

Abstract
With the goal of expanding the very limited toolkit of cross-linking agents available for nucleic acids and their protein complexes, we evaluated the merits of a wide range of bifunctional agents that may be capable of reacting with the functional groups characteristic of these types of biopolymers. The survey specifically focused on the ability of test reagents to produce desirable inter-molecular conjugates, which could reveal the identity of interacting components and the position of mutual contacts, while also considering a series of practical criteria for their utilization as viable nucleic acid probes. The survey employed models consisting of DNA, RNA, and corresponding protein complexes to mimic as close as possible typical applications. Denaturing polyacrylamide gel electrophoresis (PAGE) and mass spectrometric (MS) analyses were implemented in concert to monitor the formation of the desired conjugates. In particular, the former was used as a rapid and inexpensive tool for the efficient evaluation of cross-linker activity under a broad range of experimental conditions. The latter was applied after preliminary rounds of reaction optimization to enable full-fledged product characterization and, more significantly, differentiation between mono-functional and intra- versus inter-molecular conjugates. This information provided the feedback necessary to further optimize reaction conditions and explain possible outcomes. Among the reagents tested in the study, platinum complexes and nitrogen mustards manifested the most favorable characteristics for practical cross-linking applications, whereas other compounds provided inferior yields, or produced rather unstable conjugates that did not survive the selected analytical conditions. The observed outcomes will help guide the selection of the most appropriate cross-linking reagent for a specific task, whereas the experimental conditions described here will provide an excellent starting point for approaching these types of applications. As a whole, the results of the survey clearly emphasize that finding a universal reagent, which may afford excellent performance with all types of nucleic acid substrates, will require extending the exploration beyond the traditional chemistries employed to modify the constitutive functional groups of these vital biopolymers.
Introduction
The intimate relationship between structure and function makes structural elucidation essential to understanding the significance of biopolymers in living systems. The breadth of the functions carried out by nucleic acids and protein-nucleic acid assemblies, which include not only the storage, replication, translation and maintenance of genetic information, but also multifaceted regulatory and epigenetic activities, has been sustaining the demands for valuable structural information on these types of biomolecules. The broader application of established high-resolution techniques, such as crystallography and NMR, faces typical challenges posed by sample requirements, size limitations, and unfavorable crystallization or solubility [1]. In recent years, the advances made by mass spectrometry (MS) in biopolymer analysis have reawakened a keen interest in the utilization of chemical cross-linking for the low-resolution investigation of structures of ever increasing size and complexity [2–9]. The ability of bifunctional reagents to bridge between functional groups placed in mutual proximity has promoted their application to the identification of interfaces between bound components [10,11] and the elucidation of the organization and overall topology of complex multi-subunit assemblies [12–16]. The development of ad hoc computational approaches has realized the possibility of utilizing low-resolution data from cross-linking experiments to support full-fledged model building and refinement operations [17–21]. Further, concerted strategies combining high-resolution coordinates and cross-linking constraints have been employed to obtain atomic-resolution models for structures that were exceedingly large or flexible for NMR or crystallographic determinations, respectively [22–25].
Virtually all molecular processes that sustain life involve specific interactions between biopolymers. Therefore, their functional elucidation is not complete without comprehensive knowledge of other cellular components that may be capable of establishing mutual contacts. Early on, the introduction of the yeast two-hybrid approach [26,27], the development of reporter systems based on green fluorescent protein [28,29], and the publication of the yeast genome sequence [30] provided a very effective platform for large scale interactomics studies aimed at recognizing functional relationships between proteins, which helped identify complex signaling networks [31,32]. More recently, the combination of chemical cross-linking and MS analysis has emerged as an effective approach for revealing the identity of mutually bound components and the composition of multi-subunit assemblies [33,34]. The utilization of chemical cross-linking has been largely promoted by the need to compensate for the typically low yields of photo-cross-linking techniques based on direct UV irradiation of target substrates, which require extensive enrichment to enable conjugate characterization [35,36]. Cross-linking reactions are capable of replacing labile molecular contacts with stable covalent bridges that enable the implementation of denaturing separation techniques to characterize the ensuing conjugates, thus preventing the loss of information caused by binding dissociation. The analysis of complex mixtures produced by in vivo cross-linking has been greatly facilitated by the introduction of bifunctional reagents containing ad hoc isotope labels [37–39], or suitable tags for capturing/enriching the sought-after conjugates [40–42]. The vast majority of these techniques were developed for the analysis of protein-protein interactions, whereas very few have been introduced for nucleic acid-nucleic acid and protein-nucleic acid assemblies [21,43,44].
Selecting the most appropriate reagent for a specific purpose is essential to ensure the ability of obtaining the sought-after information. Cross-linking reactions can provide mono-functional adducts (i.e., type 0 in Scheme 1), as well as intra- and inter-molecular conjugates (i.e., type 1 and 2, respectively)[45], with distributions that are profoundly affected by both the chemistry and steric situation of the susceptible functional groups. Type 0 adducts effectively identify susceptible groups that are exposed on the surface of the target structure. In contrast, type 1 and 2 conjugates provide the coveted spatial constraints necessary to locate the position of contiguous groups for modeling operations, but only the latter can identify binding partners involved in intermolecular interactions. The partitioning between these types of conjugates, and therefore the information content provided by the analysis, depends as much on the chemistry of the regents as on the incidence and distribution of susceptible functional groups on the target substrate. In general, alkylating agents that react with specific functional groups, such as lysine-specific N-hydroxysuccinimide (NHS) esters [46,47], tend to provide fewer spatial constraints than less specific reagents, such as photoactived aryl azides [48,49], which are instead capable of attacking a broader range of functional groups, but generate more complex products mixtures for analysis. Putative reactivity profiles can be modified to a certain extent by replacing homo-bifunctional probes with hetero-bifunctional counterparts capable of conjugating different types of functional groups [50,51]. The spacing between reactive centers can be varied to bridge across different targets, which may afford different sets of distance constraints [52–55]. In addition, the ability to permeate biological membranes becomes essential for any reagent employed to study targets present in their original cellular environments [56]. In analogy with the majority of pharmaceutical compounds, these types of reagents must be sufficiently stable in biological environments to reach their target in intact, reactive form. Reagent selection should be based on a combination of these considerations, which may have significant consequences on the outcome of the investigation.
