

Abstract
Formation of signaling complexes is crucial for the orchestration of fast, efficient, and specific signal transduction. Pharmacological disruption of defined signaling complexes has the potential for specific intervention in selected regulatory pathways without affecting organism-wide disruption of parallel pathways. Signaling by epinephrine and norepinephrine through α and β adrenergic receptors acts on many signaling pathways in many cell types. Here, we initially provide an overview of the signaling complexes formed between the paradigmatic β2 adrenergic receptor and two of its most important targets, the L-type Ca2+ channel CaV1.2 and the AMPA-type glutamate receptor. Importantly, both complexes contain the trimeric Gs protein, adenylyl cyclase, and the cAMP-dependent protein kinase, PKA. We then discuss the functional implications of the formation of these complexes, how those complexes can be specifically disrupted, and how such disruption could be utilized in the pharmacological treatment of disease.
Keywords: Gs, adenylyl cyclase, PSD-95, AKAP, cAMP, norepinephrine
1. INTRODUCTION
Innumerous signaling mechanisms govern most if not all of the molecular mechanisms of cellular functions. Given that many signaling pathways are simultaneously engaged, it is crucial for the specificity and efficacy of a particular pathway that all of its individual components are in close proximity (1, 2). The last 25 years revealed a plethora of protein-protein interactions that are critical for the transfer of information between the various modules and nodes within the myriad signaling pathways at work in individual cells. With approximately 800 members, G protein–coupled receptors (GPRCs) mediate the lion’s share of cellular signaling (3). GsPCRs activate the stimulatory trimeric Gs protein and thereby cAMP production by adenylyl cyclase (AC). Although cAMP is a freely diffusible second messenger, spatial restriction of cAMP production can result in differential signaling by GsPCRs, even within the same cell (1,4, 5). For instance, stimulation of the β1 adrenergic receptor (β1 AR) in cardiomyocytes leads to phosphorylation of proteins by the cAMP-dependent protein kinase (PKA) throughout these cells, whereas stimulation of the β2 adrenergic receptor (β2 AR) is mostly restricted to the vicinity of the L-type Ca2+ channel (LTCC) CaV1.2 (4, 6).
The identification of signaling complexes formed between the β2 AR, the LTCC CaV1.2 (5, 7, 8), and the AMPA-type glutamate receptors (AMPARs) (9, 10), which also contain Gs, AC, and PKA, was a milestone that significantly promoted our understanding of cAMP signaling. Selective interception of signaling pathways by ligands that specifically disrupt the respective protein-protein interactions has the potential to restrict their effects to one rather than several signaling cascades downstream of a particular GPCR. This approach expands other recent noteworthy efforts to affect signaling by GPCRs, and especially the β2 AR, by ligands that bind to the intracellular portions of GPCRs rather than their orthosteric ligand sites (3). Pharmacological agents that disrupt binding of Gs to a GPCR impair many signaling cascades downstream of a given GPCR, whereas the specific displacement of a GPCR from its ultimate target will affect only one signaling cascade.
2. ION CHANNELS AS DRUG TARGETS
Voltage- and ligand-gated ion channels regulate neuronal, cardiovascular, endocrine, and numerous other functions. Thus, they constitute prime targets for the pharmacological treatment of a multitude of diseases, including anxiety, depression, epilepsy, hypertension, and diabetes. However, achieving selectivity is difficult given the homology and high degree of sequence conservation between members of various ion channel families. Due to their limited contact sites with their targets, this problem of selectivity is a major hurdle for the development of small organic compounds as therapeutics. Thus, biologics such as peptides and antibodies are increasingly pursued to pharmacologically target ion channels (11).
2.1. Signaling by Epinephrine and Norepinephrine via the β2 Adrenergic Receptor in the Brain
Epinephrine and norepinephrine (NE) regulate numerous physiological functions throughout our body. The fight-or-flight response is perhaps the most obvious effect of these stress hormones that exert peripheral effects such as increased heart rate mediated by downstream β1 AR signaling. In the brain, NE generally augments arousal, acuity of behavioral tasks, and learning of emotionally charged content (12–14). As detailed below, the β2 AR is required for certain forms of synaptic plasticity and forms unique signaling complexes with CaV1.2 (7, 8) and the AMPAR (9, 10). We propose that these complexes are among the most important conduits of NE signaling in the brain and thus could be important drug targets for treating conditions such as post-traumatic stress disorder (PTSD) and attention-deficit hyperactivity disorder (ADHD) (15–18).
2.2. CaV1.2 Function in Health and Disease in the Brain
Ca2+ influx through CaV1.2 governs gene expression via CREB and NFAT (19, 20) and controls neuronal excitability via Ca2+-activated K+ channels (21, 22). CaV1.2 constitutes about 80% of L-type channels in the brain (23, 24). CaV1.2 mediates a portion of long-term potentiation (LTP) induced by 200-Hz tetanic stimuli (25), particularly during aging. Function of CaV1.2 is essential for mGluR-dependent long-term depression (26) and LTP induced by a prolonged theta tetanus (PTT) (PTT-LTP) at 5–10 Hz for 90–180 s (27, 28), a rhythm naturally occurring in the brain. Both PTT-LTP and CaV1.2 are relevant for spatial learning (29, 30).
Animal studies implicated increased CaV1.2 channel activity in anxiety disorders, depression, and self-injurious behavior (24) and in the etiology of senile symptoms and Alzheimer’s disease (31–33). Haploinsufficiency in the CACNA1C gene encoding the central pore-forming α1 1.2 subunit of CaV1.2 leads to deficits in prosocial ultrasonic communication in mice, suggesting that CaV1.2 plays a significant role in regulating socio-affective communication in rodents (34). Multiple genome-wide association studies uncovered variants in the CACNA1C gene as major risk factors for schizophrenia (SCZ), bipolar disorder (BPD), and autism spectrum disorder (ASD) (35–40). Single-nucleotide polymorphisms (SNPs), including the high-risk allele s1006737, are within a 100-kb region of CACNA1C’s third intron. As this is a noncoding region, one hypothesis is that these SNPs somehow interfere with proper splicing or enhancer activity. Sequence variants in a human-specific 30-bp tandem repeat in this region that exhibit decreased enhancer activity are associated with a similar disease risk as the flanking SNPs previously linked to BPD and SCZ (41).
3. β2 ADRENERGIC RECEPTOR COMPLEXES THAT ENABLE LOCALIZED SIGNALING
3.1. The β2 Adrenergic Receptor–CaV1.2 Signaling Complex in the Brain
Earlier considerations that signaling by cAMP could be spatially restricted for the specific activation of subsets of cAMP signaling cascades by the proximity of GsPCRs with their ultimate targets inspired multiple searches for putative GsPCR signaling complexes. CaV1.2 was the first target shown to form a complex with a GsPCR that also contained all other proteins in the classic cAMP cascade. Indeed, CaV1.2 associates with the β2 AR, Gs, AC, and PKA for highly localized regulation via cAMP in the brain (7, 42). Here, the A-kinase anchor protein 5 (AKAP5) (rodent AKAP150, human AKAP79) binds to three different sites in α1 1.2, i.e., the N terminus, the loop between domains I and II, and the distal C terminus (42–44) (Figure 1). In turn, the AKAP5 N terminus interacts with different ACs (45, 46), while a short motif near its C terminus interacts with the regulatory RII subunits of PKA (Figure 1). The cytosolic C terminus of the β2 AR binds to the distal C terminus region of α1 1.2 encompassing S1928 (27). S1928 is the main PKA phosphorylation site of α 1.2 (42,47) and is essential for the upregulation of CaV1.2 activity in neurons (28) and vascular smooth muscle cells (VSMCs) (48) but, remarkably, not in the heart (49). It is unclear whether the β2 AR directly contacts S1928, potentially occluding access for PKA. Given that upregulation of CaV1.2 activity by β2 AR signaling requires not only S1928 phosphorylation but also phosphorylation of the β2 AR itself on its PKA site S261/S262 (50), it is tempting to speculate that phosphorylation of S261/S262 results in a conformational change that renders S1928 accessible. The β2 AR is displaced from CaV1.2 upon S1928 phosphorylation for about 5 min. During this period, CaV1.2 is refractory to the enhancement of its activity by a second β2 AR stimulation (27). How Gs is attached to the β2 AR–CaV1.2 complex is unclear, but it is potentially via preassociation with the β2 AR (51).
Figure 1.
