

Abstract
Objective
Alternative splicing can generate various structural and functional protein isoforms. Recently, accumulating evidence shows a relationship between alternative splicing and cancer. Cancer is a complex and chronic disease that involves malignant transformation. In this review, we consider alternative splicing events in relation to the hallmarks of cancer cells, and discuss current therapies to treat cancer-related to alternative splicing.
Data sources
Data cited in this article are from the PubMed and Embase database, primarily focusing on research published from 2000 to 2018.
Study selection
Articles were selected with the search terms “alternative splicing,” “cancer cell,” “tumor microenvironment,” and “therapy.”
Results
Alternative splicing plays an important role in tumorigenesis, development, and escape from cell death. Taking this trait of cancer cells into consideration will allow more definite diagnoses of cancer, and allow the development of more effective medicines to intervene in cancer that could focus on controlling alternative splicing or competitively binding to the final products.
Conclusions
Alternative splicing is common in cancer cells. Consideration of alternative splicing may allow different strategies for cancer therapy or the identification of novel biomarkers for cancer diagnosis.
Keywords: Alternative splicing, Cancer cell, Hallmark, Therapy, Tumor microenvironment
Introduction
Alternative splicing, a complicated but highly regulated process in human cells that was first identified by Walter in 1978,[1] allows one gene to code for multiple proteins. Recently, genome-wide applications of next-generation sequencing technology have shown that alternative splicing occurs in more than 90% of human genes.[2–7]
The splicing process is carried out by the spliceosome, which consists of five small nuclear ribonucleoprotein (snRNP) particles (U1, U2, U4, U5, and U6 snRNPs) that assemble at each intron around splice sites. Each splice site consists of a consensus sequence around each exon-intron junction that is recognized by the spliceosome.[8,9] In addition, other sequence components in exons or introns can work as enhancers or silencers and regulate the binding of splicing factors, which can either promote or inhibit the recognition of a given exon by the spliceosome. Some RNA-binding proteins may regulate splicing or the messenger RNA (mRNA) stability of genes, especially for inflammation- and tumor-related genes.[10,11] Among these RNA-binding proteins, two main nuclear RNA-binding protein families, the heterogeneous nuclear ribonucleoprotein (hnRNP) family and the serine/arginine-rich protein (SR) family, often play antagonistic roles in the regulation of exon recognition and act in combination.
After alternative splicing of pre-mRNA, the potential different modes of alternative splicing can be divided into the following categories: exon skipping, intron retention, alternative 5′/3′ donor/acceptor sites, mutually exclusive exons, alternative promoters, and alternative splicing and polyadenylation[12] [Figures 1 and 2].
Figure 1.
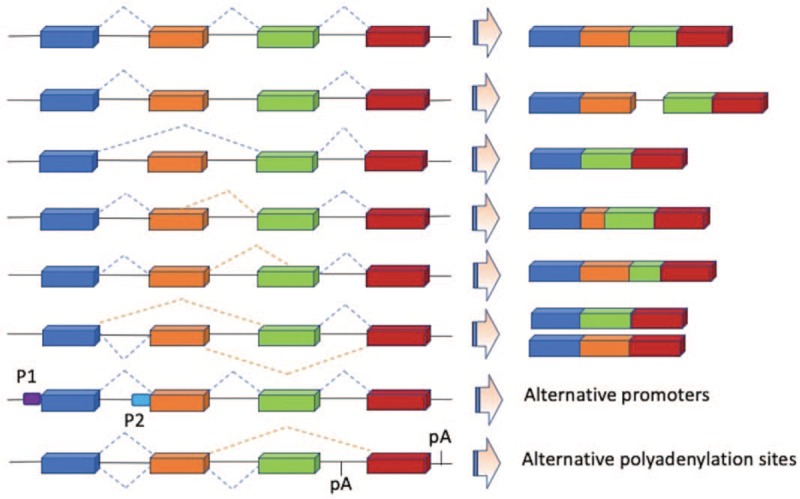
Summary of seven alternative splicing patterns from Blencowe.[12]
Figure 2.
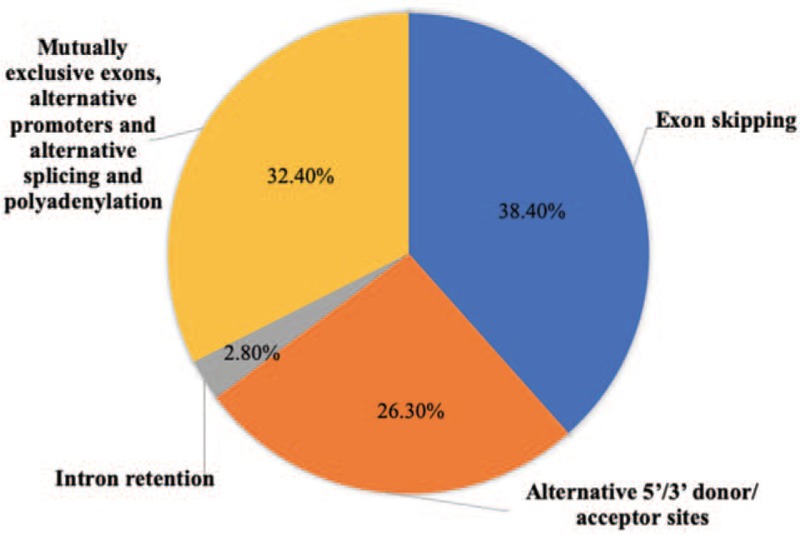
Proportion of alternative splicing events from Sugnet.[13]
Different alternative splicing patterns can result in the production of varied transcripts, and these abnormal changes in structure may influence both the gene expression level and translation of the mRNA into protein, giving different functional properties.[14–16]
However, although alternative splicing beneficially allows the production of many varied proteins from a single gene, it can also have negative effects and can play a role in cancer, posing a major challenge for modern medicine. Therefore, this review will focus on the relationship between alternative splicing and the hallmarks of cancer cells.
Alternative splicing and the hallmarks of cancer cells
Considering that the hallmarks of cancer cells are raised for several years,[17] there has been increasing recognition of the key role played by aberrant splicing in tumorigenesis, cancer progression, and resistance to therapy. The following events correlate with alternative splicing in cancer cells.
