

ABSTRACT
Effective treatment of invasive lobular carcinoma (ILC) of the breast is hampered by late detection, invasive growth, distant metastasis, and poor response to chemotherapy. Phosphoinositide 3-kinase (PI3K) signaling, one of the major druggable oncogenic signaling networks, is frequently activated in ILC. We investigated treatment response and resistance to AZD8055, an inhibitor of mammalian target of rapamycin (mTOR), in the K14-cre;Cdh1Flox/Flox;Trp53Flox/Flox (KEP) mouse model of metastatic ILC. Inhibition of mTOR signaling blocked the growth of primary KEP tumors as well as the progression of metastatic disease. However, primary tumors and distant metastases eventually acquired resistance after long-term AZD8055 treatment, despite continued effective suppression of mTOR signaling in cancer cells. Interestingly, therapeutic responses were associated with increased expression of genes related to antigen presentation. Consistent with this observation, increased numbers of tumor-infiltrating major histocompatibility complex class II-positive (MHCII+) immune cells were observed in treatment-responsive KEP tumors. Acquisition of treatment resistance was associated with loss of MHCII+ cells and reduced expression of genes related to the adaptive immune system. The therapeutic efficacy of mTOR inhibition was reduced in Rag1−/- mice lacking mature T and B lymphocytes, compared to immunocompetent mice. Furthermore, therapy responsiveness could be partially rescued by transplanting AZD8055-resistant KEP tumors into treatment-naïve immunocompetent hosts. Collectively, these data indicate that the PI3K signaling pathway is an attractive therapeutic target in invasive lobular carcinoma, and that part of the therapeutic effect of mTOR inhibition is mediated by the adaptive immune system.
KEYWORDS: Invasive lobular carcinoma, mTOR, immune system, therapy, mouse model
Introduction
Invasive lobular carcinoma (ILC) is the second most common histological type of breast cancer, representing approximately 15% of all breast cancer cases. ILCs have a specific histological growth pattern of discohesive and invasive tumor cells which typically lack the intercellular adhesion molecule E-cadherin.1 Many ILCs express ERα and are treated with endocrine therapy.2–4
Unfortunately, ILC is often relatively difficult to detect due to its indistinct margins and low radiographic opacity.5,6 Compared with the more common invasive ductal carcinoma (IDC) type, ILC is more likely to have progressed to stage III/IV at the time of diagnosis, and surgical re-excision is required more frequently.3,4 Chemo-responsiveness is generally low, and the general benefit of chemotherapy in ILC has been questioned.7–10 This underlines the need to explore new therapeutic strategies for ILC. One of the most frequently activated and druggable oncogenic pathways in breast cancer is the phosphoinositide 3-kinase (PI3K) signaling network.11 PI3K signaling is induced by various stimuli including growth factor binding to receptor tyrosine kinases (RTKs), and signals through a network of many kinases, including AKT and mammalian target of rapamycin (mTOR). mTOR acts in two complexes, mTOR complex 1 and 2 (mTORC1 and mTORC2). Effector proteins of the PI3K pathway stimulate cell growth, survival, and migration.12 Activating mutations in the PI3K signaling pathway are more common in ILC than in other breast cancer types, offering a potentially promising therapeutic target.11,13–17
In this study, we used the K14-cre;Cdh1Flox/Flox;Trp53Flox/Flox (KEP) mouse model with tissue-specific inactivation of E-cadherin (Cdh1) and p53 (Trp53) driving the formation of metastatic mouse ILC, or mILC.18 We have previously developed a KEP-based orthotopic allograft model for studying primary tumors as well as metastatic disease in mice, creating the unique and important opportunity to perform in vivo modeling of neoadjuvant (presurgical) and adjuvant (postsurgical) therapy in immunocompetent mice.19
One of the hallmarks of cancer is the escape from destruction by the immune system.20 PI3K signaling plays an important role in the survival, differentiation, proliferation, and activation of many types of immune cells.21–23 Inhibiting PI3K signaling might, therefore, influence the crosstalk between cancer cells and the host immune system. In the present work, we investigated the therapeutic benefit of targeting mTOR in ILC. We treated mice bearing primary and metastatic ILC using the mTOR inhibitor AZD8055 in a preclinical neoadjuvant and adjuvant setting. By combining protein and transcriptome analyses with in vivo experiments we identified the adaptive immune system as an important determinant of the therapeutic efficacy of mTOR inhibition in ILC.
Results
Activation of PI3K signaling is common in human and mouse ILCs
To assess the prevalence of aberrant PI3K signaling in invasive lobular carcinoma (ILC), we used publicly available data on the cBioPortal for Cancer Genomics (http://www.cbioportal.org/). Mutations in the following five genes were compared between ILC and breast cancers of other types: PIK3CA, PTEN, AKT1, AKT2, PIK3R1, and MTOR. The majority of ILCs have a mutation in a gene involved in PI3K signaling (Supplementary Table S1). This is in line with previous reports,14–17 supporting the notion that activation of PI3K signaling occurs frequently in ILC. We also assessed the presence of phosphorylated kinases belonging to the PI3K signaling pathway in an independent set of 66 human ILCs and in 30 mouse ILCs (mILCs) from K14-cre;Cdh1Flox/Flox;Trp53Flox/Flox (KEP) mice18 by immunohistochemistry (Figure 1(a)). Phosphorylated eukaryotic translation initiation factor 4E binding protein 1 (4EBP1), a marker of PI3K signaling known to correlate with pathologic grade and prognosis in breast cancer,24,25 was highly expressed in human ILCs, with an average percentage of 77% positive tumor cells. The majority of human ILCs were also found to be positive for phosphorylated S6K1-T389 (70% of the cases) and phosphorylated AKT-T308 (59%, Figure 1(a), Supplementary Figure 1). In KEP mice, the vast majority of mILCs were positive for phosphorylated 4EBP1-, AKT-S473, and phosphorylated S6-S235/236, while normal mammary gland had very low expression of these signaling markers (Figure 1(a,b)). These findings indicate that PI3K signaling is active in both human and mouse ILCs.
Figure 1.
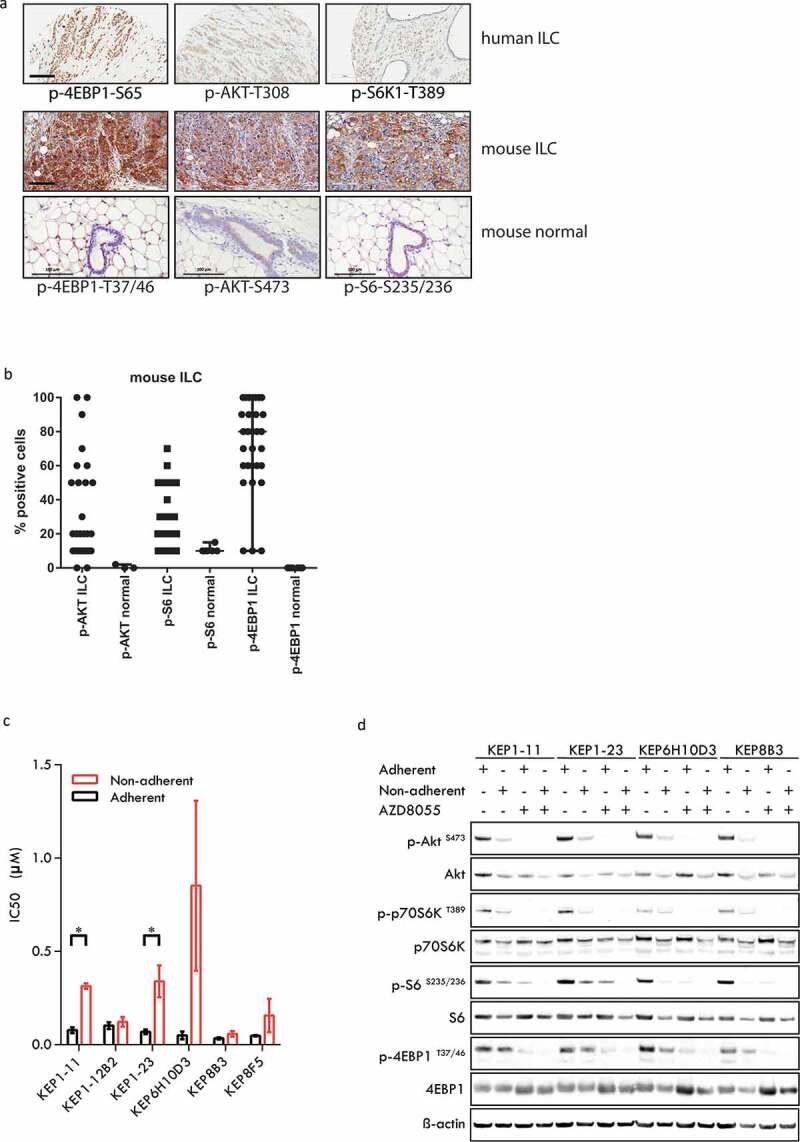
mTOR signaling in human invasive lobular carcinomas (ILCs) and mouse ILCs. (a) Upper panels: human ILC; immunohistochemistry for phospho-4EBP1 (serine 65), phospho-AKT (threonine 308) and phospho-S6K1 (threonine 389); lower panels: mouse ILC (mILCs) from K14-cre;Cdh1Flox/Flox;Trp53Flox/Flox (KEP) mice and normal mouse mammary gland; immunohistochemistry for phospho-4EBP1 (threonine 37/46), phospho-AKT (serine 473) and phospho-S6 (serine 235/236). Scale bars: 100 μm. (b) Scatter plot representing the percentage of tumor cells staining positive for mTOR signaling markers in mouse ILC (KEP) tumors and in normal mouse mammary glands. The majority of mouse ILC tumors expressed phosporylated 4EBP1 (>10% of tumor cells are positive in 27/30 cases, average 75% of tumor cells), phosphorylated AKT (>10% in 19/30 cases, average 32%) and phosphorylated S6 (>10% in 21/30 cases, average 28%). (c) IC50 values of KEP mouse mammary tumor cells for AZD8055. Cells were cultured under adherent conditions (black bars) or non-adherent conditions (red bars). (d) Immunoblot analysis of mTOR signaling markers in adherently and non-adherently growing KEP cell lines (4 clones from 3 independent tumors) in the absence or presence of AZD8055 (500nM, 24 h).
