

Abstract
A top-down proteomic strategy with semi-automated analysis of data sets has proven successful for global identification of truncated proteins without the use of chemical derivatization, enzymatic manipulation, immunoprecipitation or other enrichment. This approach provides the reliable identification of internal polypeptides formed from precursor gene products by proteolytic cleavage of both the N- and C-termini, as well as truncated proteoforms that retain one or the other termini. The strategy has been evaluated by application to the immunosuppressive extracellular vesicles released by myeloid-derived suppressor cells. More than 1000 truncated proteoforms have been identified, from which binding motifs are derived to allow characterization of the putative proteases responsible for truncation.
Keywords: truncated proteoforms, top-down mass spectrometry, cleavage motifs, protease database, extracellular vesicles, myeloid-derived suppressor cells
Graphical Abstract
INTRODUCTION
Truncation of proteins gives rise to new proteoforms that have the potential to play important roles in regulating signal transduction and cell activity.1–5 Several mechanisms can generate truncated proteins, including degradation of full-length proteins and, more rarely, alternative start and stop sites for transcription or protein translation. Studies of truncated proteoforms have been challenging because the intracellular abundances of most protease products are low, and selective enrichment has been required prior to mass spectrometric or other characterization.4, 6–8 Thus far, most analyses on the proteomic scale have depended on bottom-up strategies with tryptic cleavage, and have targeted polypeptides with new derivatized amino-termini for identification or quantitation.1–3, 6–9 In the present study top-down mass spectrometry is extended using semiautomated bioinformatic processing, and demonstrated to identify and characterize truncated polypeptides without chemical or enzymatic manipulation, immunoprecipitation or other selective enrichment. We identify polypeptide sequences released from both sides of scissile bonds in precursor proteins, and for the first time characterize internal products formed by multiple cleavages in a reliable high throughput strategy. Our approach is evaluated by analysis of the lysate of extracellular vesicles shed by cancer-induced myeloid-derived suppressor cells (MDSC). This sample was chosen for several reasons: (i) it is present in virtually all cancer patients and a major obstacle to antitumor immunity;10–13 (ii) we have previously identified many of the proteins carried by these extracellular vesicles, and demonstrated that some proteins and ubiquitinated proteoforms mediate immune suppressive functions of intact MDSC;11, 14,15 and (iii) because truncated proteins have been detected frequently in MDSC-derived extracellular vesicles.10,11 Truncated proteins have also been observed in urinary extracellular vesicles.16 One source of truncated proteins in extracellular vesicles might be polypeptides formed in parental cells and selectively removed by extracellular vesicles.17–21 An alternative or co-existing explanation is that proteins are hydrolyzed in situ by proteases active in the extracellular vesicles. This latter hypothesis is consistent with the previous identification of 49 proteases and the 37 components of the proteasome and immuno-proteasome in MDSC-derived extracellular vesicles11,22 and of 34 proteases in urinary extracellular vesicles.23 Protease, proteasome and other enzymatic activities have recently been demonstrated to occur in situ in several kinds of extracellular vesicles.23–28 Whether proteolysis occurs in one or both locations, truncated proteoforms in MDSC-derived extracellular vesicles provide novel polypeptides potentially able to impact and modulate the significant biological functions of extracellular vesicles, e.g., intercellular communication, regulation and dysregulation, immunomodulation and therapeutics.
Our first objective was to establish a successful top-down strategy to identify truncated proteoforms. Subsequent objectives included characterization of the variety of truncated polypeptides present in these biologically active extracellular vesicles, examination of cleavage sites in the precursor proteins, and consideration of the cellular and extracellular proteases responsible for the observed truncations.
EXPERIMENTAL SECTION
Myeloid-derived suppressor cells were obtained from mice carrying syngeneic primary and metastatic 4T1 mammary tumors and allowed to shed extracellular vesicles as previously reported.11, 14 Extracellular vesicles were lysed by suspension in 8 M urea in 50 mM ammonium bicarbonate (pH 8.5). A protease inhibitor cocktail (1% by volume) (Sigma Aldrich, St. Louis MO) and a deubiquitinase inhibitor PR619 (50 μM) (LifeSensors, Malvern PA) were included to control artifactual proteolysis. Samples were buffer exchanged with 50 mM ammonium bicarbonate (pH 8.5) taking final samples to 0.8 M urea. Protein content was measured using the Pierce BCA Assay Kit (Thermo Scientific, San Jose CA). Seven mouse samples were pooled to provide approximately 600 μg protein for an analysis that included fractionation by gel electrophoresis in solution. An additional biological replicate was analyzed using material from a single mouse experiment, approximately 100 μg protein.