Sustaining proliferative signaling
Compared with normal cells, a fundamental trait of cancer cells is sustaining chronic proliferation. Cancer cells can deregulate proliferative signals even without any stimulation induced by a growth factor.
Alternative splicing plays a role in this process. The RAS/RAF/extracellular regulated protein kinases (ERK) pathway, including Kirsten rat sarcoma viral (KRAS) protein, is a key element in most epithelial cell-derived tumors. A positive feedback loop coupling RAS/mitogen-activated protein kinase (MAPK) activation and CD44 variant 6 (CD44v6), which is an alternative splicing variant that includes exon v6 in the cell surface tumor marker clusters of differentiation 44 (CD44), promotes cell proliferation.[18] Once this ability of CD44v6 is utilized by cancer cells, a normal cell may be irreversibly transformed into a malignant cell. CD44v6 overexpression is strongly linked to tumorigenesis and cancer progression in colon cancer, rectal cancer, breast cancer, ovarian cancer, and pancreatic cancer.[19–21] Another conventional signaling pathway is the Wnt/β-catenin pathway. In colorectal cancers, the Wnt pathway promotes a high rate of alternative splicing events.[22] Wnt signaling can also regulate the alternative splicing factor polypyrimidine tract-binding protein 1 (PTBP1). Expression of PTBP1 is controlled by a transcriptional complex formed by β-catenin, T-cell-specific transcription factor/lymphoid enhancer-binding factor, and nuclear phospho-PKM2 (pSer37), which is phosphorylated by ERK in response to KRAS activation.[23]
Evading growth suppressors
Cancer must also circumvent growth suppression from the actions of tumor suppressor genes that negatively regulate cell proliferation. In hepatocellular tumors, RAS signaling induces AKT activation and subsequent serine/arginine-rich splicing factor 1 (SRSF1)-dependent splicing of the SV1 isoform of Krüppel-like factor 6, which is a cytoplasmic inactive variant of this tumor-suppressing transcription factor.[24] This role can further be deduced from the lack of the phosphorylation of RNA splicing factors including SRSF9, serine and arginine repetitive matrix 1 (SRRM1), SRRM2, transformer 2 homolog (TRA2B), SRSF10, and CUGBP Elav-like family member 1 in GSK3 knockout cells, which is related to 194 splicing differences in 188 genes.[25] SRSF3 overexpression counteracts p53β-mediated cell senescence by regulating alternative splicing.[26] P53β is a spliceosome of the TP53 gene, which is a key suppressor of proliferation signaling.
Resisting cell death
Apoptosis, programmed cell death, is a natural barrier to cancer development. The apoptotic machinery consists of upstream regulators and downstream effector components.[27] Caspase-9 (Casp-9) is an initial controller in this program. In lung cancer cells, hnRNPL phosphorylation by activated AKT leads to hnRNPL binding a splice site in Casp-9 pre-mRNA, generating the anti-apoptotic Casp-9b isoform[28,29] and leading to lung tumorigenesis. Casp-9b also participates in Nuclear Factor kappa-B (NF-κB) activation.[30] In hepatocellular carcinomas, SVHB, a specific splicing variant of SVH, is involved in hepatocarcinogenesis. SVHB is not only upregulated but also directly combines with p53 protein to mediate apoptosis. The suppressed expression of SVHB can accelerate the apoptotic program in hepatoma cells.[31] Therefore, there may be the potential to develop a new strategy for tumor suppression by regulating the expression of these genes.
Enabling replicative immortality
Cancer cells have the capacity to generate macroscopic tumors because of the development of unlimited replicative potential.
Telomeres participate in unlimited proliferation by protecting the ends of chromosomes.[32] In the Wnt pathway, human telomerase reverse transcriptase (hTERT), a main component of telomerase, catalyzes telomere production.[33] hTERTα and hTERTβ are the spliceosomes of hTERT.[34] hTERTα is an endogenous inhibitor of telomerase, thereby leading to cell senescence and death, while hTERTβ can trigger mRNA degradation via nonsense-mediated decay resulting from disorderly splicing of the seventh and eighth exons.[33] In myelodysplastic syndromes and melanoma, the hTERTα and hTERTβ expression levels show a substantial difference compared with controls.[35,36]
Inducing angiogenesis
In the normal physiological condition, angiogenesis is generally transient. In contrast, tumor-associated angiogenesis is immortal, can supply nutrients and oxygen, and can evacuate metabolic wastes and carbon dioxide. A well-known angiogenesis inducer is vascular endothelial growth factor (VEGF). There have been multiple studies indicating that VEGF can be regulated by alternative splicing.[37,38] Different splicing methods of the eighth exon of VEGF produce two spliceosomes with opposite functions in angiogenesis. One of these, VEGF165b, competitively binds to the VEGF receptor to inhibit angiogenesis. In human colorectal tumors, VEGF165b downregulation is a marker of poor prognosis.[39] The other VEGF splice variant, VEGF165, is proangiogenic and can be mediated by the transcription factor Wilms tumor 1 (WT1). In the absence of functional WT1, serine-arginine protein kinase 1 (SRPK1) expression and subsequent SRSF1 hyperphosphorylation increase, thereby promoting VEGF165 expression.[40] By contrast, SRPK1 inhibition can affect the progression of prostate cancer by downregulating VEGF165.[41]
Activating invasion and metastasis
Carcinomas arising from epithelial tissues progress to higher pathological grades of malignancy, as reflected by local invasion and distant metastasis. The associated cancer cells typically develop alterations in their shape and attachment to other cells and the extracellular matrix (ECM). The epithelial-mesenchymal transition program broadly regulates invasion and metastasis.[42] In this process, epithelial cells gradually lose their polarity and adhesion and transform into mesenchymal stem cells, which are multifunctional stromal cells that can differentiate into numerous cell types.[43–47] A set of studies documented that CD44 spliceosomes regulate EMT. In breast tumor tissues, the CD44 variant (CD44v) is involved in EMT activity.[48] The overexpression of the CD44 standard isoform (CD44s) is positively related to the EMT status by enhancing Akt signaling to promote the viability of cancer cells.[49] The two spliceosomes of epithelial splicing regulatory protein (ESRP), ESRP1 and ESRP2, regulate EMT.[50] ESRP1 inhibits CD44s by ectopic expression, thereby terminating EMT.[51] In lung cancer cells, decreased ESRP1 expression induces CD44s8–10 overexpression and enhances the potential ability to metastasize.[52] In prostate cancer cells, RNA binding motif 3 overexpression limits CD44s8-10 expression and allows the cells to lose the malignant phenotype and the characteristics of cancer stem cells.[53] The examples above also indicate that the proportions of CD44v and CD44s seem to determine the progress of the tumor. When the proportion of CD44s is high, tumors are always restricted to the organ. In contrast, if the CD44v proportion is high, then the occurrence of tumor invasion and metastasis will dramatically increase.
