

Abstract
In the past few decades, the field of neuroepigenetics has investigated how the brain encodes information to form long-lasting memories that lead to stable changes in behaviour. Activity-dependent molecular mechanisms, including, but not limited to, histone modification, DNA methylation and nucleosome remodelling, dynamically regulate the gene expression required for memory formation. Recently, the field has begun to examine how a learning experience is integrated at the level of both chromatin structure and synaptic physiology. Here, we provide an overview of key established epigenetic mechanisms that are important for memory formation. We explore how epigenetic mechanisms give rise to stable alterations in neuronal function by modifying synaptic structure and function, and highlight studies that demonstrate how manipulating epigenetic mechanisms may push the boundaries of memory.
Learning is the acquisition of new information, and memory is the ability to retain information in the long term for later reconstruction. In the brain, cells engaged in learning and memory processes undergo persistent changes to encode new information1,2. To consolidate new information into memory, neurons activated during learning require distinct profiles of gene expression3,4. Although the mechanisms that underlie the regulation of learning-induced gene expression are not fully characterized, researchers have turned to the epigenome as a signal-integration platform through which neurons might integrate new information at the molecular level in the service of stable changes in cell function.
Epigenome.
The collective combination of chemical modifications and proteins that interact with the human genome. The epigenome is dynamically regulated, serves as a signal-integration platform and is unique to each individual.
Histone modification.
A post-translational modification — such as acetylation, methylation or phosphorylation — of a histone, a protein that interacts with nuclear DNA and helps to condense genomic DNA into chromatin.
Epigenetic mechanisms are broadly defined as processes that regulate gene expression through the alteration of chromatin structure without changing nucleotide base sequences5,6. Five major epigenetic mechanisms that cells utilize are histone modification, histone variant exchange, nucleotide modification, non-coding RNA-mediated regulation and chromatin remodelling7. With the exception of non-coding RNAs, these mechanisms alter chromatin structure and function, adding a very complex layer of regulation to gene expression. These mechanisms are best known for their actions during cell differentiation and cell division8, including processes involved in the transgenerational passage of gene-regulatory information and the integration of environmental signals for the coordination of transcriptional responses in fully differentiated cells6.
In the last few decades, several epigenetic mechanisms have been shown to regulate learning-induced gene expression in postmitotic neurons and to establish persistent behavioural responses9–12. Exactly how these mechanisms persistently alter neuronal function to encode information into long-term memory remains unclear. Discrete cell populations within particular brain regions, such as the hippocampus13,14, have been suggested to form neuronal ensembles (known as engrams) to induce long-lasting connections that are responsible for the formation of memories. Epigenetic mechanisms are hypothesized to have a role in the acquisition and maintenance of the engram, for example by modulating the encoding process through epigenetic priming and the persistence of cell function.
The signalling mechanisms involved in the coordinated firing of neural circuits and synaptic plasticity are fairly well characterized, and the molecular events that occur dynamically at the synapse are highly complex. Various signalling cascades underlie the potentiation of synaptic responses and the structural changes of activated neurons following learning15–18. Changes in synaptic strength arise owing to the redistribution of glutamatergic receptors and changes in the activity of adhesion proteins18–22. In addition, postsynaptic dendritic spines are structurally modified through the introduction and stabilization of new actin cytoskeletal elements23,24. The learning and memory field is beginning to understand how these stable, structural changes at the synapse are regulated in response to experience and are necessary for the long-term encoding of newly learned information.
The epigenetic mechanisms involved in memory, addiction and brain disorders have previously been comprehensively reviewed25–29. Nevertheless, a common and perhaps unifying aspect among these topics is the synapse. Thus, this Review highlights pioneering studies that demonstrate how epigenetic mechanisms may be employed to encode information and regulate persistent changes at the synapse. We provide an overview of classic epigenetic mechanisms in the context of the synapse. We also discuss how epigenetic mechanisms become engaged by synaptic activity and how they may in turn lead to changes in synaptic structure and function. Last, we speculate on the conceptual avenues that could be taken to better understand how epigenetic mechanisms integrate experience into stable changes of synaptic function and ultimately cause long-lasting behavioural changes.
Histone variant exchange.
Exchange of variants of the canonical histone proteins (namely, H2A, H2B, H3 and H4). Histone variants include H2AZ and H3.3 and can generate specialized chromatin domains and alter the DNA accessibility and thus gene expression.
Nucleotide modification.
Epigenetic modification (mark) of nucleotide bases. For example, DNA methylation involves the attachment of a methyl group to the C5 position of cytosine (5mC). 5-Hydroxymethylcytosine (5hmC) seems to be more abundant in the brain.
Epigenetics in memory
Histone modifications, DNA methylation and nucleosome remodelling (FIG. 1) are three of the best-studied mechanisms that directly modulate chromatin structure to regulate the expression of genes related to learning and memory. The studies discussed below represent highlights of the initial work examining these epigenetic mechanisms in memory formation. Evidence implicating histone variant exchange30–32 and higher-order chromatin looping33,34 in learning and memory is only just emerging; these mechanisms are discussed in BOX 1. RNA modifications35,36 and non-coding RNAs37–43 regulate gene expression without altering DNA sequences (and therefore fall under the broadest definition of epigenetic phenomena) but do not directly affect chromatin structure, and are therefore not discussed below. However, they may help to establish persistent changes in neuronal function that are necessary for memory and other long-lasting changes in behaviour.
Fig. 1 |. regulation of synaptic plasticity-related gene expression through epigenetic mechanisms.
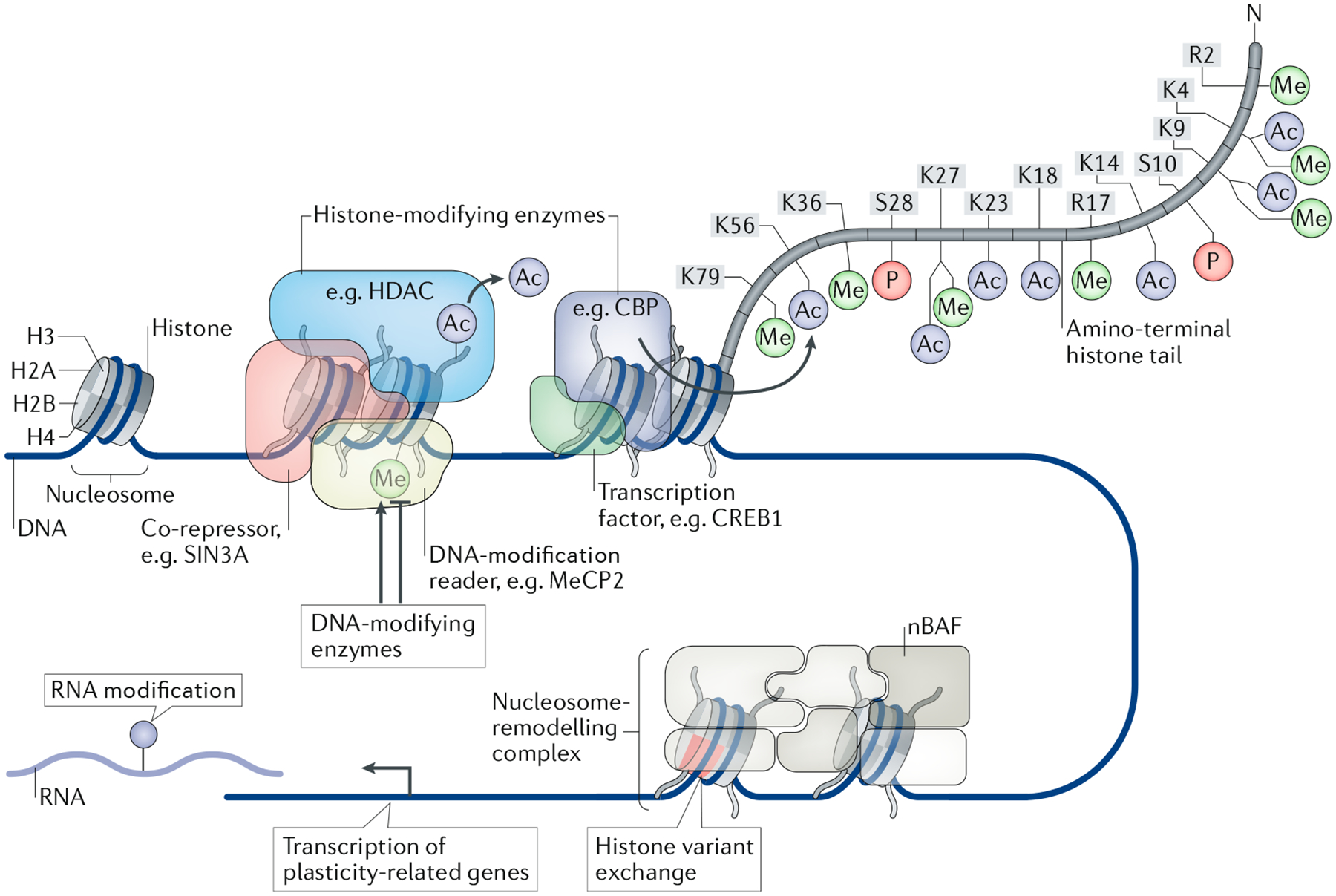
Several epigenetic mechanisms have been identified as regulators of gene expression important for synaptic plasticity and memory formation. For instance, histone acetylation mediated by the activity of histone acetyltransferases, such as CREB-binding protein (CBP), can facilitate memory-related gene expression. CBP is recruited by the transcription factor cAMP-responsive element-binding protein 1 (CREB1) and promotes a permissive transcription environment by adding acetyl groups onto the lysine tails of histones. By contrast, histone deacetylases (HDACs) remove acetyl groups from histone tails and act in concert with associated co-repressor transcription factors to reduce gene expression (for example, transcriptional co-repressor SIN3A). Gene expression can be repressed by the interaction with epigenetic enzymes, such as HDAC–repressor complexes or methyl-CpG-binding protein 2 (MeCP2), which binds to methylated DNA. DNA methylation is controlled by several DNA-modifying enzymes, including DNA methyltransferase 3A (DNMT3A) or DNMT3B and ten-eleven translocation enzymes (TETs), which reportedly repress or permit gene expression depending on the region of DNA that is methylated. Nucleosome-remodelling complexes, such as the neuronal BRG1-associated factor (nBAF) complex, interact with DNA and histones to potentially regulate chromatin structure and synapse-related gene expression through insertion of histone variants, nucleosome sliding, nucleosome eviction and chromatin looping. Although RNA-modifying enzymes do not directly affect chromatin structure, they do influence the rate of mRNA translation and the localization of RNAs, including at the synapse.
Box 1 |. Emerging aspects of neuronal chromatin regulation.
Many aspects of the regulation of chromatin structure and modification of the epigenome are not yet fully understood. although by no means new to the field of epigenetics, new work is shedding light on how histone variant exchange and higher-order chromosomal interactions regulate neuronal gene expression.
Histones are the basic proteins around which DNa is wrapped to form nucleosomes. There are five main families of histones: the core histones H2A, H2B, H3 and H4 and the linker histone H1. There also exist non-allelic and distinct histone isoforms called histone variants. The best understood variants are those in the H2A and H3 families and include H2AZ and H3.3. An ATP-dependent nucleosome-remodelling complex replaces H2A with H2AZ, which is involved in establishing transcriptional competence and nucleosome stability and is localized around transcriptional start sites225. H2AZ (and two related hypervariants H2A.Z.1 and H2A.Z.2) can also be incorporated into the nucleosome by neuronal activity to affect regulation of plasticity-related genes and the formation of fear memory30,31,226. H3.3 is involved in nucleosome assembly and usually replaces histones at active genes (reviewed elsewhere227). H3.3 accumulates in the brain from embryonic development to become the predominant H3 variant in the adult brain. Histones in neuronal chromatin seem to exhibit continuous turnover, as more than 30% of the total neuronal H3.3 pool is replaced in all mouse brain regions examined within a 4-week period32. Furthermore, neuronal activation (through various means) induced H3.3 expression, and knocking down H3.3 expression reduced the density of hippocampal dendritic spines and impaired object recognition memory and contextual fear memory. Together, these studies demonstrate a dynamic and pivotal role for histone variants in regulating the activity-dependent gene expression required for memory processes. It will be interesting to determine whether the exchange of histone variants can affect transcription in a way that influences the effects of subsequent learning or that instils permanent memory.
On a much larger dimensional scale are higher-order chromosomal interactions, which represent another level of gene regulation with emerging importance in neuroscience. One of the main approaches to studying how chromosomal regions that are separated by massive distances (tens to hundreds of kilobases) interact is called chromosomal conformation capture228. In this method, chromatin is crosslinked, isolated and then digested using specific restriction enzymes; the resulting fragments are subsequently ligated and the ligated fragments are analysed using real-time PCR. The abundance of the ligated fragments correlates with the frequency of interactions between two regions. Chromosomal conformation capture has been used to demonstrate how enhancer elements loop to make contact with promoter regions and the assembly of transcriptional complexes to coordinate gene regulation229. it is also being used to define the 3D architecture, preferential organization and specific boundaries of the genome inside a nucleus230 — all of which will further influence how gene expression is regulated. Determining how these long-range interactions and the 3D architecture of the genome regulate neuronal gene expression, as recently investigated in several studies33,231,232, will be an important area of research.
