

Abstract
Pharmacovigilance and risk minimization must be planned during drug development and forms a critical part of the regulator's decision on whether a medicinal product can be authorized. Pharmacovigilance systems should ensure proactive monitoring of all authorized medicines throughout their lifecycle in clinical use. Signal detection and management are core activities in pharmacovigilance, rapidly delivering new information on the safety of medicines in real‐world use which helps to fill knowledge gaps. The first 6 years of the European Union (EU) signal management system resulted in 453 recommendations issued by the Pharmacovigilance Risk Assessment Committee (PRAC), of which more than half were for drug labeling changes. The EU pharmacovigilance network has demonstrated its ability to detect and evaluate new drug safety signals. This has resulted in new warnings to guide the safe and effective use of medicines in Europe.
Within the European Union (EU), all authorized medicinal products have been subject to an assessment of their quality, safety, and efficacy, and a judgment of a positive benefit‐risk balance at the time of authorization. Nonetheless, not all safety issues can be addressed at the time of authorization due to the recognized limitations of clinical trials. More can be learnt of the safety of a medicine postauthorization.1
Therefore, continuous and careful monitoring of the safety profile of all medicines throughout their lifecycle is essential in identifying and minimizing risk. Proactive planning through risk management plans, marketing authorization holder (MAH) studies, and periodic safety update reports are important pharmacovigilance activities. Effective postmarketing surveillance, especially spontaneous reporting of suspected adverse drug reactions (ADRs), remains an essential part of pharmacovigilance and predominant way to detect relatively rare and unpredictable ADRs. By the end of 2018, there were ~ 14.6 million individual case safety reports (ICSRs) in EudraVigilance (EV), referring to > 8 million individual cases.2 Pharmacovigilance is paramount in promoting and protecting public health by minimizing the risk of ADRs, and optimizing the benefit‐risk balance for individual patients.
Established in 2012 by the EU Pharmacovigilance Legislation, the Pharmacovigilance Risk Assessment Committee (PRAC) plays a pivotal role in the safety monitoring and evaluation of medicinal products for human use and is at the core of the EU signal management process.3, 4, 5, 6
In this paper, we provide a brief overview of the signal detection and management process within the EU network and document its performance over the first 6 years (two mandates) of the PRAC (September 2012 to June 2018). Before 2012, the PRAC and the signal management process, as currently conducted, were not established and, therefore, comparable European data on signal evaluation are not available. This documentation of performance represents a measure of impact. Such an impact assessment can help to drive improvement, therefore, enhancing public health promotion and protection.
Key messages:
The EU signal management process is responsive with recommendations for risk minimizations made in as few as 5 days of a signal being confirmed (with a median of 5 months) and the possibility of evaluation through a referral procedure for urgent signals, which may lead when necessary to temporary measures (e.g., restriction of use).
Evidence‐based use of statistical algorithms improves the timeliness of signal detection from the spontaneous report database EV.
Spontaneous ADR data remains the most common source of drug safety signals assessed at EU level (with ~ 55% of signals identified from EV).
Review of the first 6 years of the EU signal detection and management process by the EU network for centrally and nationally authorized products showed over 26,000 potential signals reviewed resulting in 453 signals assessed by the PRAC.
About half of all signals assessed by the PRAC resulted in product information updates, including 17 following the evaluation via a referral procedure.
The science‐based signal detection and management in place in the European Union delivers robust monitoring for medicines on the market and ensures that new or changing safety issues are rapidly detected and assessed, and advice on safe and effective use reaches patients and healthcare professionals.
Overview of the Signal Management Process in The EU
A signal is defined as “information arising from one or multiple sources, including observations and experiments, which suggest a new potentially causal association, or a new aspect of a known association between an intervention and an event or set of related events, either adverse or beneficial, that is judged to be of sufficient likelihood to justify verificatory action.”6 Signal management in the European Union focuses on adverse outcomes and involves a set of activities performed to determine whether, based on an examination of ICSRs, aggregated data from active surveillance systems or studies, literature information, or other data sources, there are new risks associated with an active substance or a medicinal product or whether risks have changed.7 Safety data are continuously monitored and reviewed by regulators and the MAHs to ensure the detection and subsequent evaluation of any safety signal is rapid, robust, and reliable, and duly communicated to patients and healthcare professionals through changes to product information, where necessary.
EV is the EU system for managing and analyzing reports of suspected ADRs to medicines that are authorized or being studied in clinical trials.8, 9, 10 Monitoring of EV data is governed by EU law,5, 6 and the schematic in Figure 1 summaries the process demonstrating the six main activities (signal detection, signal validation, signal confirmation, signal analysis and prioritization, signal assessment, and recommendation) and the individual roles and interactions between the National Competent Authorities (NCAs), the European Medicines Agency (EMA), and MAHs.7
Figure 1.
