

Abstract
Photo-activatable carbon monoxide (CO)-releasing molecules (photoCORMs), have recently provided help to identify the salutary effects of CO in human pathophysiology. Among them notable is the ability of CO to sensitize chemotherapeutic-resistant cancer cells. Findings from our group have shown CO to mitigate drug resistance in certain cancer cells by the inhibition of cystathionine β-synthase (CBS), a key regulator of redox homeostasis in the cell. Diminution of the antioxidant capacity of cancer cells leads to sensitization to reactive oxygen species-producing drugs like doxorubicin and paclitaxel upon cotreatment with CO as well as in mitigating the drug effects of cisplatin. We hypothesize that the development of CO delivery techniques for coadministration with existing cancer treatment regimens may ultimately improve clinical outcomes in cancer therapy.
Keywords: : antioxidant capacity, breast cancer, carbon monoxide, chemotherapy, cystathionine β-synthase, drug resistance, glutathione, ovarian cancer, photoCORM, reactive oxygen species
Graphical abstract
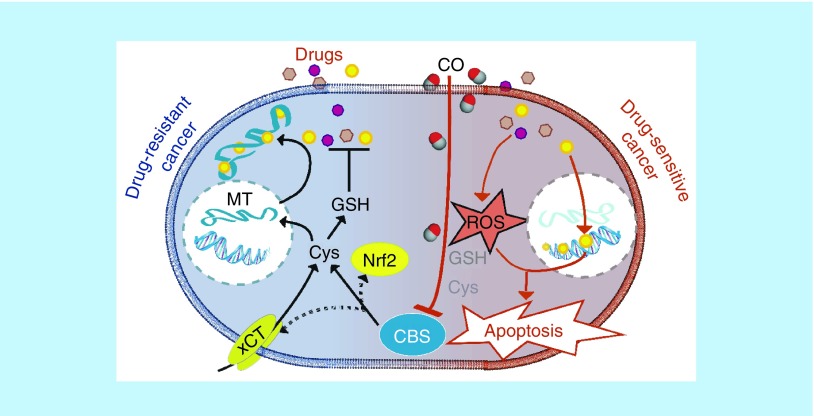
Carbon monoxide (CO), an endogenously produced gaseous molecule, has been recognized as a gasotransmitter, that elicits biological responses in mammalian pathophysiology [1,2]. Along with nitric oxide and hydrogen sulfide (H2S), gasotransmitters have recently been shown to play roles in a wide array of chemistry in biological systems. Unlike nitric oxide, a radical species, and H2S, a weak acid and reducing agent, CO is relatively inert. This weak Lewis base interacts with transition metal centers in low oxidation states. In human cells and tissues, CO reacts almost exclusively with 5-coordinate ferrous heme cofactors [3].
Historically CO is noted for its generation from incomplete hydrocarbon combustion and toxicity arising from its binding to hemoglobin, the oxygen carrier in mammals. However, endogenous production of CO as a product of heme catabolism has been known for over 70 years. This reaction is catalyzed by microsomal heme oxygenase (HO), an enzyme that exists in two isoforms namely, HO-1 (inducible) and HO-2 (constitutive) [4]. Catabolism of free heme is vital for maintaining cellular health and mitigating the toxic effects of heme-catalyzed oxidation reactions. Indeed, loss-of-function mutations in HO-1 are lethal. In the human body, micromolar amounts of CO are generated collectively by HO-1 and HO-2. Nearly all tissues are capable of HO-1 induction, though its expression in Kupffer cells and anti-inflammatory/M2-polarized macrophages suggest a role for CO in modulating immune response and inflammation. Constitutive HO-2 produces CO in the liver, testes, endothelial cells and the brain where it is reputed to play important roles in maintaining vascular tone.
Research during the past three decades has explored the potential of low doses (10–100 ppm) of CO as a therapeutic in a wide array of injury and disease models including wound healing, colitis, sepsis, cerebral malaria, diabetes, balloon angioplasty-induced stenosis, ileus/bowel immotility and organ transplantation [5,6]. The observed salutary effects of CO in these disease/injury models may be partially explained by the ability of CO to modulate intracellular levels of reactive oxygen species (ROS) in target cells, attenuate macrophage activation by cytokine and endotoxin, inhibit endothelial cell apoptosis and prevent T-cell proliferation [6]. In addition, CO has been shown to be effective in attenuating and resolving chronic inflammation, a process implicated in many diseases including cancer [7]. Upregulated and chronic inflammation is implicated as a key factor in the initiation and progression, metastatic spread and the development of therapeutic resistance in cancer [8]. In light of observations of the protective and anti-inflammatory effects of HO-1/2 and CO, recent efforts have been made to explore the possible therapeutic application of CO in cancer models.