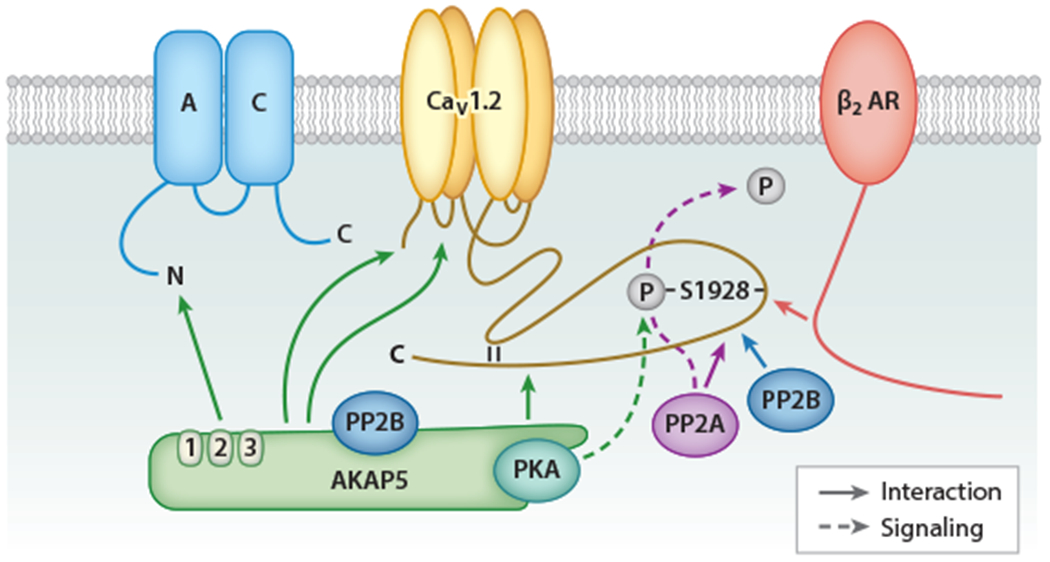
The β2 AR–AC-PKA-CaV1.2 complex. Green arrows indicate binding of the N terminus of AKAP5 to the N terminus of AC, the C terminus of AKAP5 to the distal C terminus of α1 1.2, and the so far undefined regions of AKAP5 to the N terminus and the loop between domains I and II of α1 1.2. AKAP5 links in this way AC, PKA, and PP2B to CaV1.2. The β2 AR binds with its C terminus to the region around S1928 in the distal C terminus of α1 1.2 (red arrow). PP2A and PP2B also bind directly to α1 1.2 about 40 and 50 residues downstream of S1928 (purple and blue arrows). Activation of β2 AR–Gs-AC-cAMP-PKA signaling leads to S1928 phosphorylation by PKA (dashed green line) and upregulation of CaV1.2 activity, both of which are reversed by the β11.2-associated PP2A (dashed purple line). Abbreviations: AC, adenylyl cyclase; AKAP5, A-kinase anchor protein 5; AR, adrenergic receptor; PKA, cAMP-dependent protein kinase; PP, protein phosphatase.
The CaV1.2 complex also contains the serine/threonine phosphatases PP2A and PP2B. PP2A binds with its catalytic C subunit directly to residues 1965–1971, less than 40 residues downstream of S1928, to counteract S1928 phosphorylation and the upregulation of LTCC activity (52–54). PP2B binds to α1 1.2 immediately upstream of the PP2A binding site to augment CaV1.2 activity through an unknown mechanism (55). Another PP2B molecule anchored to AKAP5 regulates α1 1.2 to curb CaV1.2 activity, in part, by governing Ca2+-dependent inactivation (44, 56, 57).
Application of the β2 AR–selective agonist albuterol in the cell-attached recording configuration increases channel open probability greater than twofold when applied inside the patch electrode but not at all when administered to the outside of the electrode. Despite the fact that over 99% of the cell surface and thus the vast majority of β2 ARs are accessible in the latter arrangement, their stimulation does not translate into upregulation of the activity of those channels that are physically occluded by the electrode (7; see also 58). Accordingly, cAMP signaling from the β2 AR to CaV1.2 is limited to less than 200 nm, the estimated dimension of the distance between β2 AR and CaV1.2 inside and outside the patch. A peptide consisting of α1 1.2 residues 1923–1942 (Pep1923–1942) displaces the β2 AR from CaV1.2 and prevents β2 AR stimulation of CaV1.2, demonstrating that β2 AR binding to CaV1.2 is required for the upregulation of channel activity and providing further evidence for the notion that this cAMP-mediated signaling is highly localized, potentially to the dimension of individual β2 AR-CaV1.2 complexes (27).
That the β2 AR and CaV1.2 colocalize within a complex at postsynaptic sites makes this signaling node a prime conduit for NE signaling (7). This notion is supported by the findings that PTT-LTP requires β2 AR and CaV1.2 activity, their association, and the phosphorylation of CaV1.2 on S1928 by PKA downstream of the β2 AR (27, 28, 59). Thus the β2 AR–CaV1.2 complex constitutes a potentially important target for the pharmacological treatment of conditions related to impaired signaling by NE or CaV1.2.
3.2. The β2 Adrenergic Receptor–CaV1.2 Signaling Complex in the Cardiovascular System
CaV1.2 controls our heart beat as well as the excitability of VSMCs, where it controls arterial diameter and thus blood pressure. Therefore, CaV1.2 has been established as a major drug target (60) in the control of cardiovascular diseases. Dihydropyridines preferentially bind to LTCCs under the depolarizing conditions that occur in VSMCs, particularly during vasospasm, and are particularly effective for the initial treatment of hypertension (61). Phenylalkylamines and benzothiazepines are use-dependent LTCC blockers. Accordingly, they inhibit cardiac LTCCs, especially during increased action potential frequencies, and are thus useful for treating arrhythmias (61). Hence, these general CaV1.2 inhibitors will likely have central and peripheral effects. To treat mental disorders, it would be important to selectively target brain-specific isoforms of CaV1.2 and largely spare CaV1.2 in the cardiovascular system. Importantly, the regulation of CaV1.2 by β ARs fundamentally differs between the brain and heart. As discussed above, the functional regulation of CaV1.2 by β ARs in neurons occurs through β2 AR and requires α1 1.2 S1928, whereas β1AR signaling regulates CaV1.2 function in the heart where α1 1.2 S1928 phosphorylation is dispensable (27, 28, 59, 62, 63). However, CaV1.2 associates with the β2 AR, Gs, AC, and PKA for localized cAMP signaling in not only the brain (7, 42) but also the heart (8, 58). During normal cardiac physiology the heart beat is controlled by the β1 AR, whereas under pathological conditions the β2 AR is upregulated and becomes more prominent in the regulation of CaV1.2 (62, 63). Thus, these mechanisms potentially constitute an additional pharmacological target site for therapeutics aimed at dampening β2 AR–CaV1.2 signaling.
Much like in neurons, macromolecular protein complexes play key roles in modulating the function of VSMCs and therefore have a major influence on vessel diameter. Control of VSMC excitability can be regulated by a myriad of signaling inputs, including protein kinases and phosphatases. The effects of these ubiquitous signaling molecules are often dependent on scaffold proteins that provide a platform for targeting and compartmentalization signaling events to specific substrates (1,2). In the next section, we describe the composition and physiological relevance of specific macromolecular complexes controlling VSMC excitability.
In VSMCs, CaV1.2, transient receptor potential (TRP) channels, and Ca2+- and voltage-gated potassium channels (BK and various KV channels) regulate cell excitability by controlling membrane potential (Em) and the magnitude of intracellular Ca2+ (64). These channels exist in complexes with signaling molecules, including PKA, protein kinase C (PKC), and cGMP-dependent protein kinase as well as protein phosphatases such as PP2B, which are organized by multivalent scaffold proteins such as AKAPs (64). AKAP5, which binds PKA, PKC, and PP2B, interacts with CaV1.2 (48, 65, 66) (Figure 2) and TRPV4 channels (67) in VSMCs. In these cells, AKAP5 may provide structural support to control the activity and location of CaV1.2 (68). Here, some CaV1.2 channels in VSMCs showed stochastic activity with low Ca2+ flux and event duration, whereas others showed persistent activity characterized by increased Ca2+ flux and events with a prolonged open time produced by the opening of two or more channels (68). The occurrence of persistently active CaV1.2 is restricted to discrete regions of the surface membrane and is highly dependent on the activity of PKC as well as the expression of AKAP5 (65, 68). Moreover, the activity of phosphatases such as AKAP5-associated PP2B counteracts anchored kinase activity and curbs persistent LTCC events (65). These results point to an important role for AKAP5-anchored PKC and PP2B activity in modulating basal persistent CaV1.2 events. The physiological significance of these findings stems from the fact that persistent CaV1.2 events account for 50% of the total dihydropyridine-sensitive (e.g., CaV1.2) Ca2+ influx at physiological Em (68), a process critical for VSMC contractility (Figure 2).
Figure 2.
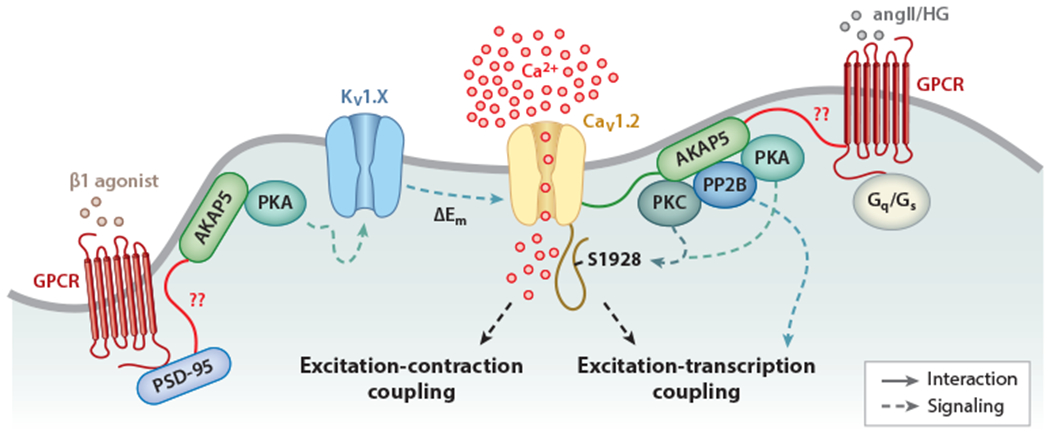
Proposed model for the regulation of vascular smooth muscle cell (VSMC) excitability by macromolecular complexes. The magnitude of Ca2+ influx via CaV1.2 is critical for the control of excitation-contraction and excitation-transcription coupling in these cells. Under physiological conditions, K+ channels oppose pressure-induced depolarization to limit CaV1.2 activity and VSMC contractility. The activity of K+ and CaV1.2 channels can be regulated by cAMP-dependent protein kinase (PKA), protein kinase C (PKC), and the protein phosphatase PP2B, which are targeted to the specific channels and G protein-coupled receptors (GPCRs) by AKAP5 and/or PSD-95, and their function may be altered during pathological conditions. Both PKA and PKC can phosphorylate CaV1.2 on S1928 (dashed blue lines) but are regulated by different GPCRs. PKA can also regulate K+ channels of the KV1 family. In turn, KV1 channels negatively control CaV1.2 activity. Whether the hypothesized interactions (solid red lines), including those involving the GPCRs that mediate angiotensin II (angII) and high glucose (HG) signaling, PSD-95, and AKAP5, occur in native VSMCs is unclear.
