

Abstract
For ~ 80 drugs, widely recognized pharmacogenetics dosing guidelines are available. However, the use of these guidelines in clinical practice remains limited as only a fraction of patients is subjected to pharmacogenetic screening. We investigated the feasibility of repurposing whole exome sequencing (WES) data for a panel of 42 variants in 11 pharmacogenes to provide a pharmacogenomic profile. Existing diagnostic WES‐data from child‐parent trios totaling 1,583 individuals were used. Results were successfully extracted for 39 variants. No information could be extracted for three variants, located in CYP2C19, UGT1A1, and CYP3A5, and for CYP2D6 copy number. At least one actionable phenotype was present in 86% of the individuals. Haplotype phasing proved relevant for CYP2B6 assignments as 1.5% of the phenotypes were corrected after phasing. In conclusion, repurposing WES‐data can yield meaningful pharmacogenetic profiles for 7 of 11 important pharmacogenes, which can be used to guide drug treatment.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Whole exome sequencing (WES) data is generated abundantly in clinical diagnostics and can potentially be repurposed for pharmacogenetics.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Can a clinically relevant pharmacogenetics profile be extracted from existing diagnostic WES? What is the added value of haplotype phasing?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Of 42 variants in 11 pharmacogenes for 1,583 individuals, genotypes and phenotypes based on existing WES data could be assigned to 70.4% of all potential genotype calls. Due to a lack of coverage and copy number variant calling, genotypes could not be assigned UGT1A1, CYP3A5, CYP2C19, and CYP2D6. Eighty‐six percent of all individuals carried at least one actionable phenotype. Haplotype phasing resulted in clinically relevant differences in phenotyping results compared with conventional, linkage‐based, assumptions for CYP2B6 genotype assignments.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Repurposing existing WES data can yield a meaningful pharmacogenetic profile, which can be used in combination with existing guidelines, without the need for additional genetic testing.
Pharmacogenetics (PGx) aims to optimize drug treatment by preventing adverse drug reactions and by increasing drug efficacy through adjustments based on one's genetic profile. For ~ 80 drugs, there is convincing evidence that PGx testing prior to prescribing leads to improved patient outcome.1, 2, 3 Hence, both the Clinical Pharmacogenetics Implementation Consortium (CPIC) and the Dutch Pharmacogenetics Working Group (DPWG) have developed widely recognized guidelines,3, 4, 5 which provide dose and drug adjustments based upon available PGx results.3, 4, 5, 6 In the Netherlands, for instance, these guidelines are integrated into the electronic prescribing and dispensing systems nationwide and are available at point of care.7
Both the CPIC and DPWG PGx guidelines originally considered patients with a known genotype and were developed in anticipation of having clinical high throughput and preemptive genotyping as the standard of care. However, many applications of PGx are still reactive, following a novel prescription or unexplained adverse drug reactions (ADRs). In contrast, preemptive testing for a panel of pharmacogenes can be used to prevent ADRs and improve treatment efficacy. The potential impact of such a preemptive panel‐based approach is high.2 Unfortunately, despite the established impact of PGx on the outcomes of drug treatment and the availability of guidelines, the number of patients with known pharmacogenetic genotypes remains limited.7
Whole exome sequencing (WES) and whole genome sequencing (WGS) have rapidly become part of the diagnostics process in the field of clinical genetics to diagnose potential genetic disorders of unknown etiology. Their application has resulted in vast amounts of sequencing data being generated. These data hold the potential to retrieve genetic information for a panel of PGx genes, which can then be repurposed to preemptively guide drug treatment. Next generation sequencing (NGS) approaches specifically designed for PGx have shown promising results, with high concordance (91–99%) with conventional PGx methods.8, 9, 10 Moreover, Cousin et al.11 have shown that extracting information on three PGx genes from existing clinical WES data can be beneficial in terms of drug dose and response in a small cohort of 94 patients. However, this study is limited by investigating a small number of PGx genes rather than extracting a full panel of genes with actionable recommendations in PGx guidelines as well as by the use of a limited sample size.
Given that PGx haplotype and phenotype assignments are currently performed based on linkage disequilibrium between known variants, it is possible that having access to phasing information may change the haplotype and phenotype assignment and potentially the clinical recommendation. Thus, trio‐based sequencing data is a particularly interesting source for the extraction of PGx variants.
In this study, we assessed the feasibility of repurposing diagnostic WES data to extract a meaningful PGx profile based on the panel used in the Ubiquitous Pharmacogenomics (U‐PGx; http://www.upgx.eu) consortium that includes all actionable genes and variants in the DPWG guidelines.12, 13 Additionally, we investigated the added value of haplotype phasing.
Results
Study cohort
The entire cohort consists of a total of 1,583 individuals from 2 different subcohorts (Figure 1). Both subcohorts consist of patients suffering from an intellectual disability (ID) and/or multiple congenital anomalies and their parents, all of whom underwent diagnostic WES and received genetic counseling during the process at the Department of Clinical Genetics at the Leiden University Medical Centre. The first, prospective, subcohort consists of individuals who were offered the opportunity to receive their PGx profile in addition to their diagnostic WES results. Between August 2016 and April 2018, 168 individuals belonging to 57 families (55 full trios, 1 trio with the exclusion of the index patient, and 1 single parent) provided informed consent and had their WES data available at the time of analysis. The second, retrospective, subcohort consists of individuals who underwent diagnostic WES prior to August 2016. This subcohort was comprised of 1,415 individuals with fully anonymized data. The protocol was approved by the Institutional Review Board of the Leiden University Medical Centre.
Figure 1.