CO in cancer therapy
Work from our group and others have provided evidence for the therapeutic potential of exogenous CO in cancer cell models in recent years [9–15]. However, the delivery of CO to malignant targets require novel delivery approaches using either CO gas or CO-releasing molecules (CORMs) [16,17]. Although some progress along this line has been achieved in recent years [5,9,11,18], therapeutic application of CO in a clinical setting is still in its infancy. Fortunately, the CORMs have provided crucial help in exploring, among others, the physiological processes that lead tumorigenic initiation, progression and ultimate management of cancer. The pathology of cancer often poses challenges arising from therapeutic drug resistance and the role of CO in mitigating such processes could be quite interesting. In this context, CO has been shown to sensitize cells to chemotherapeutics in cancer models [15]. If incorporation of CO into existing chemotherapeutic regimens could indeed improve drug efficacy, health outcomes may improve significantly. This hypothesis has prompted us to undertake research efforts to explore the effects of CO coadministration with conventional chemotherapeutics on human cancer models.
Antioxidant capacity in chemoresistant cancer
Drug resistance remains as the main impediment to the management of cancer [19,20]. Accumulating evidence suggests that the unique metabolic profile found in cancer cells is integral for imparting a drug resistant phenotype. Hallmarks of cancer cellular biology such as altered glucose metabolism, peroxisome activity, mitochondrial dysfunction and enhanced growth/signaling pathway activity alter the redox balance in cancer cells, generating higher levels of ROS and inducing chronic oxidative stress [21].
While high oxidative stress is acutely cytotoxic, the low and chronic oxidative stress within cancer cells hormetically enhances antioxidant metabolic pathways and production of antioxidants. Total cellular antioxidants are qualitatively referred to as the total antioxidant capacity of the cell, the ability of the cell to cope with oxidative stress. There is substantial evidence of the protumorigenic role for antioxidants, including the master regulator of anti-oxidative responses nuclear factor (erythroid-derived 2)-like 2 (Nrf2), antioxidant enzymes and cellular antioxidants such as nicotinamide adenine dinucleotide phosphate (NADPH), cysteine and glutathione (GSH) [22]. Cancer cells maintaining an elevated antioxidant capacity acquire resistance to future, acute stressors, including that induced by chemotherapeutic agents [19]. Many chemotherapeutics (e.g., vinca alkaloids, taxanes, anthracyclines and platinum-based drugs) and nonchemical therapies (e.g., radiation) directly generate excessive ROS in cancer cells, inducing apoptosis by interfering with processes including cell-cycle progression and DNA stability [23]. In most cases, the antineoplastic drug effects are indirectly mediated by ROS which eventually bring about the apoptotic death of the cancer cell. Furthermore, thiol-containing antioxidants and peptides, GSH and metallothionein, are known to bind and inactivate chemotherapeutics [24]. As a consequence, selective suppression of the antioxidant capacity of cancer cells by inhibiting antioxidant pathways could mitigate the incidence and intensity of therapeutic resistance and associated poor prognosis.
The chemosensitizing effects of CO observed in prostate cancer cells by Otterbein and coworkers, indicate that one mechanism by which CO exerts its salutary effect(s) is through ‘metabolic exhaustion’ a depletion of intracellular ATP combined with increased mitochondrial oxygen consumption and decreased glycolytic activity [15], which in turn has been recently reported to decrease drug efflux by ATP-binding cassette transporters [25]. Kaczara et al. have provided evidence that this occurs via uncoupling of mitochondrial respiration [26], though the nature of mitochondrial uncoupling by CO is unclear and somewhat controversial. Mechanisms involving direct/indirect interactions of CO with ion channels have also been proposed [27], though CO exerting its salutary effect(s) by ‘metabolic exhaustion’ most possibly occurs by binding to enzymes involved in energy metabolism [28,29]. CO has several demonstrated direct biological targets [3], though consensus over those that are therapeutically relevant is lacking. The elevated levels of ROS in cancer cells strongly suggest that interaction of CO with enzymes involved in maintaining their antioxidant capacity could be another important target of CO leading to drug sensitization. Therefore, we have focused on cystathionine β-synthase (CBS) as a potential target of CO in moderating the overall redox environment in cancer cells and alleviating drug resistance.
CBS
CBS, a heme-containing enzyme, catalyzes the first step of the transsulfuration pathway: the condensation of homocysteine with either serine or cysteine to generate cystathionine (CTH) and water or H2S, respectively (Figure 1) [30]. CTH is further catabolized into cysteine, catalyzed by CTH γ-lyase, the second enzyme of the transsulfuration pathway. In tissues where demand for GSH is high, including the liver and pancreas, CBS and the transsulfuration pathway provide a significant source of cysteine for GSH biosynthesis [31]. Alternatively, homocysteine can be diverted from the transsulfuration pathway and be recycled into methionine, catalyzed by the enzyme methionine synthase, a cobalamin-containing enzyme. Interestingly, methionine synthase, unlike CBS, is prone to oxidative inactivation, suggesting a prominent role for CBS in regulating methylation and transsulfuration in the cell, especially under oxidative conditions [32].
Figure 1. . Scheme of transsulfuration and glutathione biosynthesis pathways.

CBS: Cystathionine β-synthase; CGL: Cystathionine γ-lyase; GCL: Glutamate-cysteine ligase; GS: Glutathione synthase; GSH: Glutathione.
