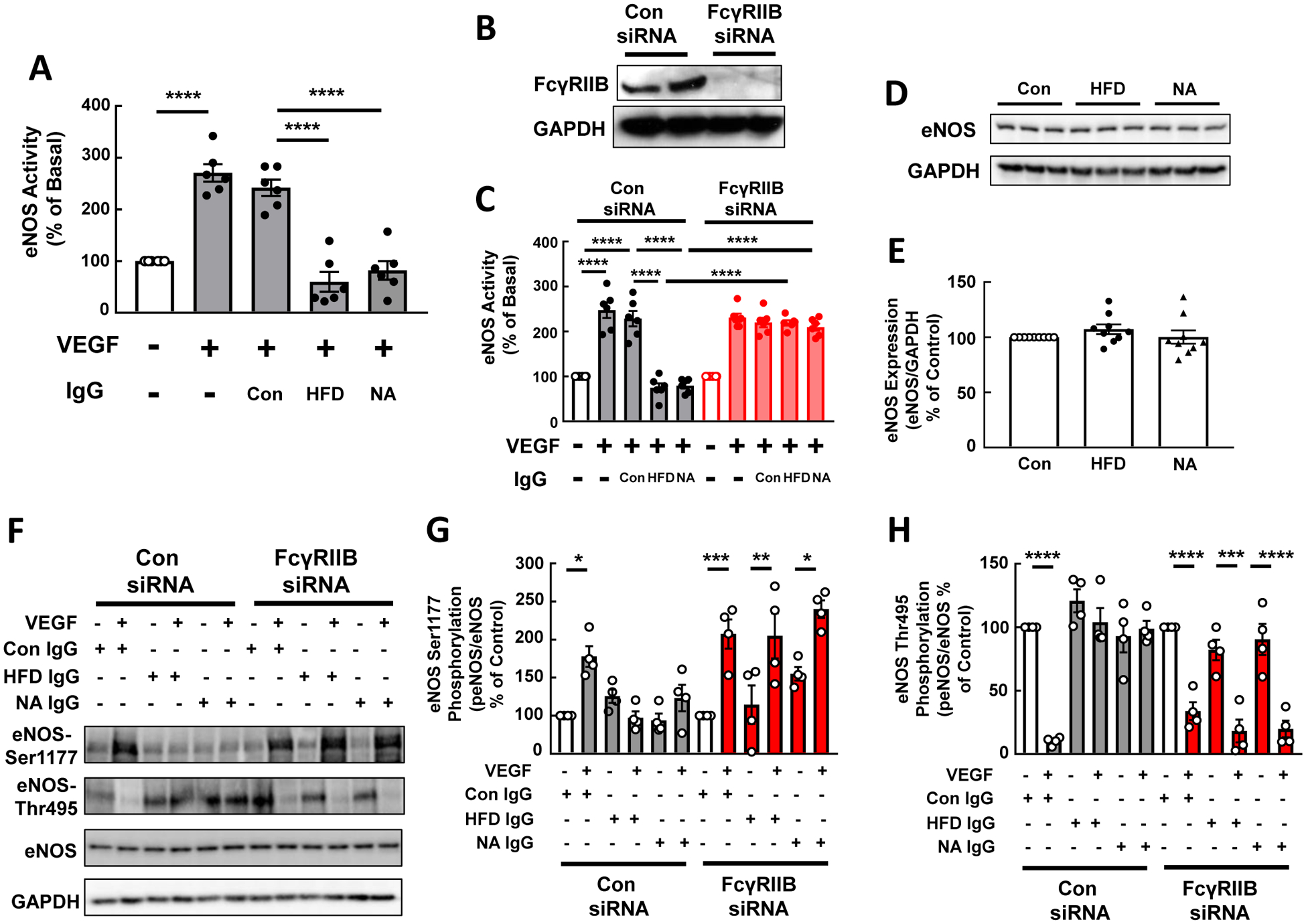
IgG from high fat diet (HFD)-fed mice and IgG hyposialylated by neuraminidase treatment inhibit eNOS activation by VEGF in human endothelial cells via FcγRIIB. A. 14C-L-arginine conversion to 14C-L-citrulline under basal conditions or in response to VEGF (100ng/ml) was measured in intact primary human aortic endothelial cells over 15min. The effects of IgG isolated from wild-type mice fed a control diet (Con IgG) or HFD (HFD IgG), or Con IgG treated with neuraminidase (NA) (NA IgG) were evaluated. B-C. Parallel experiments were performed in cells transfected with either control siRNA or FcγRIIB-targeting siRNA. Effective loss of FcγRIIB protein with siFcγRIIB transfection was confirmed by immunoblot analysis, and findings for two samples per group are shown in B. D-E. Total eNOS protein abundance was evaluated by immunoblot analysis following 48h treatment with Con IgG, HFD IgG or NA IgG. Findings for 3 samples per group are shown (D), and summary data are provided (E). F-H. Changes in eNOS Ser1177 and Thr495 phosphorylation in response to VEGF (100ng/ml) were evaluated by immunoblot analysis in cells treated with Con IgG, HFD IgG or NA IgG. Representative findings are shown (F) and summary data are provided (G,H). In bar graphs values are mean±SEM, in G and H, n=4. *p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
IgG Sialylation and BP in Humans
The human subjects ranged in age from 18 to 98, 39% were males and 61% were females, and 27% percent (928 of 3442) were on antihypertensive agents. Twenty-nine percent had a BMI less than 25, 44% were overweight with a BMI greater than or equal to 25 and less than 30, and 26% were obese with a BMI greater than or equal to 30.
