

SUMMARY
Dissecting the in vivo host-pathogen interplay is crucial to understanding the molecular mechanisms governing control or progression of intracellular infections. In this work, we explore the in vivo molecular dynamics of Mtb infection by performing dual RNA-seq on Mycobacterium tuberculosis-infected, ontogenetically distinct macrophage lineages isolated directly from murine lungs. We first define an in vivo signature of 180 genes specifically upregulated by Mtb in mouse lung macrophages, then we uncover a divergent transcriptional response of the bacteria between alveolar macrophages that appear to sustain Mtb growth through increased access to iron and fatty acids and interstitial macrophages that restrict Mtb growth through iron sequestration and higher levels of nitric oxide. We use an enrichment protocol for bacterial transcripts, which enables us to probe Mtb physiology at the host cell level in an in vivo environment, with broader application in understanding the infection dynamics of intracellular pathogens in general.
In Brief
In this study Pisu et al. performed dual RNA-seq on Mycobacterium tuberculosis-infected, ontogenetically distinct macrophage lineages isolated directly from infected murine lungs. The transcriptional response of host and bacteria diverged between alveolar macrophages that sustain Mtb growth and interstitial macrophages that restrict Mtb growth.
Graphical Abstract
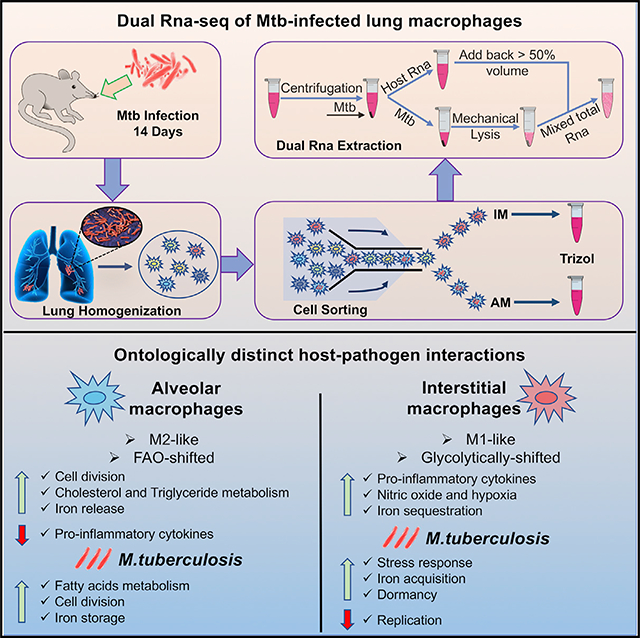
INTRODUCTION
Dual RNA sequencing (RNA-seq) is emerging as a powerful new tool that is of particular value in the dissection of the host-pathogen interplay, revealing the impact both organisms exert over each other. The majority of studies published to date have involved tissue culture models of infection conducted on cell lines or on differentiated primary cells. Although clearly of considerable value, these studies do not address the in vivo host cell heterogeneity that for many pathogens is central to the control or progression of the infection. This is of particular significance for pathogens such as Mycobacterium tuberculosis (Mtb), for which bacterial survival and growth are linked to the ontogeny and metabolism of the different macrophage lineages that co-exist in the tuberculosis granuloma (Huang et al., 2018). Dual RNA-seq would be ideally suited to determining the molecular dynamics underlying host cell phenotype and bacterial fitness among these divergent host cell lineages (Russell et al., 2019), but the challenges in generating dual RNA-seq datasets from in vivo material in which bacterial burden is low and variable, and host cell heterogeneity is high, remain daunting.
Studies undertaking dual RNA-seq on in vivo samples have been performed on total tissues rich in extracellular bacteria, such as Yersinia pseudotuberculosis-infected lymph nodes (Nuss et al., 2017), Pseudomonas aeruginosa-infected lung tissue (Damron et al., 2016), Mycobacterium leprae-infected human tissue lesions (Montoya et al., 2019), and Toxoplasma gondii-infected brain tissue (Pittman et al., 2014). Application of dual-RNA-seq to in vivo infected cell populations is in development. In particular, a new pipeline called Path-seq was recently used to recover the Mtb transcriptome from alveolar macrophages (AMs) isolated from the murine lung (Peterson et al., 2019); however, the majority of datasets in the study came from in vitro infections.
Recently we used an acute mouse Mtb challenge model with fluorescent Mtb fitness reporter strains (Sukumar et al., 2014; Tan et al., 2013) to demonstrate that bacteria in the resident AMs from the lung displayed lower stress and greater rates of replication relative to bacteria within recruited, monocyte-derived interstitial macrophages (IMs) (Huang et al., 2018). In the present study we sought to determine the host and bacterial transcriptomes associated with the different Mtb growth phenotypes (Huang et al., 2018). We performed dual RNA-seq on Mtb-infected AM and IM host cell populations isolated directly from mouse lungs. Using a modified RNA extraction protocol, together with a data analysis pipeline tailored for samples with low sequencing depth, we were able to enrich for bacterial transcripts and increase the resolution of differential gene expression (DGE). Analysis of the datasets provides novel insights into the host cell conditions that Mtb has to endure during infection of lung macrophages in vivo. Finally, the increased resolution provided by probing the transcriptomes of both host and pathogen emphasizes the significance of the metabolic interface in supporting bacterial growth in permissive host macrophages and restricting bacterial growth in controller host cells.
RESULTS
Physical Enrichment of Bacterial RNA from Total RNA in Mtb-Infected Lung Macrophages
In this work we sought to develop a streamlined RNA extraction protocol to perform in vivo dual RNA-seq on Mtb-infected lung macrophage sub-populations. We focused on an in vivo single time point, 14 days post-infection (p.i.), which enabled us to discriminate between the functional phenotypes of the resident AMs and the recruited, blood monocyte-derived IMs (Huang et al., 2018).
Our protocol (Figure 1B) is based on the differential lysis of the host and Mtb cells in Trizol/GTC. The first step involved incubation in Trizol of the sorted infected cells at room temperature (RT). This allowed complete lysis of the host cell and release of the eukaryotic RNA and intracellular bacteria. The sample was centrifuged to pellet eukaryotic cell debris and Mtb. In step 2, up to 90% of the Trizol supernatant containing the bulk of the eukaryotic RNA was separated from the pelleted Mtb and set to one side. This step achieved two goals: leaving a small amount of Trizol in the tube avoided disturbing the bacterial pellet and meant that the host RNA was not subjected to the harsh Mtb homogenization treatment (Figure 1C). In step 3, zirconia beads and fresh Trizol were added to the tube containing the bacteria, which were subjected to mechanical lysis. In step 4, we added back part of the Trizol containing the host-RNA supernatant. This step enriched bacterial transcripts while enabling sufficient recovery of total RNA for library preparation. In Figure 1D we show the relative percentage of Mtb reads recovered using preliminary optimization tissue culture test samples following the removal of differing amounts of host RNA. These were compared with the percentage of Mtb reads that were obtained from the AM and IM populations from each of the three independent in vivo experiments (Figure 1D; Table 1). The results confirm the value of this approach to manually enrich bacterial transcripts.
Figure 1. In Vivo Dual RNA-Seq Pipeline for the Enrichment of Bacterial RNA from Flow-Sorted Lung Macrophages.

(A) Flow gating strategy used to identify the two different lung macrophages lineages. CD64+ and MerTk+ macrophages were separated from the rest of the immune population and the AM and IM lineages identified by the level of expression of Siglecf. IM- and AM-infected macrophages constituted ~9% and ~2%, respectively, of the total macrophage population. The experimental infection and cellular isolation was repeated three times independently (n = 3), each time pooling and processing lung tissue from three infected mice.
(B) Diagram of the RNA extraction process. Samples were sorted in Trizol to release host RNA and pelleted, and the supernatant (containing most of the host RNA) was removed and placed aside. Fresh Trizol was added to the tube containing the bacterial pellet, and the bacteria were disrupted through mechanical lysis. A percentage of the host RNA (nominally 50%) was then transferred back to the tube containing the lysed bacteria and the resulting total RNA, now enriched for bacterial transcripts, processed following the Trizol protocol.
(C) Example of an Agilent Bioanalyzer 2100 plot shows how physical separation of the host RNA from the bacterial pellet prior Mtb homogenization preserves RNA quality and integrity, as evidenced by the intact rRNA peaks and higher quality score (RNA quality number [RQN] 7.5 versus 4.8).
(D) Bar chart illustrating the percentage of total reads that map to the Mtb Erdman genome for the test and in vivo samples.
(E) Venn diagram showing the overlap of the genes (abs[log2 FC > 1], false discovery rate [FDR] < 0.05) detected as differentially expressed on the full reference datasets by DESeq2, IHW, edgeR, and limma-voom. We nominally defined this set of genes as “true DE.”
(F) Percentage of false positives (genes detected as differentially expressed; abs[log2 FC > 1], FDR < 0.05) in the subset of 1 M reads but not part of “true DE”) for the most common used statistical approaches in RNA-seq analysis. APEGLM outperformed all other methods, with a false-positive rate of <3.5%. p values were calculated using one-way ANOVA followed by Tukey’s post hoc test.
Table 1.