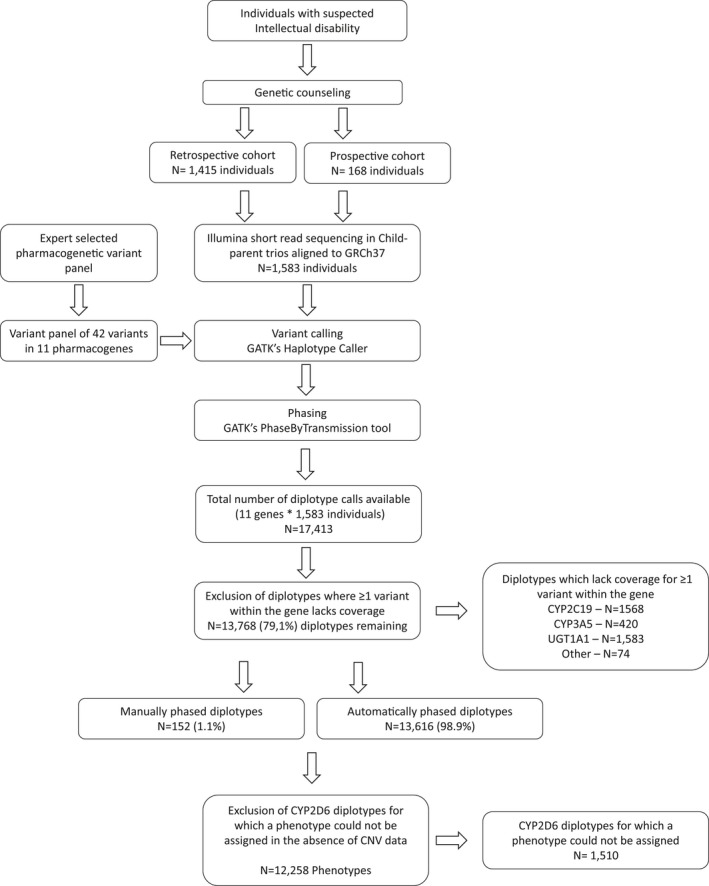
Study flowchart. Whole exome sequencing data from individuals sequenced for diagnostics was used to obtain a clinically relevant pharmacogenetics (PGx) profile. Retrospective cohort: individuals sequenced prior to August 2016; prospective cohort: individuals sequenced after August 2016 if they opted in for obtaining their PGx profile. The expert selected PGx panel was obtained from the Ubiquitous Pharmacogenomics U‐PGx consortium. Sufficient coverage was classified as haplotype quality of at least 20. Due to the absence of copy number variants (CNVs), only CYP2D6 diplotypes consisting of two null‐alleles were included as CNVs would not change the phenotype assignment. Manual phasing and phenotype assignments were based on translation tables from the U‐PGx consortium. GATK, Genome Analysis Tool Kit.
Sequencing data and variant selection
Short reads were aligned to reference genome GRCh37, followed by variant calling and haplotype phasing (Figure 1). A panel of 42 genetic variants covering 11 actionable pharmacogenes (Table S1 ) was composed. Variant and gene selection was based on the panel used in the U‐PGx (http://www.upgx.eu) and includes all actionable genes and variants in the DPWG guidelines.12, 13
Genotype and diplotype calling
The majority of diplotypes (79.1%; N = 13,768 of 17,413 potential diplotype calls) could be extracted from the available WES data. For the remaining 20.9%, one or more single nucleotide variants (SNVs) could not be determined. These sites either lacked sufficient coverage for reliable variant calling—CYP2C19 (rs12248560; g.96521657C>T in 99% of individuals; N = 1,568), CYP3A5 (rs776746; g.99270539C>T in 26% of individuals; N = 420), and 74 calls divided over 6 genes—or could not be called using the GATK HaplotypeCaller—UGT1A1 TATA‐box repeat unit for all individuals (N = 1,583; Figure 2). Haplotypes were assigned based on U‐PGx translation tables using conventional *‐alleles for cytochrome P450 enzymes and DPWG nomenclature for the remaining haplotypes14, 15 (Table S1 ).
Figure 2.
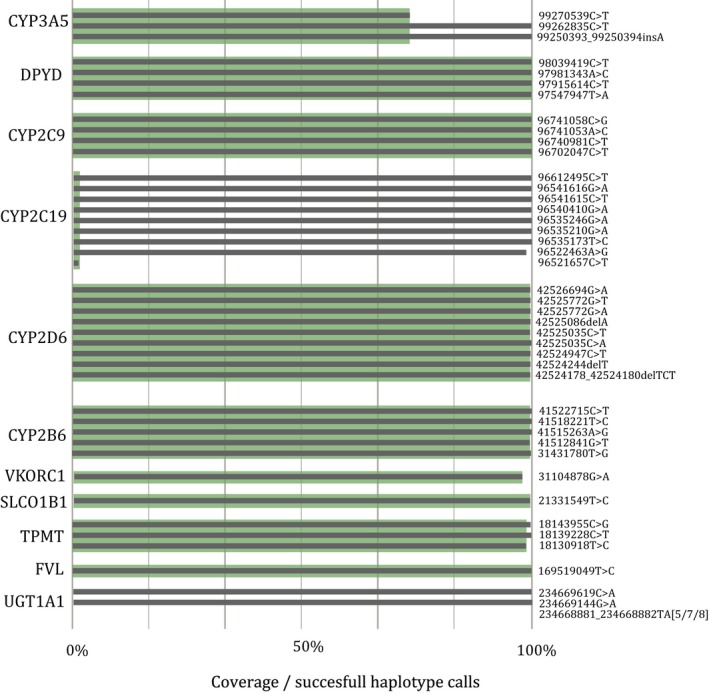
Call rate per gene. In gray: coverage per variant; in green: diplotypes available per gene. For 20.9% of all diplotype calls there was insufficient coverage (haplotype quality < 20) for ≥ 1 variant. Copy number variants could not be determined, results are based on single nucleotide variants only. In total, 13,768 diplotype calls could be included.
Phasing
In total, 13,768 diplotypes were called (Table 1 and Table S2 ), leading to the majority of individuals (70%) with complete SNV data for at least 9 genes (Figure S1 ). An automatically phased diplotype could be assigned to 13,616 calls (98.9%). For 152 calls (1.1%), a phased diplotype call could not be automatically resolved and required manual curation based on the translation table from the U‐PGx consortium. Of the 152 initially unresolved calls, 103 were in CYP2B6, for which the heterozygous presence of the g.41515263A>G and g.41512841G>T variant can lead to both a *1/*6 call as well as a *4/*9 call. Due to the high linkage disequilibrium between the g.41515263A>G and g.41512841G>T variants, it is commonly assumed that these variants occur on the same allele. Therefore, individuals carrying both the g.41515263A>G and g.41512841G>T variants are generally genotyped as CYP2B6*1/*6. This high linkage disequilibrium between g.41515263A>G and g.41512841G>T was also observed in our cohort (Figure 3 a). Wherever automatic haplotype phasing was possible, we observed an improved accuracy in diplotype calls in the CYP2B6 gene. Namely, of the heterozygous carriers of the g.41515263A>G and g.41512841G>T variants, 409 individuals with the CYP2B6*1/*6 haplotype and 6 individuals with the CYP2B6*4/*9 haplotype were identified (Figure 3 b).
Table 1.