A number of factors were found to be associated with the relative degree of IgG Fc glycan sialylation (Fig 5A). There was a trend for greater sialylation in females versus males (p=0.055), and an inverse relationship between age and IgG sialylation, with sialylation decreased by 0.025 SD with each 1 year increase in age. Of particular relevance to the present study, there was also an inverse relationship between BMI and IgG sialylation, with sialylation decreased by 0.019 SD with each increase of BMI by 1 unit. This association between obesity and lower IgG Fc glycan sialylation in humans is consistent with what we have observed in HFD-fed versus control chow-fed mice (Fig. 2). Furthermore, subjects on antihypertensive medications had 0.07 SD lower sialylation than those not on such medications. If one considers hypertension treatment as an identifier of hypertensive subjects, this finding is in agreement with prior reports in humans of an association between hyposialylation and hypertension(18–20).
Figure 5:
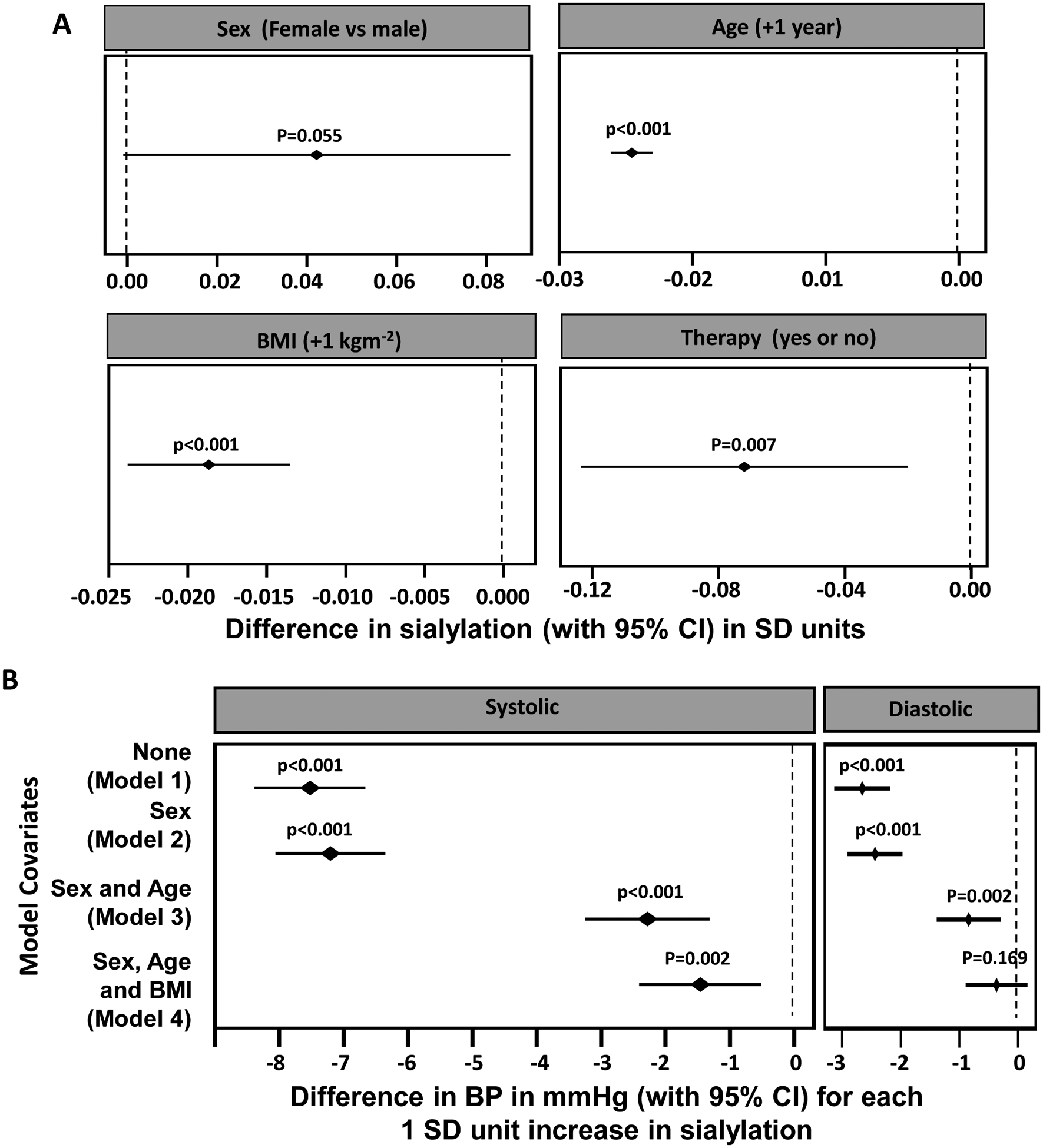
BP is negatively associated with the relative degree of IgG Fc sialylation in humans. A. Relationships between sex, age, BMI, use of antihypertensive medications and IgG Fc sialylation. The forest plots depict the change in sialylation in SD units (with 95% CI) for females compared with males, for each 1 year increase in age, for each 1 kg/m2 increase in BMI, and for the presence versus absence of antihypertensive therapy. B. The relationship between sialylation and systolic BP (SBP) and diastolic BP (DBP) was evaluated in subjects not on antihypertensive medication (n= 2514) using a stepwise mixed modeling method. The forest plots depict the change in BP in mmHg (with 95% CI) for each 1 SD unit increase in sialylation. P value is the probability of observing an effect of this size assuming the true effect is zero (indicated by dashed vertical line).