AZD8055 inhibits in vitro growth of mouse ILC cells
To evaluate mTOR signaling as a putative therapeutic target in mouse ILC, we determined the sensitivity of KEP tumor cell lines to the ATP-competitive dual mTORC1/2 inhibitor AZD8055.26 IC50 values were determined for six KEP cancer cell lines derived from three independent tumors. Because metastasis is an important problem in ILC, we also cultured KEP cancer cells under non-adherent conditions, as a simplified model for circulating cancer cells. The sensitivity of tumor cells to mTOR inhibition tended to be lower under non-adherent conditions compared to adherent growth conditions (Figure 1(c)). Expression of mTOR signaling phosphoproteins in cultured cancer cells was confirmed by immunoblot (Figure 1(d)). In line with their reduced sensitivity to AZD8055, non-adherent KEP cancer cells expressed lower levels of signaling markers than adherently growing cells. Treatment of both adherently and non-adherently growing KEP cells with 500 nM AZD8055 for 24 h caused potent reduction of phosphoprotein levels of AKT-S473, p70S6K-T389, S6-T235/236, and 4EBP1-T37/46.
Neoadjuvant mTOR inhibition blocks tumor growth
The high in vitro sensitivity of KEP cancer cells to mTOR inhibition by AZD8055 prompted us to test the anticancer efficacy of this inhibitor in vivo. We, therefore, used the previously established KEP-based mouse model of spontaneous ILC metastasis19 to perform a 28-day preclinical intervention study, modeling a neoadjuvant treatment setting. Wildtype FVB/n mice bearing an orthotopically transplanted primary KEP tumor were treated with AZD8055 for 28 days when tumors reached a diameter of 5 mm (Figure 2(a)). During treatment, mTOR inhibition effectively suppressed primary tumor growth, leading to tumor stasis. After the 28-day treatment period, tumor growth resumed immediately, with growth rates comparable to the control group (Figure 2(b)). With progression defined as a doubling in tumor size, we found that the 28-day treatment with AZD8055 extended median progression-free survival from 7 to 31 days (p < .001, Figure 2(c)). The primary KEP tumors in vehicle-treated control mice and AZD8055-treated mice were surgically removed when they reached a diameter of 15 mm, and animals were subsequently monitored for the development of metastatic disease. We defined metastasis-specific survival endpoints as either dyspnea due to lung metastases or a palpable metastasis that reached a maximum size of 15 mm. Median metastasis-specific survival of the AZD8055-treated mice was 75 days, vers47 USD days for the control animals (p = .0569, Figure 2(d)). Altogether, these results show that mTOR inhibition can block the growth of primary KEP tumors and spontaneous metastases in vivo.
Figure 2.
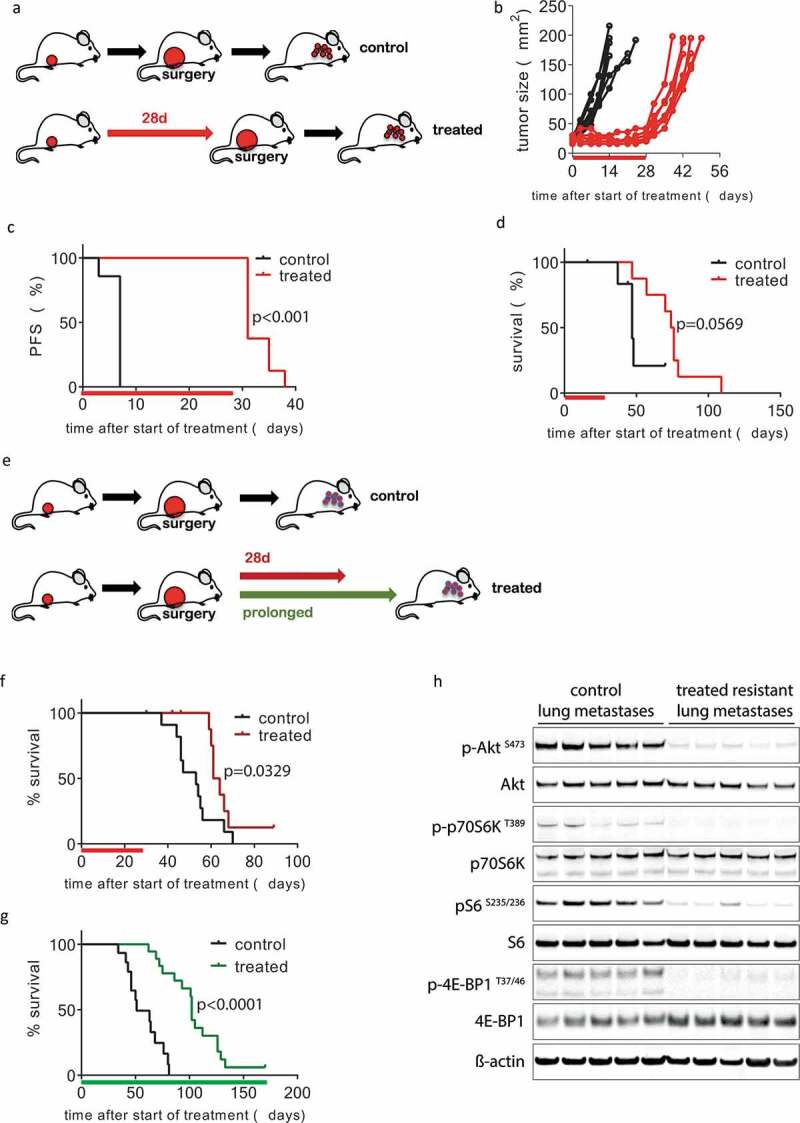
Inhibition of mTOR in vivo blocks KEP tumor growth and delays metastatic disease. (a–d) 28-day neoadjuvant treatment. (a) Schematic overview of experimental setup. Tissue fragments (1 mm3) of a mILC from a KEP donor mouse were orthotopically transplanted in recipient mice. When tumors reached a diameter of 5 mm, a 28-day treatment with daily oral AZD8055 (20 mg/kg, red arrow) or vehicle control solution (black arrow) was initiated. Tumors were surgically removed when they reached a diameter of 15 mm and mice were monitored until terminal metastatic disease developed. (b) Individual tumor growth curves in AZD8055-treated mice (red curves, n = 8) and control mice (black curves, n = 7). (c) Kaplan-Meier plot depicting progression-free survival (PFS) of AZD8055-treated mice (red curve) and control mice (black curve), with progression defined as a doubling in tumor size in mm2 (caliper measurement in two perpendicular directions) from the start of treatment (time point zero). D, Kaplan-Meier plot depicting metastasis-specific survival in AZD8055-treated mice (red curve) and control mice (black curve). Time point zero indicates start of treatment (tumor size 5 mm) in all graphs. E-H, 28-day and prolonged adjuvant treatment. E, schematic overview of experimental setup. Mice were transplanted orthotopically with a 1 mm3 fragment of a mILC from a KEP donor mouse. Tumors were surgically removed when they reached a diameter of 15 mm. After surgery, mice received treatment with daily oral vehicle control solution (black arrow) or 20 mg/kg AZD8055 (20 mg/kg) for 28 days (red arrow) or until they met one of the predefined endpoints: clinically overt metastatic disease or large locally recurrent tumors (green arrow). F, Kaplan-Meier plot depicting metastasis-specific survival of AZD8055-treated mice (red curve, n = 11) and control mice (black curve, n = 11) after 28-day adjuvant treatment (p = .0329). Time point zero indicates start of treatment (tumor size 5 mm). G, Kaplan-Meier plot depicting metastasis-specific survival in AZD8055-treated mice (green curve, n = 18) and control mice (black curve, n = 15) subjected to prolonged adjuvant treatment (p < .0001). Time point zero indicates start of treatment (tumor size 5 mm). End points due to locally recurrent tumors were censored, as well as the sacrifice of one mouse that survived for more than 150 days. H, immunoblot for mTOR signaling markers in lung metastases from AZD8055-treated and control mice from the prolonged adjuvant treatment study. Lung metastases were dissected from 5 AZD8055-treated mice and 5 control mice at the endpoint of the experiment (terminal metastatic disease with dyspnea).