During the pathological condition of angiotensin II–induced hypertension, PKC activity and persistent CaV1.2 events were increased in an AKAP5-dependent manner (65). Indeed, AKAP5 knockout mice were hypotensive and did not develop angiotensin II–induced hypertension. An increase in persistent LTCC events was also observed in VSMCs during diabetes (69), a pathological condition that unexpectedly is linked to the activation of AKAP5-anchored PKA (48). The finding that PKA induces the LTCC activity that promotes VSMC contraction in response to diabetic hyperglycemia suggests the specific engagement of pools of PKA that could be in close proximity to CaV1.2 along with a GPCR mediating its activation. Indeed, the purinergic receptor P2Y11, the only known P2Y receptor coupled to Gs, is closely associated with subpopulations of CaV1.2 and PKA and can be activated by elevations in extracellular glucose to promote CaV1.2 activity and vasoconstriction (70). Whether AKAP5 interacts with a GPCR that could mediate the angiotensin II and/or glucose effects in VSMCs is unknown.
By linking PP2B within the same signaling complex as CaV1.2, AKAP5 orchestrates a signaling module for optimal activation of the phosphatase in VSMCs during hypertension and diabetes, signaling that ultimately results in the activation of the transcription factor NFATc3 (68) (Figure 2). Among the many genes that are altered by NFATc3 activation during hypertension and diabetes, downregulation of the BK β1 and KV2.1 subunits is a prominent feature, resulting in changes in channel activity that ultimately affect Em (66, 71, 72). Importantly, the AKAP150-mediated anchoring of PP2B seems essential for activation of NFATc3 and subsequent gene expression remodeling because the aforementioned changes in BK β1 and KV2.1 subunit expression are not observed in mice expressing an AKAP150 that cannot bind PP2B (65, 66, 72). Thus, the AKAP150-mediated complex may be a distinctly critical site of action for signal transduction in VSMCs (Figure 2) that could be exploited as a potential therapeutic target for treating vascular complications associated with several pathologies, including hypertension and diabetes.
TRPV4 channels have also been found complexed with AKAP150 in VSMCs (67, 73). The association between these two proteins is critical for regulation of the ion channel by Gq-PKC signaling (67). TRPV4, AKAP150, and PKC could form a macromolecular signaling complex to regulate Ca2+ signaling. Intriguingly, optimal AKAP150-anchored PKC modulation of TRPV4 activity is highly dependent on the distance between the targeted kinase and the ion channel, with a suggested distance between them of ~200 nm (73). Whether AKAP150-anchored PKA and PP2B also regulate TRPV4 channels in VSMCs and the relevance of the AKAP150/PKC/TRPV4 complex in VSMCs during pathological conditions remain to be determined.
Although β adrenergic stimulation of PKA promotes K+ channel activity and VSMC relaxation (64) (Figure 2), the involvement of an intermediary that could link all components of the signaling complex has been unclear. Recently, the scaffold protein postsynaptic density 95 (PSD-95), which was thought to be a neuron-specific protein, was observed in VSMCs (74). Functionally, PSD-95 was necessary for the basal and isoproterenol-induced, PKA-mediated activation of KV1.x channels that promotes arterial smooth muscle relaxation (74, 75). This was due to the formation of a distinctive PSD-95-mediated signaling complex involving the β2 AR, PKA, and specifically KV1.2 channels. Although PSD-95 is associated with AKAP150 in neurons (76), whether a PSD-95-AKAP150 complex is involved in β adrenergic regulation of VSMC excitability is unknown and therefore an area for further investigation.
3.3. The β2 Adrenergic Receptor–AMPAR Signaling Complex
More than 80% of the synapses in the cortex use glutamate for fast neurotransmission. AMPARs mediate most of the basal postsynaptic response upon presynaptic glutamate release. They are potential pharmacological targets for the treatment of diseases that involve dysregulation of glutamatergic synapses, including the overexcitation that occurs during epilepsy (77) and the neuronal damage caused by ischemic conditions, nerve damage, and other insults (78–81). Like CaV1.2, AMPARs form signaling complexes with β2 ARs that also contain Gs, AC, and PKA. Accordingly, those AMPAR complexes could be additional pharmacological targets to treat disorders associated with the dysregulation of NE–β2 AR signaling such as ADHD and PTSD. Furthermore, the β2 AR has been identified as a target for the β amyloid peptide 1–42 (βAP1–42) (10), which is thought to be the main pathogen in Alzheimer’s disease (82, 83). It is tempting to speculate that βAP1–42 acts in part by dysregulating AMPAR function by the associated β2 AR, which could lead to a loss of synaptic strength and plasticity (84). Moreover, as evidence indicates that amyloid β (Aβ) oligomers bind near GluA2-containing complexes and AMPAR antagonists can inhibit Aβ oligomer binding and synaptic loss, it is quite plausible that Aβ affects AMPAR trafficking by binding directly to the GluA2 protein complex (85). Finally, the antidepressant and cognitive enhancer tianeptine augments synaptic AMPAR function and antagonizes impairment of synaptic function following stress (86), suggesting that AMPARs could be pharmacological targets for the treatment of depression and anxiety-related disorders, including PTSD.
PSD-95 is the central organizer of glutamatergic postsynaptic sites, where it anchors AMPARs by binding with its first two PDZ domains to the cytosolic C termini of auxiliary AMPAR subunits called TARPs (transmembrane AMPAR regulatory proteins) (87) (Figure 3). PSD-95 links the β2 AR to AMPARs by binding with its third PDZ domain to the C terminus of the β2 AR (9, 10); it also binds AKAP5, but it is unclear whether PSD-95 helps to recruit AC and PKA to AMPARs (76). Rather, the PSD-95 homolog synapse-associated protein 97 (SAP97) binds to the C terminus of the AMPAR GluA1 subunit (88) and is required to recruit PKA and PP2B to AMPARs, as demonstrated in HEK293 cells (76, 89). AKAP5 also links ACs to AMPARs (45, 46, 90) and binds to the SH3-GK module of PSD-95 and SAP97 (76), but the specific interaction sites have not been defined.
Figure 3.
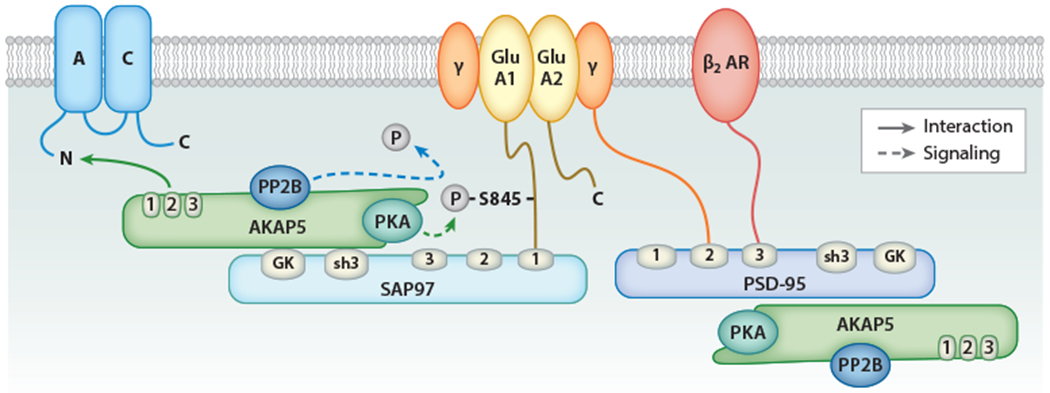
The β2 AR–Gs-AC-PKA-GluA1 complex. PSD-95 is a highly prevalent and central structural protein of excitatory glutamatergic synapses. AMPARs are linked to PSD-95 via the binding of their auxiliary TARP (γ) subunits to the first two PDZ domains of PSD-95, whereas the β2 AR binds to the third PDZ domain of PSD-95. Both PSD-95 and its homolog, SAP97, bind to AKAP5 via their SH3-GK modules. SAP97 binds to the C terminus of the GluA1 subunit of AMPAR, recruiting PKA and PP2B to the vicinity of AMPAR. Through the SAP97-AKAP5 interaction, AC is also localized close to GluA1. Stimulation of β2 AR induces cAMP increase and PKA activation, increasing S845 phosphorylation on GluA1 and, consequently, AMPAR activity. Abbreviations: AC, adenylyl cyclase; AKAP5, A-kinase anchor protein 5; AMPAR, AMPA-type glutamate receptor; AR, adrenergic receptor; PKA, cAMP-dependent protein kinase; PP, protein phosphatase; PSD-95, postsynaptic density 95; SAP97, synapse-associated protein 97; TARP, transmembrane AMPAR regulatory protein.