Haplotype frequencies
| Gene | Haplotype assignment | Number of alleles | Frequency (%) |
|---|---|---|---|
| CYP2B6 | Total | 3,154 | |
| *1 | 2,279 | 72.0 | |
| *18 | 3 | 0.09 | |
| *4 | 108 | 3.4 | |
| *6 | 733 | 23.2 | |
| *9 | 31 | 0.98 | |
| CYP2C19 | Total | 30 | |
| *1 | 21 | 70.0 | |
| *17 | 5 | 16.7 | |
| *2 | 3 | 10.0 | |
| *4A/B | 1 | 3.3 | |
| CYP2C9 | Total | 3,166 | |
| *1 | 2,561 | 80.9 | |
| *11 | 4 | 0.13 | |
| *2 | 377 | 11.9 | |
| *3 | 222 | 7.0 | |
| *5 | 2 | 0.06 | |
| CYP2D6 a | Total | 3,152 | |
| *1 | 1,966 | 62.4 | |
| *10 | 102 | 3.2 | |
| *17 | 21 | 0.7 | |
| *3 | 51 | 1.6 | |
| *4 | 554 | 17.6 | |
| *41 | 362 | 11.5 | |
| *6 | 28 | 0.9 | |
| *9 | 68 | 2.2 | |
| CYP3A5 | Total | 2,326 | |
| *1 | 299 | 12.9 | |
| *3 | 2,002 | 86.1 | |
| *6 | 21 | 0.9 | |
| *7 | 4 | 0.17 | |
| DPYD | Total | 3,162 | |
| *1 | 3,046 | 96.3 | |
| *2A | 21 | 0.66 | |
| 1236G>A | 75 | 2.2 | |
| 2846A>T | 20 | 0.63 | |
| FVL | Total | 3,166 | |
| F5 positive | 80 | 2.5 | |
| F5 negative | 3,086 | 97.5 | |
| SLCO1B1 | Total | 3,158 | |
| *5 | 443 | 14.0 | |
| wt | 2,715 | 86.0 | |
| TPMT | Total | 3,124 | |
| *3A | 122 | 3.9 | |
| *3C | 20 | 0.29 | |
| *2 | 1 | 0.03 | |
| wt | 2,981 | 95.8 | |
| VKORC1 | Total | 3,098 | |
| 1173T | 1,247 | 40.3 | |
| wt | 1,851 | 59.7 |
Frequencies based on all haplotypes, including manually phased haplotypes. Genes are included if there is sufficient coverage for all variants within that gene. Haplotype assignments are based on translation tables from the Ubiquitous Pharmacogenomics Consortium. F5: Factor V Leiden
CYP2D6 gene duplications and gene deletions could not be determined.
Figure 3.
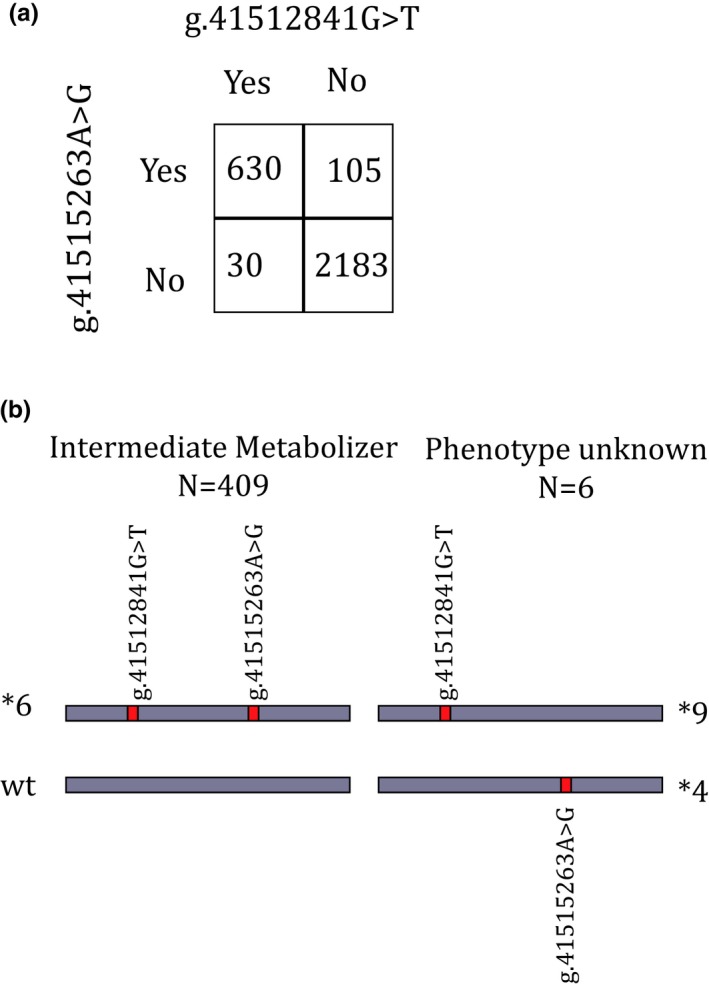
Phasing and linkage disequilibrium in CYP2B6. (a) High linkage disequilibrium is shown by the frequencies of the CYP2B6*4 and *9 variants in all automatically phased haplotypes (N = 2,948 alleles), when combined the haplotype is *6. χ2, P < 0.0001. (b) Possible configurations for the CYP2B6 variants, leading to different phenotypes. Numbers are based on all individuals who carried both variants and could be phased automatically (N = 415).
The remaining 49 diplotype calls that could not be phased automatically were distributed over CYP2C9 (N = 2), CYP2D6 (N = 28), CYP3A5 (N = 1), and TPMT (N = 18; Table S3 ). Based on the final haplotype assignments, there were no significant differences in the haplotype frequencies observed in the children compared with their parents, with the exception of VKORC1 (Table S2 ).
Phenotype calling
Diplotypes were translated into phenotypes based on the DPWG guidelines and U‐PGx translation tables (Table 2). Due to the inability to call copy number variants (CNVs) for CYP2D6, CYP2D6 phenotypes could only be called for individuals carrying two null‐alleles (e.g., CYP2D6*4/*4 or CYP2D6*3/*6) and for these a poor metabolizer (PM) phenotype was assigned (N = 66). For the remaining 1,510 individuals with sufficient coverage on all SNVs, no CYP2D6 phenotype could be assigned. In total, 20.7% of assigned phenotype calls (2,534 of 12,258) were actionable, with actionable defined as a phenotype, which is mentioned in the DPWG guidelines with at least one actionable recommendation (e.g., a dose change or change of drug; Figure 4). The phenotype observed in children did not always match the phenotype observed in either one of the parents. For example, the child can be a CYP2C9 PM whereas the parents both have an intermediate metabolizer phenotype (Figure S2 ). The majority of individuals (N = 1,360; 85.9%) carried at least one gene with an actionable phenotype (Figure 4 d).
Table 2.