GSH levels have been shown to be elevated in tumor tissues from patients with head and neck, lung, breast and ovarian cancers compared with corresponding nonmalignant tissues [22]. GSH and other antioxidants have been shown to play a key role in protecting cancer cells from a wide range of anticancer therapies, with elevated levels predictive of drug resistance and therapeutic failure. Inhibition of CBS in cancer cells exhibiting overexpression could reduce GSH levels, perturbing the balance between the generation and quenching of ROS, inducing oxidative stress and abating the drug resistant phenotype.
Inhibition of CBS by CO
CBS is unique in that it is the only pyridoxal phosphate-dependent enzyme that also contains a prosthetic heme, which renders CBS sensitive to CO. CO has a high affinity for ferrous heme in CBS [30]. The binding of CO to CBS is kinetically slow, 0.0166 s-1, as CO binds via displacement of Cys52 from the iron center. The displaced thiolate on Cys52 is stabilized by Arg266, the likely mechanism by which CO inactivates CBS. Physiological levels of CO are sufficient to inhibit CBS activity, Ki = 3 μM.
Is CBS a therapeutically relevant target of CO in cancer cells?
Substantial evidence supports the therapeutic relevance of CBS as a cancer-specific target. Previous studies have utilized RNA interference and pharmacological inhibitors to reveal the oncogenic and cytoprotective effects of CBS in ovarian, colon and breast cancers [32]. Recently, our group has found that CO, delivered from biologically compatible, photo-activatable CO-releasing molecules (photoCORMs) can induce apoptotic death in human breast and colon cancer cells [9–13]. This finding prompted us to the second phase of the project where we sought to find out the target(s) of CO in cancer cells that leads to apoptosis and whether CO binding to such targets could sensitize them to conventional chemotherapeutics. Because diminution of the antioxidant capacity could lead to drug sensitization, we hypothesized that CBS is one of the main effectors of CO-mediated sensitization of cancer cells to chemotherapeutics. We therefore selected human breast and ovarian cancer cells, two disease models where CBS is overexpressed and correlate with tumor grade, to study the drug sensitizing effects of CO.
Effects of CO in human breast cancer cells
The pathology atlas of human cancer transcriptome [33] revealed that despite absence in normal breast cells, CBS is expressed in transformed breast cancer cells to a moderate extent. This fact allowed us to study the effects of CO on CBS in a cancer cell model. The photoCORM used in our study was [Mn(CO)3(phen)(PTA)]CF3SO3, a water-soluble CORM that releases CO upon exposure to visible light (Figure 2) [10]. This CO donor allowed us to deliver CO to biological targets under the control of visible light, when desired.
Figure 2. . Structure of the cation of the photoCORM used in our study: [Mn(CO)3(phen)(PTA)]CF3SO3.

In previously published work from our group, we employed a set of three human breast cancer cell lines representing both estrogen/progesterone receptor-positive cell line MCF-7 and triple negative breast cancer cell lines MDA-MB-468 and Hs 578T to demonstrate the general applicability of our observations [34]. Quite in agreement with our hypothesis light-triggered CO delivery from 120 μM photoCORM to these cancer cells inhibited CBS enzyme activity, as measured by decreases in steady state levels of CTH (Figure 3) [34]. Incidentally, CTH and H2S, enzymatic products of CBS, enzymatic products of CBS, themselves promote endoplasmic reticulum homeostasis and positively regulate Nrf2 [35–37]. Through this H2S-Nrf2 axis, CBS promotes expression of GSH biosynthesis enzymes, as well as glucose-6-phosphate dehydrogenase, the rate-limiting enzyme in the pentose phosphate pathway, whose activity promotes production of NADPH (Figure 3), the reducing equivalent required for regeneration of GSH from glutathione disulfide (GSSG). In our previously published experiments [34], inhibition of CBS by CO led to a decreased NADPH/NADP+ and GSH/GSSG ratios indicating a reduction in the antioxidant capacity in all three breast cancer cells (Figure 3).
Figure 3. . Effect of carbon monoxide on steady state levels of transsulfuration/glutathione biosynthesis metabolites, as measured by liquid chromatography-mass spectrometry, and NADPH/NADP+ in human breast cancer cells.

CO inhibits CBS mediated production of cystathionine and H2S, downregulating Nrf2. Downregulation of Nrf2, in turn, attenuates expression of GCL, GS and G6PD, antioxidant genes downstream involved in GSH biosynthesis/regeneration of GSH from GSSG. Data representative of at least n = 3 individual experiments.
*p< 0.05.
CBS: Cystathionine β-synthase; CO: Carbon monoxide; GS: Glutathione synthase; GSH: Glutathione; GSSG: Glutathione disulfide; H2S: Hydrogen sulfide; Nrf2: Nuclear factor (erythroid-derived 2)-like 2; GCL: Glutamate-cysteine ligase; G6PD: Glucose 6-phosphate dehydrogenase.
