

Abstract
Disease-modifying therapies for proteinopathies are urgently needed yet clinical trials for the major neurodegenerative diseases, Alzheimer’s and Parkinson’s, have been failing at an alarming rate leaving patients and caregivers scrambling for any sign of hope. At the same time, for one family of proteinopathies, the rare TTR amyloidoses, disease-modifying therapy has existed for almost 3 decades and two new types of disease-modifying therapy have become available more recently. In this chapter, I discuss those therapies, examine to what extent they can be generalized for other diseases, and consider what we may learn from their relative success.
A few years ago, when talking about proteinopathies, diseases caused by protein misfolding and self-assembly into cytotoxic structures, many of us used to say something to the effect that “there are no disease-modifying therapies for any proteinopathy.” If you talked about one particular disease, for example Alzheimer’s disease or Parkinson’s disease, you were correct. Approved disease-modifying therapies for these devastating neurodegenerative disorders are not available. Only treatments of symptoms currently exist, which have limited and temporary effects. However, if you made the generalization for all the proteinopathies, as I was guilty of doing, you were wrong. Disease-modifying therapy is a treatment that at a minimum slows down the progression of the disease and at best cures the disease completely. It can achieve these goals by affecting the cause of the disease, or in some cases, the target of the disease. Such therapy has been available for the rare familial transthyretin (TTR) amyloidoses, particularly familial amyloidotic polyneuropathy (FAP), for almost 3 decades in the form of a liver transplant.1 The liver is the main organ producing disease-causing variant TTR in people who carry a mutant TTR allele. Transplant of a donor’s liver removes the main source of the offending protein and therefore modifies the course of the disease, including extension of patients’ life.2 In familial amyloidotic cardiomyopathy (FAC) caused by TTR amyloidosis, transplantation of liver and/or heart can provide disease-modifying therapy,3 whereas in cases of wild-type (WT) TTR amyloidosis, which was inappropriately named senile systemic amyloidosis though it is a cardiomyopathy, a heart transplant provides disease-modifying therapy.4 In the latter two cases, the replacement of the heart removes the target of the offending protein rather than the offending protein itself. For another type of proteinopathy, systemic amyloidosis due to aggregation of immunoglobulin light-chain (light-chain amyloidosis, AL)—a severe and rare complication of multiple myeloma— harsh chemotherapy may in some cases eradicate the cells producing the amyloidogenic variant immunoglobulin, thereby eliminating the source of the offending protein.5,6
Although the transplant options discussed above are disease-modifying therapies, they are of narrow practical, use for obvious reasons, including the limited availability of matching donated organs, the necessity of immunosuppression therapy for the rest of the affected person’s life, and the high costs involved. In addition, the benefits of these therapies have been shown to be limited for a variety of reasons, which are beyond the scope of this chapter. More generally, when we think about disease-modifying therapy, we usually refer to drugs rather than to options such as major organ transplantation or harsh chemotherapy. Interestingly, the first disease-modifying drug approved for treatment of a proteinopathy, tafamidis (Fig. 1A), was for FAP—the same disease for which disease-modifying therapy by liver transplantation already had been available. Tafamidis was first approved for human therapy in Europe in late 20117 and later in other countries, including Japan, Argentina, and Mexico.8 It failed to get the approval of the United States Food and Drug Administration (FDA) in 2012 but was accepted for review again and finally got approved in 2019. Diflunisal (Fig. 1A), an anti-inflammatory non-steroidal drug originally approved by the FDA for the treatment of arthritis, was found to work by the same mechanism as tafamidis—kinetic stabilization of the native, tetrameric form of TTR (Fig. 1B), which slows down its dissociation into monomers, misfolding, and abnormal aggregation—and has been available off-label in the United States for treatment of TTR amyloidoses.9
Fig. 1.