Proteins in the pooled preparation were precipitated at −20° C for 1 hr, resolubilized in 1% SDS, and fractionated using gradient electrophoresis in solution following the manufacturer’s instructions (Expedeon GELFrEE, San Diego CA). Twelve fractions were collected and evaluated by 1D gel electrophoresis. Fractions were pooled 1–2, 3–8, 9–10 and 11–12 according to similarities in protein sizes as visualized by 1-D gels (See Supporting Figure S-1), precipitated from the SDS solution with CHCl3:CH3OH:H2O (4:1:3), and resolubilized in H2O:ACN (97.5:2.5) with 0.1% formic acid (solvent A). Each pooled fraction was desalted and concentrated in a PepSwift RP-4H monolith trap (100 μm x 5 mm) at a flow rate of 5 μL/min, and separated on a ProSwift RP-4H monolith column 200 μm x 25 cm (Thermo Fisher) using a gradient of 5 to 55% solvent B ACN:H2O (75:25) 0.1% formic acid at a flow rate of 1.0 μL/min through 120 min. The biological replicate of smaller sample size was not fractionated. It was desalted and concentrated on a PepSwift RP-4H monolith trap into 5% solvent B and subsequently separated using a ProSwift RP-4H monolith column. A gradient of 5 to 55% solvent B was developed through 145 min. Five replicate injections were made.
Top-down LC-MS/MS analysis was carried out using an Ultimate 3000 RSLCnano system (Dionex, Sunnyvale CA) coupled via a Nanospray Flex (Thermo Fisher Scientific) to a Fusion Lumos tribrid orbitrap mass spectrometer (Thermo Fisher Scientific).29,30 Resolving power of the orbitrap was set to 120,000 at m/z 200 for both precursor and product ion analysis. Precursor spectra were acquired in intact protein mode. MS1 and MS2 spectra were acquired by averaging 2 to 5 microscans. MS/MS spectra were produced in “top-speed” data-dependent mode with a fixed duty cycle of 10s and dynamic exclusion of 60 sec. Precursor ions with a charge state of +8 and higher were selected for activation. A range of 1000 to 50,000 Da was scanned. Precursor ions were activated by: (1) electron transfer reactions (ETD, 6 ms reaction time) supplemented by collisions with nitrogen (HCD, 10% maximum energy); and/or (2) collisions with helium (CID, 25% maximum energy) in the same run.
Data sets acquired in these top-down analyses were processed using BioMarker Search in ProSight PD integrated in Proteome Discoverer 2.2. The raw data files were searched with delta M mode on, using a database constructed (ProSight PC 4.0) in XML format containing 17,013 reviewed Mus musculus protein entries from UniProt (February 2019). Precursor mass tolerance was 200 Da and fragment mass tolerance was 10 ppm. A minimum number of 5 matching fragments and P-Scores smaller than 1 × 10−5 were required for reliable identification of truncated proteoforms. Minimum C-Scores of 40 were required for identification of truncated proteoforms with additional post-translational modifications.31
Identified truncated proteoforms were manually curated based on the mass difference between each precursor gene product and associated truncated polypeptides. Mass differences between all observed and theoretical masses were in the range between −0.2 Da and +0.2 Da, with 98.8% of the truncated proteoforms observed inside the range of −0.1 Da and 0.1 Da. ProSight lite supported the assignment of fragment ions in deconvoluted product ion spectra, and the assignment of each truncated polypeptide within its precursor gene product. The latter were manually correlated to confirm the mass differences. P1 to P4 and P’1 to P’4 amino acid motifs were manually tabulated for all cleavage sites except loss of initial methionine. Putative proteases were assigned: (i) using the prediction tools PROSPER (https://prosper.erc.monash.edu.au/ 32 and iProt-Sub (http://iprot-sub.erc.monash.edu/; 33 (ii) by manually searching the P4-P4’ segments of the truncated forms against the MEROPS database release 12.1 (https://www.ebi.ac.uk/merops);34,35 and (iii) by literature review (Table S1).