Chromatin remodeling.
In general, the rearrangement and regulation of chromatin (DNA and associated proteins) by various mechanisms, including modification (for example, histone modification) and nucleosome remodelling.
Epigenetic priming.
Stable epigenetic changes (DNA modifications and exchange of transcriptional cofactors and histone variants) produced by exposure to salient stimuli that induce neuronal stimulation; these changes permit efficient transcription of memory-related genes upon re-exposure and reactivation.
Histone modifications.
One of the main entry points into understanding epigenetic mechanisms that regulate memory processes came from investigation into the role of the transcription factor cAMP-responsive element-binding protein 1 (CREB1). Transcription has long been known to be required for the formation of long-term memories44, and pioneering work in Aplysia, Drosophila and mice demonstrated that CREB1 is crucial for memory formation (reviewed in REFS45,46). In addition, the finding that phosphorylation of CREB1 at serine 133 induces recruitment of the CREB-binding protein (CBP), a histone acetyltransferase (HAT)47, prompted the hypothesis that HAT activity may be important for regulating the gene expression required for formation of long-term memory.
Indeed, early studies revealed that Cbp+/− mice — a model of rubinstein–Taybi syndrome — exhibit deficits in long-term memory that correlate with reductions in histone acetylation48–54. Later studies that dissociated the role of CBP in development from its role in the adult brain using conditional CBP-mutant mice and pharmacological inhibitors demonstrated that CBP and histone acetylation are directly involved in memory50–57
Histone acetyltransferase.
(HAT). An enzymes that catalyses the transfer of an acetyl group from acetyl-CoA to the ε-amino group of a histone lysine residue on a histone protein.
Histone deacetylases (HDACs) have the opposite molecular effects to those of HATs, and both types of enzyme interact dynamically with one another. One of the first studies to examine the bidirectional regulation of histone acetylation and its effects on gene expression was of long-term facilitation (LTF) in Aplysia11. Application of serotonin led to phosphorylation of CREB1 on the CEBP promoter and increased CBP recruitment for histone acetylation and LTF. By contrast, application of the inhibitory transmitter FMRFamide led to recruitment of HDAC5, which reduced histone acetylation and displaced CREB1 at the CEBP promoter, blocking LTF. However, HDAC5 is not is considered to be independently responsible for the deacetylase activity of histones; both HDAC5 and HDAC4 are believed to form complexes with other HDACs to regulate histone acetylation. In addition, their ability to regulate transcription has been shown to occur through their interactions with co-repressing transcription factors58.
Rubinstein–Taybi syndrome.
A condition characterized by moderate to severe intellectual disability, short stature, distinctive facial features and broad thumbs and first toes. it is often caused by CREBBP (also known as CBP) mutations.
Nevertheless, HDAC inhibition ameliorated impairments in long-term potentiation (LTP) and deficits in long-term memory in Cbp+/− mice50 and in mice expressing a HAT-inactive form of CBP52, and many subsequent studies have confirmed the function of individual HDACs in memory formation59–65. For example, expression of a deacetylase-inactive HDAC3 mutant in the dorsal hippocampus or the basal nucleus of the amygdala enhanced conditioned context fear, indicating that the deacetylase function of HDAC3 in these regions is crucial for negatively regulating the formation of context-fear memory59. In addition, several studies have identified upstream mechanisms that regulate HDAC activity to influence memory processes; for example, phosphorylation of HDAC4 or HDAC5 regulates their nuclear–cytoplasmic trafficking58,66,67, and S-nitrosylation of HDAC2 leads to its dissociation from chromatin68. Notably, HATs and HDACs have many non-histone substrates; for example, they are sometimes referred to as KATs and KDACs on the basis of their more generic lysine acetylase activity50. Their substrates also include proteins that regulate transcription and assist in chromatin remodelling, such as nuclear factor-κB (NF-κB) and oestrogen receptor-α (ERα) — some of which are known to influence memory processes69–72. Proteins that bind to acetylated histone lysine tails, such as members of the bromodomain and extra-terminal domain (BET) protein family, recruit additional protein complexes that are necessary for transcription and thus also have a role in memory formation73,74. Although we do not discuss them in detail here, bromodomain-containing protein 4 (BRD4) regulates activity-dependent expression of immediate-early genes, and chronic or acute treatment with a BRD4 inhibitor impairs novel object recognition in mice74. Thus, histone acetylation creates a permissive transcriptional state for genes required for consolidation by: increasing the accessibility of plasticity-related genes; regulating the activity of critical transcription factors through acetylation and interaction with acetylation-regulating enzymes; and recruiting histone-acetylation-recognizing proteins and, in turn, additional transcription co-activators.
Methylation of histones, primarily of their lysine and arginine residues, is another epigenetic mechanism implicated in memory formation75–78. Histone methylation can activate or repress transcription, depending on the residue that is methylated and the degree of methylation7,79,80. Over the past decade, several histone methyltransferases have been shown to be crucial for memory. For example, G9a is a histone methyltransferase that forms a complex with G9a-like protein (GLP) to catalyse the dimethylation of H3K9 (H3K9me2). Its deletion from forebrain glutamatergic neurons using CamkII-G9aflox mice resulted in abnormal locomotor activity as well as impairments in long-term contextual and cue-conditioned fear memory76. In addition, these animals showed an overall enhancement of gene expression, confirming the role of G9a as a transcriptional repressor. The cognitive defects seen in the G9a-knockout mice are suggested to be attributable to the aberrant effects on transcriptional homeostasis. For example, neuron-specific deletion of G9a led to the expression of non-neuronal genes in neurons in widespread brain regions76. Thus, reduced G9a expression or activity could lead to memory impairments through defects in the regulation of multiple genes, including cell-type-specific genes81–83.
Histone deacetylases.
(HDACs). enzymes that remove acetyl groups from lysine residues on DNA. Acetyl groups help to neutralize the positive charge of histones and/or serve as binding sites for bromodomain-containing proteins.
Long-term facilitation.
(LTF). A form of long-term synaptic plasticity observed in Aplysia californica.
Ten-eleven translocation enzymes.
(TETs). enzymes that convert 5-methylcytosine (5mC) DNA marks to 5-hydroxymethylcytosine (5hmC), which is enriched within gene bodies, promoters and transcription-factor-binding regions and may influence gene expression.
Similar to histone acetylation, histone methylation can influence the recruitment of transcription factors and the activity of other epigenetic enzymes to regulate gene expression. Contextual fear training leads to increased H3K4 trimethylation (H3K4me3), a transcriptionally permissive mark, at the Zif268 promoter in the hippocampus, and this increase is accompanied by increases in local DNA methylation and reductions in local methyl CpG-binding protein 2 (MeCP2)–DNA binding84. In addition, histone methylation can be influenced by mechanisms of histone acetylation: systemic infusion of an HDAC inhibitor reduces fear-conditioning-induced increases in H3K9me2 (which represses transcription) in the hippocampus84. The above examples demonstrate the high complexity of the effects of changes in histone methylation and methyltransferase function. Several other histone methyltransferase enzymes have been implicated in memory formation (for example, histone-lysine N-methyltransferase 2A (KMT2A)–KMT2D)85–89, as have other histone modifications (including phosphorylation, ubiquitylation, sumoylation, ribosylation and citrullination), discussed elsewhere85,90.
DNA modification.
DNA methylation is a type of DNA modification91–98 and mainly occurs symmetrically on CpG dinucleotides. Promoter methylation was originally considered to be a static mechanism to silence gene expression by recruiting methyl-CpG-binding proteins such as MeCP2 and other associated transcription-repressing protein complexes. However, DNA methylation is now considered to have more complex effects on gene expression, not only at the promoter but also in the gene body. Moreover, although DNA methylation was initially considered only to negatively regulate memory processes, neuronal activity can induce expression of several enzymes that control DNA methylation. These include DNA methyltransferases (DNMTs) and DNA-demethylating enzymes, such as growth arrest and DNA damage-inducible protein-β (GADD45B) and ten-eleven translocation enzymes (TETs)99–103.
During memory consolidation, de novo DNA methylation and demethylation occur within the CA1 region of the hippocampus and are enriched at both inter-genic and intronic regions104. These activity-induced changes in DNA methylation can correspond to the differential expression of genes that, according to gene ontology analysis, are functionally categorized under ‘ion channels’ and ‘transcription regulation’104,105. From findings such as these, DNA methylation is suggested to regulate synaptic transmission and gene transcription critical for memory formation. In support of this, pharmacological inhibition of DNMT activity blocks the induction of LTP91, and DNMT expression is upregulated following contextual fear conditioning106. Following a learning event, several memory-forming genes are transiently demethylated, and memory-suppressing genes are transiently methylated to promote synaptic plasticity94,106. Intriguingly, DNA methylation seems to mediate stable changes in the expression of memory-related genes94. For example, 30 days after fear conditioning training in rats, cortical expression of calcineurin, which negatively regulates memory formation, was reduced, and the gene encoding calcineurin was hypermethylated. Thus, epigenetic modifications can potentially induce stable changes in neuronal function that give rise to long-lasting behavioural changes. Examining epigenome modifications along time frames extending beyond the typical 1–24 hours studied by most laboratories will be very important to understand how epigenetic mechanisms may maintain long-term memories.
The site of DNA methylation within a gene is also important for memory-related gene expression. After reward learning, the expression of the immediate-early genes Egr1 and Fos is increased in the ventral tegmental area and correlates with increases in DNA methylation specifically in the 3′ ends of their gene bodies, but not in the gene promoter107. Infusion of a DNMT inhibitor into the ventral tegmental area during training was sufficient to impair the acquisition of reward-related memories. This effect may be due to the regulation of intragenic DNA methylation, as in vitro application of a DNMT inhibitor reduced KCl-induced hypermethylation of intragenic regions of the Egr1 and Fos genes and enhanced Egr1 and Fos expression. Although these results suggest that learning requires locus-specific regulation of DNA methylation, exactly how DNA methylation influences gene expression during memory formation is still being defined.
As mentioned above, mechanisms of DNA and histone modifications work in concert to regulate gene expression following learning; however, the mechanisms are not fully characterized. For example, histone methylation (H3K4me3) and DNA hydroxymethylation (5-hydroxymethylcytosine (5hmC)) were found to co-occur in intron regions of the immediate-early gene Npas4 in CA1 of the hippocampus following the retrieval of a recent fear memory108. Knockdown of the histone methyltransferase MLL1 in CA1 abolished retrieval-induced increases in DNA 5hmC levels at the Npas4 gene and prevented both fear memory and retrieval-induced increases in CA1 Npas4 mRNA108. The field of DNA methylation is still in its nascent stages, and future research will focus on understanding how gene-specific and locus-specific DNA methylation affects memory processes104,105,109,110.
Nucleosome remodelling.
Despite their known role in regulating gene expression and interactions with chromatin modifiers, the function of nucleosome-remodelling complexes (NRCs) in memory processing is somewhat understudied (FIG. 1). There are four families of NRCs (BRG1-associated factor (BAF), INO80, ISWI (imitation switch) and CHD (chromodomain helicase DNA-binding)) that regulate chromatin compaction through active sliding, ejecting or restructuring of nucleosomes111. NRCs are typically large protein complexes involving many protein subunits that probably dictate the specificity of their function for cell types and loci111. Studies in cultured Baf53b−/− hippocampal neurons revealed that the ATP-dependent BAF subunits are crucial for activity-induced dendritic outgrowth112: these neurons showed impairments in activity-dependent dendritic outgrowth and reduced expression of neurite outgrowth-related genes. Although RNAi-mediated reduction of other neuronal BAF (nBAF) subunits similarly affected activity-dependent dendritic outgrowth, BAF53B is the only subunit that has been studied within memory formation in vivo113–115. The field must further investigate how the different NRC families interact and contribute to activity-dependent gene expression and memory formation. Several mutations of NRC-encoding genes are linked to human intellectual disability disorders, including Coffin–Siris syndrome and autism spectrum disorder26, demonstrating the role of NRCs in cognitive processes.
Nucleosome-remodelling complexes.
(NrCs). Large protein complexes that, through the activity of ATP-dependent enzymes, alter histone–DNA interactions, disassemble or assemble nucleosomes, exchange histone variants or slide or reposition nucleosomes.
Histone code hypothesis.
A hypothesis that posits that specific patterns of epigenetic modifications regulate specific gene expression networks for defined cell functions.
Early-phase LTP.
(E-LTP). in this context, a form of potentiation that is dependent on covalent protein modifications, yet independent of gene expression. it is transient and short-lived (generally on the order of tens of minutes in slices).
Late-phase LTP.
(L-LTP). in this context, a form of potentiation that is dependent on transcription and translation. it is long-lasting (generally on the order of hours in slices and hours to days in vivo).
Outstanding questions.