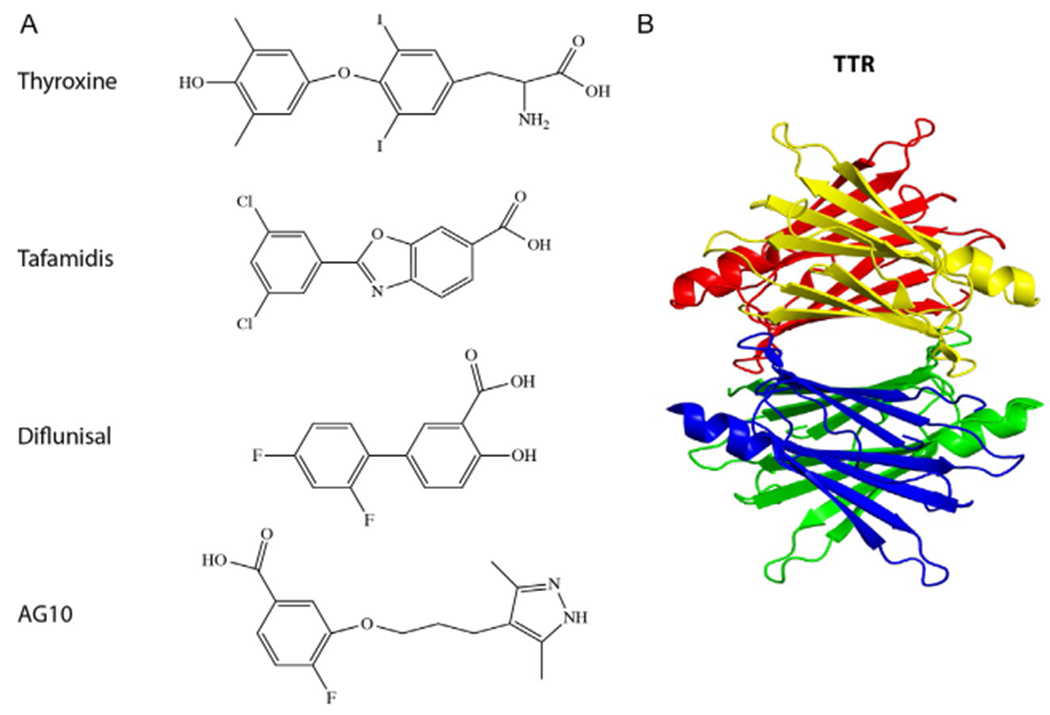
TTR and its ligands. (A) Schematic structures of the thyroid hormone, thyroxine, a natural ligand of TTR, and of the TTR ligands tafamidis, an FDA-approved drug for FAP, diflunisal, available off-label for TTR amyloidoses, and AG10, a compound in clinical trial for FAC. (B) A ribbon diagram of TTR showing the tetramer structure composed of yellow, red, blue, and green monomers.
In view of this success, one might have expected that new kinetic stabilizers would follow and become disease-modifying therapies for other proteinopathies. It was therefore somewhat surprising that the next two disease-modifying drugs were not kinetic stabilizers, and actually were again for the same disease, FAP. These drugs use a different approach, suppression of TTR gene expression, to achieve the same therapeutic goal. Patisiran, an RNAi drug that suppresses the expression of TTR, was approved by the FDA in 201810–12 and was followed shortly after by inotersen, an antisense oligonucleotide (ASO) inhibitor of TTR gene expression.13,14 Thus, currently, there are three FDA-approved, disease-modifying drugs—tafamidis, patisiran, and inotersen, and a fourth one, diflunisal, that can be used off-label in the United States, all of which target the same rare amyloidosis. In contrast, no disease-modifying drugs exist for other proteinopathies, except the chemotherapy option for AL mentioned above. This situation raises important questions: What can we learn from the success of developing disease-modifying therapy against TTR amyloidoses? Can either of these strategies—small-molecule kinetic stabilizers or nucleic-acid-based gene silencers—become a mainstream therapeutic approach for most or all other proteinopathies?