Scheme 1.
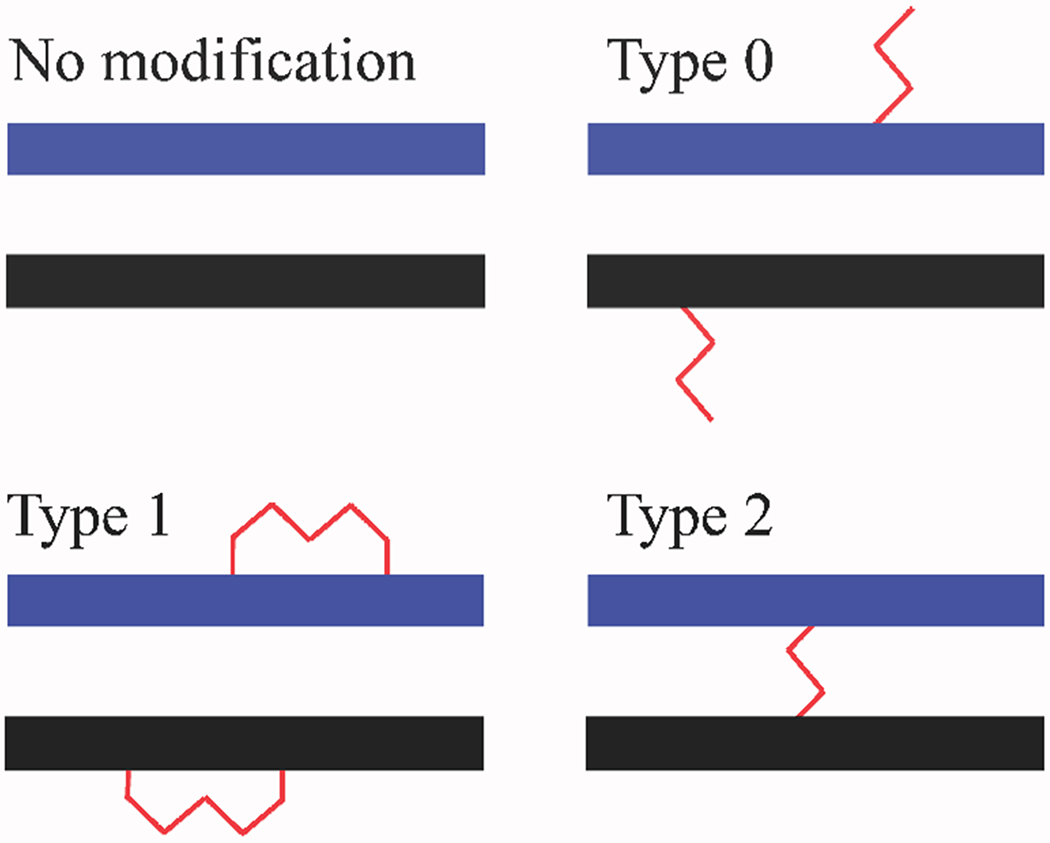
Typical products generated by treating a putative duplex substrate with a bifunctional reagent.
The marketplace offers a broad selection of bifunctional cross-linkers for protein substrates [57], whereas the choice for nucleic acids and their assemblies is much more limited. In this report, we evaluated known nucleic acid cross-linkers and compared their performance with that of other chemicals that may be capable of reacting simultaneously with the different types of biopolymers. We employed representative models for DNA, RNA, and their protein complexes to compare their reactivity with the different substrates. In particular, individual oligonucleotides were used to assess the reactivity of test reagents with nucleic acid functional groups. Double-stranded constructs were designed to evaluate the ability to produce inter- versus intra-strand conjugates, whereas protein-nucleic acid complexes were used to assess the production of hetero-conjugates. The report describes the experimental strategies employed to perform an initial screening of the types of cross-linked products, as well as those used to achieve full-fledged product characterization. The different classes of bifunctional reagents were compared according to their performance and information content. The reports offers also practical considerations regarding their application and product analysis, which may help inform reagent selection.
Experimental
Model systems.
The constructs employed in the study were selected to be as representative as possible of natural substrates. For this reason, repeating homopolymers consisting of the same type of nucleotide were deserted in favor of mixed sequences, such as (5′-TAATACGACTCACTATA-3′) and its RNA analogue 17RNAp (5′-UAAUACGACUCACUAUA-3′) (Scheme 2). Duplex constructs were obtained by annealing either strand with the corresponding complementary one, such as 17DNAc and 17RNAc (5′-TATAGTGAGTCGTATTA-3′ and 5′-UAUAGUGAGUCGUAUUA-3′, respectively). An RNA sequence replicating that of HIV-1 SL3 (5′-GGACUAGCGGAGGCUAGUCC-3′) [58] was included to represent stable secondary structures, whereas the HIV-1 nucleocapsid (NC) protein [59] was used to generate the desired complexes with the above nucleic acid substrates (Scheme 2).
Scheme 2.