RESULTS AND DISCUSSION
Identification of truncated proteoforms
Using our top-down strategy, 1020 truncated proteoforms were identified in the extracellular vesicles with high confidence, with P-scores between 1 × 10−5 and 1 × 10−145. These proteoforms were found to be derived from 225 precursor gene products (Supporting Table S-1). Gene ontology analysis of the molecular functions of the 225 precursor proteins (Figure S-2) indicates that about 16% are structural, 16% have catalytic activity, and more than half are binding proteins. Proteins in these categories have previously been reported to be abundant in extracellular vesicles shed by parental MDSC.11,14, 22 Analysis of the locations of the precursor proteins referenced to parental MDSC (Figure S-2) provides a similar distribution to that reported in a 2014 gene ontology analysis of exosomal proteins14 with the major portion assigned to cytoplasm. Removal of initial methionine was detected in 171 of the 1020 proteoforms and was accompanied by N-terminal acetylation in another 135 proteoforms. For either case the loss of initial methionine was not counted as truncation in this study of post-translational proteolysis. Figure 1A summarizes the distribution of precursors associated with different numbers of truncated proteoforms (presented in detail in Table S-1). Fifty-six percent are associated with a single truncation product. At the other end of the scale, four histones are each associated with more than 50 truncated proteoforms. As Figure 1B demonstrates, 60% of all the truncated proteoforms identified here are formed from histone precursors. Extensive histone truncation is consistent with previously documented histone clipping.4,36 Figure 1C lists the 10 histones represented by the most truncated proteoforms and the 10 non-histone precursors for which the most truncated products were identified.
Figure 1.

Summary of the truncated proteoforms identified. (A) Enumeration of truncated proteoforms formed from individual precursor proteins. (B) Distribution of identified truncated proteoforms between histone and non-histone precursors. (C) Number of truncated proteoforms formed from each of the ten most highly represented non-histone and histone precursors. (D) Distribution of N-terminal, C-terminal and internal truncated proteoforms.
Figures 2 and 3 provide illustrations of the approach. MS/MS spectra of two truncated proteoforms are presented in Figure 2., It can be observed that in both examples (1) high fragment ion density is observed, (2) mass differences with the theoretical truncated proteoform masses are lower than our set threshold of 10 ppm and (3) fragment ions explain both termini of the truncated proteoform. Based on these conservative requirements, we identified in a semi-automated fashion truncated proteoforms of the precursor gene products with high reliability.
Figure 2.

Deconvoluted product ion spectra of (A) truncated proteoform subsequently assigned to histone H2A type 1-P (C0HKE9) and (B) truncated proteoform subsequently assigned to non-histone chromosomal protein HMG-17 (P09602).
Figure 3.

Truncated proteoforms shown (highlighted in grey) against the sequences of three exemplary gene products. Red arrows indicate sites of truncation. Top: High mobility group protein B2. Six cleavage sites were identified in ten truncated proteoforms. Middle: Histone H2A type 1-P. Nineteen cleavage sites were identified in thirty-one truncated proteoforms. Bottom: Histone H3.3C. Three cleavage sites were identified in three truncated proteoforms.