To evaluate the association between IgG sialylation and BP, analysis was limited to the 2514 subjects not receiving antihypertensive therapy, thereby avoiding confounding by the influence of such treatment on BP. Stepwise mixed modeling was performed to additionally take into account the influences of sex, age and BMI, which impact IgG sialylation (Fig. 5A) and BP(21–23) (Fig. 5B). In the unadjusted model (Model 1), a 1 SD increase in IgG sialylation was associated with a 7.5 mmHg decline in SBP and a 2.7 mmHg decline in DBP. Whereas the findings changed minimally with adjustment for sex (Model 2), following adjustment for sex and age (Model 3), a 1 SD increase in IgG sialylation was associated with a 2.3 mmHg decline in SBP and a 0.8 mmHg decline in DBP. In the fully-adjusted model that additionally takes into account BMI (Model 4), a 1 SD increase in IgG sialylation was associated with a 1.5 mmHg lower SBP (p=0.001) and there was no significant association between IgG sialylation and DBP (p=0.179). Thus, the data suggest that in humans that there is an inverse relationship between obesity and IgG sialylation, and an inverse relationship between IgG sialylation and systolic BP.
Role of FcγRIIB
FcγRIIB is the sole inhibitory Fc receptor in both mice and humans(24). Its role in the BP responses to IgG isolated from HFD-fed mice was evaluated in IgG transfer studies in which the recipients were either B−/− or B−/−;FcγRIIB−/−. Following HFD IgG administration the body weights were similar in the two genotype groups (Fig. 6A). However, SBP was lower in the mice lacking FcγRIIB (Fig. 6B). In fact, the deletion of FcγRIIB in B−/− background mice yielded SBP values despite HFD IgG administration that mirrored the levels observed in B−/− recipients of control IgG (Fig. 2B). Thus, the elevation in systolic blood pressure caused by HFD IgG is mediated by FcγRIIB.
Figure 6:
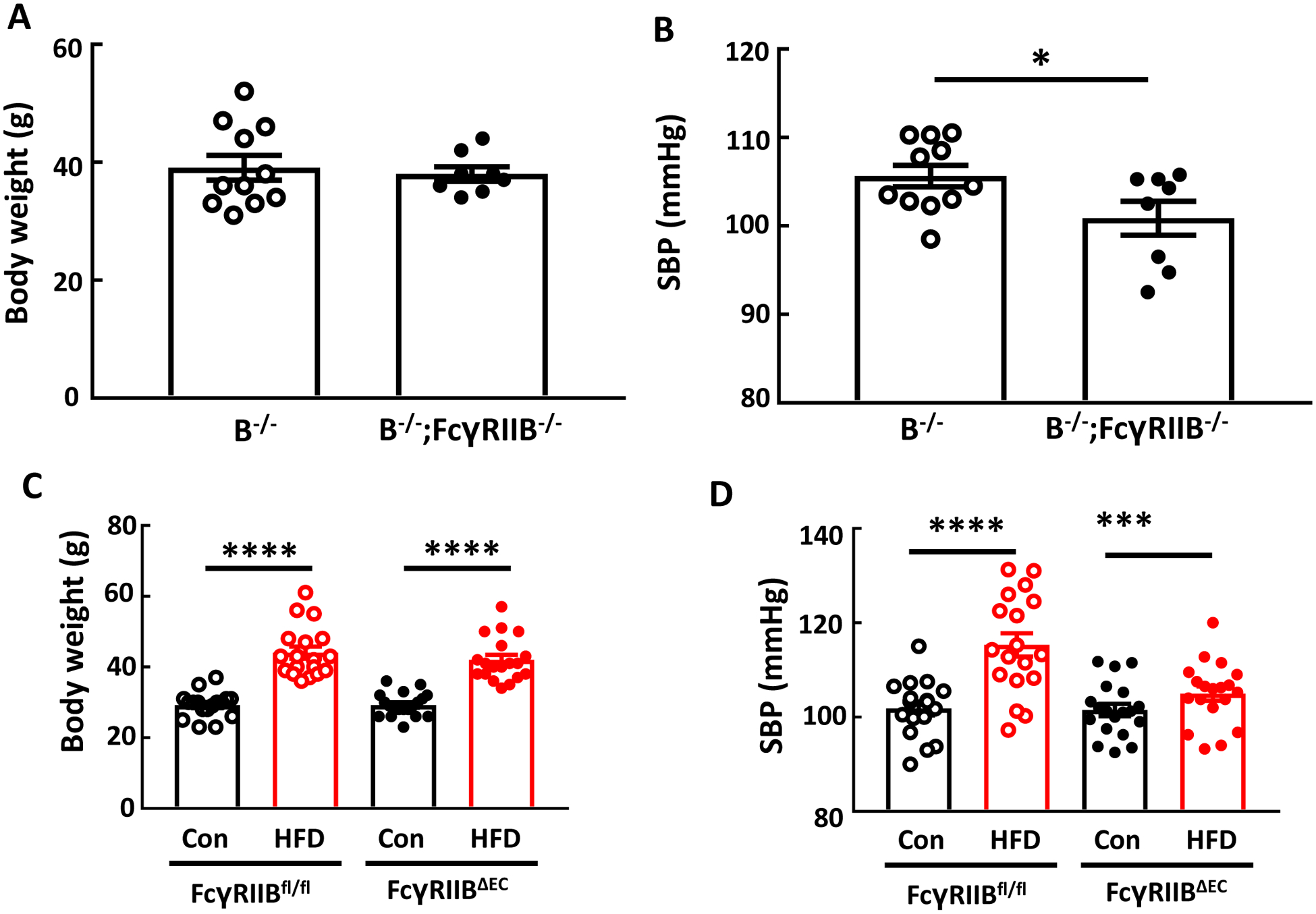
IgG isolated from high fat diet (HFD)-fed mice raises blood pressure via FcγRIIB, and endothelial FcγRIIB mediates obesity-induced hypertension. Male B−/− and B−/−;FcγRIIB−/− mice were fed a HFD for 12–14 weeks, and over the ensuing 4 weeks while remaining on HFD, they were i.p. injected with IgG (150μg/mouse, 2 times/week) isolated from wild-type mice fed a HFD. Body weight was determined (A), and systolic blood pressure (SBP) was measured by tail cuff (B). In studies of FcγRIIB in endothelium, male control floxed FcγRIIB mice (FcγRIIBfl/fl) and mice lacking the receptor selectively in endothelial cells (FcγRIIBΔEC) were placed on control chow (Con) or high fat diet (HFD) for 12 weeks, body weight was determined (C), and SBP was measured (D). Values are mean±SEM. N=8–11 per group (A,B), and n=17–19 per group (C,D). *p<0.05, *** p<0.001, **** p<0.0001.