Adjuvant mtor inhibition attenuates metastatic disease progression
To further study the therapeutic effect of mTOR inhibition on metastases, we modeled the adjuvant treatment setting in a new cohort of mice. To this end, we transplanted mice orthotopically with pieces from the same KEP tumor and monitored tumor outgrowth to a size of 15 mm, at which point we surgically removed the primary tumor and started 28 days of adjuvant treatment with AZD8055 (Figure 2(e), red arrow). In a second experiment, we tested the effects of chronic adjuvant mTOR inhibition, which was continued until the mice reached one of the pre-defined clinical endpoints (Figure 2(e), green arrow). Endpoints related to metastatic disease were dyspnea due to lung metastases or a palpable metastasis of 15 mm in diameter. Weekly X-ray computed tomography (CT) scans of the thorax in a subgroup of the mice demonstrated a slowdown in disease progression in the AZD8055-treated group compared to controls (Supplementary Fig. S2A). Dyspnea due to lung metastases was the humane end point for the majority of the mice. The 28-day adjuvant window treatment led to a median metastasis-specific survival of 62.5 days vers53 USD days in controls (p = .0329, Figure 2(f)). A more profound survival benefit of 51 days was achieved with chronic adjuvant treatment (median survival of 102 days for AZD8055-treated mice vers51 USD days for control mice, p < .0001, Figure 2(g)). One mouse in the chronic AZD8055 treatment group survived for 170 days, at which point we ended the experiment. All mice had lung metastases at postmortem examination, as confirmed by histopathology (Supplementary Fig. S2B). We harvested metastatic tumor tissue from the lungs of mice from the chronic treatment group and assessed activity of mTOR signaling by measuring the levels of phosphorylated AKT (S473), p70SK (T389), S6 (S235/235), and 4EBP1 (T37/46) (Figure 2(h)). Strikingly, signaling was still effectively inhibited in all AZD8055-treated lung metastases at the endpoint of chronic treatment. Together, these results show that adjuvant mTOR inhibition in the metastatic KEP model effectively inhibits metastatic disease. However, resistance eventually leads to disease progression despite continued suppression of mTOR signaling in lung metastases from AZD8055-treated mice.
Combined neoadjuvant and adjuvant mTOR inhibition maximizes survival
To maximize the response of KEP tumors and metastases to mTOR inhibition, we designed a new intervention study in which we combined chronic neoadjuvant and adjuvant AZD8055 treatment with surgical removal of primary KEP tumors upon progression (Figure 3(a)). Continued neoadjuvant treatment with AZD8055 resulted in prolonged growth arrest of primary KEP tumors, but eventually all tumors progressed. AZD8055-resistant KEP tumors grew fast, with growth rates comparable to untreated control tumors (Figure 3(b)). The median progression-free survival benefit was 47.5 days in the AZD8055-treated group versus control mice (54 vs 6.5 days, respectively, p < .0001, Figure 3(c)). After surgical removal of treatment-resistant KEP tumors (15 mm diameter), we continued administration of AZD8055 until terminal metastatic disease developed (dyspnea due to lung metastases or a palpable metastasis with a diameter of 15 mm). This led to a median metastasis-specific survival benefit of 63 days (survival time from treatment initiation, 116 days vers53 USD days, p < .0001, Figure 3(d)). Of all treatment regimens with AZD8055 described in this study, prolonged treatment starting in the neoadjuvant phase resulted in the longest survival (Table 1, Supplementary Fig. S3, Supplementary Table S2). To investigate the effect of mTOR inhibition on PI3K pathway signaling in the KEP tumors, we performed immunoblots on untreated control tumors, AZD8055-resistant tumors removed at 15 mm, and AZD8055-sensitive tumors from a separate mouse cohort that received neoadjuvant treatment for only 5 days. Interestingly, mTOR signaling was effectively inhibited in both the AZD8055-sensitive and AZD8055-resistant tumors (Figure 3(e)), indicating that resistance developed despite effective suppression of mTOR signaling under prolonged AZD8055 treatment.
Figure 3.
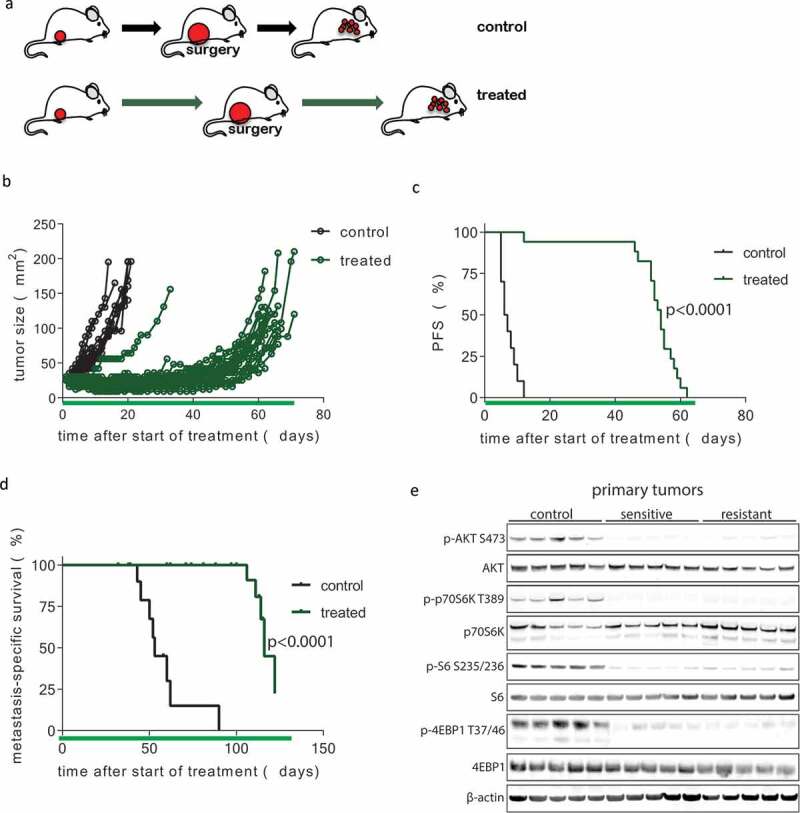
Development of resistance following prolonged neoadjuvant and adjuvant mTOR inhibition. (a) Schematic overview of experimental setup. Tissue fragments (1 mm3) of a mILC from a KEP donor mouse were orthotopically transplanted in recipient mice. Neoadjuvant treatment with daily oral AZD8055 (green arrows) or vehicle control solution (black arrows) was started when tumors reached a diameter of 5 mm. Tumors were surgically removed when they reached a diameter of 15 mm and treatment was continued in the adjuvant setting until mice were sacrificed due to terminal metastatic disease. (b) Individual tumor growth curves in AZD8055-treated mice (green curves, n = 24) and control mice (black curves, n = 10). (c) Kaplan-Meier plot depicting progression-free survival (PFS) of neoadjuvant AZD8055-treated mice (green curve) and control mice (black curve), with progression defined as a doubling in the size of the primary tumor in mm2 from the start of treatment (time point zero). (d) Kaplan-Meier plot depicting metastasis-specific survival in AZD8055-treated mice (green curve) and control mice (black curve). Time point zero indicates start of treatment (tumor size 5 mm) in all graphs. (e) Immunoblot for mTOR signaling markers in five surgically removed tumors from control mice, five therapy-sensitive tumors harvested from AZD8055-treated mice after 5 days of treatment, and five surgically removed therapy-resistant tumors from AZD8055-treated mice.
Table 1.