Number of Reads for Each Sample that Match rRNA or Either One of the Two Organisms’ Genomes
| Sample | Raw Reads | rRNA (Mtb + Mouse) | Mouse | M. tuberculosis | |||
|---|---|---|---|---|---|---|---|
| rRNA | % of Raw Reads | Aligned to GRcm38 – Rel. 94 | % of Raw Reads | Aligned to Erdman GCA000668235.1 | % of Raw Reads | ||
| AM 1 ∼25,000 infected cells | 131.5 M | 19.5 M | 14.8% | 96.2 M | 73% | 1.2 M | 0.9% |
| AM 2 ∼18,000 infected cells | 176.5 M | 41.1 M | 23.3% | 96.6 M | 55% | 1.3 M | 0.75% |
| AM 3 ∼12,000 infected cells | 309.8 M | 107.7 M | 34.8% | 51.4 M | 17% | 0.95 M | 0.3% |
| IM 1 ∼25,000 infected cells | 140.6 M | 15.3 M | 10.9% | 72 M | 51% | 1.2 M | 0.85% |
| IM 2 ∼40,000 infected cells | 47.5 M | 6.6 M | 13.9% | 33.2 M | 70% | 0.9 M | 1.9% |
| IM 3 ∼35,000 infected cells | 83.7 M | 12.2 M | 14.6% | 54.3 M | 65% | 1 M | 1.2% |
| Mtb broth 1 | 12.1 M | 1.8 M | 14.7% | – | – | 10 M | 83% |
| Mtb broth 2 | 13.7 M | 1.4 M | 10.4% | – | – | 12 M | 87.5% |
| Mtb broth3 | 15.1 M | 0.8 M | 5.4% | – | – | 12.4 M | 82% |
| Mtb – BMDM 1 | 32.6 M | 0.08 M | 0.25% | – | – | 28.5 M | 87% |
| Mtb – BMDM 2 | 35 M | 0.11 M | 0.31% | – | – | 31.6 M | 90% |
| Mtb – BMDM 3 | 25.5 M | 0.11 M | 0.47% | – | – | 23.6 M | 93% |
For the in vivo samples, the number of sorted cells is reported in the “Sample” column. As a result of the difference in the number of infected cells between AMs and IMs at 14 days post-infection, a lower bacterial RNA enrichment and higher sequencing depth were preferred in some AM samples to compensate for the reduced RNA yield.
Assessment of Different Statistical Methods for Analysis of Depth-Constrained Dual RNA-Seq Transcriptomes
The relative abundance of bacterial and host reads indicated that we achieved between 0.3% and 2% of mapped reads for Mtb from the different host populations across the three in vivo experiments (Table 1). We therefore decided to target a sequencing depth of ~1 M reads for the Mtb transcriptome from our in vivo samples, and we sought to determine the best statistical approach for analysis of our datasets that have constrained sequencing depth. Recently, a new empirical Bayes procedure (APEGLM) has been shown to be particularly robust in ranking genes by log fold change (LFC) in the presence of low counts (Zhu et al., 2019). Therefore, we tested whether this statistical procedure could also provide superior results in the analysis of our dual RNA-seq datasets with constrained read counts.
Using our Mtb reference datasets (Figure S1) that have been sequenced to saturation (Table 1) and a range of different statistical approaches for RNA-seq analysis (edgeR [Robinson et al., 2010], limma-voom [Law et al., 2014; Ritchie et al., 2015], DESeq2 [Love et al., 2014], IHW [Ignatiadis et al., 2016], Cuffdiff [Trapnell et al., 2012], and APEGLM [Zhu et al., 2019]), we performed DGE analysis on the full datasets and on five independent, randomly sampled subsets of 1 M reads (Figure S2A). We defined the differentially expressed genes using edgeR, limma-voom, DESeq2, and IHW on the full datasets as the most robust set of differentially expressed genes for analysis, nominally defined as the “true DE” (Figure 1E). We then compared the performance of the various methods on the randomly sampled subsets against this consensus list and found APEGLM to have an average power of 80.88% (Figure S2B) and an average false-positive rate of 3.5%, which was considerably more robust than the other approaches (Figure 1F). Therefore, all subsequent DGE analysis was carried out using DESeq2 and APEGLM as the means of determination of the change in expression between datasets. The data analysis pipeline is illustrated in Figure S1 and provided in STAR Methods.
Identification of an Mtb In Vivo Transcriptional Signature in Mice
Exploratory analysis of our in vivo dual RNA-seq datasets for the Mtb transcriptome revealed the presence of transcriptional signatures distinct to each of the four different environmental conditions (Figure 2A; Table S3). To identify those genes that were upregulated only by Mtb in vivo, we focused on transcripts (abs[log2 FC > 1], adjusted p < 0.05) whose expression was upregulated in one or both host macrophage populations (IMs and AMs) but not upregulated in either short-term bone marrow-derived macrophage (BMDM) or broth culture conditions. We found 274 genes that satisfied these requirements. However, in previous microarray studies, Schnappinger et al. (2003) defined a differential intraphagosomal transcriptome (454 genes induced in resting and activated BMDMs at 24 and 48 h p.i.), while Rohde et al. (2012) determined the transcriptional profile of Mtb during an extended period of infection (up to 14 days) in BMDMs. Our BMDM dataset was restricted to an early time point (6 h p.i.); therefore, to encompass a broader range of cell culture conditions, we mined the data from these earlier studies (see STAR Methods) to ensure a more robust means of identifying gene signatures unique to the in vivo infection environment. The comparison of the 274 genes from the present study with the genes identified as upregulated in BMDMs in these earlier studies identified 180 genes uniquely upregulated in macrophages in vivo (Figure 2B; Table S4). We nominally defined this set of genes as the “in vivo signature.”
Figure 2. Identification of an Mtb In Vivo Signature in Mice.

(A) Principal-component analysis (PCA) of the Mtb transcriptome from three different environmental conditions: in vivo, cell culture (BMDM), and broth (7H9-OADC).
(B) Heatmap showing relative expression levels for the “in vivo signature,” a set of 180 genes upregulated only during growth in lung macrophages.
(C) Functional characterization of the in vivo gene signature shows how many of the genes belong to the “conserved hypothetical” class with undefined function.
(D) Protein-protein network analysis of the “in vivo signature” reveals a cluster of genes involved in cholesterol degradation (Kstr2), response to nitrosative and oxidative stresses (ergothioneine), and nitrogen assimilation and export machinery (Esx-5). Only high-confidence (>0.7) interactions were used to build the network.
(E) Heatmap showing relative expression levels for the genes in the cholesterol degradation pathway Kstr2.
(F) Boxplot of the main genes related to the MMP (MutA, MutB) and MCC (PrpC, PrpD) pathways. The transcriptional profile of Mtb from in vivo samples shows upregulation of the genes related to the MMP pathway but not of those involved in the recycling of the propionyl-CoA pool through the MCC.
(G and H) Heatmaps showing relative expression levels for genes in the inorganic nitrogen (G) and ESX-5 (H) pathways in Mtb.
(I) Violin plots showing expression levels (in log-normalized counts) of genes involved in the ergothioneine biosynthesis pathway.
Where appropriate, for all plots the statistical significance is shown on the picture (*adjusted p value [p-adj.] < 0.05, **p-adj. < 0.01, ***p-adj. < 0.001, and ****p-adj. < 0.0001). q values for comparisons among the groups were calculated using the Wald test as implemented in the DESeq2 pipeline.
The assignment of function to the genes identified as upregulated in Mtb in vivo was pursued using pathway and protein-protein network analysis (Figures 2C and 2D). The pathways specifically upregulated by Mtb during the course of an in vivo infection were linked predominantly to cholesterol homeostasis, nitrogen assimilation, nitric oxide (NO) detoxification, and protein and lipid export machinery (Figure 2D). It is notable that all 15 genes within the Kstr2 regulon were strongly upregulated during in vivo infection by Mtb in both IMs and AMs (Figure 2E), suggesting that cholesterol is degraded completely to support bacterial survival in mice. This is consistent with data from both genetic analysis and chemical screening (Casabon et al., 2013; VanderVen et al., 2015). We know from previous work that cholesterol degradation leads to expansion of the propionyl-CoA pool, which induces metabolic stress on Mtb (Lee et al., 2013; Muñoz-Elías et al., 2006; Savvi et al., 2008). However, although detoxification of the propionyl-CoA pool through the methyl citrate cycle (MCC) is required for growth of Mtb in macrophages in culture, it was the methyl-malonyl pathway (MMP), leading to the synthesis of Mtb cell wall lipids, that appeared to be the preferred routing of propionyl-CoA in vivo (Figure 2F). Indeed, genes involved in the synthesis and transport of the cell wall phthiocerol dimycocerosates (PDIMs) were also upregulated in vivo specifically (fadD26, ppsA-ppsE, mmpL7, and Rv2943) (Figure S3A).
A cluster of high-confidence interactions for nodes (genes) related to nitrate/nitrite assimilation and ergothioneine (EGT) biosynthesis pathways was present in our network, suggesting that these processes may be integrated during lung macrophage infection. Genes required for inorganic nitrogen assimilation (Figure 2G) were highly expressed by Mtb in both host cell types and were implicated in either nitrate/nitrite detoxification through generation of ammonia (nirB, nirD, Rv0818, narX, narK2, and narK3) (Malm et al., 2009) or ammonia uptake (amt, glnB, and glnD) (Read et al., 2007; Williams et al., 2013). Increased intracellular levels of ammonia are essential for glutamate production, and glutamate dehydrogenase (gdh) has been shown to be indispensable for growth on glutamate or related amino acids (glutamine, aspartate) as limiting carbon sources in the presence of NO (Gallant et al., 2016). Looking at possible organic nitrogen sources, our dataset revealed upregulation of genes linked to the import of amino acids that can easily be converted to glutamate: proX, proZ, proW, and proV (glycine betaine transport), gabP (arginine and GABA), rv0072 and rv0073 (glutamine), and rocE (ornithine) (Figure S3B). Glutamate, together with cysteine, is also the substrate of egtA, the enzyme of the EGT pathway responsible for the production of the end product γ-glutamyl cysteine, that has been shown to be required in the detoxification of reactive oxidative and nitrosative species in Mtb. A knockout (KO) mutant of egtA led to a 4 log10 reduction in bacterial burden in mice infection (Saini et al., 2016; Sao Emani et al., 2018). Intriguingly, although all the genes of the EGT biosynthetic pathway were upregulated in our in vivo datasets, the magnitude of this response differed between the two host macrophage lineages, with the induction being elevated in IMs (Figure 2L), which are known to present a more hostile intracellular environment with increased levels of NO (Huang et al., 2018) (Figure 5C). Therefore, in lung macrophages, generation of glutamate appears to be a link between detoxification of NO via EGT biosynthesis and nitrogen metabolism for energy production. EGT genes (Figure S3C) are regulated by the 4Fe-4S cluster DNA-binding protein WhiB3, which maintains redox homeostasis (Cumming et al., 2018). WhiB3 also regulates expression of the molybdate transport system modA, modB, modC, which was also highly expressed in vivo (Figure S4B), providing an additional rationale for the significance of nitrogen assimilation and metabolism in supporting Mtb growth in a reducing environment (Cumming et al., 2017).
