

Abstract
Many secretory proteins are activated by cleavage at specific sites. The proprotein convertases (PCs) form a family of nine secretory subtilisin-like serine proteases, seven of which cleave at specific basic residues within the trans-Golgi network, granules, or at the cell surface/endosomes. The seventh member, PC7, is a type-I transmembrane (TM) protein with a 97-residue–long cytosolic tail (CT). PC7 sheds human transferrin receptor 1 (hTfR1) into soluble shTfR1 in endosomes. To better understand the physiological roles of PC7, here we focused on the relationship between the CT-regulated trafficking of PC7 and its ability to shed hTfR1. Deletion of the TMCT resulted in soluble PC7 and loss of its hTfR1 shedding activity. Extensive CT deletions and mutagenesis analyses helped us zoom in on three residues in the CT, namely Glu-719, Glu-721, and Leu-725, that are part of a novel motif, EXEXXXL725, critical for PC7 activity on hTfR1. NMR studies of two 14-mer peptides mimicking this region of the CT and its Ala variants revealed that the three exposed residues are on the same side of the molecule. This led to the identification of adaptor protein 2 (AP-2) as a protein that recognizes the EXEXXXL725 motif, thus representing a potentially new regulator of PC7 trafficking and cleavage activity. Immunocytochemistry of the subcellular localization of PC7 and its Ala variants of Leu-725 and Glu-719 and Glu-721 revealed that Leu-725 enhances PC7 localization to early endosomes and that, together with Glu-719 and Glu-721, it increases the endosomal activity of PC7 on hTfR1.
Keywords: shedding, trafficking, protease, protein dynamic, convertase, adaptor protein 2 (AP-2), cell sorting, cytosolic tail (CT), proprotein convertase type 7, transferrin receptor 1(TfR1)
Introduction
Many secretory proteins are synthesized as precursors that are subsequently activated or inactivated by limited proteolysis at specific sites (1, 2). Most of these cleavages are achieved along the secretory pathway and/or at the cell surface by the proprotein convertases (PCs),2 serine proteases related to subtilisin/kexin (PCSKs) (1). The nine members of the mammalian PC family are subdivided into three groups. (i) The first seven basic amino acid (aa)-specific PCs include the soluble PC1/3, PC2, PC4, PC5/6-A, PACE4, and the type-1 membrane-bound furin, PC5/6-B and PC7. These proteases cleave their substrates at basic aa within the general motif (K/R)-2(Xn)-(K/R)↓, where Xn = 0, 1, 2, or 3 spacer aa (1). (ii) The type-I transmembrane protease SKI-1/S1P activates a number of membrane-bound transcription factors such as the sterol regulatory element-binding proteins 1 and 2 and the activating transcription factor 6 (1, 3, 4). (iii) The soluble PCSK9 is the last member of the family that is implicated in low-density lipoprotein (LDL)–cholesterol regulation via its nonenzymatic ability to enhance the degradation of the hepatic LDL receptor (5).
All PCs are first synthesized as inactive zymogens (proPC) that, except for PC2, are subsequently auto-catalytically cleaved in the endoplasmic reticulum (ER) at the C terminus of the inhibitory prodomain. The resulting noncovalent heterodimeric complex (pro-catalytic) then exits the ER and traffics either to the cis/medial-Golgi (SKI-1) or trans-Golgi network (TGN)/secretory granules/cell surface/endosomes where, upon sorting to their final destination, most PCs are activated. Zymogen activation of the basic aa-specific PCs occurs by either a second autocatalytic cleavage within the prodomain, thereby liberating this segment from the active protease (e.g. for furin) (6), or by following separation of the catalytic subunit from the inhibitory prodomain (e.g. for PC4 and PC7).
PC7 is the most ancient and highly-conserved secretory protease of the basic aa-specific PC-family members (7). Recent behavioral tests showed that PC7 knockout (KO) mice are healthy and have a normal lifespan but exhibit anxiolytic and novelty-seeking behaviors (8, 9). In contrast, PC7 knockdown is lethal in nonmammalian species such as Xenopus (10) and zebrafish (11). Although mammalian PC7 may exert redundant cleavage activities with some PCs such as furin (12), it uniquely sheds, in endosomal compartments, human transferrin receptor 1 (hTfR1) into soluble shTfR1 (13). It may also participate in the cleavage/activation of pro-epidermal growth factor (proEGF) (14). Cell biology studies revealed that the cytosolic tail (CT; human aa 689–785; Fig. 1A) of PC7 likely contains critical motifs for its sorting to endosomes, wherein it is presumably activated, and cleaves hTfR1 and proEGF (13–15). These could be either early or recycling endosomes, from which hTfR1 recycles back to the plasma membrane via a fast (early endosomes) (16) or a slow (recycling endosomes) (17) route. In that context, it was also shown that the aa motif PLC726 in the CT of PC7 is essential for its endosomal internalization from the cell surface (18), but whether it also regulates the shedding of hTfR1 by PC7 was not studied. In addition, the same group later showed that a cluster of five basic aa (HRSRKAK714) close to two reversible Cys-palmitoylated residues (Cys-699,704) in the CT of PC7 are essential for the TGN localization and endocytic trafficking of PC7 (19). Like furin, PC7 is mostly localized to the TGN and may recycle from the cell surface to the TGN, through endosomes (12, 15, 18, 20). However, the site of activation where the prodomain of PC7 separates from the catalytic domain and where PC7 gets activated and subsequently cleaves its substrates is not defined, but it is thought to occur in acidic early endosomes (13, 15). In that context, it was recently shown that palmitoylation of Cys-699,704 in the CT of PC7 is critical for its processing of anthrax toxin by promoting their co-association in uncharacterized plasma membrane microdomains (21). However, palmitoylation was not essential for cleavage of other PC7 substrates such as hTfR1 (13), E-cadherin, and insulin-growth factor receptor 1 (21).
Figure 1.

Common critical residues in the CT of PC7 of different species. A, sequence alignment of PC7 from different species: human, rat, mouse, and X. laevis. The TM and the CT are indicated by two different horizontal lines. Conserved amino acids between species are noted with stars (identical) and either single dots (similar) or double dots (similar functionality). B, Western blot analysis of cell lysates (cells) and media (med) after overexpression of hTfR1-V5 with empty vector, human PC7 (hPC7), rat PC7 (rPC7), Xenopus PC7 (xPC7) or its chimeric forms (Xenopus/human transmembrane domain and cytosolic tail (xTMCT/hTMCT) and Xenopus/human cytosolic tail (xCT/hCT)) in HEK293 cells. Normalization was performed using the hTfR1-V5 shed by hPC7 as a reference. These results are representative of three independent experiments. C, Western blot analysis of cell lysates after overexpression of mouse proEGF (mproEGF) with empty vector, hPC7, rPC7, xPC7, or its truncated forms (Xenopus/human transmembrane domain + cytosolic tail (xTMCT/hTMCT) and Xenopus/human cytosolic tail (xCT/hCT)) in HEK293 cells. Normalization was performed using the mproEGF-V5 cleavage by hPC7 as a reference in HEK293 cells. These results are representative of three independent experiments. Error bars indicate averaged values ± S.D. ns, not significant (Student's t test).
In this work, we present evidence that the shedding of hTfR1 by human PC7 depends on the integrity of a specific motif EXEXXXL725 present in its CT and that Leu is the only critical aa in the PLC726 motif. We also demonstrated that the PC7-mediated hTfR1 shedding is positively regulated by the cytosolic adaptor protein complex AP-2, as evidenced by overexpression and knockdown experiments of its μ-adaptin subunit.
Results
Critical CT residues for the PC7-induced cleavage of hTfR1
To better appreciate the critical aa in the CT of PC7 that may regulate its subcellular trafficking and hence its ability to shed hTfR1, we first aligned the transmembrane (TM) and CT segments of human (aa 668–785), rat, mouse, and Xenopus laevis PC7 (Fig. 1A). Although the TM is relatively well-conserved between these species (85% aa identity between human and mouse/rat and 55% for X. laevis), the CT is less so with 58, 46, and 35% aa identity between human, rat, mouse, and X. laevis PC7, respectively. To further delineate the critical aa in the CT, we generated chimeric constructs of human (hPC7) or X. laevis (xPC7) where their TMCT or CT was interchanged. Co-expression of the C-terminally V5-tagged hTfR1-V5 with native hPC7, rat PC7 (rPC7), xPC7, or four selected chimeras was achieved in HEK293 cells (Fig. 1B). Western blot analysis of cell lysates and media were analyzed for hTfR1 using a V5 monoclonal antibody (mAb) (13), and in lysates for PC7 using an in-house mammalian PC7 polyclonal antibody recognizing its catalytic subunit (22). The data show that PC7 derived from all constructs was able to shed hTfR1-V5 into the medium (shTfR1-V5) to a similar extent as evidenced from the ratio of media levels of shTfR1 to cellular levels normalized to β-actin (Fig. 1B). This suggests that some conserved residues in the CT of these species may be critical for this processing. We also note that the zymogen processing of proPC7 into PC7 occurs, but it seems somewhat less efficient for xPC7, hPC7-xCT, and hPC7-xTMCT chimeras. Because our antibody is specific for human, rat, and mouse PC7, this may explain why it poorly recognizes xPC7. Finally, similar conclusions were also drawn from co-expression of the above constructs with mouse pro-epidermal growth factor (mproEGF-V5) (Fig. 1C) (14), suggesting that the above results are applicable to other PC7 substrates.