Also, treatments with the slow H2S-releasing drug (p-methoxyphenyl)morpholino-phosphinodithioic acid (GYY 4137) and CTH were able to restore the elevated antioxidant capacity of cancer cell line MCF-7, showing that inhibition of CBS by CO was indeed responsible for the observed lowering of NADPH/NADP+ and GSH/GSSG ratios [34]. To further confirm the role of CBS in these measurements, we checked whether expression of CBS activity in a CBS-free cell model could increase the NADPH/NADP+ and GSH/GSSG ratios through H2S-mediated stabilization of Nrf2. In this attempt, we overexpressed CBS in the normal human breast cell line MCF-10A (normally lacking CBS expression/activity) and observed increased ratios of these redox pairs (Figure 4) [34]. Together these findings published from our group confirm that CBS is an important player in maintaining an elevated antioxidant capacity in human breast cancer cells [34].
Figure 4. . Overexpression of cystathionine β-synthase in normal human breast cell line MCF-10A enhances the antioxidant capacity, as measured by the increase in ratios of NADPH/NADP+ and GSH/GSSG.

Data representative of at least n = 3 individual experiments.
*p< 0.05.
CBS: Cystathionine β-synthase; GSH: Glutathione; GSSG: Glutathione disulfide.
Finally, we wanted to check whether CBS inhibition by CO, and the subsequent lowering of the antioxidant capacity of the cancer cells, sensitizes them toward chemotherapeutics. In such attempt, we pretreated MCF-7 cells with CO (delivered from the photoCORM) for 30 min followed by addition of doxorubicin. The results from cell viability assays 48 h post-treatment clearly showed that pretreatment of the cells with CO significantly enhanced the cytotoxic effects of doxorubicin compared with doxorubicin treatment alone (Figure 5) [34]. Similar results were obtained with paclitaxel as the chemotherapeutic (Figure 5) [34]. In conclusion, our results from this previous study strongly suggest that CO has the ability to lower the antioxidant capacity of breast cancer cells by inhibition of CBS, and consequently sensitize the cells to chemotherapeutics such as doxorubicin and paclitaxel [34].
Figure 5. . Annexin V-FITC/propodium iodide staining and cell viability by trypan blue exclusion, demonstrating cotreatment with exogenous carbon monoxide sensitizes human breast cancer cell line MCF-7 to conventional chemotherapeutics, doxorubicin and paclitaxel.

Data representative of at least n = 3 individual experiments.
*p< 0.05.
CO: Carbon monoxide.
Effects of CO in human ovarian cancer cells
In neoplasms of the ovaries, CBS expression correlates with carcinogenesis and tumor grade; we therefore hypothesized that CO could inhibit CBS in an ovarian cancer model. Recent work published by our group, involving careful screening of the human ovarian cancer cells OVCAR-5 and SKOV-3 and their cisplatin-resistant variants, revealed that the expression of the transsulfuration pathway enzymes CBS and cystathionine γ-lyase in the cisplatin-resistant cell lines are both profoundly higher compared with the parent cell lines [38]. This important finding suggested a direct role for CBS and the transsulfuration pathway in imparting cisplatin-resistance. One largely elucidated mechanism of cisplatin resistance is the binding and inactivation of cisplatin by cellular thiols GSH and metallothionein, both of which are highly dependent on the bioavailability of cysteine (a product of CBS activity) [39]. In initial measurements the cisplatin-resistant ovarian cancer cells, as expected, exhibited nearly twofold higher steady state levels of the transsulfuration metabolites (CTH and cysteine), GSH and its biosynthesis metabolite γ-Glu-Cys (Figure 6), and the expression of metallothionein compared with the cisplatin-sensitive variants [38].
Figure 6. . Steady state levels of transsulfuration/glutathione biosynthesis metabolites in cisplatin-resistant ovarian cancer cell lines compared with respective parent cell lines OVCAR-5 and SKOV-3.

Cisplatin-resistant ovarian cancer cells exhibit elevated levels of transsulfuration pathway metabolites (cystathionine and Cys) and GSH biosynthesis pathway metabolites (γ-Glu-Cys and GSH). Data representative of at least n = 3 individual experiments.
*p< 0.05.
Cys: Cysteine; Glu: Glutamic acid; GSH: Glutathione.
Furthermore, N-acetyl-cysteine, a bioavailable cysteine donor, further increased the levels of both GSH and metallothionein in these cells [38]. It is important to note that GSH and metallothionein are the two clinically relevant markers and predictors of cisplatin resistance in ovarian cancer patients [39]. Treatment of the cisplatin-resistant ovarian cancer cells with CO attenuated all the steady state levels of these CBS-derived products (Figure 7). Also, the effect of CO was mimicked by lentiviral-mediated silencing of CBS in these cisplatin-resistant cells, supporting the hypothesis that CO-derived attenuation of the metabolite levels was mediated by CBS [38].
Figure 7. . Effect of carbon monoxide on transsulfuration/glutathione biosynthesis pathway metabolites, assayed by liquid chromatography-mass spectrometry, in cisplatin-resistant ovarian cancer cell lines, 24 h post-treatment.

