

Abstract
Rationale:
Mechanical forces are transduced to nuclear responses via the Linkers of the Nucleo- and Cytoskeleton (LINC) complex, which couples the cytoskeleton to the nuclear lamina and associated chromatin. While disruption of the LINC complex can cause cardiomyopathy, the relevant interactions that bridge the nucleo- to cytoskeleton are poorly understood in the cardiomyocyte, where cytoskeletal organization is unique. Further, while microtubules and desmin intermediate filaments associate closely with cardiomyocyte nuclei, the importance of these interactions is unknown.
Objective:
Here we sought to determine how cytoskeletal interactions with the LINC complex regulate nuclear homeostasis in the cardiomyocyte.
Methods and Results:
To this end, we acutely disrupted the LINC complex, microtubules, actin, and intermediate filaments and assessed the consequences on nuclear morphology and genome organization in rat ventricular cardiomyocytes via a combination of super-resolution imaging, biophysical, and genomic approaches. We find that a balance of dynamic microtubules and desmin intermediate filaments is required to maintain nuclear shape and the fidelity of the nuclear envelope and lamina. Upon depletion of desmin (or nesprin-3, its binding partner in the LINC complex), polymerizing microtubules collapse the nucleus and drive infolding of the nuclear membrane. This results in DNA damage, a loss of genome organization, and broad transcriptional changes. The collapse in nuclear integrity is concomitant with compromised contractile function and may contribute to the pathophysiological changes observed in desmin-related myopathies.
Conclusions:
Disrupting the tethering of desmin to the nucleus results in a loss of nuclear homeostasis and rapid alterations to cardiomyocyte function. Our data suggest that a balance of forces imposed by intermediate filaments and microtubules is required to maintain nuclear structure and genome organization in the cardiomyocyte.
Keywords: Cytoskeleton, desmin, mechanobiology, genome organization, nucleus, cytoskeletal dynamics, cardiac, microtubule
Subject Terms: Cardiomyopathy, Cell Biology/Structural Biology, Cell Signaling/Signal Transduction, Contractile Function, Myocardial Biology
Graphical Abstract

INTRODUCTION
Myocytes generate the contractile force required to pump blood throughout the body. They experience cyclic stress and strain with each heartbeat, and alterations in these mechanical stressors can lead to cardiomyocyte hypertrophy and eventually heart failure. While several force-dependent signaling cascades that regulate cardiomyocyte growth have been described,1 our understanding of how mechanical forces are sensed and transmitted to the nucleus remains poor. Recent work, primarily conducted in non-muscle cells, has demonstrated that, alongside force-dependent signaling cascades, mechanical forces can also be transmitted directly to the nucleus via the Linkers of the Nucleo- and Cytoskeleton (LINC) complex to modulate force-dependent gene expression.
The LINC complex is composed of the nuclear envelope spectrin repeat proteins (nesprins) 1–4 in the outer nuclear membrane and the Sad1p-UNC-84 (SUN) domain 1 and 2 proteins in the inner nuclear membrane. This complex anchors actin filaments, microtubules, and intermediate filaments to the nucleus. SUN proteins link to the nuclear lamina, which in turn directly binds chromatin in lamina-associated chromatin domains (LADs). Hence, the LINC complex forms a contiguous route for force transmission from the cytoskeleton to chromatin.2,3
In studies of non-muscle cells, a particular emphasis has been placed on the interactions between nesprin 1+2 and the actin cytoskeleton, as actin stress fibers or transmembrane actin-associated nuclear (TAN) lines4,5 regulate strain sensing in adherent cells. There is comparatively less research exploring the role of microtubules and intermediate filaments, and the LINC complex remains poorly understood in cardiomyocytes, which exhibit divergent cytoarchitecture from that of adherent cells. Along with a unique, grid-like organization of microtubules and intermediate filaments, actin stress fibers or TAN lines are undetected in mature cardiomyocytes, as actin is predominantly sequestered into sarcomeres. Thus, the precise cytoskeletal connections to the nucleus that are relevant to cardiomyocyte biology remain largely mysterious. This area of study is of particular clinical importance, as mutations in many cytoskeletal, LINC complex, and lamin proteins are associated with cardiomyopathy,6–8 and rodent studies demonstrate that loss of LINC proteins is sufficient to cause cardiomyopathy.9
Desmin, a type-III muscle-specific intermediate filament, is the predominant intermediate filament in muscle. Desmin forms a honeycomb-like scaffold that wraps around the sarcomere at the z-disk, coupling it to the sarcolemma, intercalated disk and various organelles including the mitochondria and the nucleus.10 Desmin is involved in transmitting strain through muscle,11 for the proper maintenance of mitochondrial morphology and function,12–14 and recent work suggests desmin may function as a signaling platform for mechanosensing.15 Mutations in desmin or desmin chaperones lead to “desminopathies,” 6,16 a broad spectrum of muscle diseases that include myofibrillar myopathy, arryhthmogenic cardiomyopathy and dilated cardiomyopathy,7 and both loss17 and gain18–20 of desmin function are sufficient to induce heart disease.
With respect to its interactions with the nucleus, desmin remains understudied. Desmin has been shown to be in close proximity to the nuclear envelope and to co-immunoprecipitate with plectin and nesprin-3,21,22 but the importance of this interaction is unknown.
Here we sought to determine how desmin, microtubules, and actin maintain nuclear morphology in the adult cardiomyocyte. We utilized genetic and pharmacological tools to disrupt cytoskeletal interactions with the nucleus and interrogated structural and functional consequences using a combination of high-resolution microscopy, transcriptomic and genomic assays, and functional tests. We find that desmin is required to maintain nuclear shape and genome architecture in the cardiomyocyte via its interaction with the LINC complex. Our data further suggest a push-pull balance between microtubules and intermediate filaments that preserves nuclear homeostasis and cardiomyocyte function.
METHODS
Data availability.
The genomic datasets generated and analyzed for Figure 7 and Online Figure VI of this study have been deposited in GEO and will be made publicly accessible upon publication of this manuscript.
Detailed experimental procedures, animal protocols and approvals, and statistical methods are described in the supplementary material online.
RESULTS
Modulation of the cytoskeleton alters nuclear morphology.
The cytoarchitecture and proximity of cytoskeletal elements to the nucleus differs in mature cardiomyocytes from that of non-muscle cells. While non-sarcomeric actin is present in the cardiomyocyte,23 actin stress fibers or TAN lines have not been observed in mature cardiomyocytes. In contrast, both desmin and microtubules exist in close proximity to the cardiomyocyte nucleus.24,25 Structured illumination microscopy (SIM) images (Figure 1A) demonstrate microtubules running predominantly along the long axis of the cardiomyocyte and forming a cage around the nucleus, which resides in the interior of the myocyte. Desmin intermediate filaments run predominantly along the short axis of the cardiomyocyte at the level of the sarcomeric Z-disk and form lateral connections at the nuclear membrane (Figure 1A). To assess if any of these components (actin, desmin, microtubules) are important for regulating baseline nuclear morphology, we used pharmacological and genetic tools to disrupt each of these individually and assessed nuclear dimensions in primary, adult, terminally differentiated rat ventricular cardiomyocytes. Inhibition of actomyosin tension with ROCK/Rho inhibitors (Y16 and Y27632), which often leads to nuclear rounding in non-muscle cells,26,27 had no effect on baseline nuclear morphology (Figure 1B–C). Depolymerization of actin with latrunculin A (Lat A, 10 μM for 24hrs) mildly increased nuclear width, with no significant differences detected in length, height, or volume (Figure 1C).
Figure 1: Acute desmin depletion causes involution of cardiomyocyte nuclei.