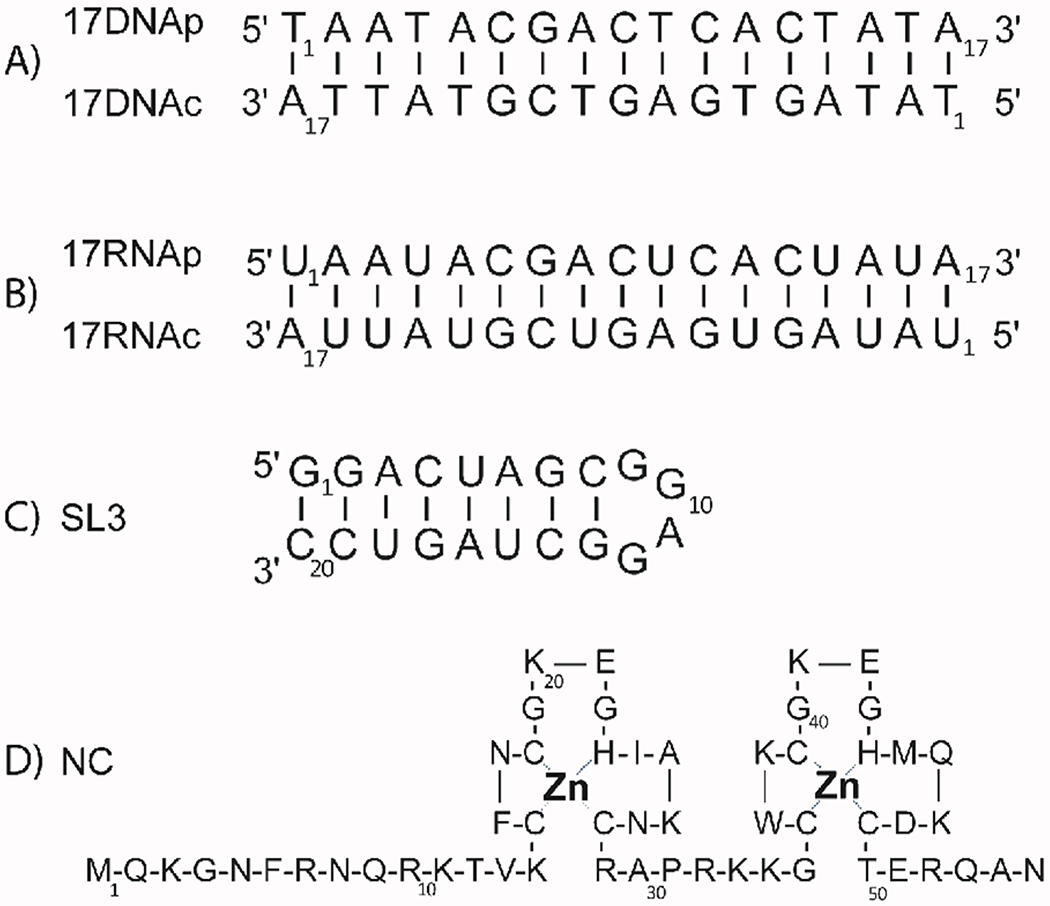
Model substrates employed in the study. A) 17DNA(p:c) DNA duplex; B) 17RNA(p:c) RNA duplex; C) HIV-1 stemloop domain 3 (SL3 RNA); D) HIV-1 nucleocapsid protein (NC). Duplex samples were obtained by annealing corresponding complementary strands, whereas protein-nucleic acid complexes were obtained by simply mixing the desired components and monitoring the outcome by ESI-MS analysis under non-denaturing conditions (see Experimental).
All DNA and RNA oligonucleotides were obtained from commercial sources (i.e., Integrated DNA Technologies, Coralville, IO). Initial samples were extensively desalted by buffer-exchange against 150 mM ammonium acetate (pH adjusted to 7.0) in Millipore (North Bend, OH) Centricon devices with 3 kDa molecular weight cut-off (MWCO). Duplex constructs were obtained by mixing complementary strands in equimolar amounts (50-100 μM range), heating the solution at 95 °C for 5 min., and then slow-cooling to room temperature in 2 h. The SL3 construct was heat-refolded at 95 °C for 5 min., followed by fast cooling on ice for 5 min. to promote intramolecular folding into a stable stemloop structure. All samples were re-equilibrated at room temperature for 2 h. The outcome of the annealing procedure was monitored by MS analysis (vide infra). Recombinant NC was expressed in E. coli BL21 (DE3)-pLysE, purified under non-denaturing conditions, and extensively desalted against 100 mM ammonium acetate (pH adjusted to 7.0), as previously described [60]. Protein complexes were obtained by mixing equimolar amounts (5-20 μM range) of NC and the desired nucleic acid construct, and incubating at room temperature for 2 h. The outcome was monitored by MS analysis, which enabled the detection of intact complexes including also the two Zn(II) ions coordinated by the metalloprotein component (vide infra).
Bifunctional cross-linkers.
Different families of bifunctional reagents were selected on the basis of their known or putative reactivity with functional groups present in nucleic acid biopolymers (Table 1). When available, these chemicals were obtained from commercial sources and utilized as received, without further purification. 2-chloro-N-(2-chloroethyl)-N-methylethanamine (mechlorethamine, NM), bis(2-chloroethyl)amine hydrochloride, 4-(4-[Bis(2-chloroethyl)amino]phenyl)butyric acid (chlorambucil), 1,3-bis(2-chloroethyl)-1-nitrosourea (carmustine), trans-diamminedichloroplatinum(II) (transplatin), cis-diamminedichloroplatinum(II) (cisplatin), potassium trichloro(ethylene)platinate(II) hydrate (Zeise’s salt), potassium tetrachloroplatinate(II) (tetrachloroplatin), 8-methoxypsoralen (8-MOP), trioxsalen, 4′-aminomethyltrioxsalen hydrochloride (AMT), formaldehyde, 1,4-Butanediol dimethanesulfonate (busulphan), p-azidophenyl isothiocyanate, 2-iminothiolane, epibromohydrin, 1,4 butanediol diglycidil ether and ethidium bromide were purchased from Sigma-Aldrich (St. Louis, MO) and used as received (Table 1). Sodium nitrite was obtained from Mallinckrodt Inc. (Paris, KY). Bis(sulfosuccinimidyl) suberate (BS3), succinimidyl-[4-(psoralen-8-yloxy)]-butyrate (SPB) and succinimidyl 4,4′-azipentanoate (SDA) were obtained from Thermo Fisher Scientific (Waltham, MA) (Table 1).