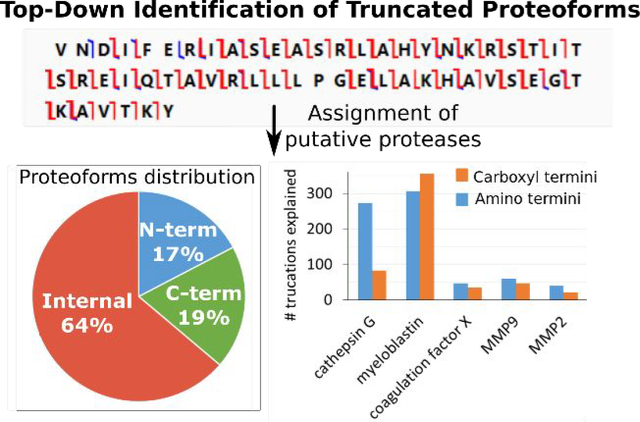
In Figure 3 multiple proteolytic sites and truncated polypeptides are mapped onto the sequences of three representative precursor gene products. In the top map, 19 cleavage sites in High mobility group protein B2 resulted in 31 truncated proteoforms, all of which carry neither terminus of the gene product precursor. The middle map shows 3 cleavage sites in Histone H2A type 1-P that led to 3 proteoforms. None of these carry the C-terminus and several of the proteoforms also do not contain the N-Terminus. In the bottom map 6 proteolytic sites in Histone H3.3C resulted in 10 proteoforms, none of which carry the N-terminus. Specific assignments may be found in Table S-1. Based on such two-step analyses, 17% (176) of the 1020 proteoforms identified contain the N-terminus of the precursor as listed in UniProt (see Experimental Methods), 19% (194) the C-terminus, and 64% (650) are internal polypeptides formed from their precursors by two or more proteolytic events (Figure 1D). The considerable number of internal proteoforms with neither of the original termini present is of special interest, since bottom-up strategies are reported not to provide reliable identifications of internal truncation products.
Despite the successful application of our strategy for an informative global analysis, we propose that the 1020 truncated proteoforms identified here constitute only a portion of the truncated polypeptides present in the MDSC extracellular vesicles studied. Several reasons for a shortfall can be identified. The molecular masses reported here are distributed between 1380 to 17,102 Da and favor the range 5000 to 8000 Da (Figure S-3). Likely, statistically significant identifications of heavier proteoforms are limited because concentrations are too low to meet the instrumental requirement for higher ion counts as the mass increases. Second, no polypeptides are characterized as being modified by both truncation and additive modifications beyond N-terminal acetylation. It should be noted that the inclusion of multiple modifications in a database search using BioMarker Search mode with delta M mode decreases the statistical power of the search and can result in missed identifications. Additive modifications of intact proteins have been reported in previous, untargeted proteomic studies of MDSC extracellular vesicles. 10, 11,14, 15, 22, 30 In one of these studies 20 out of 75 intact proteoforms identified carried multiple modifications including acetylation, phosphorylation, cysteinylation, cysteine sulfonic acid, citrullination, mono-, di- and tri-methylation and oxidation.10 Thus, the list of truncated proteoforms that also carry additive modifications is likely to be expanded when improved search software becomes available. Lastly, 225 precursor proteins are represented in this study, while more than 1200 gene products have been identified in MDSC extracellular vesicles.
Assignment of putative proteases
An analysis of the frequency with which amino acid residues are found on each side of a cleaved amide bond is presented in Figure 4. The P1 and P’1 positions are noted by orange and blue bars, respectively. It is immediately apparent that many proteases contribute. A more complete analysis captures the four amino acids on each side of each cleaved amide bond (P1 to P4 and P’1 to P’4) to provide motifs that enable identification of putative catalytic proteases. These motifs were used to carry out protease assignments using the MEROPS database query by specificity. “What peptidase can cleave this bond?” Queries of “What are the known cleavage sites in this protein?” were also performed. We selected this database because MEROPS is manually curated and offers information on experimentally characterized proteases and a large number of naturally occurring substrate cleavages.34,35 Considering that MEROPS is a database in expansion and does not contain all substrate cleavages, we also utilized the web servers PROSPER and iProt-Sub that based on a substrate protein predict the targeting protease and its cleavage sites through a machine learning approach, which was trained with datasets from the MEROPS database.32,33 Note that our approach does not consider the possibility of consecutive proteolysis. The observed motifs of each truncated proteoform are provided in Supplementary Table S-1, along with the assigned proteases.
Figure 4.

Analysis of amino acid residues within the amide bonds broken to form truncated proteoforms. Blue bars enumerate residues carrying the carboxyl moiety (P1) of each scissile bond. Orange bars enumerate residues that contribute the amino group (P1’) to each scissile bond.