In summary, there is little doubt that histone modifications, DNA methylation and NRCs have a role in learning and memory. The field has yet to answer many questions about the nature of their functions. First, are any epigenetic mechanisms cell type-specific, and if so, what are their consequences? This may be especially relevant to NRCs, which exhibit cell-type-specific subunit expression. Second, do enzymes that may have histone-modifying functions in the nucleus have subcellular roles (for example, non-histone-modifying functions in the cytoplasm), and is there any crosstalk between these enzymes in different cellular compartments? Third, what are the patterns established by histone and DNA modifications, and how do they coordinate gene expression for specific cell functions (that is, in line with the histone code hypothesis116)? Fourth, do histone modifications and other epigenomic modifications represent a molecular substrate of memory in a way that is relevant to the concept of the engram25?
Extinction.
Weakening of a conditioned response owing to long or repeated trials of memory retrieval in which the conditioned stimulus is removed. extinction is hypothesized to result from the formation of new memories.
Reconsolidation.
The re-encoding and re-stabilization of a memory after reactivation, during which time the memory is hypothesized to be labile and vulnerable to manipulation.
Remote memories.
Here, memories that were encoded a long time previously and that have since become independent of the hippocampus and dependent on cortical regions of the brain (through a process sometimes termed systems consolidation).
Pushing the boundaries of memory
Epigenetic mechanisms have an important role in forming long-term memories, altering the chromatin landscape in a way that leads to a more permissive transcriptional environment for the expression of memory-promoting genes. In parallel to the identification of various epigenetic enzymes as regulators of memory processes55,95,106,117–119, researchers have asked whether these enzymes could be exploited to expand the type or amount of information normally acquired and/or stored after a learning event or to extend the time for which it is stored.
Targeting histone modifications.
Several early studies demonstrated that histone acetylation could be observed during memory consolidation, a process during which gene expression is necessary for long-term memory formation75,120,121. One study demonstrated that blocking histone deacetylation using the nonspecific HDAC inhibitor trichostatin A (TSA) changed the response to a single tetanus stimulation from a transcription-independent form of LTP known as early-phase LTP (E-LTP) to transcription-dependent, late-phase LTP (L-LTP), similar to that typically observed following multiple high-frequency tetani122. This finding raised the intriguing question of whether a similar phenomenon could be observed at the behavioural level when examining memory.
In an object recognition memory task, mice given the HDAC inhibitor sodium butyrate (NaB; intraperitoneally) after a single subthreshold training session that was normally insufficient to induce lasting memory exhibited robust long-term memory 24 hours later123. These effects seem to be persistent, as mice exhibited long-term memory after 7 days, a time point at which even mice trained over a longer session will fail to show long-term memory123. These results were corroborated with additional techniques using genetic focal deletions and pharmacological inhibition of HDAC3 (REFS117,124–126). Manipulations of HDAC3-dependent acetylation can affect the formation of not just contextual memory but also auditory memory. Rats treated with the HDAC3-selective inhibitor RGFP966 not only exhibited more robust memory for the learned association between a sound and a water reward127 but also encoded additional, highly specific features of sounds associated with reward into memory128. These studies demonstrate that histone-modification mechanisms may ultimately alter the kind of information encoded during memory consolidation. These mechanisms could potentially be manipulated to increase the amount and alter the types of information being consolidated, as well as to increase information persistence in memory.
Other studies have investigated the influence of targeting histone modifications on other memory processes, including the extinction or reconsolidation of recent memories, as well as other memory types, such as remote memories. Pharmacological agents that target histone-modifying enzymes to transform these memory processes might thus have therapeutic potential for individuals suffering from traumatic long-term memories (that is, in post-traumatic stress disorder)27. During normal fear extinction training, histone acetylation increases on H3 and H4 histones (for example, H3K14, H3K9 and H4K8) in the hippocampus129,130, lateral amygdala59,61 and infralimbic prefrontal cortex131. Studies involving systemic or region-specific infusions of HDAC inhibitors revealed that histone acetylation can enhance the extinction of recent long-term memories, including fear-associated or drug-associated memories129–134. Similar studies have examined the role of histone acetylation in reconsolidation; however, the findings are conflicting, suggesting that the effects of manipulating reconsolidation by altering histone acetylation may be specific to the type of memory or learning process109. For example, the retrieval or reactivation of contextual memory is reported to induce acetylation of H3 and H4 in the hippocampus and lateral amygdala. Moreover, HDAC inhibitors (such as TSA) promote, and HAT inhibitors impair57,61,135, the reconsolidation of fear conditioning, whereas HDAC inhibitors have no effect on inhibitory avoidance136. This difference in effect could be explained by the fact that these tasks depend on different neural systems; inhibitory avoidance tasks require an instrumental response whereas fear conditioning is a Pavlovian association. However, a more selective approach may have yielded a different result, as virus-mediated reduction of HDAC3 activity in the dorsal striatum was sufficient to accelerate the formation of habit behaviour in instrumental learning tasks137. Thus, there is still much to discover regarding the role of HDACs in different brain regions and in the acquisition of different types of memory.
Epigenetic hypothesis of age-related cognitive impairments.
A hypothesis proposing that the repression of chromatin and alterations in the expression of synapse-related genes lead to cognitive impairment in ageing brains.
Remote memories are thought to be more resistant than recent memories to manipulations (such as extinction paradigms) during reconsolidation after reactivation. However, administration of an HDAC2-selective inhibitor during the reconsolidation phase after spaced or massed extinction of a conditioned fear memory attenuated remote memories68, by enhancing histone acetylation (specifically H3K9 or H3K14) and promoting the expression of neuroplasticity-related genes and LTP. Moreover, in non-treated animals, the recall of remote memory led to less histone acetylation than that observed in animals that recalled recently encoded memories. Thus, changes in histone acetylation may alter mechanisms of synaptic plasticity to make remote memories more labile and ‘recent-like’. In line with this idea is the epigenetic hypothesis of age-related cognitive impairments and preclinical evidence demonstrating that manipulating histone acetylation enzymes can rescue disease-related and age-related memory impairments138. However, the exact epigenetic mechanisms underlying age-related memory impairments are unknown121,139,140; which target genes are affected and how acetylation of their histones enhances and induces the persistence of memory processes beyond the capacity of normal memory are yet to be understood. The current progress and limitations of HDAC inhibitors are discussed in BOX 2.
Box 2 |. Histone deacetylase inhibitors.
Pharmacological histone deacetylase inhibitors (HDACis) have been used often to study the role of histone acetylation in learning and memory. there are two categories of HDACs: zinc-dependent HDACs and NAD-dependent sirtuins (SIRTs). All zinc-dependent HDACs are expressed in the brain, primarily by neurons, and are categorized on the basis of sequence similarity into class I (HDAC1–HDAC3 and HDAC8), classes IIa and IIb (HDAC4–HDAC7, HDAC9 and HDAC10) and class IV (HDAC11). Among SIRT1–SIRT7, only SIRT1–SIRT3 and SIRT5 have deacetylase activity. Several HDACis were developed for treating cancer, and several potent HDACis have shown promising preclinical effects in promoting the histone acetylation required for memory processes.
Initially, pan-HDACis demonstrated the ability of HDACs to regulate learning-induced gene expression, long-term potentiation and memory consolidation52,122 before the roles of specific HDAC classes and isoforms were dissected. Of the class I HDACs, HDAC2 and HDAC3 strongly regulate learning and memory. Chronic treatment with SAHA (an inhibitor of class I and IIb HDACs) enhances fear conditioning memory in wild-type mice but does not further enhance fear-conditioned freezing in HDAC2-deficient mice118, suggesting that SAHA enhances fear memory through inhibition of HDAC2. Such findings have prompted efforts to develop HDAC2-selective inhibitors for improving memory233. Similarly, the most abundant class I HDAC in the brain, HDAC3, also negatively regulates long-term memory59,117,124,125,234. Mice treated with the HDAC3-selective inhibitor RGFP966 showed enhanced long-term object location memory and object recognition memory and increased H4K8 acetylation117,124. Chronic administration of HDAC-isoform-selective inhibitors has, however, produced mixed results. For example, chronic homecage RGFP966 treatment did not alter dendritic spine density in the hippocampus or ameliorate memory impairments in a mouse model of alzheimer disease235,236. Some evidence suggests that activation of SIRT1 deacetylase activity may reduce neurodegenerative processes: in CK-p25 mice, a model of neurodegeneration, oral administration of the SIRT1 activator SRT3657 recapitulated the neuroprotective effects of caloric restriction, delaying the onset of neuronal death237. Although the roles of different HDAC isoforms still need to be fully characterized, such studies indicate that HDACs are pivotal regulators of neural plasticity and behaviour. For more studies using HDACis, see reFs238,239.
The (mostly) pro-mnemonic effects of HDACis in animal models instilled high hopes for their therapeutic potential to ameliorate cognitive impairment. However, several points must be borne in mind when considering HDACis for possible therapeutic treatment. For example, although the inhibitors were developed against specific enzymes and tested for their in vitro activity on purified enzymes, they were often not tested against purified protein complexes. An inhibitor may thus disrupt protein–protein interactions among HDACs, negating much of its claimed HDAC specificity67. these inhibitors also show low cell-type specificity; although many inhibitors penetrate the blood–brain barrier (BBB), they do not exclusively act in particular brain regions or on specific gene sets in the brain. Nevertheless, systemic off-target effects could be reduced by increasing the BBB permeability of these inhibitors. Soon, pharmacological agents will be able to be targeted to specific cell types.
Another important consideration is whether chronic and acute treatment may differentially affect memory processes. HDACis can induce differential effects depending on the behavioural context, therefore different treatments may need to be designed for specific cognitive impairments in different disease states. We are still in the early stages of understanding how to target HDACs and their complex roles in memory. However, given their dynamic roles and powerful effects, efforts dedicated to characterizing and manipulating HDAC mechanisms promise to be of therapeutic value.
Histone methylation has been implicated in the initial consolidation of memories77,85,86,89,90,141 and seems to be induced by and to regulate other memory processes (such as extinction) and memory reactivation90,108,141,142. The importance of histone methylation in age-related and disease-related cognitive impairments has also been explored. For example, H3K9me3 is increased in the hippocampus of aged animals compared with young-adult mice, and systemic administration of an inhibitor of the histone methyltransferase SUV39H1 improved object location and fear conditioning memory performance in aged, but not young-adult mice78. SUV39H1 inhibition in aged animals also restored levels of hippocampal H3K9me3, baseline levels of the synaptic glutamate receptor subunit GluR1 and the density of thin and stubby spines in the hippocampus to young adult levels. These initial findings support the hypothesis that dysregulated epigenetic mechanisms contribute to age-related cognitive dysfunction. In addition, these results demonstrate the potential therapeutic value of targeting histone methylation for rescuing age-related cognitive impairments.
Targeting DNA methylation.
DNA methylation regulates memory processes, including consolidation, extinction103,143,144 and reconsolidation61, as well as synaptic processes such as synaptic scaling (BOX 3), and could be targeted to enhance memory formation. For example, in mice, overexpression of DNMT3A2 before training, similar to HDAC inhibition, resulted in the long-term memory of normally subthreshold training. In addition, inducible overexpression of DNMT3A2 in the hippocampus specifically before extinction training facilitated extinction145. Moreover, reducing endogenous DNMT3A2 activity in the mouse dorsal hippocampus using short hairpin RNAs abrogated extinction memories145. Endogenous DNMT3A2 also declines with age, and restoring DNMT3A2 levels in aged mice reduces age-related memory impairments98. Thus, it will be crucial to identify the loci targeted by DNMT3A2 that are relevant for hippocampus-dependent memory to better understand its mechanism in the context of memory.
Box 3 |. DNA modification and cell-wide synaptic plasticity.
Although DNA methylation enzymes have been implicated in learning and memory, the underlying cellular mechanisms remain fairly unclear. A new perspective was provided by recent studies identifying a relationship between DNa methylation and cell-wide synaptic plasticity. In one study in cultured hippocampal neurons, treatment with the sodium channel tetradotoxin (TTX), which reduces synaptic activity and triggers global synaptic upscaling, increased the expression of ten-eleven translocation enzyme 3 (TET3)240, which oxidizes 5-methylcytosine (5mC) into 5-hydroxymethylcytosine (5hmC) to initiate DNA demethylation. Indeed, TET3 was crucial for homeostatic synaptic upscaling, a cell-wide synaptic plasticity mechanism (distinct from synapse-specific, Hebbian forms of plasticity)241. Short hairpin RNA-mediated knockdown of TET3 considerably increased the amplitude of miniature glutamatergic excitatory postsynaptic currents (mEPSCs) measured by whole-cell patch clamp240. By contrast, neurons overexpressing TET3 exhibited smaller mEPSC amplitudes, suggesting that increases and decreases in the levels of TET3 bidirectionally affect excitatory synaptic transmission. Importantly, these effects on synaptic transmission were attributable to changes in DNA oxidation, but not to changes in oxidation-independent functions that TET enzymes are known to have.