Phenotype frequencies and actionability
| Gene | Phenotype | Number of subjects | Frequency | Actionable |
|---|---|---|---|---|
| CYP2B6 | — | 1,577 | — | — |
| PM | 105 | 6.7% | Yes | |
| IM | 528 | 33.5% | Yes | |
| EM | 944 | 59.9% | — | |
| CYP2C19 | — | 15 | — | — |
| PM | — | Yes | ||
| IM | 4 | 26.7% | Yes | |
| EM | 11 | 73.3% | — | |
| UM | — | — | Yes | |
| CYP2C9 | 1,583 | — | — | |
| PM | 59 | 3.7% | Yes | |
| IM | 487 | 30.2% | Yes | |
| EM | 1,037 | 65.5% | — | |
| CYP2D6 | — | 1,576 | — | — |
| PMa | 66 | 4.2% | Yes | |
| Not assigned | 1,510 | 95.8% | — | |
| CYP3A5 | — | 1,163 | — | — |
| PM | 882 | 75.8% | — | |
| IM | 263 | 22.6% | Yes | |
| EM | 18 | 1.5% | Yes | |
| DPYD | — | 1,581 | — | — |
| AS: 0 | — | — | Yes | |
| AS: 0.5 | — | — | Yes | |
| AS: 1 | 21 | 1.3% | Yes | |
| AS: 1.5 | 95 | 6.0% | Yes | |
| AS: 2 | 1,465 | 92.7% | — | |
| F5L | — | 1,583 | — | — |
| F5 Absent | 1,504 | 95.0% | — | |
| F5 Heterozygous | 78 | 4.9% | Yes | |
| F5 Homozygous | 1 | 0.06% | Yes | |
| SLCO1B1 | — | 1,579 | — | — |
| Normal function | 1,172 | 74.2% | ||
| Decreased function | 371 | 23.5% | Yes | |
| Poor function | 36 | 2.3% | Yes | |
| TPMT | — | 1,562 | — | — |
| PM | 1 | 0.06% | Yes | |
| IM | 139 | 8.9% | Yes | |
| EM | 1,422 | 91.0% | — | |
| VKORC1 | — | 1,549 | — | — |
| Normal function (1173CC) | 564 | 36.4% | — | |
| Decreased function (1173CT) | 723 | 46.7% | — | |
| Poor function (1173TT) | 262 | 16.9% | Yes |
Phenotypes are based on the Ubiquitous Pharmacogenomics consortium translation tables, actionability is based on the Dutch Pharmacogenetic Working Group guidelines, whereas actionable is defined as a phenotype accompanied by at least one dosing advise.
AS, Activity Score; EM, extensive metabolizer; F5, Factor V Leiden; IM, intermediate metabolizer; PM, poor metabolizer; UM, ultra‐rapid metabolizer.
Poor metabolizer phenotype assigned based on diplotype consisting of two null‐alleles. For all other diplotypes no CYP2D6 phenotype could be assigned as copy number variants could not be determined.
Figure 4.
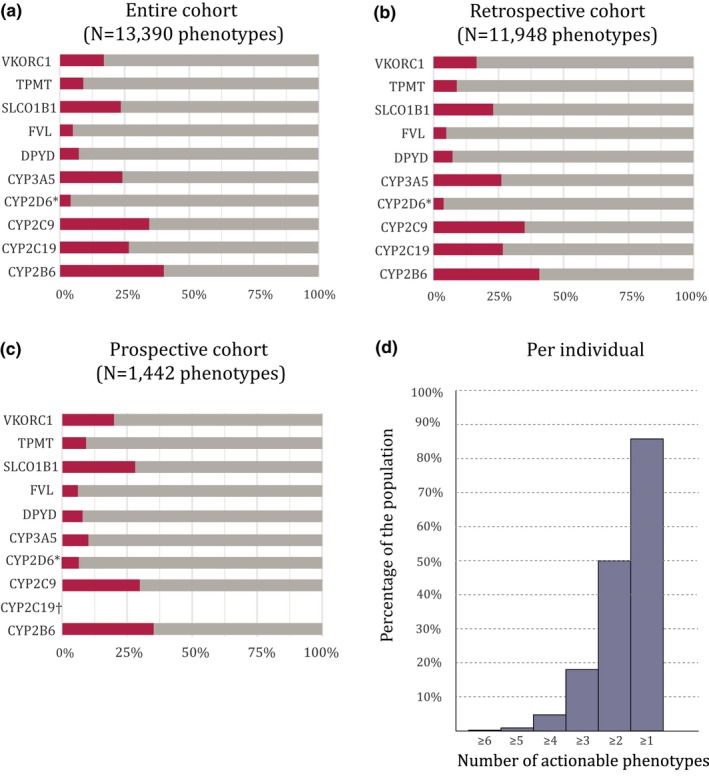
Actionable phenotypes. (a) Actionable phenotypes of the entire cohort, (b) the retrospective cohort, and (c) the prospective cohort. Red: actionable; gray: not actionable. (d) The total number of actionable phenotypes per individual. Actionable is classified as: any phenotype with a dosing advice available in the Dutch Pharmacogenetics Working Group guidelines. An unknown phenotype is categorized as not actionable. Results are based on all genotypes with sufficient coverage (haplotype quality > 20). *Due to an inability to call copy number variants, only CYP2D6 diplotypes consisting of two null‐alleles were assigned a phenotype (poor metabolizer), no phenotype was assigned for other phenotypes classifying them as unknown. †CYP2C19 phenotypes could not be determined for any of the individuals in the prospective cohort due to a lack of coverage for one of the variants.
Diplotype to phenotype translations based on CPIC guidelines yield similar results (Table S4 , Figure S3 ), with 85.1% (N = 1,347) of the population carrying at least one gene with an actionable phenotype and 2,459 actionable phenotypes.
Comparison of genotyping
To assess the correctness of assigned phenotypes based on WES data, seven trios (21 individuals) were randomly selected for orthogonal genotyping on a commercial platform (the pharmacoscan from Thermo Fisher Scientific, Waltham, MA). Due to an inability to call CNVs for CYP2D6 based on WES data, no CYP2D6 phenotype was assigned for the majority of individuals. Nonetheless, a comparison of SNVs identified in the WES pipeline with the SNVs identified on the commercial platform was possible. Of the diplotypes that could be called on both platforms (N = 161), the concordance was 96.9% (N = 156). Due to insufficient genotype quality, 49 diplotype calls had to be excluded from the WES data. These calls were located in the UGT1A1, CYP2C19, CYP3A5, and VKORC1 genes. On the commercial platform, calls were available for all individuals and all genes with the exception of Factor V Leiden (FVL), which was not present on the array.