PTT-LTP depends not only on the activation of the β2 AR and CaV1.2 but also on AKAP5-mediated PKA anchoring and phosphorylation of the AMPAR subunit on S845 (59, 90). S845 phosphorylation augments the channel activity of AMPARs (91), amplifying depolarization at postsynaptic sites. β2 ARs, AMPARs, and CaV1.2 are colocalized at postsynaptic sites of glutamatergic neurons (7, 9). There, they likely form a functional unit such that, upon presynaptic glutamate release, the local AMPAR-driven depolarization can activate CaV1.2 (28) (Figure 4).
Figure 4.
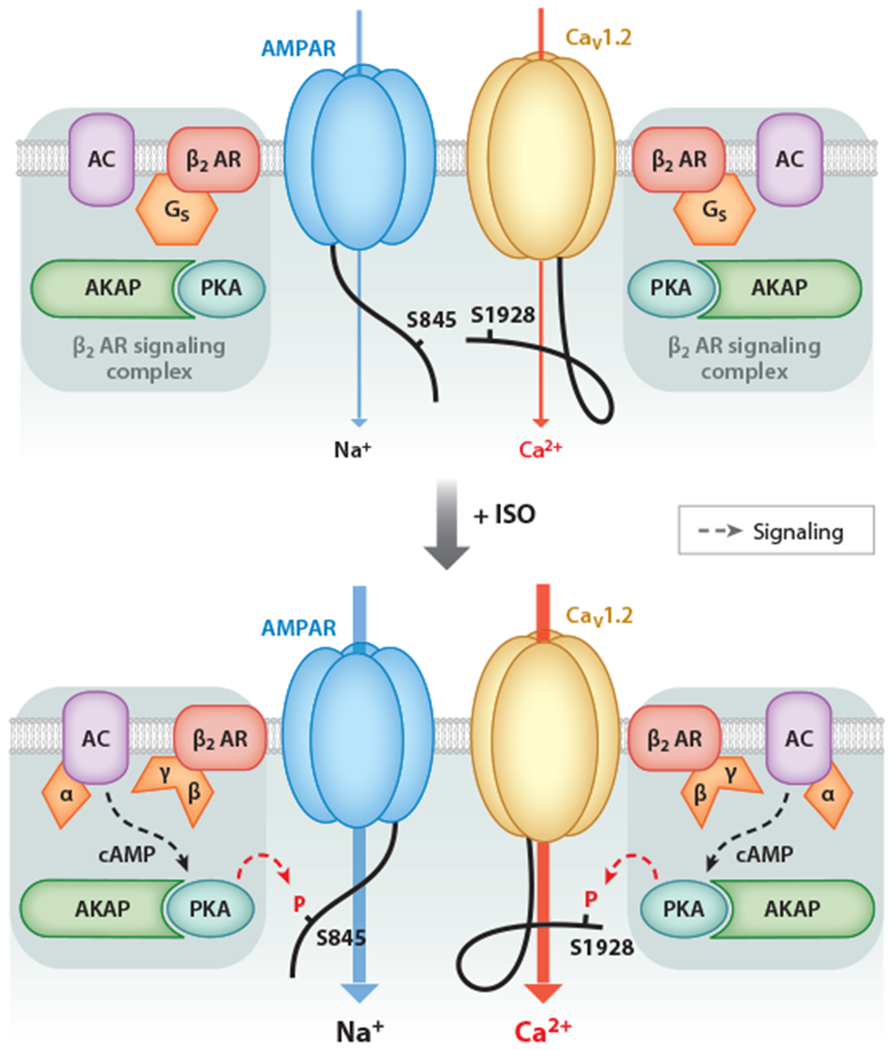
The β2 adrenergic receptor (β2 AR)–CaV1.2 and β2 AR–GluA1 signaling complexes participate in prolonged theta tetanus long-term potentiation (PTT-LTP). β2 AR activation is required in the induction of PTT-LTP. Stimulation of β2 AR augments CaV1.2 and AMPA-type glutamate receptor (AMPAR) channel activity via phosphorylation of CaV1.2 on S1928 and GluA1 on S845, respectively, by A-kinase anchor protein (AKAP)-anchored cAMP-dependent protein kinase (PKA). The upregulation in AMPAR activity increases depolarization upon synaptic transmission and thereby increases CaV1.2 activation, in addition to the increased open probability of CaV1.2 due to S1928 phosphorylation. Phosphorylation of both S845 and S1928 is required for the induction of PTT-LTP. Adapted from Reference 28 with permission from AAAS.
4. PHARMACOLOGICAL TARGETING OF β2 ADRENERGIC RECEPTOR PROTEIN-PROTEIN INTERACTIONS
4.1. Cell-Penetrating Peptides as Biologics
The use of biologically active peptides targeting protein-protein interactions has greatly expanded into a broad range of therapeutic areas (11, 92–94). Importantly, the potential for small organic compounds to disrupt protein-protein interactions is generally thought to be low because the interaction surfaces between proteins are typically much larger than their interaction surfaces with small drugs (11, 92, 93). Peptides have larger interaction surfaces and can acutely disrupt protein interactions in a highly effective and specific manner. However, the hydrophilic nature of many peptides results in low membrane permeability and prevents their access to intracellular targets, which would, without additional modifications, limit their therapeutic value. In the specific case of using peptides for the treatment of brain disorders, delivery of the hydrophilic peptides across the blood–brain barrier (BBB) must be achieved.
The HIV-1 protein transactivator of transcription (TAT) and the Drosophila melanogaster Antennapedia protein (Ant) were found to efficiently cross the plasma membrane (94–97). The translocation properties of Ant were narrowed down to a short, 16-amino-acid peptide corresponding to the third helix of the Antennapedia homeodomain, called pAntp or Penetratin (98). In the case of TAT, a minimal 9-residue-long basic sequence (residues 49–57) was found to mediate its cellular uptake (99). Many short cell-penetrating peptides (CPPs) varying from 5 to 30 residues in length have been identified (92, 93).
CPPs can pass through cell membranes via energy-dependent and energy-independent mechanisms but do not seem to require specific receptors (92). Passive, energy-independent entry of peptides into cells occurs by transient membrane disruption or spontaneous translocation. Energy-dependent internalization of a CPP occurs via endocytosis. CPPs can essentially be categorized into three main classes: mostly cationic (e.g., TAT- and pAntp-derived), amphipathic [with alternating regions of hydrophilic (here, cationic lysine and arginine residues) and hydrophobic residues (valine, leucine, isoleucine, and alanine)], and mostly hydrophobic (92).
TAT peptide conjugates have been found to disrupt protein binding in many systems (100). Disruption of postsynaptic interactions is accomplished in cultured hippocampal neurons (101), acute brain slices (102), and in vivo (103). Even full-length recombinant proteins are typically membrane-permeant when carrying the TAT sequence (94, 100, 104). Many TAT fusion proteins have been expressed in Escherichia coli, purified and successfully used to disrupt protein-protein interactions in mammalian cells (94, 104, 105). TAT proteins can also penetrate various tissues, including an intact BBB (94,105, 106). An alternative to the actual TAT sequence is a stretch of 11 arginine residues, which is as effective in rendering peptides membrane-permeant as TAT itself (9, 107, 108). In addition, the attachment of lipid moieties, e.g., myristoylation (27, 109) or stearylation (92), can also facilitate the penetration of macromolecules into cells. In fact, myristoylation of an already TAT-tagged peptide makes the peptide more membrane-permeant (110).
Numerous biochemical entities have been developed for targeting protein-protein interactions. Below, we describe in detail the paradigmatic pepducins before discussing the nature and potential use of nanobodies (Nbs)/intrabodies (Ibs) and nucleic acid aptamers. Finally, we discuss the use of peptides conjugated to the TAT and related poly-arginine segments that can disrupt β2 AR complexes with CaV1.2 and AMPARs.
4.2. GPCR Signaling and Biased Agonism: Paradigmatic Targets for Biologics Affecting Protein-Protein Interactions
The intracellular regions of GPCRs have been identified as new target sites for drugs that modify interactions between downstream effectors and modulators such as Gs, GPCR kinases (GRKs), and arrestins. Recent examples include peptides (pepducins), cellularly expressed antibody derivatives (Ibs, Nbs), and RNA- or DNA-based aptamers. Ligand binding to GPCRs can activate signaling that depends on G proteins and arrestins (111). Agonist binding to β2 AR causes a conformational change that promotes the exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) on Gαs within the heterotrimeric stimulatory Gs protein, causing Gαs to dissociate from the obligate heterodimeric Gβγ subunit. Gαs activates ACs, and Gβγ modulates effectors such as the K+ channel Kir3, phospholipase C, AC, and voltage-gated Ca2+ channels via direct interaction with these proteins (112).