Similar results were reported in a study examining DNA methyltransferases (DNMTs)242. The small-molecule DNMT competitive inhibitor RG108 drove synaptic upscaling in cultures of cortical pyramidal neurons, and knockdown of Dnmt1 and Dnmt3a blocked this effect. In contrast to the study in hippocampal neurons described above240, in which TTX increased the expression of TET3, but not TET1 or TET2, this study in cortical neurons242 showed that chronic TTX treatment increased the expression of TET1, but not TET3 (or DNMT1 or DNMT3). This discrepancy could be attributable to many factors but does suggest that cell-type specific mechanisms may be in play. Together, these two studies, and earlier studies implicating methyl CpG-binding protein 2 (MeCP2), which binds methylated DNa243–245, reveal a more causal relationship between DNA methylation mechanisms and synaptic scaling.
What these synaptic upscaling observations imply about normal learning and memory processes in the brain is not clear. Nevertheless, these results provide new and unusual insights into the regulation of cell-wide synaptic plasticity by epigenetic mechanisms and potentially provide new insight into understanding how manipulations of histone modification or DNA methylation could drive long-term memory for otherwise subthreshold learning events; promote formation of long-term memories that persist beyond the normal lifespan of other memories; enable more neurons than normal to be engaged during learning; or gate the encoding of additional features during learning (observations discussed in the main text).
The above examples demonstrate the ability of several epigenetic enzymes to push the boundaries of memory processes. Although several target genes that are necessary for these memory-enhancing effects have been identified117,146, the precise mechanisms underlying their effects are unknown. Manipulations targeting epigenetic processes may potentially enhance memory by inducing persistent changes in the structure and function of synapses.
Epigenetics and the synapse
Over the past decade, there has been an emphasis on identifying epigenetic modifiers and remodellers involved in memory processes and synaptic plasticity. Given their important effects, epigenetic mechanisms have more recently begun to be integrated into the framework of cellular theories of memory. To better incorporate epigenetics into this framework, researchers have begun to examine the role of epigenetics in molecular processes that regulate synaptic structure and function. Here, we review theories of synaptic tagging and mechanisms of synapse-to-nucleus signalling in relation to memory formation and postulate the potential role that epigenetic mechanisms may have in these cellular processes. We highlight reports that begin to address how altering the chromatin landscape affects the expression and activity of synapse-related proteins to affect synaptic plasticity and memory formation. Other epigenetic mechanisms that do not alter chromatin structure but that have been implicated in memory processes are discussed in BOX 4. Understanding the functional relationship between the epigenome and synaptic structure and function will be necessary to better understand fundamental aspects of memory and disorders associated with memory dysfunction.
Box 4 |. Additional epigenetic mechanisms in memory processes to explore.
There is still much to be discovered regarding the finer details of the epigenetic mechanisms discussed in this article, and new epigenetic mechanisms involved in memory processes are still being identified.
For example, accumulating evidence suggests that several types of RNA species, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), can regulate synapse-related gene expression and memory formation35,143,246–249, with their ability to regulate neural gene expression at least partly attributable to their epigenetic functions. For example, the expression of extra-coding rNAs transcribed from DNa overlapping with the boundaries of the immediate-early gene Fos is necessary for formation of fear memory37. Fos extra-coding RNA molecules may interact with DNMTs and thus direct site-specific DNa methylation and Fos gene regulation. In addition, although not yet seen in the context of memory processing, some evidence from embryonic mouse fibroblasts suggests that certain enhancer rNAs interact with CREB-binding protein (CBP) to stimulate histone acetylation and transcription250.
Moving forward, focus should be placed on understanding how gene regulators interact with one another and coordinate their influence on transcription. in one example of such efforts, deletion of Hdac3 (encoding histone deacetylase 3) from the hippocampus ameliorated impairments in both memory and synaptic plasticity that were caused by mutations of the gene encoding the BAF53B subunit of the BRG1-associated factor (BAF) nucleosome-remodelling complex251. One possible explanation for this rescue is that HDAC3 deletion could have led to enhanced histone acetylation at memory-related genes that may have promoted a permissive chromatin structure despite the absence of normal nucleosome remodelling. Alternatively, BAF53B disruption might have impaired memory by preventing histone-acetyltransferase-dependent histone acetylation, which was restored by the deletion of HDAC3.
The complexity of gene regulation by epigenetic mechanisms such as these must be thoroughly investigated; these research pursuits will be critical to elucidating how cells integrate information to induce long-lasting changes in behaviour and should point to novel therapeutic avenues for human disorders associated with mutations in genes encoding neuronal BAF complex subunits, including BAF53B.
Activity-dependent changes that promote long-lasting forms of synaptic strength — for example, LTP — are crucial for encoding and maintaining information17,24. LTP is initiated by the activity of postsynaptic NMDA receptors (NMDARs) and calcium/calmodulin-dependent protein kinase II (CAMKII). LTP is stabilized through the activation of cell surface receptors on a potentiated subpopulation of dendritic spines, which induces changes in actin polymerization23,147–151. These proteins include several cell adhesion proteins, such as integrins22,152, cadherins21,153–155 and neurexins18,156, which mediate the cell–cell and cell–extracellular matrix interactions that are crucial for synaptic plasticity and memory formation (FIG. 2). In addition, cadherin adhesion molecules form trans-synaptic interactions and, following neuronal activity, become increasingly localized at the synaptic membrane and promote AMPA receptor (AMPAR) stabilization157. Signalling pathways downstream from these surface receptors, together with the activity of ionotropic glutamate receptors such as AMPARs, promote the local disassembly of the cytoskeleton and trafficking of additional 4 network and enable synaptic proteins, such as CAMKII, to alter dendritic spine activity.
Fig. 2 |. synaptic plasticity and interactions between the epigenome and synapse.
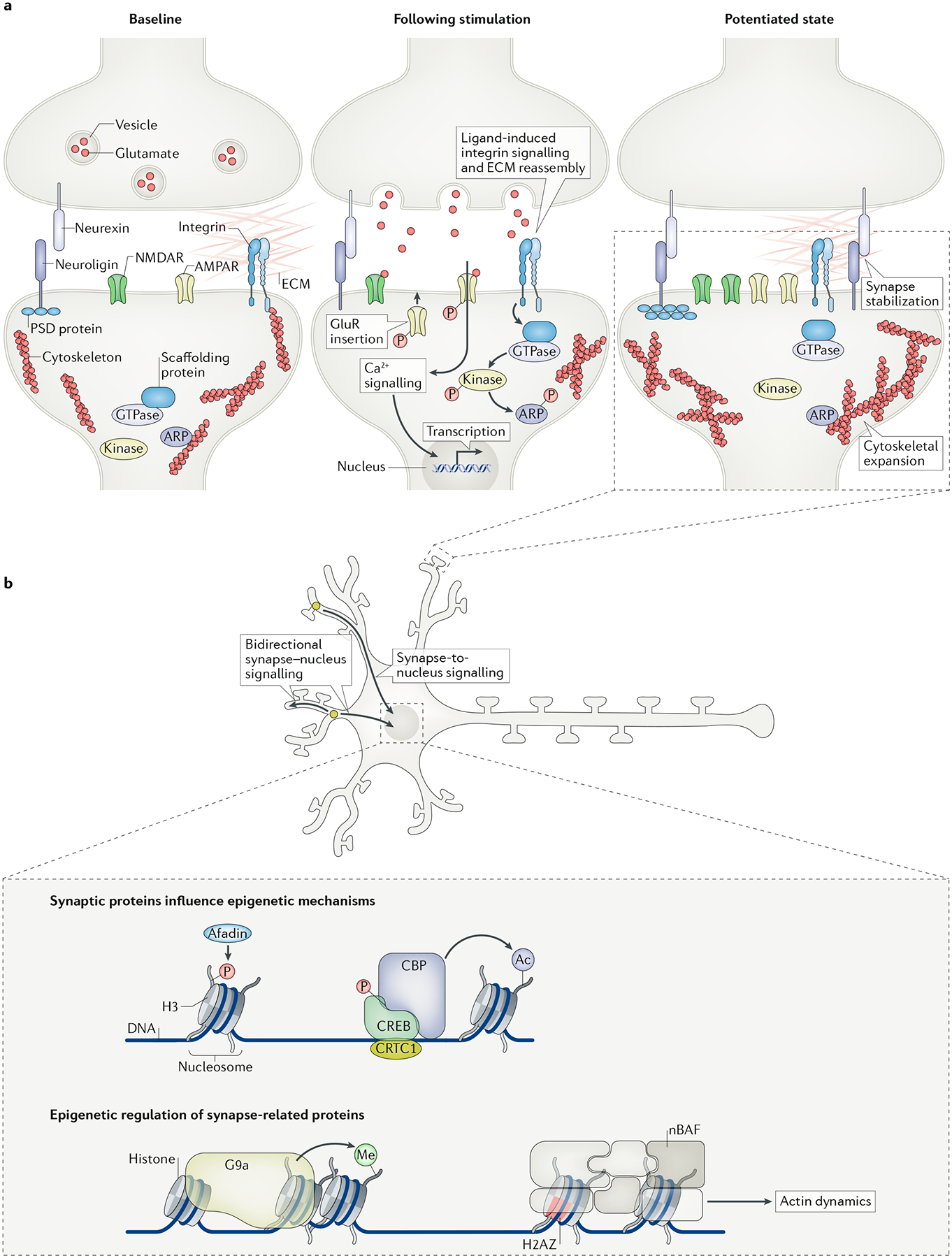
a | Dendritic spines are filopodia-actin (F-actin)-rich protrusions that receive information from neighbouring cells via several types of surface receptor (left). The strength of synaptic transmission correlates with the size of dendritic spines. Synapses undergo changes in actin polymerization to rapidly expand the dendritic spine head and translocate synaptic proteins (middle). Multiple signalling cascades are activated to facilitate different aspects of synaptic plasticity. First, the activation of the ionotropic NMDA receptor (NMDAR) initiates several calcium-dependent signalling cascades important for regulation of synaptic protein activity and nuclear transcription. Second, the interaction of presynaptic neurexin and postsynaptic neuroligin cell adhesion molecules stabilizes transient synaptic contacts for synapse specification. Third, integrin receptors detect extracellular matrix (ECM) signals and promote the disassembly of cytoskeleton proteins. One downstream integrin mechanism is the activation of cofilin or other actin-related proteins (ARPs), which leads to the depolymerization and reorganization of actin filaments. Fourth, AMPA receptors (AMPARs) are trafficked to the postsynaptic density (PSD). Together, these mechanisms rebuild the dendritic spine head, increase the concentration of glutamatergic receptors and regulate the translocation of synaptic proteins (right). b | Following neuronal stimulation, synaptic proteins can translocate to the nucleus or induce signalling cascades to promote transcription. Although the time points are not fully characterized, epigenetic regulation of transcription is proposed to regulate memory and synaptic plasticity. It is hypothesized that epigenetic mechanisms alter chromatin structure to permit transcription of genes crucial for immediate cellular responses (such as immediate-early genes) and synaptic potentiation. Evidence suggests that synaptic proteins can translocate and interact with epigenetic modifiers to potentially also induce long-lasting changes in gene regulation. Along these same lines, epigenetic mechanisms regulate the expression of synapse-related genes (for example, genes important for cytoskeleton polymerization) and thus influence synaptic structure and function. In order to fully understand how changes to the epigenome and synapse lead to long-lasting changes in behaviour, it is critical to further explore how bidirectional interactions between the synapse and nucleus occur to persistently alter neuronal function. CBP, CREB-binding protein; CREB, cAMP-responsive element-binding protein; CRTC1, CREB-regulated transcriptional co-activator 1; GluR, glutamate receptor; nBAF, neuronal BRG1-associated factor.
The cytoskeleton of the dendritic spine head is rebuilt and expanded through the polymerization of filopodia-actin (F-actin). Signalling cascades initiated by calcium-dependent GTPases, including the RHOA and PAK pathways, prevent depolymerization and drive the organization of actin filaments163. Cell adhesion molecules facilitate activity-dependent reorganization of actin filaments to prevent potentiated spines from returning to their pre-potentiated state. For example, integrins help to link actin filament bundles to the plasma membrane and the extracellular matrix164. Newly synthesized proteins are required to further reconstruct the cytoskeleton and thus to consolidate and maintain the potentiation of synapses23,147,159,165–171.
A key open question is whether epigenetic mechanisms, which can induce stable changes in cell function, initiate and maintain the learning-induced potentiation of synapses through the molecular processes outlined above. Below, we probe the relationship between epigenetics and mechanisms affecting synaptic structure and function.
Epigenetic mechanisms in synaptic tagging.
Long-lasting forms of synaptic plasticity are thought to require gene expression, protein synthesis and the formation of new synaptic connections172,173. However, exactly how changes in gene expression and protein synthesis give rise to synapse-specific alterations is less clear. Frey and Morris hypothesized that synapses are ‘tagged’ following stimulation and then capture newly synthesized gene products that are functionally relevant for plasticity174. This capture of nearby plasticity-related proteins (PRPs) facilitates molecular interactions between neighbouring dendritic spines to transform short-term plasticity into long-term plasticity, which are mediated by E-LTP and L-LTP, respectively175–179.
Extra-coding RNAs.