1. Kinetic stabilizers
TTR is a native homotetramer of a 127-amino acid-long polypeptide that acts as a carrier of thyroxine (Fig. 1A) and retinol-binding protein in the blood. The tetramer structure is a “dimer of dimers” (Fig. 1B), containing a binding site for the ligands at the dimer—dimer interface (Fig. 2). Mutations in the TTR gene that cause amino acid substitutions in the polypeptide chain can destabilize the structure, leading to dissociation of the tetramer first into dimers and then into monomers, followed by misfolding of the monomers and self-assembly of the misfolded protein into toxic oligomers and aggregates.16
Fig. 2.
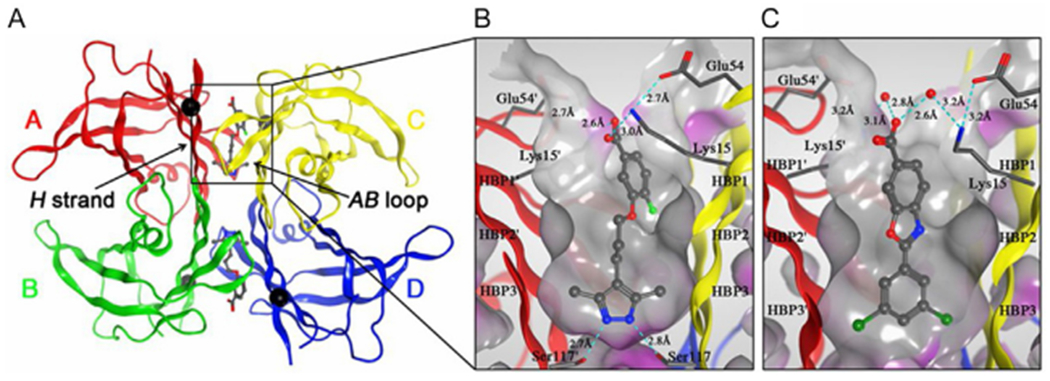
Crystal structures of V122I-TTR ligand complexes. (A) Quaternary structure of AG10 bound to V122I-TTR shown as a ribbon representation with monomers colored individually and positions of each of the V122I mutations shown as black spheres located on the H β-strand, which interacts with the adjacent AB-loop on the AC/BD interface. (B) AG10 in complex with V122I-TTR. (C) Tafamidis in complex with V122I-TTR. Close-up views of one of the two identical thyroxine binding sites with different colored ribbons for the two monomers of the tetramer composing the binding site. A Connolly molecular surface15 was applied to residues within 10 Å of ligand in the thyroxine binding pocket and colored gray for hydrophobic and purple for polar residues. Reproduced from reference Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc. Natl. Acad. Sci. USA 2013;110:9992–9997.
The most common form of FAP is caused by the substitution V30 M in TTR. Interestingly, a subset of carriers of this mutation were found to develop a much milder disease than the typical form, leading to the discovery that patients with mild disease had another mutation, T119 M, on the other allele of the TTR gene.17 The T119 M substitution was found to stabilize the tetramer structure and protect it against the destabilizing effect of V30 M. 18 The discovery that the de-stabilizing V30 M substitution could be compensated for by another structural change in the TTR tetramer structure prompted Kelly and co-worker to search for small molecules that could achieve a similar stabilizing effect to the T119 M substitution, leading to the discovery of tafamidis,18,19 a small-molecule kinetic stabilizer that binds to TTR at the thyroxin binding site.
Importantly, the two thyroxin-binding sites in TTR are at the weaker dimer—dimer interface16 and because two other carrier proteins are the main transporters of thyroxin in the blood, whereas TTR is only a minor transporter, these sites in TTR are rarely occupied in humans (<1% thyroxin bound).20 Thus, if tafamidis, diflunisal, or other kinetic stabilizers occupy these sites and protect the tetramer from dissociation, the impact on thyroxin transportation is minimal. Using the same principle, another kinetic stabilizer with substantially improved potency, AG10 (Figs. 1A and 2),21 was discovered and is currently in clinical trials.22 Although both tafamidis and AG10 bind to the thyroxine binding site at the dimer—dimer interface of TTR (Fig. 2), unlike tafamidis, AG10 is effective against V122I-TTR, a variant that causes FAC in 3—4% of African-Americans (~1.3 million people) and is hypothesized to contribute to the increased prevalence of heart failure in this population.23 It will be interesting to compare the efficacy of AG10 to that of the other FDA-approved drugs. In particular, if it is indeed more efficacious yet comes into the market relatively late, it will be instructive for other drug developers to watch the rate of adoption of the new drug among clinicians.