Next the cell type in which FcγRIIB contributes to obesity-induced hypertension was elucidated using FcγRIIB floxed mice. In FcγRIIBfl/fl controls there was a predictable increase in bodyweight with HFD feeding versus control chow feeding, and mice lacking FcγRIIB exclusively in endothelial cells (FcγRIIBΔEC) displayed comparable weight gain on HFD (Fig. 6C). However, whereas diet-induced obesity caused systolic hypertension in the FcγRIIBfl/fl controls, despite identical weight gain the development of systolic hypertension was absent in FcγRIIBΔEC (Fig. 6D). Thus, FcγRIIB in endothelium is critically involved in obesity-induced hypertension.
Prevention of Obesity-induced Hypertension
Having revealed a key role for IgG hyposialyation in obesity-induced hypertension (Fig. 2,3), it was then determined how the normalization of IgG sialylation would impact the development of the disorder. This was accomplished by providing supplementation with the sialic acid precursor N-acetyl-D-mannosamine (ManNAc). Mice administered ManNAc had weight gain on HFD that mirrored that obtained in mice receiving drinking water alone (Fig. 7A), and the supplementation caused an increase in the sialylation of the IgG glycan (Fig. 7B). Most importantly, ManNAc caused full prevention of obesity-induced systolic hypertension (Fig. 7C). Thus, supplementation with the sialic acid precursor ManNAc breaks the link between obesity and hypertension in mice.
Figure 7:
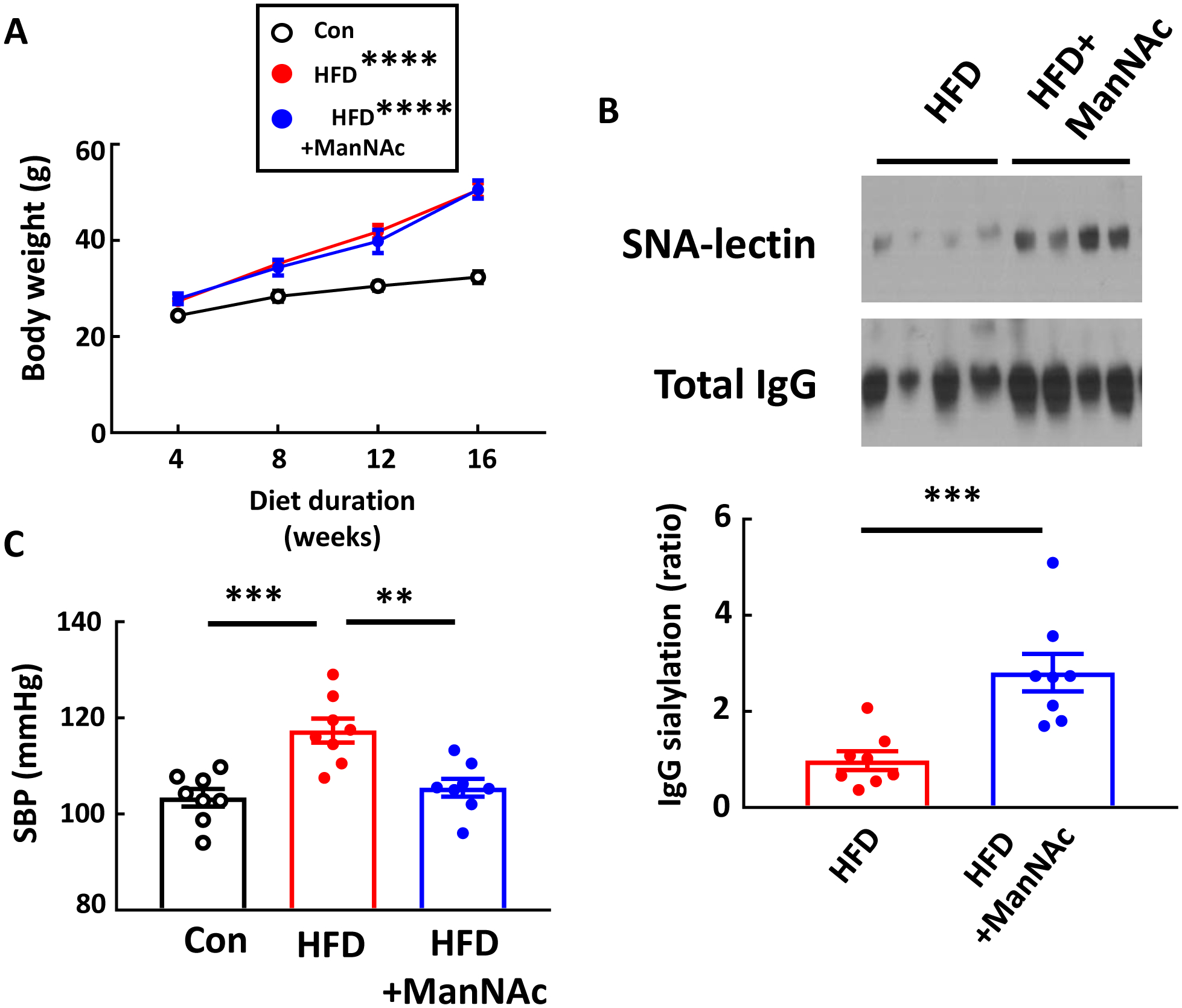
ManNAc supplementation protects mice from obesity-induced hypertension. Male wild-type mice were placed on control chow (Con) or high fat diet (HFD) for 16 weeks, and during that time the HFD-fed mice received either regular drinking water or ManNAc-supplemented drinking water. Body weight was measured every 4 weeks (A). Plasma IgG was isolated, and its sialylation was evaluated by SNA-lectin blotting (B). Graph depicts the relative sialylation. Systolic blood pressure (SBP) was measured by tail cuff (C). Values are mean±SEM, n=8 per group. In A, comparisons with the Control group were made by two-way ANOVA and Tukey’s multiple comparison test. ** p<0.01, *** p<0.001, **** p<0.0001.