Reprogramming energy metabolism
Since Otto Warburg first observed that cancer cells have abnormal energy metabolism, the idea that neoplastic disease reprograms energy metabolism for fuel cell growth and division has been increasingly accepted. Even in the presence of oxygen, these cells can refine their glucose metabolism and energy production to glycolysis by limiting energy metabolism, thereby leading to a state called aerobic glycolysis.[54]
Pyruvate kinase (PKM) is the key enzyme in aerobic glycolysis. The two different splicing variants of PKM in enzyme kinetics, PKM1 and PKM2, contain the mutually exclusive exons 10 and 9, respectively.[11,55] PKM1 expression accelerates oxidative phosphorylation in the brain and muscle, while PKM2 expression improves the accumulation of upstream glycolytic regulators to pulse the anabolic metabolism and tumor proliferation.[56,57] PKM2 overexpression and the excessive accumulation of lactic acid are observed in glioblastoma, lung cancer, multiple myeloma (MM), and hepatocellular carcinoma.[11,58–60] Additionally, increased PTBP1 levels play a role in tumorigenesis, and are associated with a shift in the alternative splicing of the transcript encoding PKM.[61]
Glycolytic fueling is associated with activated oncogenes and mutant tumor suppressors. A recent study revealed an mammalian target of rapamycin complex 1/S6 kinase pathway, leading to the phosphorylation of kinase SRPK2 and subsequent activation of SR protein. This pathway is linked to the U1-70K spliceosome component, and can improve lipogenesis-related transcript splicing to fuel cancer metabolism.[62] In solid tumors, hypoxic regions frequently originate because of a decrease in oxygen availability. Hypoxia-inducible transcription factors (HIFs) can mediate cellular responses to hypoxia.[63] Hypoxia functions in a similar way to oncoproteins, and independently increases the HIF1α and HIF2α levels.[64,65] Parkin can inhibit breast tumor progression by targeting HIF-1α for ubiquitination and degradation.[66]
Evading immune destruction
Cells and tissues are actively and constantly monitored by the immune system, which recognizes and eliminates numerous incipient cancer cells and nascent tumors.[67] Nevertheless, the invasion of immune cells can induce immunoassociated inflammation and subsequent tumorigenesis.[68]
The immune response is classified into innate immunity and acquired immunity. Interferon (IFN) is a pivotal member of the innate immune pathway. Interferon regulatory factor-1 (IRF-1) is a main regulator of IFN transcription, but transcriptome sequencing showed that IRF-1 is also associated with alternative splicing in the regulation of growth and differentiation. For instance, carcinoembryonic antigen-related cell adhesion molecule 1 generates variants whenever hnRNP proteins combined with a variable exon 7 can form a complex with promoter-bound IRF-1.[69] hnRNP A1/A2 or SF2/ASF knockdown decreases the inclusion of exons 2 and 3 in IRF-3 pre-mRNA and affects the immunomodulatory functions of human non-small cell lung cancer (NSCLC) cells.[70]
The main effectors of acquired immunity are lymphocytes, which include two main groups, B cells and T cells. T cells are also regulated by alternative splicing of CD45.[71] The exclusion of exon cassettes 4, 5, and 6, and the generation of CD45RO,[72,73] also attenuate T cell activation via strong dimerization.[74] hnRNPL-like is directly related to immunoreactive growth hormone mRNA and is more highly expressed in plasma cells than in B cells.[75]
Alternative splicing and the tumor microenvironment
An adverse tissue microenvironment may also cause alternative splicing to become tumorigenic. Mutations and genetic changes alone may not be sufficient to drive cancer as a clinical disease. The tissue microenvironment provides crucial signaling to initiated tumor cells.[76]
As mentioned above, hypoxia is a common situation in solid tumors, and the presence of hypoxia has been linked to malignant progression, metastasis, resistance to therapy, and poor clinical outcomes following treatment. When hepatocellular carcinoma cells were cultivated under hypoxia-mimicking conditions, exon array analysis showed 3059 alternative splicing events in 2005 genes.[77] HIF activation can act through increased expression of CDC-like kinase 1 (CLK1) kinase leading to global hyperphosphorylation of SR proteins and the activation of hypoxia-dependent splice sites in HeLa cells.[78] To some extent, hypoxia also means glucose deprivation. Lack of glucose can cooperate with hypoxia to activate the HIF1α pathway.
Reactive oxygen species (ROS) can have both anti-cancer and tumorigenic effects. Low production of ROS can promote apoptosis, whereas excessive generation of ROS can interfere with signaling pathways and be involved in several pathological conditions, including cancer.[79] In a human gastric cancer cell line (AGS), oxidative stress led to phosphorylation and translocation of splicing factor TRA2B from the nucleus to the cytoplasm. As a consequence, alternative splicing of several variable exons in CD44, related to invasiveness, was observed.[80]
Another trait of the tumor microenvironment is hyperosmosis. Stress signals emanating from osmotic shock activate the p38-MAPK pathway via the upstream kinases MKK3 and MKK6 (mitogen-activated protein kinase 3 and 6). Activation of the p38-MAPK pathway induces hnRNPA1 phosphorylation in the nucleus, which is then exported into the cytoplasm and can affect many endogenous alternative splicing events.[81–83]
Growth factors are major regulators of tumor progression, including clonal expansion, invasion across tissue barriers, angiogenesis, and colonization of distant niches.[84] Epidermal growth factor,[85] hepatocyte growth factor,[86] transforming growth factor-β,[87] insulin growth factor,[88] and VEGF are all involved in various alternative splicing events.