Figure 5. Transcriptional Signatures Specific to Mtb in AM and Enrichment map for the Infected versus Uninfected IMs.

(A) (I) Heatmaps showing relative expression levels for genes related to cell growth and division, ribosomal protein synthesis, cell wall synthesis, and TCA cycle and beta oxidation pathways for Mtb in AM. (II) Boxplots showing the expression levels (in log normalized counts) of the sigD and anti-sigma factor sigD (rv3413c) in Mtb.
(B) Enrichment map comparing pathways upregulated in infected versus uninfected IMs. Only GSEA gene sets with enrichment p values < 0.01 were considered in building the network.
(C) Violin plots showing the expression levels (in log-normalized counts) of the host genes Nos2 and Hif1a.
Statistical significance is provided for each plot (*p-adj. < 0.05, **p-adj. < 0.01, ***p-adj. < 0.001, and ****p-adj. < 0.0001). q values for comparisons among the groups were calculated using the Wald test as implemented in the DESeq2 pipeline.
Previous work has shown that the ESX-5 secretion system enhances bacterial virulence in vivo, although the specific mechanism(s) involved are undefined (Bottai et al., 2012; Sayes et al., 2012). The Esx-5 locus is thought to be regulated by the Pst/SenX3-RegX3 transcriptional regulatory system that responds to inorganic phosphate limitation (Elliott and Tischler, 2016a, 2016b). Although we did not specifically observe upregulation of the genes involved in response to phosphate starvation (data not shown), we note that genes encoding the subunits of the transporter were highly upregulated in vivo, with the induction being more prominent for Mtb in IMs (Figures 2H and S4A). This differential expression is consistent with reports demonstrating that IL-1β and inflammasome activation via NALP3 is dependent on a functional ESX-5 secretion system (Abdallah et al., 2011). Our data from the host transcriptome analysis reinforce this conclusion, as both the inflammasome and IL-1β pathways were more strongly upregulated in the IM lineage (Figure S3C).
Analysis of Different Host-Pathogen Transcriptional Patterns in Mtb-Infected and Uninfected AMs and IMs
PCA analysis of the mouse transcripts (Figure 3D) shows a very clear separation between ontologically linked host responses (PC1) and infection-associated responses (PC2). In previous studies we had found that the resident AM lineage was more supportive of bacterial growth than the blood monocyte-derived IM lineage (Huang et al., 2018). Therefore, in this work we sought to identify those ontology-specific responses involved in the differential growth dynamics reported previously. In order to generate a detailed appreciation of the transcriptional differences between the two host cell lineages, we directly compared both bacterial and host RNA-seq profiles in infected AMs versus IMs. A total of 319 Mtb genes (223 up in IMs, 96 up in AMs, adjusted p < 0.05) were found to be differentially expressed between the two bacterial populations (Figure 3A; Table S3), while among the host cell transcripts 3,732 genes were differentially expressed in IMs compared with AMs, and 4,097 genes in AMs compared with IMs, with adjusted p < 0.05 (Figure 3B; Table S5).
Figure 3. Analysis of Different Host-Pathogen Transcriptional Patterns in Mtb-Infected and Uninfected AMs and IMs.

(A) MA plot for the transcriptome of Mtb in the AM and IM lineages. The top 50 genes ordered by log2 fold change are highlighted. Genes with adjusted p < 0.05 are considered statistically significant.
(B) Volcano plot showing differential expression of macrophage genes in the IM- and AM-infected populations. The top 50 genes ordered by p-adj. are shown. Genes with FDRs < 0.05 are considered statistically significant.
(C) Heatmap showing relative expression levels of monocyte and AM-lineage-associated genes.
(D) PCA comparing the transcriptomes of the macrophage populations. A clear separation between ontologically linked (PC1) and infection-associated (PC2) responses is observed.
(E) Enrichment map comparing pathways upregulated in infected AMs versus infected IMs. Only GSEA gene sets with enrichment p values < 0.01 were considered in the network.
In order to identify host transcriptional signatures specific to infection versus those associated with macrophage ontogeny, we also performed DGE analysis on bystander, uninfected macrophage populations recovered from Mtb-infected mouse lungs. In summary, within the bystander cell populations, 4,707 genes were upregulated in IMs and 4,035 genes in AMs, with adjusted p < 0.05 (Table S5). Furthermore, to identify the ontogenically specific responses to Mtb infection, we also compared the transcriptional profiles of infected and uninfected cells in each macrophage subset. In AMs, 3,453 genes were upregulated and 3,119 downregulated following infection (Table S5), while in IMs, 3,614 genes were upregulated and 3,298 genes downregulated (adjusted p < 0.05) (Table S5).
To acquire greater insights into the biological processes of significance that characterize each macrophage subtype and their infection status, we performed gene set enrichment analysis (GSEA) on the ranked lists of genes (Merico et al., 2010) and scored ~26,000 gene sets for enrichment in each of our transcriptional comparisons. When considering the infected populations, a total of 205 gene sets were enriched in IMs and 375 in AMs (p < 0.01), while among the uninfected cells, 201 and 236 gene sets were enriched, respectively (p < 0.01). As previously stated, we also analyzed the transcriptional differences between infected and uninfected populations in each macrophage subtype: for AMs, 156 gene sets were enriched in the infected population and 161 in the uninfected one (p < 0.01), while for IMs, 109 and 63 were enriched for each population, respectively (p < 0.01). From this GSEA we then constructed network enrichment maps (Reimand et al., 2019) to provide a broad overview of the main biological themes associated with each host cell lineage and infection condition (Figures 3E, 4B, 5B, and S5).
Figure 4. Identification of Host and Bacterial Transcriptional Responses in Mtb Growth-Permissive AMs.

(A) Boxplots showing relative expression levels (in log-normalized counts) of genes involved in cholesterol and triglyceride metabolism in the host and genes involved in lipid import, degradation, and biosynthesis in Mtb. Our dataset reveals a transcriptional signature correlated with high availability of lipid carbon sources for the bacteria in AM and consequent use of these nutrients by Mtb during growth in this macrophage lineage.
(B) Enrichment map comparing pathways upregulated in infected versus uninfected AMs.
(C) Heatmap showing relative expression levels for the genes in the NRF2-oxidative stress response pathway.
(D) Extracellular flux analysis on AMs and IMs sorted from the mouse lung after 2 weeks of infection with 103 Mtb. (I) Oxygen consumption rate (OCR) was measured using the Cell Mito Stress Test Kit (Agilent). Oligo, oligomycin; FCCP, fluoro-carbonyl cyanide phenylhydrazone; eto, etomoxir; Rot/AA, rotenone and antimycin A. (II) Spare respiratory capacity (SRC) was calculated by normalizing maximum OCR to the basal level. (III) Reduction of maximum OCR after injection of etomoxir (Eto), a fatty acid oxidation inhibitor. p values were calculated using Student’s t test.
(E) (I) GSEA enrichment plot of infected versus uninfected AM for the “organic acid transport” pathway. (II) Boxplots showing relative expression levels (in log-normalized counts) of genes involved in the transport of molecules needed to prevent inflammation-associated intracellular damages: Slc7A11, which promotes cystine uptake and glutathione biosynthesis (Bannai, 1986), and Abcc1, involved in the extrusion of reduced glutathione (Cole, 2014).
Unless otherwise specified, the statistical significance is provided for each plot (*p-adj. < 0.05, **p-adj. < 0.01, ***p-adj. < 0.001, and ****p-adj. < 0.0001). q values for comparisons among the groups were calculated using the Wald test as implemented in the DESeq2 pipeline.
Identification of Host and Bacterial Transcriptional Responses in Mtb Growth-Permissive AMs
The enrichment maps that compare the two different macrophage ontologies (Figures 3E and S5) show marked upregulation of genes associated with pathways related to cell division (E2F targets, DNA replication, microtubule reorganization, mitotic spindle, and G/M checkpoints), fatty acid metabolism, oxidative phosphorylation, and mitochondrial function in both the infected and uninfected resident AM populations, therefore indicating an ontogeny-specific metabolic bias for this population of host cells.
Research into macrophage immune function has revealed a linkage between fatty acid metabolism and an optimal response to type I interferon (Wu et al., 2016), a pathway that is well known to promote Mtb pathogenesis (McNab et al., 2015). Intriguingly, the enrichment maps for AM showed upregulation of pathways related to triglyceride and cholesterol metabolic processes. In-depth analysis of the most highly expressed genes associated with these pathways revealed a small subset in AM (Figure 4A), most specifically Mgl, a monoacylglycerol lipase that hydrolyzes intracellular triglyceride stores to free fatty acids and glycerol; Lpl, a lipoprotein lipase that completes hydrolysis of monoglycerides from degradation of lipoprotein triglycerides; LipA, lysosomal acid lipase, the primary enzyme that hydrolyzes cholesteryl esters derived from low-density lipoprotein (LDL); 7-dehydrocholesterol (7-DHC) reductase (Dhcr7), which converts 7-DHC to cholesterol; the peroxisome proliferator-activated receptor gamma (Ppar-γ), which regulates lipid uptake; and Lpin1, the co-activator of PPAR-γ, which encodes an enzyme that catalyzes the penultimate step in triglyceride synthesis and is involved in lipid sequestration and foamy macrophage formation. This clear metabolic shift to cholesterol and fatty acid metabolism is consistent with previous data that linked the metabolic state of the AM population with the enhanced growth phenotype observed for Mtb in these cells in vivo (Huang et al., 2018).