A total of 25 putative proteases were assigned which could account for 85% of the N-terminal and 73% of the C-terminal truncations observed (Table 1). Interestingly, 14 out of the 25 proteases proposed have already been shown to be present in MDSC-derived extracellular vesicles and also in parental MDSC. Seven of these proteases have been found to be more abundant in extracellular vesicles than in parental MDSC, with enrichments ranging between 2.3- and 39-fold.22 Thirty-four proteoforms (3.3 %) were recognized for whose production no proteases could be matched (Figure S-4). The majority of cleavages with unassigned proteases occur after tyrosine or phenylalanine, Y/F-Xaa, which could be explained by a chymotrypsin-like protease such as found in the immunoproteasome.11,37,38
Table 1.
Putative proteases assigned by MEROPS and the web servers PROSPER and iProt-Sub to produce the amino termini and carboxyl termini of the identified truncated forms. *
| Assigned Proteases | Number of truncated proteoforms cleaved |
|
|---|---|---|
| Amino termini | Carboxyl termini | |
| calpain-1 | 10 | 6 |
| calpain-2 | 0 | 1 |
| cathepsin B | 0 | 4 |
| cathepsin D | 26 | 5 |
| cathepsin E | 28 | 11 |
| cathepsin G | 273 | 83 |
| cathepsin K | 15 | 20 |
| cathepsin L | 24 | 10 |
| cathepsin S | 8 | 4 |
| cathepsin Z | 0 | 22 |
| coagulation factor X | 46 | 35 |
| dipeptidyl peptidase 1 | 1 | 0 |
| elastase-2 | 67 | 101 |
| legumain | 2 | 31 |
| matriptase-3 | 0 | 1 |
| Meprin A subunit alpha | 0 | 1 |
| Methionine aminopeptidase-1 and -2 | 171 | 0 |
| MMP2 | 39 | 20 |
| MMP3 | 10 | 30 |
| MMP7 | 1 | 0 |
| MMP9 | 60 | 47 |
| Myeloblastin | 306 | 355 |
| plasmin | 92 | 61 |
| thrombin | 33 | 36 |
| tripeptidyl-peptidase-1 and -2 | 24 | 0 |
In some cases, more than one protease could be responsible for the production of a truncated form. Previously identified proteases in MDSC and MDSC-derived extracellular vesicles are shown in italics.
Eight cathepsins (B, D, E, G, K, L, S and Z) are assigned as putative proteases, four of which (cathepsins B, D, G and Z) have previously been identified in MDSC-derived extracellular vesicles.11,22,30 Cathepsins are known to play a role in the endosomal pathway, but can have diverse functions depending on their cellular and extracellular location.41 From these eight putative proteases, about 20% of the truncations are assigned to cathepsin G by the prediction tools. Additionally, 26 truncated proteoforms (originating from neutrophilic granule protein; HMG-17; histone H2A type-1K, −1P, −2A, −2C, −3 and -J; and histone H2B types −1B and −1H) showed the loss of one or two C-terminal residues that could be explained by the carboxydipeptidase and carboxymonopeptidase activity of cathepsin B and cathepsin Z, respectively. A third of the truncations observed (34%) correspond to cleavages at sites with small hydrophobic P1-P1’ residues such as Ala-Xaa and Val-Xaa. Many of these cleavages are assigned by the prediction tools and/or MEROPS to proteases such as elastase-2; matrix metalloprotease-2, −3 and −9; or cathepsin D, E, G, L and S. Additionally, myeloblastin a protease present in MDSC and their derived extracellular vesicles could help explain these cleavages based on its high sequence similarity (68%) to human myeloblastin.39,40 The assignment of plasmin, thrombin and coagulation factor X as putative proteases is made based on MEROPS search by specificity and on the proteolytic specificity reported in Gasalia et al.42 However, it is relevant to note that for the proposed proteases plasmin and thrombin, only the precursor proteins have been identified in MDSC-derived extracellular vesicles (exosomes). In the case of prothrombin, the precursor of thrombin, we also identified its activating proteins coagulation factor Xa and factor V in extracellular vesicles.11, 22 In the case of plasminogen, the precursor of plasmin, the activating proteins—tissue-type plasminogen activator, urokinase plasminogen activator, factor XIIa and kallikrein--have not yet been detected. The loss of the N-terminal tripeptide “MSE” from histone H1.1, H1.2, H1.3 and H1.4, is observed in 25 of the truncated proteoforms identified. Additionally, one truncated proteoform of H1.5 showed the loss of the N-terminal dipeptide “MS”. These truncations could be supported by the presence of the N-terminal proteases tripeptidyl peptidases-1 and −2 and dipeptidyl peptidases-1, −3 and −9 which have been previously reported in MDSC-derived extracellular vesicles.11, 22 Interestingly, these extracellular vesicles also carry protease inhibitors such as stefin-2, α2-macroglobulin, kininogen-1, leukocyte elastase inhibitor A and serine protease inhibitors, which could regulate proteolysis if active. Protease inhibitors have also been reported in urinary extracellular vesicles23 and it is notable that in situ catalytic activity has been observed in urinary extracellular vesicles known to carry cathepsins and myeloblastin.23
In this analysis, we found a 56 % overlap between proteases assigned by database search or prediction and those previously identified in both MDSC extracellular vesicles and their parental cells. The cleavage pattern does not allow us to discern if proteolysis occurs uniquely in one entity and appears to support the conclusion that both serve as sources for truncated proteoforms in the extracellular vesicles Additional experiments are needed to verify in situ activities of proteases carried by extracellular vesicles, as well as to screen truncated proteoforms for biological activity.