The ECM has an important structural support function for cells but is not a static entity. The ECM can be modulated by tumor cells or stromal cells in response to wounding, inflammation, or cancer cell-derived stimuli. Changes in matrix composition, three-dimensional organization, or matrix stiffness communicate with many cell surface receptors[89,90] and result in a signaling response,[91] including changes in alternative splicing. An experiment that remodeled the ECM through activation of extracellular matrix metalloproteinase 3 in mouse mammary epithelial cells induced the expression of splice variant Ras-related C3 botulinum toxin substrate 1b (RAC1b), primarily through release of the repressor hnRNPA1 from an alternative exon.[92] In these cells, RAC1b caused an increase in cellular ROS and stimulated the expression of the transcription factor Snail, which induced epithelial-mesenchymal transition.[93]
Cytokines released by immune cells in the tumor microenvironment can be received by other immune cells and tumor cells of epithelial origin. However, the relationship between them remains to be explored. Interleukin-6 or granulocyte macrophage-colony stimulating factor modulated alternative splicing of BCL2L1 in K562 leukemia cells in favor of the anti-apoptotic splice variant BCL-x(L). Both cytokines required different intronic sequences for their responses, but the underlying molecular mechanisms remained unclear.[94]
Alternative splicing and therapy in cancer
The previous sections of this review describe how both the misregulation of alternative splicing and specific alternative splicing are highly associated with the specificity and severity of disease. Therefore, modulating this process might prevent cancer development and/or alter the course of disease. This could be an exciting strategy for therapy and allow the identification of novel biomarkers for cancer diagnosis.
Common conventional therapeutics involve targeting protein isoforms, expression, and alternative splicing through transacting elements. For example, X-box binding protein 1 (XBP1) is a basic region/leucine zipper transcription factor of the cAMP responsive element binding protein-activation transcription factor (CREB-ATF) family that plays an important prosurvival role in MM cells. Toyocamycin inhibits Inositol-requiring kinase 1a (IRE1a)-induced ATP-dependent XBP1 mRNA cleavage in vitro, with no apparent effect on IRE1a autophosphorylation. Therefore, this agent can be used to modulate multiple myeloma (MM) cell death.[95]
However, the therapeutic targeting of splicing factors might affect multiple transcripts, thereby disrupting normal intra-cellular function and generating undesirable side effects. To overcome this challenge, oligonucleotide and RNA-based gene therapies have been proposed. One of the approaches frequently adopted to target splicing is the use of anti-sense oligonucleotides (ASOs). These can be used to target a splice site by blocking it and thereby altering its recognition by the spliceosome, redirecting splicing to an adjacent site.[96] ASOs can also be used to prevent the binding of trans-acting regulatory splicing factors by targeting their binding sites.[97,98]
Some new conceptions in therapy are gradually emerging. Designing a splicing factor to intentionally act on the anti-apoptotic gene BCL-x leads to a high level of its splicing variant, thereby promoting apoptosis and enhancing sensitivity to chemotherapy drugs.[99] In addition, spliceosome inhibitory drugs such as Spliceostatin A or Sudemycins, which target the U2 snRNP component SF3B1,[100] have shown some tumor-cell-specific cytotoxic effects in leukemia that were associated with specific changes to alternative splicing.[101]
Conclusions
Alternative splicing plays a significant role in cancer, allowing the malignant progression of initiated tumor cells and contributing specifically to tumor progression. In this article, we reviewed alternative splicing in relation to the hallmarks of cancer cells. In cancer, alternative splicing remains to be comprehensively explored and understood, from tumorigenesis to cancer progression, from intercellular changes to extracellular variation, and from treatment to prevention.
This review describes the improper regulation of alternative splicing and its correlation with disease specificity and severity. Therefore, modifying this process may block the course of disease. This may be an exciting strategy for therapy and the identification of biomarkers for cancer diagnosis, as shown by an attempt to consider the spliceosome as a biomarker in prostate cancer.[102] Common conventional therapeutics involves targeting protein isoforms, expression, and alternative splicing through trans-acting elements [Figure 3]. Although studies on ASO therapies for spinal muscular atrophy and Duchenne muscular dystrophy are still in clinical trials[103,104] and these diseases are unrelated to cancer, this mode of therapy may also prove applicable to the treatment of cancer.
Figure 3.
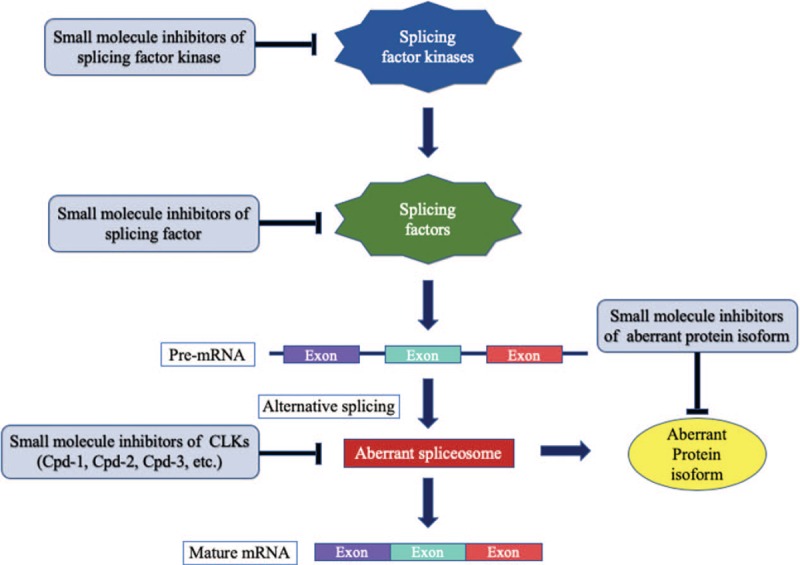
Common alternative splicing therapy methods.
Funding
This review was supported by the Nature Science Foundation of Beijing Municipality (No. 7151011).
Conflicts of interest
None.