CO inhibits cystathionine β-synthase, decreasing cystine uptake via the xCT and lowering steady state levels of cystathionine, the substrate for cystathionine γ-lyase. Consequently, CO decreases intracellular cysteine levels to inhibit glutathione biosynthesis as measured by steady state levels of γ-Glu-Cys and GSH, products of GCL and GS, respectively. Data representative of at least n = 3 individual experiments.
*p< 0.05.
CBS: Cystathionine β-synthase; CO: Carbon monoxide; Cys: Cysteine; GCL: Glutamate–cysteine ligase; Glu: Glutamic acid; GS: Glutathione synthase; GSH: Glutathione; H2S: Hydrogen sulfide; xCT: Glutamate–cystine antiporter.
In addition to the transsulfuration pathway, H2S generated by CBS has been shown to increase the activity of the cystine–glutamate antiporter xCT which imports, from the extracellular milieu, a significant portion of cystine into cancer cells that is converted into cysteine. This cysteine is also utilized in the synthesis of GSH and metallothionein [40]. In our experiment, the cisplatin-resistant ovarian cancer cells exhibited nearly twofold increase in the uptake of cystine-D4 (compared with the cisplatin-sensitive cells) in cell culture conditions, as indicated by the intracellular cysteine-D2 concentrations within the cancer cells (Figure 8) [38]. CO treatment of the same cells led to a significant drop in the concentration of cysteine-D2 showing that inhibition of CBS by CO (with concomitant decrease in H2S) downregulates the activity of the xCT antiporter (Figure 3).
Figure 8. . Glutamate–cystine antiporter activity assayed by extracellular D4-cystine uptake and determined by relative steady state levels of intracellular D2-cysteine in cisplatin-resistant and wild-type ovarian cancer cell lines OVCAR-5 and SKOV-3.

Cisplatin-resistant ovarian cancer cells exhibit significantly greater uptake of extracellular D4-cystine compared with their wild-type counterparts. Data representative of at least n = 3 individual experiments.
*p< 0.05.
Addition of H2S by the use of the slow H2S-releasing drug GYY 4137 enhanced the cystine-D4 uptake, an observation that further confirms the role of CBS in positively regulating the activity of the xCT antiporter (Figure 9) [38].
Figure 9. . Relative glutamate–cystine antiporter activity assayed by extracellular D4-cystine uptake, determined by steady state levels of intracellular D2-cysteine in cisplatin-resistant ovarian cancer cell lines silenced for cystathionine β-synthase and treated with H2S for 24 h.

Silencing CBS expression decreases D4-cystine uptake, which is mitigated by the donation of extracellular H2S. Data representative of at least n = 3 individual experiments.
*p< 0.05.
CBS: Cystathionine β-synthase; H2S: Hydrogen sulfide.
Finally, we tested the hypothesis that CO does sensitize the cisplatin-resistant ovarian cancer cells and makes them susceptible to cisplatin. Indeed, when the cisplatin-resistant ovarian cancer cells were cotreated with cisplatin and CO, we observed a much higher extent of apoptotic cell death of cells compared with cells that were treated only with cisplatin (Figure 10) [38]. Together, these results published by our group elucidate a distinct role for CBS in imparting cisplatin resistance and provide preliminary evidence for CO as a novel adjuvant therapeutic for ovarian cancer [38].
Figure 10. . Trypan blue exclusion cell viability assay of cisplatin-resistant ovarian cancer cells 24 h post-treatment with carbon monoxide, cisplatin or coadministration of carbon monoxide and cisplatin.

Cells cotreated with CO and cisplatin exhibit greater cell death compared with cells treated with cisplatin alone. Data representative of at least n = 3 individual experiments.
*p< 0.05.
CO: Carbon monoxide.
Future perspective
Breast cancer is the leading cancer diagnosed in women and the second leading cause of cancer-related death in women. It is estimated that one in eight women will be diagnosed with breast cancer in their lifetime and over 40,000 women deaths will occur each year due to lack of or poor response to treatment [41]. Incidentally, chemotherapeutic drug resistance is a major impediment to effective treatment of breast cancer. The cancer cells often acquire resistance to cisplatin, paclitaxel and doxorubicin, and leads to poor clinical outcomes. Because elevated levels of ROS and alteration of redox balance are common hallmarks of cancer progression and drug resistance [42], we focused on CBS, an enzyme intimately involved with redox homeostasis [35]. Although role of CBS in cancer has been examined to find out the role of H2S in the pathophysiology of cancer [43], we focused on the interactions of CO with CBS and the utility of this gasotransmitter in cancer therapy. In particular, we are interested in the role of CBS in promoting an elevated antioxidant capacity and drug resistance. Our results, described above, strongly suggest that CBS inhibition by CO could be one effective strategy for enhancing the efficacy of drugs such as doxorubicin and paclitaxel which exert their tumoricidal effect(s) via induction of ROS [44,45].