A, SIM image showing the close association between microtubules (yellow), desmin (magenta) and the nucleus (blue). Right panel shows zoom merge from dotted box. Scale bar = 1 μm. B, Representative Hoechst-stained live adult rat CM nuclei. C, Nuclear size parameters represented by percent change from time-matched control mean. DMSO (Y27632): N= 3 hearts, n= 68 nuclei; Y27632 (10 μM 24 hrs): N= 3, n= 69; Y16 (10 μM 24 hrs): N= 3, n= 66; DMSO (LatA): N=3, n= 58; LatA (10 μM 24 hrs): N= 3, n= 56; DMSO (Colch): N= 3, n= 82; Colch (1 μM for 24 hrs): N= 3, n= 81; Y27632 (10 μM 24 hrs) + Colch (1 μM 24 hrs): N= 3, n= 67; scramble (48 hrs): N= 3, n= 85, Desmin KD (48 hrs): N= 3, n= 98. Statistical significance determined via One-Way ANOVA with post-hoc Bonferroni correction. D, Transmitted light images (left) of identical cardiomyocytes at different time points in culture; Quantification (right) of cardiomyocyte length and width over time in culture. No virus N=2, n=157, scram N=2, n=138, desmin KD N=2, n=128. Data presented as mean +/− 1 SD. For statistical significance, * = p<0.05, ** = p<0.01, *** = p<0.001 vs. control.
In contrast, disruption of the microtubule network via colchicine (Figure 1C, Colch, 1 μM for 24hrs) caused an increase in nuclear length, consistent with previous descriptions of microtubules providing compressive force on the nucleus.28 This increase in length was met with a corresponding decrease in height and width and a modest decrease in nuclear volume. In non-muscle cells, microtubule depolymerization can activate RhoA signaling to increase actomyosin-dependent compression on the nucleus.29 To determine if activated RhoA caused the nuclear elongation we observed with microtubule depolymerization, we inhibited RhoA with Y27632 and again treated cells with colchicine. We observed no prevention of the colchicine-dependent changes to nuclear morphology (Figure 1C, lower middle panel), indicating that Rho signaling was unlikely causing the compression we observed, and instead implicating a direct compressive force applied to the nucleus by microtubules.
To assess the role of intermediate filaments, we developed genetic tools to acutely knockdown desmin. We generated shRNAs against desmin (“desmin KD”), introduced them into adenovirus, and transduced adult rat cardiomyocytes in vitro. After 48 hours, approximately 50–75% of desmin protein was depleted (Online Figure IA). Desmin RNA was reduced Δ20 fold and was comfortably the most downregulated gene in the transcriptome (as shown later in Figure 7). To our surprise, acute desmin depletion led to a Δ50% decrease in nuclear volume, predominantly driven by a decrease in nuclear height and width (Figure 1B–C). Taken together, this data suggests desmin maintains nuclear size, perhaps via tension applied along the short axis of the nucleus, while microtubules provide compression along the long axis, and non-sarcomeric actin plays a minimal basal role.
Importantly, these changes in nuclear size were independent of any change in cell size upon desmin or microtubule manipulation. Under our culture conditions, cardiomyocytes are stable morphologically and functionally for at least 72 hours, and only rod-shaped (aspect ratio >3:1) myocytes with preserved membrane morphology (striations) are used for analysis. Cardiomyocyte length, width, or viability were unchanged by the addition of adenovirus encoding a scramble construct or desmin shRNA (Figure 1D), and colchicine treatment similarly had no effect on myocyte size (Online Figure IB). Thus, desmin depletion causes a specific nuclear involution within a static cell frame.
Acute desmin depletion causes nuclear infolding and chromatin compaction.
We next evaluated this nuclear involution more closely by examining nuclear architecture using super-resolution imaging and electron microscopy. First, we performed immunofluorescence on the intermediate filaments that compose the nuclear lamina: lamin A/C and lamin B1. Desmin depleted cells showed severe lamina wrinkling and infolding (Figure 2A), which was present in greater than 95 percent of nuclei observed (lower panel, Figure 2B) as assessed by our blinded scoring system (upper panel, Figure 2B). To characterize lamina infolding in live cells, we developed an adenovirus encoding lamin B1-mCherry and co-transduced the virus with either scrambled or desmin KD constructs. At approximately 30 hours post-transduction with shRNA, we performed 10 hr time lapse imaging of infected cardiomyocyte nuclei (Figure 2C, Online Movie I and II). In desmin-depleted cardiomyocytes, discrete sections of the lamina appear to fold inward as the nucleus itself progressively collapses over several hours (white arrows, Figure 2C). To further assess the nature of the folds, we performed electron microscopy (Figure 2D). In desmin-depleted cardiomyocytes, infolds ran deep into the nucleus and were accompanied by the double-membrane nuclear envelope (see high magnification inset on the right-hand side). Quantification of EM images (see Online Movie III and IV) revealed a four-fold increase in the infolded area in desmin KD cells (Fig 2E, top). Additionally, while electron-dense heterochromatin was typically restricted to the nuclear periphery and nucleolus in scramble cells (Figure 2D), we observed a considerable extension of the electron-dense chromatin layer – interpreted as heterochromatin - deep into the nuclear interior with desmin depletion (Figure 2D–E). This may reflect chromatin compaction as a result of nuclear involution, or a proliferation of heterochromatin.
Figure 2: Acute desmin or nesprin-3 depletion causes lamina infolding.

A, Representative Lamin A/C (upper) or Lamin B1 (lower) immunostaining in cardiomyocyte nuclei upon scramble or desmin KD. Scale = 1 μm. B, Grading scale developed to assess the severity of lamina defects. Upper panel shows examples of “1” or nearly perfect nuclei (≤1 fold or malformation), “2” or nuclei with mild defects (2–3 folds or malformations), and “3” or highly disrupted lamina architecture (3+ folds or malformations). Lower panel shows result of blinded grading as represented by percent nuclei. Control (48 hr scramble): N= 3, n= 68. Desmin KD (48 hrs): N= 3, n= 85. Statistical significance determined using Chi-squared test. C, Time-lapse images of cardiomyocytes co-infected with LaminB1-mCherry and scramble (left) or desmin KD (right) starting at approximately 30 hours post-infection. White arrows emphasize discrete regions of lamina infolding. D, Electron microscopy representative images of nuclei from longitudinal sections of either scramble (top) or desmin KD (middle, bottom) cardiomyocytes. The right panels show higher magnification images of the boxed regions at left. Left panel scale = 2 μm. Right panel scale = 200 nm. E, Upper panel quantifying the fraction of infolded area per nucleus. Lower panel quantifies the proliferation of electron-dense material (interpreted as heterochromatin) and its distance dependence tangential to the nuclear envelope. Gray bar on the left denotes the uncertainty in the precise location of the inner nuclear membrane due to the resolution limit. scramble: N= 2, n= 18. Desmin KD: N= 2, n= 23. Statistical significance determined using two-sided t-test. Line graph represents the mean with whiskers representing the standard error. F, Top, cartoon schematic of the cardiomyocyte LINC complex. Bottom, Western blot showing nesprin-3 KD in response to shRNA-mediated KD using variant 1. Quantification can be found in Online Figure IIB. G, Immunofluorescence (left) of Hoechst and laminB1 in CM nuclei 48hrs after infection with scramble or shNesprin-3 variant 1. Scale = 1 μm. Nuclear grading (right) of Nesprin-3 KD nuclei. scramble: N= 2, n=39. Nesp-3 KD: N=2, n=31. Statistical significance determined via Chi-squared test. For statistical significance, * = p<0.05, ** = p<0.01, *** = p<0.001 vs. control.
Depletion of nesprin-3 phenocopies the nuclear involution seen upon desmin depletion.