Footnotes
How to cite this article: Qi F, Li Y, Yang X, Wu YP, Lin LJ, Liu XM. Significance of alternative splicing in cancer cells. Chin Med J 2019;133:221–228. doi: 10.1097/CM9.0000000000000542
References
- 1.Gilbert W. Why genes in pieces? Nature 1978; 271:501.doi: 10.1038/271501a0. [DOI] [PubMed] [Google Scholar]
- 2.Vania Goncalves V, Joana JFS, Jordan P. Signaling pathways driving aberrant splicing in cancer cells. Genes 2018; 9:9.doi: 10.3390/genes9010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee Y, Rio DC. Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem 2015; 84:291–323. doi: 10.1146/annurev-biochem-060614-034316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu Y, Wang X, Forouzmand E, Joshua J, Feng Q, Sowd GA, et al. Molecular mechanisms for CFIm-mediated regulation of mRNA alternative polyadenylation. Mol Cell 2018; 69:62–74. doi: 10.1016/j.molcel.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holdt LM, Kohlmaier A, Teupser D. Molecular roles and function of circular RNAs in eukaryotic cells. Cell Mol Life Sci 2018; 75:1071–1098. doi: 10.1007/s00018-017-2688-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aebersold R, Agar JN, Amster IJ, Baker MS, Bertozzi CR, Boja ES, et al. How many human proteoforms are there? Nat Chem Biol 2018; 14:206–214. doi: 10.1038/nchembio.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun S, Enculescu M, Setty ST, Cortés-López M, de Almeida BP, Sutandy FXR, et al. Decoding a cancer-relevant splicing decision in the RON proto-oncogene using high-throughput mutagenesis. Nat Commun 2018; 9:3315.doi: 10.1038/s41467-018-05748-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol 2014; 15:108–121. doi: 10.1038/nrm3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X, Yan C, Zhan X, Li L, Lei J, Shi Y. Structure of the human activated spliceosome in three conformational states. Cell Res 2018; 28:307–322. doi: 10.1038/cr.2018.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Huang Q, Liu W, Zhu Q, Cui CP, Xu L, et al. Mutually exclusive acetylation and ubiquitylation of the splicing factor SRSF5 control tumor growth. Nat Commun 2018; 9:2464.doi: 10.1038/s41467-018-04815-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morita M, Sato T, Nomura M, Sakamoto Y, Inoue Y, Tanaka R, et al. PKM1 confers metabolic advantages and promotes cell-autonomous tumor cell growth. Cancer Cell 2018; 33:355–367e7. doi: 10.1016/j.ccell.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Blencowe BJ. Alternative splicing: new insights from global analyses. Cell 2006; 126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 13.Sugnet CW, Kent WJ, Ares M, Jr, Haussler D. Transcriptome and genome conservation of alternative splicing events in humans and mice. Pac Symp Biocomput 2004; 66–77. doi: 10.1142/9789812704856_0007. [DOI] [PubMed] [Google Scholar]
- 14.Climente-González H, Porta-Pardo E, Godzik A, Eyras E. The functional impact of alternative splicing in cancer. Cell Rep 2017; 20:2215–2226. doi: 10.1016/j.celrep.2017.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Zhou J, Zhao S, Dunker AK. Intrinsically disordered proteins link alternative splicing and post-translational modifications to complex cell signaling and regulation. J Mol Biol 2018; 430:2342–2359. doi: 10.1016/j.jmb.2018.03.028. [DOI] [PubMed] [Google Scholar]
- 16.Weatheritt RJ, Sterne-Weiler T, Blencowe BJ. The ribosome-engaged landscape of alternative splicing. Nat Struct Mol Biol 2016; 23:1117–1123. doi: 10.1038/nsmb.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Mortensen AC, Spiegelberg D, Haylock AK, Lundqvist H, Nestor M. Preclinical evaluation of a novel engineered recombinant human anti-CD44v6 antibody for potential use in radio-immunotherapy. Int J Oncol 2018; 52:1875–1885. doi: 10.3892/ijo.2018.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu XJ, Li XD, Zhang H, Zhang X, Ning ZH, Yin YM, et al. Clinical significance of CD44s, CD44v3 and CD44v6 in breast cancer. J Int Med Res 2015; 43:173–179. doi: 10.1177/0300060514559793. [DOI] [PubMed] [Google Scholar]
- 20.Liu HG, Lv L, Shen H. Intratumoral heterogeneity of CD44v6 in rectal cancer. Clin Transl Oncol 2017; 19:425–431. doi: 10.1007/s12094-016-1542-9. [DOI] [PubMed] [Google Scholar]
- 21.Matzke-Ogi A, Jannasch K, Shatirishvili M, Fuchs B, Chiblak S, Morton J, et al. Inhibition of tumor growth and metastasis in pancreatic cancer models by interference with CD44v6 signaling. Gastroenterology 2016; 150:513–525.e10. doi: 10.1053/j.gastro.2015.10.020. [DOI] [PubMed] [Google Scholar]
- 22.Aydemir TB, Cousins RJ. The multiple faces of the metal transporter ZIP14 (SLC39A14). J Nutr 2018; 148:174–184. doi: 10.1093/jn/nxx041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aguilera O, Muñoz-Sagastibelza M, Torrejón B, Borrero-Palacios A, Del Puerto-Nevado L, Martínez-Useros J, et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016; 7:47954–47965. doi: 10.18632/oncotarget.10087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S, Venkatraman V, Crowgey EL, Liu T, Fu Z, Holewinski R, et al. Protein S-nitrosylation controls glycogen synthase kinase 3β function independent of its phosphorylation state. Circ Res 2018; 122:1517–1531. doi: 10.1161/CIRCRESAHA.118.312789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shinde MY, Sidoli S, Kulej K, Mallory MJ, Radens CM, Reicherter AL, et al. Phosphoproteomics reveals that glycogen synthase kinase-3 phosphorylates multiple splicing factors and is associated with alternative splicing. J Biol Chem 2017; 292:18240–18255. doi: 10.1074/jbc.M117.813527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gautrey H, Jackson C, Dittrich AL, Browell D, Lennard T, Tyson-Capper A. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol 2015; 12:1139–1151. doi: 10.1080/15476286.2015.1076610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C, Liu TS, Zhao SC, Yang WZ, Chen ZP, Yan Y. XIAP impairs mitochondrial function during apoptosis by regulating the Bcl-2 family in renal cell carcinoma. Exp Ther Med 2018; 15:4587–4593. doi: 10.3892/etm.2018.