CONCLUSIONS
A top-down strategy has been successfully demonstrated for the high throughput proteomic characterization of truncated proteoforms, in which bioinformatic identifications have been supplemented by supervised bioinformatics and manual processing (readily automatable). The sequences of more than 1000 truncated proteins have been characterized in extracellular vesicles shed by myeloid-derived suppressor cells. Identifications were made without enrichment or chemical derivatization and are linked to 225 murine gene products. Of considerable analytical interest is the reliable identification of a significant number of internal polypeptides (64% of the total), which carry neither of the precursor termini. We point out that our list of proteoforms is incomplete for this sample and suggest that MDSC-derived extracellular vesicles collectively contain more than 3000 truncated proteoforms. Identification of truncated proteoforms was further combined with automated searching of a (evolving) protease database using observed motifs and evaluated as a new strategy for predicting causal proteases. In the present study the profile developed for putative proteolytic enzymes accounted for 96.7 % of the observed cleavage sites and supports the conclusion that truncated proteoforms were formed both in parental MDSC cells and within the extracellular vesicles. We propose that the tools combined and demonstrated here provide a new and effective strategy to study proteolytic events in cells and tissue, characterizing precursors, products and putative proteases.
Supplementary Material
ACKNOWLEDGMENTS
Support is acknowledged by the following grants from the US National Institutes of Health: GM21248, OD019938, CA84232, CA115880. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
Table S-1 – Identified truncated proteoforms per gene product and assigned putative proteases.
Figure S-1—GelFrEE fractions visualized by 1-D PAGE.
Figure S-2 – Gene ontology analysis of gene product precursors of the truncated proteoforms identified.
Figure S-3 – Distribution of molecular masses of the truncated proteoforms identified.
Figure S-4 – Number of truncated proteoforms for which a putative protease could be assigned to explain the produced amino termini and/or carboxyl termini of truncated forms.