We next generated a number of membrane-bound human PC7 constructs with successively shortened C-terminally–truncated segments of its CT, as well as a soluble form (shPC7) lacking the TMCT. These included the wild type (WT) sequence with a CT composed of 97 aa and truncated forms containing 88 (L774X), 67 (L753X), and 29 (G717X) residues and ending at Ser-689 (Q690X; ΔCT) of the CT (Fig. 2A). We then co-expressed hTfR1 with each of these constructs in hepatic HuH7 (Fig. 2B) and HEK293 (Fig. 2C) cells. As shown previously (13), soluble PC7 does not shed hTfR1, reinforcing the importance of the membrane association of PC7 for such activity (Fig. 2, B and C). The lack of either 98% of the CT (ΔCT) or aa 717–785 (G717X) results in an ∼80% (±4.1%) reduction of the PC7-shedding of hTfR1, whereas the lack of aa 753–785 (L753X) or aa 774–785 (L774X) does not significantly affect such processing (Fig. 2, B and C). We conclude that the most critical information regulating the activity of PC7 on hTfR1 resides in the segment comprising aa 717–752 in the CT (Fig. 2A; ♦), with some remaining information in the membranous segment comprising aa 668–689 present in the ΔCT construct.
Figure 2.

Cytosolic tail of PC7 is essential for its cleavage activity. A, schematic representation of hPC7 and its truncated forms. The different domains of the protein are emphasized (SP, signal peptide; Pro, prosegment; TM, transmembrane domain; CT, cytosolic tail). The soluble form of PC7 (shPC7) is devoid of the TM and CT domain. Schematic representation of hTfR1-V5 shed by hPC7. B and C, Western blot analysis of cell lysates and media after overexpression of hTfR1-V5, empty vector and PC7, or its truncated forms in HuH7 cells (B) or HEK293 cells (C). Normalization was performed using the hTfR1-V5 shed by hPC7 as a reference. These results are representative of three independent experiments. Error bars indicate averaged values ± S.D. ***, p < 0.001; ns, not significant (Student's t test).
New EXEXXXL motif in the CT of human PC7 is important for its cleavage activity
Alignment of human aa 717–753 with the equivalent sequences in the CT of rat, mouse, and Xenopus PC7 revealed that the stretch ESXPL725 is absolutely conserved (Fig. 3A). The human PLC726 motif was previously reported to regulate endosomal entry of PC7 (18), but the importance of this sequence in the regulation of the enzymatic activity of PC7 was not investigated. Accordingly, Ala replacement of PLC (P724A,L725A,C726A) and PL (P724A,L725A) resulted in ∼60% (±3.7%) and ∼70% (±8.5%) reduction of PC7 activity on hTfR1 (Fig. 3B), suggesting that Cys-726 is not critical. Indeed, it is replaced by Phe in Xenopus, and the C726F and C726A mutations do not significantly influence the PC7 activity (Fig. 3C). Interestingly, although the P724A mutant exhibits similar activity to WT PC7, the mutant L725A lost ∼60% (±3.7%) of the activity, similar to the P724A,L725A and P724A,L725A,C726A mutants (Fig. 3C). Thus, Leu-725 seems to be the only aa in the PLC motif that is critical for PC7 ability to shed hTfR1.
Figure 3.

New EXEXXXL motif in the CT of human PC7 is important for its cleavage activity. A, alignment of the CT between aa 717 and 753 (vertical lines) from different species: human, rat, mouse, and Xenopus. Conserved amino acids between species are noted with stars (identical) and either single dots (similar) or double dots (similar functionality). B and C, Western blot analysis of cell lysates (cells) and media (med) after overexpression of hTfR1-V5 with empty vector, hPC7, its truncated form without the CT (ΔCT) or different CT mutants in HEK293 cells. Normalization was performed using the hTfR1-V5 shed by hPC7 as a reference. The main mutants composing the EXEXXXL motif are annotated by a star. These results are representative of three independent experiments. Error bars indicate averaged values ± S.D. *, p < 0.1; **, p < 0.01; ***, p < 0.001, ns, not significant (Student's t test).
We next focused on the possible presence of other critical aa preceding Leu-725 in the sequence ELESVPL725. Because Ala mutation of Leu-720, Ser-722, Val-723 or the sequence ESV723 did not affect the PC7 activity (Fig. 4A), this suggests that they are not critical. In addition, individually the E719A or E721A mutations also did not significantly affect the PC7 activity (Fig. 3C). Interestingly Glu-719 is replaced by Ala in mouse and rat and Gly in Xenopus PC7 (Fig. 3A), in line with a minor role of this aa residue. In contrast, the double E719A,E721A and the multiple E719A,E721A,P724A,L725A,C726A or E719A,E721A,L725A mutations reduced by ≥50% (±4.9–11.1%) the PC7 activity, similar to the L725A mutation (Fig. 3, B and C). Note that the bar graph values represent an average of three experiments. However, we only show Western blotting data of one representative experiment. As controls, our data showed that E733A,E735A did not affect PC7 activity (Fig. 3B). Similar results and conclusions were reached from experiments in HeLa and HuH7 cells (Fig. 4, B and C). Overall, this suggests that EXEXXXL725 is the critical motif in the CT of human PC7 that regulates its activity on hTfR1. Because mutations of the EXE721 or Leu-725 result in similar reductions of activity, this may indicate that EXEXXXL725 is a binding motif recognized by a common putative cytosolic partner.
Figure 4.

PC7 EXEXXXL motif is critical in multiple cell lines. A, Western blot analysis of cell lysates (cells) and media (med) after overexpression of hTfR1-V5, empty vector, hPC7, L720A, E721A/S722A/V723A (ESV723-AAA), S722A, and V723A mutants in HEK293 cells. Normalization was performed using the hTfR1-V5 shed by hPC7 as a reference. These results are representative of three independent experiments. B and C, Western blot analysis of cell lysates (cells) and media (med) after overexpression of hTfR1-V5, empty vector, hPC7, its truncated form without the CT (ΔCT), and different CT mutants in HeLa cells (B) or in HuH7 cells (C). Normalization was performed using the hTfR1-V5 shed by hPC7 as a reference. The main mutants composing the EXEXXXL motif are annotated by a star. These results are representative of three independent experiments. All error bars indicate averaged values ± S.D. *, p < 0.1; **, p < 0.01; ***, p < 0.001; ns, not significant (Student's t test).
NMR secondary structure prediction of the EXEXXXL motif in the CT of human PC7
Because of the theoretically higher mobility of a truncated peptide compared with the same sequence in the native protein, it is difficult to predict the native structure of a motif in the protein from a short peptide structure. However, sequence information of local motifs orchestrate the secondary structure and folding of proteins and give some idea to the native structure of the motif in the protein (23, 24). In fact, the only crystal structure of any basic aa-specific PC known is that of soluble furin lacking the TMCT (25). Thus, the unavailability of a crystal structure of PC7 or its CT led us to solve the structure of a chemically-synthesized CT 14-mer peptide (aa 717–730) using solution NMR methods to investigate the structure of this part of the CT in human PC7. The results suggested that the side chains of the key Glu and Leu residues in the EXEXXXL725 motif are lying on the same side of the molecule, where a likely protein–protein interaction (PPI) could happen (Fig. 5). As anticipated for a short peptide, the two last aa at both ends exhibit more mobility than the rest of the residues, which, however, finely converge in the ensemble. Furthermore, it was found that the peptide does not have a regular secondary structural element (α-helix, β-sheet, or turns), supporting the role of this segment as part of an accessible loop, because nonregular secondary structures are found in half of all PPIs as deduced from a survey of protein data banks (26). The data support the notion that the three key residues Glu-719,721 and Leu-725 lie on the same side of the molecule (Fig. 5A) and hence are well-poised to possibly interact with a partner/adaptor protein, as identified below.
Figure 5.
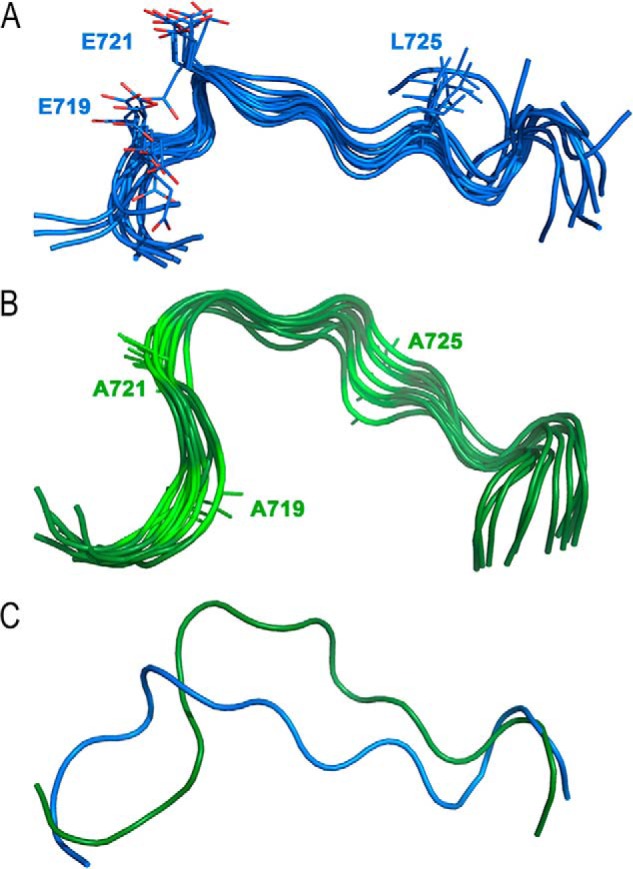
Cartoon representation of solution NMR structure of the human PC7 14-mer CT peptide (aa 717–730) and its Ala mutant. A, ensemble of 10 water-refined structures of CT peptide (aa 717–730). The side chains of Glu-719, Glu-721, and Leu-725 are displayed as lines with C atoms in blue and O atoms in red. B, ensemble of 10 water-refined structures of Ala-719,721,725–mutated CT peptide (aa 717–730). The side chain of mutated residues are shown as green lines. C, alignment of average energy minimized structures of both peptides with the same color code.