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bates DO, Morris JC, Oltean S, Donaldson LF. Pharmacology of modulators of alternative splicing. Pharmacol Rev 2017; 69:63–79. doi: 10.1124/pr.115.011239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet 2016; 135:851–867. doi: 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vu NT, Park MA, Shultz MD, Bulut GB, Ladd AC, Chalfant CE. Caspase-9b interacts directly with cIAP1 to drive agonist-independent activation of NF-κB and lung tumorigenesis. Cancer Res 2016; 76:2977–2989. doi: 10.1158/0008-5472.CAN-15-2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serrat R, Mirra S, Figueiro-Silva J, Navas-Pérez E, Quevedo M, López-Doménech G, et al. The Armc10/SVH gene: genome context, regulation of mitochondrial dynamics and protection against Aβ-induced mitochondrial fragmentation. Cell Death Dis 2014; 5:e1163.doi: 10.1038/cddis.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaspar TB, Sá A, Lopes JM, Sobrinho-Simões M, Soares P, Vinagre J. Telomere maintenance mechanisms in cancer. Genes 2018; 9:241.doi: 10.3390/genes9050241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barthel FP, Wesseling P, Verhaak RGW. Reconstructing the molecular life history of gliomas. Acta Neuropathologica 2018; 135:649–670. doi: 10.1007/s00401-018-1842-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu X, Wang Y, Chang G, Wang F, Geng X. Alternative splicing of hTERT pre-mRNA: a potential strategy for the regulation of telomerase activity. Int J Mol Sci 2017; 18:567.doi: 10.3390/ijms18030567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong W, Qian Y, Yang L. Telomerase, hTERT and splice variants in patients with myelodysplastic syndromes. Leuk Res 2014; 38:830–835. doi: 10.1016/j.leukres.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Lincz LF, Mudge LM, Scorgie FE, Sakoff JA, Hamilton CS, Seldon M. Quantification of hTERT splice variants in melanoma by SYBR green real-time polymerase chain reaction indicates a negative regulatory role for the beta deletion variant. Neoplasia 2008; 10:1131–1137. doi: 10.1593/neo.08644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics. Nat Rev Cancer 2008; 8:880–887. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biselli-Chicote PM, Oliveira AR, Pavarino EC, Goloni-Bertollo EM. VEGF gene alternative splicing: pro- and anti-angiogenic isoforms in cancer. J Cancer Res Clin Oncol 2012; 138:363–370. doi: 10.1007/s00432-011-1073-2. [DOI] [PubMed] [Google Scholar]
- 39.Błochowiak KJ, Sokalski J, Bodnar MB, Trzybulska D, Marszałek AK, Witmanowski H. Expression of VEGF165b, VEGFR1, VEGFR2 and CD34 in benign and malignant tumors of parotid glands. Adv Clin Exp Med 2018; 27:83–90. doi: 10.17219/acem/64876. [DOI] [PubMed] [Google Scholar]
- 40.Stevens M, Oltean S. Modulation of VEGF-A alternative splicing as a novel treatment in chronic kidney disease. Genes 2018; 9:98.doi: 10.3390/genes9020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mavrou A, Oltean S. SRPK1 inhibition in prostate cancer: a novel anti-angiogenic treatment through modulation of VEGF alternative splicing. Pharmacol Res 2016; 107:276–281. doi: 10.1016/j.phrs.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell 2017; 168:670–691. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol 2018; 13:395–412. doi: 10.1146/annurev-pathol-020117-043854. [DOI] [PubMed] [Google Scholar]
- 44.Wang D, Rai B, Qi F, Liu T, Wang J, Wang X, et al. Influence of the Twist gene on the invasion and metastasis of colon cancer. Oncol Rep 2018; 39:31–44. doi: 10.3892/or.2017.6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yadav AS, Pandey PR, Butti R, Radharani NNV, Roy S, Bhalara SR, et al. The biology and therapeutic implications of tumor dormancy and reactivation. Front Oncol 2018; 8:72.doi: 10.3389/fonc.2018.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elghonaimy EA, Ibrahim SA, Youns A, Hussein Z, Nouh MA, El-Mamlouk T, et al. Secretome of tumor-associated leukocytes augment epithelial-mesenchymal transition in positive lymph node breast cancer patients via activation of EGFR/Tyr845 and NF-kB/p65 signaling pathway. Tumour Biol 2016; 37:12441–12453. doi: 10.1007/s13277-016-5123-x. [DOI] [PubMed] [Google Scholar]
- 47.Zhang M, Song S, Yi Z, Zhao X, Fu L, Wang L, et al. Human biliverdin reductase promotes EMT through the ERK1/2 signal pathway in breast cancer. Eur J Pharmacol 2016; 788:45–53. doi: 10.1016/j.ejphar.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 48.Lin CH, Chiang MC, Chen YJ. MicroRNA-328 inhibits migration and epithelial–mesenchymal transition by targeting CD44 in nasopharyngeal carcinoma cells. Onco Targets Ther 2018; 11:2375–2385. doi: 10.2147/OTT.S151665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trowbridge IS, Thomas ML. CD45: an emerging role as a protein tyrosine phosphatase required for lymphocyte activation and development. Annu Rev Immunol 1994; 12:85–116. doi: 10.1146/annurev.iy.12.040194.000505. [DOI] [PubMed] [Google Scholar]
- 50.Yun Z, Weinberg RA. Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front Med 2018; 12:361–373. doi: 10.1007/s11684-018-0656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walser TC, Jing Z, Tran LM, Q Lin Y, Yaoblan N, Wang G, et al. Silencing the snail-dependent RNA splice regulator ESRP1 drives malignant transformation of human pulmonary epithelial cells. J Med Case Rep 2018; 78:1986–1999. doi: 10.1186/s13256-017-1493-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamauchi T, Fernandez JRE, Imamura CK, Yamauchi H, Jinno H, Takahashi M, et al. Dynamic changes in CD44v-positive cells after preoperative anti-HER2 therapy and its correlation with pathologic complete response in HER2-positive breast cancer. Oncotarget 2018; 9:6872–6882. doi: 10.18632/oncotarget.23914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hagiwara M, Kikuchi E, Tanaka N, Kosaka T, Mikami S, Saya H, et al. Variant isoforms of CD44 involves acquisition of chemoresistance to cisplatin and has potential as a novel indicator for identifying a cisplatin-resistant population in urothelial cancer. BMC Cancer 2018; 18:113.doi: 10.1186/s12885-018-3988-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warburg OH. The metabolism of tumours: investigations from the Kaiser Wilhelm Institute for Biology, Berlin-Dahlem. JAMA 1931; 96:1982.doi: 10.1001/jama.1931.02720490062043. [Google Scholar]