Data Repository
https://chorusproject.org/pages/authentication.html#/login
Username: cdpumd@gmail.com
Password: Fenselau2019
REFERENCES
- 1.Gevaert K, Goethals M, Martens L, van Damme J, Staes A, Thomas GR, Vandekerckhove J Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides. Nature Biotech. 2003,121, 566–571. [DOI] [PubMed] [Google Scholar]
- 2.Mahrus S, S.; Trinidad JC; Barkan DT; Sali A, Burlingame AL; Wells JA Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell, 2008,134, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fortelny N; Pavlidis P; Overall CM The path of no return—truncated protein N-termini and current ignorance of their genesis. Proteomics 2015, 15, 2547–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tvardovskiy A, Wrzesinski K, Sidoli S, Fey SJ, Rogowska-Wrzesinska A, Jensen ON Top-down and middle-down protein analysis reveals that intact and clipped human histones differ in post-translational modification patterns. Molec. Cell. Prot 2015,14, 3142–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marshall NC, Finlay BB, Overall CM Sharpening host defenses during infection: proteases cut to the chase. Molec. Cell. Prot 2017,16, S161–S171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleifeld O; Doucet A; Keller U; Prudova A; Schilling O; Kainthan RK; Starr AE; Foster LJ; Kishakedathu JN; Overall CM Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nature Biotech. 2010, 28, 281–288. [DOI] [PubMed] [Google Scholar]
- 7.Jakoby T; van den Berg BH; Tholey A Quantitative protease cleavage site profiling using tandem-mass-tag labeling and LC-MALDI-TOF/TOF MS/MS analysis. J.Proteome Res 2012,11,1812–1820. [DOI] [PubMed] [Google Scholar]
- 8.Marino G; Eckhard U; Overall CM Protein termini and their modifications revealed by positional proteomics. Chem. Biol 2015, 2015,1754–1764. [DOI] [PubMed] [Google Scholar]
- 9.Tholey A; Becker A Top-down proteomics for the analysis of proteolytic events—methods, applications and perspectives. Biochim. Biophys. Acta 2017, 1864, 2191–2199. [DOI] [PubMed] [Google Scholar]
- 10.Geis-Asteggiante L; Dhabaria A; Edwards N; Ostrand-Rosenberg S; Fenselau C Top-down analysis of low mass proteins in exosomes shed by murine myeloid-derived suppressor cells. Internat. J. Mass Spectrom 2015, 378, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams KR; Chauhan S; Patel D; Clements V; Wang Y; Jay S; Edwards N; Ostrand-Rosenberg S; Fenselau C Ubiquitin conjugation probed by inflammation in MDSC extracellular vesicles. J. Proteome Res 2018, 17,315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostrand-Rosenberg S; Fenselau C Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol 2018, 200,422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker KH; Beury DW; Ostrand-Rosenberg S Myeloid-Derived Suppressor Cells: Critical Cells Driving Immune Suppression in the Tumor Microenvironment. Adv. Cancer Res 2014,128,95–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke M; Choksawangkarn W; Edwards N; Ostrand-Rosenberg S; Fenselau C Exosomes from myeloid derived suppressor cells carry biologically active proteins. J. Proteome Res 2014. 13, 863–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chauhan S; Danielson S; Clements V; Edwards NJ; Ostrand-Rosenberg S; Fenselau C Surface glycoproteins of exosomes shed by myeloid-derived suppressor cells contribute to function. J. Proteome Res 2017,16, 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huebner AR; Cheng L; Somparn P; Knepper MA; Fenton RA; Pisitkun T Deubiquitylation of protein cargo is not an essential step in exosome formation. Mol. Cell. Prot 2016,15,1556–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boing AN; Stap J; Hau CM; Afink GB; Ris-Stalpers C; Reits EA; Sturk A; van Noorden CJ; Nieuwland R Active caspase-3 is removed from cells by release of caspase-3-enriched vesicles. Biochem Biophys Acta 2013,1833:1844–1852. [DOI] [PubMed] [Google Scholar]
- 18.Baisauli F; Lopez-Otin C; Mittelbrunn M Exosomes and Autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol 2014, 5, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harding C; Heuser J; Stahl P Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol 1983, 97, 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan B; Teng K; Wu C; Adam M; Johnstone RM Electron Microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol 1985, 101, 942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnstone RM; Adam M; Hammond JR; Orr L; Turbide C Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem 1987, 262, 9412–9520. [PubMed] [Google Scholar]