Analysis of the pathways upregulated by AM following infection (Figure 4B) unveiled a small gene network involved in response to oxidative stress. Recent work by Rothchild et al. (2019) revealed that at early time points (up to 10 days p.i.), an NRF2-mediated oxidative stress response is upregulated in AM-infected cells compared with bystander and naive macrophages, and overexpression of the NRF2 signature is associated with reduced pro-inflammatory responses and impaired control of bacterial growth. Our data agree with these findings, as we show that at 14 days p.i., the same NRF2 pathway is upregulated in infected AMs (Figure 4C), while pro-inflammatory cytokines (Figure S6) and molecules needed to prevent inflammation-associated intracellular damage are expressed at lower levels than infected IMs (Figure 4E). Among the pathways that are downregulated in AMs following infection are those related to oxidative phosphorylation, cholesterol, and fatty acid biosynthesis, but the levels of expression of the genes in these pathways remain higher than in IMs (Figure S4C).
We used the Seahorse Extracellular Flux analyzer to examine the cell metabolism of AMs and IMs isolated from Mtb-infected mouse lungs as an independent validation for our RNA-seq analysis. Consistently, AMs exhibited much higher basal and maximal oxygen consumption rates (OCRs) compared with IMs, indicating the engagement of oxidative phosphorylation and mitochondrial activities (Figure 4DI). Notably, AMs also revealed a higher spare respiratory capacity (SRC) (Figure 4DII). Last, the maximal OCR in AM was reduced to the basal level after inhibition of the fatty acid oxidation pathway with etomoxir (Eto), whereas Eto had no effect on OCR in IMs (Figures 4DI and 4DIII), indicating that AMs are engaged in fatty acid oxidation. This observed disparity between the mitochondrial activity in AMs and IMs validates the RNA-seq data indicating that fatty acid metabolism is more active in AMs.
These results support the contention that at 14 days p.i., the infected AMs represent a host cellular niche more permissive for bacterial growth, an interpretation also supported by the transcriptional profile of Mtb isolated from AMs. These bacteria exhibited clear upregulation of several genes known to be involved in cell division and growth (ftsK, sepF, cdpC, rv1215c, ripA, and ripD), ribosomal protein synthesis (fusA1, rpsG, tuf, rpmG2, ppiA, and ppiB), and cell wall synthesis and remodeling (embB, rv0648, rv3258c, and fbpC) (Figure 5AI). Moreover, sigD and the anti-sigma factor SigD rv3413c, which had previously been shown to modulate the expression of ribosomal genes, were also upregulated (Figure 5AII) (Calamita et al., 2005). In addition, in context with the AM host cell transcriptional profile indicating an abundance of free fatty acids, glycerol, and triglycerides, the bacterial transcriptome showed upregulation of genes known to be involved in fatty acid import (mce1A, mce1B, mce1C, and mce1F) (Nazarova et al., 2017, 2019), mycolic acid biosynthesis (hadA, hadB, and hadC), lipid degradation (rv1075c, lipU, plcB, and plcC), and TCA cycle and β oxidation (acn, rv0111, icl2, fixA, icd2, echA8, and accE5) (Figures 4A and 5AI), suggesting that fatty acid oxidation may be the preferred route of energy production for growth of Mtb in the AM lineage.
Pro-inflammatory Host Gene Signatures in IMs Are Associated with an Enhanced Mtb Bacterial Stress Response
Transcriptional profiling of both infected and uninfected IM populations revealed a marked induction of pro-inflammatory pathways such as NF-κB activation and Th1 immune response (IL-1β, TNF-α, and IFN-γ) (Figures 3E and S5). Interestingly, one of the most robust networks upregulated in these cells are those genes associated with adhesion and chemotaxis, in agreement with the demonstration that this host cell population is recruited to the granuloma site from the peripheral blood (Huang et al., 2018). Looking at pathways differentially expressed upon infection (Figure 5B), IMs showed marked upregulation of gene sets related to ubiquitination, NO and reactive oxygen species (ROS) biosynthesis, secreted factors, glycolysis, and inflammatory responses. In addition, pathways related to potassium and chloride transport, two inorganic ions necessary for Mtb survival (MacGilvary et al., 2019; Tan et al., 2013), were downregulated. These host cell signatures are consistent with the IM lineage’s posing a much harsher environment for Mtb. The data indicate that monocyte-derived macrophages, recruited to the site of infection from the blood, are already activated toward a Th1 immune response potentially geared to contain Mtb growth.
Turning to the bacterium, analysis of the 226 genes upregulated by Mtb in the IM host cell population indicated that the bacterium copes with numerous different environmental stresses. Indeed, the dosR operon that mediates the transcriptional response of Mtb to oxygen limitation and NO (Bagchi et al., 2005) and whose induction is required by the bacteria to enter into a non-replicative state was upregulated (dosT, dosR, dosS, and Rv3134c) (Figure 6AI). This is also consistent with the bacterial fitness reporter strain (smyc’::mCherry;hspX’::gfp), which has been shown to express higher GFP levels in IMs following a controlling immune response (Huang et al., 2018; Sukumar et al., 2014) that led to activation of a dosR regulon promoter element (Figure 6AIII). The type II toxin-antitoxin-chaperon module higA1-higB1-secB that has been shown to be induced under stress conditions and hypothesized to be involved in bacterial persistence (Fivian-Hughes and Davis, 2010) was also upregulated, along with many genes related to DNA repair and protein folding (Figure 6AII). Finally, while most of the genes in the “in vivo signature” are common to Mtb in both AMs and IMs, we do observe further elevation of expression of genes involved in EGT biosynthesis, molybdate transport, and nitrogen assimilation pathways by Mtb in IMs, consistent with the analysis of the mouse transcriptome from the infected cells, which reveals that both Hif1A (hypoxia inducible factor) and Nos2 (NO synthase) are upregulated in this host cell population (Figure 5C). These data suggest that host-derived NO and a reducing environment are significant environmental pressures encountered by Mtb in IMs.
Figure 6. Transcriptional Signatures Specific to Mtb in IMs and Iron Metabolism.

(A) (I) Boxplots showing the expression levels (in log-normalized counts) of genes related to the dosR operon and HigA1 toxin/antitoxin system. (II) Heatmap showing the relative expression levels of genes involved in DNA repair and protein-folding processes in Mtb. (III) Violin plots showing the relative expression levels (in log-normalized counts) of the hspX and ppe37 genes for Mtb in the AM and IM lineages.
(B) Heatmaps showing the relative expression levels of iron-related genes in both the host and Mtb. The divergent transcriptional response to iron metabolism in the AM and IM populations correlate with a different iron-response phenotype in Mtb.
(C) Boxplot showing the expression levels (in log-normalized counts) of the host Nramp1 gene involved in the transport of iron into the cytoplasm and suppression of bacterial growth.
(D) Boxplots showing the expression levels (in log-normalized counts) of the Mtb genes irtA, mbtN, and eccA3, involved in carboxymycobactin uptake, synthesis, and ESX-3 secretion system (Serafini et al., 2013), respectively.
Where appropriate, statistical significance is associated with each plot (*p-adj. < 0.05, **p-adj. < 0.01, ***p-adj. < 0.001, and ****p-adj. < 0.0001). q values for comparisons among the groups were calculated using the Wald test as implemented in the DESeq2 pipeline.
Iron Metabolism Is a Prominent Transcriptional Signature Differentiating Mtb Infection in AMs and IMs
A robust transcriptional signature related to genes associated with iron metabolism was observed in both the host and pathogen (Figure 6B). Specifically, AM-derived bacteria overexpressed genes associated with iron storage, suggesting that they are in an iron-replete environment. These bacteria upregulated expression of BfrB (ferritin), a storage protein essential for iron sequestration during oxidative stress conditions (Pandey and Rodriguez, 2012) and Rv0452 a transcriptional repressor of the mmpS4 and mmpL4 siderophore exporter proteins involved in iron scavenging (Chen et al., 2018; Kahramanoglou et al., 2014).
Conversely, IM-derived Mtb showed a transcriptional signature normally associated with low-iron conditions. Mtb synthesizes the Fe3+-specific siderophores mycobactin and carboxymycobactin to scavenge insoluble and the protein-bound iron directly from the host. Genes involved in carboxymycobactin synthesis are organized into two clusters, mbt1 and mbt2 (Krithika et al., 2006), and both loci were strongly upregulated in Mtb isolated from IM (Figure 6B). In addition, genes encoding the iron-regulated ABC transporter IrtAB, which is required for Fe+3-carboxymycobactin uptake, were also overexpressed (Rodriguez and Smith, 2006). Recently, Tullius et al. (2019) described a new means of iron acquisition, heme-iron acquisition (HIA), and showed that PPE37, an iron-regulated PPE family member, was essential for HIA. PPE37 expression was strongly upregulated in Mtb in IMs, suggesting that this pathway of iron acquisition may also be important for Mtb survival in IMs in vivo (Figure 6AIII). This is a broad transcriptional signature indicating that the bacteria in the IM host cell lineage are competing to acquire iron from their host, in marked contrast to the iron-replete environment that is experienced by Mtb in AMs.