Of the five discordant calls, one was due to a DPYD variant (1236G>A), which was not included in the commercial platform. Two additional calls could not be resolved by the commercial platform due to the absence of phasing information, both of which were TPMT *1/*3A. The fourth discrepant call concerned a gene duplication of CYP2D6 that could not be identified in the WES data due to limitations in CNV calling. Given the observed diplotype of this last call (CYP2D6 *1/*4), the predicted phenotype does not change in the presence of a duplication based on the U‐PGx translation tables and is classified as intermediate metabolizer both with and without duplication. Last, there was a discrepant CYP2D6 diplotype call. The WES pipeline is called a CYP2D6*4/*10, whereas the commercial platform is called a CYP2D6*4/*4 without duplication. Looking closer at the WES data revealed that this individual was homozygous for the CYP2D6*10 (g.42526694G>A) variant. This variant is also part of the CYP2D6*4 haplotype. The CYP2D6*4 variant (g.42524947C>T) was found in 87% of all CYP2D6 reads, indicating heterozygosity (Figure S4 ). Given the presence of multiple wildtype calls for the CYP2D6*4 (g.42524947C>T) variant in the WES data, it not likely that this individual is homozygous for this variant, and the CYP2D6*4/*10 assignment seems most probable.
Exploratory analysis of the heterozygosity ratio to assess CYP2D6 deletions
Due to limited consent, only genotypes for the selected SNVs in the CYP2D6 locus (Table S1 ) were available and it was not possible to use microsatellites to determine CYP2D6 deletions.16 Therefore, we explored if it is possible to use the CYP2D6 heterozygosity ratio to assess potential CYP2D6 deletions. For each individual with complete SNV data (N = 1,576), a heterozygosity ratio was calculated. A high proportion of heterozygous variants indicate the presence of two different CYP2D6 alleles and, therefore, a low probability of a CYP2D6 deletion. However, a low ratio does not confirm the presence of a deletion, as an individual can be homozygous for all variants in the CYP2D6 locus. The distribution of the heterozygosity ratio (Figure 5) shows that approximately half of the individuals (47.4%) are heterozygous for all variants observed, thereby excluding the possibility of a CYP2D6 deletion. A heterozygosity ratio cutoff set at < 0.25 resulted in 78 individuals (5.3%) for whom a CYP2D6 deletion could not be ruled out. Moreover, of these 78 individuals, 7.7% were genotyped as heterozygous for at least one of the SNVs in our panel (Table S1 ). As these variants are all located in the exons of CYP2D6, a full gene deletion is highly unlikely for these individuals. To decrease the risk of false‐negative results, a more conservative heterozygosity ratio cutoff was set at 0.4 resulting in 173 individuals (11%) for whom a CYP2D6 deletion could not be ruled out.
Figure 5.
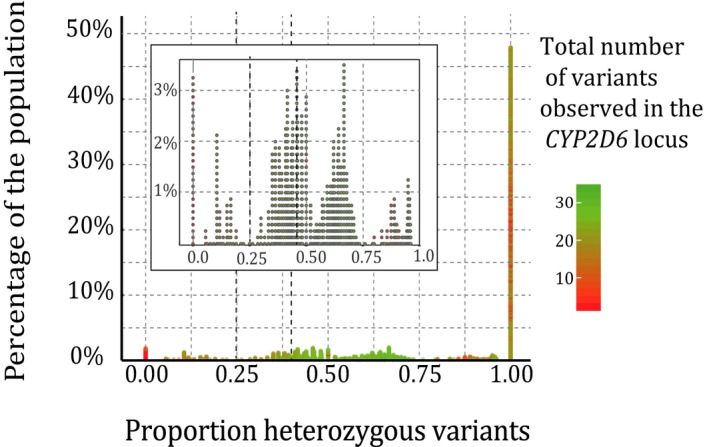
Heterozygosity ratios in CYP2D6. All variants in the CYP2D6 locus were included. The higher the proportion of heterozygous variants, the less likely there is a gene deletion.
Discussion
We have shown that repurposing existing diagnostic WES data for PGx yields successful results for a large proportion of diplotype calls (13,768 of 17,413 potential calls; 76.9%). Unfortunately, inherent to the use of WES data, several phenotypes could not be accurately determined due to a lack of coverage and missing CNV information, resulting in 12,258 reliable phenotype calls (70.4%). Our data also show that 86% of the studied population carried at least one actionable phenotype. This number is lower compared with the 91–99% indicated in previous studies.17, 18 Because frequencies of the haplotypes that could be identified in this study were comparable to frequencies reported in literature this is not expected to be the cause of the lower number of individuals with an actionable phenotype.19 However, the lack of coverage for several genes and inability to identify CYP2D6 CNVs, can be the cause of this discrepancy. These limitations in coverage and CNV calling were also reported previously when using WES data for PGx.8, 10
A novelty of our study is the use of haplotype phasing for resolving pharmacogenetic genotypes and phenotypes. For six individuals, the haplotype phasing proved to be valuable in genotyping CYP2B6. For CYP2B6, it is known that the *4 and *9 variants can occur separately even though they are in strong linkage disequilibrium. Conventionally, haplotype assignments use linkage disequilibrium in the assignment. For CYP2B6, this means that most laboratories assume the *4 (g.41515263A>G) and *9 (g.41512841G>T) variants to be located on the same allele, resulting in a *6 assignment. We have now shown that in 1.5% of the observations (N = 6 of 415), where an individual is heterozygous for both the *4 and the *9 variant, the variants are located in trans‐conformation (CYP2B6*4/*9). Animal and tissue studies have shown conflicting results in regard to the impact of these individual variants on enzyme function, resulting in uncertainty as to what the effect is on enzymatic function and, therefore, what phenotype should be assigned.20, 21, 22, 23, 24 In the DPWG guidelines, CYP2B6*6 is designated as nonfunctional. However, this assignment is only for the combination of the two SNVs (CYP2B6*6) and not for the individual variants (CYP2B6*4 and CYP2B6*9).3
Although we applied haplotype phasing based on child‐parent trios, alternative methods are available. Haplotype phasing methods can be roughly categorized into direct and inferential approaches. Direct methods include single‐cell sequencing and paired read sequencing, which are accurate but also costly.25 Inferential methods are either pedigree or population based. Population‐based methods use estimated probabilities based on population frequencies and pedigree‐based methods use the shared alleles between two or more individuals.26
Assessing CNVs from WES data is challenging, which is particularly limiting for CYP2D6. A previous study used eXome Hidden‐Markov Model as a tool to identify CNVs in WES data, leading to discrepancies in CNV in 7% of CYP2D6 calls, compared with orthogonal testing.8 Furthermore, multiple approaches determining CNVs based on sequencing depth have been developed over the past years. Unfortunately, these methods have been shown to be unreliable for individual patient‐level CNV calling, leading to a limited value of these algorithms in the diagnostic setting.27 Another widely recognized approach is the use of microsatellite markers with a high degree of heterozygosity.16 However, due to consent limited to the predefined SNVs (Table S1 ) we were not able to use this method. Therefore, we explored the use of the heterozygosity ratio of the entire CYP2D6 locus to assess CYP2D6 deletions. A heterozygosity ratio cutoff set at 0.25 (25% heterozygous variants) resulted in 5.3% of the individuals for whom a CYP2D6 deletion could not be ruled out. This frequency seems concordant with the 1–7% CYP2D6 gene deletions reported in literature for the Dutch population.28 However, 6 individuals in this group were genotyped heterozygous (e.g., CYP2D6*1/*4) for at least one of the selected important SNVs in CYP2D6 (Table S1 ), all of which are exonic variants. The presence of heterozygous variants in the CYP2D6 exons indicates that a CYP2D6 gene deletion is highly unlikely indicating that a low ratio of heterozygosity does not necessarily confirm a gene deletion. Additionally, the presence of heterozygous variants in the upstream and downstream regions captured in our aggregated data could result in a high ratio even in the presence of a gene deletion. A more conservative cutoff was set at 0.4, to decrease the change of excluding individuals with false high ratios. With this cutoff, a CYP2D6 deletion could not be ruled out for 11% of the individuals, indicating a lower risk for false‐negative results. In case of limited consent, an approach based on the heterozygosity ratio could potentially be used to indicate individuals for whom a gene deletion is highly unlikely. Obtaining reliable data in regard to CNVs in CYP2D6 is of importance for clinical practice, as the presence of a deletion or duplication can both increase and decrease CYP2D6 enzyme function. Only for individuals with two null‐alleles based on SNV data (e.g., *3/*3) a CNV will not change the assigned phenotype (PM). In our cohort, this led to reliable phenotype calls for 4.2% of the individuals, for the remainder of the cohort it is expected that 2–9% of the individuals will carry either a gene deletion or duplication, which will affect the phenotype assigned.19 Therefore, we argue that due to the inability to accurately phenotype for the majority of the population, phenotypes based on our pipeline should not be used in clinical practice until there are more accurate methods for CNV calling available.