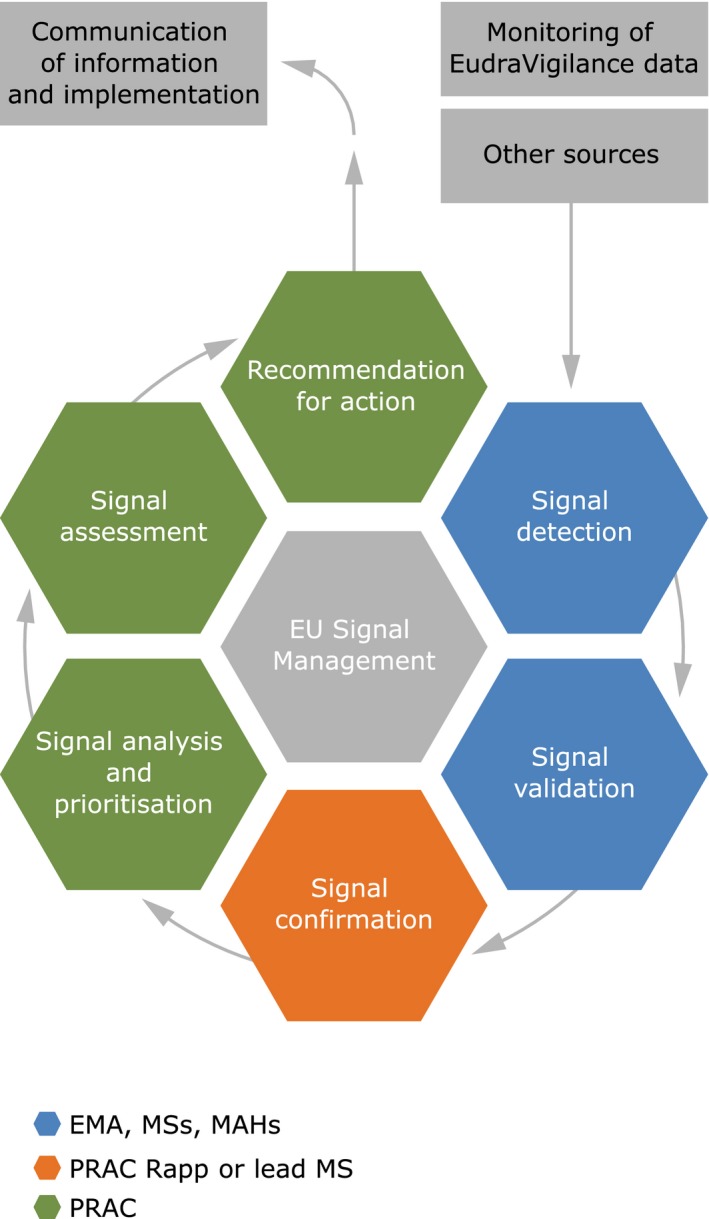
Simplified schematic of signal management process in the European Union. EMA, European Medicines Agency; EU, European Union; MAH, Marketing Authorisation Holder; MSs, Member States; PRAC, Pharmacovigilance Risk Assessment Committee.
Signal Detection
Signal detection within the EU network is a collaboration among the EMA, NCAs, and MAHs and builds on the engagement of patients and healthcare professionals in reporting suspected ADRs and participating studies.3 The EMA is responsible for the detection and monitoring of safety issues relating to centrally authorized medicinal products (CAPs), together with the PRAC rapporteurs, while nationally authorized products are monitored by specific member states in line with work‐sharing arrangements.7 Best practice in signal management is supported by clear guidance, including the Good Pharmacovigilance Practices and Strengthening Collaboration for Operating Pharmacovigilance in Europe (SCOPE).11, 12
Early detection and prompt evaluation of any new or changing safety issue is paramount to successful signal management. At the EMA, signals are identified from a variety of sources and an integrated approach to signal detection is followed.
Once a potential signal is detected, further evaluation is undertaken to validate the signal in order to ensure there is enough evidence to justify further analysis. Line listings of ICSRs are initially reviewed and provide an overview of the signal, including demographics of the patients, whether the outcome was fatal, the use of concomitant medicines, and associated adverse reactions. Although this may be sufficient to refute the potential signal in some cases, many potential signals require a more detailed review of the individual case reports. Clinical relevance, exposure, temporal association, biological plausibility, dechallenge and rechallenge, severity of reaction, and outcome are important considerations, as well as the novelty of the suspected reaction and previous awareness of the association. Other information sources, such as literature and experimental findings, may help in supporting or disproving an association.