Median metastasis-specific survival in five experimental groups.
| Group | Median survival (days) |
|---|---|
| Surgery only | 52 |
| 28d neoadjuvant + surgery | 75 |
| Surgery + 28d adjuvant | 62.5 |
| Prolonged neoadjuvant + surgery + prolonged adjuvant | 116 |
| Surgery + prolonged adjuvant | 102 |
Therapeutic response to AZD8055 correlates with activation of immunological processes
Since development of resistance to AZD8055 in KEP tumors was not associated with reactivation of mTOR signaling, we set out to explore which other biological processes might play a role in the dynamics of therapy response and resistance after long-term mTOR inhibition. We harvested vehicle-treated control tumors, AZD8055-sensitive tumors after 5 days of treatment and AZD8055-resistant tumors that progressed during prolonged treatment (Figure 4(a), Supplementary Fig. S2C) in order to compare treatment-sensitive tumors to treatment-resistant tumors. Immunohistochemical quantification of Ki-67 and cleaved caspase 3 (CC3) positive cells in tumor sections showed that after 5 days of AZD8055 treatment, mTOR inhibition suppressed proliferation of cancer cells, while the number of apoptotic cells was unchanged compared to control tumors (Figure 4(b,c)). Next, we analyzed AZD8055-sensitive, -resistant and untreated control tumor samples using reverse phase protein array (RPPA) to identify (phospho)protein expression patterns that correlate with therapy resistance. Unsupervised hierarchical clustering using the expression of 136 epitopes separated AZD8055-treated tumors from untreated control tumors but did not separate AZD8055-resistant tumors from AZD8055-sensitive tumors (Supplementary Fig. S4). Low expression of known markers of mTOR activity in all AZD8055-treated tumor samples confirmed effective inhibition of mTOR signaling, even in the AZD8055-resistant tumor samples (Figure 4(d)). Because receptor tyrosine kinase (RTK) activation has been described as a mechanism of resistance to AZD8055,27,28 we complemented the RPPA analysis with a phospho-RTK array, which did not reveal activation of any RTKs in AZD8055-resistant tumor samples (data not shown).
Figure 4.
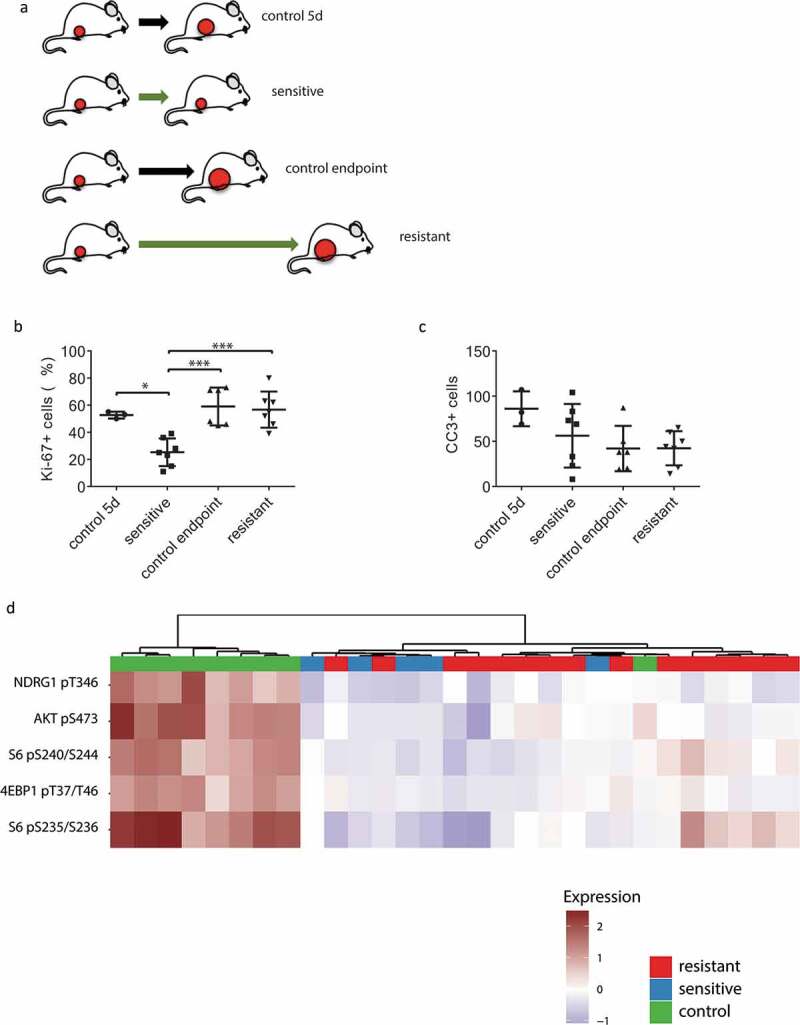
Characterization of AZD8055-sensitive and -resistant mILCs by immunohistochemistry and RPPA analysis. (a) Schematic overview of experimental setup to generate the different tumor groups. Mice were transplanted orthotopically with a 1 mm3 fragment of a mILC from a KEP donor mouse. When tumors reached a diameter of 5 mm, daily treatment with AZD8055 (green arrows) or vehicle control solution (black arrows) was started. AZD8055-sensitive tumors (n = 10) were harvested after 5 days of AZD8055 treatment. AZD8055-resistant tumors (n = 20) were harvested when they progressed on AZD8055 treatment to a diameter of 15 mm. Vehicle-treated control tumors were harvested after 5 days (n = 4) or when they reached a diameter of 15 mm (n = 6). (b) Immunohistochemical quantification of percentages of Ki-67 positive tumor cells in peripheral tumor parts. * p < .05; *** p < .001. (c) Immunohistochemical quantification of number of cleaved caspase 3 (CC3) positive tumor cells per 10 high magnification fields of view. (d) Unsupervised hierarchical clustering analysis of Reverse Phase Protein Array (RPPA) data from 29 KEP tumors (9 control tumors, 5 AZD8055-sensitive tumors, and 15 AZD8055-resistant tumors). The heatmap shows expression levels of selected epitopes representing known PI3K signaling markers. The complete heatmap is shown in Supplementary Fig. S4.
We next performed transcriptome analysis using RNA sequencing (RNA-seq) data from untreated control tumors and AZD8055-sensitive and -resistant tumors. Since gene expression profiles of untreated control tumors from both time points (day 5 and endpoint) were indistinguishable by principle component analysis (data not shown), we pooled RNA-seq data from all control tumors into a single group to increase statistical power. To find biological traits associated with therapy response and resistance, we performed gene ontology (GO) enrichment analysis using untreated control tumors, AZD8055-sensitive tumors, and AZD8055-resistant tumors (Table 2, Supplementary Fig. S5). Compared to untreated control tumors and AZD8055-resistant tumors, AZD8055-sensitive tumors showed reduced transcript levels of genes related to cell proliferation. Intriguingly, the GO enrichment analysis also showed upregulation of immunological processes in AZD8055-sensitive tumors compared to control tumors, and downregulation of other immunological processes in AZD8055-resistant tumors compared to AZD8055-sensitive tumors, thus pointing to the immune system as a possible player in the response of mouse ILC to mTOR inhibition. The top enriched gene ontologies that were upregulated in AZD8055-sensitive tumors versus control tumors are related to antigen presentation via major histocompatibility complex class II (in bold, Table 2). We plotted the expression of the genes in these ontologies as a combined metagene RNA expression score for antigen presentation through MHCII for AZD8055-sensitive, -resistant and control tumors. This plot visualizes that transcription of this gene set related to antigen presentation is upregulated in AZD8055-sensitive tumors after 5 days of treatment but this is lost in AZD8055-resistant tumors (Figure 5(a), Supplementary Fig. S6).
Table 2.
Gene ontology analysis of differential transcript expression for control tumors, treatment-sensitive tumors and treatment-resistant tumors (top enriched GO IDs with p-value <0.01).
| top 5 gene ontologies by fold enrinchment | GO ID | fold enrichment | p-value | |
|---|---|---|---|---|
|
upregulated sensitive versus control |
antigen processing and presentation of peptide or polysaccharide antigen via MHC class II | 0002504 | 20.20 | 8.67E-05 |
| antigen processing and presentation of peptide antigen via MHC class II | 0002495 | 20.20 | 8.67E-05 | |
| antigen processing and presentation of exogenous peptide antigen via MHC class II | 0019886 | 20.20 | 7.31E-04 | |
| lymphocyte chemotaxis | 0048247 | 15.77 | 1.65E-10 | |
| T cell migration | 0072678 | 15.71 | 3.90E-03 | |
|
downregulated sensitive versus control |
mitotic cytokinesis | 0000281 | 20.83 | 5.49E-04 |
| cytoskeleton-dependent cytokinesis | 0061640 | 17.30 | 1.91E-03 | |
| spindle assembly | 0051225 | 13.48 | 9.92E-03 | |
| mitotic spindle organization | 0007052 | 13.48 | 9.92E-03 | |
| sister chromatid segregation | 0000819 | 13.10 | 1.93E-06 | |
|
upregulated resistant versus sensitive |
response to hypoxia | 0001666 | 7.72 | 7.82E-03 |
| response to decreased oxygen levels | 0036293 | 7.51 | 9.99E-03 | |
| positive regulation of multicellular organismal process | 0051240 | 2.59 | 8.01E-03 | |
| animal organ development | 0048513 | 2.40 | 2.41E-05 | |
| system development | 0048731 | 2.08 | 4.16E-05 | |
|
downregulated resistant versus sensitive |
T cell chemotaxis | 0010818 | 21.41 | 5.02E-04 |
| negative thymic T cell selection | 0045060 | 15.73 | 9.01E-05 | |
| T cell migration | 0072678 | 14.95 | 3.57E-06 | |
| negative T cell selection | 0043383 | 14.68 | 1.62E-04 | |
| antigen processing and presentation of exogenous peptide antigen | 0002478 | 13.98 | 1.22E-06 | |
|
upregulated resistant versus control |
regulation of multicellular organismal process | 0051239 | 2.23 | 3.91E-03 |
| system development | 0048731 | 1.97 | 4.57E-03 | |
|
downregulated resistant versus control |
adhesion of symbiont to host | 0044406 | 33.58 | 3.19E-04 |
| cellular response to interferon-beta | 0035458 | 31.71 | 2.38E-16 | |
| response to interferon-beta | 0035456 | 28.47 | 1.20E-16 | |
| defense response to protozoan | 0042832 | 22.73 | 3.76E-07 | |
| response to protozoan | 0001562 | 22.23 | 5.05E-08 |
Figure 5.