Table 1:
Names and chemical structures of test compounds included in the study.
 |
Ethidium diazide was synthetized in house by following the method described by Woolley and Dohrmann [61]. The product was characterized by UV-vis spectroscopy [62] and MS analysis. Bis-2 iodoethyl amine was prepared according to the Finkelstein reaction [63]. Briefly, 0.507 g (2.8 mmol) of bis-2 chloroethyl amine hydrochloride (Sigma-Aldrich) were dissolved in 50 mL of acetone. 4.2 g (28 mmol) of sodium iodide (Sigma-Aldrich) were added in two portions and refluxed for two days. The precipitated sodium chloride was eliminated and the liquid was stored at −80 °C overnight to promote further precipitation of insoluble salts. The product was finally precipitated by addition of acetic acid, washed with acetone, and characterized by MS analysis.
Cross-linking reactions.
A broad range of reaction conditions were explored, including different probe to substrate ratios, incubation times, temperatures, and types of buffers. Typical reaction mixtures consisted of 20 μl of 20 μM solution of substrate in either 100 mM ammonium acetate (pH adjusted to 7.0), or ad hoc buffer indicated in the text. An appropriate amount of cross-linker was added to each sample to achieve final probe to substrate ratios ranging from 5:1 to 100:1. Each reaction mixture was subsequently incubated at room temperature overnight (~14 h), unless otherwise noted. Photo-reactive reagents were incubated with substrate for 2 h in the dark and then irradiated at 365 nm for up to 60 min. In addition to ammonium acetate, we explored also phosphate and borate buffers, as well as water, as possible reaction media to test whether low cross-linking yields may be due to side reactions with other nucleophilic groups in solution [64,65]. Each reaction was quenched either by ethanol precipitation, or by performing immediate MS analysis. In general, ethanol precipitation was preferred when initial experiments had revealed the need for a desalting step, which was typically made necessary by the presence of non-volatile cations present in cross-linker stocks. Each sample was diluted in 150 mM ammonium acetate prior to analysis to achieve a final 1-5 μM concentration of total substrate.
Gel electrophoretic analysis of reaction mixtures.
An initial assessment of the ability to produce inter-molecular conjugates (type 2) was readily obtained by submitting reaction mixtures to denaturing polyacrylamide gel electrophoresis (PAGE). In the absence of conjugation, multi-component assemblies tend to dissociate under denaturing conditions and their non-covalently bound subunits are detected as discrete bands on the gel. In contrast, cross-linked complexes tend to resist dissociation and migrate as intact, whole species stabilized by inter-molecular covalent bridging. Therefore, this approach can offer a very convenient option for evaluating reaction yields and driving the optimization process, especially when sample availability is not a concern. The analysis of samples consisting of DNA and RNA oligonucleotides and their protein complexes was performed by using 20% polyacrylamide gels containing 7.5 M of urea. Separation was carried out in TBE buffer (89 mM Tris, 89 mM boric acid, and 2 mM EDTA adjusted to pH 8.0) at a constant voltage of 80 V. It should be noted that, owing to an isoelectric point of 9.9 units, free NC protein that was neither bound nor conjugated to a cognate nucleic acid construct consistently failed to migrate into the gel under the selected conditions, thus eluding detection. A 0.04% Stains-All (Sigma-Aldrich) solution in 40% formamide capable of staining both nucleic acid and protein substrates was employed to effectively stain the gels in the study, which were subsequently scanned for information safe-keep.
Mass spectrometric analysis.
Cross-linked products were either analyzed intact according to a typical top-down strategy [66,67], or upon digestion with proteases and nucleotide specific nucleases according to a bottom up approach [68,69]. For the latter, RNase T1 (Thermo Fisher Scientific), RNase A (Sigma-Aldrich), and trypsin (Thermo Fisher Scientific) were desalted as explained above for the initial substrates, and then stored at −20°C before use. Enzymatic digestion of cross-linked product was carried out in 150 mM of ammonium acetate (pH adjusted to 7.0) by adding nuclease at 1:10 enzyme to substrate ratio and trypsin at 1:40 enzyme to substrate ratio, followed by 1 to 3-h incubation at 37°C prior to MS analysis. Unless otherwise indicated, the digestion mixtures were analyzed without further treatment. As indicated above, all salts and buffers based on alkaline and alkaline-earth cations were replaced with ammonium equivalents to minimize the incidence of salt adducts, which may lead to signal suppression and resolution degradation [70,71 and references therein]. In addition to representing the first choice for MS analysis, the ammonium acetate system employed here provided also the neutral environment necessary to prevent degradation of rather labile RNA samples and preserve the activity of the digestion enzymes.
All samples were analyzed by direct infusion electrospray ionization (ESI) on Thermo Fisher Scientific (Waltham, MA) LTQ-Orbitrap Velos mass spectrometer. The analyses were performed in nanoflow mode by using quartz emitters produced in house by utilizing a Sutter Instruments Co. (Novato, CA) P 2000 laser pipette puller. Samples with concentrations ranging from 1 to 5μM of total substrate in 150 mM ammonium acetate were analyzed in either negative or positive ion mode, as indicated in the text. Up to 5-μL samples were typically loaded onto each emitter by using a gel-loader pipette tip. A stainless steel wire was inserted through the back-end of the emitter to supply an ionizing voltage that ranged between 0.8 and 1.2 kV. Source temperature and desolvation conditions were adjusted by closely monitoring the incidence of ammonium adducts and water clusters [72]. Typical source temperature was 200°C, whereas desolvation voltage was in the 35-40 V range. Tandem mass spectrometry (MS/MS) experiments involved isolating the precursor ion of interest in the LTQ element of the instrument, activating fragmentation in either the LTQ or the C-trap, and finally performing fragment detection in the Orbitrap. Duplex dissociation was typically achieved by using a nominal activation voltage of 20 V, whereas strand fragmentation for sequencing purposes was achieved at voltages exceeding 50 V. Alternatively, duplex dissociation was carried out in source by increasing the desolvation voltages greater than 70 V, which far exceeded typical voltages employed to achieve efficient declustering of ammonium adducts (e.g., 35-40 V). The reported parameters were those employed to dissociate the 17nt duplex DNA in the study. Other constructs required voltage settings that were characteristically dependent on size and GC content. The instrument was calibrated by using a 0.5 mg/mL solution of CsI in 50/50 water/methanol, which provided a typical 2 ppm mass accuracy. Data interpretation was accomplished with the aid of Mongo Oligo Calculator [73], Peptide mass [74], MS-Product (http://prospector.ucsf.edu/prospector/mshome.htm), and ad hoc software developed in house.