The main signal detection analysis in this paper is for CAPs, as the EMA hold data for all signal steps for EMA activities and only for validated signals for NCA activities. In contrast, signal management through PRAC is reviewed for CAPs and nationally authorized products. In the first 6 years of the PRAC, the EMA's signal management team reviewed in‐depth information on 13,550 potential signals (i.e., drug‐event pairs from screening of the EV database, scientific literature, or information received from other regulatory authorities, etc.). Of these, 283 signals (2.1%) were validated for further analysis, with the majority (85%) confirmed, prioritized, and assessed by the PRAC.
Signal Management
Rapid communication of any safety information among the EMA, NCAs, and committees is critical to ensure rapid and robust analysis of signals and to support timely action to protect public health. All confirmed signals are brought to the attention of the PRAC for prioritization and assessment, with a recommendation for action to optimize safe and effective use of the product (Figure 2). The evaluation normally takes the form of an assessment of all available data within a signal procedure but, in some situations, for example, when there are questions on the benefit‐risk balance of a medicine, this may take the form of a referral procedure evaluation (in accordance with Articles 31 or 107i of Directive 2001/83/EC, or Article 20 of Regulation (EC) No. 726/20044, 5).
Figure 2.
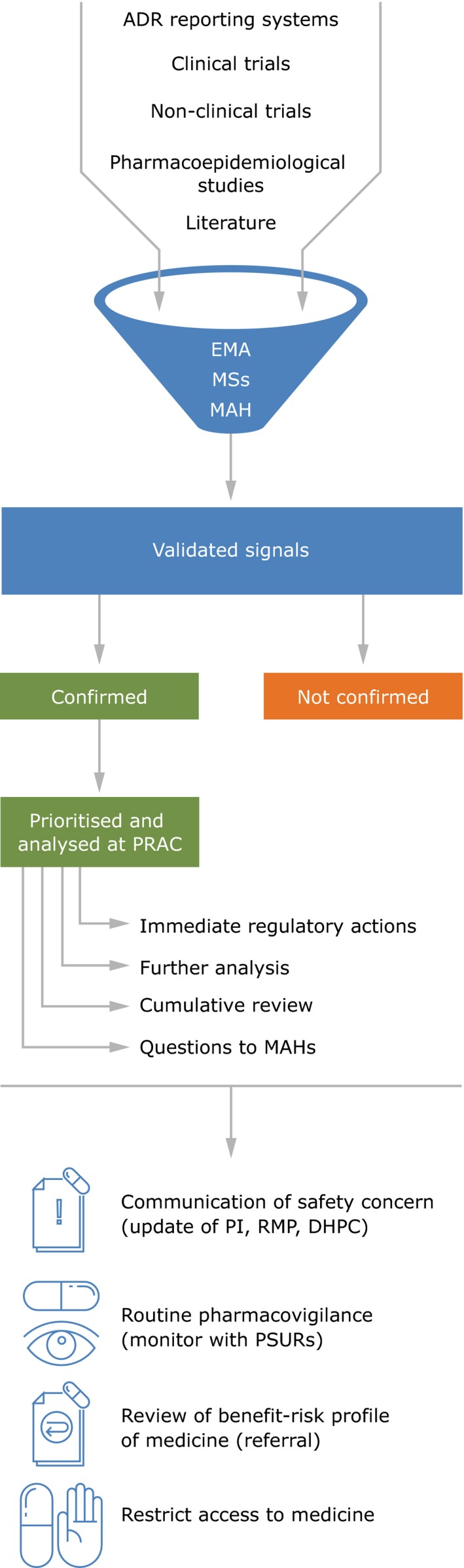
Summary of the signal management process within the European Union and consequent recommendations of PRAC. ADR, Adverse Drug Reaction; DHPC, Direct Healthcare Professional Communication; EMA, European Medicines Agency; MAH, Marketing Authorisation Holder; MSs, Member States; PI, Product Information; PRAC, Pharmacovigilance Risk Assessment Committee; PSUR, Periodic Safety Update Report; RMP, Risk Management Plan.
From July 2012, during the first 6‐year mandate of the PRAC, the EU network has reviewed a combined total of over 26,000 potential signals (that is Drug‐Event Combinations, which require further screening). This, in turn, has led to 563 potential signals validated, 283 by the EMA and 279 by NCAs of the member states (in addition to one received from an MAH in the context of the pilot period, which started on February 22, 2018, whereby MAHs of selected active substances have to monitor them in EV and inform EMA and NCAs of validated signals with their medicines). The reduction from 26,000 to 563 validated signals is based on the well‐established signal validation approaches, including whether something is already labeled in the product information. Of the 563 validated signals, 465 (82.6%) were confirmed. This has resulted in a total of 774 signal discussions at PRAC (initial and follow‐up counted separately), involving 453 individual signals (Figures 3 and 4). Due to the cutoff date for inclusion in the PRAC agenda, 12 signals confirmed in this period were assessed by the PRAC in the following month (July 2018).
Figure 3.