We next conducted studies to explore potential mechanisms of desmin’s protection of nuclear morphology. Multiple interactions between desmin and the nucleus have been proposed: nesprin-3, encoded by SYNE3, binds to plectin,30,31 which binds to intermediate filaments including desmin (Figure 2F),30,32 but LINC-independent interactions between desmin and the nucleus have also been suggested.32 We hypothesized that if desmin interacted with the nucleus via nesprin-3, then depletion of nesprin-3 would phenocopy desmin KD and cause nuclear involution. We generated three shRNA constructs targeted against different regions of nesprin-3 (“nesp-3 KD”, Online Figure IIA). After 48hrs of adenoviral transduction all three shRNA variants robustly reduced nesprin-3 protein expression by Δ80–90%, and we confirmed transcript knockdown via RT-qPCR and western blot (Figure 2F, Online Figure IIB–C). Indeed, all three nesprin-3 KD variants drove a similar nuclear collapse and severe lamina infolding as we observed upon desmin depletion (Figure 2G and Online Figure III), consistent with desmin interacting with the nucleus via nesprin-3 to protect against involution.
Nuclear infolding is driven by dynamic microtubules.
We next sought to determine the cause of nuclear involution. Microtubules can provide compressive forces on nuclei (Fig. 1B–C), and previous work has mapped multiple interactions between microtubules and nesprins 1/2 in muscle cells33,34. As we also noted that microtubules were often present at sites of nuclear infolding (Figure 3A), we hypothesized that lamina infolding may be driven by microtubules compressing the nucleus, which is exacerbated in the absence of desmin/nesprin-3. To test this hypothesis, we treated desmin and nesprin-3 KD cardiomyocytes with colchicine to depolymerize microtubules prior to the onset of overt nuclear infolding (see design schematic in Figure 3B and effect of colchicine on microtubules in Figure 3C). Despite confirmation that shRNA-mediated KD was not disrupted by colchicine treatment (Online Figure IID–E), nuclear infolding and involution was largely prevented when microtubules were depolymerized in desmin or nesprin-3 KD cardiomyocytes (Figure 3D). This result was confirmed for each of the 3 nesprin-3 shRNA variants (Online Figure III).
Figure 3: Disrupting dynamic microtubules prevents nuclear defects arising from desmin or nesprin-3 depletion.
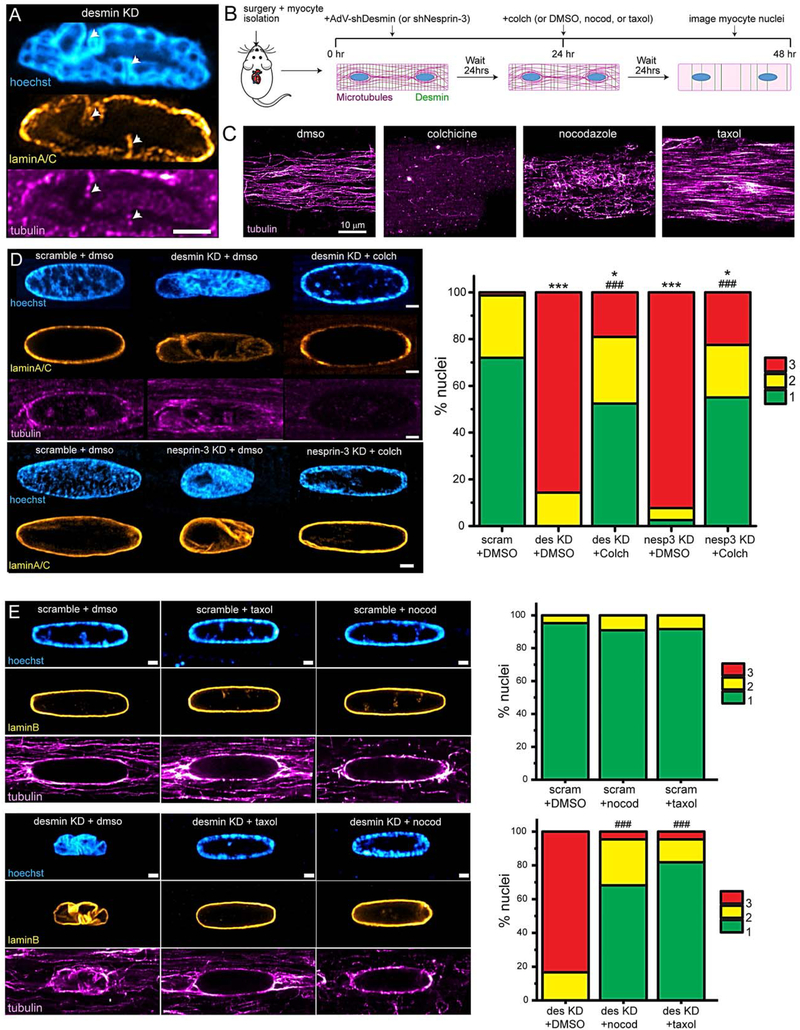
A, Representative image of desmin KD nucleus displaying microtubules present in nuclear infolds (white arrows). Scale = 2 μm. B, Experimental schematic for microtubule and desmin disruption. C, Representative images of microtubules in cardiomyocytes following treatment with microtubule-targeting drugs. D, Representative images (left) of nuclei from experiment depicted in B. Scale bar is 1 μm. Blinded nuclear grading (right) of nuclei. scramble + DMSO: N= 2, n= 38. Des KD + DMSO: N=2, n= 28. Des KD + Colch (10 μM): N=2, n= 42. Nesp-3 KD + DMSO: N= 2, n= 39. Nesp-3 KD + Colch (10 μM): N= 2, n= 40. Statistical significance determined via Chi-square test. E, Representative images (left) and nuclear grading (right) from experiment depicted in B. Scale bar is 2 μm. scramble + DMSO: N= 2, n= 21. scramble + nocod (0.5 μM): N=2, n= 22. scramble + taxol (10 μM): N=2, n= 24. Desmin KD + DMSO: N= 2, n= 24. Desmin KD + nocod (0.5 μM): N= 2, n= 22. Desmin KD + taxol (10 μM): N=2, n= 22. Statistical significance determined via Chi-squared test. For statistical significance, * = p<0.05, ** = p<0.01, *** = p<0.001 vs. control; # = p<0.05, ## = p<0.01, ### = p<0.001 vs. desmin KD + DMSO or nesprin-3 KD + DMSO.
Treatment with Latrunculin A did not prevent infold formation (Online Figure IVA), and treatment with colchicine after the folds had formed (Online Figure IVB) did not reverse the infolding phenotype, suggesting that folds are microtubule specific and, once formed, are not readily reversible. Further, neither desmin depletion nor colchicine treatment altered the expression of lamin A/C or lamin B1 (Online Figure IVC), suggesting that a changing composition of the nuclear lamina is unlikely to explain nuclear infolding (or prevention thereof).
We also sought visual confirmation of microtubule-dependent nuclear infolding. To this end, we triple-transduced cells with lamin B1-mCherry, EMTB-GFP (to demarcate microtubules), and scramble or desmin KD adenovirus. We observed concurrent formation of lamina infolds with microtubule protrusion into desmin KD nuclei, but no such infolding in scramble nuclei, despite the presence of the perinuclear microtubule cage (Online Movies V–VI).
These visual observations suggested that growing, dynamic microtubules are driving nuclear infolding upon loss of desmin/nesprin-3 tethering to the nucleus. To further test this hypothesis, and to control for off-target consequences of gross microtubule loss upon colchicine treatment, we performed additional experiments with the microtubule-targeting drugs taxol and nocodazole. Nocodazole (at low concentrations) sequesters free tubulin to selectively reduce dynamic microtubule populations, while largely preserving stable, long-lasting microtubules. In contrast, taxol stabilizes and polymerizes microtubules, but also eventually reduces microtubule dynamicity by forcing tubulin into the polymerized pool. The different effects of these three pharmacologic agents on cardiac microtubules can be visualized in Figure 3C. Strikingly, both taxol and nocodazole conferred robust protection from nuclear infolding in desmin-KD cardiomyocytes, despite the remaining presence of a stable cage of microtubules encircling the nucleus, as seen in Figure 3E. This suggests that it is the dynamic growth of microtubules – and not the density of the network per se – that underlies nuclear involution upon desmin depletion.