The success of the kinetic-stabilizers approach for generating disease-modifying drugs for TTR amyloidosis raises the question whether a similar approach could be used for other amyloidogenic proteins. However, although small-molecule stabilizers of protein—protein interaction have been studied for many proteins,24,25 their use in the proteinopathy filed is rare. When considering this question, an immediate realization is that for this approach to be successful, the protein under consideration must have a stable structure. Unfortunately, this is not the case for the proteins implicated in the major proteinopathies—Aβ and tau in Alzheimer’s disease, α-synuclein in Parkinson’s disease, and islet amyloid polypeptide in type-2 diabetes. These proteins are natively unstructured and thus lack a native fold amenable to stabilization by small molecules. For these protein, kinetic stabilizers would work only in the unlikely event that they bind to, and stabilize, ordered elements of these proteins, which comprise a small fraction of the conformational space the proteins sample. However, in several dozen other proteinopathies, the offending protein is natively structured. Can we expect to see new kinetic stabilizers in the near future as therapeutic candidates for these diseases?
A first example of such an attempt directed at AL amyloidosis has been published recently by the Kelly group26 who screened 650,000 small molecules and discovered four structural classes of compounds that protected recombinant or cell-secreted antibody light chains from endoproteolysis, which is a key step in transforming these proteins into aggregation-prone fragments. The choice of AL as the target for this screening may reflect the relatively low pharmacokinetic demands of the compounds, which do not have to cross cellular membranes or the blood-brain barrier to reach their target, as would be the case for many proteinopathies. A similar strategy could be used for other systemic amyloidoses, such as dialysis-related amyloidosis, in which β2-microglobulin β2m) forms amyloid leading to carpal tunnel syndrome in patients on chronic dialysis, or inflammation-induced systemic amyloidosis caused by serum amyloid A. Screening of small molecules against β2m has been reported,27, 28 yet it is not entirely clear whether the compounds identified stabilize the native structure or prevent formation of amyloid after partial misfolding of the native protein.
The success in identifying kinetic stabilizers for TTR suggests that proteins whose native structures are oligomeric, such as superoxide dismutase 1 (SOD1), the product of a gene in which mutations cause familial amyotrophic lateral sclerosis (ALS), or TP53, mutations in which cause cancer, might also be targets for this strategy. This assumes that small molecules could find a binding site at the interface between two monomers and thus stabilize the oligomer against dissociation. It is not clear, however, if this could be a general tactic or if the success with TTR is due to the location of the natural ligand binding site at the dimer—dimer interface, where the kinetic stabilizers bind.
SOD1 is a homodimeric, antioxidant enzyme that catalyzes highly reactive superoxide radical anions formed during respiration into less reactive hydrogen peroxide (Fig. 3). Similar to TTR, the structure of SOD1 is characterized by a high β-sheet content, and in both cases, over a hundred point-mutations in the cognate gene lead to amino acid substitutions that destabilize the protein’s structure and cause it to misfold and aggregate. However, compared to TTR’s ligand, thyroxine (Fig. 1A), superoxide radical anions are too small to be mimicked successfully by small molecule ligands that could act as kinetic stabilizers (Fig. 3). An additional major challenge for developing kinetic stabilizers for SOD1 that would be effective in vivo is the need for the compounds to pass through the blood—brain barrier, the neuronal membrane, and the mitochondrial membranes to reach their target. Wright et al. have reported identifying isoproterenol and 5-fluorouridine as lead compounds that inhibited SOD1 aggregation, though both compounds bound a region at the core of the SOD1 fibrillar aggregates rather than the proposed dimer interface site, suggesting that they did not actually act as kinetic stabilizers.29
Fig. 3.