A form of non-coding, sense-strand rNA that is non-polyadenylated, encoded by a portion of DNA that overlaps the boundaries of another gene.
Recent evidence suggests that epigenetic mechanisms may promote long-term plasticity through synaptic tagging. In a recent study in aged animals, which show hippocampal L-LTP deficits, the HDAC3 inhibitor RGFP966 re-established synaptic tagging and capture and restored L-LTP180. This study investigated whether HDAC3 affects synaptic tagging by using a two-pathway ‘weak-before-strong’ experiment. In this experiment, two stimulating electrodes in the stratum radiatum of the hippocampus induce E-LTP at one synaptic input 1 (S1) of a neural pathway using a weak stimulation, and L-LTP at another synaptic input (S2) of the same neural pathway using a stronger stimulation, while a recording electrode in between the inputs records from distal apical dendrites. Unlike the stimulation at S1, the stronger stimulation at S2 synapses is hypothesized to tag synapses and to promote the expression of PRPs to stabilize potentiation. The application of RGFP966 to these hippocampal slices transformed E-LTP at the weakly stimulated S1 input into L-LTP180. This finding suggests that HDAC3 inhibition may promote the expression of PRPs that are typically expressed following the stronger stimulation as at S2. However, whether HDACs in aged or young neurons deacetylate transcription factors and/or co-regulators, and/or histones, is currently unclear. Nevertheless, this work raises the possibility that epigenetic enzymes have a role in synaptic tagging and capture processes, potentially by regulating nuclear gene expression.
Enhancer RNAs.
A form of non-coding rNA transcribed from active enhancers. They can control mrNA transcription, challenging the idea that enhancers are merely sites of transcription factor assembly.
Notably, local translation in dendrites can be affected by RNA modifications, which represent another type of epigenetic mechanism181–183. Epigenetic mechanisms are therefore hypothesized to regulate local translation in dendrites to assist synaptic tagging and potentiation and to facilitate the transcription of PRP-encoding mRNAs in the nucleus. Further experiments — in particular, looking at the effects of manipulating the localization of epigenetic enzymes in the cell — should shed light on these potential mechanisms.
From the synapse to the epigenome.
With the induction of potentiation, synapses are hypothesized to induce signalling cascades to alter gene expression to enable synapse-specific plasticity. One proposed mechanism for synapse-to-nucleus signalling is the activity-dependent nuclear translocation of synaptic proteins, which then alter transcription160,169,184–189. Consistent with this proposal, synaptic stimulation induces the translocation of the CREB-regulated transcriptional co-activator 1 (CRTC1) from dendrites to the nucleus, where it assists in the regulation of a set of CREB target genes190. However, few studies have examined how synaptic proteins that shuttle to the nucleus may induce long-lasting changes in chromatin structure186,190–192.
Activity-induced synaptic proteins may influence epigenetic mechanisms. For example, expression of brain-specific fibroblast growth factor 1B (FGF1B), which is required for CA3–CA1 LTP and hippocampus-dependent learning, depends on CRTC1 in the nucleus. Whereas weak memory training induces only transient expression of FGF1B (mRNA and protein), strong training leads to sustained FGF1B expression. Following (weak or strong) training, the HDAC3–nuclear receptor co-repressor (NCOR) complex is removed from the Fgf1b promoter, and phosphorylated CREB and CBP are recruited in its place to induce transient expression of Fgf1b (0.5–1 hour following training). Unlike weak training, however, strong training leads to subsequent CRTC1-mediated exchange of CBP for the HAT KAT5 on the Fgf1b promoter, which in turn induces persistent expression of Fgf1b (2 hours following training)193. This KAT5 substitution was required for hippocampal synaptic plasticity and memory enhancement.
E3 ubiquitin ligase.
A protein that facilitates the interaction of a target (or substrate) protein with an ubiquitin-conjugating e2 enzyme to enable the transfer of ubiquitin to the target protein.
CRTC1 is not the only synaptic protein with a potential role in regulating epigenetics. Following neuronal stimulation, the synaptic protein afadin shuttles to the nucleus to promote the phosphorylation of H3S10, a histone modification that transforms condensed heterochromatin to provide a more transcriptionally permissive, euchromatin state194,195, and this epigenetic mechanism is required for dendritic spine remodelling196.
Synaptophysin.
A synaptic vesicle membrane protein that is ubiquitously expressed throughout the brain and has a role in synapse formation.
These are some of the first findings to demonstrate that nuclear translocation of synaptic proteins may underlie epigenetic processes required for memory. Further research examining how information is transported from the synapse to the nucleus will be needed to understand how synaptic signals induce learning-related gene expression.
INTACT.
(isolation of nuclei tagged in specific cell types). A method to isolate nuclei tagged in specific cell types for further examination for specific proteins or rNAs or high-throughput sequencing.
Epigenetic regulation of the synapse: transmembrane proteins.
Integrins are transmembrane adhesion receptors that modulate dendritic morphology by mediating signals from the extracellular matrix and interacting with diffusible factors such as oestrogen and brain-derived neurotrophic factor (BDNF)152,197,198. Integrin activation induces postsynaptic RHO GTPase signalling, thus activating LIM domain kinase (LIMK), which in turn deactivates the actin-filament-severing cofilin, to regulate cytoskeleton dynamics22,152,199–202. The induction of this cytoskeleton-regulating pathway promotes LTP via the insertion of glutamatergic receptors and the expansion of the postsynaptic spine densities160,203–206. Although actin-related proteins and their upstream effectors have been heavily implicated in synaptic plasticity, there is less information regarding the epigenetic regulation of the expression and activity of actin-regulating proteins.
TRAP.
(Translating ribosome affinity purification). A ribosome-tagging method in which a fusion protein binds ribosomal proteins and immunoprecipitation purification processes isolate biologically relevant mrNA transcripts.
Assay for transposase-accessible chromatin using sequencing.
(ATAC-seq). A method for mapping genome-wide chromatin accessibility. A transposase inserts sequencing adaptors into accessible regions of chromatin, before adaptor-ligated DNA fragments are sequenced.
The nBAF subunit of the BAF53B NRC may have a role in the activation of actin-regulating pathways and the stabilization of potentiated synapses following the induction of learning115. In the hippocampus of wild-type mice, theta-burst stimulation (TBS) induces LTP and alters synaptic morphology by affecting actin regulation: TBS promotes increases in spines containing phosphorylated p21-activated kinase (PAK) and downstream cofilin204. However, compared with their wild-type counterparts, Baf53b+/− mice do not express TBS-induced LTP, and TBS-induced inactivation (that is, phosphorylation) of the actin-severing protein cofilin in the postsynaptic density is reduced. Baf53b+/− mice also fail to show activity-dependent increases in hippocampal expression of genes involved in the postsynaptic cell membrane and cytoskeleton115. Thus, the structural and functional LTP deficits in Baf53b+/− mice may result from altered activity-dependent-expression of synapse-related proteins. In a follow-up study, deficits in hippocampus-dependent memory and hippocampal LTP in mice lacking the BAF53B subdomain 2 were rescued by overexpression of a phosphomimetic of cofilin in the hippocampus114. Consistent with this work, overexpression of BAF53B within the lateral amygdala led to enhanced memory formation and thin-spine density, whereas Baf53b knockdown within the lateral amygdala impaired fear memory formation113. Together, these studies demonstrate that NRCs have a key role in synaptic plasticity and memory, perhaps by regulating actin dynamics.
Zinc-finger proteins.
(ZFPs). A large family of transcription factors with finger-like DNA-sequence-specific domains. Fusion of a DNA-binding domain specific for an 18–20 bp genomic locus to a chromatin-modifying enzyme enables targeted epigenetic regulation.
From the epigenome to the synapse: GluRs.
The reorganization of the cytoskeleton expands dendritic spine heads and is associated with the trafficking of additional AMPARs to the synapse. The trafficking of AMPARs and NMDAR-dependent increases in Ca2+ lead to long-lasting forms of LTP19,20,207–211. Epigenetic regulation of GluR subunit expression occurs during critical periods of synaptic remodelling, including development212, stress213,214 and following drug exposure215. Understanding how extracellular signals influence the epigenetic regulation of synaptic GluR expression in these contexts is necessary.
After exposure to stress, AMPARs are ubiquitylated for degradation, and recent evidence suggests that this ubiquitylation results from epigenetic changes214. In rats, repeated stress led to glucocorticoid-receptor-dependent increases in expression of Hdac2 and increased occupancy of HDAC2 on the G9a promoter, reducing G9a expression. Normally, G9a methylates the promoter of the gene encoding an e3 ubiquitin ligase, reducing its expression; thus, in stressed animals, upregulated HDAC2 reduces G9a expression and indirectly promotes the expression of the E3 ubiquitin ligase. In stressed rats, knockdown of Hdac2 prevented the increases in E3 and the degradation of AMPARs in the prefrontal cortex and ameliorated the stress-induced disruption of AMPAR-mediated plasticity216,217. These and other studies suggest that stress hormone signalling affects epigenetic regulators (including neural-restrictive silencer factor (NRSF) as well as HDAC2) that then persistently alter synaptic receptor expression, either directly or indirectly213,218,219. The precise molecular mechanisms giving rise to the persistence of these effects on receptor expression, and whether these effects are established and/or maintained by long-lasting epigenomic changes, remain to be understood.
From the epigenome to the synapse: presynaptic proteins.
Neurexins are presynaptic cell adhesion molecules that interact with various postsynaptic ligands to govern the connectivity of synaptic circuits (FIG. 2). Each of the thousands of expressed neurexin isoforms may show different specificities for various postsynaptic ligands (for example, different neuroligins) and thus have different effects156. For example, the inclusion of alternative splicing sequence 4 (SS4) in neurexin 1 interferes with postsynaptic AMPAR trafficking and represses long-term synaptic plasticity220. Histone modifications can regulate exon splicing by affecting either the recruitment of splice machinery or mechanisms of transcriptional elongation221,222, although, to date, little evidence demonstrates a definitively causal role for epigenetic mechanisms in regulating mRNA splicing and isoform expression in memory.
Nevertheless, shedding new light on this topic, a recent study in the mouse dentate gyrus reported that neuronal activity drives the recruitment of the HDAC2–p66a complex onto exon 22 of Nrxn1 to promote the inclusion of SS4 in neurexin 1 (REF.223). Subsequently, the histone methyltransferase SUV39H1 preserves SS4 inclusion by trimethylating H3K9 on exon 22 of Nrxn1. NRXN1 that contained SS4 showed reduced binding affinity for postsynaptic neuroligin 1B (NLGN1B) and synaptophysin clustered on NLGN1B-expressing cells. However, these reductions in trans-synaptic interactions were not seen in neurons from Suv39h1+/− mice, supporting the hypothesis that mechanisms downstream from histone methylation regulate synapse formation. Moreover, in mice that underwent context-specific fear conditioning followed by distraction training (that is, exposure to a neutral context following the initial recall test), knockdown of Suv39h1 in the dentate gyrus reduced freezing in the shock-associated context compared with wild-type controls. Thus, SUV39H1 activity is crucial for memory preservation, possibly by preventing synaptic rewiring that could interfere with mechanisms of memory preservation. These results provide an example of how epigenetic mechanisms can regulate gene expression, through splicing, to preserve memories through the stabilization of synaptic structure. This study also outlines a possible mechanism by which a histone modification at a specific locus is necessary for memory persistence. Future studies — for example, using CRISPR-targeted approaches (BOX 5) — will be needed to establish a causal relationship between the histone modification and splicing, and to determine whether splicing regulation is unique to memory persistence or occurs in other memory processes.
Box 5 |. Tools to study epigenetic processes.
The advent of recent genetic tools will assist in the pursuit of answering the open questions outlined in this article. For example, deep-sequencing methods such as single-cell RNA sequencing will be useful to analyse how subpopulations within cell types of the brain are uniquely altered during memory formation and updating252. Methods that allow for isolation or manipulation of specific cell types, such as iNTACT (isolation of nuclei tagged in specific cell types) and TrAP (translating ribosome affinity purification), will also prove useful. The best implementation of these techniques would be to overlay sequencing results (for example, RNA sequencing with chromatin immunoprecipitation (ChIP) sequencing) to obtain a more accurate representation of changes in gene expression. Researchers can also turn to Hi-C and ATAC-seq (assay for transposase-accessible chromatin using sequencing) to examine the long-term changes in chromatin structure that enable a memory to be sustained and recalled. To provide more causal evidence of how epigenetic mechanisms regulate cellular and behavioural plasticity, researchers could use tools such as zinc-finger proteins (ZFPs) and CRISPR–Cas9 to induce locus-specific epigenetic modulations139,249,253,254. Furthermore, temporally specific manipulations such as optogenetic control of transcription factors and epigenetic enzymes will enable the control of gene expression within specific brain regions255.
To further advance this work, the field must use techniques that control locus-specific epigenetic mechanisms in a temporally specific manner. Given the sophisticated techniques available, the role of epigenetics in cellular and behavioural functions can be examined simultaneously. For example, changes in gene expression can be induced by CRISPR–dCas9, and changes in the function of individual cells can be assessed using calcium imaging. Ultimately, understanding the epigenetic regulation of synaptic plasticity may reveal gene regulation mechanisms that, when disrupted, lead to abnormalities in cellular function that are observed in cognitive disorders.