- 55.Shi X, Ran L, Liu Y, Zhong SH, Zhou PP, Liao MX, et al. Knockdown of hnRNP A2/B1 inhibits cell proliferation, invasion and cell cycle triggering apoptosis in cervical cancer via PI3K/AKT signaling pathway. Oncol Rep 2018; 39:939–950. doi: 10.3892/or.2018.6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wiese EK, Taro H. Tyrosine kinase signaling in cancer metabolism: PKM2 paradox in the Warburg Effect. Front Cell Dev Biol 2018; 6:79.doi: 10.3389/fcell.2018.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang C, Huang Z, Bai P, Luo G, Zhao X, Wang X, et al. Expression of pyruvate kinase M2 in human bladder cancer and its correlation with clinical parameters and prognosis. Onco Targets Ther 2018; 11:2075–2082. doi: 10.2147/OTT.S152999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao X, Zhao L, Yang H, Li J, Min X, Yang F, et al. Pyruvate kinase M2 interacts with nuclear sterol regulatory element-binding protein 1a and thereby activates lipogenesis and cell proliferation in hepatocellular carcinoma. J Biol Chem 2018; 293:6623–6634. doi: 10.1074/jbc.RA117.000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, et al. Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev 2016; 30:1020–1033. doi: 10.1101/gad.278549.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gu Z, Xia J, Xu H, Frech I, Tricot G, Zhan F. NEK2 promotes aerobic glycolysis in multiple myeloma through regulating splicing of pyruvate kinase. J Hematol Oncol 2017; 10:17.doi: 10.1186/s13045-017-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takahashi H, Nishimura J, Kagawa Y, Kano Y, Takahashi Y, Wu X, et al. Significance of polypyrimidine tract-binding protein 1 expression in colorectal cancer. Mol Cancer Ther 2015; 14:1705–1716. doi: 10.1158/1535-7163.MCT-14-0142. [DOI] [PubMed] [Google Scholar]
- 62.Lee G, Zheng Y, Cho S, Jang C, England C, Dempsey JM, et al. Post-transcriptional regulation of de novo lipogenesis by mTORC1-S6K1-SRPK2 signaling. Cell 2017; 171:1545–1558. doi: 10.1016/j.cell.2017.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 2013; 123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jia D, Park JH, Jung KH, Levine H, Kaipparettu BA. Elucidating the metabolic plasticity of cancer: mitochondrial reprogramming and hybrid metabolic states. Cells 2018; 7:21.doi: 10.3390/cells7030021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Myung-Chul K, Sung-Hyun H, Na-Yon K, Hong-Seok L, Sumin J, Yeseul Y, et al. Hypoxia promotes acquisition of aggressive phenotypes in human malignant mesothelioma. BMC Cancer 2018; 18:819.doi: 10.1186/s12885-018-4720-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu J, Zhang C, Zhao Y, Yue X, Wu H, Huang S, et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat Commun 2017; 8:1823.doi: 10.1038/s41467-017-01947-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ribatti D. The concept of immune surveillance against tumors: the first theories. Oncotarget 2017; 8:7175–7180. doi: 10.18632/oncotarget.12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Slobodin G, Rimar D. Regulatory T cells in systemic sclerosis: a comprehensive review. Clin Rev Allergy Immunol 2017; 52:194–201. doi: 10.1007/s12016-016-8563-6. [DOI] [PubMed] [Google Scholar]
- 69.Dery KJ, Silver C, Yang L, Shively J. Interferon regulatory factor 1 and a variant of heterogeneous nuclear ribonucleoprotein L coordinately silence the gene for adhesion protein CEACAM1. J Biol Chem 2018; 293:9277–9291. doi: 10.1074/jbc.RA117.001507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo R, Li Y, Ning J, Sun D, Lin L, Liu X. HnRNP A1/A2 and SF2/ASF regulate alternative splicing of interferon regulatory factor-3 and affect immunomodulatory functions in human non-small cell lung cancer cells. PLos One 2013; 8:e62729.doi: 10.1371/journal.pone.0062729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mustelin T, Rahmouni S, Bottini N, Alonso A. Role of protein tyrosine phosphatases in T cell activation. Immunol Rev 2003; 191:139–147. doi: 10.1034/j.1600-065x.2003.00014.x. [DOI] [PubMed] [Google Scholar]
- 72.Kuwabara T, Matsui Y, Ishikawa F, Kondo M. Regulation of T-cell signaling by post-translational modifications in autoimmune disease. Int J Mol Sci 2018; 19:819.doi: 10.3390/ijms19030819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang W, Niu C, Fu RY, Peng ZY. Mycobacterium tuberculosis H37Rv infection regulates alternative splicing in Macrophages. Bioengineered 2018; 9:203–208. doi: 10.1080/21655979.2017.1387692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wiljan H, Annika B, William L, Rafael P. Proteinaceous regulators and inhibitors of protein tyrosine phosphatases. Molecules 2018; 23:e395.doi: 103390/molecules23020395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aitor A, Antonio C, Chetan C, Stuart C, Dimitrios D, Brian L, et al. Endoplasmic reticulum stress signaling- from basic mechanisms to clinical applications. FEBS J 2018; 286:241–278. doi: 10.1111/febs.14608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang CP, Chen CC, Shyr CR. Xenogeneic cell therapy provides a novel potential therapeutic option for cancers by restoring tissue function, repairing cancer wound and reviving anti-tumor immune responses. Cancer Cell Int 2018; 18:9.doi: 10.1186/s12935-018-0501-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sena JA, Wang L, Heasley LE, Hu CJ. Hypoxia regulates alternative splicing of HIF and non-HIF target genes. Mol Cancer Res 2014; 12:1233–1243. doi: 10.1158/1541-7786.MCR-14-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jakubauskiene E, Vilys L, Makino Y, Poellinger L, Kanopka A. Increased serine-arginine (SR) protein phosphorylation changes Pre-mRNA splicing in hypoxia. J Biol Chem 2015; 290:18079–18089. doi: 10.1074/jbc.M115.639690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prasad S, Gupta SC, Pandey MK, Tyagi AK, Deb L. Oxidative stress and cancer: advances and challenges. Oxidative Med Cell Longev 2016; 2016:5010423.doi: 10.1155/2016/5010423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kajita K, Kuwano Y, Satake Y, Kano S, Kurokawa K, Akaike Y, et al. Ultraconserved region-containing transformer 2β4 controls senescence of colon cancer cells. Oncogenesis 2016; 5:e213.doi: 10.1038/oncsis.