Turning to the host, both macrophage subsets upregulated genes in a manner similar to the iron-response profiles reported for M1 versus M2 macrophages (Recalcati et al., 2010; Sukhbaatar and Weichhart, 2018) (Figure 6B). In IMs, which are restrictive for Mtb growth, one of the most highly differentially expressed genes was the transcriptional regulator Spi-C. Spi-C has been shown to be upregulated in a heme-dependent manner in splenic red pulp macrophages that are required to degrade senescent erythrocytes and recycle heme-associated iron (Haldar et al., 2014). IMs also exhibited increased levels of expression of mRNA for Scara5, a ferritin-binding scavenger receptor (Li et al., 2009), Trfc, the transferrin receptor, haptoglobin, which binds hemoglobin, and most significantly the intracellular iron transporter Nramp1 (Figure 6C), which has been shown to control intracellular bacterial infections by limiting availability of divalent cations in the phagosome (Cherayil, 2011; Van Zandt et al., 2008). C57BL/6 mice have a loss-of-function mutation in the Slc11a1 gene (Nramp1), associated with inability to clear some types of intracellular infections (Vidal et al., 1996). However, this lesion does not affect the rate of import/export of iron from cells, indicating that it is part of a larger physiological program (Kuhn et al., 1999). The iron starvation phenotype observed by Mtb in IM supports the hypothesis that these genes are involved in the generation of an iron sequestration phenotype consistent with M1 macrophage activation and increased microbicidal activity (Gaetano et al., 2010).
In contrast, AMs exhibited enhanced expression of the transcription factor Myc, and ectopic expression of Myc has been shown to drive polarization of macrophages toward a M2-like phenotype (Pello, 2016; Zhong et al., 2018), which is linked to iron release (Gaetano et al., 2010; Soares and Hamza, 2016). As illustrated in Figure 6B, AMs showed relatively higher levels of expression of transcripts for CD163, the hemoglobin scavenger receptor, Hrg1, the endosomal heme transporter, and Ngal, the lipocalin 2 receptor (Gaetano et al., 2010; Jung et al., 2012; Soares and Hamza, 2016). In addition, they showed increased transcripts for Aco1/Irp1, the dual aconitase/cytosolic iron response protein that regulates transcription of several IRE-containing genes (Sanchez et al., 2011; Wang and Pantopoulos, 2011) including Aco2, the mitochondrial aconitase, Fech, ferrochelatase required for heme biosynthesis, Hmox2, a hemoxygenase involved in breakdown of heme and release of iron, and Trf, transferrin. Moreover, mRNAs encoding several iron-containing, iron-utilizing, and iron-regulating mitochondrial proteins were upregulated indicative of the enhanced mitochondrial activity know to occur in these cells (namely, Fxn, the mitochondrial iron regulator frataxin; Alas1, aminolevulinate synthase; fdx1, a mitochondrial electron transfer protein adrenodoxin; and Sfxn1 & 2, iron-regulated mitochondrial serine transporters) (Kelly and O’Neill, 2015; Mills and O’Neill, 2016). This profile is consistent with the M2-like polarization profile and enhanced mitochondrial respiration of the AM population.
DISCUSSION
Successful execution of dual RNA-seq from in vivo-derived infected host cell populations remains a considerable technical challenge. However, in the present study we were able to generate robust datasets on both host and bacterium from two distinct host myeloid lineages isolated directly from infected mouse lungs using a simple bacterial RNA enrichment protocol. Moreover the use of fluorescent bacterial fitness reporter strains in an in vivo infection model that incorporates host cell heterogeneity and provides an adequate source of infected host cells for flow sorting and isolation increases the resolution of such studies (Russell et al., 2019). The data presented here detail the transcriptional responses of both host and pathogen during in vivo infection of two ontologically distinct macrophage lineages.
The dual RNA-seq datasets in this study provide further elucidation of why AMs represent a preferred niche for Mtb replication. The data are also consistent with IMs’ being recruited to the site of infection and fulfilling the major role in the containment of bacterial growth. Previous work showed that depletion of the AM population resulted in an 80% reduction in bacterial burden, while depletion of IMs resulted in almost a 10-fold increase in bacterial burden (Huang et al., 2018). Our analysis of both host and bacterial transcriptomes re-emphasizes the significance of nutritional immunity (Kochan, 1973) for in vivo control of infection. The key role played by iron metabolism in tuberculosis infection is not a new theme and is backed by an extensive body of data from genome-wide susceptibility studies (Gallant et al., 2007; Govoni and Gros, 1998; Vidal et al., 1996) to experimental in vitro infections (Abreu et al., 2018; Dahl et al., 2018; De Voss et al., 2000; Olakanmi et al., 2000, 2013; Wagner et al., 2005). Moreover, many of the host genes involved in the iron transcriptional signature have been reported to be responsive to heme-associated iron, tempting one to speculate that the hemorrhaging and red blood cell damage, frequently observed in infected mouse tissue, may drive this aspect of the host cell response. This interpretation is consistent with a recent report that ferroptosis, or iron-mediated cell death, is a robust signature in Mtb-infected mouse lung tissue (Amaral et al., 2019).
Finally, although both Mtb populations are shifted to cholesterol use in vivo, the Mtb in the AM population exhibits a more marked upregulation of genes involved in acquisition and use of fatty acids. Metabolic flux analysis of host macrophages provided experimental verification of the increased oxidative phosphorylation and mitochondrial function in AMs compared with IMs, providing further support that the growth advantage experienced by Mtb in AMs is also a consequence of the metabolic interface between host cell and pathogen.
In the present study we examine a single time point during mouse infection, and although such analysis needs to be extended to later time points following the development of acquired immunity, it holds considerable value as a proof of principle and technical advance that stresses the importance of interrogating in vivo-derived host cell and pathogen populations to unravel the molecular basis of infection and the disease process.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David G. Russell (dgr8@cornell.edu).
This study did not generate new unique reagents. Plasmids used in this study will be available through contact information provided in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD64 | Biolegend | Cat# 139307, RRID:AB_2561962 |
| Mertk | Thermo Fisher | Cat# 12–5751–82, RRID:AB_257262 |
| SiglecF | BD Bioscience | Cat# 564514, RRID:AB_2738833 |
| Bacterial and Virus Strains | ||
| M.tuberculosis Erdman mCherry | Originally from Tanya Parish lab https://dx.plos.org/10.1371/journal.pone.0009823 | N/A |
| Critical Commercial Assays | ||
| Ribo-Zero Gold rRNA Removal Kit (Epidemiology) | Illumina | Cat# MRZE724 |
| Glycoblue | Thermo Fisher | Cat# AM9515 |
| Seahorse XFp Cell Mito Stress Test Kit | Agilent | Cat# 103010–100 |
| Deposited Data | ||
| Bystander AM and IM populations | GEO: GSE108844 | N/A |
| MicroArray studies | Rohde et al., 2012; Schnappinger et al., 2003 | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | The Jackson Laboratory | Cat# JAX:000664, RRID:IMSR_JAX:000664 |
| Software and Algorithms | ||
| DESeq2 | Love et al., 2014 | N/A |
| ApeGLM | Zhu et al., 2019 | N/A |
| GSEA | Broad Institute http://software.broadinstitute.org/gsea/index.jsp | N/A |
| EnrichmentMap | Reimand et al., 2019 | N/A |
| FastQC (v. 0.11.5) | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | N/A |
| Flexbar (v. 3.4) | Roehr et al., 2017 | N/A |
| Bowtie2 | Langmead and Salzberg, 2012 | N/A |
| Hisat2 (v. 2.1.0) | Kim et al., 2015 | N/A |
| HTSeq (v. 0.11.0) | Anders et al., 2015 | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mtb Strains
Mycobacterium tuberculosis Erdman ATCC 35801 was the parental strain used for all experiments. The fluorescent reporter smyc’::mCherry was previously described (Sukumar et al., 2014). Bacteria were grown at 37°C to mid-log phase in MiddleBrook 7H9 broth supplemented with 10% oleic acid/albumin/dextrose/catalase (OADC Enrichment - Becton, Dickinson and Company), 0.2% glycerol, 0.05% Tyloxapol (Sigma-Aldrich). Hygromycin B (50 μg/ml) was used as a selection marker for the fluorescent strain smyc’::mCherry. For mice infection, aliquots were frozen in 10% glycerol, titered and stored at −80°C until use, while fresh bacterial cultures were used for RNA extraction from tissue and broth cultures (reference samples).
Mice
C57BL/6J WT mice were purchased from The Jackson Laboratory. The mice used in this study were 6–8 weeks old. All mice were maintained in a specific pathogen free animal biosafety level 3 facility at Cornell University. Animal care was in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care. All animal procedures were approved by the Institutional Animal Care and Use Committee of Cornell University.
METHOD DETAILS
BMDM culture and infection
BMDM cells were isolated from femur and tibia of euthanized C57BL/6J WT mice (Jackson Laboratories). Briefly, femur and tibia were dissected from the mice and surrounding tissues and muscles removed. Both ends of the bones were cut, bone-marrow flushed with Dulbecco’s modified Eagle’s medium (DMEM) using a 21-gage needle, then centrifuged at 1000 rpm for 5′ and resuspended in DMEM supplemented with 10% fetal bovine serum (Thermo Scientific), 2 mM L-glutamine, 1 mM sodium pyruvate, 10% L-cell conditioning media and 1% penicillin/streptomycin (Corning cellgro). Cells were then grown at 37°C for 7 days in culture dishes while media was replenished every 3 days. At day 7, non-adherent cells together with red blood cells were removed using PBS washes. BMDM were infected with Mtb smyc’::mCherry at a multiplicity of infection of 2:1 for 4 hours (test samples) or 6 hours (reference samples). Briefly, macrophages were seeded in T75 vented tissue culture flasks at a density of ~2×107 per flask. The next day, mid-log phase Mtb cultures (OD600 = 0.5 to 0.6) were harvested by centrifugation, resuspended in basal uptake buffer (PBS with 4.5 mg/ml glucose, 5 mg/ml BSA, 0.1 mg/ml CaCl2, 0.1 mg/ml MgCl2, and 1 mg/ml gelatin), passaged 10 times through a 21-gauge needle, and then used to infect macrophages. Extracellular bacteria were removed after 2 hours and the media replaced. At 4h or 6h post-infection, RNA was extracted as described in the corresponding sections of this paper.