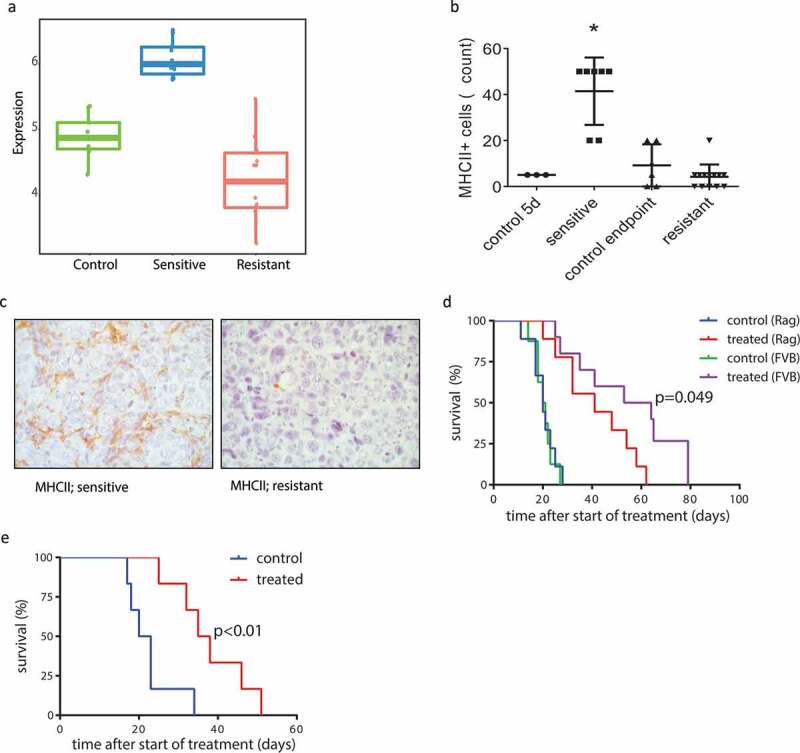
Involvement of the adaptive immune system in response and acquired resistance to AZD8055. (a) Metagene score for RNA expression levels (relative numbers of transcript reads) of genes involved in antigen presentation via MHCII, of vehicle-treated control tumors (green), AZD8055-sensitive tumors (blue), and AZD8055-resistant tumors (red). Sensitive vs control p < .0001, resistant vs control p = .0023, resistant vs sensitive p < .0001. (b) Immunohistochemical quantification of MHCII positive cells in control tumors and AZD8055-sensitive and -resistant tumors. Shown are numbers of MHCII positive cells per 5 high magnification fields of view. Asterisk (*) indicates a significant difference between sensitive tumors and all three other groups (p < .0001). (c) Representative microphotographs of immunohistochemical detection of cells expressing MHCII in the stroma of AZD8055-sensitive and AZD8055-resistant tumors. (d) Transplantation and treatment of a treatment-naïve KEP tumor in Rag1−/- mice lacking mature T and B lymphocytes and immunocompetent FVB/N mice. Orthotopic transplantation of a 1 mm mILC tumor fragment was performed and the mice received 28 days of neoadjuvant treatment with AZD8055 or vehicle control. An event was recorded in a Kaplan-Meier analysis when the primary tumor reached a diameter of 15 mm. Median latency: 20 days (control, Rag1−/- mice, n = 9), 20.5 days (control, FVB/N mice, n = 7), 41 days (treated, Rag1−/- mice, n = 9), and 58.5 days (treated, FVB/N mice, n = 10). Treated Rag1−/- mice vs FVB/N mice: p = .049. Treated FVB/N mice vs control FVB/N mice: p < .0001. Treated Rag1−/- mice vs control Rag1−/- mice: p = .0004. (e) Serial transplantation and treatment of an AZD8055-resistant tumor in a naïve cohort of immunocompetent FVB/N mice. FVB/N mice received an orthotopic transplantation with a 1 mm piece taken from a resistant tumor (treated until endpoint), and received 28 days of neoadjuvant treatment with AZD8055 or vehicle control. An event was recorded in a Kaplan-Meier analysis when the primary tumor reached a diameter of 15 mm. Median latency: 36.5 (treated) and 21.5 days (control), p = .0053. Control: n = 6. Treated: n = 6.
To further investigate changes in the immune system induced by mTOR inhibition, we performed flow cytometry analysis on tumor tissue and blood from untreated control animals and neoadjuvant-treated animals with resistant primary tumors (15 mm diameter), for a panel of immune cell markers (CD45, B220, CD3, CD4, CD8, γδTCR, FOXP3, CD11b, Ly6G, Ly6C, c-KIT, and F4/80). In blood, fewer Ly6G+ cells (neutrophils) were detected in treated animals. In tumor tissue however, no significant differences were detected (Supplementary Fig. S7A). In addition, we applied the following panel of immunohistochemistry markers on paraffin-embedded tissues from sensitive, resistant, and control tumors: MHCII, CD3, CD4, CD8, B220, FOXP3, F4/80, granzyme B, and phosphorylated STAT1. While most of these markers did not identify consistent differences between sensitive tumors, resistant tumors, and controls, AZD8055-sensitive tumors contained significantly more MHCII positive cells compared to control tumors and AZD8055-resistant tumors (p < .0001, Figure 5(b,c), Supplementary Fig. S7B). Importantly, the immunohistochemical signal for MHCII was not seen in the cancer cells, but in cells in the tumor microenvironment (Figure 5(c)). Indeed, KEP cells have very low expression of MHCII, and do not show any upregulation of MHCII after treatment with AZD8055 in vitro (Supplementary Fig. S8A, S8B).
These results show that the response of primary KEP tumors to treatment with AZD8055 is associated with MHCII upregulation in the primary tumor immune environment, as well as upregulated transcription of genes related to antigen presentation. Because the dendritic cell is an important antigen-presenting cell, we quantified RNA expression of the dendritic cell marker CD11c (Itgax), and, in addition, we quantified the expression of a 9-gene set of dendritic cell markers that we composed, based on literature.29 Indeed, in sensitive tumors, there is a significant increase in the expression of dendritic cell markers compared to control and resistant tumors (Supplementary Fig. S8C).
Subsequently, we performed immunohistochemical analysis of lung metastases from untreated and AZD8055-treated mice with metastatic disease (prolonged neoadjuvant treatment, primary tumor removal at 15 mm diameter, followed by prolonged adjuvant treatment) for CD4, CD8, NKp46, Granzyme B, FOXP3, F4/80, MHCII, and Ly6G. This revealed that in treated mice, compared with untreated mice, fewer CD8 + T cells were present in lung metastases. No statistically significant differences were detected for other the other markers (Supplementary Fig. S7C). With the same panel, we analyzed the lung tissue itself, including mice from both early and late time points in the experiment. This revealed that in untreated control mice, CD4 + T cells increased between 5 days and the experimental endpoint (metastatic disease). FOXP3+ cell counts in lung tissue tended to be higher in mice with metastatic disease, but the only significant difference was between endpoint control mice and 28-day treated mice. Lung tissue from untreated mice with metastatic disease (endpoint) contained more Ly6G+ cells and fewer F4/80+ cells, compared to all other groups (Supplementary Fig. S7D).
Response of mILC to AZD8055 is partly mediated by the adaptive immune system
The association between the treatment response and the increased numbers of MHCII-positive cells, as well as transcriptomic evidence of activated antigen presentation processes, suggest that the effect of mTOR inhibition on KEP tumor growth is in part influenced by the immune system, and not solely driven by cancer cell-intrinsic processes. To test the contribution of the adaptive immune system to treatment efficacy of AZD8055, we performed parallel intervention studies in cohorts of immunocompetent wildtype mice and T and B cell deficient Rag1−/- mice engrafted with fragments of a treatment-naïve KEP tumor. As reported previously,30 the absence of the adaptive immune system did not affect KEP tumor outgrowth in untreated control animals (Figure 5(d)). Both treatment cohorts of mice were dosed daily with 50 mg/kg AZD8055 when tumors reached a diameter of 5 mm. mTOR inhibition slowed down tumor growth in both cohorts, but the median tumor-related survival (time until tumors reached a diameter of 15 mm) was 17.5 days shorter for the AZD8055-treated Rag1−/- cohort compared to the AZD8055-treated wildtype mice (p = .049, Figure 5(d)). Thus, therapeutic efficacy of AZD8055 was significantly reduced in the absence of a functional adaptive immune system.