It was shown that removal of the side chains from these triad residues (i.e. Ala mutations of Glu-719,721 and Leu-725) resulted in a substantial reduction in the activity of PC7 on hTfR1, which is likely due to eliminated interactions of these residues with one or more cytosolic adaptor protein(s). To discover more structural elements of the motif, an Ala-719,721,725–mutated 14-mer peptide was synthesized, and the structure was solved by solution NMR. The data revealed that these mutations affected the backbone structure of the peptide (Fig. 5B). Although the mutated peptide still has a coiled-coil structure, alignments of the two structures showed that the mutated peptide has more curvature compared with the native PC7-derived peptide (Fig. 5C). Altered curvature causes a re-orientation and re-positioning of Ala-725 compared with Leu-725, demonstrating a significantly-altered peptide structure. In conclusion, our NMR data demonstrated clear differences between the native CT fragment and its Ala mutant. We showed that the Ala mutant is more compact compared with the peptide representing the native CT, which translates into the greater accessibility of the Glu side chains to interact with potential binding partners.
Subcellular localization of human PC7 and its CT mutants
The intracellular localization of human PC7 and its CT mutants was best analyzed by fluorescence immunocytochemistry in HeLa cells, compared with HEK293 or HuH7 cells, because HeLa cells have a more visible subcellular distribution of organelles, including its Golgi architecture (27). Furthermore, we reached similar conclusions regarding the effect of the PC7 mutants on hTfR1 cleavage activity in HeLa cells (Fig. 4B). Immunocytochemistry of C-terminally V5-tagged human PC7, its tagged ΔCT variant, or mutants of the critical aa in the EXEXXXL motif was analyzed in permeabilized HeLa cells (Figs. 6 and 7). Accordingly, we visualized the localization of PC7 in these cells and defined its co-localization with a TGN marker Golgin-97 (Fig. 6) or an early endosomal marker EEA1 (Fig. 7). Collectively, the data showed that ∼53% of WT PC7 localizes to the TGN (25.9 ± 19.0%) or early endosomes (27.4 ± 8.2%).
Figure 6.

Localization of human PC7 and its CT mutants with trans-Golgi network. Immunofluorescence of hPC7 or its CT mutants (red labeling) on permeabilized HeLa cells. Cell compartments Golgin-97 marker are labeled in green. Cell nuclei are marked by DAPI (blue labeling). Quantification of co-localization between hPC7 and its mutants with Golgin-97 using IMARIS software. These results are representative of a minimum of three independent experiments, and quantification represents an average of n = 15 cells. Error bars indicate averaged values ± S.D. ***, p < 0.001; ns, not significant (Student's t test). Bar = 1 μm.
Figure 7.

Localization of human PC7 and its CT mutants with early endosomes. Immunofluorescence of hPC7 or its CT mutants (red labeling) on permeabilized HeLa cells. The early endosomal marker (EEA1) is labeled in green. Cell nuclei are marked by DAPI (blue labeling). Quantification of co-localization between hPC7 and its mutants with EEA1 using IMARIS software. These results are representative of a minimum three independent experiments, and quantification represents an average of n = 15 cells. Error bars indicate averaged values ± S.D. **, p < 0.01; ***, p < 0.001 (Student's t test). Bar = 1 μm.
The data show that compared with WT PC7, the loss of the CT (ΔCT) or the mutation L725A that decreased by ∼80% of the PC7 activity on hTfR1 (Fig. 2, B and C) also significantly reduced the PC7 localization in the TGN by ∼70–75% (±5.6–14.1%) (Fig. 6). In addition, the EEA1 co-localization of PC7-ΔCT and PC7-L725A is reduced by ∼80% (±6%) and ∼50% (±14.6%), respectively (Fig. 7).
The localization of the remaining ∼47% immunoreactivity of WT PC7 has yet to be defined. To unravel the cell-surface localization of PC7 and its mutants, immunocytochemistry under nonpermeabilizing conditions was used (Fig. 8, A and B). The low-density lipoprotein receptor (LDLR) was selected as a cell-surface marker. These data showed that only ∼8.4% (±0.1%) of WT PC7 co-localized with the LDLR at the cell surface (Fig. 8B). In contrast, the three PC7 mutants co-localized ∼1.3-fold (±0.3) more with LDLR (28), suggesting a preferred re-localization of PC7 EXEXXXL mutants at or close to the cell surface (Fig. 8, A and B).
Figure 8.

Localization of human PC7 and its CT mutants at the cell surface. A, immunofluorescence of hPC7 or its CT mutants (red) and LDLR (green) in nonpermeabilized HeLa cells. Cell nuclei are marked by DAPI (blue). B, quantification of the immunoreactivity of hPC7 or its mutants with the cell-surface marker LDLR using IMARIS software. These results are representative of a minimum three independent experiments, and quantification represents an average of n = 15 cells per condition. C, Western blot analysis of PC7 WT and mutants, biotinylated and immunoprecipitated with streptavidin beads. Error bars indicate averaged values ± S.D. *, p < 0.1, (Student's t test). Bar = 1 μm.
It was previously shown that in T-cells PC7 mostly localizes to vesicles that line up immediately beneath the plasma membrane (29). Indeed, WT PC7 seems to localize to discrete vesicular areas close to the cell surface, whereas the immunoreactivity of the PC7 mutants spreads all along the plasma membrane (Fig. 8A). These results agree with those from a previous report using permeabilizing conditions on a PC7 Tac-CT-L725A membrane-bound chimera that is also enriched at the cell surface (18). As an orthogonal approach, we used cell-surface biotinylation assays (15). Upon normalization of PC7 levels to the red Ponceau staining, the data confirmed that the EXEXXXL mutants are ∼10–15% more biotinylated compared with WT PC7 (Fig. 8C), supporting the notion that this motif regulates the entry of PC7 into vesicles close to the cell surface.
Interestingly, the P724A mutant is ∼50% (±6.7%) less enriched in the TGN (Fig. S1A). The soluble PC7 is not endocytosed, and both PC7-WT and its P724A mutant similarly co-localize with EEA1 (Fig. S1B). These data agree with our observation of the similar hTfR1 shedding activity of PC7-WT or its P724A mutant (Fig. 3C). This suggests that PC7 is active if transported into early endosomes and that it is comparably much less active in the TGN or the cell surface, as suggested previously (13). Furthermore, our results using the full-length PC7 agree with those of an earlier report where the mutant P724A in a Tac-CT-chimera is also enriched at or close to the cell surface (18).
Notably, L725A (Fig. 6), P724A,L725A,C726A, P724A,L725A, and P724A (Fig. S1A) are ∼75% (±5.6%), ∼50% (±9.7%), ∼90% (±4.8%), and ∼65% (±6.7%) less co-localized with Golgin-97, respectively. This suggests that the PL725 plays a major role for the localization of PC7 in the TGN. In addition, similar to L725A (Fig. 7), but different from P724A (Fig. S1B), both P724A,L725A,C726A and P724A,L725A are ∼50% (±3–9.8%) less co-localized with EEA1 (Fig. S1B). These data and those of Fig. 7 demonstrate that Leu-725 is the critical residue in the PLC motif for the localization of PC7 in early endosomes, and hence for its activity on hTfR1 (Figs. 3 and 4).
Notably, the absence of Glu-719,721 predominates over that of Leu-725 in keeping PC7 in the TGN, because E719A,E721A and E719A,E721A,L725A are similarly localized to the TGN as WT (Fig. 6). Both Leu-725 and Glu-719,721 are critical for the EEA1 localization of PC7, because their Ala mutants are ∼40% (±7.2–14.6%) less co-localized with EEA1 than WT PC7 (Fig. 7), befitting their ∼50% lower activity on hTfR1 (Fig. 2C). These results suggest that Glu-719,721 and Leu-725 are critical for the efficient localization of PC7 in early endosomes, and hence for PC7 activity on hTfR1 (Figs. 3 and 4). In sum, we conclude that the motif EXEXXXL725 modulates the efficient entry of PC7 into early endosomes and its cleavage activity on hTfR1.
Finally, compared with WT PC7, the PC7-ΔCT exhibits a much lower localization in early endosomes than the L725A and E719A,E721A,L725A mutants (Fig. 7), and yet the hTfR1 shedding activities of the above mutants are ∼2-fold (±0.4) higher than that of PC7-ΔCT (Fig. 3). This suggests that the CT of PC7 contains motifs other than the EXEXXXL725 that modulate its efficient entry into early endosomes from the cell surface, its activity and retrograde endocytic trafficking to the TGN via endosomes. Indeed, a basic aa cluster HRSRKALK714 and two palmitoylated Cys-699,704 seem to be critical for this trafficking (19).