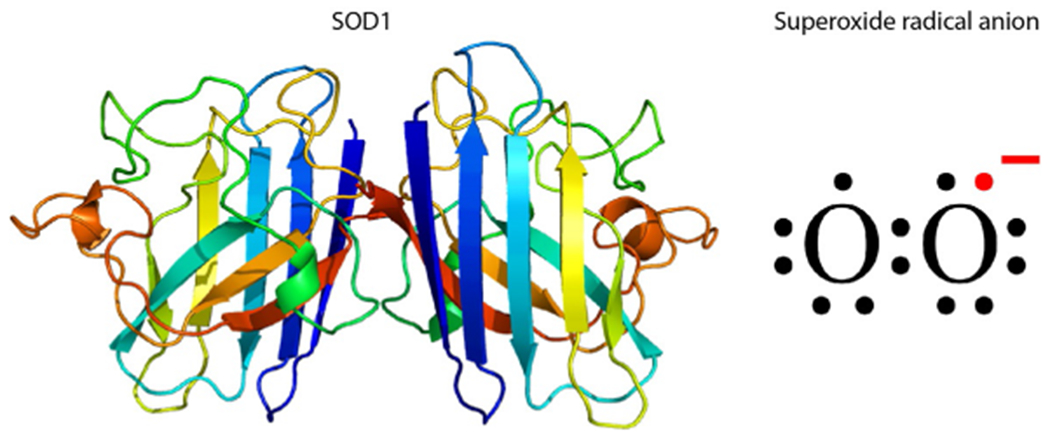
Cu/Zn-Superoxide dismutase 1 (SOD1) and its natural substrate. Left—a ribbon diagram of SOD1. Right—an electronic diagram of superoxide radical anion.
The tumor-suppressor protein TP53 is another attractive oligomeric protein candidate for kinetic stabilizers, many of which have been explored through cell-based or in vitro screening approaches.30–32 A subset of these stabilizers might inhibit the amyloid-like aggregation of certain TP53 variants.33 Targeting monomeric proteins with kinetic stabilizers may be more challenging and depends on the availability of a suitable binding site for the stabilizer that could negate the de-stabilizing effects of amino acid substitutions or environmental insults.
2. RNAi and antisense gene silencers
Patisiran and inotersen hopefully will show positive results in the treatment of patients now that they are approved for this purpose by the FDA.34 The literature extant on these two compounds still comprises mostly data generated in the clinical trials leading to FDA approval.35,36 New studies are needed for full evaluation of the efficacy and safety of each drug and how they compare to the kinetic stabilizers. According to its website, Alnylam Pharmaceuticals, the creator of patisiran, is not currently developing similar products for additional proteinopathies. In contrast, Ionis Pharmaceuticals, the company that developed inotersen, has several products in different stages of development for proteinopathies, including Huntington’s disease (targeting huntingtin), ALS (targeting SOD1 or C9ORF72), and Alzheimer’s disease and frontotemporal dementia (targeting tau). Development of antisense drugs for these diseases is based on the assumption that reducing the concentration of the endogenous protein can be accomplished without causing severe side effects.