If nuclear involution requires microtubule interaction with nesprins 1/2, then disrupting this interaction should also prevent nuclear involution. Acute depletion of the genes encoding nesprins 1/2 is challenging due to their very large size and high degree of splicing. As an alternative approach, we generated adenovirus over-expressing a dominant-negative KASH peptide (DN-KASH), which disrupts the interaction between nesprins and SUN proteins3 to disrupt all cytoskeletal-LINC interactions (Online Figure VA). Consistent with previous reports3, we confirmed that the typical perinuclear ring localization of nesprins 1, 2, and 3 was disrupted upon DN-KASH expression (Online Figure VB–C). If involution requires microtubule interaction with nesprins 1/235, involution should be prevented by DN-KASH (even in the absence of desmin or nesprin-3. Alternatively, if involution does not occur through nesprins 1/2, then DN-KASH should itself cause involution due to the disruption of nesprin-3 and desmin. We found that DN-KASH did not cause involution, but instead nuclei expanded in all axes and increased in volume (Online Figure VD). Moreover, the nuclear lamina did not display infolding, and in fact was even less distorted than control nuclei (Online Figure VE). Further, nuclear infolding caused by either desmin or nesprin-3 depletion was largely prevented by co-expression of DN-KASH (Online Figure VF). Together, these results implicate a model where microtubules interacting with nesprin 1 and/or 2 drive nuclear infolding, which is normally resisted by desmin and nesprin-3. However, given the reliance on the DN-KASH construct, additional experiments will be required to precisely define any interactions between microtubules and the various spliciforms of nesprins 1 and 2.
Desmin depletion leads to DNA damage.
Given the gross disruption of nuclear morphology upon desmin depletion, we sought to determine if this was associated with DNA damage. γH2AX, an indicator of double-stranded DNA breaks, dramatically increased upon desmin depletion, and this DNA-damage was significantly prevented by microtubule depolymerization, as quantified via both immunofluorescence and western blot (Figure 4A–C). The shape change that occurred upon desmin KD was also rescued with colchicine treatment (Figure 4B). We noted that several desmin KD nuclei with preserved size and morphology after colchicine treatment still had elevated levels of γH2AX, suggesting that the DNA damage observed in this model did not require involution. However, there was a strong positive correlation between nuclear collapse and increased DNA damage, as the γH2AX signal increased exponentially with a decrease in nuclear size in desmin depleted myocytes (Figure 4D, left). No such correlation was seen in scramble nuclei (Figure 4D, right). Taken together, these data are consistent with a model where desmin-nesprin-3 tethering of the nucleus protects it from microtubule-dependent infolding, collapse, and DNA damage.
Figure 4: Desmin KD causes DNA damage that is partially prevented by microtubule depolymerization.

A, Representative immunofluorescence images of yH2AX, an indicator of DNA damage and repair. B, yH2AX intensity, nuclear length, width, and area measured from immunofluorescence imaging. Scrambled (48 hrs) + DMSO: N= 2, n= 226. Desmin KD (48 hrs) + DMSO N= 2, n= 128. Scrambled (48 hrs) + colch (10 μM 32 hrs): N= 2, n= 157. Desmin KD (48 hrs) + colch (10 μM 32 hrs): N= 2, n= 160. C, Western blot (left) and quantification (right) for yH2AX (green) and H3 (red, loading control). Treatment conditions are identical as above. Repeat lanes in each group are 5 and 7 μl of lysate, and the quantification always pulled from the average of 5 and 7 μl of lysate and normalized to H3. Data is from 3 independent rat CM isolations. H3 is unaltered with either desmin KD or colch treatment (data not show). Statistical significance determined via two-way ANOVA with post-hoc Bonferroni. D, Correlation between yH2AX and nuclear area from the same experiments. yH2AX signal was plotted on a logarithmic axis to demonstrate the exponential relationship between nuclear area and DNA damage, and so that the distribution of scramble data could be visualized on the same scale. Log10 transformed data for Desmin KD + colch group was fit to linear regression analysis, with Pearson’s correlation coefficient listed and p-value indicating that slope is significantly different than zero. For statistical significance, * = p<0.05, ** = p<0.01, *** = p<0.001 vs. control; # = p<0.05, ## = p<0.01, ### = p<0.001 vs. desmin KD + DMSO or nesprin-3 KD + DMSO.
Desmin depletion disrupts nuclear homeostasis in situ.
As the forces imposed on cardiomyocyte nuclei will differ in beating tissue compared to isolated cardiomyocytes, we next used two approaches to investigate desmin’s nuclear protection in situ. Per the original characterizations by Milner et al. and Li et al., desmin knock-out mice exhibit severe defects in myocardial architecture and cardiomyopathy17,52, making it difficult to interpret any nuclear phenotype36 as primary to the loss of desmin or secondary to disease pathology. We thus sought to deplete desmin in a minority of cardiomyocytes in vivo, which was accomplished via the generation and transduction of a low-dose of AAV9 encoding shRNA against desmin. We injected both male and female P4/5 rats with AAV9-shDesmin-BFP (KD) or saline (sham) at a viral dosage that achieved 30.1 +/− 4.8% transduction efficiency of cardiomyocytes. After 5 weeks, we extracted the hearts from these rats and performed tissue sectioning and immunofluorescence imaging. As expected, we observed desmin expression only in cardiac myocytes, and KD hearts demonstrated mild tissue disorganization. Fortuitously, most cardiomyocytes could be identified as either clearly positive or clearly negative for desmin immuno-reactivity (Figure 5A–B). We thus compared nuclei in desmin-negative myocytes to two controls – myocyte nuclei from sham-injected hearts, and from internal, tissue-matched desmin-positive controls. Using cTNT labeling to identify cardiomyocyte nuclei, nuclei were traced to obtain size parameters, and scored blindly by two independent observers to quantify morphology defects. Upon desmin depletion, nuclei were shorter, rounder, and had significantly more morphological defects (Figure 5B–C). In KD hearts, cardiomyocytes still containing desmin had nuclei that were morphologically more similar to their sham counterparts than their desmin-depleted neighbors (Figure 5B–C), supporting a primary role of desmin in maintaining nuclear architecture in vivo.
Figure 5: Desmin KD causes nuclear malformation in vivo.

A, Sham (left) or desmin KD (right) rat left ventricular sections stained with desmin (orange) and DAPI (blue). The extra-nuclear blue signal is background fluorescence in the DAPI channel. Green and red arrows point to desmin-positive and desmin-negative cardiomyocyte nuclei, respectively. Scale = 10 μm. Cardiomyocyte nuclei were distinguished from non-myocyte nuclei via cTNT counterstaining (not shown). B, Zoomed-in images of representative desmin-positive and desmin-negative nuclei in tissue. Scale = 1 μm. C, Nuclear length (left), roundness (middle) and nuclear grading (right) of nuclei in tissue. *= compared to sham, #= compared to des+ cardiomyocytes. Sham: N=6 hearts, n= 188 nuclei. Desmin KD (des+): N=4, n= 75. Desmin KD (des-): N= 4, n= 100. Statistical significance determined via one-way ANOVA with post-hoc Bonferroni correction for length and roundness, and via Chi-squared test for blinded scoring. For statistical significance, * = p<0.05, ** = p<0.01, *** = p<0.001; # = p<0.05, ## = p<0.01, ### = p<0.001.