AP-2 is necessary for PC7 endosomal sorting and shedding activity on hTfR1
Selective transport of transmembrane proteins to different intracellular organelles often involves the recognition of sorting signals in their CT by one or more members of a family of heterotetrameric AP complexes named AP-1 through AP-5, each composed of four subunits (μ, γ, σ, and β) (30). These cytosolic adaptor complexes mediate sorting of cargos from the TGN, cell surface, endosomes, and lysosomes. AP-1 is a clathrin-associated complex that, in most cell types, mediates sorting between the TGN and endosomes, whereas AP-2 is involved in clathrin-dependent cell-surface endocytosis in which cargo proteins are incorporated into vesicles destined for fusion with the early endosome (31).
Both AP-1 and AP-2 complexes usually recognize the motifs YXX-(bulky hydrophobic aa) or (D/E)XXXL(L/I) (31). Because two aa in the deduced critical motif EXEXXXL725 in the CT of PC7 are absolutely conserved between species (Fig. 1A) and resemble partially the (D/E)XXXL(L/I) motif recognized by AP-1 as well as AP-2, we investigated their possible involvement in the sorting and/or sheddase activity of PC7. The hPC7 EXEXXXL725-sorting motif appears to belong to the family of sorting motifs recognized by the three σ1-adaptins of the various AP-1 complexes and by the σ2-adaptin subunit of the AP-2 complex (32–34). To investigate the contribution of AP-1 and AP-2 to hPC7 sorting, and thus hTfR1 shedding, we decided to alter the expression levels of AP-1 and AP-2 via overexpression and knockdown of their μ1 and μ2 adaptin subunits, because the absence of μ-adaptin renders the remaining trimeric adaptin complex nonfunctional (35, 36). Accordingly, we either overexpressed or silenced the μ subunit of either AP-1 or AP-2 in HeLa and HEK293 cells (Figs. 9–12).
Figure 9.
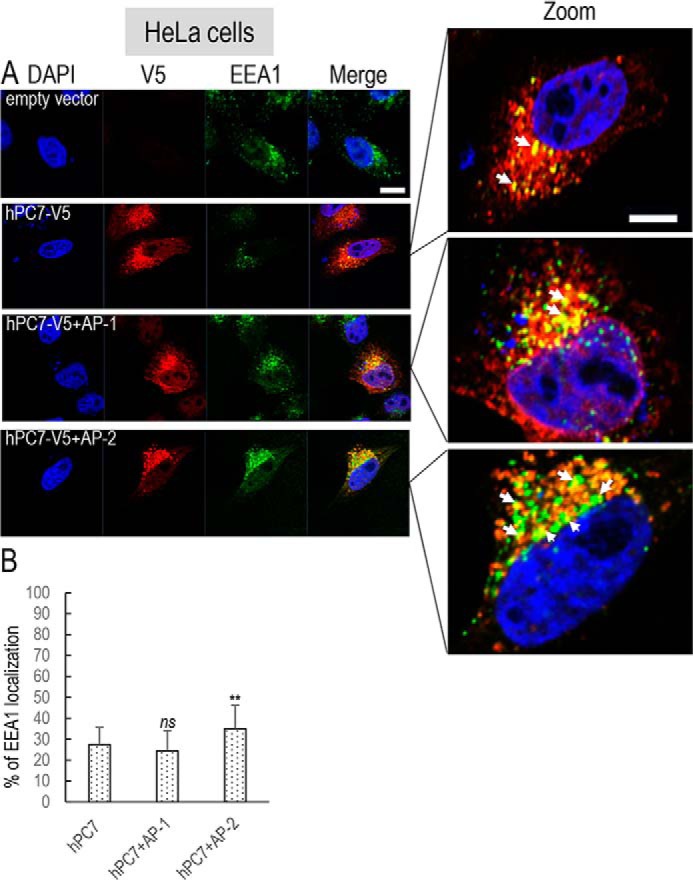
Overexpression of AP and localization of human PC7 with early endosomes. A, immunofluorescence of hPC7 (red labeling) in the presence of AP proteins, on permeabilized HeLa cells. The early endosomal marker (EEA1) is labeled in green. Cell nuclei are marked by DAPI (blue labeling). The right panels were expanded 2.5-fold (notice the scale bar size) to better visualize the co-localizations (yellow). Bar = 1 μm. B, quantifications were performed using IMARIS software. Co-localization of EEA1 with WT upon overexpression of AP-1μ or AP-2μ. These results are representative of a minimum three independent experiments, and quantification represents an average of n = 15 cells. Error bars indicate averaged values ± S.D. **, p < 0.01, ns, not significant (Student's t test).
Figure 10.

Adaptor-protein 2 regulates the hTfR1 cleavage activity of PC7. A, Western blot analysis of cell lysates (cells) and media (med) after overexpression of hTfR1-V5 with hPC7 and AP-1μ or AP-2μ in HEK293 cells. Normalization was performed using hTfR1-V5 cleavage by hPC7 as a reference. These results are representative of three independent experiments. B, Western blot analysis of cell lysates (cells) and media (med) after overexpression of hTfR1-V5 and hPC7 with siRNA control (scramble) or with siRNA against AP-1μ or AP-2μ in HEK293 cells. siRNA silencing was quantified by quantitative PCR. These results are representative of three independent experiments. C, co-immunoprecipitation assay of HEK293 cells transfected with a plasmid expressing Flag-tagged AP-2μ and hPC7-V5 or empty vector as a negative control. Immunoprecipitation (IP) was performed with beads coupled to an anti-Flag antibody. Cell lysates (input) and immunoprecipitates were analyzed by Western blotting (WB) with an anti-V5 antibody or anti-Flag antibody. These results are representative of three independent experiments. Error bars indicate averaged values ± S.D. *, p < 0.05; ***, p < 0.001; ns, not significant (Student's t test).
Figure 11.

Overexpression of AP and localization of human PC7 CT mutants with early endosomes. Top panel, immunofluorescence of hPC7 or its CT mutants (red labeling) in the presence of AP-2 protein on permeabilized HeLa cells. The early endosomal marker (EEA1) is labeled in green. Cell nuclei are marked by DAPI (blue labeling). Quantifications were performed using IMARIS software. Co-localization of EEA1 with WT hPC7, E719A,E721A mutant (2nd panel), L725A mutant (3rd panel), E719A,E721A,L725A (4th panel) mutant upon overexpression of AP-2μ. These results are representative of a minimum of three independent experiments, and quantification represents an average of n = 15 cells. Error bars indicate averaged values ± S.D. Bottom panel: **, p < 0.01, ***, p < 0.001, ns, not significant (Student's t test). Bar = 1 μm.
Figure 12.

Overexpression of AP and localization of human PC7 or its CT mutant with TGN. A, immunofluorescence of hPC7 or its CT mutant (red labeling) in the presence of AP proteins on permeabilized HeLa cells. Cell compartments Golgin-97 marker (TGN marker) are labeled in green. Cell nuclei are marked by DAPI (blue labeling). Quantification, using IMARIS software, of the co-localization between hPC7 (B), and its mutant E719A,E721A,L725A (C) and Golgin-97 in the presence of overexpressed AP-1m (B) or AP-2m (B and C). These results are representative of minimum three independent experiments, and quantification is representative of n = 15 cells. Error bars indicate averaged values ± S.D. ns, not significant (Student's t test). Bar = 1 μm.
In view of the critical importance of the localization of PC7 in early endosomes for its activity on hTfR1, we first concentrated on the co-localization of PC7 (Fig. 9) and its mutants (Fig. 11) with EEA1 following overexpression of the μ subunit of either AP-1 or AP-2 in HeLa cells (Figs. 9–11). The data show that overexpression of the AP-2μ subunit, but not that of AP-1μ (even at higher concentration), results in a significant ∼1.3-fold (±0.1) increased localization of WT PC7 with EEA1 (white arrows) in HeLa cells (Fig. 9), as well as an ∼1.6-fold (±0.2) higher activity of PC7 on hTfR1 in HEK293 cells (Fig. 10A). Moreover, these results are supported by a 17% (±10%) increased co-localization between PC7 and AP-2 (Fig. S2). In agreement, only siRNA treatment (88% efficacy) against AP-2μ resulted in a significant ∼30% (±10%) reduction of PC7 activity, whereas siRNA against AP-1μ (96% efficacy) had no significant effect (Fig. 10B). We conclude that AP-2 is implicated in the localization of WT PC7 in early endosomes, similar to its reported effect on its hTfR1 substrate (37), and hence in the up-regulation of the hTfR1 cleavage by PC7. The reverse would be expected in cells lacking AP-2 (Fig. 10B). This result was supported by the co-immunoprecipitation of the membrane-bound protein PC7 and the cytosolic one AP-2μ in HEK293 cells overexpressing the AP-2μ–Flag and hPC7-V5 (Fig. 10C). However, an ∼70% lower co-immunoprecipitation was observed upon mutation of the EXEXXXL725 motif to AXAXXXA725 (Fig. 10C). These results suggest a direct binding of AP-2 to the exposed EXEXXXL725 motif in the CT or an indirect binding via a “bridging” molecule that has a lower affinity for the mutant motif.