Gs-independent signaling involves arrestins and GRKs (113, 114). Upon GPCR activation, the PH domain of GRKs binds to isoprenylated, membrane-anchored Gβγ, leading to GRK translocation to the plasma membrane (115). GRKs phosphorylate serine/threonine residues on the GPCR, creating binding sites for arrestins (116). Arrestin binding causes homologous desensitization of the activated GPCR by sterically hindering Gαs binding (117), whereas heterologous desensitization of other GPCRs occurs through inhibition of Gαs coupling by PKA and PKC (113). β-Arrestin-2 recruits phosphodiesterase 4D to phosphorylated β2 AR to dampen the increase in cAMP levels induced by the restimulation of β2 AR (118). In fact, heterologous phosphorylation allows for the occurrence of β-arrestin-dependent effects in the absence of homologous receptor activation (119). β-Arrestins also function as adapters for clathrin-mediated endocytosis, with clathrin, AP-2, ARF-6, and NSF facilitating GPCR internalization and recycling and trafficking of the internalized GPCR (113, 114). Finally, arrestins recruit MAP kinases (120, 121) and Src (122). Binding of Src to arrestin upon β2 AR stimulation activates Src, which in turn stimulates activation of ERK1/2 (122). In addition, β-arrestins can transactivate EGFR in a GRK5- and GRK6-dependent manner, leading to ERK activation (123). Arrestins also serve as adapters for a complex consisting of JNK1/2 and upstream kinases MKK4 and MKK7 to facilitate the phosphorylation of JNKs (124).
Ligands can bias GPCR signaling to either Gαs or arrestin (113). GPCRs exist in their active and inactive forms in multiple dynamically interconverting conformations (125). The conformational differences induced by certain ligands are propagated to transducers and regulatory proteins to impart differing signaling consequences (126–130). It has been suggested that GRKs recognize the different conformations of ligand-receptor complexes and endow specific phosphorylation patterns onto GPCRs, leading to distinct conformations of transducers and regulatory proteins through these apparent phosphorylation barcodes (131, 132). The phenomenon of biased signaling has therapeutic value because defined GPCR ligands can be designed to tweak signaling to a desired pathway by inducing a specific GPCR conformation that yields the desired pharmacological effect and minimizes unwanted side effects.
4.2.1. Pepducins.
Pepducins are short, lipidated (e.g., palmitoyl- or myristoyl-conjugated) CPPs with sequences derived from intracellular loops (ICLs) of GPCRs. They can act as agonists, antagonists, or biased agonists of the cognate GPCR. A systematic analysis identified four classes of pepducins targeting β2 AR signaling with differential signaling properties: (a) partial agonists promoting both Gαs signaling and β-arrestin recruitment; (b) β-arrestin-biased pepducins, mainly derived from ICL1; (c) receptor-dependent Gαs-biased pepducins (e.g., pepducin ICL3–9); and (d) receptor-independent, Gαs-biased pepducins (e.g., pepducin ICL3–8) (133). ICL3–8 and ICL3–9 are derived, respectively, from the proximal and central portions of the third ICL of the β2 AR and do not promote β-arrestin recruitment and β2 AR internalization. ICL3–8 is receptor-independent, i.e., it increases cAMP production by binding to and directly activating G«s. ICL3–9 stimulates cAMP production by stimulating Gαs recruitment to β2 AR. ICL3–9 induces a β2 AR conformation different from that induced by the full β2 AR agonist isoproterenol but allows interaction with Gαs, albeit with a binding mode distinct from that stimulated by isoproterenol.
The β-arrestin-biased pepducin ICL1–9 increases cardiomyocyte contractility (134). The efficacy of β-arrestin recruitment to the β2 AR by ICL1–9 is about 50%, relative to isoproterenol, but it does not induce cAMP production or bind to the orthosteric site. Although ICL1–9 triggers less GRK-dependent β2 AR phosphorylation and receptor internalization than isoproterenol, it induces more prolonged ERK activation with higher efficacy than isoproterenol and with faster kinetics than the β-arrestin-biased orthosteric agonist carvedilol (134). By stabilizing a β2 AR conformation that is favorable for β-arrestin binding, ICL1–9 increases contractility in murine cardiomyocytes in an unconventional cAMP-dependent manner that does not require Ca2+ mobilization (134). The specificity or promiscuity of signaling modulation by pepducins depends on their mechanism of action. For example, ICL3–8 functions by directly activating Gαs and increasing cAMP production in conjunction with several GsPCRs (133). On the other hand, ICL3–9 functions by inducing a distinct β2 AR conformation, which stimulates Gαs in conjunction with β2 AR and the closely related β1AR but not the prostaglandin E2 receptor PGE2R (133).
4.2.2. Nanobodies and intrabodies.
Camelids produce antibodies lacking light chains. The variable region of these heavy-chain antibodies (VHH domain) can be recombinantly expressed. The ~15-kDa products are Nbs (135), which can be expressed exogenously in mammalian or insect cells as Ibs. Different Nbs were developed that specifically bind to the intracellular domains of active (agonist-bound) or inactive (antagonist-bound) β2 AR, stabilizing the corresponding conformations (136). When coexpressed with β2 AR in HEK293 cells as Ibs, Nbs specific to inactive β2 AR inhibit cAMP production as well as β-arrestin recruitment and reduce β2 AR expression. Several active conformation-stabilizing Ibs also inhibit cAMP production and β-arrestin recruitment by stabilizing a conformation that is not conducive to the binding of Gαs and GRKs, the steric hinderance of β-arrestin access to the β2 AR, and enhanced β2 AR–Gi coupling by active conformation-stabilizing Ibs.
Antibody antigen-binding fragments (Fabs) can be converted to single-chain antibodies (135) and expressed as Ibs. Fabs that bind to the active conformation of β-arrestin coupled to the vasopressin receptor modulate interactions of β-arrestin with ERK and clathrin with variable and biased effects (137). One of them, Fab9, augments the binding of β-arrestin to ERK but not to clathrin (137), demonstrating that Fabs can exert an overall stimulatory effect. Fab5/Ib5 inhibits clathrin-mediated receptor internalization without affecting ERK (137). Steric hinderance of clathrin binding to β-arrestin and allosteric modulation of β-arrestin could mediate this effect. However, Ib5 nonselectively inhibits β-arrestin-mediated endocytosis of a wide range of GPCRs, including the β2 AR; muscarinic M2 receptor; dopaminergic D1, 2, 3, and 4; and μ-opioid receptor (137).
4.2.3. Aptamers.
Aptamers are DNA or RNA strands exhibiting secondary structures that facilitate their binding to target molecules with high specificity and affinity. Aptamers are typically identified through an iterative screening process called SELEX (systematic evolution of ligands by exponential enrichment) (138, 139). Nucleic acids can be readily subjected to chemical modification (140), including myristoylation, to render them membrane-permeant (141). As aptamers that specifically bind to the active β2 AR conformation inhibit isoproterenol-stimulated cAMP production in β2 AR–expressing HEK293 cells and inactive conformation-specific aptamers have no effect, it is thought that active conformation-specific aptamers act by blocking the access of Gαs (142). The effect of the stabilized active conformation on other arms of the signaling cascade and the functional consequences of this remain to be established.
Pepducins, intrabodies, and aptamers intercepting β2 AR signaling may find their therapeutic use in the treatment of a broad range of diseases, including congestive heart failure (CHF), hypertension, asthma, chronic obstructive pulmonary disorder, and ASD. They are predicted to exhibit fewer side effects compared to the β-blockers currently in clinical use. For example, although β1AR is an important therapeutic target in CHF, β2 AR may also contribute to cardiac pathology due to the upregulation of heterotrimeric Gi, to which the β2 AR is also coupled (143, 144). Conventional β2 AR blockade inhibits Gi to restore inotropy, but one undesirable consequence is that this also inhibits the prosurvival effects mediated by Gβγ (145). Specific targeting of β-arrestin signaling, as demonstrated for ICL1–9, is likely to stimulate both contractility and cell survival and may prove to be a promising therapeutic approach in the future. Nevertheless, as β-arrestin signaling activates diverse pathways in different cell types, the signaling consequences of its targeting must be examined in each case.
4.3. Pharmacological Targeting of the Interactions of the β2 Adrenergic Receptor and Phosphatases with CaV1.2 and AMPA-Type Glutamate Receptors
The β2 AR–Gs-AC-PKA-CaV1.2 and β2 AR-Gs-AC-PKA-GluA1 complexes are so far the only known complexes that assemble a GsPCR (i.e., β2 AR) with all intermediaries (Gs, AC, PKA) and the final target (CaV1.2, GluA1). Nearly all interactions have been mapped out except for Gs association (1, 7–9, 27, 28, 42–44, 90, 146) (Figures 1 and 3). However, most interactions are not unique to the CaV1.2 or AMPAR complex. For instance, AKAP5 links PKA to a number of different proteins (1, 2). Accordingly, using peptides that displace either AC or PKA from AKAP5 will affect various signaling cascades. Displacing AKAP5 from PSD-95 or SAP97 could potentially provide a more selective effect; however, their interactions are currently not defined in sufficient detail for designing peptides that would accomplish such displacements. Rather, targeting the direct binding of the β2 AR to residues 1923–1942 in the α1 1.2 C terminus has the potential to be quite selective, as no analogous interaction is currently known (27). In fact, a membrane-permeant myristoylated peptide derived from residues 1923–1942 in α1 1.2 displaces the β2 AR from α1 1.2 and not from the AMPAR complex when applied to acute forebrain slices (27). Consistently, it only affects β2 AR–triggered phosphorylation of α1 1.2 but not the AMPAR GluA1 subunit at the respective PKA site in the slices. The converse is true for a peptide that displaces the β2 AR from the AMPAR complex and is alternatively either 11-Arg-conjugated (9, 27) or myristoylated (B. Lee & J.W. Hell, unpublished data). However, this peptide might affect some other function related to PSD-95 because it interferes with the binding of β2 AR with the third PDZ domain of PSD-95, its link to the AMPAR, and this PDZ domain mediates binding of other proteins as well (9). Still, an 11-Arg-conjugated version of this peptide effectively prevents upregulation of GluA1 phosphorylation and its otherwise consequent surface accumulation in hippocampal cultures (9) and spike-time-dependent plasticity in acute cortical slices (14).