Results and Discussion
Rapid assessment of mono-functional vs. bridging outcomes by gel electrophoresis.
Reaction mixtures obtained by incubating the various substrates with each reagent were initially analyzed by denaturing polyacrylamide gel electrophoresis (PAGE), which afforded a very convenient approach for evaluating reaction yields and driving the optimization process. These capabilities are exemplified by the analysis of the products of DNA and RNA duplexes with the photo-activated reagent 8-methoxypsoralen (8-MOP, Table 1). This reagent is known for its ability to intercalate within double-stranded regions and produce both intra- and inter-strand products [75]. The duplex substrates were obtained by annealing complementary strands through a classic heat-refolding procedure (see Experimental). Reaction mixtures obtained upon irradiation with a 365 nm light for up to 30 min. were analyzed by denaturing 20% PAGE in the presence of 7.5 M of urea. The results showed a band corresponding to intact DNA duplex, which migrated more slowly than the individual single-stranded components produced by dissociation of the initial substrate (Figure 1A, lane 4). Although these data confirmed the formation of desired inter-molecular crosslinks (type 2 products in Scheme 1), the resolution afforded by this technique was not sufficient to unambiguously differentiate intra-molecular products in which 8-MOP bridged nucleotides on the same strand (type 1) from mono-functional products in which the cross-linker had failed to complete the bridging reaction (type 0). Nevertheless, these types of determinations can support the effective optimization of the reaction conditions by monitoring the ratio of conjugated versus non-conjugated species in the sample. In this case, it also allowed us to immediately appreciate the different susceptibility to 8-MOP cross-linking manifested by DNA and RNA samples (vide infra).
Figure 1.

Denaturing PAGE analysis of cross-linking reaction mixtures. A) Reaction of 8-MOP with 17DNA(p:c) and 17RNA(p:c) (see Experimental for details). Irradiated DNA sample displayed a new band (xl-DS) with a relative mobility (Rf) of 0.69 versus the corresponding single-strands (SS) (lane 4), consistent with inter-strand conjugation. In contrast, no new bands were detected for the irradiated RNA sample, consistent with the lack of inter-strand conjugation. B) Reaction of NM with SL3, NC, and NC•SL3 complex. Crosslinked NC•SL3 complexes showed a new band (xl-NC•SL3) Rf = 0.34 versus SL3, which is consistent with the formation of a cross-linking product (lanes 6 and 9) (see Experimental). All gels were stained by using Stains-All, which can effectively stain both nucleic acid and protein moieties. It must be noted, however, that an isoelectric point of 9.9 units prevented free NC protein, which was neither bound nor conjugated to the cognate nucleic acid substrate, from migrating into the gel, thus eluding detection. No multimeric nucleic acids artifacts were ever detected under the denaturing conditions employed in these experiments.
In similar fashion, denaturing PAGE can support also the optimization of protein-nucleic acid reagents, as exemplified by the reaction of nitrogen mustard (NM) with a model system consisting of the assembly of the HIV-1 nucleocapsid (NC) protein with the SL3 domain of viral RNA (Scheme 2) [76]. NM is a classic homo-bifunctional cross-linker with excellent reactivity towards nucleophilic functional groups of both proteins and nucleic acids [77–79]. This type of reagent can provide the coveted inter-molecular conjugates between protein and RNA, but also intra-molecular conjugates within either component of the complex (Scheme 1). In addition, however, NM can also produce monofunctional products in which one arm reacts with either component, while the other may be promptly hydrolyzed before completing the bridging reaction. Analysis by denaturing PAGE allowed the immediate discrimination of type 2 conjugates, which migrated as tethered undissociated species, against corresponding dissociation products (Figure 1B, lane 6 and 9). Also in this case, the electrophoretic resolution was insufficient to differentiate unreacted species from mono-functional type 0 and intra-molecular type 1 products. Modest band shifts were observed for reaction mixtures obtained at higher NM concentrations (Figure 1B, lane 7), which could be ascribed to the small mass increments associated with these covalent adducts. Increasing percentages of bis-acrylamide in the gel (e.g., from 20 to 25%) could be employed to improve the resolving power of this technique. However, these types of efforts are frequently unnecessary for reaction optimization purposes.
Evaluation of bridging outcomes by top-down MS analysis.
Direct MS analysis of reaction mixtures can immediately reveal the stoichiometry and yield of adduct formation and, in many cases, the partitioning between mono-functional and cross-linked bifunctional products. The representative data in Figure 2 illustrate these capabilities. In panel 2A), abundant adducts produced by 8-MOP reaction with the 17DNA(p:c) duplex were immediately recognized with up to two equivalents of cross-linker. In panel 2B), only unreacted 17RNA(p:c) could be detected in a sample obtained under identical experimental conditions (see Experimental), in agreement with the data obtained by denaturing PAGE analysis (Figure 1A, lane 8). When the sample obtained by treating NC•SL3 with NM was analyzed, signals could be detected corresponding to mono-functional type 0 adducts, as well as type 1 or 2 crosslinks (panel 2C). It is important to note that the photo-activated reaction of 8-MOP takes place without change of mass [75,80] and, thus, these data could not immediately reveal whether the observed products consisted of mono- or species. In contrast, NM reaction takes place with a formal elimination of HCl (i.e., ΔM = 35.977 u) per reacting 2-chloroethyl function. Therefore, the observed loss of 71.944 u was safely ascribable to the formation of stable bifunctional crosslinks. In contrast, hydrolysis of unreacted 2-chloroethyl function results in a characteristic ΔM of 101.08 u, which can be employed as diagnostic for mono-functional type 0 products.