We next investigated the effect of overexpression of AP-2μ on the co-localization of the PC7 EXEXXXL725 mutants with EEA1 (Fig. 11). In all cases, we found that overexpression of AP-2μ does not significantly affect the degree of co-localization of the PC7 mutants E719A,E721A, L725A, or E719A,E721A,L725A (Fig. 11, lower panel) with EEA1. These results support the notion that AP-2 interacts with the critical EXEXXXL725 motif in the CT of PC7 and regulates its entry into early endosomes and hence its activity on hTfR1. As a control experiment, we used an siRNA against AP-2 and verified the impact of hTfR1 localization in early endosomes and TGN (Fig. S3). Coherent with the literature (37), in our knockdown experiments, we observed no change in hTfR1 co-localization with Golgin-97, but a significant decrease in co-localization with EEA1, as AP-2 is necessary for hTfR1 recycling to EEA1 but is not involved in its trafficking to the Golgi.
Finally, because the EXEXXXL725 motif does not play a major role in the TGN localization of PC7 (Fig. 6), it was of interest to test the possible roles of AP-1 and AP-2 in this process. Our results showed that neither overexpression of AP-2μ nor AP-1μ affect the TGN localization of WT PC7 nor its E719A,E721A,L725A mutant (Fig. 12). Similar results were also obtained with the E719A,E721A and L725A mutants (data not shown). We conclude that the TGN localization of PC7 requires Leu-725 but is not regulated by either AP-2 or AP-1, suggesting an alternative mechanism.
Discussion
Although PC7 is the most conserved member of the secretory basic amino acid–specific PC family (7), very little is known about its physiological functions (9), subcellular trafficking, activation, and processing of specific substrates (1, 13–15, 18, 19). The viability of Pcsk7 KO mice and the absence of overt pathological phenotype(s) suggests that this widely-expressed convertase is either redundant with other members of the family, such as furin, or that its absence may actually be beneficial in adult mice (8, 9). Indeed, epidemiological data suggested that PCSK7 rare gain–of–function SNPs or overexpression may be associated with a higher triglyceride phenotype (38), and biochemical data further provided a possible link between PC7 and cardiovascular diseases (39, 40). In addition, gene duplication or deletion of PCSK7 is associated with a rare congenital heart anomaly known as total anomalous pulmonary venous connection, where pulmonary veins of the left atrium abnormally connect to the right atrium or systemic venous system (41). These epidemiological data suggested that PC7 may play a critical role in the activation of the vascular endothelial growth factor receptor 2 (VEGFR-2) (41). Coincidentally, the latter type-1 membrane-bound receptor exhibits a conserved EXEXXXL1330 motif in its CT sequence.
To better understand the biology of the secretory membrane-bound PC7, herein we undertook an extensive analysis of the role of its CT in the regulation of its intracellular trafficking and ability to enhance the shedding of hTfR1 in endosomes, the only validated human specific substrate of PC7 (13).
Our extensive mutagenesis and deletion analyses of the CT led to the conclusion of the existence of a specific EXEXXXL725 motif that modulates the steady-state concentration of PC7 in early endosomes and its cleavage activity on hTfR1 (Figs. 1–5). This was further supported by NMR studies that demonstrated that in this exposed motif the Glu-719,721 and Leu-725 reside on the same side of the molecule (Fig. 5), suggesting that they may interact with a specific cytosolic protein. The similarity of the conserved EXXXL part of the critical motif to the di-leucine–based recognition sequences for AP-2 led us to demonstrate that AP-2 indeed enhances the subcellular trafficking of PC7 into early endosomes, where it may get activated and thus becomes apt to shed hTfR1 (Figs. 9 and 10). The same applied to the processing of mouse proEGF (data not shown).
It was suggested that in addition to furin, mouse PC7 mediates the processing of mouse proNotch-1 in the TGN, based on the fact that the mouse equivalent P723A,L724A,C725A mutation in the CT did not affect such processing in mouse melanoma B16F1 cells (12). In contrast, we demonstrated that the TGN localization of human PC7 P724A,L725A,C726A is ∼50% lower than WT PC7 (Fig. S1A). Thus, it would be informative if the results of human and mouse proNotch-1 processing at RRRR1664↓EL and RQRR1654↓EL, respectively, by human and mouse PC7 were compared.
The present results do not exclude the possibility that, aside from hTfR1 or proEGF cleavage, activated PC7 recycles back to the TGN from early endosomes and performs other cleavages therein (12, 19). Indeed, it was shown that the human CT segment proximal to the TM (aa 689–714; Fig. 1A) contains a nonconserved basic sequence HRSRKAK714, close to two reversible Cys-palmitoylated conserved residues (Cys-699,704) that are essential for the TGN localization and endocytic trafficking of human PC7 (19). Until recently, the importance of the Cys-palmitoylation in the regulation of the cleavage of physiological substrates of PC7 remained obscure. Indeed, it is dispensable in the processing of many of its substrates, such as hTfR1 (13), mproEGF (14), and the redundant cleavage of E-cadherin and IGF-1 by PC7 and furin (21, 42). Interestingly, it was recently shown that the Cys-palmitoylation of PC7 and of the anthrax toxin protective antigen (PA) is performed by the same protein acyltransferase ZDHHC5 (21). These Cys-palmitoylations promote the association of both PC7 and PA with plasma membrane microdomains favoring subsequent cleavage of PA by PC7 (21). It is thus plausible that the Cys-palmitoylation of PC7 mediates the concentration of PC7 and some of its Cys-palmitoylated substrates in membrane microdomains.
Although autocatalytic cleavage of proPC7 to PC7 occurs in the ER (15), the noncovalent inactive complex of the prodomain PC7 (22) exits the ER, crosses the TGN, and reaches the cell surface, and then endosomes. Gradual acidification and/or changes in calcium concentration along the late secretory pathway may lead to the separation of active PC7 from its inhibitory prodomain (13).
The exact PC7 zymogen activation mechanism and the fate of the prodomain are still unresolved. In cells overexpressing vaccinia virus recombinants of the prodomain of PC7, this segment was secreted intact in the medium but not that of furin (22). We presume that the prodomain separates from the main catalytic subunit before or after entry of the prodomain–PC7 complex into early endosomes from the cell surface, whereupon PC7 gets activated and sheds hTfR1. Future studies should address this critical question in detail, as it may lead to the identification of a specific mechanism and/or partners implicated in the activation of PC7.
Finally, it should be mentioned that besides the conventional ER to TGN route, PC7 is the only convertase known to reach the cell surface by an alternative faster unconventional pathway directly from the ER to the cell surface (15). The TM of PC7 (but not that of furin) seems to be critical for the ability of PC7 to traffic through this unconventional route (15). We still do not know whether zymogen activation of PC7 traversing this unconventional pathway takes place or whether this is a source of readily available and enzymatically inactive PC7 for other functions that are independent of its catalytic activity, as proposed for the R504H mutant of PC7 in the regulation of triglycerides (39, 43).
In conclusion, the data presented in this work better defined the critical elements in the CT of PC7 that regulate its ability to traffic to early endosomes where it is able to shed hTfR1 and also to cleave mouse proEGF. It emerged that the sheddase activity of PC7 critically depends on this sorting motif recognized by AP-2 but that additional proteins are taking part in the regulation of PC7 trafficking kinetics (Fig. 13).
Figure 13.
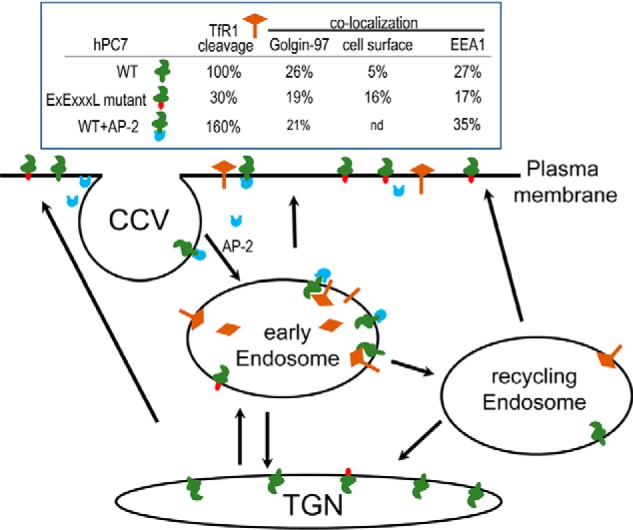
Schematic representation of PC7 trafficking and cleavage activity. The table summarizes the cleavage activity and trafficking of hPC7 and its mutants to the TGN (Golgin-97), cell surface, and early endosomes (EEA1). Activity cleavage results are semi-quantitative and normalized to the WT hPC7 (100%). The cartoon summarizes the localization and cleavage activity of PC7 WT (green CT) and its EXEXXXL mutant (red CT) and the regulation of hPC7 in the presence of AP-2 (blue). CCV, clathrin-coated vesicles; nd, not determined.
Experimental procedures
Plasmids
All cDNA mutants hPC7, rPC7, and xPC7 were cloned, as reported, on pIRES-2–EGFP vector (Clontech) (13). Some of them were subcloned at the C terminus with a V5, Flag, or HA tag. Human hTfR1-V5, mouse AP-1, mouse AP-2, and mouse proEGF-V5 were cloned as reported (13, 14). Table S1 recapitulates all oligonucleotides used in mutant constructions.
Cell culture and transfections
HuH7, HEK293, and HeLa cells lines were cultivated in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen). Cells were incubated in a 6-well plates (500,000 cells/well) at 37 °C under 5% CO2. HeLa and HuH7 cells were transfected in an equimolar quantity of plasmid with a final quantity of 2 μg of cDNA with Polyplus reagent (jetPRIME®). HEK293 cells were transfected in equimolar quantity of plasmid with a final quantity of 1 μg of cDNA using Polyplus reagent. Twenty hours after transfection, cells were washed in a serum-free medium and incubated for another 20 h, whereupon the cells were collected and lysed with 1× RIPA in order to prepare protein for Western blot analysis.
siRNA and quantitative RT-PCR
A pool of four siRNA against human AP-1μ, AP-2μ, and a scrambled siRNA (Dharmacon; siGENOME SMARTpool) were transfected with a final 50 nm concentration, using DhamaFECT1 transfection reagent (Dharmacon), as recommended by the manufacturer's protocol.