Due to practical limitations of organ level experiments, we also sought to establish a parallel, more flexible platform to interrogate the interplay between microtubules and intermediate filaments in loaded, beating cardiac syncytia. To this end we co-cultured neonatal rat ventricular myocytes (NRVMs) and cardiac fibroblasts on aligned, nano-patterned substrates that promote tissue maturation and syncytial formation (Figure 6A, upper panel). Within 48hrs of plating on patterned substrates, the tissue is robustly aligned along the pattern axis and exhibits improved maturation compared to un-patterned cells. This is demonstrated by improved myofibril alignment, uniaxial beating, and, importantly, cytoarchitecture reminiscent of mature cardiac tissue (Figure 6A, lower panels). Of particular note, the organization of desmin into a striated, transverse structure is strongly promoted by tissue patterning. Three days post-plating tissues were infected with adenovirus encoding either control shRNA or shRNA targeted toward desmin and nuclear phenotypes were assessed. Upon desmin KD, α-actinin organization was still evident but slightly misaligned, and transverse desmin filaments were no longer observed (Figure 6B). Despite the modest change in sarcomeric architecture, nuclear morphology was significantly compromised (Figure 6C). As in unloaded adult myocytes, desmin KD drove involution and infolding of nuclei in beating NRVM syncytia, as well as a robust increase in DNA damage. Colchicine treatment to depolymerize microtubules reduced DNA damage and fully protected from the morphological abnormalities induced by desmin depletion (Figure 6C–D). Together with the above, this data indicates that desmin protects against microtubule-dependent disruption of nuclear homeostasis in situ.
Figure 6: Desmin protects from microtubule-dependent nuclear disruption in beating cardiac syncytia.

A, Top, experimental design for nano-patterning cardiac syncytia. Bottom, immunostaining of a-actinin, desmin, and tubulin in unpatterned (left) and patterned (right) NRVM syncytia. Scale = 10 μm. B, Immunostaining of patterned NRVM syncytia in scramble or desmin KD infected cultures. Transverse desmin filaments are no longer evident upon desmin KD. C, Representative field of NRVM nuclei in nano-patterned syncytia immunostained for LaminB1 and yH2AX (top), and 4x zoom in of representative nuclei (bottom inset). D, Quantification of nuclear dimensions and yH2AX intensity in NRVM nuclei from nano-patterned syncytia. scram: N=3 NRVM isolations, n= 419 nuclei. Desmin KD: N=3, n= 388. Scramble + colch: N= 3, n= 357. Desmin KD + colch: N=3, n= 260. α-actinin (not shown) was used to distinguish myocytes from non-myocytes. Statistical significance determined via two-way ANOVA with post-hoc Bonferroni comparison. For statistical significance, * = p<0.05, ** = p<0.01, *** = p<0.001 vs. control; # = p<0.05, ## = p<0.01, ### = p<0.001 vs. desmin KD + DMSO.
Desmin depletion drives large-scale changes to gene expression and compromises lamina-associated chromatin.
As DNA damage is a well-established response to a loss of genome-organization,37–39 we next assessed the genomic and transcriptomic consequences of desmin depletion. RNA sequencing revealed a large number of differentially expressed genes between scramble and desmin KD cardiomyocytes (Figure 7A), with approximately 20% of the genome differentially regulated following desmin depletion (at a cutoff of > 2-fold change, adj. p-value <0.05). Fortuitously, desmin KD did not significantly affect the expression of any of the Nesprin or SUN isoforms detected in cardiomyocytes, simplifying the interpretation of experiments manipulating the LINC complex. Bioinformatic analysis of these differentially expressed genes indicated that the most significantly altered gene ontology (GO) groups encoded transcripts involved in ion handling, contractility and mitochondrial function and metabolism, functional groups previously noted to be altered with misregulation of desmin. Moreover, acute desmin depletion produced a genetic signature highly associated with cardiac dysfunction, hypertrophy and dilated cardiomyopathy (Figure 7B), consistent with known phenotypes caused by desmin mutations.
Figure 7: Desmin KD results in large-scale changes to gene expression and compromises lamina-bound chromatin.

A, RNAseq was performed on scramble/null and Desmin KD cardiomyocytes. Volcano plot showing differentially expressed genes upon desmin depletion compared to scramble. Genes were considered significantly different with a log2 fold change >=|1| and false discovery rate of 0.05. B, Gene Ontology (GO) Analysis of differentially expressed genes via ToppGene, showing the most significantly altered groups of genes upon desmin depletion, as well as associated phenotypes or diseases associated with the desmin KD transcriptome. C, Representative LaminB ChIP-seq track from entire chromosome 10 (rn6). LADs are labeled with black boxes immediately above track. Area in red box is magnified in bottom track (1–18,894,761 bp of chromosome 10). D, Stacked bar chart indicating percentage of genes which changed expression upon desmin KD (log2 fold change >=|1| and adjusted p-value <0.05), categorized as whether they resided inside or outside a LAD in control conditions. Number of genes indicated in bar chart. The distribution of up and down regulated genes in and out of LADs is significantly different as determined via Chi-squared comparison (Chi-squared statistic = 32.2). E, PantherDB Gene Ontology analysis of the 93 genes which resided in LADs in control cells and were upregulated upon desmin depletion.
Given the disruption of lamina architecture (Figure 2), we hypothesized that the desmin-dependent loss of genome organization may arise from disruption of lamina-associated chromatin domains. We thus assessed LADs by performing lamin B chromatin immunoprecipitation followed by sequencing (LB ChIP-seq). We first confirmed our ability to specifically ChIP lamin B from rat cardiomyocytes (Online Figure VIA). We then performed LB ChIP-seq from three independent pools of scramble-treated cardiomyocytes. After confirming high correlation between the input and ChIP replicates (Online Figure VIB), we merged replicate data, input normalized LB ChIP-seq data, and defined LADs using an unbiased algorithm40 (Figure 7C, Online Figure VIC, Supplemental Table). Similar to LADs in cardiomyocytes and other cell types from different model species,41–44 rat cardiomyocyte LADs encompass approximately 40% of the genome and house 7561 genes and features (ensembl, Online Figure VIC, Supplemental Table).
We further confirmed lamin B expression and our ability to ChIP lamin B in desmin-depleted cells (Online Figure VIB); however, LB-ChIP in desmin-depleted cells resulted in only a fraction of enriched chromatin compared to scramble control, despite normal levels of LaminB1. Thus, three biological LB ChIP-seq replicates resulted in fewer than 8.6M uniquely mapping reads, compared to over 40M for the control samples using the same amount of starting material for ChIP. This suggests that desmin-depletion compromises normal chromatin enrichment to lamin B. Given the low number of reads, it was not feasible to define LADs in KD cardiomyocytes; however, we note that the input-normalized LB ChIP-seq reads from desmin-KD cells are localized in LAD regions (as defined in scramble cardiomyocytes; Figure 7C – compare bottom vs. top tracks), further supporting the hypothesis that LAD structure and LaminB1-chromatin interactions are compromised upon desmin depletion.
Finally, we determined the impact of the potential “LAD loss” on gene transcription. Genes in LADs are preferentially transcriptionally repressed compared to those not in LADs;41 consistently, we observed that genes found in control cardiomyocyte LADs tended to be up-regulated when “lost” from LADs upon desmin KD, while genes not found in LADs were significantly more likely to be down-regulated upon desmin depletion (Figure 7D–E). Together, this data indicates that acute loss of desmin rapidly compromises genome organization in the cardiomyocyte, with broad consequences on the cardiac transcriptome.
Acute Desmin Depletion Disrupts Excitation-Contraction Coupling.
Given the marked genomic and transcriptomic changes, particularly in genes involved in calcium handling and contractility, we next assessed Excitation-Contraction (EC) coupling and cell shortening in desmin KD myocytes. As desmin may regulate EC coupling through a variety of mechanisms, we also interrogated these metrics in nesprin-3 depleted cells to better isolate an effect of disrupting the desmin-nesprin3 axis. Cardiomyocytes depleted of either desmin or nesprin-3 displayed altered calcium transients (Figure 8A) and contractility (Figure 8C) upon electrical stimulation. Consistent with the differential expression of multiple transcripts integral to the maintenance of a normal calcium transient (e.g. SERCA2A, RYR2, PLN), desmin KD myocytes demonstrated briefer cytosolic calcium transients, with a faster decay of the calcium transient, and this was accompanied by briefer contractions and less cell shortening (Figure 8A–D, red). Nesprin-3 depleted myocytes exhibited very similar changes to calcium handling and contractility (Figure 8A–D, blue), suggesting that at least part of desmin’s regulation of EC coupling likely arises from its role at the nucleus. Together, these data demonstrate significant and rapid alterations in cardiomyocyte function with acute disruption of desmin or nesprin-3, suggesting that cytoskeletal coupling to the LINC complex helps maintain normal EC coupling.