CaV1.2 also has different phosphatases linked to it. Remarkably, PP2B directly interacts with residues 1943–1971 immediately downstream of the PKA phosphorylation site S1928 and of the β2 AR binding site in the C terminus of α1 1.2 (55) (Figure 1). This PP2B does not dephosphorylate S1928 but rather augments CaV1.2 activity, possibly by dephosphorylating a hypothetical inhibitory phosphorylation site. A second PP2B attachment site is provided by AKAP5, which is important for Ca2+-dependent inactivation of CaV1.2 (44, 57). Finally, the PP2A catalytic C subunit, rather than one of its targeting B-type subunits, directly binds immediately downstream of PP2B to residues 1965–1971 (52–54). PP2A dephosphorylates S1928, and displacing PP2A with a corresponding peptide augments channel activity, consistent with a role of PP2A in counteracting cAMP-mediated upregulation of CaV1.2 (54). Although phosphatase targeting to CaV1.2 is complex, some interactions such as the direct binding of the PP2A C subunit to α1 1.2 have the potential to be unique and thus constitute prospective drug targets for controlling CaV1.2 activity.
5. CONCLUSION AND PERSPECTIVE
Biologics are rapidly emerging as promising therapeutics under development and in the clinic. Membrane-permeant peptides have vast pharmacological potential given their ability to specifically and selectively target physiologically relevant protein-protein interactions among the multitude of protein signaling complexes in the cellular milieu. We envision the development of a number of peptides that can precisely target and disrupt protein-protein interactions in defined complexes that modulate the function of CaV1.2 and glutamate receptors in specific subcellular compartments. As the target ion channels serve widespread and multiple functions, such peptides possibly could exert their effects at a quasi-microsurgery molecular level and thus limit the off-target side effects elicited by many of the existing small-molecule drugs in the clinic.
ACKNOWLEDGMENTS
Research by the authors was funded by NIH grants R01HL098200 and R01HL121059 (to M.F.N.) and R01NS078792, R01MH097887, and R01AG055357 (to J.W.H.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Dai S, Hall DD, Hell JW. 2009. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol. Rev 89:411–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langeberg LK, Scott JD. 2015. Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol 16:232–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaturvedi M, Schilling J, Beautrait A, Bouvier M, Benovic JL, Shukla AK. 2018. Emerging paradigm of intracellular targeting of G protein-coupled receptors. Trends Biochem. Sci 43:533–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinberg SF, Brunton LL. 2001. Compartmentation of G protein-coupled signaling pathways in cardiac myocytes. Annu. Rev. Pharmacol. Toxicol 41:751–73 [DOI] [PubMed] [Google Scholar]
- 5.Patriarchi T, Buonarati OR, Hell JW. 2018. Postsynaptic localization and regulation of AMPA receptors and Cav1.2 by β2 adrenergic receptor/PKA and Ca2+/CaMKII signaling. EMBO J. 37:e99771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao RP, Cheng H, Zhou YY, Kuschel M, Lakatta EG. 1999. Recent advances in cardiac β2-adrenergic signal transduction. Circ. Res 85:1092–100 [DOI] [PubMed] [Google Scholar]
- 7.Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, et al. 2001. A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293:98–101. Erratum. 2001. Science 293(5531):804 [DOI] [PubMed] [Google Scholar]
- 8.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. 2006. Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. PNAS 103:7500–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joiner ML, Lise MF, Yuen EY, Kam AY, Zhang M, et al. 2010. Assembly of a β2-adrenergic receptor-GluR1 signalling complex for localized cAMP signalling. EMBO J. 29:482–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D, Govindaiah G, Liu R, De Arcangelis V, Cox CL, Xiang YK. 2010. Binding of amyloid β peptide to β2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. Faseb J. 24:3511–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wulff H, Christophersen P, Colussi P, Chandy KG, Yarov-Yarovoy V. 2019. Antibodies and venom peptides: new modalities for ion channels. Nat. Rev. Drug Discov 18:339–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berman DE, Dudai Y. 2001. Memory extinction, learning anew, and learning the new: dissociations in the molecular machinery of learning in cortex. Science 291:2417–19 [DOI] [PubMed] [Google Scholar]
- 13.Cahill L, Prins B, Weber M, McGaugh JL. 1994. β-Adrenergic activation and memory for emotional events. Nature 371:702–4 [DOI] [PubMed] [Google Scholar]
- 14.He K, Huertas M, Hong SZ, Tie X, Hell JW, et al. 2015. Distinct eligibility traces for LTP and LTD in cortical synapses. Neuron 88:528–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brookes K, Xu X, Chen W, Zhou K, Neale B,et al. 2006. The analysis of 51 genes in DSM-IV combined type attention deficit hyperactivity disorder: association signals in DRD4, DAT1 and 16 other genes. Mol. Psychiatry 11:934–53 [DOI] [PubMed] [Google Scholar]
- 16.Lasky-Su J, Neale BM, Franke B, Anney RJ, Zhou K, et al. 2008. Genome-wide association scan of quantitative traits for attention deficit hyperactivity disorder identifies novel associations and confirms candidate gene associations. Am. J. Med. Genet. B Neuropsychiatr. Genet 147B:1345–54 [DOI] [PubMed] [Google Scholar]
- 17.Liberzon I, King AP, Ressler KJ, Almli LM, Zhang P,et al. 2014. Interaction of the ADRB2 gene polymorphism with childhood trauma in predicting adult symptoms of posttraumatic stress disorder. JAMA Psychiatry 71:1174–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Dell TJ, Connor SA, Guglietta R, Nguyen PV. 2015. β-Adrenergic receptor signaling and modulation of long-term potentiation in the mammalian hippocampus. Learn. Mem 22:461–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Pink MD, Murphy JG, Stein A, Dell’Acqua ML, Hogan PG. 2012. Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat. Struct. Mol. Biol 19:337–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, et al. 2012. Cav1 and Cav2 channels engage distinct modes of Ca2+ signaling to control CREB-dependent gene expression. Cell 149:1112–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marrion NV,Tavalin ST. 1998. Selective activation of Ca2+-activated K+ channels byco-localized Ca2+ channels in hippocampal neurons. Nature 395:900–5 [DOI] [PubMed] [Google Scholar]
- 22.Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, et al. 2006. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 314:615–20 [DOI] [PubMed] [Google Scholar]
- 23.Hell JW,Westenbroek RE,Warner C,Ahlijanian MK, Prystay W, et al. 1993. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. J. Cell Biol 123:949–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, et al. 2004. Isoform-specific regulation of mood behavior and pancreatic β cell and cardiovascular function by L-type Ca2+ channels. J. Clin. Investig 113:1430–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, et al. 2005. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J. Neurosci 25:9883–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolshakov VY, Siegelbaum SA. 1994. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science 264:148–52 [DOI] [PubMed] [Google Scholar]
- 27.Patriarchi T, Qian H, Di Biase V, Malik ZA, Chowdhury D, et al. 2016. Phosphorylation of Cav1.2 on S1928 uncouples the L-type Ca2+ channel from the β2 adrenergic receptor. EMBO J. 35:1330–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian H, Patriarchi T, Price JL, Matt L, Lee B, et al. 2017. Phosphorylation of Ser1928 mediates the enhanced activity of the L-type Ca2+ channel Cav1.2 by the β2-adrenergic receptor in neurons. Sci. Signal. 10:eaaf9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu H, Real E, Takamiya K, Kang MG, Ledoux J, et al. 2007. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell 131:160–73 [DOI] [PubMed] [Google Scholar]
- 30.White JA, McKinney BC, John MC, Powers PA, Kamp TJ, Murphy GG. 2008. Conditional forebrain deletion of the L-type calcium channel Cav1.2 disrupts remote spatial memories in mice. Learn. Mem 15:1–5 [DOI] [PubMed] [Google Scholar]
- 31.Davare MA, Hell JW 2003. Increased phosphorylation of the neuronal L-type Ca2+ channel Cav1.2 during aging. PNAS 100:16018–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deyo RA, Straube KT, Disterhoft JF. 1989. Nimodipine facilitates associative learning in aging rabbits. Science 243:809–11 [DOI] [PubMed] [Google Scholar]
- 33.Nunez-Santana FL, Oh MM, Antion MD, Lee A, Hell JW, Disterhoft JF. 2014. Surface L-type Ca2+ channel expression levels are increased in aged hippocampus. Aging Cell 13:111–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kisko TM, Braun MD, Michels S, Witt SH, Rietschel M, et al. 2018. Cacna1c haploinsufficiency leads to pro-social 50-kHz ultrasonic communication deficits in rats. Dis. Model. Mech 11:dmm034116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, et al. 2004. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119:19–31 [DOI] [PubMed] [Google Scholar]
- 36.Ferreira MA, O’Donovan MC, Meng YA,Jones IR, Ruderfer DM, et al. 2008. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet 40:1056–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nyegaard M, Demontis D, Foldager L, Hedemand A, Flint TJ, et al. 2010. CACNA1C (rs1006737) is associated with schizophrenia. Mol. Psychiatry 15:119–21 [DOI] [PubMed] [Google Scholar]
- 38.Green EK, Grozeva D, Jones I, Jones L, Kirov G, et al. 2010. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol. Psychiatry 15:1016–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, et al. 2012. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog. Neurobiol 99:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smoller JW. 2013. Disorders and borders: psychiatric genetics and nosology. Am. J. Med. Genet. B Neuropsychiatr. Genet 162B:559–78 [DOI] [PubMed] [Google Scholar]