To verify the siRNA efficacy, quantitative RT-PCRs were performed on HEK293 cells to measure AP-1μ and AP-2μ mRNA levels. Before quantification, total RNA extraction was performed using 1 ml of TRIzol reagent (Invitrogen) according to the manufacturer's instructions. To verify the RNA extraction quality, Superscript II reverse-transcriptase amplification was done, as recommended by Invitrogen. The quality of newly-extracted RNA was visualized on a 1% agarose gel.
Real-time PCR was carried out using Viia7 System (Applied Biosystems). Reactions were run in duplicate in two independent experiments. Human TATA-box–binding protein (TBP) gene was used as an internal control to normalize the variability in expression levels. The sets of primers were as follows: hAP1M1 forward, GAGATCGTGTGGTCCATCAAGTC, and reverse AAGTGGGCCCGCATCA; hAP2M1 forward, CAAGGCCAGCGAGAATGC, and reverse, GCGCTGATCTGCGATTCC. TBP gene was used to normalize gene expression and quantify the variability of the expression level of AP proteins in the presence of siRNA. Expressions were analyzed using the ΔCT method (44).
Western blot analysis
Cell lysates and media were separated on an 8% SDS-polyacrylamide gel. After migration, gels were transferred onto a nitrocellulose membrane (GE Healthcare) overnight and blocked 1 h at room temperature with a 1:1 blocking buffer (Mandel), 1× PBS. Membranes were incubated overnight at 4 °C in a 1:1 blocking buffer (Mandel), 1× PBS solution with first antibody (Ab): anti-PC7 polyclonal Ab that recognizes the N-terminal prosegment and the active form (1:10,000), anti-V5 Ab (Invitrogen) (1:3000), and β-actin (Sigma) (1:3000). Proteins were revealed with a secondary fluorescent anti-mouse Ab 680 (Mandel) (1:10,000) or anti-rabbit Ab 800 (Mandel). Revelation of tagged proteins occurred on an Odyssey Li-Cor imaging machine, and quantification was performed with Image Studio Lite version 5.2 software. To verify the expression of AP-1μ–Flag and AP-2μ–Flag, anti-Flag M2 HRP (1:3000) (Sigma) was used.
Co-immunoprecipitation
HEK293 cells were plated in 100-mm2 plates and transfected the next day in an equimolar quantity of plasmid with a final quantity of 4 μg of cDNA using Polyplus reagent. Forty eight hours post-transfection, cells were lysed in 0.5% CHAPS buffer (5 m NaCl, 1 m Tris, pH 7.5, 0.5% CHAPS), and protein concentration was calculated using Bradford assay (Bio-Rad). 1 mg of protein was incubated with anti-Flag M2 antibody (Sigma) for 2 h prior to incubation with protein A/G PLUS-agarose (Santa Cruz Biotechnology) for 1 h. The samples were washed three times with 0.5% CHAPS buffer. The immunoprecipitates were resolved on an 8% SDS-polyacrylamide gel and revealed by a V5-HRP antibody (1:10,000; Invitrogen).
Peptide synthesis
Two 14-mer peptides were synthesized: WT PC7 GTELESVPLCSSKD, and its triple mutant GTALASVPACSSKD. The peptides were synthesized either on 2-chlorotritylchloride or TentaGel S RAM resin on an automated peptide synthesizer. The peptide chains' elongation was carried out by standard Fmoc solid-phase peptide synthesis. Fmoc-protected aa were coupled using 5 eq of protected aa, 5 eq of 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5]pyridinium 3-oxide hexafluorophosphate (HATU), and 15 eq of DIPEA in DMF. Fmoc group was removed with 20% piperidine in DMF. For synthesis on 2-chlorotritylchloride resin, the first aa was loaded on the resin using 1.2 eq of aa and 4 eq of DIPEA in DMF for 3 h. The unreacted groups were capped with a mixture of DCM/MeOH/DIPEA, 85:10:5, and N-terminal acetylation was achieved by a solution of DCM/DIPEA/Ac2O, 85:10:5, for 1 h. The global deprotection was achieved using the mixture trifluoroacetic acid/thioanisole/H2O/phenol/ethanedithiol, 82.5:5:5:5:2.5 for 2 h. After evaporation of 50% of cleaving mixture, the crude peptide was triturated with diethyl ether (Et2O) and centrifuged, and the supernatant was discarded. The resulting solid was purified on a Waters preparative HPLC system (autosampler 2707, quaternary gradient module 2535, UV detector 2489 (λ = 214 and 230 nm), fraction collector WFCIII) equipped with an ACE5 C18 column (250 × 21.2 mm, 5-μm spherical particle size). The purity of the fractions was determined (more than 95%) by an Agilent Technologies 1100 analytical HPLC system equipped with a diode array detector (λ = 210, 214, 230, and 254 nm) and C18 columns of Phenomenex Jupiter (5 mm, 4.6 μm, 250 mm). Electrospray ionization–high-resolution mass spectrometry (TripleTOF 5600, ABSciex; Foster City, CA) was used to confirm the identity of the pure peptides.
NMR studies
Samples were prepared by solubilizing the peptides in a 10% D2O/H2O mixture (2–3 mm). For the Cys-containing peptide, fully deuterated 1,4-dithiothreitol was added at the maximum concentration of 10 mm. The NMR experiments were performed on a Agilent Varian (600 MHz for 1H) spectrometer at ambient temperature. 13C chemical shift was assigned by a 1H–13C HSQC and 1H chemical shifts using a 1H–1H TOCSY experiment (mixing time of 50 ms). The interproton distances were assigned based on the 1H–1H NOESY (mixing time of 50 ms) and 1H–1H ROESY (mixing time 250 ms) experiments.
The program DANGLE was used to predict the Φ and Ψ dihedral angles on the basis of backbone and 13Cb chemical shift values for both peptides (45). For the native truncated peptide, all NOEs were assigned manually, divided into weak, medium, and strong, and converted into distance restraints using CcpNmr Analysis version 2.4 (46). Structures were calculated using the ARIA 2.2 and CNS programs on the CCPN grid server (47, 48). After removing the violated NOE, the structures were recalculated on the CCPN grid server (https://www.ccpn.ac.uk/v2-software/software/web-apps-general). This server also refined the structures in water. The refined structures were further validated with the program Procheck (Fig. 3) (49).
For the Ala-mutated peptide 3D structure, calculations were achieved with AMBER 14.0 package (http://ambermd.org). The starting structure of the peptide has been generated with LEaP program embedded in AMBER 14.0. For the solvated system, a truncated octahedral box of water around the peptide structure has been added (2245 TIP3P water residues). The side chains of one lysine and one aspartic acid were defined as positively and negatively charged, respectively, leading to a total peptide charge of 0. The entire system was subjected to dynamics under the constant pressure for 10 ns with time-averaged distance restraints and dihedral restraints derived from NMR spectroscopy. A total of 97 interproton distance restraints (64 intraresidual, 26 sequential, and 7 medium range) and 17 dihedral angles restraints were applied for structure solution. The calibration peaks' volume–to–distance constraints were performed with CALIBA program included in CYANA 2.1 (50) software. The interproton distances and dihedral angles were introduced in the molecular dynamics simulations with the force constants f = 50 kcal/(mol × Å2) and 10 kcal/(mol × rad2), respectively. The geometry of the peptide groups was kept fixed according to NMR data (all-trans) with the force constant f = 50 kcal/(mol × rad2). MD simulations were performed with a 2-fs time step and a 10-Å cutoff radius. Isotropic position scaling was used to maintain the pressure. The system was heated from 10 to 303 K during the first 20 ps of MD and then the temperature was maintained at 303 K. The coordinates were recorded at each 4 ps (2000th step), and 20 conformations obtained in the last steps of MD simulations were chosen for final structure analysis and validated with program Procheck.
Immunofluorescence
HeLa cells were transfected with V5-tagged WT hPC7 or its Ala mutants. Cells were transfected with 0.5 μg final of cDNA and 1.8 μl of FuGENE reagent (Promega). Twenty hours post-transfection, cells were washed in a serum-free medium. Twenty hours post-washing, all treatments, until the first antibody incubation, occurred on ice. First, cells were washed three times with cold PBS one time and fixed with 4% fresh formaldehyde during 10 min. After two more washes, the cells were permeabilized with 0.1% Triton X-100 for 7 min. After three more washes using cold PBS, blocking was done with PBS containing 1% BSA for 30 min. Cells were incubated with primary antibody overnight at 4 °C: anti-V5 Ab (1:500, Invitrogen) or anti-V5 Ab (1:500, Abcam), anti-Golgin-97 Ab (1:500, Santa Cruz Biotechnology), anti-EEA1 Ab (1:200, Abcam), anti-HA Ab (1:2000, Cell Signaling Technology), anti-PC7 Ab (1:500, Cell Signaling Technology), and anti-LDLR (1:200, R&D Systems). Antigen–antibody complexes were revealed by 1-h incubations with the corresponding species-specific Alexa Fluor (488, 555, or 647)-tagged antibodies (Molecular Probes). The nuclei were stained with Prolong DABCO with DAPI (Thermo Fisher Scientific). Immunofluorescence analyses were performed on a confocal microscope (Zeiss LSM-710).