Discussion
Obesity-related hypertension is a world-wide medical challenge that greatly increases the risk of stroke, coronary heart disease and chronic renal failure(5). Its pathogenesis has been primarily attributed to mechanisms involving the kidney and sympathetic nervous system(1; 6). Based on our prior work implicating the inhibitory IgG receptor FcγRIIB in the disorder and our more recent discovery of key participation of IgG in obesity-induced insulin resistance(7; 8), the present investigation set out to determine if alterations in IgG mediate obesity-induced hypertension. We reveal that mice deficient in mature B cells, which thereby do not produce IgG, are protected from the disorder, and IgG transfer experiments then directly implicated immunoglobulins, with IgG from obese mice causing an increase in SBP when transferred to recipients lacking endogenous IgG. Hyposialylation of the Fc glycan on IgG was determined to be the causal process, partnership between IgG and FcγRIIB in disease pathogenesis was shown, and the endothelium was determined to be the cell type in which FcγRIIB is operative in obesity–induced hypertension. We additionally demonstrated inverse relationships between BMI and IgG sialylation and between IgG sialylation and BP in humans. Finally, the discovery regarding IgG hypsialylation was leveraged to reveal an entirely new approach to prevent the disorder by supplementation with the sialic acid precursor ManNAc.
Fc receptors, including FcγRIIB, mediate cellular responses to IgG and also to pentraxins. CRP is the acute phase reactant pentraxin in humans, and mice express CRP but their acute phase reactant pentraxin is the highly homologous serum amyloid P component, or SAP(25). We previously found that neither CRP nor SAP increase with diet-induced obesity in mice, and total IgG levels are also not elevated(7). In mice others have observed either no change or an increase in total IgG with obesity, and in humans total IgG may rise with obesity and it falls after weight loss following gastric banding(26–28). In studies of obesity-related insulin resistance, we and others showed that B cell-deficient mice are protected from the disorder, and whereas IgG from lean control mice has no effect on insulin sensitivity when transferred to obese B cell-deficient recipients, IgG from HFD-fed mice causes insulin resistance(8; 11). In the present work we took the same two approaches and identified IgG as a pathogenetic factor in obesity-induced hypertension. In the broader category of essential hypertension, it has been known for many years that individuals with hypertension of unknown primary etiology have elevated IgG levels(29). In addition, in mice B cell and T cell deficiency results in blunted BP elevation in response to angiotensin II(30; 31), the pharmacologic depletion of B cells has the same impact, and the BP rise with angiotensin II returns with adoptive transfer of naïve B cells, suggesting that responses to angiotensin II are influenced by T cell regulation by B cells(32). There is additionally evidence that autoantibodies influence BP, with autoantigens that include receptors and ion channels, such as a1-adrenoreceptors, β1-adrenoreceptors, AT1 receptors and L-type voltage-operated calcium channels(29; 33). In contrast, we now implicate a post-translational modification of the Fc region of IgG in BP modulation, demonstrating by gain-of-function and loss-of-function in mice that there is a role for IgG hyposialylation in the genesis of obesity-induced hypertension.
In the present work we have additionally evaluated the relationship between IgG Fc sialylation and BP in human subjects. Prior studies have observed associations between IgG N-glycans and hypertension(18; 19), and a recent report comparing hypertensive and healthy subjects (630 total subjects) in four northwestern Chinese minority populations found relative Fc hyposialylation in the hypertensive individuals(20). In 3442 adult volunteers we have determined that the relative degree of IgG Fc glycan sialylation declines with greater age and greater BMI, and there was a trend for greater sialylation in females versus males. Consistent with prior studies of normotensive versus hypertensive subjects(18–20), we found that individuals receiving hypertension treatment have lower Fc sialylation than subjects not on antihypertensive therapy. Importantly, to evaluate the association between IgG sialylation and BP in a manner that avoids confounding by the influence of antihypertensive therapy, we then performed stepwise mixed modeling with data from the 2514 subjects not receiving such treatment. These analyses demonstrated that after associations related to sex, age, and BMI are taken into account, there is an inverse relationship between IgG sialylation and systolic BP. Each 1 SD increase in IgG sialylation was associated with a 1.5 mmHg decrease in SBP. Across the spectrum of IgG sialylation from 2SD below to 2SD above the mean for the population this equates to a 6 mmHg difference in SBP. Thus, we provide evidence that the connection between IgG hyposialylation and obesity-induced hypertension that we have identified in mice may be operative in human hypertension.