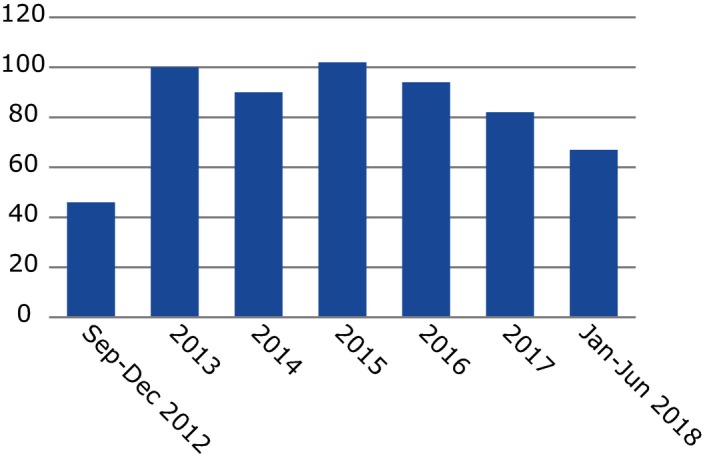
Number of signals assessed by the PRAC in last 6 years. Signals may be assessed (and thus counted in the chart) in >1 year. Although the graph looks like there were a high number of signals in 2012, this is because there were ongoing safety issues before the PRAC was formally established, which were ongoing and fed into the PRAC. Therefore, 2013 would be a better considered baseline.
Figure 4.

Trends in adverse event reports and signals in the last 6 years. 1. Established EMA signal validation processes, including strength of evidence (such as biological mechanism, confounding by disease, causal relationship, etc.), clinical relevance, and previous awareness of an association. 2. Reasons for not confirming a validated signal include: signals already adequately handled through a different procedure, unsubstantial new evidence since a previous review, or available data assessed does not warrant further analysis. *This includes one CAP signal from an MAH in the context of the pilot, which started on February 22, 2018. CAP, Centrally Authorized Product; DHPC, Direct Healthcare Professional Communication; EMA, European Medicines Agency; EU, European Union; EV, EudraVigilance; ICSRs, Individual Case Safety Reports; NAP, Nationally Authorized Product; NCA, National Competent Authorities; PASS, Post Authorization Safety Study; PhV, pharmacovigilance; PI, product information; PRAC, Pharmacovigilance Risk Assessment Committee; RMP, risk management plan.
The main source of information was ADR data from EV, which contributed to 55% of detected signals, with EV data alone leading to the detection of 77 signals (17%) and to a further 171 signals (38%) in combination with other sources.2 During this period, unless already counted in the EV category, potential signals also came from: national reviews 66 (15%), scientific literature 59 (13%), studies 24 (5%), and communication of signals from other regulatory authorities 13 (3%; Figure 5). Just over half of these assessments (50.3%) led to a recommendation for an update of the product information, demonstrating the value of signal detection in providing up‐to‐date safety information to healthcare professionals and patients to promote the safe and effective use of medicines. Other recommendations during this period included routine pharmacovigilance and monitoring within the periodic safety update report (34%), referral evaluations (3.8%), or updates to risk management plans (2%). Figure 6 gives an overview of signal outcomes during the first 6 years of the PRAC, which shows its continued positive role in delivering recommendations, in particular those for labeling changes and routine pharmacovigilance. Additional means of communication, such as Direct Healthcare Professional Communications (DHPCs), played a significant role in highlighting new, important information on safety concerns, which require a change to the current use of a medicinal product. A total of 29 DHPCs were considered necessary by the PRAC ranging from important recommendations to prescribers on the restriction of a medicine's use in women planning pregnancy to raising awareness and minimizing the risk of medication errors. Such DHPCs have the potential to deliver healthcare professionals in the European Union with clear, reliable, and prompt information on the safe use of medicines, protecting patients, and promoting public health.
Figure 5.