Mice infection and lung cells isolation
Mice were infected intranasally with Mtb. Mice were anesthetized and inoculated with ~1000 CFU of the Erdman strain smyc’:: mCherry, resuspended in 25ul of PBS containing 0.05% Tween 80. Inoculum dosage was confirmed by plating different dilutions of the bacterial stock in 7H10 agar plates supplemented with OADC, glycerol and hygromycin B. Plates were incubated at 37°C and colonies enumerated 3 weeks after. Two weeks post-infection mice were sacrificed, the lungs aseptically removed and placed in PBS containing 5% FBS. To minimize unwanted changes in the gene expression profile of both host and bacteria, samples were kept on ice and immediately processed using a GentleMACS tissue dissociator (Miltenyi Biotec). The dissociated lung material was then passed through a 70 μM cell strainer and red blood cells were lysed with ACK lysis buffer (Lonza).
Sorting of AM and IM populations
Staining of the lung phagocytes was streamlined to minimize processing time and unwanted changes in the gene expression profile. Briefly, lung cell suspensions were incubated for 20 minutes in the dark with fluorophore-conjugated antibodies against mouse CD64 (X54–5/7.1), MerTK (DS5MMER) and SiglecF (E50–2440) from BioLegend, Thermo Fisher or BD Biosciences. The panel of antibodies used in this study to separate the two phagocytic lung populations has been described and validated in a previous study (Huang et al., 2018). Stained samples have then been washed in PBS, resuspended in sorting buffer (PBS, 5% FBS, 5mM EDTA, 25mM HEPES) and sorted according to the gating strategy depicted in Figure 1A. Samples were maintained at 4°C during sorting and collected directly into Trizol (Ambion).
Recovery of Mtb-infected cells from the lung tissues for subsequent processing varied from 1.2 – 2.5×104 for AM and 2.5 – 4×104 for IM, and a full description of these samples and subsequent RNA yields is provided in Table 1. In those cases where the number of recovered infected cells were lower than 40k, we chose a trade-off between enrichment and sequencing depth in order to have enough input RNA for library preparation (Figure 1D; Table 1)
Extracellular Flux Analysis (Agilent Seahorse)
Real-time cell metabolism of AM and IM was determined by using a XFp Extracellular Flux Analyzer. FACS sorted AM and IM were plated in 8-well Seahorse plates. Mitochondrial function was determined by using the Seahorse XFp Cell Mito Stress Test Kit (Agilent Technologies, Santa Clara, CA), according to the manufacturer’s instruction. Three or four measurements were obtained under basal conditions and after the sequential addition of oligomycin, FCCP, etomoxir and Rotenone/antimycin A.
Development of the Dual RNA-seq protocol
Previous experiments informed us that 4 × 104 was the maximum number of infected AM recoverable following sorting from 3 pooled mouse lungs. Therefore, since the number of infected host cells and consequently the amount of total mixed RNA recovered from an in vivo infection was likely to be limited, we performed preliminary experiments to assess if a modified rRNA depletion step worked efficiently for low-input samples and if the amount of total RNA left after bacterial enrichment would be sufficient for library preparation: our library preparation kit (see “Library preparation and sequencing”) requires at least 2ng of input RNA after rRNA depletion. From previous experiments, we estimated to recover ~90ng of total RNA from 40k eukaryotic cells, which after rRNA subtraction (95% of total RNA) would result in ~5ng of input RNA. Therefore, we tested the protocol using 2 biological replicates of BMDM, infected with fluorescent Mtb Erdman (smyc’::mCherry) at an MOI of 2:1 for 4h. 4×104 Mtb-infected cells were sorted, collected and incubated in Trizol to lyse the host macrophages (Figure 1B). The samples were processed for RNA isolation as detailed in the following section, following removal of 30% and 50% of the volume of the supernatant containing host RNA. In both cases we were able to recover enough mRNA (after rRNA depletion) for library preparation and we achieved an enrichment where 5% or 9% of the total reads aligned to the Mtb reference genome when discarding 30% or 50% of the volume of Trizol containing host RNA, respectively. Moreover, the modified rRNA depletion step was able to efficiently deplete ribosomal RNA from all the samples (Table S1).
RNA extraction
Bacterial RNA extraction for reference samples (BMDM at 6h of infection and broth cultures) was performed using the protocol previously described (Rohde et al., 2012). Extraction of total RNA (eukaryotic + bacterial) from lung phagocytic populations was performed using the modified protocol to optimize the pathogen/host RNA ratio.
In brief, sorted samples were collected in Trizol, mixed and incubated for 5 minutes at RT to allow complete dissociation of the nucleoprotein complexes of the host cells. Specimens were then centrifuged at max speed for 20’ to pellet intact mycobacterial cells. ~80% of the Trizol (containing host RNA) was removed and placed in an RNase-free tube for later use. 0.1mm zirconia/silica beads (Biospec) together with fresh Trizol (~400ul) were then added to the Mtb containing tube and bacilli were lysed using a BeadBeater. Subsequently, based on the number of sorted cells, a portion of the host RNA (usually > 50% of the volume) was added back to the tube containing bacterial RNA, mixed and the extraction carried out following the Trizol protocol. (Chomczynski and Sacchi, 1987). Because of the limited number of infected phagocytes recovered from the sorting procedure, we used Glycoblue (Invitrogen) during the precipitation step to maximize RNA recovery and have sufficient quantity for library preparation. RNA samples were resuspended in nuclease-free water (Ambion) and genomic DNA contamination removed using the commercially available Turbo-DNA free kit (Invitrogen).
Library preparation and sequencing
rRNA removal was performed using 50–100ng total RNA input and a modified protocol for the Ribo-Zero Epidemiology Gold rRNA removal kit (Illumina). Briefly, 90 μL bead stock was used per sample, together with 2 μL each of reaction buffer and removal solution in a 20 μL reaction volume, as detailed in the manufacturer’s protocol. The rRNA-depleted samples were purified by precipitating the RNA. Sequencing libraries were generated using the NEB Next Ultra II Directional RNA Library Prep Kit for Illumina (New England Bio-Labs). Libraries were sequenced on a Next Seq 500 (Illumina) in multiple rounds until the desired sequencing depth for bacterial reads was reached (target 1M 85nt reads).
QUANTIFICATION AND STATISTICAL ANALYSIS
Rationale for the use of APEGLM – Approximate posterior estimation for generalized linear models
Estimation of LFC (log fold change) represents a continuing challenge for RNA-seq analysis of datasets with low sequencing depth and limited numbers of biological replicates. In presence of low or high variable counts, the maximum likelihood estimates (MLE) for the LFCs have high variance leading to poor ranking of genes by effect sizes. Because ranking of genes by LFC is a common approach in the analysis of transcriptional data and ranked lists of genes are used in downstream applications such as network and pathway analysis, different approaches have been proposed to stabilize the estimation of LFC for genes with low counts and high variance, including filtering out low count genes and using scaled pseudocounts (edgeR, limma-voom) or through the use of Bayesian modeling. For example DESeq2, one of the most popular packages for differential gene expression analysis, (Love et al., 2014) uses an adaptive normally distributed prior for shrinkage of LFC.
However recently, a new empirical Bayes procedure (APEGLM) that makes use of a heavy-tailed Cauchy prior distribution has been proposed to stabilize the estimator of LFC. (Zhu et al., 2019) Briefly, using the same framework of DESeq2 (a Negative Binomial (NB) generalized linear model (GLM)), apeglm exchanges the normal distribution with a heavy-tailed Cauchy distribution on the effect sizes, with fixed shape and scale adapted to the distribution of observed MLE of the effect sizes for all genes. For each gene, it then uses a LaPlace approximation to provide shrinkage estimates and corresponding SD (Zhu et al., 2019), This removes the needs for filtering rules or pseudocounts and maximizes the power of the current data to estimate the effect size for each gene. In particular, it has been shown that this statistical approach preserves true, large differences in LFC across conditions and is superior to common methods in ranking of genes by LFC in presence of low counts (Zhu et al., 2019).
Dual RNA-seq Data analysis
Raw sequencing reads were analyzed using FastQC (v. 0.11.5) for quality control. Flexbar (v. 3.4)(Roehr et al., 2017) has been used to remove low quality reads and trim Illumina adapters. rRNA reads have been removed using Bowtie2 (Langmead and Salzberg, 2012) (–sensitive mode) and a custom GTF file. For the processing of in vivo datasets, filtered fastq files were split using Bowtie2 (–very-sensitive mode) into species-specific files using the two reference genomes, GRCm38.94 for Mus musculus and NCBI assembly GCA_00668235.1 for Mtb Erdman. Hisat2 (v. 2.1.0) (Kim et al., 2015) was used to align reads to the respective transcriptomes and raw read counts for each sample were obtained using HTSeq (v. 0.11.0) (Anders et al., 2015). Two separate raw gene-count matrixes were obtained: one for the Mtb samples and one for the Mus musculus samples. Unless noted, exploratory, visualization and differential gene expression analysis was carried out in R using the DeSeq2 pipeline and APEGLM for LFC estimation. Genes with less than 10 raw counts across all samples were excluded from downstream analysis.