In this context, it is interesting to note that the choice the companies made has been to develop gene silencers that would target both the wild-type and mutant forms of the genes encoding the offending proteins rather than only the mutant forms (I have no knowledge whether this is the case for products currently in development). This choice likely is based more on business strategies than on scientific rationale. Silencing only the mutant allele of a gene that produces an aggregation-prone, disease-linked protein while leaving the wild-type protein unaffected likely would achieve superior therapeutic results because the wild-type form would be available at 50% of the normal level, or higher if compensatory mechanisms to increase gene expression spring into action. This would decrease the likelihood of side effects due to loss of function of the protein. The strategy of suppressing both the mutant and the wild-type allele leads to overall lower levels of the protein, slowing down the aggregation process, but half of the protein that is still produced is the mutant form. The advantage of using a “one size fits all” RNAi or ASO is that only one drug needs to be approved by regulatory authorities, which results in substantial savings of development costs. Moreover, even if specific RNAi/ASO were developed for individual mutations and received regulatory approval, the market for these therapeutics might not be profitable because of the small number of patients with each specific mutation. Nonetheless, in view of the advantage such drugs would offer to patients, I hope that in the era of personalized medicine, governments and regulatory agencies will pick up the ball and help both fund the development and facilitate the approval of unique drugs tailored to specific mutations.
3. Final thoughts
As mentioned above, for the abundant proteinopathies, such as Alzheimer’s and Parkinson’s diseases, kinetic stabilizers are unlikely to be a viable option because the associated proteins are natively unstructured. The leading approach for developing disease-modifying therapy for these diseases has been immunotherapy, which to date has yielded disappointing results but may still be a viable approach.37−39 Small molecules, peptides, and various other compounds targeting the production, clearance, and self-assembly of the offending proteins also have been explored as potential disease-modifying therapy for these diseases. A few made it to clinical trials, but those trials were not successful. Here, I chose to focus on recent approaches that have yielded successful disease-modifying therapy for one group of rare diseases—TTR amyloidoses, in hope that the lessons learned can be applied in the near future to other diseases. This will be, no doubt, a major challenge, yet if academic scientists, the biotechnology and pharmaceutical industries, and government agencies cooperate, I believe the likelihood of success is high.
Acknowledgments
The work was supported by NIH/NIA grants R01AG050721 and RF1AG054000, and by the UCLA Mary S. Easton Center for Alzheimer’s Disease Research Endowment.
References
- 1.Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40:242–246. [DOI] [PubMed] [Google Scholar]
- 2.Benson MD. Liver transplantation and transthyretin amyloidosis. Muscle Nerve. 2013;47:157–162. [DOI] [PubMed] [Google Scholar]
- 3.Nelson LM, Penninga L, Sander K, et al. Long-term outcome in patients treated with combined heart and liver transplantation for familial amyloidotic cardiomyopathy. Clin Transpl. 2013;27:203–209. [DOI] [PubMed] [Google Scholar]
- 4.Sousa M, Monohan G, Rajagopalan N, Grigorian A, Guglin M. Heart transplantation in cardiac amyloidosis. Heart Fail Rev. 2017;22:317–327. [DOI] [PubMed] [Google Scholar]
- 5.Dispenzieri A, Gertz MA, Buadi F. What do i need to know about immunoglobulin light chain (AL) amyloidosis? Blood Rev. 2012;26:137–154. [DOI] [PubMed] [Google Scholar]
- 6.Gertz MA. Immunoglobulin light chain amyloidosis: 2013 update on diagnosis, prognosis, and treatment. Am J Hematol. 2013;88:416–425. [DOI] [PubMed] [Google Scholar]
- 7.Said G, Grippon S, Kirkpatrick P. Tafamidis. Nat Rev Drug Discov. 2012;11:185–186. [DOI] [PubMed] [Google Scholar]
- 8.Scott LJ. Tafamidis: a review of its use in familial amyloid polyneuropathy. Drugs. 2014;74:1371–1378. [DOI] [PubMed] [Google Scholar]
- 9.Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:11–21. [DOI] [PubMed] [Google Scholar]
- 11.Hoy SM. Patisiran: first global approval. Drugs. 2018;78:1625–1631. [DOI] [PubMed] [Google Scholar]