Cell-surface biotinylation
For detection of cell-surface biotinylation of PC7, HEK293 cells were transiently transfected with either PC7 WT, PC7 (E719A and E721A), PC7 (L725A), or PC7 (E719A, E721A, and PC7 L725A). At 48 h post-transfection, cells were washed twice with ice-cold PBS and incubated on ice with 0.5 mg/ml sulfosuccinimidyl-6-(biotin-amido)hexanoate (sulfo-NHS-LC-biotin, Thermo Fisher Scientific) for 20 min. Following biotinylation, cells were washed once with PBS/BSA (0.1 mg/ml) (Sigma) and twice with ice-cold PBS. Cells were then scraped in lysis buffer (20 mm Tris (pH 7.5), 5 mm EDTA, 5 mm EGTA, 0.5% maltoside (Cayman Chemical Co.)), and lysates were homogenized with a mortar and pestle. Homogenates were kept on ice for 10 min and spin at 3000 rpm for 10 min at 4 °C. Supernatants were collected, and 30 μl of streptavidin (Pierce) were added to 200 μg of protein and left overnight on a rotating wheel. Following overnight incubation, samples were spun for 2 min at 7000 rpm, and supernatants were collected (corresponding intracellular pool). Pellets were washed three times with 200 μl of lysis buffer. The pellets were finally resuspended in 20 μl of 6× Laemmli before being heated and loaded on an 8% SDS-polyacrylamide gel with their corresponding intracellular pool. The % of PC7 at the cell surface was normalized to the total amount of PC7 (surface + intracellular pool).
Statistical immunofluorescence analysis
Co-localization of fluorescently-labeled protein was quantified with IMARIS analysis software (8.2.1) along with aXTension script named Colocalize Spots. We used the same approach as mentioned in Rajan et al. (51). Fluorescent background was first normalized using empty vector. Positive signals were found using the Imaris function spots from each fluorescent marker images. The spot diameter used was 1.2 μm with the same quality factor for each image. The Colocalize Spots script considers co-localization between two spots when their center–to–center distance is equal of inferior to 0.8 μm. WT hPC7-V5 co-localization with each marker was used for normalization. Quantification is based on the analysis of 15 cells from a minimum of three independent experiments in each condition. Student's t test was used for estimation of the statistical significance.
Statistical analysis
Data are reported as mean ± S.D. Statistical significance was determined using the Student's t test.
Author contributions
L. D. and N. G. S. conceptualization; L. D. data curation; L. D. and N. G. S. validation; L. D. investigation; L. D. and N. G. S. writing-original draft; L. D. writing-review and editing; S. D., V. D., E. S., P. S., and R. D. formal analysis; A. E. resources; J. G. methodology; N. G. S. supervision; N. G. S. funding acquisition; N. G. S. visualization; N. G. S. project administration.
Supplementary Material
Acknowledgments
We thank Sandrine Lacoste for cell biology, Annik Prat for the generation of the Fig. 9 model of PC7 trafficking and functions, Dominic Fillion for microscopy, and Brigitte Mary for editorial assistance.
This work was supported by Canadian Institutes of Health Research Foundation Scheme 148363 and Canada Research Chair 950–231335. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3 and Table S1.
- PC
- proprotein convertase
- CT
- cytosolic tail
- TM
- transmembrane
- x
- Xenopus
- h
- human
- r
- rat
- mproEGF
- mouse proEGF
- aa
- amino acid
- DAPI
- 4′,6-diamidino-2-phenylindole
- LDL
- low-density lipoprotein
- LDLR
- low-density lipoprotein receptor
- AP
- adaptor protein
- TGN
- trans-Golgi network
- PPI
- protein–protein interaction
- ER
- endoplasmic reticulum
- HRP
- horseradish peroxidase
- PA
- protective antigen
- Ab
- antibody
- DIPEA
- diisopropylethylamine
- DMF
- N,N-dimethylformamide
- DCM
- dichloromethane
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- MD
- molecular dynamics
- TBP
- TATA-box–binding protein.
References
- 1. Seidah N. G., and Prat A. (2012) The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug. Discov. 11, 367–383 10.1038/nrd3699 [DOI] [PubMed] [Google Scholar]
- 2. Seidah N. G., Sadr M. S., Chrétien M., and Mbikay M. (2013) The multifaceted proprotein convertases: their unique, redundant, complementary and opposite functions. J. Biol. Chem. 288, 21473–21481 10.1074/jbc.R113.481549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ye J., Rawson R. B., Komuro R., Chen X., Davé U. P., Prywes R., Brown M. S., and Goldstein J. L. (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 10.1016/S1097-2765(00)00133-7 [DOI] [PubMed] [Google Scholar]
- 4. Brown M. S., and Goldstein J. L. (1999) A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. U.S.A. 96, 11041–11048 10.1073/pnas.96.20.11041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seidah N. G., Awan Z., Chrétien M., and Mbikay M. (2014) PCSK9: a key modulator of cardiovascular health. Circ. Res. 114, 1022–1036 10.1161/CIRCRESAHA.114.301621 [DOI] [PubMed] [Google Scholar]
- 6. Seidah N. G. (2017) The PCSK9 revolution and the potential of PCSK9-based therapies to reduce LDL-cholesterol. Glob. Cardiol. Sci. Pract. 2017, e201702 10.21542/gcsp.2017.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seidah N. G., Hamelin J., Mamarbachi M., Dong W., Tardos H., Mbikay M., Chretien M., and Day R. (1996) cDNA structure, tissue distribution, and chromosomal localization of rat PC7, a novel mammalian proprotein convertase closest to yeast kexin-like proteinases. Proc. Natl. Acad. Sci. U.S.A. 93, 3388–3393 10.1073/pnas.93.8.3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Besnard J., Ruda G. F., Setola V., Abecassis K., Rodriguiz R. M., Huang X. P., Norval S., Sassano M. F., Shin A. I., Webster L. A., Simeons F. R., Stojanovski L., Prat A., Seidah N. G., Constam D. B., et al. (2012) Automated design of ligands to polypharmacological profiles. Nature 492, 215–220 10.1038/nature11691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wetsel W. C., Rodriguiz R. M., Guillemot J., Rousselet E., Essalmani R., Kim I. H., Bryant J. C., Marcinkiewicz J., Desjardins R., Day R., Constam D. B., Prat A., and Seidah N. G. (2013) Disruption of the expression of the proprotein convertase PC7 reduces BDNF production and affects learning and memory in mice. Proc. Natl. Acad. Sci. U.S.A. 110, 17362–17367 10.1073/pnas.1314698110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Senturker S., Thomas J. T., Mateshaytis J., and Moos M. Jr. (2012) A homolog of subtilisin-like proprotein convertase 7 is essential to anterior neural development in Xenopus. PLoS ONE 7, e39380 10.1371/journal.pone.0039380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Turpeinen H., Oksanen A., Kivinen V., Kukkurainen S., Uusimäki A., Rämet M., Parikka M., Hytönen V. P., Nykter M., and Pesu M. (2013) Proprotein convertase subtilisin/kexin type 7 (PCSK7) is essential for the zebrafish development and bioavailability of transforming growth factor β1a (TGFβ1a). J. Biol. Chem. 288, 36610–36623 10.1074/jbc.M113.453183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ginefra P., Filippi B. G. H., Donovan P., Bessonnard S., and Constam D. B. (2018) Compartment-specific biosensors reveal a complementary subcellular distribution of bioactive furin and PC7. Cell Rep. 22, 2176–2189 10.1016/j.celrep.2018.02.005 [DOI] [PubMed] [Google Scholar]
- 13. Guillemot J., Canuel M., Essalmani R., Prat A., and Seidah N. G. (2013) Implication of the proprotein convertases in iron homeostasis: proprotein convertase 7 sheds human transferrin receptor 1 and furin activates hepcidin. Hepatology 57, 2514–2524 10.1002/hep.26297 [DOI] [PubMed] [Google Scholar]
- 14. Rousselet E., Benjannet S., Marcinkiewicz E., Asselin M. C., Lazure C., and Seidah N. G. (2011) The proprotein convertase PC7 enhances the activation of the EGF receptor pathway through processing of the EGF precursor. J. Biol. Chem. 286, 9185–9195 10.1074/jbc.M110.189936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rousselet E., Benjannet S., Hamelin J., Canuel M., and Seidah N. G. (2011) The proprotein convertase PC7: unique zymogen activation and trafficking pathways. J. Biol. Chem. 286, 2728–2738 10.1074/jbc.M110.192344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chamberland J. P., Antonow L. T., Dias Santos M., and Ritter B. (2016) NECAP2 controls clathrin coat recruitment to early endosomes for fast endocytic recycling. J. Cell Sci. 129, 2625–2637 10.1242/jcs.173708 [DOI] [PubMed] [Google Scholar]