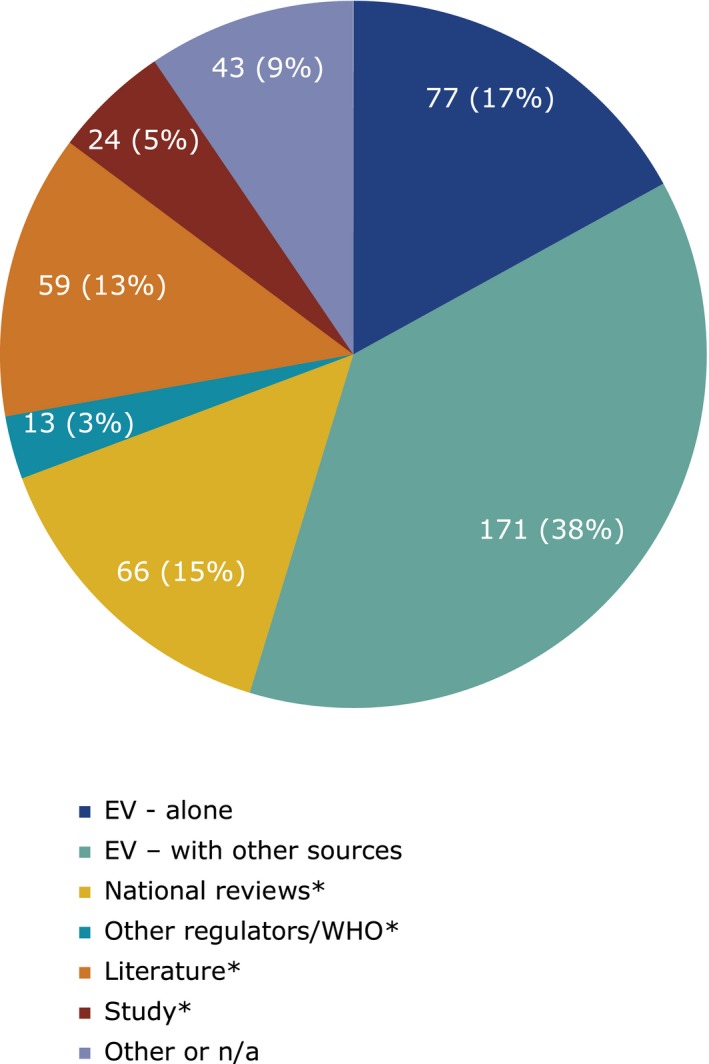
Sources of information of the signals prioritized and analyzed at the Pharmacovigilance Risk Assessment Committee from September 2012 to June 2018. EV, EudraVigilance; n/a, not applicable; PRAC, Pharmacovigilance Risk Assessment Committee; WHO, World Health Organization. *Excluding those counted already in categories above.
Figure 6.
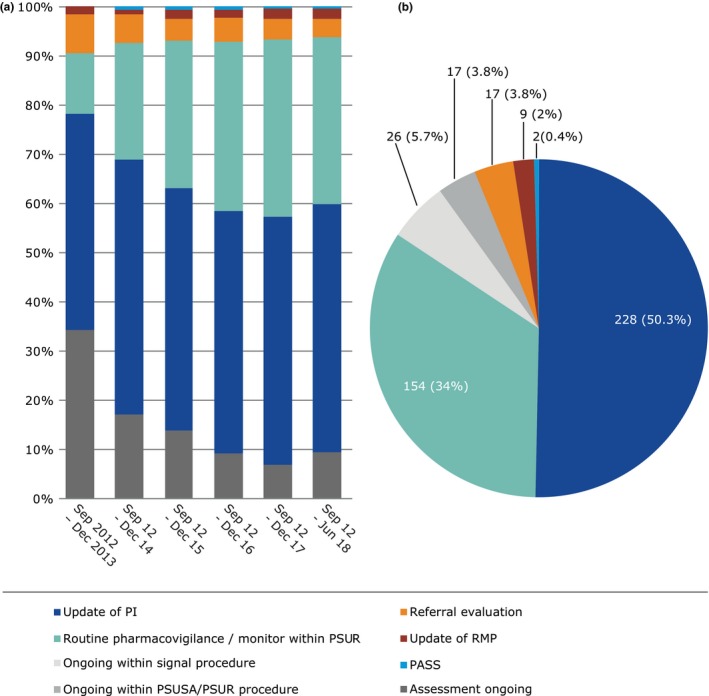
Outcomes of signals prioritized and analyzed at the Pharmacovigilance Risk Assessment Committee in the past 6 years. (a) Per year (b) September 2012 to June 2018. Note for a, 2012 data is merged with 2013 due to the low numbers. PASS, Post Authorization Safety Study; PI, Product Information; PSUR, Periodic Safety Update Report; PSUSA, Periodic Safety Update Single Assessment; Referral, in accordance with the Articles 31 and 107i of Directive 2001/83/EC(4) and Article 20 of Regulation (EC) No. 726/2004; RMP, Risk Management Plan.
Impact of Signal Detection in Improving Patient Care
The strengthened signal management system established in the EU in 2012 has marked a new era of safety and transparency underpinned by collaboration. The timeliness of the detection of safety issues and assessment and communication of safety concerns through PRAC recommendations are critically important outcomes of the signal management process and the speed in which any known risk or change in safety information is communicated to patients is a key determinant in minimizing harm. From July 2012, the time taken from confirmation of a validated signal to the adoption of a PRAC recommendation has been as quick as 5 days, with a median of 5 months. This often includes time for processes such as requests for and assessment of supplementary information from MAHs, data from authors of studies published in the literature, or expansion of the scope of the assessment, for example, the inclusion of other substances in a class. In some cases, an immediate communication (such as DHPC) might be warranted for a serious safety concern, enabling healthcare professionals to be aware of new or emerging safety and efficacy information, and able to take the necessary actions in a timely manner. Table 1 provides examples of signals brought to the attention of the PRAC and the time taken to issue a recommendation, ensuring prescribers and patients are aware of safety warnings related to their medicines as quickly as possible.