- 12.Wood H FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat Rev Neurol. 2018;14:570. [DOI] [PubMed] [Google Scholar]
- 13.Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keam SJ. Inotersen: first global approval. Drugs. 2018;78:1371–1376. [DOI] [PubMed] [Google Scholar]
- 15.Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221:709–713. [DOI] [PubMed] [Google Scholar]
- 16.Foss TR, Wiseman RL, Kelly JW. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry. 2005;44:15525–15533. [DOI] [PubMed] [Google Scholar]
- 17.Coelho T Familial amyloid polyneuropathy: new developments in genetics and treatment. Curr Opin Neurol. 1996;9:355–359. [PubMed] [Google Scholar]
- 18.Hammarstreöm P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–716. [DOI] [PubMed] [Google Scholar]
- 19.Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109:9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci U S A. 2001;98:5566–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penchala SC, Connelly S, Wang Y, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci U S A. 2013;110:9992–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Judge DP, Falk RH, Maurer MS, et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol. 2019;74:285–295. [DOI] [PubMed] [Google Scholar]
- 23.Buxbaum JN, Ruberg FL. Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med. 2017;19:733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarzycka B, Kuenemann MA, Miteva MA, Nicolaes GAF, Vriend G, Sperandio O. Stabilization of protein-protein interaction complexes through small molecules. Drug Discov Today. 2016;21:48–57. [DOI] [PubMed] [Google Scholar]
- 25.Andrei SA, Sijbesma E, Hann M, et al. Stabilization of protein-protein interactions in drug discovery. Expert Opin Drug Discovery. 2017;12:925–940. [DOI] [PubMed] [Google Scholar]
- 26.Morgan GJ, Yan NL, Mortenson DE, et al. Stabilization of amyloidogenic immunoglobulin light chains by small molecules. Proc Natl Acad Sci U S A. 2019;116:8360–8369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carazzone C, Colombo R, Quaglia M, et al. Sulfonated molecules that bind a partially structured species of beta2-microglobulin also influence refolding and fibrillogenesis. Electrophoresis. 2008;29:1502–1510. [DOI] [PubMed] [Google Scholar]
- 28.Regazzoni L, Colombo R, Bertoletti L, et al. Screening of fibrillogenesis inhibitors of beta2-microglobulin: integrated strategies by mass spectrometry capillary electrophoresis and in silico simulations. Anal Chim Acta. 2011;685:153–161. [DOI] [PubMed] [Google Scholar]
- 29.Wright GS, Antonyuk SV, Kershaw NM, Strange RW, Samar Hasnain S. Ligand binding and aggregation of pathogenic SOD1. Nat Commun. 2013;4:1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selivanova G, Wiman KG. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene. 2007;26:2243–2254. [DOI] [PubMed] [Google Scholar]
- 31.Wiman KG. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene. 2010;29:4245–4252. [DOI] [PubMed] [Google Scholar]
- 32.Wilcken R, Liu X, Zimmermann MO, et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J Am Chem Soc. 2012;134:6810–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J, Reumers J, Couceiro JR, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7:285–295. [DOI] [PubMed] [Google Scholar]
- 34.Solomon SD, Adams D, Kristen A, et al. Effects of Patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation. 2019;139:431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mickle K, Lasser KE, Hoch JS, Cipriano LE, Dreitlein WB, Pearson SD. The effectiveness and value of Patisiran and Inotersen for hereditary transthyretin amyloidosis. J Manag Care Spec Pharm. 2019;25:10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gertz MA, Scheinberg M, Waddington-Cruz M, et al. Inotersen for the treatment of adults with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis. Expert Rev Clin Pharmacol. 2019;65:1–11. [DOI] [PubMed] [Google Scholar]
- 37.Piton M, Hirtz C, Desmetz C, et al. Alzheimer’s disease: advances in drug development. J Alzheimers Dis. 2018;65:3–13. [DOI] [PubMed] [Google Scholar]
- 38.Herline K, Drummond E, Wisniewski T. Recent advancements toward therapeutic vaccines against Alzheimer’s disease. Expert Rev Vaccines. 2018;17:707–721. [DOI] [PubMed] [Google Scholar]
- 39.Zella SMA, Metzdorf J, Ciftci E, et al. Emerging immunotherapies for Parkinson disease. Neurol Ther. 2019;8:29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]