- 17. Gu J., Faundez V., and Werner E. (2010) Endosomal recycling regulates anthrax toxin receptor 1/tumor endothelial marker 8-dependent cell spreading. Exp. Cell Res. 316, 1946–1957 10.1016/j.yexcr.2010.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Declercq J., Meulemans S., Plets E., and Creemers J. W. (2012) Internalization of the proprotein convertase PC7 from the plasma membrane is mediated by a novel motif. J. Biol. Chem. 287, 9052–9060 10.1074/jbc.M111.306407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Declercq J., Ramos-Molina B., Sannerud R., Brouwers B., Pruniau V. P. E. G., Meulemans S., Plets E., Annaert W., and Creemers J. W. M. (2017) Endosome to trans-Golgi network transport of proprotein convertase 7 is mediated by a cluster of basic amino acids and palmitoylated cysteines. Eur. J. Cell Biol. 96, 432–439 10.1016/j.ejcb.2017.03.010 [DOI] [PubMed] [Google Scholar]
- 20. van de Loo J. W., Creemers J. W., Bright N. A., Young B. D., Roebroek A. J., and Van de Ven W. J. (1997) Biosynthesis, distinct post-translational modifications, and functional characterization of lymphoma proprotein convertase. J. Biol. Chem. 272, 27116–27123 10.1074/jbc.272.43.27116 [DOI] [PubMed] [Google Scholar]
- 21. Sergeeva O. A., and van der Goot F. G. (2019) Anthrax toxin requires ZDHHC5-mediated palmitoylation of its surface-processing host enzymes. Proc. Natl. Acad. Sci. U.S.A. 116, 1279–1288 10.1073/pnas.1812588116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhong M., Munzer J. S., Basak A., Benjannet S., Mowla S. J., Decroly E., Chrétien M., and Seidah N. G. (1999) The prosegments of furin and PC7 as potent inhibitors of proprotein convertases. In vitro and ex vivo assessment of their efficacy and selectivity. J. Biol. Chem. 274, 33913–33920 10.1074/jbc.274.48.33913 [DOI] [PubMed] [Google Scholar]
- 23. Baldwin R. L., and Rose G. D. (1999) Is protein folding hierarchic? I. Local structure and peptide folding. Trends Biochem. Sci. 24, 26–33 10.1016/S0968-0004(98)01346-2 [DOI] [PubMed] [Google Scholar]
- 24. Voelz V. A., Shell M. S., and Dill K. A. (2009) Predicting peptide structures in native proteins from physical simulations of fragments. PLoS Comput. Biol. 5, e1000281 10.1371/journal.pcbi.1000281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Henrich S., Cameron A., Bourenkov G. P., Kiefersauer R., Huber R., Lindberg I., Bode W., and Than M. E. (2003) The crystal structure of the proprotein processing proteinase furin explains its stringent specificity. Nat. Struct. Biol. 10, 520–526 10.1038/nsb941 [DOI] [PubMed] [Google Scholar]
- 26. Gavenonis J., Sheneman B. A., Siegert T. R., Eshelman M. R., and Kritzer J. A. (2014) Comprehensive analysis of loops at protein–protein interfaces for macrocycle design. Nat. Chem. Biol. 10, 716–722 10.1038/nchembio.1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tie H. C., Chen B., Sun X., Cheng L., and Lu L. (2017) Quantitative localization of a Golgi protein by imaging its center of fluorescence mass. J. Vis. Exp. 2017, 10.3791/55996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Susan-Resiga D., Girard E., Kiss R. S., Essalmani R., Hamelin J., Asselin M. C., Awan Z., Butkinaree C., Fleury A., Soldera A., Dory Y. L., Baass A., and Seidah N. G. (2017) The proprotein convertase subtilisin/kexin type 9-resistant R410S low density lipoprotein receptor mutation: a novel mechanism causing familial hypercholesterolemia. J. Biol. Chem. 292, 1573–1590 10.1074/jbc.M116.769430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Leonhardt R. M., Fiegl D., Rufer E., Karger A., Bettin B., and Knittler M. R. (2010) Post-endoplasmic reticulum rescue of unstable MHC class I requires proprotein convertase PC7. J. Immunol. 184, 2985–2998 10.4049/jimmunol.0900308 [DOI] [PubMed] [Google Scholar]
- 30. Guardia C. M., De Pace R., Mattera R., and Bonifacino J. S. (2018) Neuronal functions of adaptor complexes involved in protein sorting. Curr. Opin. Neurobiol. 51, 103–110 10.1016/j.conb.2018.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dacks J. B., and Robinson M. S. (2017) Outerwear through the ages: evolutionary cell biology of vesicle coats. Curr. Opin. Cell Biol. 47, 108–116 10.1016/j.ceb.2017.04.001 [DOI] [PubMed] [Google Scholar]
- 32. Kelly B. T., McCoy A. J., Späte K., Miller S. E., Evans P. R., Höning S., and Owen D. J. (2008) A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature 456, 976–979 10.1038/nature07422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Doray B., Lee I., Knisely J., Bu G., and Kornfeld S. (2007) The γ/σ1 and α/σ2 hemicomplexes of clathrin adaptors AP-1 and AP-2 harbor the dileucine recognition site. Mol. Biol. Cell 18, 1887–1896 10.1091/mbc.e07-01-0012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baltes J., Larsen J. V., Radhakrishnan K., Geumann C., Kratzke M., Petersen C. M., and Schu P. (2014) σ1B adaptin regulates adipogenesis by mediating the sorting of sortilin in adipose tissue. J. Cell Sci. 127, 3477–3487 10.1242/jcs.146886 [DOI] [PubMed] [Google Scholar]
- 35. Meyer C., Zizioli D., Lausmann S., Eskelinen E. L., Hamann J., Saftig P., von Figura K., and Schu P. (2000) mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 19, 2193–2203 10.1093/emboj/19.10.2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Medigeshi G. R., Krikunova M., Radhakrishnan K., Wenzel D., Klingauf J., and Schu P. (2008) AP-1 membrane-cytoplasm recycling regulated by mu1A-adaptin. Traffic 9, 121–132 10.1111/j.1600-0854.2007.00672.x [DOI] [PubMed] [Google Scholar]
- 37. Nesterov A., Carter R. E., Sorkina T., Gill G. N., and Sorkin A. (1999) Inhibition of the receptor-binding function of clathrin adaptor protein AP-2 by dominant-negative mutant mu2 subunit and its effects on endocytosis. EMBO J. 18, 2489–2499 10.1093/emboj/18.9.2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peloso G. M., Auer P. L., Bis J. C., Voorman A., Morrison A. C., Stitziel N. O., Brody J. A., Khetarpal S. A., Crosby J. R., Fornage M., Isaacs A., Jakobsdottir J., Feitosa M. F., Davies G., Huffman J. E., et al. (2014) Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am. J. Hum. Genet. 94, 223–232 10.1016/j.ajhg.2014.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ashraf Y., Sachan V., Essalmani R., Duval S., Marcinkiewicz J., Hamelin J., Roubtsova A., Prat A., and Seidah N. G. (2018) The proprotein convertase 7 regulates triglyceride levels via enhanced Apoa5 degradation. Atheroscler. Suppl. 32, 56 10.1016/j.atherosclerosissup.2018.04.169 [DOI] [Google Scholar]
- 40. Dongiovanni P., Meroni M., Baselli G., Mancina R. M., Ruscica M., Longo M., Rametta R., Cespiati A., Pelusi S., Ferri N., Ranzani V., Nobili V., Pihlajamaki J., Fracanzani A. L., Badiali S., et al. (2019) PCSK7 gene variation bridges atherogenic dyslipidemia with hepatic inflammation in NAFLD patients. J. Lipid Res. 60, 1144–1153 10.1194/jlr.P090449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shi X., Cheng L., Jiao X., Chen B., Li Z., Liang Y., Liu W., Wang J., Liu G., Xu Y., Sun J., Fu Q., Lu Y., and Chen S. (2018) Rare copy number variants identify novel genes in sporadic total anomalous pulmonary vein connection. Front. Genet. 9, 559 10.3389/fgene.2018.00559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khatib A. M., Siegfried G., Prat A., Luis J., Chrétien M., Metrakos P., and Seidah N. G. (2001) Inhibition of proprotein convertases is associated with loss of growth and tumorigenicity of HT-29 human colon carcinoma cells: importance of insulin-like growth factor-1 (IGF-1) receptor processing in IGF-1-mediated functions. J. Biol. Chem. 276, 30686–30693 10.1074/jbc.M101725200 [DOI] [PubMed] [Google Scholar]
- 43. Guillemot J., Essalmani R., Hamelin J., and Seidah N. G. (2014) Is there a link between proprotein convertase PC7 activity and human lipid homeostasis? FEBS Open Bio 4, 741–745 10.1016/j.fob.2014.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 45. Cheung M. S., Maguire M. L., Stevens T. J., and Broadhurst R. W. (2010) DANGLE: a Bayesian inferential method for predicting protein backbone dihedral angles and secondary structure. J. Magn. Reson. 202, 223–233 10.1016/j.jmr.2009.11.008 [DOI] [PubMed] [Google Scholar]
- 46. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 47. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol. Crystallogr 54, 905–921 10.1107/S0907444998003254 [DOI] [PubMed] [Google Scholar]
- 48. Rieping W., Habeck M., Bardiaux B., Bernard A., Malliavin T. E., and Nilges M. (2007) ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 23, 381–382 10.1093/bioinformatics/btl589 [DOI] [PubMed] [Google Scholar]
- 49. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 10.1007/bf00228148 [DOI] [PubMed] [Google Scholar]
- 50. Güntert P., Mumenthaler C., and Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 10.1006/jmbi.1997.1284 [DOI] [PubMed] [Google Scholar]
- 51. Rajan S. G., Gallik K. L., Monaghan J. R., Uribe R. A., Bronner M. E., and Saxena A. (2018) Tracking neural crest cell cycle progression in vivo. Genesis 56, e23214 10.1002/dvg.23214 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.