Table 1.
Example of the variety of signals brought to the PRAC leading to different recommendations
| Signal | What was the signal and what evidence supported it? | What action was taken | Communication of riska |
|---|---|---|---|
| Dolutegravir (TIVICAY) – Birth outcomes in HIV‐infected women |
Dolutegravir is an integrase strand transfer inhibitor indicated for the treatment of HIV‐1 infection in adults and children over 6 years of age. It is available in the EU as well as over 80 further countries. Preliminary data from an observational study (Tsepamo) on birth outcomes in HIV‐infected women conducted in Botswana, suggested a potential increased risk of neural tube defects associated with the use of dolutegravir containing medicines at the time of conception. In May 2018, it was brought to the attention of the EMA as an ESI, confirmed as a signal, prioritized, and assessed by PRAC, which issued a recommendation within 5 days. |
|
5 days |
| Sodium‐glucose transporter protein 2 inhibitors – diabetic ketoacidosis | SGLT2 inhibitors are used together with diet and exercise in patients with type 2 diabetes, either alone or in combination with other diabetes medicines. In 2015, as part of routine signal monitoring activities, a search in EV retrieved 148 cases related to diabetic ketoacidosis in association with the SGLT2 inhibitors canagliflozin, dapagliflozin, or empagliflozin. Several relevant literature articles were also deemed supportive for the signal. The signal was confirmed, prioritized, and assessed by the PRAC. |
|
10 days |
| Cladribine (LITAK) –PML |
Cladribine is a chemotherapy drug used to treat hairy cell leukemia, a certain type of chronic lymphocytic leukemia, as well as multiple sclerosis. In 2017, as part of routine signal monitoring activities, a review of case reports in EV identified seven well‐documented cases of PML associated with cladribine; five of which were published in the literature. In five cases, the diagnosis could be confirmed with a reliable level of certainty, which led to the signal confirmation for further analysis by PRAC. |
|
5.5 months |
| Insulins – Risk of medication error leading to dysglycemia |
Medication errors leading to potentially serious dysglycemia have long been an acknowledged risk for all insulin‐containing products. In 2016, a UK National Health Service patient safety alert on the risk of withdrawing insulin from prefilled pen devices or reusable cartridges leading to dysglycemia was published in line with the EMA guidance on risk minimization for insulin products. A review of the PI for all centrally authorized insulin‐containing products in the EU revealed that not all contained warnings against insulin extraction and administration other than using the pen device. The signal was confirmed and prioritized and assessed by the PRAC. |
|
7 months |
| Levonorgestrel intrauterine device – Anxiety, panic attacks, mood changes, sleep disorders, and restlessness | The signal was initiated following a publication of a petition by a German patient organization requesting the inclusion of possible side effects, including panic attacks, anxiety, mood changes, sleep disorders, and restlessness, which may be associated with the use of IUDs containing levonorgestrel medicines. The signal was confirmed for further analysis by the PRAC. |
|
9 months |
DHPC, Direct Healthcare Professional Communication; EMA, European Medicines Agency; ESI, emerging safety issue; EU, European Union; EV, EudraVigilance; FDA, US Food and Drug Administration; HCP, healthcare professional; ICSRs, Individual Case Safety Reports; IUD, intrauterine device; MAH, marketing authorization holder; PI, product information; PL, package leaflet; PML, progressive multifocal leukoencephalopathy; PRAC, Pharmacovigilance Risk Assessment Committee; RMP, risk management plan; SGLT2, sodium‐glucose transport protein 2; WHO, World Health Organization.
Time from signal confirmation to first public communication of risk.