Figure 2.
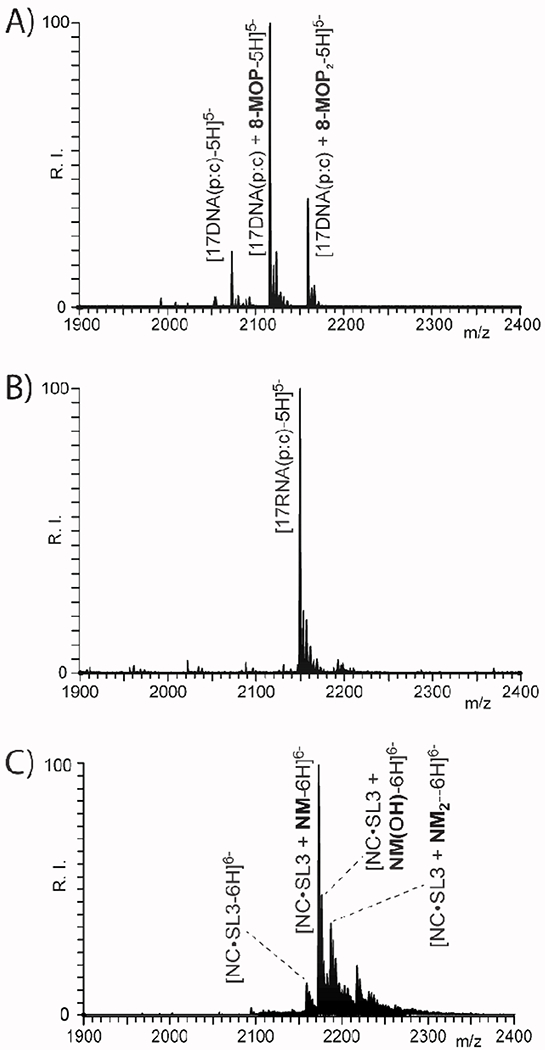
ESI-MS spectra obtained from reaction mixtures of A) 8-MOP and 17DNA(p:c); B) 8-MOP and 17RNA(p:c); and C) NM and NC•SL3 complex (see Experimental). The irradiated DNA sample displayed adducts with up to two 8-MOP equivalents, whereas its RNA counterpart did not react under identical conditions. The NM-treated sample displayed crosslinked products, together with type 0 adducts with a hydrolyzed 2-chloroethyl arm. The masses of the species displayed in this figure are provided in Table S1 of Supporting Information.
Alternative top-down approaches could be implemented to assess the partitioning between intra- and inter-molecular products (i.e., type 1 and 2) by employing gas-phase dissociation to mimic the intrinsic denaturing effects of PAGE. Indeed, denaturation of weakly bound complexes can be obtained either by increasing the desolvation voltage employed in the analysis (i.e., in-source dissociation [81]), or by performing collision induced dissociation (CID) during classic tandem MS experiments [82]. As observed in Figure 2, typical source conditions employed in our analyses were sufficiently gentle to enable the detection of intact duplex and protein-nucleic acid complexes (see Experimental) [60,72]. However, progressively higher desolvation voltages were capable of inducing their dissociation, thus enabling the discrimination of substrates devoid of bridging crosslinks from inter-molecular conjugates, which remain unaffected. For example, the representative data in Figure 3 were obtained from the 8-MOP-17DNA(p:c) sample at a desolvation voltage of either 50 or 100 V. These conditions produced signals corresponding to the individual strands of the original duplex, which contained both type 0 and 1 8-MOP adducts. At the same time, bridged type 2 products were detected in intact duplex form, due to the stabilizing inter-strand crosslinks. The observed masses enabled the correct assignment of unreacted strands versus type 0 and 1 counterparts (see Table 1S of Supporting Information), but the latter were correctly recognizable only upon full-fledged fragmentation in MS/MS experiments (vide infra).
Figure 3.
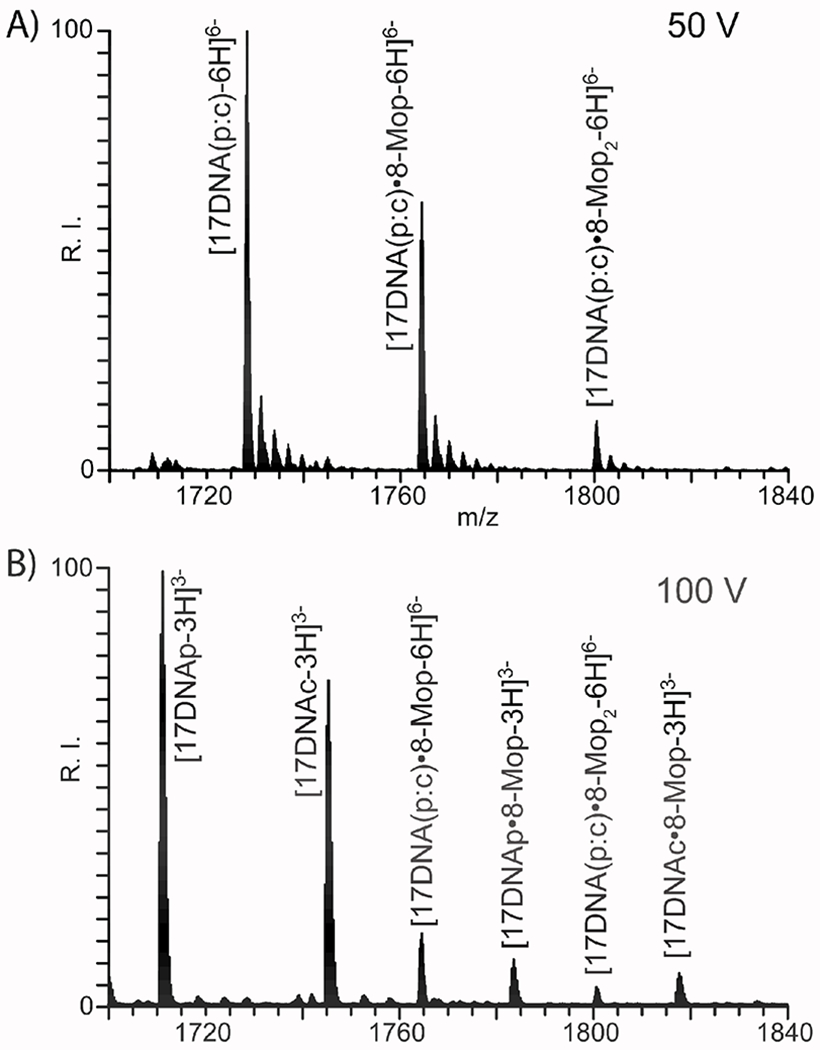
ESI-MS analysis of 8-MOP adducts of 17DNA(p:c) obtained at A) 50V and b) 100V desolvation voltages to induce in-source dissociation (see Experimental). The formation of inter-strand crosslinks was revealed by the presence of intact type 2 products, which persisted at even higher desolvation voltages capable of inducing backbone fragmentation. The masses of the species displayed in this figure are provided in Table S1 (see Supporting Information).