Because difficulties with coverage and CNV calling are inherent limitations to the use of WES, it is difficult to solve these problems without resorting to other technologies like WGS or array‐based techniques. These techniques have been suggested by other groups as more suitable for PGx profiling.8, 29 However, both these technologies have not been routinely implemented as extensively as WES in a diagnostic setting. Applying these technologies will, therefore, lead to additional testing costs, whereas repurposing existing WES data does not. Moreover, a relatively simple solution to increase the performance of WES for PGx is to expand the WES capture kit used with relevant intronic sites. One downside to this, compared with WGS, is that the need might arise for additional intronic regions, which are not yet included in the capture kits. Additionally, increasing the capture kit will still not resolve CNV calling problems and a new capture kit will need to be tested thoroughly. Nonetheless, the main limitations of WES are gene‐specific and do not apply to all pharmacogenes. Therefore, we argue that despite the inherent limitations of WES data for PGx, a reliable profile can be extracted from WES data for the majority of clinically relevant pharmacogenes.
Currently, multiple efforts are ongoing to develop tools that can assist in extracting PGx profiles from NGS data. One such approach is the Stargazer tool.30, 31 Stargazer incorporates haplotype calling for 28 pharmacogenes and CNV calling for CYP2D6 based on NGS data in a user‐friendly algorithm. Stargazer's ability to call CYP2D6 diplotypes was evaluated on a sample consisting of WGS data of 32 trios, showing a 99.0% concordance with conventional SNV‐typing. Although results are very promising, validation in a larger sample is needed. Moreover, any approach with WES data is still restricted to the limitations inherent to the use of WES, as described above.
The potential impact of implementing a preemptive PGx panel‐based test for a substantial number of individuals, as can be done by repurposing diagnostic sequencing data, is large. In the Netherlands, there are 3,628,597 incident prescriptions per year (1 in every 19) for drugs that interact with the genes included in our study (excluding only FVL). Based on simulations, it was estimated that 23.6% of these prescriptions would lead to an actionable gene–drug interaction.32 By testing preemptively, these drug–gene interactions can be managed in a timely manner, potentially reducing the number of ADRs. More specifically, the individuals included in this study were originally sequenced to diagnose the cause of ID in the children. Several studies have shown that polypharmacy is more common among patients with ID.33, 34, 35 More important, patients with ID more often use antipsychotics, anticonvulsants, and/or antidepressants that frequently result in gene–drug interactions making the value of a PGx profile for this population even more meaningful. Unfortunately, many of these drugs are metabolized by either CYP2D6 or CYP2C19, both of which have shown to be difficult to determine based on existing WES data in our study.
The results of our actionability analysis are based on a panel of 11 genes designed by the U‐PGx consortium to cover all actionable pharmacogenes in the DPWG guidelines.12 However, many groups implementing PGx also use CPIC guidelines.36 The CPIC guidelines currently provide recommendations for genetic variants in 19 genes.5 Due to limited consent, covering only the DPWG genes, it was not possible to determine the phenotypes for all genes covered by the CPIC guidelines. However, for the 11 genes for which SNV data were available, translations based on CPIC guidelines showed that the number of individuals with at least one actionable phenotype was comparable to the DPWG guidelines (85.1% and 85.9%, respectively).37 The slight difference was due to the FVL gene, which is not included in the CPIC guidelines and, therefore, not actionable.
Conclusion
Despite the inherent limitations in regard to coverage of intronic variants and CNVs, this study shows that it is possible to repurpose existing diagnostic WES data to extract a PGx profile for 7 of 11 clinically actionable PGx genes. Additionally, the availability of trio data with phased haplotype information allows more accurate phenotype predictions, particularly for CYP2B6.
Methods
At the Leiden University Medical Center, WES for diagnostic purposes has been used since 2013. When possible, the index patient and both of the parents are sequenced to allow for haplotype phasing and the discovery of de novo variants. From August 2016 onward, individuals were asked if they wanted to retrieve their PGx profile from their WES data. Individuals who consented were included in the prospective subcohort of this study (Figure 1). The retrospective subcohort consisted of individuals sequenced prior to August 2016 who were assigned anonymous study identifications before inclusion (N = 1,415). All individuals received genetic counselling during the diagnostic WES process. The study was approved by the Institutional Review Board of the Leiden University Medical Center.
Variant selection
The gene and variant panel used was based on the panel designed for the U‐PGx consortium's PREPARE study (version June 2017), with the exclusion of the HLA genes due to their high complexity and the lack of tagging single nucleotide polymorphisms in the white population.13, 38 In brief, variants were selected based on the availability of a corresponding DPWG guideline, the effect of the variant on protein function, and the frequency of the variant.12 The final panel consisted of 42 variants located in 11 pharmacogenes.