- 22.Geis-Asteggiante L; Belew AT; Clements VK; Edwards NJ; Ostrand-Rosenberg S; El-Sayed NM; Fenselau C Differential content of proteins, mRNAs and miRNAs suggests that MDSC and their exosomes may mediate distinct immune suppressive functions. J. Proteome Res 2018, 16, 486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Musante L; Tataruch D; Gu D; Liu S; Forsblom C; Groop P; Holthofer H Proteases and protease Inhibitors of Urinary Extracellular Vesicles in Diabetic Nephropathy. J. Diabetes Res 2015, ID 289734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buschow SI; van Balkom BW; Alberts M; Heck AJ; Wauben J; Stoorvogel W MHC class II-associated proteins in B-cell exosomes and potential functional implications for exosome biogenesis. Immunol. Cell Biol 2010, 88, 851–856. [DOI] [PubMed] [Google Scholar]
- 25.Lai RC; Tan SS; Teh BJ; Sze SK; Arslan F; de Kleijn DP; Choo A; Lim SK Proteolytic Potential of the MSC Exosome Proteome: Implications for an Exosome-Mediated Delivery of Therapeutic Proteasome. Int. J. Proteomics 2012. 2012,1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ronquist KG; Sanchez C; Dubois L; Chioureas D; Fonseca P; Larsson A; Ullén A; Yachnin J; Ronquist G; Panaretakis T Energy-requiring uptake of prostasomes and PC3 cell-derived exosomes into non-malignant and malignant cells. J. Extracell.Vesicles 2016, 5,1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu Y; Chen X; Pan Q; Wang Y; Su S; Jiang C; Li Y; Xu N; Wu L; Lou X; Lui S A Comprehensive Proteomics Analysis Reveals a Secretory Path- and Status-Dependent Signature of Exosomes Released from Tumor-Associated Macrophages. J. Proteome Res 2015, 14, 4319–4331. [DOI] [PubMed] [Google Scholar]
- 28.Ronquist G Extracellular vesicles and energy metabolism. Clinica Chimica Acta 2019, 488,116–121. [DOI] [PubMed] [Google Scholar]
- 29.Chen DP; Gomes F; Abeykoon D; Lemma B; Wang Y; Fushman D; Fenselau C Top-down analysis of branched proteins using mass spectrometry. Anal. Chem. 2018, 90, 4032–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geis-Asteggiante L; Ostrand-Rosenberg S; Fenselau C; Edwards NJ Evaluation of spectral counting for relative quantitation of proteoforms in top-down proteomics. Anal. Chem 2016, 88, 10900–10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LeDuc RD; Fellers RT; Early BP; Greer JB; Thomas PM; Kelleher NL The C-score: a Bayesian framework to sharply improve proteoform scoring in high-throughput top down proteomics. J. Proteome Res 2014,13, 3231–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song J; Tan H; Perry AJ; Akutsu T; Webb I; Whistock JC; Pike RN PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PLoS ONE 2012, 7, e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song J; Wang Y; Li F; Akutsu T; Rawlings ND; Webb GI; Chou KC iProt-Sub: a comprehensive package for accurately mapping and predicting protease-specific substrates and cleavage sites. Briefings in Bioinformatics. 2019, 20, 638–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rawlings ND; Waller M; Barrett AJ; Bateman A MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rawlings ND; Barrett AJ; Thomas PD; Huang X; Bateman A; Finn RD The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhaenens M; Gilbert P; Meert P; Vossaert K; Deforce D Histone proteolysis: a proposal for categorization into clipping and degradation. Bioessays 2014, 37, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winter MB; La Greca F; Arastu-Kapur S; Caiazza F; Cimermancic P; Bucchholz TJ; Anderl J; Ravalin M; Bohn MF; Sali A; O’Donoghue AJ; Craik CS Immunoproteasome functions explained by divergence in cleavage specificity and regulation. eLife 2017, 6, e27364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oberdorf J; Carlson EJ; & Skach WR Uncoupling proteasome peptidase and ATPase activities results in cytosolic release of an ER polytopic protein. J. Cell Science, 2006,119, 303–313. [DOI] [PubMed] [Google Scholar]
- 39.Rao NV; Wehner NG; Marshall BC; Gray WR; Gray BH; Hoidal JR Characterization of proteinase-3 (PR-3), a neutrophil serine proteinase. Structural and functional properties. J. Biol. Chem 1991, 266, 9540–9548. [PubMed] [Google Scholar]
- 40.Hajjar E; Broemstrup T; Kantari C; Witko-Sarsat V; Reuter N Structures of human proteinase 3 and neutrophil elastase–so similar yet so different. The FEBS Journal 2010, 277, 2238–2254. [DOI] [PubMed] [Google Scholar]
- 41.Vidak E; Javoršek U; Vizovišek M; Turk B Cysteine cathepsins and their extracellular roles: shaping the microenvironment. Cells 2019, 8, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gosalia DN, Salisbury CM, Maly DJ, Ellman JA, & Diamond SL Profiling serine protease substrate specificity with solution phase fluorogenic peptide microarrays. Proteomics 2005, 5, 1292–1298. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.