To test whether acquired resistance of KEP tumors to AZD8055 is dependent on the host environment, we serially transplanted fragments of three resistant KEP tumors into treatment-naïve syngeneic wildtype mice, and treated daily with 50 mg/kg AZD8055 when tumors reached a diameter of 5 mm. The median survival benefit of AZD8055 treatment in the three cohorts was 15 days, 20 days, and 26 days (all p < .01, Figure 5(e), Supplementary Fig. S9). The observation that AZD8055 treatment has a significant effect on transplanted AZD8055-resistant tumors in treatment-naïve host mice indicates that resistance to mTOR inhibition is either a partially reversible cancer cell-intrinsic process (such as DNA methylation) and/or in part mediated by the host environment.
Taken together, our findings suggest a role of the adaptive immune system in the response of mouse ILC to mTOR inhibition. Activation of the adaptive immune system is induced by AZD8055 in therapy-responsive tumors and eventually lost upon acquisition of resistance.
Discussion
In this work, we studied the effects of mTORC1/2 inhibition in the KEP mouse model of metastatic invasive lobular carcinoma (ILC) of the breast. Metastasis is responsible for approximately 90% of cancer-related deaths.31 Unfortunately, there is a relative paucity of preclinical models that reflect cancer metastasis. The transplantable KEP model offers a unique opportunity to study the primary tumor as well as the metastatic cascade of invasive lobular breast cancer in an immunocompetent host.19 We combined surgical intervention with neoadjuvant and adjuvant treatment with the dual mTORC1/2 inhibitor AZD8055, monitored progression of primary tumors and metastatic disease, and investigated traits associated with therapeutic response and resistance to AZD8055. We found that AZD8055 effectively suppressed mTOR signaling in KEP tumors, and that activation of the adaptive immune system contributed to the therapeutic response to mTOR inhibition. In contrast, in the case of chemotherapy, response of mammary tumors in mouse models does not depend on the adaptive immune system.30 While suppression of mTOR signaling continued to be effective during AZD8055 treatment, therapy-associated activation of the adaptive immune system seemed to be transient, and its decline coincided with the development of therapy resistance in mouse ILC. In the relatively poorly immunogenic KEP model that was used in the current study, there is little immunogenic cell death, and we did not observe severe necrosis in the transplanted tumors, suggesting that the transient activation of the immune system should not simply be explained by immunogenic cell death.30
Pharmacological inhibition of mTOR is applied clinically to suppress the immune system in patients who receive an organ transplant. Known effects of mTOR inhibition in immune cells include reduced functions of T cells and dendritic cells, including antigen presentation, and stimulation of regulatory T cells, which in turn inhibit effector T cells.32–36 In the current study, however, mTOR inhibition with AZD8055 led to an increase in MHCII expression and activation of transcriptional programs related to antigen presentation through MHCII. In line with our findings, others have reported increased expression of MHCII on macrophages and dendritic cells after a combination treatment with AZD8055 and an agonist CD40 antibody.37 In addition, mTORC2-deficient Rictor−/- dendritic cells have been shown to display increased pro-inflammatory activity and can inhibit tumor growth by promoting CD8+ effector T cells.38,39
Eventually, we observed that most tumors became resistant within a narrow time window (visualized by the steep decline in progression-free survival in Figure 3(c)), suggesting that resistance might not be explained by stochastic events, but rather by a single time-dependent biological process. While our study does not provide detailed insight into the mechanisms underlying the development of resistance, it could be envisioned that this process involves some type of immune cell exhaustion. Previous studies indicate that mTOR inhibition, on the one hand, enhances immune-stimulatory function in existing differentiated DCs, but on the other hand impairs dendritic cell development, maturation, and survival.40–42
The interplay between the immune system and neoplastic cells is an important topic in the biology and treatment of cancer. Activated PI3K signaling in cancer may help tumor cells to escape from immunosurveillance.22 The reduced efficacy of AZD8055 treatment in T and B cell deficient mice indicates the contribution of the adaptive immune system to the therapeutic efficacy of mTOR inhibition in mILC (Figure 5(d)). Based on these findings, it would be interesting to study whether combining mTOR inhibition with cancer immunotherapy will convert the relatively short-term therapeutic benefit into a long-lasting tumor control. Combining targeted therapy with immunotherapeutics is currently a topic of investigation for various types of cancer.43–45 Immunotherapy could possibly improve the success of mTOR inhibition in cancer treatment. Indeed, others have combined AZD8055 with an agonist CD40 antibody in a model of metastatic renal cell carcinoma, resulting in an improved antitumor immune response which included increased numbers of dendritic cells.37 In a syngeneic model of oral cavity cancer, combining PD-L1 blockade with mTOR inhibition also led to an enhanced immune response.46 Also in diffuse-type gastric cancer, which, interestingly, is another disease where E-cadherin is involved, susceptibility to mTOR inhibition and checkpoint inhibition is a topic of investigation.47
In summary, mTOR inhibition in the metastatic KEP mouse ILC model leads to transient tumor growth arrest and activation of immunological processes related to the adaptive immune system. Loss of this activation is associated with acquired resistance to therapy, and the therapeutic efficacy of mTOR inhibition is partially determined by the host’s adaptive immune system. Future research may be directed at a better understanding of the temporal dynamics and mechanisms by which mTOR inhibition impacts the immune system, and how to prolong its antitumor effect, possibly in combination with immunotherapy.
Materials and methods
Analysis of publicly available datasets
Mutation and clinical information files were downloaded from the cBioPortal for Cancer Genomics (http://www.cbioportal.org/) for eleven breast cancer studies: Breast Cancer – METABRIC,48,49 Breast Invasive Carcinoma – British Columbia,50 Breast Invasive Carcinoma – Broad,51 Breast Invasive Carcinoma – Sanger,52 Breast cancer patient xenografts – British Columbia,53 Mutational profiles of metastatic breast cancer – France,54 The Metastatic Breast Cancer Project (Provisional, April 2018), Breast Invasive Carcinoma – TCGA,15 Breast Invasive Carcinoma – TCGA,11 Breast Invasive Carcinoma (TCGA, PanCancer Atlas) and Breast Invasive Carcinoma (TCGA, Provisional). Duplicate samples and samples of cancer type ‘Breast Mixed Ductal and Lobular Carcinoma’ were excluded. This left 1,759 samples of which 200 were of type Breast Invasive Lobular Carcinoma (ILC). Mutations in the following five genes were compared between ILC and non-ILC samples: PIK3CA, PTEN, AKT1, AKT2, PIK3R1, and MTOR. A Fisher’s exact test was performed to evaluate statistical significance.
In vitro experiments
Cdh1−/-;Trp53−/- cancer cell lines (KEP cells), generated from de novo mammary tumors from K14-cre;Cdh1Flox/Flox;Trp53Flox/Flox (KEP) mice, were exposed to a range of concentrations of AZD8055, and the 50% inhibitory concentration of AZD8055 was calculated. We used CellTiter-Glo to measure cell viability. For analysis of signaling inhibition by AZD8055, KEP cells were exposed to 500 nM of AZD8055 for 24 hours, after which they were lysed for immunoblotting. For low adherent culture conditions, we used Costar ultra-low attachment surface culture plates (Corning Incorporated, NY, USA).
Mouse studies
Mouse ILC (mILC) tumors were harvested from the established K14-cre;Cdh1Flox/Flox;Trp53Flox/Flox (KEP) model,18 bred to an FVB/N genetic background. Small (1 mm3) fragments of KEP tumor tissue were surgically transplanted into the right fourth mammary gland of female FVB/NCrl mice (Charles River). Neoadjuvant treatment started once tumor diameters reached 5 mm. AZD8055 (AstraZeneca) was formulated with 0.5% hydroxypropylmethylcellulose (Fluka BioChemika) and 0.1% polysorbate 80 (TWEEN 80), and administered by oral gavage, 20 mg/kg, once daily. For untreated control mice, the vehicle solution without AZD8055 was used. Tumor sizes were calculated as the product of 2 perpendicular caliper-measured diameters. To model the clinical course of metastatic breast cancer, surgical removal of large primary tumors at a diameter of 15 mm was performed as described previously.19 Mice were subsequently monitored for metastatic disease and treated with AZD8055 in case of adjuvant therapy experiments. All tumor samples from in vivo studies were harvested 1 h after the final dose administration. In Kaplan-Meier analyses for metastasis-specific survival, clinical endpoints related to metastatic disease were dyspnea due to lung metastasis, or a palpable metastasis of 15 mm in diameter. Censored events were recorded in cases where mice had to be sacrificed due to other reasons (e.g. local tumor recurrence after surgical removal). Timepoint 0 was defined as the day when the primary tumor transplant reached a diameter of 5 mm. The results in Kaplan-Meier analyses were tested for significance using a Log-rank (Mantel-Cox) test in GraphPad Prism® 7. Multiple hypothesis testing correction (Bonferroni method) was applied for analysis of the immunohistochemical quantification of immune cell populations, and for the comparison of median survival in all treatment groups (Supplementary Table S2). For experiments with mice lacking T and B cells, we used Rag1−/- mice with an FVB genetic background. All animal studies in this work have been approved by the Animal Ethics Committee of the Netherlands Cancer Institute and performed in accordance with national and institutional laws and guidelines for animal care and use.