Variant calling
Sequencing was performed on Illumina HiSeq4000 (Illumina, San Diego, CA) using 150 bp reads, from 2015 onward. Samples analyzed prior to 2015 were sequenced on HiSeq2500 (100 bp reads) or HiSeq2000 (100 bp reads). Paired‐end sequencing technology was used. Agilent SureSelect V5 was used for enrichment. Short reads were aligned to reference genome GRCh37, using the bwa tool with the BWA‐MEM algorithm,39 followed by variant calling using the GATK's HaplotypeCaller.40 In order to accurately phase the reads, the analysis was performed in child–parent trios. Data from individuals who did not consent to retrieve their PGx results were used for phasing and disregarded thereafter. Variant call format (VCF) files were phased using the GATK's PhaseByTransmission tool41 resulting in two fully phased alleles for each individual in the trio. Variants that could not be phased were reported separately. For each locus of interest, FASTA format sequences were generated for each allele of each individual by applying the variants in each locus to the reference sequence by using Mutalyzer.42, 43 The VCF files were then used to create a coverage track in BED format for each individual for each locus of interest. A haplotype quality of at least 20 is required to be considered “covered.” The phased VCF file per individual is additionally used to enumerate the total number and heterozygous number of variants per locus per individual.
Genotyping and phenotyping
The FASTA sequences were used for genotype assignments. Haplotype assignments were done according to U‐PGx translation tables.12, 13 A “No call” was assigned when at least one variant in the gene lacked coverage. If an unphased variant was present and no other variants were observed, the individual was haplotyped as being heterozygous for the unphased variant. In the case of multiple variants in the gene of which at least one unphased, genotype calling was done manually based on linkage, the most likely combination of variants by using the U‐PGx assignments for these variant combinations were used. Phenotypes were assigned according to the U‐PGx translation tables based on the DPWG guidelines.12, 13 For CYP2D6, only when the assigned diplotype consisted of two null‐alleles (e.g., CYP2D6*4/*4 or CYP2D6*3/*6) a PM phenotype was assigned, as a duplication or deletion would not change the assignment. All other CYP2D6 diplotypes were excluded from further analysis as the presence of a CNV could change the phenotype. A phenotype was considered actionable when it was described in the DPWG guidelines with advice in regard to a dose adjustment, drug change, or intensive monitoring. Additionally, genotype to phenotype translations were also performed based on CPIC guidelines for all 11 genes in our panel.5
Comparison
For comparison, 21 samples retrieved from 7 trios, were randomly selected from the prospective cohort and genotyped on a commercial platform (the pharmacoscan from Thermo Fisher Scientific).44
This platform identifies 4,627 variants in 1,191 pharmacogenes, among which are all genes from the panel used in this study with the exception of FVL. Genotype calls from the WES‐pipeline were compared with the results obtained with the commercial platform.
Regions of heterozygosity in CYP2D6
For all genes in the panel, consent did not extend to the entire gene locus but only to the specific pharmacogenetic variants in the selected genes (Table S1 ). Additionally, per individual, per gene locus aggregated data containing the number of all heterozygous and homozygous variants were available. To assess possible deletions in CYP2D6, a heterozygosity ratio was calculated for each individual. The number of heterozygous variants in the CYP2D6 locus was divided by the total number of variants within this locus, resulting in the heterozygosity ratio. A high proportion of heterozygous variants indicate the presence of two different alleles and, therefore, a low to nonexisting chance of a deletion. As this locus also includes upstream and downstream sequences, which are not included in a CYP2D6 gene deletion, the proportion of heterozygous variants can be higher than zero even in the presence of a deletion due to variants in these upstream and downstream regions. As exact locations of the variants could not be obtained, the impact of variants in these regions could not be determined.
Based on the distribution of the ratio of heterozygosity, both a strict (0.25) and conservative (0.4) cutoff in the ratio were examined. The conservative cutoff decreases the change of falsely excluding the presence of a deletion based on a high ratio of heterozygosity due to upstream and downstream variants. As all the SNVs selected in this study (Table S1 ) are located in CYP2D6 exons, which are part of the deletion region, heterozygosity for these variants would automatically rule out the presence of a deletion. Genotypes of individuals below either cutoff were assessed to determine the heterozygosity for the selected SNVs. Heterozygous calls for any of these SNVs will indicate the presence of two CYP2D6 alleles and, therefore, the absence of a CYP2D6 deletion despite a low ratio of heterozygosity, providing an estimate of the number of falsely low ratios of heterozygosity. This approach is exploratory, as there is no detailed information available regarding all variant locations, definite answers regarding the presence of a CYP2D6 gene deletion cannot be provided.
Funding
The research leading to these results has received funding from the European Community's Horizon 2020 Program under grant agreement no. 668353 (U‐PGx).
Conflict of Interest
All authors declared no competing interests for this work.
Author Contributions
M.L., M.K., H.J.G., S.A., and J.S. wrote the manuscript. G.S., M.K., H.J.G., and J.S. designed the research. C.R., M.H, S.B., M.L., and W.A. performed the research. M.L., M.K., J.S., and S.A. analyzed the data.
Supporting information
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Tables S1–S4.
References
- 1. Relling, M.V. & Evans, W.E. Pharmacogenomics in the clinic. Nature 526, 343–350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schildcrout, J.S. et al. Optimizing drug outcomes through pharmacogenetics: a case for preemptive genotyping. Clin. Pharmacol. Ther. 92, 235–242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koninklijke Nederlandse Maatschappij ter bevordering van de Pharmacie. Pharmacogenetics <http://www.knmp.nl/farmacogenetica> (2019). [Google Scholar]
- 4. PharmGKB . DPWG: Dutch Pharmacogenetics Working Group <https://www.pharmgkb.org/page/dpwg>. Accessed April 10, 2017.