Continuous Process Improvement and Future Research
Optimization of the EU signal management system is based on three main “pillars”: continuous process evaluation, exploration of new methodologies, and enhanced transparency. Continuous evaluation of the EU signal detection and management process is essential to maintain and improve its effective and efficient operation. Identifying and managing elements where there are opportunities for improvement and reinforcing elements that work well strengthens the functioning of the process and, thus, its success in improving public health. Realizing the fundamental importance of this work, the PRAC established a subgroup, the Signal Management Review Technical Working Group (SMART). This includes experts from the NCAs, the EMA, PRAC members, and academic leaders in the field of signal detection, and its purpose is to review process improvements and foster innovations of new methodologies in signal detection and management. Recent process improvements include the implementation of enhanced monitoring of the pediatric and geriatric populations, greater visualization of abuse, misuse, overdose, medication errors, and occupational exposure, and quicker identification of new signals triggered by an unexpected increase in the total count of cases received over a particular timeframe.13 Disproportionality metrics were developed to support prompt detection of new safety concerns and have been demonstrated to detect about 50% of ADRs before other currently used methods of signal detection.14
The second area is methodological improvement. Evidence from the major research project PROTECT,15 which tested existing methodologies and their practical applications in real‐life studies, has led to evidence‐based process improvement in signal detection. The introduction of new statistical methodology for signal detection in EV was achieved through careful analysis of the utility of the reporting odds ratio instead of the proportional reporting ratio to detect ADRs in pharmacovigilance,16, 17 in line with PROTECT recommendations on ease of implementation and interpretation, as well as efficient use of resources. Furthermore, subgroup analysis, enabled by the size of the EV database, was proven to improve sensitivity and precision while reducing false‐positives, positively impacting on the safety monitoring of medicinal products.18
The third area of process improvement is the EMA's continuous effort to enhance the transparency of relevant information on medicines.11 The publication on the EMA website of the PRAC agendas, highlights, and minutes, as well as the individual PRAC recommendations for signals, allows everyone, from interested patients and healthcare professionals to MAHs, to obtain up‐to‐date and detailed information on the medicines they use.19 Furthermore, the recent improvement in 2015 to translate and publish the PRAC recommendations into a total of 25 languages (official EU languages as well as Norwegian and Icelandic), speeds up conversion of PRAC recommendations into warnings in product information and reduces unnecessary duplication of effort for NCAs and MAHs. This has also ensured a harmonized approach to product information and reduced the risk of interpretation error.
Although the data provided in our analysis shows the important contributory influence the signal management system has had on communicating safety information to medicine users via product labeling, it is important to recognize limitations that could direct further research to better validate its impact. Repeating the analysis in the future with a larger dataset would provide an insight into the validity of the data and enable examination of trends over time. This would also allow for informative comparisons on the nature and distribution of the signals, for example, between different drug classes or Medical Dictionary for Regulatory Activities terms.
Data from 2015 onward has shown that the proportion of signals that arise from ICSRs has remained consistent at approximately two‐thirds of signals. Nonetheless, a further detailed evaluation of the influence of the data source would be an area of significant interest for future research. Exploring whether increasing interest in real‐world data, such as databases of electronic health records and social media is contributing more to signal detection, would provide helpful insights into the strengths and weaknesses of spontaneous reporting systems and the complementarity of these new approaches.
Finally, as this analysis was descriptive, looking at absolute numbers of new product information warnings, there was no comparator arm. A comparison of the EU signal management system alongside other regulators’ outputs might be considered an interesting direction for future research.
Conclusions and Future Perspective
A new era of patient protection and transparency in EU medicines safety has been delivered. The implementation of the EU pharmacovigilance legislation in 2012 was the most significant change in the regulation of human medicines in the EU since 1995.3, 20, 21 Over its first 6 years of operation, the strengthened pharmacovigilance process has resulted in over 26,000 potential signals reviewed and 453 confirmed signals assessed by the PRAC. More than half of the PRAC recommendations have resulted in changes to medicine product information supporting safe and effective use of medicines, demonstrating that the EU signal management process reliably detects, assesses, and deals with safety issues and enables the risk of ADRs to be minimized. At the heart of this process is a strong collaboration based on the expertise from the NCAs and the EMA, with the contribution of MAH pharmacovigilance systems, and combined with a culture of continuous improvement. Enhanced transparency, including the publication of PRAC recommendations and their translations in all EU languages, ensures rapid updating of product information and provides healthcare professionals and patients across Europe with robust, reliable, and rapid information on the safety of their medicines.
The EU network remains dedicated to the ongoing improvement of the efficacy and efficiency of the EU signal detection and management process. Several initiatives are ongoing to optimize the effectiveness and efficiency of signal management, including use of real‐world data and a focus on monitoring challenges, such as misuse and abuse of medicines, safety in the elderly, and drug–drug interactions, as well as the impact on pharmacovigilance systems of nonserious suspected ADRs. Recognizing, evaluating, and improving the existing system will ensure a better and more rapid analysis and understanding of safety concerns and highlight areas of missing information, which can be managed through risk management planning. In this way, safe and effective medicines can reach patients who need them and the confidence of patients in the medicines they use is supported.
Disclaimer
The views expressed in this paper are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the agencies or organizations with which the authors are affiliated.
Funding
No funding was received for this work.
Conflict of Interest
J.P., G.G., A.S., and P.A. are employees of the EMA. J.R. is an employee of the UK Medicines and Healthcare products Regulatory Agency and was the Chair of the PRAC from September 2012 until September 2018. S.S. is an employee of the Dutch Medicines Evaluation Board and has been a member of the PRAC since September 2012 (and Chair since September 2018).
Supporting information
Table S1. Signals between September 2012 and June 2018 resulting in Direct Healthcare Professional Communications providing action, advice or changes to current practice.