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Douglas JN, Gardner LA, Salapa HE, Levin MC. Antibodies to the RNA binding protein heterogeneous nuclear ribonucleoprotein A1 colocalize to stress granules resulting in altered RNA and protein levels in a model of neurodegeneration in multiple sclerosis. J Clin Cell Immunol 2016; 7:402.doi: 10.4172/2155-9899.1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kooshapur H, Choudhury NR, Simon B, Mühlbauer M, Jussupow A, Fernandez N, et al. Structural basis for terminal loop recognition and stimulation of pri-miRNA-18a processing by hnRNP A1. Nat Commun 2018; 9:2479.doi: 10.1038/s41467-018-04871-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakka K, Ghigna C, Gabellini D, Dilworth FJ. Diversification of the muscle proteome through alternative splicing. Skelet Muscle 2018; 8:8.doi: 10.1186/s13395-018-0152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res 2017; 7:1016–1036. [PMC free article] [PubMed] [Google Scholar]
- 85.Wang YC, Chang KC, Lin BW, Lee JC, Lai CH, Lin LJ, et al. The EGF/hnRNP Q1 axis is involved in tumorigenesis via the regulation of cell cycle-related genes. Exp Mol Med 2018; 50:70.doi: 10.1038/s12276-018-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Munoz U, Puche JE, Hannivoort R, Lang UE, Cohen-Naftaly M, Friedman SL. Hepatocyte growth factor enhances alternative splicing of the Krüppel-like factor 6 (KLF6) tumor suppressor to promote growth through SRSF1. Mol Cancer Res 2012; 10:1216–1227. doi: 10.1158/1541-7786.MCR-12-0213. [DOI] [PubMed] [Google Scholar]
- 87.Horiguchi K, Sakamoto K, Koinuma D, Semba K, Inoue A, Inoue S, et al. TGF-β drives epithelial-mesenchymal transition through EF1-mediated downregulation of ESRP. Oncogene 2012; 31:3190–3201. doi: 10.1038/onc.2011.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Malakar P, Chartarifsky L, Hija A, Leibowitz G, Glaser B, Dor Y, et al. Insulin receptor alternative splicing is regulated by insulin signaling and modulates beta cell survival. Sci Rep 2016; 6:31222.doi: 10.1038/srep31222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leight JL, Drain AP, Weaver VM. Extracellular matrix remodeling and stiffening modulate tumor phenotype and treatment response. Annu Rev Cancer Biol 2017; 1:313–334. doi: 10.1146/annurev-cancerbio-050216-034431. [Google Scholar]
- 90.Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci 2018; 25:20.doi: 10.1186/s12929-018-0426-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rubashkin MG, Ou G, Weaver VM. Deconstructing signaling in three dimensions. Biochemistry 2014; 53:2078–2090. doi: 10.1021/bi401710d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pelisch F, Khauv D, Risso G, Stallings-Mann M, Blaustein M, Quadrana L, et al. Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J Cell Biochem 2012; 113:2319–2329. doi: 10.1002/jcb.24103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005; 436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li CY, Chu JY, Yu JK, Huang XQ, Liu XJ, Shi L, et al. Regulation of alternative splicing of Bcl-x by IL-6, GM-CSF and TPA. Cell Res 2004; 14:473–479. doi: 10.1038/sj.cr.7290250. [DOI] [PubMed] [Google Scholar]
- 95.Ri M, Tashiro E, Oikawa D, Shinjo S, Tokuda M, Yokouchi Y, et al. Identification of toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress-induced XBP1 mRNA splicing. Blood Cancer J 2012; 2:e79.doi: 10.1038/bcj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res 2016; 44:6549–6563. doi: 10.1093/nar/gkw533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Havens MA, Duelli DM, Hastings ML. Targeting RNA splicing for disease therapy. Wiley Interdiscip Rev RNA 2013; 4:247–266. doi: 10.1002/wrna.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McClorey G, Wood MJ. An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr Opin Pharmacol 2015; 24:52–58. doi: 10.1016/j.coph.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 99.Lucie C, Julie L, Audrey V, Nicole P, Pascal P. Messenger RNA life-cycle in cancer cells: emerging role of conventional and non-conventional RNA-binding proteins? Int J Mol Sci 2018; 19:650.doi: 10.3390/ijms19030650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Convertini P, Shen M, Potter PM, Palacios G, Lagisetti C, de la Grange P, et al. Sudemycin E influences alternative splicing and changes chromatin modifications. Nucleic Acids Res 2014; 42:4947–4961. doi: 10.1093/nar/gku151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xargay-Torrent S, López-Guerra M, Rosich L, Montraveta A, Roldán J, Rodríguez V, et al. The splicing modulator sudemycin induces a specific antitumor response and cooperates with ibrutinib in chronic lymphocytic leukemia. Oncotarget 2015; 6:22734–22749. doi: 10.18632/oncotarget.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014; 371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fletcher S, Bellgard MI, Price L, Akkari AP, Wilton SD. Translational development of splice-modifying antisense oligomers. Expert Opin Biol Ther 2017; 17:15–30. doi: 10.1080/14712598.2017.1250880. [DOI] [PubMed] [Google Scholar]
- 104.Aartsma-Rus A, Krieg AM. FDA approves eteplirsen for duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther 2017; 27:1–3. doi: 10.1089/nat.2016.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]