- 5. Clinical Pharmacogenetics Implementation Consortium . CPIC‐Guidelines <https://cpicpgx.org/> (2017). Accessed April 4, 2017. [Google Scholar]
- 6. Whirl‐Carrillo, M. et al. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 92, 414–417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bank, P.C.D. , Swen, J.J. & Guchelaar, H.J. Implementation of pharmacogenomics in everyday clinical settings. Adv. Pharmacol. 83, 219–246 (2018). [DOI] [PubMed] [Google Scholar]
- 8. Yang, W. et al. Comparison of genome sequencing and clinical genotyping for pharmacogenes. Clin. Pharmacol. Ther. 100, 380–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bielinski, S.J. et al. Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time‐using genomic data to individualize treatment protocol. Mayo Clin. Proc. 89, 25–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Londin, E.R. , Clark, P. , Sponziello, M. , Kricka, L.J. , Fortina, P. & Park, J.Y. Performance of exome sequencing for pharmacogenomics. Pers. Med. 12, 109–115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cousin, M.A. et al. Pharmacogenomic findings from clinical whole exome sequencing of diagnostic odyssey patients. Mol. Genet. Genom. Med. 5, 269–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Wouden, C.H. et al. Development of the PGx‐Passport: a panel of actionable germline genetic variants for pre‐emptive pharmacogenetic testing. Clin. Pharmacol. Ther. 106, 866–873 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Wouden, C.H. et al. Implementing pharmacogenomics in Europe: design and implementation strategy of the ubiquitous pharmacogenomics consortium. Clin. Pharmacol. Ther. 101, 341–358 (2017). [DOI] [PubMed] [Google Scholar]
- 14. Dutch Pharmacogenetics Working Group . Transparency document pharmacogenetics <https://www.knmp.nl/downloads/g-standaard/transparantiedocumenten/transparantiedocument-farmacogenetica.pdf> [in Dutch]. Accessed June 13, 2017. [Google Scholar]
- 15. Gaedigk, A. , Ingelman‐Sundberg, M. , Miller, N.A. , Leeder, J.S. , Whirl‐Carrillo, M. & Klein, T.E. The Pharmacogene Variation (PharmVar) Consortium: incorporation of the human cytochrome P450 (CYP) allele nomenclature database. Clin. Pharmacol. Ther. 103, 399–401 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dezentje, V.O. et al. CYP2D6 genotype‐ and endoxifen‐guided tamoxifen dose escalation increases endoxifen serum concentrations without increasing side effects. Breast Cancer Res. Treat. 153, 583–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dunnenberger, H.M. et al. Preemptive clinical pharmacogenetics implementation: current programs in five US medical centers. Ann. Rev. Pharmacol. Toxicol. 55, 89–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Driest, S.L. et al. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin. Pharmacol. Ther. 95, 423–431 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou, Y. , Ingelman‐Sundberg, M. & Lauschke, V.M. Worldwide distribution of cytochrome P450 alleles: a meta‐analysis of population‐scale sequencing projects. Clin. Pharmacol. Ther. 102, 688–700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lang, T. et al. Extensive genetic polymorphism in the human CYP2B6 gene with impact on expression and function in human liver. Pharmacogenetics 11, 399–415 (2001). [DOI] [PubMed] [Google Scholar]
- 21. Kirchheiner, J. et al. Bupropion and 4‐OH‐bupropion pharmacokinetics in relation to genetic polymorphisms in CYP2B6. Pharmacogenetics 13, 619–626 (2003). [DOI] [PubMed] [Google Scholar]
- 22. Tsuchiya, K. et al. Homozygous CYP2B6 *6 (Q172H and K262R) correlates with high plasma efavirenz concentrations in HIV‐1 patients treated with standard efavirenz‐containing regimens. Biochem. Biophys. Res. Commun. 319, 1322–1326 (2004). [DOI] [PubMed] [Google Scholar]
- 23. Hofmann, M.H. et al. Aberrant splicing caused by single nucleotide polymorphism c.516G>T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CYP2B6 in liver. J. Pharmacol. Exp. Ther. 325, 284–292 (2008). [DOI] [PubMed] [Google Scholar]
- 24. Watanabe, T. et al. Functional characterization of 40 CYP2B6 allelic variants by assessing efavirenz 8‐hydroxylation. Biochem. Pharmacol. 156, 420–430 (2018). [DOI] [PubMed] [Google Scholar]
- 25. Snyder, M.W. , Adey, A. , Kitzman, J.O. & Shendure, J. Haplotype‐resolved genome sequencing: experimental methods and applications. Nat. Rev. Genet. 16, 344–358 (2015). [DOI] [PubMed] [Google Scholar]
- 26. Browning, S.R. & Browning, B.L. Haplotype phasing: existing methods and new developments. Nat. Rev. Genet. 12, 703–714 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yao, R. et al. Evaluation of three read‐depth based CNV detection tools using whole‐exome sequencing data. Mol. Cytogenet. 10, 30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bank, P.C.D. , Swen, J.J. , Schaap, R.D. , Klootwijk, D.B. , Baak‐Pablo, R. & Guchelaar, H.J. A pilot study of the implementation of pharmacogenomic pharmacist initiated pre‐emptive testing in primary care. Eur. J. Hum. Genet. 27, 1532–1541 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reisberg, S. et al. Translating genotype data of 44,000 biobank participants into clinical pharmacogenetic recommendations: challenges and solutions. Genet. Med. 21, 1345–1354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee, S.B. et al. Stargazer: a software tool for calling star alleles from next‐generation sequencing data using CYP2D6 as a model. Genet. Med. 21, 361–372 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee, S.B. , Wheeler, M.M. , Thummel, K.E. & Nickerson, D.A. Calling star alleles with stargazer in 28 pharmacogenes with whole genome sequences. Clin. Pharmacol. Ther. 106, 1328–1337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bank, P.C.D. , Swen, J.J. & Guchelaar, H.J. Estimated nationwide impact of implementing a preemptive pharmacogenetic panel approach to guide drug prescribing in primary care in The Netherlands. BMC Med 17, 110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peklar, J. et al. Medication and supplement use in older people with and without intellectual disability: an observational, cross‐sectional study. PLoS One 12, e0184390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salomon, C. , Britt, H. , Pollack, A. & Trollor, J. Primary care for people with an intellectual disability ‐ what is prescribed? An analysis of medication recommendations from the BEACH dataset. BJGP Open 2, bjgpopen18X101541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O'Dwyer, M. , McCallion, P. , McCarron, M. & Henman, M. Medication use and potentially inappropriate prescribing in older adults with intellectual disabilities: a neglected area of research. Ther. Adv. Drug Saf. 9, 535–557 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Relling, M.V. & Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 89, 464–467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bank, P.C.D. et al. Comparison of the Guidelines of the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group. Clin. Pharmacol. Ther. 103, 599–618 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Osanlou, O. , Pirmohamed, M. & Daly, A.K. Pharmacogenetics of adverse drug reactions. Adv. Pharmacol. 83, 155–190 (2018). [DOI] [PubMed] [Google Scholar]
- 39. Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv preprint arXiv:13033997 <https://arxiv.org/abs/1303.3997 (2013).
- 40. Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv <https://www.biorxiv.org/content/10.1101/201178v3> (2017).
- 41. Broad institute PhaseByTransmission <https://software.broadinstitute.org/gatk/documentation/tooldocs/3.8-0/org_broadinstitute_gatk_tools_walkers_phasing_PhaseByTransmission.php> (2018).
- 42. Wildeman, M. , van Ophuizen, E. , den Dunnen, J.T. & Taschner, P.E. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mut. 29, 6–13 (2008). [DOI] [PubMed] [Google Scholar]
- 43. Vis, J.K. , Vermaat, M. , Taschner, P.E. , Kok, J.N. & Laros, J.F. An efficient algorithm for the extraction of HGVS variant descriptions from sequences. Bioinformatics 31, 3751–3757 (2015). [DOI] [PubMed] [Google Scholar]
- 44. ThermoFisher . Pharmacoscan Assay <https://www.thermofisher.com/order/catalog/product/903010TS> (2017). Accessed December 21, 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Figure S4.
Tables S1–S4.