Pathway enrichment analysis was performed to compare the host conditions (AM versus IM, Infected versus Uninfected). A ranked list of genes was generated and GSEA analysis performed using the gene-sets provided by the BaderLab (http://download.baderlab.org/EM_Genesets/) excluding electronically annotated gene-sets. As of December 2018 this list contained around 26000 pathways from different sources (GO, Reactome, Panther, IOB, NetPath) (Reimand et al., 2019). GSEA analysis was carried out with the following settings: 1) number of permutations: 2000; 2) Enrichment statistic: weighted; 3) Min and max gen-sets size: 10 and 500; 4) Normalization mode: meandiv. Gene-sets enriched in both populations at a nominal p value < 0.01 were selected to build an Enrichment Map in Cytoscape (Merico et al., 2010; Reimand et al., 2019). Because of the technical differences in the mRNA enrichment strategy between infected (rRNA depletion) and uninfected (polyA selection - Huang et al., 2018) samples, for the host cell infected versus uninfected comparisons (Table S5) genes known to be non-polyadenylated have been excluded from the downstream analysis with GSEA and Cytoscape. The protein-protein interaction network for the Mtb “in vivo signature” was created in Cytoscape using the STRING app (1.4.1). Only high-confidence interactions (co-expression, experiments, neighborhood, co-occurrence with a score > 0.7) relative to query proteins were considered. The entire data pipeline (Linux commands, software settings and R code) is provided in Supplementary Materials.
Data mining to define the Mtb in vivo gene-signature
Using our own datasets (14 days AM and IM in vivo, 6hr BMDM cultures and 7H9-OADC broth cultures) we performed differential gene expression to identify a list of 274 genes that were specifically upregulated in one or both in vivo host macrophage populations (IM and AM) but not in either short-term BMDM or broth culture conditions. After that, we mined the microarray datasets from previous studies (Rohde et al., 2012; Schnappinger et al., 2003) to generate another list of genes that have been reported to be specifically upregulated in BMDM cultures (at different time points – 24h, 48h up to 14 days of cell culture infection in the Rohde et al., 2012, or under different conditions – activated versus resting BMDM in Schnappinger et al., 2003) but not in broth cultures (7H9-OADC). Finally, we compared our 274 gene-list with this microarray-derived list and all the genes that overlapped were excluded from the “in vivo signature.” We ended up with 180 genes specific to the in vivo infection environment. This certainly represents a conservative approach, however it allowed us to identify Mtb expression signatures unique to the in vivo lung macrophage environment.
Statistical Analysis
Statistical testing for the differential gene expression analysis (DGE) was performed as described (Love et al., 2014). Shrinkage of effect sizes (LFC estimates) has been performed using the APEGLM method (Zhu et al., 2019). Unless specified otherwise, genes having a false discovery rate (FDR) < 0.05 and a fold change > 1.5 were considered significant. Visualization and clustering (PCA, heatmaps of sample-to-sample distances) were performed on variance stabilized counts (vst) (Anders and Huber, 2010) with the option “blind = TRUE” in the DESeq2 package in order to compare samples in an unbiased manner. Heat-maps for specific groups of genes were generated using the normalized counts obtained from the DESeq2 analysis, which have been log-transformed and Z-scaled using the package heatmap2 in R.
DATA AND CODE AVAILABILITY
The datasets supporting the conclusions of this article are available in GEO: GSE132354 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132354
The data analysis pipeline (Linux code and R scripts) is provided as a zip file in the supplemental materials.
The datasets for the uninfected AM and IM samples are available in GEO: GSE108844 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108844
Supplementary Material
Dual RNA-seq analysis of M. tuberculosis infected macrophages
Infected macrophages were isolated directly from mouse lung
Transcriptional signatures of host and pathogen varied with macrophage ontogeny
Data highlighted a key role for nutritional immunity in limiting bacterial growth
ACKNOWLEDGMENTS
All animal procedures were approved by the Institutional Animal Care and Use Committee of Cornell University. The work was supported by grants AI118582 and AI134183 to D.G.R. from the National Institutes of Health, USA. We wish to thank Linda Bennett for technical and organizational support. We also wish to thank Brian C. VanderVen and Saikat Boliar for critical reading of the manuscript.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.033.
REFERENCES
- Abdallah AM, Bestebroer J, Savage ND, de Punder K, van Zon M, Wilson L, Korbee CJ, van der Sar AM, Ottenhoff TH, van der Wel NN, et al. (2011). Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J. Immunol 187, 4744–4753. [DOI] [PubMed] [Google Scholar]
- Abreu R, Essler L, Loy A, Quinn F, and Giri P (2018). Heparin inhibits intracellular Mycobacterium tuberculosis bacterial replication by reducing iron levels in human macrophages. Sci. Rep 8, 7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, Mayer-Barber KD, Andrade BB, and Sher A (2019). A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J. Exp. Med 216, 556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi G, Chauhan S, Sharma D, and Tyagi JS (2005). Transcription and autoregulation of the Rv3134c-devR-devS operon of Mycobacterium tuberculosis. Microbiology 151, 4045–4053. [DOI] [PubMed] [Google Scholar]
- Bannai S (1986). Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem 261, 2256–2263. [PubMed] [Google Scholar]
- Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, Sayes F, Bitter W, Brennan MJ, Leclerc C, Batoni G, et al. (2012). Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol. Microbiol 83, 1195–1209. [DOI] [PubMed] [Google Scholar]
- Calamita H, Ko C, Tyagi S, Yoshimatsu T, Morrison NE, and Bishai WR (2005). The Mycobacterium tuberculosis SigD sigma factor controls the expression of ribosome-associated gene products in stationary phase and is required for full virulence. Cell. Microbiol 7, 233–244. [DOI] [PubMed] [Google Scholar]
- Casabon I, Zhu SH, Otani H, Liu J, Mohn WW, and Eltis LD (2013). Regulation of the KstR2 regulon of Mycobacterium tuberculosis by a cholesterol catabolite. Mol. Microbiol 89, 1201–1212. [DOI] [PubMed] [Google Scholar]
- Chen Y, Chen J, Zhang S, Shi W, Zhang W, Zhu M, and Zhang Y (2018). Novel mutations associated with clofazimine resistance in Mycobacterium abscessus. Antimicrob. Agents Chemother. 62, e00544–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherayil BJ (2011). The role of iron in the immune response to bacterial infection. Immunol. Res 50, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, and Sacchi N (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem 162, 156–159. [DOI] [PubMed] [Google Scholar]
- Cole SP (2014). Multidrug resistance protein 1 (MRP1, ABCC1), a “multi-tasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem 289, 30880–30888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming BM, Rahman MA, Lamprecht DA, Rohde KH, Saini V, Adamson JH, Russell DG, and Steyn AJC (2017). Mycobacterium tuberculosis arrests host cycle at the G1/S transition to establish long term infection. PLoS Pathog. 13, e1006389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumming BM, Chinta KC, Reddy VP, and Steyn AJC (2018). Role of ergothioneine in microbial physiology and pathogenesis. Antioxid. Redox Signal. 28, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl SL, Woodworth JS, Lerche CJ, Cramer EP, Nielsen PR, Moser C, Thomsen AR, Borregaard N, and Cowland JB (2018). Lipocalin-2 functions as inhibitor of innate resistance to Mycobacterium tuberculosis. Front. Immunol 9, 2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damron FH, Oglesby-Sherrouse AG, Wilks A, and Barbier M (2016). Dual-seq transcriptomics reveals the battle for iron during Pseudomonas aeruginosa acute murine pneumonia. Sci. Rep. 6, 39172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, and Barry CE 3rd. (2000). The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U S A 97, 1252–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SR, and Tischler AD (2016a). Phosphate responsive regulation provides insights for ESX-5 function in Mycobacterium tuberculosis. Curr. Genet 62, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SR, and Tischler AD (2016b). Phosphate starvation: a novel signal that triggers ESX-5 secretion in Mycobacterium tuberculosis. Mol. Microbiol 100, 510–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fivian-Hughes AS, and Davis EO (2010). Analyzing the regulatory role of the HigA antitoxin within Mycobacterium tuberculosis. J. Bacteriol 192, 4348–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaetano C, Massimo L, and Alberto M (2010). Control of iron homeostasis as a key component of macrophage polarization. Haematologica 95, 1801–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant CJ, Malik S, Jabado N, Cellier M, Simkin L, Finlay BB, Graviss EA, Gros P, Musser JM, and Schurr E (2007). Reduced in vitro functional activity of human NRAMP1 (SLC11A1) allele that predisposes to increased risk of pediatric tuberculosis disease. Genes Immun. 8, 691–698. [DOI] [PubMed] [Google Scholar]
- Gallant JL, Viljoen AJ, van Helden PD, and Wiid IJ (2016). Glutamate dehydrogenase is required by Mycobacterium bovis BCG for resistance to cellular stress. PLoS ONE 11, e0147706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govoni G, and Gros P (1998). Macrophage NRAMP1 and its role in resistance to microbial infections. Inflamm. Res 47, 277–284. [DOI] [PubMed] [Google Scholar]
- Haldar M, Kohyama M, So AY, Kc W, Wu X, Briseño CG, Satpathy AT, Kretzer NM, Arase H, Rajasekaran NS, et al. (2014). Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell 156, 1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Nazarova EV, Tan S, Liu Y, and Russell DG (2018). Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med 215, 1135–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatiadis N, Klaus B, Zaugg JB, and Huber W (2016). Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 13, 577–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Weigert A, Tausendschön M, Mora J, Ören B, Sola A, Hotter G, Muta T, and Brüne B (2012). Interleukin-10-induced neutrophil gelatinase-associated lipocalin production in macrophages with consequences for tumor growth. Mol. Cell. Biol. 32, 3938–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahramanoglou C, Cortes T, Matange N, Hunt DM, Visweswariah SS, Young DB, and Buxton RS (2014). Genomic mapping of cAMP receptor protein (CRP Mt) in Mycobacterium tuberculosis: relation to transcriptional start sites and the role of CRPMt as a transcription factor. Nucleic Acids Res. 42, 8320–8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly B, and O’Neill LA (2015). Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 25, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochan I (1973). The role of iron in bacterial infections, with special consideration of host-tubercle bacillus interaction. Curr. Top. Microbiol. Immunol 60, 1–30. [DOI] [PubMed] [Google Scholar]