Immunohistochemistry
For the generation of a human tissue microarray (TMA), 79 human ILCs were selected from the NKI pathology archive based on the diagnostic pathology report. Central revision was done to confirm the diagnosis of invasive lobular carcinoma. In 66 of these cases, there was sufficient tissue to be used in a TMA. Triplicate cores from these 66 invasive lobular carcinomas were incorporated into the TMA, and immunohistochemically stained with the following antibodies: Cell Signaling 9206 (phospho-S6K1 T389), 2965 (phospho-AKT T308), and 9456 (phospho-4EBP1 S65). For mouse tissues, the following antibodies were used: Cell Signaling 4060 (phospho-AKT S473), 2855 (phospho-4EBP1 T37/46), 2211 (phospho-S6 S235/236), Abcam ab25333 (MHCII), ab10558 (CD45), Thermo Fisher Scientific RM-9107 (CD3), eBioscience 14–9766 (CD4), 14–0808 (CD8a), 14–5773 (FOXP3), NKI internally produced anti-B220, AbD Serotec MCA497 (F4/80), and Novusbio NB100-684 (granzyme B). Further details are available in the Supplementary Methods.
Immunoblot analysis
For immunoblot analysis of mouse tumors, frozen tumor tissues were cut, while cooled on ice, into pieces of approximately 2 mm, and lysed on ice in RIPA buffer with phosphatase and protease inhibitors. NuPAGE 4–12% Bis-Tris Midi Protein Gels were used to run the protein lysates. The following primary antibodies were used: Cell Signaling 4060 (phospho-AKT S473, clone D9E), 9272 (AKT), 9234 (phospho-p70S6K T389, clone 108D2), 2708 (p70S6K, clone 49D7), 4858 (phospho-S6 S235/236, clone D57.2.2E), 2217 (S6, clone 5G10), 2855 (phospho-4EBP1 T37/46, clone 236B4), 9644 (4EBP1, clone 53H11), and Sigma A1978 (β-actin, clone AC-15).
Reverse phase protein array (RPPA)
Frozen tumor samples were submitted to the RPPA Core Facility of the MD Anderson Cancer Center, Houston, TX, United States. In short, frozen tumors were lysed and protein was extracted. Lysates were serial-diluted and printed on nitrocellulose-coated slides. Slides were probed with 136 validated primary antibodies followed by detection with appropriate biotinylated secondary antibodies. The signal obtained was amplified using an avidin-biotinylated peroxidase. Signals were visualized by a secondary streptavidin-conjugated HRP and DAB colorimetric reaction. The slides were scanned, analyzed, and quantified, with estimation of relative protein levels. Further details are available online at https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core/rppa-process.html. RPPA data were clustered using Ward’s hierarchical clustering method. Control: n = 9. Sensitive (treated 5 days): n = 5. Resistant (treated until endpoint): n = 15.
RNA-seq analysis
Frozen tissue samples were submitted for RNA-seq analysis to the Genomics Core Facility of the Netherlands Cancer Institute, Amsterdam, The Netherlands. Control: n = 10 (4 at 5 days, 6 at endpoint). Sensitive (treated 5 days): n = 10. Resistant (treated until endpoint): n = 18. We normalized RNA-seq raw transcript counts with the DESeq2 R package using the trimmed mean of M-values.55 With the normalized expressions, we performed differential gene expression analysis using the limma R package.56 GO term enrichment was computed using Panther.57 The metagene scores for RNA expression levels of gene sets (relative numbers of transcript reads) were compared using the ROAST gene set test.58
Acknowledgments
This work is part of the Oncode Institute, which is partly financed by the Dutch Cancer Society.
Research in the Jonkers laboratory is funded by the Netherlands Organisation for Scientific Research (NWO; Vici 91814643) and the European Research Council (ERC SyG 319661 Combat Cancer).
Research in the De Visser laboratory is funded by the Dutch Cancer Society (KWF10083; KWF10623) and a European Research Council Consolidator grant (InflaMet 615300). The authors thank AstraZeneca for providing the AZD8055 compound, and the following core facilities of The Netherlands Cancer Institute (NKI) for their contributions: the Core Facility Molecular Pathology and Biobanking (CFMPB) for their contribution to the analysis of tumors from patients, the Flow Cytometry Facility, the Experimental Animal Pathology Facility for the processing and staining of mouse tissues, and the Laboratory Animal Facility for animal care and support of the animal experiments. The authors thank Lennart Kester for bioinformatics support, and Cheei-Sing Hau for supporting experiments.
Disclosure statement
AstraZeneca provided the mTOR inhibitor AZD8055.
Supplementary material
Supplemental data for this article can be accessed publisher’s website.
References
- 1.Lakhani SR, Ellis IO, Schnitt SJ, Tan PH, van de Vijver MJ.. WHO classification of tumours of the breast. Vol. 4, Lyon: IARC Press; 2012. [Google Scholar]
- 2.Arpino G, Bardou VJ, Clark GM, Elledge RM. Infiltrating lobular carcinoma of the breast: tumor characteristics and clinical outcome. Breast Cancer Res. 2004;6(3):R149–R56. doi: 10.1186/bcr767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biglia N, Maggiorotto F, Liberale V, Bounous VE, Sgro LG, Pecchio S, D’Alonzo M, Ponzone R. Clinical-pathologic features, long term-outcome and surgical treatment in a large series of patients with invasive lobular carcinoma (ILC) and invasive ductal carcinoma (IDC). Eur J Surg Oncol. 2013;39(5):455–14. doi: 10.1016/j.ejso.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Li CI, Uribe DJ, Daling JR. Clinical characteristics of different histologic types of breast cancer. Br J Cancer. 2005;93(9):1046–1052. doi: 10.1038/sj.bjc.6602787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter AJ, Evans EB, Foxcroft LM, Simpson PT, Lakhani SR. Mammographic and ultrasound features of invasive lobular carcinoma of the breast. J Med Imaging Radiat Oncol. 2014;58(1):1–10. doi: 10.1111/1754-9485.12080. [DOI] [PubMed] [Google Scholar]
- 6.Hadjiminas DJ, Zacharioudakis KE, Tasoulis MK, Hu JC, Lanitis S, Bright-Thomas R, Dimopoulos NG, Hornzee G, Cunningham DA, Cleator SJ, et al. Adequacy of diagnostic tests and surgical management of symptomatic invasive lobular carcinoma of the breast. Ann R Coll Surg Engl. 2015;97(8):578–583. doi: 10.1308/rcsann.2015.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farese SA, Aebi S. Infiltrating lobular carcinoma of the breast: systemic treatment. Breast Dis. 2008;30:45–52. doi: 10.3233/BD-2009-0281. [DOI] [PubMed] [Google Scholar]
- 8.Joh JE, Esposito NN, Kiluk JV, Laronga C, Khakpour N, Soliman H, Catherine Lee M. Pathologic tumor response of invasive lobular carcinoma to neo-adjuvant chemotherapy. Breast J. 2012;18(6):569–574. doi: 10.1111/tbj.2012.18.issue-6. [DOI] [PubMed] [Google Scholar]
- 9.Katz A, Saad ED, Porter P, Pusztai L. Primary systemic chemotherapy of invasive lobular carcinoma of the breast. Lancet Oncol. 2007;8(1):55–62. doi: 10.1016/S1470-2045(06)71011-7. [DOI] [PubMed] [Google Scholar]
- 10.Truin W, Vugts G, Roumen RM, Maaskant-Braat AJ, Nieuwenhuijzen GA. van der Heiden-van der Loo M. Differences in response and surgical management with neoadjuvant chemotherapy in invasive lobular versus ductal breast cancer. Ann Surg Oncol. 2016;23:51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas N . Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 13.Barbareschi M, Buttitta F, Felicioni L, Cotrupi S, Barassi F, Del Grammastro M, Ferro A, Dalla Palma P, Galligioni E, Marchetti A, et al. Different prognostic roles of mutations in the helical and kinase domains of the PIK3CA gene in breast carcinomas. Clin Cancer Res. 2007;13(20):6064–6069. doi: 10.1158/1078-0432.CCR-07-0266. [DOI] [PubMed] [Google Scholar]
- 14.Christgen M, Noskowicz M, Schipper E, Christgen H, Heil C, Krech T, Länger F, Kreipe H, Lehmann U. Oncogenic PIK3CA mutations in lobular breast cancer progression. Genes, Chromosomes Cancer. 2013;52(1):69–80. doi: 10.1002/gcc.v52.1. [DOI] [PubMed] [Google Scholar]
- 15.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163(2):506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desmedt C, Zoppoli G, Gundem G, Pruneri G, Larsimont D, Fornili M, Fumagalli D, Brown D, Rothé F, Vincent D, et al. Genomic characterization of primary invasive lobular breast cancer. J Clinical Oncol. 2016;34(16):1872–1881. doi: 10.1200/JCO.2015.64.0334. [DOI] [PubMed] [Google Scholar]