The concerted application of denaturing PAGE and MS analysis constitutes an excellent platform for completing initial optimization operations, which tend to involve monitoring the outcome of reactions performed upon systematic variation of essential parameters. Denaturing PAGE can provide a rapid and inexpensive evaluation of the yield of inter-molecular cross-linking, whereas MS analysis can differentiate mono- from bifunctional products. The ability to multiplex sample analysis afforded by the former enables one to screen very rapidly the effects of probe to substrate ratio, pH, reaction temperature and duration, which can have profound effects on cross-linking yields and distribution. The latter instead can identify the factors that most affect the competition between inter-molecular bridging and either intra-molecular closing or hydrolysis. Once the optimization phase is complete, the MS platform affords the ability to characterize the conjugates, identify the bridged components, and locate the sequence position of the crosslinks.
Characterization of conjugated species.
In interactomics and structural applications, the sought-after information consists of either the identity of the conjugate components, or the sequence position of cross-linked residues, which can be translated into valuable spatial constraints for model-building operations [12]. Acquiring these types of information requires completing the characterization of cross-linked products, which can be carried out through either bottom-up procedures involving digestion, mass mapping, and MS/MS analysis of the hydrolytic products, or top-down approaches based on direct gas-phase fragmentation of mass-selected samples with no hydrolytic steps. These strategies are routinely employed for protein analysis in proteomics [83,84], but are readily applicable in analogous fashion also to nucleic acid-containing samples [15,67]. Bottom-up approaches rely on the ability to generate unique hydrolytic mixtures that facilitate the recognition of conjugated species. In the case of protein-nucleic acid conjugates, the different elemental compositions of their fundamental units can lead to readily distinguishable products with characteristic masses and fragmentation patterns. In addition, conjugation may inhibit the activity of the specific cleaving enzymes, which may result in maps that are significantly different from those obtained in the absence of crosslinks.
These concepts can be exemplified by the case of the products of NM reaction with NC•SL3 described above. The heterogeneous nature of this system required that the reaction material be digested with both protease and endonuclease to enable the comprehensive mapping of all components of the complex (see Experimental). Figure 4 provides representative data obtained by direct infusion analysis in negative ion mode (see Experimental). The spectrum was dominated by relatively short peptide and oligonucleotide products generated by the enzymatic activity, which were made negatively charged by the presence of acidic carboxylate and phosphate groups, respectively. In addition, weaker signals were recognized as consisting of short peptide-oligonucleotides conjugates bridged by the NM reagent (Figure 4). When the polarity was reversed, additional peptides and heteroconjugates were readily detected as positively charged ions (not shown). The masses provided by these determinations matched very closely those obtained from the sequences of the biopolymers in the complex, which were calculated by taking in account the incremental mass of NM adducts and the position of residues/nucleotides susceptible to specific enzymatic attack (Table 2S of Supporting Information).
Figure 4.
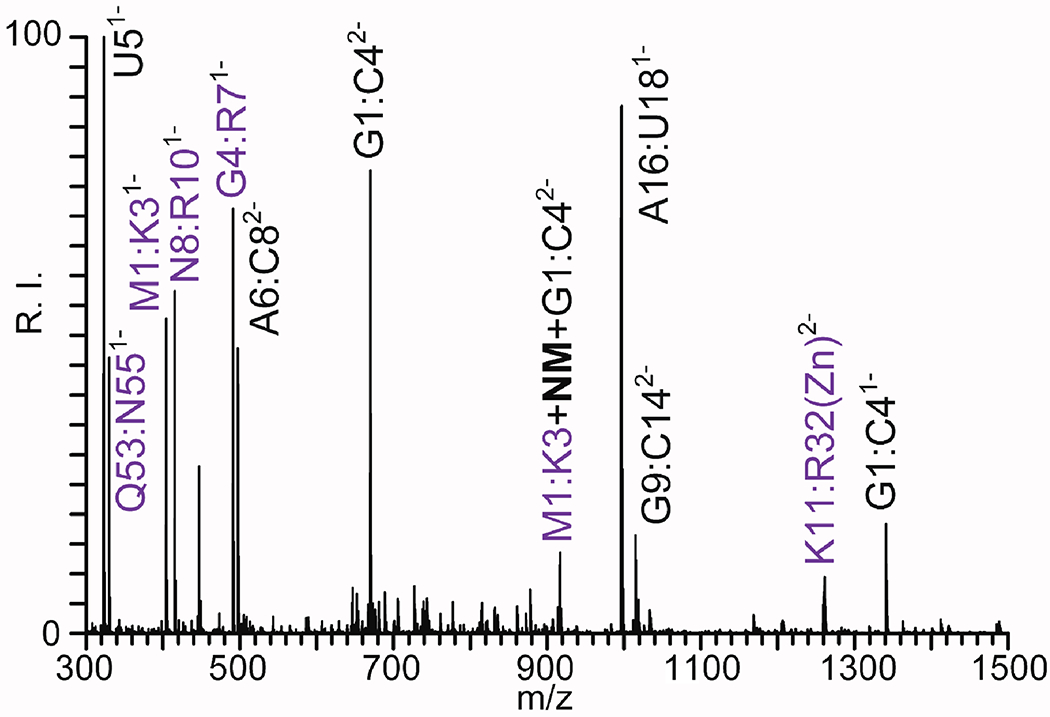
ESI-MS analysis of a digestion mixture of NC•SL3 complex treated with NM. The product mixture was digested with both RNase A and trypsin (see Experimental). The concerted digestion procedure produced both peptide (purple) and oligonucleotide (black) products, as well as their conjugates bridged by the NM cross-linker. The masses of the species displayed in this figure are provided in Table 2S of Supporting Information.