- Krithika R, Marathe U, Saxena P, Ansari MZ, Mohanty D, and Gokhale RS (2006). A genetic locus required for iron acquisition in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U S A 103, 2069–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn DE, Baker BD, Lafuse WP, and Zwilling BS (1999). Differential iron transport into phagosomes isolated from the RAW264.7 macrophage cell lines transfected with Nramp1Gly169 or Nramp1Asp169. J. Leukoc. Biol. 66, 113–119. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, VanderVen BC, Fahey RJ, and Russell DG (2013). Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J. Biol. Chem 288, 6788–6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Paragas N, Ned RM, Qiu A, Viltard M, Leete T, Drexler IR, Chen X, Sanna-Cherchi S, Mohammed F, et al. (2009). Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev. Cell 16, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGilvary NJ, Kevorkian YL, and Tan S (2019). Potassium response and homeostasis in Mycobacterium tuberculosis modulates environmental adaptation and is important for host colonization. PLoS Pathog. 15, e1007591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm S, Tiffert Y, Micklinghoff J, Schultze S, Joost I, Weber I, Horst S, Ackermann B, Schmidt M, Wohlleben W, et al. (2009). The roles of the nitrate reductase NarGHJI, the nitrite reductase NirBD and the response regulator GlnR in nitrate assimilation of Mycobacterium tuberculosis. Microbiology 155, 1332–1339. [DOI] [PubMed] [Google Scholar]
- McNab F, Mayer-Barber K, Sher A, Wack A, and O’Garra A (2015). Type I interferons in infectious disease. Nat. Rev. Immunol 15, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, and O’Neill LA (2016). Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 46, 13–21. [DOI] [PubMed] [Google Scholar]
- Montoya DJ, Andrade P, Silva BJA, Teles RMB, Ma F, Bryson B, Sadanand S, Noel T, Lu J, Sarno E, et al. (2019). Dual RNA-seq of human leprosy lesions identifies bacterial determinants linked to host immune response. Cell Rep. 26, 3574–3585.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Elías EJ, Upton AM, Cherian J, and McKinney JD (2006). Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol. Microbiol 60, 1109–1122. [DOI] [PubMed] [Google Scholar]
- Nazarova EV, Montague CR, La T, Wilburn KM, Sukumar N, Lee W, Caldwell S, Russell DG, and VanderVen BC (2017). Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. eLife 6, e26969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarova EV, Montague CR, Huang L, La T, Russell D, and VanderVen BC (2019). The genetic requirements of fatty acid import by Mycobacterium tuberculosis within macrophages. eLife 8, e43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss AM, Beckstette M, Pimenova M, Schmühl C, Opitz W, Pisano F, Heroven AK, and Dersch P (2017). Tissue dual RNA-seq allows fast discovery of infection-specific functions and riboregulators shaping host-pathogen transcriptomes. Proc. Natl. Acad. Sci. U S A 114, E791–E800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olakanmi O, Britigan BE, and Schlesinger LS (2000). Gallium disrupts iron metabolism of mycobacteria residing within human macrophages. Infect. Immun. 68, 5619–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olakanmi O, Kesavalu B, Pasula R, Abdalla MY, Schlesinger LS, and Britigan BE (2013). Gallium nitrate is efficacious in murine models of tuberculosis and inhibits key bacterial Fe-dependent enzymes. Antimicrob. Agents Chemother. 57, 6074–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R, and Rodriguez GM (2012). A ferritin mutant of Mycobacterium tuberculosis is highly susceptible to killing by antibiotics and is unable to establish a chronic infection in mice. Infect. Immun 80, 3650–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pello OM (2016). Macrophages and c-Myc cross paths. OncoImmunology 5, e1151991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson EJ, Bailo R, Rothchild AC, Arrieta-Ortiz ML, Kaur A, Pan M, Mai D, Abidi AA, Cooper C, Aderem A, et al. (2019). Path-seq identifies an essential mycolate remodeling program for mycobacterial host adaptation. Mol. Syst. Biol 15, e8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman KJ, Aliota MT, and Knoll LJ (2014). Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection. BMC Genomics 15, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read R, Pashley CA, Smith D, and Parish T (2007). The role of GlnD in ammonia assimilation in Mycobacterium tuberculosis. Tuberculosis (Edinb.) 87, 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, Zammataro L, Girelli D, and Cairo G (2010). Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol 40, 824–835. [DOI] [PubMed] [Google Scholar]
- Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, Wadi L, Meyer M, Wong J, Xu C, et al. (2019). Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc 14, 482–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GM, and Smith I (2006). Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J. Bacteriol 188, 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehr JT, Dieterich C, and Reinert K (2017). Flexbar 3.0—SIMD and multicore parallelization. Bioinformatics 33, 2941–2942. [DOI] [PubMed] [Google Scholar]
- Rohde KH, Veiga DF, Caldwell S, Balázsi G, and Russell DG (2012). Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog. 8, e1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothchild AC, Olson GS, Nemeth J, Amon LM, Mai D, Gold ES, Diercks AH, and Aderem A (2019). Alveolar macrophages generate a noncanonical NRF2-driven transcriptional response to Mycobacterium tuberculosis in vivo. Sci. Immunol. 4, eaaw6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DG, Huang L, and VanderVen BC (2019). Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol 19, 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini V, Cumming BM, Guidry L, Lamprecht DA, Adamson JH, Reddy VP, Chinta KC, Mazorodze JH, Glasgow JN, Richard-Greenblatt M, et al. (2016). Ergothioneine maintains redox and bioenergetic homeostasis essential for drug susceptibility and virulence of Mycobacterium tuberculosis. Cell Rep. 14, 572–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez M, Galy B, Schwanhaeusser B, Blake J, Bähr-Ivacevic T, Benes V, Selbach M, Muckenthaler MU, and Hentze MW (2011). Iron regulatory protein-1 and −2: transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins. Blood 118, e168–e179. [DOI] [PubMed] [Google Scholar]
- Sao Emani C, Williams MJ, Van Helden PD, Taylor MJC, Wiid IJ, and Baker B (2018). Gamma-glutamylcysteine protects ergothioneine-deficient Mycobacterium tuberculosis mutants against oxidative and nitrosative stress. Biochem. Biophys. Res. Commun 495, 174–178. [DOI] [PubMed] [Google Scholar]
- Savvi S, Warner DF, Kana BD, McKinney JD, Mizrahi V, and Dawes SS (2008). Functional characterization of a vitamin B12-dependent methyl-malonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J. Bacteriol. 190, 3886–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayes F, Sun L, Di Luca M, Simeone R, Degaiffier N, Fiette L, Esin S, Brosch R, Bottai D, Leclerc C, and Majlessi L (2012). Strong immunogenicity and cross-reactivity of Mycobacterium tuberculosis ESX-5 type VII secretion: encoded PE-PPE proteins predicts vaccine potential. Cell Host Microbe 11, 352–363. [DOI] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, and Schoolnik GK (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med 198, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini A, Pisu D, Palù G, Rodriguez GM, and Manganelli R (2013). The ESX-3 secretion system is necessary for iron and zinc homeostasis in Mycobacterium tuberculosis. PLoS ONE 8, e78351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MP, and Hamza I (2016). Macrophages and iron metabolism. Immunity 44, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhbaatar N, and Weichhart T (2018). Iron regulation: macrophages in control. Pharmaceuticals (Basel) 11, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar N, Tan S, Aldridge BB, and Russell DG (2014). Exploitation of Mycobacterium tuberculosis reporter strains to probe the impact of vaccination at sites of infection. PLoS Pathog. 10, e1004394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Sukumar N, Abramovitch RB, Parish T, and Russell DG (2013). Mycobacterium tuberculosis responds to chloride and pH as synergistic cues to the immune status of its host cell. PLoS Pathog. 9, e1003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullius MV, Nava S, and Horwitz MA (2019). PPE37 is essential for Mycobacterium tuberculosis heme-iron acquisition (HIA), and a defective PPE37 in Mycobacterium bovis BCG prevents HIA. Infect. Immun. 87, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zandt KE, Sow FB, Florence WC, Zwilling BS, Satoskar AR, Schlesinger LS, and Lafuse WP (2008). The iron export protein ferroportin 1 is differentially expressed in mouse macrophage populations and is present in the mycobacterial-containing phagosome. J. Leukoc. Biol 84, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, Crowe AM, Eltis LD, Perola E, Deininger DD, et al. (2015). Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog. 11, e1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal SM, Pinner E, Lepage P, Gauthier S, and Gros P (1996). Natural resistance to intracellular infections: Nramp1 encodes a membrane phosphoglycoprotein absent in macrophages from susceptible (Nramp1 D169) mouse strains. J. Immunol 157, 3559–3568. [PubMed] [Google Scholar]
- Wagner D, Maser J, Lai B, Cai Z, Barry CE 3rd, Höner Zu Bentrup K, Russell DG, and Bermudez LE (2005). Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell’s endosomal system. J. Immunol 174, 1491–1500. [DOI] [PubMed] [Google Scholar]
- Wang J, and Pantopoulos K (2011). Regulation of cellular iron metabolism. Biochem. J. 434, 365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KJ, Bennett MH, Barton GR, Jenkins VA, and Robertson BD (2013). Adenylylation of mycobacterial Glnk (PII) protein is induced by nitrogen limitation. Tuberculosis (Edinb.) 93, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, Patterson A, Smith AM, Chang CH, Liu Z, et al. (2016). Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 44, 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Lee HN, Kim SH, Park SA, Kim W, Cha YN, and Surh YJ (2018). Myc-nick promotes efferocytosis through M2 macrophage polarization during resolution of inflammation. FASEB J. 32, 5312–5325. [DOI] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, and Love MI (2019). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this article are available in GEO: GSE132354 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132354
The data analysis pipeline (Linux code and R scripts) is provided as a zip file in the supplemental materials.
The datasets for the uninfected AM and IM samples are available in GEO: GSE108844 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108844