Conclusions and outstanding questions
As discussed above, epigenetic mechanisms are critical modulators of synaptic plasticity and memory. In turn, activity-dependent synaptic changes engage epigenetic mechanisms. Not only are enzymes that modify the epigenome necessary for various memory processes, but pharmacologically or genetically altering the function of these enzymes dramatically changes the ability of neurons to encode information. Altering epigenetic mechanisms can affect the molecular and cellular mechanisms that establish the usual limits of synaptic plasticity, memory formation and memory persistence, and may perhaps even affect how many neurons are engaged in learning and memory. In turn, the relationship between the epigenome and the synapse is probably an important factor in these processes: synaptic proteins are reported to affect epigenetic mechanisms, and vice versa. Despite correlational demonstrations of these interactions, we are still lacking a fundamental understanding of how they regulate memory.
One clear question is whether there are direct and causal mechanisms of information transfer from the synapse to the epigenome that are necessary for stable changes in neuronal function. That is, are there bona fide synaptic proteins (or other molecules, such as RNA) that are locally translated or released from the synapse upon activity-dependent stimulation that travel to the nucleus? In addition, do synaptic proteins that translocate to the nucleus directly participate in the epigenetic regulation of genes that stabilize changes in synaptic structure and function at the originally engaged synapses? To answer these questions, the coordinated cell-type-specific and locus-specific interactions between synaptic and epigenetic proteins must be examined (BOX 5).
Another key open question is whether persistent changes in the epigenome at specific synaptic protein-encoding genes establish and/or maintain stable changes in synaptic structure and function. Some of the studies discussed above begin to answer this question; however, the field has just begun to investigate how epigenetic control of synapse-related genes is regulated during memory processes. Moreover, we must examine how epigenetic regulation of established memory-associated genes affects synaptic function. Relatedly, whether the epigenome represents a form of molecular memory for past experiences, and whether learning-induced epigenomic modifications directly participate in the encoding of information that is required for the engram, are unknown224. Addressing questions such as these will provide insight into the dynamic relationship between the epigenome and synapse and thus will substantially advance our understanding of the molecular mechanisms underlying the formation, persistence and limits of memory.
Acknowledgements
This work was supported by the US National Institutes of Health (National Institute on Aging grants AG051807, AG050787 and AG054349; National Institute of Mental Health grant MH101491; and National Institute on Drug Abuse grant DA025922). Initial illustrations were designed by P. Schiffmacher of Schiffmacher Illustration & Design. The authors thank A. López and T. Hemstedt for their intellectual contributions to this Review.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Neuroscience thanks S. Bonn and F. Lubin, and the other anonymous reviewer(s), for their contribution to the peer review of this work.
References
- 1.Kandel ER, Dudai Y & Mayford MR The molecular and systems biology of memory. Cell 157, 163–186 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Johansen JP, Cain CK, Ostroff LE & LeDoux JE Molecular mechanisms of fear learning and memory. Cell 147, 509–524 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alberini CM Transcription factors in long-term memory and synaptic plasticity. Physiol. Rev 89, 121–145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hernandez PJ & Abel T The role of protein synthesis in memory consolidation: progress amid decades of debate. Neurobiol. Learn. Mem 89, 293–311 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Day JJ & Sweatt JD Epigenetic mechanisms in cognition. Neuron 70, 813–829 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldberg AD, Allis CD & Bernstein E Epigenetics: a landscape takes shape. Cell 128, 635–638 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Kouzarides T Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Reik W Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Swank MW & Sweatt JD Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning. J. Neurosci 21, 3383–3391 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levenson JM & Sweatt JD Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci 6, 108–118 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Guan Z et al. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 111, 483–493 (2002). [DOI] [PubMed] [Google Scholar]
- 12.Barrett RM & Wood MA Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn. Mem 15, 460–467 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frankland PW & Josselyn SA Neuroscience: in search of the memory molecule. Nature 535, 41–42 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Josselyn SA & Frankland PW Memory allocation: mechanisms and function. Annu. Rev. Neurosci 41, 389–413 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Penzes P & Rafalovich I Regulation of the actin cytoskeleton in dendritic spines. Adv. Exp. Med. Biol 970, 81–95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lynch G, Kramár EA & Gall CM Protein synthesis and consolidation of memory-related synaptic changes. Brain Res. 1621, 62–72 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Malenka RC Synaptic plasticity in the hippocampus: LTP and LTD. Cell 78, 535–538 (1994). [DOI] [PubMed] [Google Scholar]
- 18.Südhof TC Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carroll RC, Beattie EC, von Zastrow M & Malenka RC Role of AMPA receptor endocytosis in synaptic plasticity. Nat. Rev. Neurosci 2, 315–324 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Malinow R & Malenka RC AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci 25, 103–126 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Riehl R et al. Cadherin function is required for axon outgrowth in retinal ganglion cells in vivo. Neuron 17, 837–848 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Park YK & Goda Y Integrins in synapse regulation. Nat. Rev. Neurosci 17, 745–756 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Konietzny A, Bär J & Mikhaylova M Dendritic actin cytoskeleton: structure, functions, and regulations. Front. Cell. Neurosci 11, 147 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lynch G, Rex CS & Gall CM LTP consolidation: substrates, explanatory power, and functional significance. Neuropharmacology 52, 12–23 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Bastle RM & Maze IS Chromatin regulation in complex brain disorders. Curr. Opin. Behav. Sci 25, 57–65 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.López AJ & Wood MA Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front. Behav. Neurosci 9, 100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwapis JL & Wood MA Epigenetic mechanisms in fear conditioning: implications for treating post-traumatic stress disorder. Trends Neurosci 37, 706–720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White AO & Wood MA Does stress remove the HDAC brakes for the formation and persistence of long-term memory? Neurobiol. Learn. Mem 112, 61–67 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker DM & Nestler EJ Neuroepigenetics and addiction. Handb. Clin. Neurol 148, 747–765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zovkic IB, Paulukaitis BS, Day JJ, Etikala DM & Sweatt JD Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature 515, 582–586 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stefanelli G et al. Learning and age-related changes in genome-wide H2A.Z binding in the mouse hippocampus. Cell Rep. 22, 1124–1131 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maze I et al. Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87, 77–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bharadwaj R et al. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron 84, 997–1008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watson LA & Tsai L-H In the loop: how chromatin topology links genome structure to function in mechanisms underlying learning and memory. Curr. Opin. Neurobiol 43, 48–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Widagdo J et al. Experience-dependent accumulation of N6-methyladenosine in the prefrontal cortex is associated with memory processes in mice. J. Neurosci 36, 6771–6777 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bero AW et al. Early remodeling of the neocortex upon episodic memory encoding. Proc. Natl Acad. Sci. USA 111, 11852–11857 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savell KE et al. Extra-coding RNAs regulate neuronal DNA methylation dynamics. Nat. Commun 7, 12091 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Q et al. MicroRNA-mediated disruption of dendritogenesis during a critical period of development influences cognitive capacity later in life. Proc. Natl Acad. Sci. USA 114, 9188–9193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sim S-E et al. The brain-enriched microRNA miR-9–3p regulates synaptic plasticity and memory. J. Neurosci 36, 8641–8652 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heyer MP & Kenny PJ Corticostriatal microRNAs in addiction. Brain Res 1628, 2–16 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Lee K et al. An activity-regulated microRNA, miR-188, controls dendritic plasticity and synaptic transmission by downregulating neuropilin-2. J. Neurosci 32, 5678–5687 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woldemichael BT et al. The microRNA cluster miR-183/96/182 contributes to long-term memory in a protein phosphatase 1-dependent manner. Nat. Commun 7, 12594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qureshi IA & Mehler MF Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci 13, 528–541 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agranoff BW, Davis RE, Casola L & Lim R Actinomycin D blocks formation of memory of shock-avoidance in goldfish. Science 158, 1600–1601 (1967). [DOI] [PubMed] [Google Scholar]
- 45.Silva AJ, Kogan JH, Frankland PW & Kida S CREB and memory. Annu. Rev. Neurosci 21, 127–148 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Yin JC & Tully T CREB and the formation of long-term memory. Curr. Opin. Neurobiol 6, 264–268 (1996). [DOI] [PubMed] [Google Scholar]
- 47.Chrivia JC et al. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365, 855–859 (1993). [DOI] [PubMed] [Google Scholar]
- 48.Bourtchouladze R et al. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc. Natl Acad. Sci. USA 100, 10518–10522 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oike Y et al. Truncated CBP protein leads to classical Rubinstein-Taybi syndrome phenotypes in mice: implications for a dominant-negative mechanism. Hum. Mol. Genet 8, 387–396 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Alarcon J et al. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein–Taybi syndrome and its amelioration. Neuron 42, 947–959 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Korzus E Rubinstein-Taybi syndrome and epigenetic alterations. Adv. Exp. Med. Biol 978, 39–62 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korzus E, Rosenfeld MG & Mayford M CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42, 961–972 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates the importance of histone acetylation in memory formation: transgenic mice with deficient CBP HAT activity exhibit impaired long-term memory, which was rescued by administration of an HDAC inhibitor.
- 53.Wood MA, Attner MA, Oliveira AMM, Brindle PK & Abel T A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn. Mem 13, 609–617 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wood MA et al. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn. Mem 12, 111–119 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barrett RM et al. Hippocampal focal knockout of CBP affects specific histone modifications, long-term potentiation, and long-term memory. Neuropsychopharmacology 36, 1545–1556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malvaez M, Mhillaj E, Matheos DP, Palmery M & Wood MA CBP in the nucleus accumbens regulates cocaine-induced histone acetylation and is critical for cocaine-associated behaviors. J. Neurosci 31, 16941–16948 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maddox SA, Watts CS & Schafe GE p300/CBP histone acetyltransferase activity is required for newly acquired and reactivated fear memories in the lateral amygdala. Learn. Mem 20, 109–119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sando R et al. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151, 821–834 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kwapis JL et al. Context and auditory fear are differentially regulated by HDAC3 activity in the lateral and basal subnuclei of the amygdala. Neuropsychopharmacology 42, 1284–1294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gräff J & Tsai L-H Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci 14, 97–111 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Maddox SA & Schafe GE Epigenetic alterations in the lateral amygdala are required for reconsolidation of a Pavlovian fear memory. Learn. Mem 18, 579–593 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li B, Carey M & Workman JL The role of chromatin during transcription. Cell 128, 707–719 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Zhang T, Cooper S & Brockdorff N The interplay of histone modifications — writers that read. EMBO Rep 16, 1467–1481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peixoto L & Abel T The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 38, 62–76 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morris MJ, Mahgoub M, Na ES, Pranav H & Monteggia LM Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. J. Neurosci 33, 6401–6411 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taniguchi M et al. HDAC5 and its target gene, Npas4, function in the nucleus accumbens to regulate cocaine-conditioned behaviors. Neuron 96, 130–144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lahm A et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl Acad. Sci. USA 104, 17335–17340 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gräff J et al. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell 156, 261–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Although a reconsolidation-updating paradigm fails to alter remote memories, administration of an HDAC2-targeting inhibitor during reconsolidation persistently attenuates remote memories.
- 69.Zocchi L & Sassone-Corsi P SIRT1-mediated deacetylation of MeCP2 contributes to BDNF expression. Epigenetics 7, 695–700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Federman N et al. Nuclear factor κB-dependent histone acetylation is specifically involved in persistent forms of memory. J. Neurosci 33, 7603–7614 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lopez-Atalaya JP & Barco A Can changes in histone acetylation contribute to memory formation? Trends Genet. 30, 529–539 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Rashid AJ, Cole CJ & Josselyn SA Emerging roles for MEF2 transcription factors in memory. Genes Brain Behav. 13, 118–125 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Sartor GC, Powell SK, Brothers SP & Wahlestedt C Epigenetic readers of lysine acetylation regulate cocaine-induced plasticity. J. Neurosci 35, 15062–15072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Korb E, Herre M, Zucker-Scharff I, Darnell RB & Allis CD BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat. Neurosci 18, 1464–1473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fischer A, Sananbenesi F, Wang X, Dobbin M & Tsai L-H Recovery of learning and memory is associated with chromatin remodelling. Nature 447, 178–182 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Schaefer A et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron 64, 678–691 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; Knockdown of GLP–G9a leads to derepression of genes expressed in adult neurons and leads to deficits in learning.