Clinically cisplatin is the standard of care for the management of human ovarian cancer [46]. Unfortunately, the majority of patients develop resistance to cisplatin and succumb to the disease. Our results indicate that modulation of CBS activity in ovarian cancer cells might provide a strategy for mitigating drug-resistance observed frequently with ovarian cancer patients. While our work focused on the effects of CO-mediated inhibition of CBS on the transsulfuration pathway, notable work by Suematsu and coworkers has focused on the transmethylation pathway [47]. In this work, stress-induced CO production in human leukemia cell line U937 suppressed CBS activity, in contrast to our studies in breast and ovarian cancer models, to promote generation of cellular antioxidants via decreased phosphofructokinase/fructose bisphosphatase type-3 methylation. This resulted in a shift in glucose biotransformation toward the pentose phosphate pathway, promoting generation of cellular reductants to impart resistance to chemotherapeutics [47]. The paradoxical effect(s) of CO-mediated inhibition of CBS in leukemia [47] versus breast/ovarian cancer tissues [34,38] highlight the context-dependent role of CBS in tumor growth and progression, a concept strongly emphasized in a recent review on the role of CBS in different human cancers [48]. It is very apparent that CBS has a functional role in chemotherapeutic resistance and together, these findings suggest that CO, a relatively unreactive gaseous molecule once thought of as a toxic byproduct of heme catabolism, exerts profound biological effects in cancer cells. It is quite possible that smart CO delivery to cancer cells could sensitize them to conventional chemotherapeutics and mitigate the grim statistics of cancer-related death arising from drug resistance.
A close scrutiny of the literature reveals that most antitumor therapeutics are aimed at proteins that drive cancer proliferation. Blocking of such pathways almost always triggers the evolution of escape routes or alternative pathways to meet the continuous demand for proliferation in cancer cells [49]. However little is known about whether cancer cells are equally efficient in adapting alternative routes when it comes to the inhibition of ‘cancer maintenance/homeostatic’ functionalities. Redox homeostasis is one such function and the CBS-CO inhibitory axis essentially perturbs this crucial pathway. Because upregulation of antioxidant capacity in adaptation of intrinsic oxidative stress in cancer cells often leads to drug resistance, disruption of the homeostatic rather than proliferative potential of the cancer cells through inhibition of the redox regulatory effects of CBS by CO might be an effective strategy to eliminate these cells [50]. The results of our studies on CBS inhibition by CO in relation to cancer pathogenesis and therapeutic resistance, presented here, support another perspective to the therapeutic potential of CO.
Executive summary.
Carbon monoxide (CO) has recently been identified as a gasotransmitter, a diffusible molecule that plays many roles in physiological processes.
Concomitant development of improved CO donor molecules, including photo-activatable CO-releasing molecules (photoCORMs), has been helpful in identifying the beneficial effects of CO in human pathophysiology.
Elucidation of these effects has strengthened the potential for CO-based therapeutics. Detailed studies in cell culture and animal models have also demonstrated promise for the development of CO-based cancer therapies, notably the ability of CO to sensitize chemotherapeutic-resistant cancer cells.
Chemoresistance is one of the leading causes of therapeutic failure and poor clinical outcomes in cancer. This perspective summarizes the findings on the ability of CO to mitigate drug resistance in certain cancer cells through inhibition of cystathionine β-synthase (CBS), a key regulator of redox homeostasis in the cell.
In human breast cancer cells, the heme-containing enzyme CBS maintains the antioxidant capacity by maintaining increased ratios of reduced/oxidized glutathione (GSH/GSSG), reduced/oxidized NADPH (NADPH/NADP+) compared with normal breast cells lacking CBS. Inhibition of CBS by CO (delivered from a photoCORM) lowers these antioxidant ratios considerably. Because H2S, a product of CBS activity, positively regulates Nrf2, inhibition of CBS by CO downregualtes enzymes regulate by Nrf2, including those involved in GSH biosynthesis and the pentose phosphate pathway. Genetic silencing of CBS mimics the effect of CO treatment in these same cells. Overexpression of CBS and addition of H2S reverse such diminution of the levels of the antioxidants. Collectively, diminution of the antioxidant capacity of human breast cancers leads to sensitization to reactive oxygen species producing drugs like doxorubicin and paclitaxel upon cotreatment with CO.
In human ovarian malignancy, CBS is overexpressed only in those ovarian cancer cells resistant to cisplatin. CO treatment of these cells results in reduction of intracellular cysteine, GSH and nuclear metallothionein expression; features implicated in mitigating the drug effects of cisplatin. Cell viability studies clearly demonstrate sensitization of human ovarian cancer cells to cisplatin upon cotreatment with CO, indicating the role of CBS in the emergence of drug resistance in ovarian cancer cells.
Collectively the results suggest that improved delivery techniques for coadministration of CO with existing cancer chemotherapeutics could improve clinical outcomes in cancer therapy.