Acknowledgments
The authors gratefully acknowledge Radoslav Zhelev and Vladimira Yalmanova for their contribution to the graphics in the paper.
References
- 1. Vandenbroucke, J.P. Observational research, randomised trials, and two views of medical science. PLoS Med. 5, e67 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. 2018 Annual Report on EudraVigilance for the European Parliament, the Council and the Commission. EUROPEAN MEDICINES AGENCY <https://www.ema.europa.eu/en/documents/report/2018-annual-report-eudravigilance-european-parliament-council-commission-reporting-period-1-january_en.pdf> (2019). Accessed November 27, 2019.
- 3. Santoro, A. , Genov, G. , Spooner, A. , Raine, J. & Arlett, P. Promoting and protecting public health: how the European Union pharmacovigilance system works. Drug Safety 40, 855–869 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. EUROPEAN COMMISSION <https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf> (2012). Accessed November 27, 2019.
- 5. Regulation (EC) No. 726/2004 of the European Parliament and of the Council of 31 March 2004 laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. EUROPEAN COMMISSION <https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02004R0726-20190128> (2019). Accessed November 27, 2019.
- 6. Commission Implementing Regulation (EU) No. 520/2012 of 19 June 2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) No. 726/2004 of the European Parliament and of the Council and Directive 2001/83/EC of the European Parliament and of the Council. EUROPEAN COMMISSION <https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2012_520/reg_2012_520_en.pdf> (2012). Accessed November 27, 2019.
- 7. Guideline on good pharmacovigilance practices (GVP): module IX: signal management (Rev 1). EUROPEAN MEDICINES AGENCY <https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-module-ix-signal-management-rev-1_en.pdf> (2017). Accessed November 27, 2019.
- 8. European Medicines Agency policy on access to EudraVigilance data for medicinal products for human use: revision 4. EUROPEAN MEDICINES AGENCY <https://www.ema.europa.eu/en/documents/other/european-medicines-agency-policy-access-eudravigilance-data-medicinal-products-human-use-revision-4_en.pdf> (2019). Accessed November 27, 2019.
- 9. EudraVigilance <https://www.ema.europa.eu/humanregulatory/research-development/pharmacovigilance/eudravigilance>. Accessed November 27, 2019.
- 10. Postigo, R. et al EudraVigilance medicines safety database: publicly accessible data for research and public health protection. Drug Safety 41, 665–675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Signal Management: Strengthening Collaboration for Operating Pharmacovigilance in Europe (SCOPE) joint action <http://www.scopejointaction.eu/> (2019). Accessed November 27, 2019.
- 12. Screening for adverse reactions in EudraVigilance. EUROPEAN MEDICINES AGENCY <https://www.ema.europa.eu/en/documents/other/screening-adverse-reactions-eudravigilance_en.pdf> (2016). Accessed November 27, 2019.
- 13. Pinheiro, L.C. , Candore, G. , Zaccaria, C. , Slattery, J. & Arlett, P. An algorithm to detect unexpected increases in frequency of reports of adverse events in EudraVigilance. Pharmacoepidemiol. Drug Safety 27, 38–45 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alvarez, Y. , Hidalgo, A. , Maignen, F. & Slattery, J. Validation of statistical signal detection procedures in EudraVigilance post‐authorization data: a retrospective evaluation of the potential for earlier signalling. Drug Safety 33, 475–487 (2010). [DOI] [PubMed] [Google Scholar]
- 15. The pharmacoepidemiological research on outcomes of therapeutics by a European consortium. THE INNOVATIVE MEDICINES INITIATIVE (IMI) <http://www.imi-protect.eu/>. Accessed November 27, 2019.
- 16. Wisniewski, A.F. et al Good signal detection practices: evidence from IMI PROTECT. Drug Safety 39, 469–490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Candore, G. et al Comparison of statistical signal detection methods within and across spontaneous reporting databases. Drug Safety 38, 577–587 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Slattery, J. , Alvarez, Y. & Hidalgo, A. Choosing thresholds for statistical signal detection with the proportional reporting ratio. Drug Safety 36, 687–692 (2013). [DOI] [PubMed] [Google Scholar]
- 19. Lane, S. , Lynn, E. & Shakir, S. Investigation assessing the publicly available evidence supporting postmarketing withdrawals, revocations and suspensions of marketing authorisations in the EU since 2012. BMJ Open 8, e019759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Legal framework: Pharmacovigilance <https://www.ema.europa.eu/en/human-regulatory/overview/pharmacovigilance/legal-framework>. Accessed January 15, 2019.
- 21. Arlett, P. et al Proactively managing the risk of marketed drugs: experience with the EMA Pharmacovigilance Risk Assessment Committee. Nat. Rev. Drug Discov. 13, 395–397 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Signals between September 2012 and June 2018 resulting in Direct Healthcare Professional Communications providing action, advice or changes to current practice.