- 41.Song JHT, Lowe CB, Kingsley DM. 2018. Characterization of a human-specific tandem repeat associated with bipolar disorder and schizophrenia. Am. J. Hum. Genet 103:421–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davare MA, Dong F, Rubin CS, Hell JW. 1999. The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J. Biol. Chem 274:30280–87 [DOI] [PubMed] [Google Scholar]
- 43.Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, et al. 2007. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry 46:1635–46 [DOI] [PubMed] [Google Scholar]
- 44.Oliveria SF,Dell’Acqua ML,Sather WA. 2007AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55:261–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willoughby D, Masada N, Wachten S, Pagano M, Halls ML, et al. 2010. AKAP79/150 interacts with AC8 and regulates Ca2+-dependent cAMP synthesis in pancreatic and neuronal systems. J. Biol. Chem 285:20328–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, et al. 2010. AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and −6 to a-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J. Biol. Chem 285:14450–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeJongh KS,Murphy BJ,Colvin AA,Hell JW,Takahashi M, Catterall WA. 1996. Specific phosphorylation of a site in the full length form of the a1 subunit of the cardiac L-type calcium channel by adenosine 3/,5/-cyclic monophosphate-dependent protein kinase. Biochemistry 35:10392–402 [DOI] [PubMed] [Google Scholar]
- 48.Nystoriak MA, Nieves-Cintron M, Patriarchi T, Buonarati OR, Prada MP, et al. 2017. Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel Cav1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci. Signal. 10:eaaf9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, et al. 2008. Unchanged β-adrenergic stimulation of cardiac L-type calcium channels in Cav1.2 phosphorylation site S1928A mutant mice. J. Biol. Chem 283:34738–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen A, Nieves-Cintron M, Deng Y, Shi Q, Chowdhury D, et al. 2018. Functionally distinct and selectively phosphorylated GPCR subpopulations co-exist in a single cell. Nat. Commun 9:1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dupre DJ, Robitaille M, Ethier N, Villeneuve LR, Mamarbachi AM, Hebert TE. 2006. Seven transmembrane receptor core signaling complexes are assembled prior to plasma membrane trafficking. J. Biol. Chem 281:34561–73 [DOI] [PubMed] [Google Scholar]
- 52.Davare MA, Horne MC, Hell JW. 2000. Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J. Biol. Chem 275:39710–17 [DOI] [PubMed] [Google Scholar]
- 53.Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, et al. 2006. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry 45:3448–59 [DOI] [PubMed] [Google Scholar]
- 54.Xu H, Ginsburg KS, Hall DD, Zimmermann M, Stein IS, et al. 2010. Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel Cav1.2. Biochemistry 49:10298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tandan S, Wang Y, Wang TT,Jiang N, Hall DD, et al. 2009. Physical and functional interaction between calcineurin and the cardiac L-type Ca2+ channel. Circ. Res 105:51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dittmer PJ, Dell’Acqua ML, Sather WA. 2014. Ca2+/calcineurin-dependent inactivation of neuronal L-type Ca2+ channels requires priming by AKAP-anchored protein kinase A. Cell Rep. 7:1410–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oliveria SF, Dittmer PJ, Youn DH, Dell’Acqua ML, Sather WA. 2012. Localized calcineurin confers Ca2+-dependent inactivation on neuronal L-type Ca2+ channels. J. Neurosci 32:15328–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen-Izu Y, Xiao R-P, Izu LT, Cheng H, Kuschel M, et al. 2000. Gi-dependent localization of β2-adrenergic receptor signaling to L-type Ca2+ channels. Biophys. J 79:2547–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qian H, Matt L, Zhang M, Nguyen M, Patriarchi T,et al. 2012. β2-Adrenergic receptor supports prolonged theta tetanus-induced LTP. J. Neurophysiol 107:2703–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zamponi GW, Striessnig J, Koschak A, Dolphin AC. 2015. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev 67:821–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Striessnig J 2008. Ca2+ channel blockers In Encyclopedia of Molecular Pharmacology, ed. Offermanns S, Rosenthal W, pp. 295–300. Berlin: Springer-Verlag [Google Scholar]
- 62.Steinberg SF. 1999. The molecular basis for distinct β-adrenergic receptor subtype actions in cardiomyocytes. Circ. Res 85:1101–11 [DOI] [PubMed] [Google Scholar]
- 63.Kamp TJ, Hell JW. 2000. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res 87:1095–102 [DOI] [PubMed] [Google Scholar]
- 64.Tykocki NR, Boerman EM,Jackson WF. 2017. Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Compr. Physiol 7:485–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Navedo MF, Nieves-Cintron M, Amberg GC, Yuan C, Votaw VS, et al. 2008. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ. Res 102:e20–35 [DOI] [PubMed] [Google Scholar]
- 66.Nystoriak MA,Nieves-Cintron M,Nygren PJ,Hinke SA,Nichols CB,et al. 2014AKAP150 contributes to enhanced vascular tone by facilitating large-conductance Ca2+-activated K+ channel remodeling in hyperglycemia and diabetes mellitus. Circ. Res 214:607–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mercado J, Baylie R, Navedo MF, Yuan C, Scott JD, et al. 2014. Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle. J. Gen. Physiol 143:559–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Navedo MF, Amberg GC. 2013. Local regulation of L-type Ca2+ channel sparklets in arterial smooth muscle. Microcirculation 20:290–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Navedo MF, Takeda Y, Nieves-Cintron M, Molkentin JD, Santana LF. 2010. Elevated Ca2+ sparklet activity during acute hyperglycemia and diabetes in cerebral arterial smooth muscle cells. Am. J. Physiol. Cell Physiol 298:C211–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prada MP, Syed AU, Buonarati OR, Reddy GR, Nystoriak MA, et al. 2019. A GS-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. eLife 8:e42214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nieves-Cintron M, Amberg GC, Nichols CB, Molkentin JD, Santana LF. 2007. Activation of NFATc3 down-regulates the β1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension. J. Biol. Chem 282:3231–40 [DOI] [PubMed] [Google Scholar]
- 72.Nieves-Cintron M, Syed AU, Nystoriak MA, Navedo MF. 2018. Regulation of voltage-gated potassium channels in vascular smooth muscle during hypertension and metabolic disorders. Microcirculation 25:e12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tajada S, Moreno CM, O’Dwyer S, Woods S, Sato D, et al. 2017. Distance constraints on activation of TRPV4 channels by AKAP150-bound PKCα in arterial myocytes. J. Gen. Physiol 149:639–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moore CL, Nelson PL, Parelkar NK, Rusch NJ, Rhee SW. 2014. Protein kinase A-phosphorylated KV1 channels in PSD95 signaling complex contribute to the resting membrane potential and diameter of cerebral arteries. Circ. Res 114:1258–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moore CL, McClenahan SJ, Hanvey HM, Jang DS, Nelson PL, et al. 2015. β1-Adrenergic receptor-mediated dilation of rat cerebral artery requires Shaker-type KV1 channels on PSD95 scaffold. J. Cereb. Blood Flow Metab 35:1537–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. 2000. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron 27:107–19 [DOI] [PubMed] [Google Scholar]
- 77.Chen CY,Matt L, Hell JW, Rogawski MA. 2014. Perampanel inhibition of AMPA receptor currents in cultured hippocampal neurons. PLOS ONE 9:e108021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grooms SY, Opitz T, Bennett MV, Zukin RS. 2000. Status epilepticus decreases glutamate receptor 2 mRNA and protein expression in hippocampal pyramidal cells before neuronal death. PNAS 97:3631–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu SJ, Zukin RS. 2007. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30:126–34 [DOI] [PubMed] [Google Scholar]
- 80.Spaethling JM, Klein DM, Singh P, Meaney DF. 2008. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J. Neurotrauma 25:1207–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Henley JM,Wilkinson KA. 2016. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci 17:337–50 [DOI] [PubMed] [Google Scholar]
- 82.Walsh DM, Selkoe DJ. 2007. Aβ oligomers—a decade of discovery. J. Neurochem 101:1172–84 [DOI] [PubMed] [Google Scholar]
- 83.Hampel H, Prvulovic D, Teipel S,Jessen F, Luckhaus C,et al. 2011. The future of Alzheimer’s disease: the next 10 years. Prog. Neurobiol 95:718–28 [DOI] [PubMed] [Google Scholar]
- 84.Shankar GM,Li S,Mehta TH, Garcia-Munoz A, Shepardson NE,et al. 2008Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med 14:837–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao WQ, Santini F, Breese R, Ross D, Zhang XD, et al. 2010. Inhibition of calcineurin-mediated endocytosis and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid β oligomer-induced synaptic disruption. J. Biol. Chem 285:7619–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang H, Etherington LA, Hafner AS, Belelli D, Coussen F, et al. 2013. Regulation of AMPA receptor surface trafficking and synaptic plasticity by a cognitive enhancer and antidepressant molecule. Mol. Psychiatry 18:471–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. 2002. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. PNAS 99:13902–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. 1998. SAP97 is associated with the α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J. Biol. Chem 273:19518–24 [DOI] [PubMed] [Google Scholar]
- 89.Tavalin SJ, Colledge M, Hell JW, Langeberg LK, Huganir RL, Scott JD. 2002. Regulation of GluR1 by the A-kinase anchoring protein 79 (AKAP79) signaling complex shares properties with long-term depression. J. Neurosci 22:3044–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang M, Patriarchi T, Stein IS, Qian H,Matt L, et al. 2013Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic β-adrenergic signaling. J. Biol. Chem 288:17918–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. 2000. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci 20:89–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guidotti G, Brambilla L, Rossi D. 2017. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol. Sci 38:406–24 [DOI] [PubMed] [Google Scholar]