Figure 8: Excitation-contraction coupling is similarly altered upon acute desmin or nesprin-3 depletion.

A, Left, change in cytosolic calcium (F/F0), and, right, amplitude-normalized F/F0 time course of Fluo-3 loaded adult rat cardiomyocytes electrically stimulated at 1 Hz. B, Quantification of [Ca2+]i transient amplitude (left), rise time (middle), and decay tau (right). Scramble: N=3, n= 52 cells. Desmin KD: N=3, n=45 cells. Nesprin-3 KD: N=3, n=45 cells. C, Left, average contractility trace (sarcomere shortening) and, right, amplitude-normalized shortening of the identical adult rat cardiomyocytes from experiment in A. D, Quantification of contractile amplitudes (left) shortening time (middle) and relaxation tau (right). Statistical significance determined via One-Way ANOVA with Bonferroni correction. For statistical significance, * = p<0.05, ** = p<0.01, *** = p<0.001 vs. control.
DISCUSSION
From this work we conclude that the intermediate filament desmin preserves nuclear homeostasis in cardiac muscle cells via its interactions with the LINC complex. Desmin and microtubules form a filamentous meshwork around the cardiomyocyte nucleus, largely consisting of axial microtubules and transverse intermediate filaments. Here we find that they maintain nuclear shape and size and help preserve the architecture of the nuclear lamina and its associated chromatin-interacting domains. Acute depletion of desmin causes rapid nuclear involution, infolding of the nuclear envelope and lamina, and DNA damage that are driven by dynamic microtubules. Further, desmin depletion drives the loss of chromatin enrichment at the nuclear lamina and large transcriptomic changes that are concomitant with impaired cardiomyocyte function.
Multiple, non-exclusive mechanisms may underlie the desmin and microtubule-mediated maintenance of nuclear integrity. First, our data support a model where desmin and microtubules maintain a force-balance on the cardiomyocyte nucleus. Desmin, via attachments to the myofilaments and the nucleus (via nesprin-3), may provide resting tension on the nucleus, resisting compressive loads. Microtubules, on the other hand, compress and contain the nucleus in a cage-like structure, and connect to the outer nuclear membrane via nesprin 1 and/or 2. Upon acute loss of desmin, this force-balance is disrupted, allowing dynamic microtubules to drive infolding of the nuclear envelope. Alternatively, desmin may function as a scaffold to protect the cardiomyocyte nucleus from aberrant microtubule compression. Desmin is required for proper microtubule network organization,25 and under normal conditions desmin may sequester microtubules or guide microtubule growth peripheral to the nucleus, mitigating microtubule compression. A third hypothesis involves the activation of signaling cascades that may structurally weaken the nucleus upon release of desmin. Cytoskeletal tension on the nucleus can stiffen the nuclear lamina both via increasing levels of lamin A/C45 and via emerin-dependent signaling cascades.46 Release of desmin or desmin-LINC tension may soften the lamina47, thus allowing microtubule-dependent forces to more readily involute the nucleus. In the future, direct measurements of the magnitude and directionality of forces imposed on the cardiomyocyte nucleus using discrete force sensors may help discriminate between these models, although they likely are not independent. More work is needed to establish the precise molecular mechanism by which microtubules interact with the outer nuclear membrane and the LINC complex, and whether this interaction is direct or requires specific microtubule associated proteins to drive nuclear infolding.
The situation is further complicated in the mechanical environment of cardiomyocytes in situ, which experience a complex mix of compression, strain, torsion, and shear. In both in situ and in vitro experiments, we observed decreased nuclear length and increased nuclear morphology defects upon desmin depletion. Yet in vivo we observed rounding of desmin-depleted nuclei, a phenotype not consistently observed in isolated cardiomyocytes. This could arise from several contextual differences, such as continuous stretch and contraction in vivo leading to the rounding of nuclei lacking attachment to the desmin network or the increased time lacking desmin in these hearts.
The differential effects of acute and chronic desmin depletion are also worth consideration. It is unclear if disruption of desmin (or nesprins) is compensated for over time or during development. While germline desmin knockout (KO) mice have been reported to have altered nuclear morphology in skeletal48 and cardiac muscle,14 underlying mechanisms were not determined, and whether this was a direct or indirect consequence of the loss of desmin remained unclear. Our data provide a novel, direct mechanism for desmin’s nuclear protection. Desmin KO mice display muscle weakness, exercise intolerance49 and prominent alterations to their transcriptome, particularly in relation to metabolic and mitochondrial genes,10,13,14 and desmin depleted or mutant zebrafish exhibit defective EC coupling and contractility in skeletal and cardiac muscle.50,51 Intriguingly, we see differential regulation of a variety of metabolic, calcium handling and contractility-associated genes within 24hrs of desmin depletion in adult mammalian cardiomyocytes, and disruption of nesprin-3 produces similar contractile defects. While whole-animal phenotypes are likely multi-factorial due to desmin’s promiscuous interactions, disrupting its association with the nucleus should be considered as a potential driver of diverse phenotypic consequences seen upon desmin loss or gain of function.
While several mutations in nesprins 1 and 2 are associated with cardiomyopathy, we are unaware of myopathy-related mutations in nesprin-3. Nesprin-3 KO mice are viable52, but cardiac phenotypes have not been explored and warrant further study, particularly upon application of mechanical stress. It is also worth noting that nesprin isoforms may compensate for one another during development, as supported by mouse models of nesprin ablation9.
Considerable remodeling of both the desmin and microtubule networks occur in human heart failure.53,54 Desmin protein levels are increased 3–5 fold, and desmin is phosphorylated, cleaved and aggregated.18,19 Microtubules also proliferate and are highly stabilized and detyrosinated, particularly those enriched around the cardiomyocyte nucleus. Future work should interrogate how this cytoskeletal proliferation and post-translational modification affects the relationship between desmin, microtubules and nucleus, and whether this may contribute to altered genome organization and pathology in heart failure. Of note, disruption of lamin A/C, which also causes cardiomyopathy, leads to disorganization of desmin filaments at the nucleus.55 Together with our work, this indicates bi-directional interactions between the cytoskeleton and nucleoskeleton that are required to preserve nuclear integrity in the heart.
Cytoskeletal attachments to the nucleus have received considerable attention in recent years as potential sites of mechanotransduction. While much of the work has focused on the coupling of actomyosin stress fibers to the nucleus, little is known regarding the roles of microtubules and intermediate filaments. Microtubule-LINC interactions are required for nuclear positioning in skeletal muscle,34,35 where desmin also plays a role,56 yet the regulation of nuclear mechanotransduction is unexplored. Given their intriguing ability to modulate lamina-associated chromatin, future studies should explore whether microtubules and intermediate filaments are involved in cardiomyocyte mechanosensing and response or merely provide structural reinforcement.
In sum, our data indicate that desmin and microtubules form a dense, interconnected network that surrounds the cardiomyocyte nucleus, and, when properly balanced, maintains nuclear homeostasis. Disruption of endogenous desmin increases the susceptibility of cardiomyocyte nuclei to microtubule-dependent collapse, a loss of genome architecture and DNA damage, and this disruption of nuclear homeostasis is associated with impaired cardiomyocyte function. This work provides mechanistic insight into the role of desmin intermediate filaments and suggests that improper regulation of nuclear integrity may contribute to pathology in desmin-related diseases.
Supplementary Material
{kind=link}
NOVELTY AND SIGNIFICANCE.
What Is Known?