Footnotes
Financial & competing interests disclosure
Financial support from the NSF grant DMR-1409335 and the Cancer Research Coordinating Committee (UC) grant CTR-19-580346 is gratefully acknowledged. Research in UCLA was supported by the NIH U01 grant HD087221. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Wang R. (). Evolution of gasotransmitter biology and medicine. : Signal Transduction and the Gasotransmirtters. NO, CO and H2S in Biology and Medicine. Humana Press, NJ, USA, 3–32 (2004). [Google Scholar]
- 2.Kim HP, Ryter SW, Choi AMK. CO as a cellular signaling molecule. Ann. Rev. Pharmacol. Toxicol. 46, 411–449 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Motterlini R, Foresti R. Biological signaling by carbon monoxide and carbon monoxide-releasing molecules. Am. J. Physiol.-Cell Physiol. 312(3), C302–C313 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Kikuchi G, Yoshida T, Noguchi M. Heme oxygenase and heme degradation. Biochem. Biophys. Res. Commun. 338(1), 558–567 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 9(9), 728–743 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Motterlini R, Haas B, Foresti R. Emerging concepts on the anti-inflammatory actions of carbon monoxide-releasing molecules (CO-RMs). Med. Gas Res. 2(1), 28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coussens LM, Werb Z. Inflammation and cancer. Nature 420(6917), 860–867 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raposo TP, Beirao BCB, Pang LY, Queiroga FL, Argyle DJ. Inflammation and cancer: till death tears them apart. Vet. J. 205(2), 161–174 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Chakraborty I, Jimenez J, Mascharak PK. CO-induced apoptotic death of colorectal cancer cells by a luminescent photoCORM grafted on biocompatible carboxymethyl chitosan. Chem. Commun. 53(40), 5519–5522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakraborty I, Carrington SJ, Roseman G, Mascharak PK. Synthesis, structures, and CO release capacity of a family of water-soluble photoCORMs: assessment of the biocompatibility and their phototoxicity toward human breast cancer cells. Inorg. Chem. 56(3), 1534–1545 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Chakraborty I, Carrington SJ, Hauser J, Oliver SRJ, Mascharak PK. Rapid eradication of human breast cancer cells through trackable light-triggered CO delivery by mesoporous silica nanoparticles packed with a designed photoCORM. ACS Chem. Mater. 27(24), 8387–8397 (2015). [Google Scholar]
- 12.Chakraborty I, Carrington SJ, Mascharak PK. Design strategies to improve the sensitivity of photoactive metal carbonyl complexes (photoCORMs) to visible light and their potential as CO-donors to biological targets. Acc. Chem. Res. 47(8), 2603–2611 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Carrington SJ, Chakraborty I, Mascharak PK. Rapid CO release from a Mn(I) carbonyl complex derived from azopyridine upon exposure to visible light and its phototoxicity toward malignant cells. Chem. Commun. 49(96), 11254–11256 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Kourti M, Jiang WG, Cai J. Aspects of carbon monoxide in form of CO-releasing molecules used in cancer treatment: more light on the way. Oxid. Med. Cell. Longev. 2017, 9326454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wegiel B, Gallo D, Csizmadia E. et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 73(23), 7009–7021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zobi F. CO and CO-releasing molecules in medicinal chemistry. Future Med. Chem. 5(2), 175–188 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Schatzschneider U. Novel lead structures and activation mechanisms for CO-releasing molecules (CORMs). Br. J. Pharmacol. 172(6), 1638–1650 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinto MN, Chakraborty I, Sandoval C, Mascharak PK. Eradication of HT-29 colorectal adenocarcinoma cells by controlled photorelease of CO from a CO-releasing polymer (photoCORP-1) triggered by visible light through an optical fiber-based device. J. Control. Rel. 264, 192–202 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Housman G, Byler S, Heerboth S. et al. Drug resistance in cancer: an overview. Cancers 6(3), 1769–1792 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lippert TH, Ruoff H-J, Volm M. Intrinsic and acquired drug resistance in malignant tumors. Arzneimittelforschung 58(6), 261–264 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Grek CL, Tew KD. Redox metabolism and malignancy. Curr. Opin. Pharmacol. 10(4), 362–368 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hawk MA, McCallister C, Schafer ZT. Antioxidant activity during tumor progression: a necessity for the survival of cancer cells? Cancers 8(10), 92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Discusses the protumorigenic role of antioxidant activity in facilitating tumor progression.
- 23.Yokoyama C, Sueyoshi Y, Ema M, Mori Y, Takaishi K, Hisatomi H. Induction of oxidative stress by anticancer drugs in the presence and absence of cells. Oncol. Lett. 14(5), 6066–6070 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galluzzi L, Senovilla L, Vitale I. et al. Molecular mechanisms of cisplatin resistance. Oncogene 31(15), 1869–1883 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Li YJ, Dang JJ, Liang QJ, Yin LC. Thermal-responsive carbon monoxide (CO) delivery expedites metabolic exhaustion of cancer cells toward reversal of chemotherapy resistance. ACS Cent. Sci. 5(6), 1044–1058 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaczara P, Motterlini R, Rosen GM. et al. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: a role for mitoBK(Ca) channels. Biochim. Biophys. Acta 1847(10), 1297–1309 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Wilkinson WJ, Kemp PJ. Carbon monoxide: an emerging regulator of ion channels. J. Physiol. 589(13), 3055–3062 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo Iacono L, Boczkowski J, Zini R. et al. A carbon monoxide-releasing molecule (CORM-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species. Free Radic. Biol. Med. 50(11), 1556–1564 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Long R, Salouage I, Berdeaux A, Motterlini R, Morin D. CORM-3, a water soluble CO-releasing molecule, uncouples mitochondrial respiration via interaction with the phosphate carrier. BBA-Bioenergetics 1837(1), 201–209 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Banerjee R, Zou CG. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: a PLP-dependent hemesensor protein. Arch. Biochem. Biophys. 433(1), 144–156 (2005). [DOI] [PubMed] [Google Scholar]; • Summarizes the work of Banerjee and coworkers identifying cystathionine β-synthase (CBS) as a direct binding partner of carbon monoxide (CO) and subsequent inhibition of the enzyme activity.