- 17.Michaut M, Chin SF, Majewski I, Severson TM, Bismeijer T, de Koning L, Peeters JK, Schouten PC, Rueda OM, Bosma AJ, et al. Integration of genomic, transcriptomic and proteomic data identifies two biologically distinct subtypes of invasive lobular breast cancer. Sci Rep. 2016;6:18517. doi: 10.1038/srep18517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, van Beijnum JR, Griffioen AW, Vink J, Krimpenfort P, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10(5):437–449. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Doornebal CW, Klarenbeek S, Braumuller TM, Klijn CN, Ciampricotti M, Hau CS, Hollmann MW, Jonkers J, de Visser KE. A preclinical mouse model of invasive lobular breast cancer metastasis. Cancer Res. 2013;73(1):353–363. doi: 10.1158/0008-5472.CAN-11-4208. [DOI] [PubMed] [Google Scholar]
- 20.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Dituri F, Mazzocca A, Giannelli G, Antonaci S. PI3K functions in cancer progression, anticancer immunity and immune evasion by tumors. Clin Dev Immunol. 2011;2011:947858. doi: 10.1155/2011/947858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xue G, Zippelius A, Wicki A, Mandala M, Tang F, Massi D, Hemmings BA. Integrated Akt/PKB signaling in immunomodulation and its potential role in cancer immunotherapy. J Natl Cancer Inst. 2015;107:7. doi: 10.1093/jnci/djv171. [DOI] [PubMed] [Google Scholar]
- 23.Koyasu S. The role of PI3K in immune cells. Nat Immunol. 2003;4(4):313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]
- 24.Karlsson E, Perez-Tenorio G, Amin R, Bostner J, Skoog L, Fornander T, Sgroi DC, Nordenskjöld B, Hallbeck A-L, Stål O, et al. The mTOR effectors 4EBP1 and S6K2 are frequently coexpressed, and associated with a poor prognosis and endocrine resistance in breast cancer: a retrospective study including patients from the randomised Stockholm tamoxifen trials. Breast Cancer Res. 2013;15(5):R96. doi: 10.1186/bcr3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rojo F, Najera L, Lirola J, Jimenez J, Guzman M, Sabadell MD, Baselga J, Cajal SRY. 4E-binding protein 1, a cell signaling hallmark in breast cancer that correlates with pathologic grade and prognosis. Clin Cancer Res. 2007;13(1):81–89. doi: 10.1158/1078-0432.CCR-06-1560. [DOI] [PubMed] [Google Scholar]
- 26.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70(1):288–298. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 27.Wei F, Zhang Y, Geng L, Zhang P, Wang G, Liu Y. mTOR inhibition induces EGFR feedback activation in association with its resistance to human pancreatic cancer. Int J Mol Sci. 2015;16(2):3267–3282. doi: 10.3390/ijms16023267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujishita T, Kojima Y, Kajino-Sakamoto R, Taketo MM, Aoki M. Tumor microenvironment confers mTOR inhibitor resistance in invasive intestinal adenocarcinoma. Oncogene. 2017;36:6480–6489. [DOI] [PubMed] [Google Scholar]
- 29.Gardner A, Ruffell B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016;37(12):855–865. doi: 10.1016/j.it.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciampricotti M, Hau CS, Doornebal CW, Jonkers J, de Visser KE. Chemotherapy response of spontaneous mammary tumors is independent of the adaptive immune system. Nat Med. 2012;18(3):344–346. author reply 6. doi: 10.1038/nm.2652. [DOI] [PubMed] [Google Scholar]
- 31.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 32.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9(5):324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stallone G, Infante B, Di Lorenzo A, Rascio F, Zaza G, Grandaliano G. mTOR inhibitors effects on regulatory T cells and on dendritic cells. J Transl Med. 2016;14(1):152. doi: 10.1186/s12967-016-0916-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaoul N, Fayolle C, Desrues B, Oberkampf M, Tang A, Ladant D, Leclerc C. Rapamycin impairs antitumor CD8+ T-cell responses and vaccine-induced tumor eradication. Cancer Res. 2015;75(16):3279–3291. doi: 10.1158/0008-5472.CAN-15-0454. [DOI] [PubMed] [Google Scholar]
- 36.Verbrugge I, Gasparini A, Haynes NM, Hagekyriakou J, Galli M, Stewart TJ, Abrams SI, Yagita H, Verheij M, Johnstone RW, et al. The curative outcome of radioimmunotherapy in a mouse breast cancer model relies on mTOR signaling. Radiat Res. 2014;182(2):219–229. doi: 10.1667/RR13511.1. [DOI] [PubMed] [Google Scholar]
- 37.Jiang Q, Weiss JM, Back T, Chan T, Ortaldo JR, Guichard S, Wiltrout RH. mTOR kinase inhibitor AZD8055 enhances the immunotherapeutic activity of an agonist CD40 antibody in cancer treatment. Cancer Res. 2011;71(12):4074–4084. doi: 10.1158/0008-5472.CAN-10-3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raich-Regue D, Fabian KP, Watson AR, Fecek RJ, Storkus WJ, Thomson AW. Intratumoral delivery of mTORC2-deficient dendritic cells inhibits B16 melanoma growth by promoting CD8(+) effector T cell responses. Oncoimmunology. 2016;5(6):e1146841. doi: 10.1080/2162402X.2016.1146841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raich-Regue D, Rosborough BR, Watson AR, McGeachy MJ, Turnquist HR, Thomson AW. mTORC2 deficiency in myeloid dendritic cells enhances their allogeneic Th1 and Th17 stimulatory ability after TLR4 ligation in vitro and in vivo. J Immunol. 2015;194:4767–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hackstein H, Taner T, Zahorchak AF, Morelli AE, Logar AJ, Gessner A, Thomson AW. Rapamycin inhibits IL-4–induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood. 2003;101(11):4457–4463. doi: 10.1182/blood-2002-11-3370. [DOI] [PubMed] [Google Scholar]
- 41.Taner T, Hackstein H, Wang Z, Morelli AE, Thomson AW. Rapamycin-treated, alloantigen-pulsed host dendritic cells induce ag-specific T cell regulation and prolong graft survival. Am J Transplant. 2005;5(2):228–236. doi: 10.1046/j.1600-6143.2004.00673.x. [DOI] [PubMed] [Google Scholar]
- 42.Zeng H. mTOR signaling in immune cells and its implications for cancer immunotherapy. Cancer Lett. 2017;408:182–189. doi: 10.1016/j.canlet.2017.08.038. [DOI] [PubMed] [Google Scholar]
- 43.Keller HR, Zhang X, Li L, Schaider H, Wells JW. Overcoming resistance to targeted therapy with immunotherapy and combination therapy for metastatic melanoma. Oncotarget. 2017;8(43):75675–75686. doi: 10.18632/oncotarget.v8i43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colli LM, Machiela MJ, Zhang H, Myers TA, Jessop L, Delattre O, Yu K, Chanock SJ. Landscape of combination immunotherapy and targeted therapy to improve cancer management. Cancer Res. 2017;77(13):3666–3671. doi: 10.1158/0008-5472.CAN-16-3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karachaliou N, Gonzalez-Cao M, Sosa A, Berenguer J, Bracht JWP, Ito M, Rosell R. The combination of checkpoint immunotherapy and targeted therapy in cancer. Ann Transl Med. 2017;5(19):388. doi: 10.21037/atm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore EC, Cash HA, Caruso AM, Uppaluri R, Hodge JW, Van Waes C, Allen CT. Enhanced tumor control with combination mTOR and PD-L1 inhibition in syngeneic oral cavity cancers. Cancer Immunol Res. 2016;4(7):611–620. doi: 10.1158/2326-6066.CIR-15-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukamachi H, Kim SK, Koh J, Lee HS, Sasaki Y, Yamashita K. A subset of diffuse-type gastric cancer is susceptible to mTOR inhibitors and checkpoint inhibitors. J Exp Clin Cancer Res. 2019;38(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, Gelmon K, Chia S C, Wan A, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2015;518(7539):422–426. doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC. Mutational profile of metastatic breast cancers: a retrospective analysis. PLoS Med. 2016;13(12):e1002201. doi: 10.1371/journal.pmed.1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45(D1):D183–D9. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu D, Lim E, Vaillant F, Asselin-Labat ML, Visvader JE, Smyth GK. ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics. 2010;26(17):2176–2182. doi: 10.1093/bioinformatics/btq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.