The nucleus is an important site for the integration of mechanical signals in the cell.
The cytoskeleton has extensive attachments to the nucleus via the LINC complex.
The LINC complex serves as a bridge directly from the cytoskeleton to chromatin.
What New Information Does This Article Contain?
Acute loss of desmin, a type-III intermediate filament, or its LINC-binding partner, nesprin-3, leads to rapid shrinkage and infolding of cardiomyocyte nuclei in a microtubule-dependent manner, concomitant with defects in calcium cycling and contractility.
Acute loss of desmin also leads to DNA damage and the disruption of key chromatin organizational structures known as lamina associated domains (LADs).
Our data suggest that nuclear homeostasis in the cardiomyocyte is maintained by a push-pull balance between microtubules and intermediate filaments at the nucleus.
Accumulating evidence indicates that cytoskeletal connections to the nucleus are important for the integration of mechanical signals, yet little is known about these connections in the cardiomyocyte. In this study, we aimed to characterize the role of microtubule and intermediate filament connections to the nucleus. We found that acute loss of desmin intermediate filaments or their binding partner nesprin-3 causes severe and rapid nuclear involution, which is driven by the polymerization of microtubules. This nuclear involution is also associated with DNA damage, the loss of lamina-associated chromatin domains, and a decrease in cardiomyocyte function. Our study provides new insight into how the LINC complex and its attachments to the surrounding cytoskeleton affect nuclear homeostasis and cardiomyocyte function. Our findings suggest a novel push-pull cytoskeletal mechanism that mediates nuclear homeostasis in the cardiomyocyte and adds to our understanding of how the misregulation of cytoskeletal connections to the nucleus may contribute to the development and progression of myopathy.
ACKNOWLEDGEMENTS
Additional thanks to Drs. Dan Conway and Gant Luxton for kindly providing AdV-DN-KASH and nesprin antibodies, respectively.
SOURCES OF FUNDING
We would like to thank the University of Pennsylvania’s Cardiovascular Institute. Funding for this work came from the National Institutes of Health (NIH) R01-HL133080 to BLP, T32 R05346–09 to PR, T32 GM7229–41 and F31HL142238 to JH, and for RJ NIH DP2 HL147123, Gilead Sciences Research Scholar Award, Burroughs Wellcome Fund Career Award for Medical Scientists.
Support for BP, JH, PR and RJ was also provided by the Center for Engineering MechanoBiology through a grant from the National Science Foundation’s Science and Technology Center program: 15–48571.
Nonstandard Abbreviations and Acronyms:
- DN
dominant negative
- KD
knock down
- LAD
lamina associated domain
- LINC
linkers of nucleo-cytoskeleton
- NRVM
neonatal rat ventricular cardiomyocyte
- SIM
structured illumination microscopy
- SUN1/2
Sad1p-UNC-84 domain 1 and 2 proteins
- TAN
transmembrane actin-associated nuclear lines
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, Wang D, York AJ, Wu H, Houser SR, Seidman CE, Seidman JG, Regnier M, Metzger JM, Wu JC, Molkentin JD. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell. 2016;165:1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szczesny SE, Mauck RL. The Nuclear Option: Evidence Implicating the Cell Nucleus in Mechanotransduction. J Biomech Eng. 2017;139:021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lombardi ML, Jaalouk DE, Shanahan CM, Burke B, Roux KJ, Lammerding J. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J Biol Chem. 2011;286:26743–26753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luxton GWG, Gomes ER, Folker ES, Vintinner E, Gundersen GG. Linear arrays of nuclear envelope proteins harness retrograde actin flow for nuclear movement. Science. 2010;329:956–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luxton GWG, Gomes ER, Folker ES, Worman HJ, Gundersen GG. TAN lines: a novel nuclear envelope structure involved in nuclear positioning. Nucleus. 2011;2:173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsikitis M, Galata Z, Mavroidis M, Psarras S, Capetanaki Y. Intermediate filaments in cardiomyopathy. Biophys Rev. 2018;10:1007–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stroud MJ. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiomyopathy. Biophys Rev. 2018;10:1033–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee I, Zhang J, Moore-Morris T, Pfeiffer E, Buchholz KS, Liu A, Ouyang K, Stroud MJ, Gerace L, Evans SM, McCulloch A, Chen J. Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet. 2014;10:e1004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capetanaki Y, Bloch RJ, Kouloumenta A, Mavroidis M, Psarras S. Muscle intermediate filaments and their links to membranes and membranous organelles. Experimental Cell Research. 2007;313:2063–2076. [DOI] [PubMed] [Google Scholar]
- 11.Boriek AM, Capetanaki Y, Hwang W, Officer T, Badshah M, Rodarte J, Tidball JG. Desmin integrates the three-dimensional mechanical properties of muscles. Am J Physiol, Cell Physiol. 2001;280:C46–52. [DOI] [PubMed] [Google Scholar]
- 12.Diokmetzidou A, Soumaka E, Kloukina I, Tsikitis M, Makridakis M, Varela A, Davos CH, Georgopoulos S, Anesti V, Vlahou A, Capetanaki Y. Desmin and αB-crystallin interplay in the maintenance of mitochondrial homeostasis and cardiomyocyte survival. Journal of Cell Science. 2016;129:3705–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fountoulakis M, Soumaka E, Rapti K, Mavroidis M, Tsangaris G, Maris A, Weisleder N, Capetanaki Y. Alterations in the heart mitochondrial proteome in a desmin null heart failure model. Journal of Molecular and Cellular Cardiology. 2005;38:461–474. [DOI] [PubMed] [Google Scholar]
- 14.Capetanaki Y, Milner DJ, Weitzer G. Desmin in Muscle Formation and Maintenance: Knockouts and Consequences. Cell Struct Funct. 1997;22:103–116. [DOI] [PubMed] [Google Scholar]
- 15.Palmisano MG, Bremner SN, Hornberger TA, Meyer GA, Domenighetti AA, Shah SB, Kiss B, Kellermayer M, Ryan AF, Lieber RL. Skeletal muscle intermediate filaments form a stress-transmitting and stress-signaling network. Journal of Cell Science. 2015;128:219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JDH, de Walle HEK, Capetanaki Y, van der Kooi AJ, van Langen IM, van den Berg MP, van Tintelen JP. Desmin-related myopathy. Clin Genet. 2011;80:354–366. [DOI] [PubMed] [Google Scholar]
- 17.Milner DJ, Weitzer G, Tran D, Bradley A, Capetanaki Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol. 1996;134:1255–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agnetti G, Halperin VL, Kirk JA, Chakir K, Guo Y, Lund L, Nicolini F, Gherli T, Guarnieri C, Caldarera CM, Tomaselli GF, Kass DA, Van Eyk JE. Desmin modifications associate with amyloid-like oligomers deposition in heart failure. Cardiovascular Research. 2014;102:24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rainer PP, Dong P, Sorge M, Fert-Bober J, Holewinski RJ, Wang Y, Foss CA, An SS, Baracca A, Solaini G, Glabe CG, Pomper MG, Van Eyk JE, Tomaselli GF, Paolocci N, Agnetti G. Desmin Phosphorylation Triggers Preamyloid Oligomers Formation and Myocyte Dysfunction in Acquired Heart Failure. Circulation Research. 2018;122:e75–e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLendon PM, Robbins J. Desmin-related cardiomyopathy: an unfolding story. AJP: Heart and Circulatory Physiology. 2011;301:H1220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konieczny P, Fuchs P, Reipert S, Kunz WS, Zeöld A, Fischer I, Paulin D, Schröder R, Wiche G. Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J Cell Biol. 2008;181:667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reipert S, Steinböck F, Fischer I, Bittner RE, Zeöld A, Wiche G. Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Experimental Cell Research. 1999;252:479–491. [DOI] [PubMed] [Google Scholar]