- 77.Gupta-Agarwal S et al. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J. Neurosci 32, 5440–5453 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Snigdha S et al. H3k9me3 inhibition improves memory, promotes spine formation, and increases BDNF levels in the aged hippocampus. J. Neurosci 36, 3611–3622 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jenuwein T & Allis CD Translating the histone code. Science 293, 1074–1080 (2001). [DOI] [PubMed] [Google Scholar]
- 80.Zhang Y & Reinberg D Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343–2360 (2001). [DOI] [PubMed] [Google Scholar]
- 81.Maze I et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Covington HE et al. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron 71, 656–670 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Anderson EM et al. Overexpression of the histone dimethyltransferase G9a in nucleus accumbens shell increases cocaine self-administration, stress-induced reinstatement, and anxiety. J. Neurosci 38, 803–813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gupta S et al. Histone methylation regulates memory formation. J. Neurosci 30, 3589–3599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kerimoglu C et al. KMT2A and KMT2B mediate memory function by affecting distinct genomic regions. Cell Rep. 20, 538–548 (2017). [DOI] [PubMed] [Google Scholar]
- 86.Kerimoglu C et al. Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J. Neurosci 33, 3452–3464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Damez-Werno DM et al. Histone arginine methylation in cocaine action in the nucleus accumbens. Proc. Natl Acad. Sci. USA 113, 9623–9628 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Benevento M et al. Histone methylation by the Kleefstra syndrome protein EHMT1 mediates homeostatic synaptic scaling. Neuron 91, 341–355 (2016). [DOI] [PubMed] [Google Scholar]
- 89.Jakovcevski M et al. Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J. Neurosci 35, 5097–5108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jarome TJ & Lubin FD Histone lysine methylation: critical regulator of memory and behavior. Rev. Neurosci 24, 375–387 (2013). [DOI] [PubMed] [Google Scholar]
- 91.Levenson JM et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem 281, 15763–15773 (2006). [DOI] [PubMed] [Google Scholar]
- 92.Monsey MS, Ota KT, Akingbade IF, Hong ES & Schafe GE Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLOS ONE 6, e19958 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maddox SA, Watts CS & Schafe GE DNA methyltransferase activity is required for memory-related neural plasticity in the lateral amygdala. Neurobiol. Learn. Mem 107, 93–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miller CA et al. Cortical DNA methylation maintains remote memory. Nat. Neurosci 13, 664–666 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; Associative learning induces gene-specific cortical hypermethylation in rats, and DNMT inhibition 1 month after learning disrupted remote memory.
- 95.Lubin FD, Roth TL & Sweatt JD Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci 28, 10576–10586 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Biergans SD, Jones JC, Treiber N, Galizia CG & Szyszka P DNA methylation mediates the discriminatory power of associative long-term memory in honeybees. PLOS ONE 7, e39349 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brueckner B et al. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res. 65, 6305–6311 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Oliveira AMM, Hemstedt TJ & Bading H Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci 15, 1111–1113 (2012). [DOI] [PubMed] [Google Scholar]
- 99.Sultan FA, Wang J, Tront J, Liebermann DA & Sweatt JD Genetic deletion of Gadd45b, a regulator of active DNA demethylation, enhances long-term memory and synaptic plasticity. J. Neurosci 32, 17059–17066 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ma DK et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323, 1074–1077 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leach PT et al. Gadd45b knockout mice exhibit selective deficits in hippocampus-dependent long-term memory. Learn. Mem 19, 319–324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gavin DP, Chase KA & Sharma RP Active DNA demethylation in post-mitotic neurons: a reason for optimism. Neuropharmacology 75, 233–245 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rudenko A et al. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 79, 1109–1122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Halder R et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat. Neurosci 19, 102–110 (2016). [DOI] [PubMed] [Google Scholar]; Rapid de novo DNA methylation and demethylation occur in the hippocampus in response to fear conditioning.
- 105.Oliveira AMM DNA methylation: a permissive mark in memory formation and maintenance. Learn. Mem 23, 587–593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Miller CA & Sweatt JD Covalent modification of DNA regulates memory formation. Neuron 53, 857–869 (2007). [DOI] [PubMed] [Google Scholar]
- 107.Day JJ et al. DNA methylation regulates associative reward learning. Nat. Neurosci 16, 1445–1452 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Webb WM et al. Dynamic association of epigenetic H3K4me3 and DNA 5hmC marks in the dorsal hippocampus and anterior cingulate cortex following reactivation of a fear memory. Neurobiol. Learn. Mem 142, 66–78 (2017). [DOI] [PubMed] [Google Scholar]
- 109.Hemstedt TJ, Lattal KM & Wood MA Reconsolidation and extinction: Using epigenetic signatures to challenge conventional wisdom. Neurobiol. Learn. Mem 142, 55–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Spruijt CG & Vermeulen M DNA methylation: old dog, new tricks? Nat. Struct. Mol. Biol 21, 949–954 (2014). [DOI] [PubMed] [Google Scholar]
- 111.Hargreaves DC & Crabtree GR ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu JI et al. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56, 94–108 (2007). [DOI] [PubMed] [Google Scholar]
- 113.Yoo M et al. BAF53b, a neuron-specific nucleosome remodeling factor, is induced after learning and facilitates long-term memory consolidation. J. Neurosci 37, 3686–3697 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vogel Ciernia A et al. Mutation of neuron-specific chromatin remodeling subunit BAF53b: rescue of plasticity and memory by manipulating actin remodeling. Learn. Mem 24, 199–209 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vogel-Ciernia A et al. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat. Neurosci 16, 552–561 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; Genetic manipulation of the nBAF nucleosome-remodelling subunit BAF53B alters hippocampal gene expression and leads to impairments in both long-term memory and long-lasting forms of synaptic plasticity.
- 116.Borrelli E, Nestler EJ, Allis CD & Sassone-Corsi P Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McQuown SC et al. HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci 31, 764–774 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Guan J-S et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Feng J et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci 13, 423–430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Levenson JM et al. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem 279, 40545–40559 (2004). [DOI] [PubMed] [Google Scholar]
- 121.Peleg S et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328, 753–756 (2010). [DOI] [PubMed] [Google Scholar]
- 122.Vecsey CG et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J. Neurosci 27, 6128–6140 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stefanko DP, Barrett RM, Ly AR, Reolon GK & Wood MA Modulation of long-term memory for object recognition via HDAC inhibition. Proc. Natl Acad. Sci. USA 106, 9447–9452 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Malvaez M et al. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl Acad. Sci. USA 110, 2647–2652 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rogge GA, Singh H, Dang R & Wood MA HDAC3 is a negative regulator of cocaine-context-associated memory formation. J. Neurosci 33, 6623–6632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haettig J et al. HDAC inhibition modulates hippocampus-dependent long-term memory for object location in a CBP-dependent manner. Learn. Mem 18, 71–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bieszczad KM & Weinberger NM Representational gain in cortical area underlies increase of memory strength. Proc. Natl Acad. Sci. USA 107, 3793–3798 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bieszczad KM et al. Histone deacetylase inhibition via RGFP966 releases the brakes on sensory cortical plasticity and the specificity of memory formation. J. Neurosci 35, 13124–13132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bredy TW et al. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn. Mem 14, 268–276 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stafford JM, Raybuck JD, Ryabinin AE & Lattal KM Increasing histone acetylation in the hippocampus-infralimbic network enhances fear extinction. Biol. Psychiatry 72, 25–33 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fujita Y et al. Vorinostat, a histone deacetylase inhibitor, facilitates fear extinction and enhances expression of the hippocampal NR2B-containing NMDA receptor gene. J. Psychiatr. Res 46, 635–643 (2012). [DOI] [PubMed] [Google Scholar]
- 132.Bredy TW & Barad M The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn. Mem 15, 39–45 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Itzhak Y, Liddie S & Anderson KL Sodium butyrate-induced histone acetylation strengthens the expression of cocaine-associated contextual memory. Neurobiol. Learn. Mem 102, 34–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lattal MK, Barrett RM & Wood MA Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav. Neurosci 121, 1125–1131 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lubin FD & Sweatt JD The IκB kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron 55, 942–957 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Blank M et al. TrkB blockade in the hippocampus after training or retrieval impairs memory: protection from consolidation impairment by histone deacetylase inhibition. J. Neural Transm 123, 159–165 (2016). [DOI] [PubMed] [Google Scholar]
- 137.Malvaez M et al. Habits are negatively regulated by histone deacetylase 3 in the dorsal striatum. Biol. Psychiatry 84, 383–392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Penner MR, Roth TL, Barnes CA & Sweatt JD An epigenetic hypothesis of aging-related cognitive dysfunction. Front. Aging Neurosci 2, 9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kwapis JL et al. Epigenetic regulation of the circadian gene Per1 contributes to age-related changes in hippocampal memory. Nat. Commun 9, 3323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Spiegel AM, Sewal AS & Rapp PR Epigenetic contributions to cognitive aging: disentangling mindspan and lifespan. Learn. Mem 21, 569–574 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jarome TJ, Perez GA, Hauser RM, Hatch KM & Lubin FD EZH2 methyltransferase activity controls Pten expression and mTOR signaling during fear memory reconsolidation. J. Neurosci 38, 7635–7648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Balemans MCM et al. Hippocampal dysfunction in the Euchromatin histone methyltransferase 1 heterozygous knockout mouse model for Kleefstra syndrome. Hum. Mol. Genet 22, 852–866 (2013). [DOI] [PubMed] [Google Scholar]
- 143.Baker-Andresen D et al. Persistent variations in neuronal DNA methylation following cocaine self-administration and protracted abstinence in mice. Neuroepigenetics 4, 1–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li X et al. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc. Natl Acad. Sci. USA 111, 7120–7125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; Fear extinction induces a redistribution of 5hmC that is mediated by TET3 in the infralimbic prefrontal cortex.
- 145.Oliveira AMM, Hemstedt TJ, Freitag HE & Bading H Dnmt3a2: a hub for enhancing cognitive functions. Mol. Psychiatry 21, 1130–1136 (2016). [DOI] [PubMed] [Google Scholar]
- 146.Hawk JD et al. NR4A nuclear receptors support memory enhancement by histone deacetylase inhibitors. J. Clin. Invest 122, 3593–3602 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Berry KP & Nedivi E Spine dynamics: are they all the same? Neuron 96, 43–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Meyer D, Bonhoeffer T & Scheuss V Balance and stability of synaptic structures during synaptic plasticity. Neuron 82, 430–443 (2014). [DOI] [PubMed] [Google Scholar]
- 149.Caroni P, Donato F & Muller D Structural plasticity upon learning: regulation and functions. Nat. Rev. Neurosci 13, 478–490 (2012). [DOI] [PubMed] [Google Scholar]
- 150.Rex CS et al. Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron 67, 603–617 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Young EJ et al. Selective, retrieval-independent disruption of methamphetamine-associated memory by actin depolymerization. Biol. Psychiatry 75, 96–104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Babayan AH et al. Integrin dynamics produce a delayed stage of long-term potentiation and memory consolidation. J. Neurosci 32, 12854–12861 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tamura K, Shan WS, Hendrickson WA, Colman DR & Shapiro L Structure-function analysis of cell adhesion by neural (N-) cadherin. Neuron 20, 1153–1163 (1998). [DOI] [PubMed] [Google Scholar]
- 154.Bozdagi O, Shan W, Tanaka H, Benson DL & Huntley GW Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron 28, 245–259 (2000). [DOI] [PubMed] [Google Scholar]
- 155.Huntley GW, Gil O & Bozdagi O The cadherin family of cell adhesion molecules: multiple roles in synaptic plasticity. Neuroscientist 8, 221–233 (2002). [DOI] [PubMed] [Google Scholar]
- 156.Südhof TC Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171, 745–769 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Brigidi GS et al. Palmitoylation of δ-catenin by DHHC5 mediates activity-induced synapse plasticity. Nat. Neurosci 17, 522–532 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Paoletti P, Bellone C & Zhou Q NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 14, 383–400 (2013). [DOI] [PubMed] [Google Scholar]
- 159.Lee S-JR, Escobedo-Lozoya Y, Szatmari EM & Yasuda R Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458, 299–304 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Murakoshi H, Wang H & Yasuda R Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472, 100–104 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rudy JW Variation in the persistence of memory: an interplay between actin dynamics and AMPA receptors. Brain Res. 1621, 29–37 (2015). [DOI] [PubMed] [Google Scholar]
- 162.Ye X & Carew TJ Small G protein signaling in neuronal plasticity and memory formation: the specific role of ras family proteins. Neuron 68, 340–361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cingolani LA & Goda Y Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci 9, 344–356 (2008). [DOI] [PubMed] [Google Scholar]
- 164.DeMali KA, Wennerberg K & Burridge K Integrin signaling to the actin cytoskeleton. Curr. Opin. Cell Biol 15, 572–582 (2003). [DOI] [PubMed] [Google Scholar]
- 165.Chen LY, Rex CS, Casale MS, Gall CM & Lynch G Changes in synaptic morphology accompany actin signaling during LTP. J. Neurosci 27, 5363–5372 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim Y et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 442, 814–817 (2006). [DOI] [PubMed] [Google Scholar]
- 167.Yang EJ, Yoon J-H, Min DS & Chung KC LIM kinase 1 activates cAMP-responsive element-binding protein during the neuronal differentiation of immortalized hippocampal progenitor cells. J. Biol. Chem 279, 8903–8910 (2004). [DOI] [PubMed] [Google Scholar]
- 168.Matsuzaki M, Honkura N, Ellis-Davies GCR & Kasai H Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Harvey CD & Svoboda K Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 450, 1195–1200 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rudy JW Actin dynamics and the evolution of the memory trace. Brain Res. 1621, 17–28 (2015). [DOI] [PubMed] [Google Scholar]
- 171.Mirisis AA, Alexandrescu A, Carew TJ & Kopec AM The contribution of spatial and temporal molecular networks in the induction of long-term memory and its underlying synaptic plasticity. AIMS Neurosci. 3, 356–384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Goelet P, Castellucci VF, Schacher S & Kandel ER The long and the short of long-term memory — a molecular framework. Nature 322, 419–422 (1986). [DOI] [PubMed] [Google Scholar]
- 173.Bailey CH, Bartsch D & Kandel ER Toward a molecular definition of long-term memory storage. Proc. Natl Acad. Sci. USA 93, 13445–13452 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Frey U & Morris RG Synaptic tagging and long-term potentiation. Nature 385, 533–536 (1997). [DOI] [PubMed] [Google Scholar]
- 175.Martin KC in Encyclopedia of Neuroscience (ed. Squire LR) 719–723 (Academic Press, 2009). [Google Scholar]
- 176.Martin KC & Kosik KS Synaptic tagging — who’s it? Nat. Rev. Neurosci 3, 813–820 (2002). [DOI] [PubMed] [Google Scholar]
- 177.Wang DO et al. Synapse-and stimulus-specific local translation during long-term neuronal plasticity. Science 324, 1536–1540 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Martin KC Synaptic tagging during synapse-specific long-term facilitation of Aplysia sensory-motor neurons. Neurobiol. Learn. Mem 78, 489–497 (2002). [DOI] [PubMed] [Google Scholar]
- 179.Rogerson T et al. Synaptic tagging during memory allocation. Nat. Rev. Neurosci 15, 157–169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sharma M, Shetty MS, Arumugam TV & Sajikumar S Histone deacetylase 3 inhibition re-establishes synaptic tagging and capture in aging through the activation of nuclear factor kappa B. Sci. Rep 5, 16616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper examines the role of histone acetylation in synaptic tag and capture and finds that inhibition of HDAC3 augments L-LTP and re-establishes synaptic tag and capture in the hippocampus of aged rats.