Having found a key role for relative IgG sialylation in obesity-induced hypertension, mechanistic linkage of IgG and FcγRIIB was then queried. Recognizing that VEGF is critically involved in endothelial regulation of vasomotor tone, as exemplified by the hypertension that complicates VEGF inhibitor therapy(34), the linkage was first evaluated in studies of eNOS activation by VEGF in primary human endothelial cells. VEGF stimulation of eNOS enzymatic activity was potently inhibited by both IgG isolated from HFD-fed mice and NA-treated IgG, it was related to an attenuation of VEGF-induced eNOS Ser1177 phosphorylation and eNOS Thr 495 dephosphorylation and not changes in eNOS expression, and the inhibition was entirely dependent on FcγRIIB. In vivo linkage was demonstrated in IgG transfer experiments in which the elevation of SBP in response to HFD IgG was negated in mice deficient in FcγRIIB. Since FcγRIIB is expressed in numerous cell types, classically mediating immune responses in B cells, T cells, dendritic cells and monocytes/macrophages(35), it was important to identify the cell type in which FcγRIIB is operative in obesity-induced hypertension. Only recently has FcγRIIB expression and modulation of cell function been demonstrated in endothelial cells(24,36). Using floxed FcγRIIB mice, we found that obesity-induced hypertension is mediated by the receptor in endothelium. This observation is consistent with our prior results in wild-type mice versus mice globally deficient in FcγRIIB. In that work we demonstrated that FcγRIIB participates in the hypertension but not in the tachycardia observed with obesity, with the heartrate change likely resulting from sympathetic nervous system activation(37), suggesting a peripheral (non-CNS, non-sympathetic nervous system) site of action of FcγRIIB(7). In parallel, we now demonstrate that B cells participate in the hypertension but not the tachycardia observed with obesity, likely reflecting a peripheral, non-nervous system location of B cell involvement. Thus, not only an important new ligand-receptor pair operative in obesity-induced hypertension, but also the cellular site of their participation, has been elucidated.
To date there is limited data regarding the potential involvement of FcγRIIB in hypertension in humans. We previously identified a loss-of-function variant of human FcγRIIB that lacks the capacity to antagonize eNOS in cultured endothelial cells. Prior to the current knowledge regarding IgG sialylation and BP, a potential influence of the FcγRIIB variant on BP was interrogated in subjects in the Dallas Heart Study with CRP levels greater than 2.0mg/L (n=2069). The loss-of-function variant was associated with lower systolic BP(24). Thus, a link between FcγRIIB and human hypertension may be emerging, and further interrogation of IgG sialylation, FcγRIIB, and their genetic determinants in the context of hypertension in humans is now warranted.
Having demonstrated that the hyposialylation of the Fc glycan on IgG plays an important role in obesity-induced hypertension, we tested if the promotion of sialylation prevents the hypertension. Without impacting weight gain, supplementation of the drinking water with the sialic acid precursor ManNAc normalized IgG sialylation and systolic blood pressure in mice fed a HFD for 12 weeks. ManNAc supplementation is presently being considered in individuals with the neuromuscular disorder hereditary inclusion body myopathy (HIBM), which is due to mutations in the GNE gene that encodes ManNAc kinase, which catalyzes the first two rate-limiting steps in sialic acid synthesis(38). ManNAc has been found to be safe and well-tolerated by HIBM clinical study participants(39). In the present work in mice, the prevention of obesity-induced hypertension by ManNAc normalization of IgG sialylation strengthens the evidence that IgG hyposialylation underlies the disorder. As importantly, it indicates that it may be possible to employ such supplementation to combat the hypertension independent of efforts to target the underlying obesity.
There are numerous strengths of the present work. Multiple approaches were employed in mice to implicate IgG hyposialylation in obesity-induced hypetension, and in cell culture detailed studies were performed to determine how altered IgG impacts eNOS activation. In the human studies the factors that influence IgG sialylation and the relationship between IgG sialylation and BP were interrogated in a large number of subjects, and the glycan analysis was focused on the Fc region of IgG. One limitation is the requirement to use tail cuff measurements in the mouse experiments with IgG transfer because radiotelemetry was not feasible in that context. In addition, the observations in humans were made only in a Croatian population that is primarily Caucasian. The present findings prompt the need for future studies in other ethnic and racial groups.
In light of our recent report that hyposialylated IgG activates endothelial FcγRIIB to promote obesity-induced insulin resistance(8), the current findings indicate that the hypertension and insulin resistance that complicate obesity likely have shared mechanistic origins. Our observations with endothelial FcγRIIB deletion and also with ManNAc supplementation further reveal that the common pathogenetic processes can potentially be targeted therapeutically with a single intervention to improve both cardiovascular and metabolic health. As such, while continuing to pursue strategies to combat the obesity that frequently underlies type 2 diabetes and hypertension, it may be possible to break the link between the obesity and these two woefully common disorders.
Supplementary Material
Clinical Perspective.
What is New?
Hyposialylation of the Fc glycan on IgG is identified as a key contributing factor in obesity-induced hypertension.
FcγRIIB, the inhibitory receptor for IgG, in endothelial cells is implicated in obesity-induced hypertension.
In mice, administration of the sialic acid precursor N-acetyl-D-mannosamine (ManNAc) increases IgG sialylation and prevents obesity-induced hypertension.