- 93.Kauffman WB, Fuselier T, He J,Wimley WC. 2015Mechanism matters: a taxonomy of cell penetrating peptides. Trends Biochem. Sci 40:749–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schwarze SR, Dowdy SF. 2000. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol. Sci 21:45–48 [DOI] [PubMed] [Google Scholar]
- 95.Frankel AD, Pabo CO. 1988. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55:1189–93 [DOI] [PubMed] [Google Scholar]
- 96.Green M, Loewenstein PM. 1988. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 55:1179–88 [DOI] [PubMed] [Google Scholar]
- 97.Joliot A, Pernelle C, Deagostini-Bazin H, Prochiantz A.1991. Antennapedia homeobox peptide regulates neural morphogenesis. PNAS 88:1864–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Derossi D, Joliot AH, Chassaing G, Prochiantz A. 1994. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem 269:10444–50 [PubMed] [Google Scholar]
- 99.Park J, Ryu J, Kim KA, Lee HJ, Bahn JH, et al. 2002. Mutational analysis of a human immunodeficiency virus type 1 Tat protein transduction domain which is required for delivery of an exogenous protein into mammalian cells. J. Gen. Virol 83:1173–81 [DOI] [PubMed] [Google Scholar]
- 100.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. 1999. In vivo protein transduction: delivery of a biological active protein into the mouse. Science 285:1569–72 [DOI] [PubMed] [Google Scholar]
- 101.Passafaro M, Sala C, Niethammer M, Sheng M. 1999Microtubule binding by CRIPT and its potential role in the synaptic clustering of PSD-95. Nat. Neurosci 2:1063–69 [DOI] [PubMed] [Google Scholar]
- 102.Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, et al. 2011. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J. Neurosci 31:9170–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, et al. 2002. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298:846–50 [DOI] [PubMed] [Google Scholar]
- 104.Nagahara H,Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, et al. 1998. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med 4:1449–52 [DOI] [PubMed] [Google Scholar]
- 105.Schwartz MA, Schaller MD, Ginsberg MH. 1995. Integrins: emerging paradigms of signal transduction. Ann. Rev. Cell Dev. Biol 11:549–99 [DOI] [PubMed] [Google Scholar]
- 106.Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, et al. 2011. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat. Med 17:822–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, et al. 2007. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 26:4879–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Matsushita M, Tomizawa K, Moriwaki A, Li ST, Terada H, Matsui H. 2001. A high-efficiency protein transduction system demonstrating the role of PKA in long-lasting long-term potentiation. J. Neurosci 21:6000–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nelson AR, Borland L, Allbritton NL, Sims CE. 2007. Myristoyl-based transport of peptides into living cells. Biochemistry 46:14771–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Francois-Moutal L, Wang Y, Moutal A, Cottier KE, Melemedjian OK, et al. 2015. A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain 156:1247–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wootten D, Christopoulos A,Marti-Solano M, Babu MM, Sexton PM. 2018. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol 19:638–53 [DOI] [PubMed] [Google Scholar]
- 112.Dupre DJ, Robitaille M, Rebois RV, Hebert TE. 2009. The role of Gβγ subunits in the organization, assembly, and function of GPCR signaling complexes. Annu. Rev. Pharmacol. Toxicol 49:31–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lefkowitz RJ. 2013. Arrestins come of age: a personal historical perspective. Prog. Mol. Biol. Transl. Sci 118:3–18 [DOI] [PubMed] [Google Scholar]
- 114.Smith JS, Rajagopal S. 2016. The β-arrestins: multifunctional regulators of G protein-coupled receptors. J. Biol. Chem 291:8969–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lodowski DT, Pitcher JA, Capel WD, Lefkowitz RJ, Tesmer JJ. 2003. Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gβγ. Science 300:1256–62 [DOI] [PubMed] [Google Scholar]
- 116.Bouvier M, Hausdorff WP, De Blasi A, O’Dowd BF, Kobilka BK, et al. 1988. Removal of phosphorylation sites from the β2-adrenergic receptor delays onset of agonist-promoted desensitization. Nature 333:370–73 [DOI] [PubMed] [Google Scholar]
- 117.Hausdorff WP, Caron MG, Lefkowitz RJ. 1990. Turning off the signal: desensitization of β-adrenergic receptor function. Faseb J. 4:2881–89 [PubMed] [Google Scholar]
- 118.Shi Q, Li M, Mika D, Fu Q, Kim S, et al. 2017. Heterologous desensitization of cardiac β-adrenergic signal via hormone-induced βAR/arrestin/PDE4 complexes. Cardiovasc. Res 113:656–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Toth AD, Prokop S, Gyombolai P, Varnai P, Balla A, et al. 2018. Heterologous phosphorylation-induced formation of a stability lock permits regulation of inactive receptors by β-arrestins. J. Biol. Chem 293:876–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. 2007. β-Arrestins and cell signaling. Annu. Rev. Physiol 69:483–510 [DOI] [PubMed] [Google Scholar]
- 121.Patel PA, Tilley DG, Rockman HA. 2008. β-Arrestin-mediated signaling in the heart. Circ. J 72:1725–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, et al. 1999. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science 283:655–61 [DOI] [PubMed] [Google Scholar]
- 123.Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, et al. 2007. β-Arrestin-mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J. Clin. Investig 117:2445–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kook S, Zhan X, Kaoud TS, Dalby KN, Gurevich VV, Gurevich EV. 2013. Arrestin-3 binds c-Jun N-terminal kinase 1 (JNK1) and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J. Biol. Chem 288:37332–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hilger D,Masureel M, Kobilka BK. 2018. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol 25:4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Furness SGB, Liang YL, Nowell CJ, Halls ML, Wookey PJ, et al. 2016. Ligand-dependent modulation of G protein conformation alters drug efficacy. Cell 167:739–49.e11 [DOI] [PubMed] [Google Scholar]
- 127.Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, et al. 2017. Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature 547:68–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee M-H, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, et al. 2016The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 531:665–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liang YL, Khoshouei M, Glukhova A, Furness SGB, Zhao P, et al. 2018. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555:121–25 [DOI] [PubMed] [Google Scholar]
- 130.Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, et al. 2016. β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531:661–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, et al. 2011. Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal 4:ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Butcher AJ, Prihandoko R, Kong KC, McWilliams P, Edwards JM, et al. 2011. Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem 286:11506–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Carr R 3rd, Du Y, Quoyer J, Panettieri RA Jr.,Janz JM, et al. 2014. Development and characterization of pepducins as Gs-biased allosteric agonists. J. Biol. Chem 289:35668–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Carr R 3rd, Schilling J, Song J, Carter RL, Du Y, et al. 2016. β-Arrestin-biased signaling through the β2-adrenergic receptor promotes cardiomyocyte contraction. PNAS 113:E4107–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vincke C, Muyldermans S. 2012. Introduction to heavy chain antibodies and derived nanobodies. Methods Mol. Biol 911:15–26 [DOI] [PubMed] [Google Scholar]
- 136.Staus DP,Wingler LM, Strachan RT, Rasmussen SG,Pardon E,et al. 2014. Regulation of β2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol. Pharmacol 85:472–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ghosh E, Srivastava A, Baidya M, Kumari P, Dwivedi H, et al. 2017A synthetic intrabody-based selective and generic inhibitor of GPCR endocytosis. Nat. Nanotechnol 12:1190–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Daniels DA, Sohal AK, Rees S, Grisshammer R. 2002. Generation of RNA aptamers to the G-protein-coupled receptor for neurotensin, NTS-1. Anal. Biochem 305:214–26 [DOI] [PubMed] [Google Scholar]
- 139.Tuerk C, Gold L. 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–10 [DOI] [PubMed] [Google Scholar]
- 140.Ni S, Yao H, Wang L, Lu J, Jiang F, et al. 2017. Chemical modifications of nucleic acid aptamers for therapeutic purposes. Int. J. Mol. Sci 18:1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Youn P, Chen Y, Furgeson DY. 2014. A myristoylated cell-penetrating peptide bearing a transferrin receptor-targeting sequence for neuro-targeted siRNA delivery. Mol. Pharm 11:486–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kahsai AW, Wisler JW, Lee J, Ahn S, Cahill TJ 3rd, et al. 2016. Conformationally selective RNA aptamers allosterically modulate the β2-adrenoceptor. Nat. Chem. Biol 12:709–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bohm M, Eschenhagen T, Gierschik P, Larisch K, Lensche H, et al. 1994. Radioimmunochemical quantification of Giα in right and left ventricles from patients with ischaemic and dilated cardiomyopathy and predominant left ventricular failure. J. Mol. Cell Cardiol 26:133–49 [DOI] [PubMed] [Google Scholar]
- 144.Feldman AM, Cates AE, Veazey WB, Hershberger RE, Bristow MR, et al. 1988. Increase of the 40,000-mol wt pertussis toxin substrate (G protein) in the failing human heart. J. Clin. Investig 82:189–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP. 2001. Dual modulation of cell survival and cell death by β2-adrenergic signaling in adult mouse cardiac myocytes. PNAS 98:1607–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Oliveria SF, Gomez LL, Dell’Acqua ML. 2003. Imaging kinase-AKAP79-phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J. Cell Biol 160:101–12 [DOI] [PMC free article] [PubMed] [Google Scholar]