- 181.Merkurjev D et al. Synaptic N6-methyladenosine (m6A) epitranscriptome reveals functional partitioning of localized transcripts. Nat. Neurosci 21, 1004–1014 (2018). [DOI] [PubMed] [Google Scholar]
- 182.Leighton LJ et al. Experience-dependent neural plasticity, learning, and memory in the era of epitranscriptomics. Genes Brain Behav. 17, e12426 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Cajigas IJ et al. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74, 453–466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tang S & Yasuda R Imaging ERK and PKA activation in single dendritic spines during structural plasticity. Neuron 93, 1315–1324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Cohen SM et al. Calmodulin shuttling mediates cytonuclear signaling to trigger experience-dependent transcription and memory. Nat. Commun 9, 2451 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cohen S & Greenberg ME Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu. Rev. Cell Dev. Biol 24, 183–209 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Karpova A et al. Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell 152, 1119–1133 (2013). [DOI] [PubMed] [Google Scholar]
- 188.Harvey CD, Yasuda R, Zhong H & Svoboda K The spread of Ras activity triggered by activation of a single dendritic spine. Science 321, 136–140 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhai S, Ark ED, Parra-Bueno P & Yasuda R Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science 342, 1107–1111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ch’ng TH et al. Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell 150, 207–221 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ch’ng TH & Martin KC Synapse-to-nucleus signaling. Curr. Opin. Neurobiol 21, 345–352 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Jordan BA & Kreutz MR Nucleocytoplasmic protein shuttling: the direct route in synapse-to-nucleus signaling. Trends Neurosci. 32, 392–401 (2009). [DOI] [PubMed] [Google Scholar]
- 193.Uchida S et al. CRTC1 nuclear translocation following learning modulates memory strength via exchange of chromatin remodeling complexes on the Fgf1 gene. Cell Rep. 18, 352–366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; A strong learning event leads to an exchange of CBP to HAT KAT5 on the Fgf1b promoter to induce persistent expression; these events are mediated by transcriptional activity of the synaptic protein CRTC1.
- 194.Maze I, Noh K-M & Allis CD Histone regulation in the CNS: basic principles of epigenetic plasticity. Neuropsychopharmacology 38, 3–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kumar A et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48, 303–314 (2005). [DOI] [PubMed] [Google Scholar]
- 196.VanLeeuwen J-E et al. Coordinated nuclear and synaptic shuttling of afadin promotes spine plasticity and histone modifications. J. Biol. Chem 289, 10831–10842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kramár EA, Babayan AH, Gall CM & Lynch G Estrogen promotes learning-related plasticity by modifying the synaptic cytoskeleton. Neuroscience 239, 3–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rex CS et al. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J. Neurosci 27, 3017–3029 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wiggins A, Smith RJ, Shen H-W & Kalivas PW Integrins modulate relapse to cocaine-seeking. J. Neurosci 31, 16177–16184 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kramár EA, Lin B, Rex CS, Gall CM & Lynch G Integrin-driven actin polymerization consolidates long-term potentiation. Proc. Natl Acad. Sci. USA 103, 5579–5584 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Charrier C et al. A crosstalk between β1 and β3 integrins controls glycine receptor and gephyrin trafficking at synapses. Nat. Neurosci 13, 1388–1395 (2010). [DOI] [PubMed] [Google Scholar]
- 202.Mitra SK & Schlaepfer DD Integrin-regulated FAK–Src signaling in normal and cancer cells. Curr. Opin. Cell Biol 18, 516–523 (2006). [DOI] [PubMed] [Google Scholar]
- 203.Cingolani LA et al. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by β3 integrins. Neuron 58, 749–762 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rex CS et al. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J. Cell Biol 186, 85–97 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Briz V et al. Activity-dependent rapid local RhoA synthesis is required for hippocampal synaptic plasticity. J. Neurosci 35, 2269–2282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Golden SA et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat. Med 19, 337–344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wolf ME & Tseng KY Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front. Mol. Neurosci 5, 72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ju W et al. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat. Neurosci 7, 244–253 (2004). [DOI] [PubMed] [Google Scholar]
- 209.Nicoll RA & Roche KW Long-term potentiation: peeling the onion. Neuropharmacology 74, 18–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Huganir RL & Nicoll RA AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lin JW et al. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat. Neurosci 3, 1282–1290 (2000). [DOI] [PubMed] [Google Scholar]
- 212.Rodenas-Ruano A, Chávez AE, Cossio MJ, Castillo PE & Zukin RS REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat. Neurosci 15, 1382–1390 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Singh-Taylor A et al. NRSF-dependent epigenetic mechanisms contribute to programming of stress-sensitive neurons by neonatal experience, promoting resilience. Mol. Psychiatry 23, 648–657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wei J et al. Histone modification of Nedd4 ubiquitin ligase controls the loss of AMPA receptors and cognitive impairment induced by repeated stress. J. Neurosci 36, 2119–2130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Jayanthi S et al. Methamphetamine downregulates striatal glutamate receptors via diverse epigenetic mechanisms. Biol. Psychiatry 76, 47–56 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Francis TC et al. Molecular basis of dendritic atrophy and activity in stress susceptibility. Mol. Psychiatry 22, 1512–1519 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Krugers HJ, Hoogenraad CC & Groc L Stress hormones and AMPA receptor trafficking in synaptic plasticity and memory. Nat. Rev. Neurosci 11, 675–681 (2010). [DOI] [PubMed] [Google Scholar]
- 218.Chen Y et al. Impairment of synaptic plasticity by the stress mediator CRH involves selective destruction of thin dendritic spines via RhoA signaling. Mol. Psychiatry 18, 485–496 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Andres AL et al. NMDA receptor activation and calpain contribute to disruption of dendritic spines by the stress neuropeptide CRH. J. Neurosci 33, 16945–16960 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Aoto J, Martinelli DC, Malenka RC, Tabuchi K & Südhof TC Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell 154, 75–88 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Luco RF, Allo M, Schor IE, Kornblihtt AR & Misteli T Epigenetics in alternative pre-mRNA splicing. Cell 144, 16–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hu Q, Greene C & Heller E Specific histone modifications associate with alternative exon selection during mammalian development. Preprint at https://www.biorxiv.org/content/early2018/07/18/361816 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ding X et al. Activity-induced histone modifications govern neurexin-1 mRNA splicing and memory preservation. Nat. Neurosci 20, 690–699 (2017). [DOI] [PubMed] [Google Scholar]; This paper shows that stabilization of memories requires a shift in expression of the Nrxn1 splice isoform within the hippocampus, which occurs through accumulating a repressive histone marker, H3K9me3, at the splicing site.
- 224.Ripoli C Engrampigenetics: Epigenetics of engram memory cells. Behav. Brain Res 325, 297–302 (2017). [DOI] [PubMed] [Google Scholar]
- 225.Weber CM, Ramachandran S & Henikoff S Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell 53, 819–830 (2014). [DOI] [PubMed] [Google Scholar]
- 226.Dunn CJ et al. Histone hypervariants H2A.Z.1 and H2A.Z.2 play independent and context-specific roles in neuronal activity-induced transcription of Arc/Arg3.1 and other immediate early genes. eNeuro 4, ENEURO.0040–17.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Henikoff S & Smith MM Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol 7, a019364 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dekker J, Rippe K, Dekker M & Kleckner N Capturing chromosome conformation. Science 295, 1306–1311 (2002). [DOI] [PubMed] [Google Scholar]
- 229.Li Y, Hu M & Shen Y Gene regulation in the 3D genome. Hum. Mol. Genet 27, R228–R233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Quinodoz SA et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mitchell AC et al. Longitudinal assessment of neuronal 3D genomes in mouse prefrontal cortex. Nat. Commun 7, 12743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Engmann O et al. Cocaine-induced chromatin modifications associate with increased expression and three-dimensional looping of Auts2. Biol. Psychiatry 82, 794–805 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wagner FF et al. Kinetically selective inhibitors of histone deacetylase 2 (HDAC2) as cognition enhancers. Chem. Sci 6, 804–815 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.McQuown SC & Wood MA HDAC3 and the molecular brake pad hypothesis. Neurobiol. Learn. Mem 96, 27–34 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bowers ME, Xia B, Carreiro S & Ressler KJ The Class I HDAC inhibitor RGFP963 enhances consolidation of cued fear extinction. Learn. Mem 22, 225–231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Rumbaugh G et al. Pharmacological selectivity within class I histone deacetylases predicts effects on synaptic function and memory rescue. Neuropsychopharmacology 40, 2307–2316 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Gräff J et al. A dietary regimen of caloric restriction or pharmacological activation of SIRT1 to delay the onset of neurodegeneration. J. Neurosci 33, 8951–8960 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Volmar C-H & Wahlestedt C Histone deacetylases (HDACs) and brain function. Neuroepigenetics 1, 20–27 (2015). [Google Scholar]
- 239.Gräff J & Tsai L-H The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev. Pharmacol. Toxicol 53, 311–330 (2013). [DOI] [PubMed] [Google Scholar]
- 240.Yu H et al. Tet3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat. Neurosci 18, 836–843 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Guzman-Karlsson MC, Meadows JP, Gavin CF, Hablitz JJ & Sweatt JD Transcriptional and epigenetic regulation of Hebbian and non-Hebbian plasticity. Neuropharmacology 80, 3–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Meadows JP et al. DNA methylation regulates neuronal glutamatergic synaptic scaling. Sci. Signal 8, ra61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Blackman MP, Djukic B, Nelson SB & Turrigiano GG A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J. Neurosci 32, 13529–13536 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Nelson ED, Kavalali ET & Monteggia LM MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr. Biol 16, 710–716 (2006). [DOI] [PubMed] [Google Scholar]
- 245.Qiu Z et al. The Rett syndrome protein MeCP2 regulates synaptic scaling. J. Neurosci 32, 989–994 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Bredy TW, Lin Q, Wei W, Baker-Andresen D & Mattick JS MicroRNA regulation of neural plasticity and memory. Neurobiol. Learn. Mem 96, 89–94 (2011). [DOI] [PubMed] [Google Scholar]
- 247.Wong HH-W et al. RNA docking and local translation regulate site-specific axon remodeling in vivo. Neuron 95, 852–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Briz V et al. The non-coding RNA BC1 regulates experience-dependent structural plasticity and learning. Nat. Commun 8, 293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Walters BJ et al. The role of the RNA demethylase FTO (fat mass and obesity-associated) and mRNA methylation in hippocampal memory formation. Neuropsychopharmacology 42, 1502–1510 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Bose DA et al. RNA binding to CBP stimulates histone acetylation and transcription. Cell 168, 135–149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Shu G et al. Deleting HDAC3 rescues long-term memory impairments induced by disruption of the neuron-specific chromatin remodeling subunit BAF53b. Learn. Mem 25, 109–114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Tang F et al. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat. Protoc 5, 516–535 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Konermann S et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Heller EA et al. Locus-specific epigenetic remodeling controls addiction-and depression-related behaviors. Nat. Neurosci 17, 1720–1727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Zhou H et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat. Neurosci 21, 440–446 (2018). [DOI] [PubMed] [Google Scholar]