- 31.Belalcázar AD, Ball JG, Frost LM, Valentovic MA, Wilkinson J. Transsulfuration is a significant source of sulfur for glutathione production in human mammary epithelial cells. ISRN Biochem. 2013, 637897 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu HR, Blake S, Chan KT, Pearson RB, Kang J. Cystathionine beta-synthase in physiology and cancer. Biomed. Res. Int. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uhlen M, Zhang C, Lee S. et al. A pathology atlas of the human cancer transcriptome. Science 357(6352), pii: eaan2507 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Kawahara B, Moller T, Hu-Moore K. et al. Attenuation of antioxidant capacity in human breast cancer cells by carbon monoxide through inhibition of cystathionine β-synthase activity: implications in chemotherapeutic drug sensitivity. J. Med. Chem. 60(19), 8000–8010 (2017). [DOI] [PubMed] [Google Scholar]; •• Findings from this study connected the chemosensitizing effects of CO with modulation of cancer cellular antioxidants via modulation of CBS activity in three breast cancer cell lines.
- 35.Sen S, Kawahara B, Gupta D. et al. Role of cystathionine beta-synthase in human breast cancer. Free Radic. Biol. Med. 86, 228–238 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Sen S, Kawahara B, Mahata SK. et al. Cystathionine: a novel oncometabolite in human breast cancer. Arch. Biochem. Biophys. 604, 95–102 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Hourihan JM, Kenna JG, Hayes JD. The gasotransmitter hydrogen sulfide induces Nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal. 19(5), 465–481 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Kawahara B, Ramadoss S, Chaudhuri G, Janzen C, Sen S, Mascharak PK. Carbon monoxide sensitizes cisplatin-resistant ovarian cancer cell lines toward cisplatin via attenuation of levels of glutathione and nuclear metallothionein. J. Inorg. Biochem. 191, 29–39 (2019). [DOI] [PubMed] [Google Scholar]; •• Establishes that CO sensitizes cisplatin-resistant ovarian cancer cells through attenuation of biomarkers of cisplatin-resistance (glutathione, metallothionein) via inhibition of CBS.
- 39.Galluzzi L, Senovilla L, Vitale I. et al. Molecular mechanisms of cisplatin resistance. Oncogene 31(15), 1869–1883 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Verschoor ML, Singh G. Ets-1 regulates intracellular glutathione levels: key target for resistant ovarian cancer. Mol. Cancer 12(1), 138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.National Breast Cancer Foundation I. Breast cancer facts 2015 (2016) www.nationalbreastcancer.org/breast-cancer-facts
- 42.Kumari S, Badana AK, Mohan GM, Shailender G, Malla R. Reactive oxygen species: a key constituent in cancer survival. Biomark. Insights 13, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hellmich MR, Szabo C. Hydrogen sulfide and cancer. Handb. Exp. Pharmacol. 230, 233–241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramanathan B, Jan KY, Chen CH, Hour TC, Yu HJ, Pu YS. Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res. 65(18), 8455–8460 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Yokoyama C, Sueyoshi Y, Ema M, Mori Y, Takaishi K, Hisatomi H. Induction of oxidative stress by anticancer drugs in the presence and absence of cells. Oncol. Lett. 14(5), 6066–6070 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsuo K, Lin YG, Roman LD, Sood AK. Overcoming platinum resistance in ovarian carcinoma. Expert Opin. Investig. Drugs 19(11), 1339–1354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamamoto T, Takano N, Ishiwata K. et al. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat. Commun. 5, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu HR, Blake S, Chan KT, Pearson RB, Kang J. Cystathionine beta-synthase in physiology and cancer. Biomed. Res. Int. 2018, 3205125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frantz S. Drug approval triggers debate on future direction for cancer treatments. Nat. Rev. Drug Discov. 5(2), 91–91 (2006). [DOI] [PubMed] [Google Scholar]; • Highlights the unmet need to specifically identify and understand which cellular processes impart chemotherapeutic resistance; therapeutic CO may fill this unmet need.
- 50.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 8(7), 579–591 (2009). [DOI] [PubMed] [Google Scholar]; • Proposes pharmacological oxidative insult could selectively kill cancer cells; CO may be an ideal candidate to treat cancer by this approach.