In humans, obesity is associated with lower sialylation of the Fc glycan on IgG, and systolic BP is inversely related to IgG Fc glycan sialylation.
What are the Clinical Implications?
Low levels of IgG Fc glycan sialylation may identify individuals at greater risk of developing hypertension.
The degree of sialylation of IgG may predict the relative response of an individual to antihypertensive therapy.
ManNAc supplementation and endothelial FcγRIIB blockade/inhibition warrant study as potential interventions to break the link between obesity and hypertension.
Acknowledgements
The authors thank Zhongyun Wang for valuable technical assistance.
Sources of Funding
The work was supported by NIH grants HL115122 (P.W.S.), DK110127 (C.M.), HL133179 (W.V.) and AG057571 (W.V.), and by the Crystal Charity Ball Center for Pediatric Critical Care Research (P.W.S.), the Hartwell Foundation (P.W.S.), the Pak Center of Mineral Metabolism and Clinical Research (W.V.), and the George M. O’Brien Kidney Research Core Center (NIH P30-DK079328). Additional support was provided by the European Structural and Investment Funds IRI (grant #KK.01.2.1.01.0003) (G.L.), and the Croatian National Centre of Research Excellence in Personalized Healthcare (grant #KK.01.1.1.01.0010) (G.L.). The 10,001 Dalmatians project was supported by the MRC Human Genetics Unit, the Croatian Ministry of Science, Education and Sports (grant 216-1080315-0302), the European Union framework program 6 EUROSPAN project (contract no. LSHG-CT-2006-018947), and the Croatian Science Foundation (grant 8875).
Non-standard Abbreviations and Acronyms:
- eNOS
endothelial NO synthase
- FcγRIIB
Fc γ receptor IIB
- FcγRIIBfl/fl
FcγRIIB floxed mouse
- FcγRIIBΔEC
endothelial FcγRIIB-deficient mouse
- HFD
high fat diet
- HIBM
hereditary inclusion body myopathy
- LC-ESI-MS
liquid chromatography-electrospray ionization-tandem mass spectrometry
- ManNAc
N-acetyl-D-mannosamine
- NA
neuraminidase
Footnotes
Disclosures
None.
References
- 1.Hall JE, do Carmo JM, da Silva AA, Wang Z and Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res 2015;116:991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 3.Kotsis V, Jordan J, Micic D, Finer N, Leitner DR, Toplak H, Tokgozoglu L, Athyros V, Elisaf M, Filippatos TD, et al. Obesity and cardiovascular risk: a call for action from the European Society of Hypertension Working Group of Obesity, Diabetes and the High-risk Patient and European Association for the Study of Obesity: part A: mechanisms of obesity induced hypertension, diabetes and dyslipidemia and practice guidelines for treatment. J Hypertens 2018;36:1427–1440. [DOI] [PubMed] [Google Scholar]
- 4.Gupta AK, Nasothimiou EG, Chang CL, Sever PS, Dahlof B and Poulter NR. Baseline predictors of resistant hypertension in the Anglo-Scandinavian Cardiac Outcome Trial (ASCOT): a risk score to identify those at high-risk. J Hypertens 2011;29:2004–2013. [DOI] [PubMed] [Google Scholar]
- 5.He J and Whelton PK. Elevated systolic blood pressure and risk of cardiovascular and renal disease: overview of evidence from observational epidemiologic studies and randomized controlled trials. Am Heart J 1999;138:211–219. [DOI] [PubMed] [Google Scholar]
- 6.Landsberg L, Aronne LJ, Beilin LJ, Burke V, Igel LI, Lloyd-Jones D and Sowers J. Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment: a position paper of The Obesity Society and the American Society of Hypertension. J Clin Hypertens (Greenwich) 2013;15:14–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sundgren NC, Vongpatanasin W, Boggan BM, Tanigaki K, Yuhanna IS, Chambliss KL, Mineo C and Shaul PW. IgG receptor FcgammaRIIB plays a key role in obesity-induced hypertension. Hypertension 2015;65:456–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanigaki K, Sacharidou A, Peng J, Chambliss KL, Yuhanna IS, Ghosh D, Ahmed M, Szalai AJ, Vongpatanasin W, Mattrey RF, et al. Hyposialylated IgG activates endothelial IgG receptor FcgammaRIIB to promote obesity-induced insulin resistance. J Clin Invest 2018;128:309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anthony RM, Wermeling F and Ravetch JV. Novel roles for the IgG Fc glycan. Ann N Y Acad Sci 2012;1253:170–180. [DOI] [PubMed] [Google Scholar]
- 10.Tanigaki K, Chambliss KL, Yuhanna IS, Sacharidou A, Ahmed M, Atochin DN, Huang PL, Shaul PW and Mineo C. Endothelial Fcgamma receptor IIB activation blunts insulin delivery to skeletal muscle to cause insulin resistance in mice. Diabetes 2016;65:1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 2011;17:610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vongpatanasin W, Thomas GD, Schwartz R, Cassis LA, Osborne-Lawrence S, Hahner L, Gibson LL, Black S, Samols D and Shaul PW. C-reactive protein causes downregulation of vascular angiotensin subtype 2 receptors and systolic hypertension in mice. Circulation 2007;115:1020–1028. [DOI] [PubMed] [Google Scholar]
- 13.Tanigaki K, Mineo C, Yuhanna IS, Chambliss KL, Quon MJ, Bonvini E and Shaul PW. C-reactive protein inhibits insulin activation of endothelial nitric oxide synthase via the immunoreceptor tyrosine-based inhibition motif of FcgammaRIIB and SHIP-1. Circ Res 2009;104:1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michel T and Vanhoutte PM. Cellular signaling and NO production. Pflugers Arch 2010;459:807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falck D, Jansen BC, de HN and Wuhrer M. High-Throughput Analysis of IgG Fc Glycopeptides by LC-MS. Methods Mol Biol 2017;1503:31–47. [DOI] [PubMed] [Google Scholar]