- 23.Kee AJ, Gunning PW, Hardeman EC. Diverse roles of the actin cytoskeleton in striated muscle. J Muscle Res Cell Motil. 2009;30:187–197. [DOI] [PubMed] [Google Scholar]
- 24.Bloom S, Lockard VG, Bloom M. Intermediate filament-mediated stretch-induced changes in chromatin: a hypothesis for growth initiation in cardiac myocytes. Journal of Molecular and Cellular Cardiology. 1996;28:2123–2127. [DOI] [PubMed] [Google Scholar]
- 25.Robison P, Caporizzo MA, Ahmadzadeh H, Bogush AI, Chen CY, Margulies KB, Shenoy VB, Prosser BL. Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science. 2016;352:aaf0659–aaf0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khatau SB, Hale CM, Stewart-Hutchinson PJ, Patel MS, Stewart CL, Searson PC, Hodzic D, Wirtz D. A perinuclear actin cap regulates nuclear shape. Proc Natl Acad Sci USA. 2009;106:19017–19022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Versaevel M, Grevesse T, Gabriele S. Spatial coordination between cell and nuclear shape within micropatterned endothelial cells. Nat Commun. 2012;3:671. [DOI] [PubMed] [Google Scholar]
- 28.Kim D-H, Li B, Si F, Phillip JM, Wirtz D, Sun SX. Volume regulation and shape bifurcation in the cell nucleus. Journal of Cell Science. 2015;128:3375–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol. 2016;17:914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilhelmsen K, Litjens SHM, Kuikman I, Tshimbalanga N, Janssen H, van den Bout I, Raymond K, Sonnenberg A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J Cell Biol. 2005;171:799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ketema M, Wilhelmsen K, Kuikman I, Janssen H, Hodzic D, Sonnenberg A. Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. Journal of Cell Science. 2007;120:3384–3394. [DOI] [PubMed] [Google Scholar]
- 32.Staszewska I, Fischer I, Wiche G. Plectin isoform 1-dependent nuclear docking of desmin networks affects myonuclear architecture and expression of mechanotransducers. Hum Mol Genet. 2015;24:7373–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gimpel P, Lee YL, Sobota RM, Calvi A, Koullourou V, Patel R, Mamchaoui K, Nédélec F, Shackleton S, Schmoranzer J, Burke B, Cadot B, Gomes ER. Nesprin-1α-Dependent Microtubule Nucleation from the Nuclear Envelope via Akap450 Is Necessary for Nuclear Positioning in Muscle Cells. Curr Biol. 2017;27:2999–3009.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson MH, Holzbaur ELF. Nesprins anchor kinesin-1 motors to the nucleus to drive nuclear distribution in muscle cells. Development. 2015;142:218–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stroud MJ, Feng W, Zhang J, Veevers J, Fang X, Gerace L, Chen J. Nesprin 1α2 is essential for mouse postnatal viability and nuclear positioning in skeletal muscle. J Cell Biol. 2017;216:1915–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thornell L, Carlsson L, Li Z, Mericskay M, Paulin D. Null mutation in the desmin gene gives rise to a cardiomyopathy. Journal of Molecular and Cellular Cardiology. 1997;29:2107–2124. [DOI] [PubMed] [Google Scholar]
- 37.Graziano S, Kreienkamp R, Coll-Bonfill N, Gonzalo S. Causes and consequences of genomic instability in laminopathies: Replication stress and interferon response. Nucleus. 2018;9:258–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irianto J, Xia Y, Pfeifer CR, Athirasala A, Ji J, Alvey C, Tewari M, Bennett RR, Harding SM, Liu AJ, Greenberg RA, Discher DE. DNA Damage Follows Repair Factor Depletion and Portends Genome Variation in Cancer Cells after Pore Migration. Curr Biol. 2017;27:210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu T, MacPhail SH, Banáth JP, Klokov D, Olive PL. Endogenous expression of phosphorylated histone H2AX in tumors in relation to DNA double-strand breaks and genomic instability. DNA Repair (Amst). 2006;5:935–946. [DOI] [PubMed] [Google Scholar]
- 40.Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poleshko A, Shah PP, Gupta M, Babu A, Morley MP, Manderfield LJ, Ifkovits JL, Calderon D, Aghajanian H, Sierra-Pagán JE, Sun Z, Wang Q, Li L, Dubois NC, Morrisey EE, Lazar MA, Smith CL, Epstein JA, Jain R. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell. 2017;171:573–587.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, Gregory BD, Adams PD, Berger SL. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SWM, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, Reinders M, Wessels L, van Steensel B. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohwi M, Lupton JR, Lai S-L, Miller MR, Doe CQ. Developmentally regulated subnuclear genome reorganization restricts neural progenitor competence in Drosophila. Cell. 2013;152:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PCDP, Pinter J, Pajerowski JD, Spinler KR, Shin J-W, Tewari M, Rehfeldt F, Speicher DW, Discher DE. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guilluy C, Osborne LD, Van Landeghem L, Sharek L, Superfine R, Garcia-Mata R, Burridge K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat Cell Biol. 2014;16:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cho S, Vashisth M, Abbas A, Majkut S, Vogel K, Xia Y, Ivanovska IL, Irianto J, Tewari M, Zhu K, Tichy ED, Mourkioti F, Tang H-Y, Greenberg RA, Prosser BL, Discher DE. Mechanosensing by the Lamina Protects against Nuclear Rupture, DNA Damage, and Cell-Cycle Arrest. Dev Cell. 2019;49:920–935.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shah SB, Davis J, Weisleder N, Kostavassili I, McCulloch AD, Ralston E, Capetanaki Y, Lieber RL. Structural and functional roles of desmin in mouse skeletal muscle during passive deformation. Biophysj. 2004;86:2993–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C, Gerdes AM, Capetanaki Y. The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. Journal of Molecular and Cellular Cardiology. 1999;31:2063–2076. [DOI] [PubMed] [Google Scholar]
- 50.Li M, Andersson-Lendahl M, Sejersen T, Arner A. Knockdown of desmin in zebrafish larvae affects interfilament spacing and mechanical properties of skeletal muscle. J Gen Physiol. 2013;141:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramspacher C, Steed E, Boselli F, Ferreira R, Faggianelli N, Roth S, Spiegelhalter C, Messaddeq N, Trinh L, Liebling M, Chacko N, Tessadori F, Bakkers J, Laporte J, Hnia K, Vermot J. Developmental Alterations in Heart Biomechanics and Skeletal Muscle Function in Desmin Mutants Suggest an Early Pathological Root for Desminopathies. Cell Rep. 2015;11:1564–1576. [DOI] [PubMed] [Google Scholar]
- 52.Ketema M, Kreft M, Secades P, Janssen H, Sonnenberg A. Nesprin-3 connects plectin and vimentin to the nuclear envelope of Sertoli cells but is not required for Sertoli cell function in spermatogenesis. Mol Biol Cell. 2013;24:2454–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen CY, Caporizzo MA, Bedi K, Vite A, Bogush AI, Robison P, Heffler JG, Salomon AK, Kelly NA, Babu A, Morley MP, Margulies KB, Prosser BL. Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat Med. 2018;260:682–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heling A, Zimmermann R, Kostin S, Maeno Y, Hein S, Devaux B, Bauer E, Klövekorn WP, Schlepper M, Schaper W, Schaper J. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circulation Research. 2000;86:846–853. [DOI] [PubMed] [Google Scholar]
- 55.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. Journal of Clinical Investigation. 2004;113:357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roman W, Martins JP, Carvalho FA, Voituriez R, Abella JVG, Santos NC, Cadot B, Way M, Gomes ER. Myofibril contraction and crosslinking drive nuclear movement to the periphery of skeletal muscle. Nat Cell Biol. 2017;19:1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genomic datasets generated and analyzed for Figure 7 and Online Figure VI of this study have been deposited in GEO and will be made publicly accessible upon publication of this manuscript.
Detailed experimental procedures, animal protocols and approvals, and statistical methods are described in the supplementary material online.