- 16.Benedetti E, Pucic-Bakovic M, Keser T, Wahl A, Hassinen A, Yang JY, Liu L, Trbojevic-Akmacic I, Razdorov G, Stambuk J, et al. Network inference from glycoproteomics data reveals new reactions in the IgG glycosylation pathway. Nat Commun 2017;8:1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruhns P Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012;119:5640–5649. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Klaric L, Yu X, Thaqi K, Dong J, Novokmet M, Wilson J, Polasek O, Liu Y, Kristic J, et al. The association between glycosylation of immunoglobulin G and hypertension: a multiple ethnic cross-sectional study. Medicine (Baltimore) 2016;95:e3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao Q, Dolikun M, Stambuk J, Wang H, Zhao F, Yiliham N, Wang Y, Trbojevic-Akmacic I, Zhang J, Fang H, et al. Immunoglobulin G N-glycans as potential postgenomic biomarkers for hypertension in the kazakh population. OMICS 2017;21:380–389. [DOI] [PubMed] [Google Scholar]
- 20.Liu JN, Dolikun M, Stambuk J, Trbojevic-Akmacic I, Zhang J, Wang H, Zheng DQ, Zhang XY, Peng HL, Zhao ZY, et al. The association between subclass-specific IgG Fc N-glycosylation profiles and hypertension in the Uygur, Kazak, Kirgiz, and Tajik populations. J Hum Hypertens 2018;32:555–563. [DOI] [PubMed] [Google Scholar]
- 21.Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension 2001;37:1199–1208. [DOI] [PubMed] [Google Scholar]
- 22.Pinto E Blood pressure and ageing. Postgrad Med J 2007;83:109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Droyvold WB, Midthjell K, Nilsen TI and Holmen J. Change in body mass index and its impact on blood pressure: a prospective population study. Int J Obes (Lond) 2005;29:650–655. [DOI] [PubMed] [Google Scholar]
- 24.Tanigaki K, Sundgren N, Khera A, Vongpatanasin W, Mineo C and Shaul PW. Fcgamma receptors and ligands and cardiovascular disease. Circ Res 2015;116:368–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pepys MB, Baltz M, Gomer K, Davies AJ and Doenhoff M. Serum amyloid P-component is an acute-phase reactant in the mouse. Nature 1979;278:259–261. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez-Quintela A, Alende R, Gude F, Campos J, Rey J, Meijide LM, Fernandez-Merino C and Vidal C. Serum levels of immunoglobulins (IgG, IgA, IgM) in a general adult population and their relationship with alcohol consumption, smoking and common metabolic abnormalities. Clin Exp Immunol 2008;151:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakcak I, Avsar MF, Hamamci EO, Bostanoglu S, Sonisik M, Bostanoglu A, Erdem NZ and Cosgun E. Comparison of early and late changes in immunoglobulins and acute phase reactants after laparoscopic adjustable gastric banding in patients with morbid obesity. Obes Surg 2010;20:610–615. [DOI] [PubMed] [Google Scholar]
- 28.Arai S, Maehara N, Iwamura Y, Honda S, Nakashima K, Kai T, Ogishi M, Morita K, Kurokawa J, Mori M, et al. Obesity-associated autoantibody production requires AIM to retain the immunoglobulin M immune complex on follicular dendritic cells. Cell Rep 2013;3:1187–1198. [DOI] [PubMed] [Google Scholar]
- 29.Chan CT, Lieu M, Toh BH, Kyaw TS, Bobik A, Sobey CG and Drummond GR. Antibodies in the Pathogenesis of Hypertension. Biomed Res Int 2014:504045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C and Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 2007;204:2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS and Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 2010;298:R1089–R1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, Kim HA, Krishnan SM, Lewis CV, Salimova E, et al. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension 2015;66:1023–1033. [DOI] [PubMed] [Google Scholar]
- 33.Selvaraj UM, Poinsatte K, Torres V, Ortega SB and Stowe AM. Heterogeneity of B cell functions in stroke-related risk, prevention, injury, and repair. Neurotherapeutics 2016;13:729–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Touyz RM, Herrmann SMS and Herrmann J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J Am Soc Hypertens 2018;12:409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravetch JV and Lanier LL. Immune inhibitory receptors. Science 2000;290:84–89. [DOI] [PubMed] [Google Scholar]
- 36.Tanigaki K, Vongpatanasin W, Barrera JA, Atochin DN, Huang PL, Bonvini E, Shaul PW and Mineo C. C-reactive protein causes insulin resistance in mice through Fcgamma receptor IIB-mediated inhibition of skeletal muscle glucose delivery. Diabetes 2013;62:721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lohmeier TE, Iliescu R, Liu B, Henegar JR, Maric-Bilkan C and Irwin ED. Systemic and renal-specific sympathoinhibition in obesity hypertension. Hypertension 2012;59:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galeano B, Klootwijk R, Manoli I, Sun M, Ciccone C, Darvish D, Starost MF, Zerfas PM, Hoffmann VJ, Hoogstraten-Miller S, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest 2007;117:1585–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishino I, Carrillo-Carrasco N and Argov Z. GNE myopathy: current update and future therapy. J Neurol Neurosurg Psychiatry 2015;86:385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.