The peptide-oligonucleotide conjugates detected by mass mapping are typically located at the interface between bound components, which can reveal the overall organization of the assembly. Further, the sequence position of the cross-linked residues/nucleotides in the hetero-conjugate, which is attainable by MS/MS analysis, can identify the actual contacts between bound components. This type of experiment is exemplified in Figure 5, which displays the fragmentation data obtained by gas-phase activation of the m/z 914.23 precursor ion observed in Figure 4. Based on the mapping information, this species was tentatively assigned as a putative M1:K3 + NM + G1:C4 hetero-conjugate. Consistent with its hybrid nature, a large number of fragments were observed in negative ion mode (Figure 5a), which were part of the characteristic ion series produced by backbone dissociation of peptides or oligonucleotides [85]. A similar pattern was observed in positive ion mode, although fragments produced by the peptide moiety were featured more prominently (Figure 5b). Although these ions series were not complete, they still provided strong indications that the conjugation sites were located on M1 and G2, thus illustrating the value of performing analyses in both polarities to obtain complementary information and increase the confidence in the assignment of hetero-conjugate crosslinks.
Figure 5.
MS/MS analysis of the precursor ion detected at m/z 914.23 in Figure 4. Panel A) was obtained in negative ion mode with 43 V activation voltage. Panel B) was obtained in positive ion mode with 51 V activation (see Experimental). Panel C) displays the fragmentation pattern produced by the putative structure assigned to this precursor ion.
Cleavage of the N-glycosidic bond is a prominent process observed upon gas-phase activation of oligonucleotides [86–88], which is typically enhanced by covalent modification of the nucleobase system, especially at the N7 position of purines. In the context of the M1:K3 + NM + G1:C4 product, this rather facile cleavage led to the detection of abasic oligonucleotide fragments and peptide ions cross-linked to guanine nucleobases, with the latter pointing toward M1 as the site of conjugation on the M1:K3 peptide (Figure 5a). Additionally, a fragment consisting of guanine linked to a CH3SH moiety corroborated the participation of the methionine residue in conjugation. Considering that NM engages in typical nucleophilic substitutions, the more likely outcome should have been the alkylation of one of the multiple thiolates present in NC•SL3, rather than the effective modification of a much less reactive thioether function. It could be argued that all Cys residues in the ribonucleoprotein are involved in Zn(II) coordination and may be thus prevented from engaging in nucleophilic substitution. Over the years, however, it has been unambiguously demonstrated that metal-coordinating Cys can still effectively engage in alkylation reactions [89–91]. For these reasons, the surprising reactivity of the M1 residue could be ascribed to its spatial situation in the ribonucleoprotein assembly, which could place the thioether within reach of the 2-chloroethyl function of a NM molecule that had already used the other function to alkylate the G2 nucleotide of the RNA component.
Among the challenges posed by these types of analyses, the typically low abundance of inter-molecular type 2 products makes careful reaction optimization a necessity. Rash attempts to increase yields by simply increasing probe concentrations can often lead to probe-induced conformational changes and kinetic traps. Coupling liquid chromatography with MS analysis can help facilitate the characterization of low-abundance conjugates in the presence of prominent unreacted species [92]. Alternative strategies have been developed to enable the enrichment of cross-linked products by using affinity tags included in the cross-linker’s structure, or capitalizing on specific physical-chemical properties possessed by the conjugates. Among the former, numerous biotinylated cross-linkers have been introduced to enable the capture of conjugated products before MS analysis [93]. Among the latter, the strong interaction between TiO2 and phosphate groups has been employed to setup two-dimensional chromatographic systems for the specific enrichment of peptide-RNA conjugates [94]. Another challenge consists of the interpretation of these types of data, which still tend to involve extensive user intervention [43]. Any ad hoc software cannot count solely on the known reactivity profile of a certain reagent to predict possible cross-linking patterns, but must also consider the possibility that such reactivity may be profoundly affected by steric factors, which are still poorly understood. Nevertheless, in spite of the challenges, the combination of bifunctional cross-linking and MS detection represents a still growing approach for obtaining information that may be otherwise inaccessible.
Evaluation of putative cross-linking reagents.
The techniques described above were employed in concert to evaluate the ability of different classes of nucleic acid reagents to produce inter-molecular crosslinks in both nucleic acid-nucleic acid and protein-nucleic acid assemblies. The results obtained for each class are presented below with a brief discussion of our findings in the context of prior information on chemical properties and analytical behavior. Table 2 provides a summary of the survey, which ranks the reagents in the study according to selected of the criteria discussed above.
Table 2.
Evaluation of putative cross-linking reagents by using selected nucleic acid and protein-nucleic acid substrates (see Experimental). The production of inter-molecular crosslinks was assessed by using both denaturing PAGE and MS analysis. The observed performance was rated as excellent (✓ ✓ ✓), acceptable (✓ ✓), limited (✓), or negligible (✖).
| Name | Class | DNA type 0 | RNA type 0 | Protein type 0 | DNA-DNA crosslink | RNA-RNA crosslink | Prot.-RNA crosslink |
|---|---|---|---|---|---|---|---|
| 8-methoxypsoralen | Psoralens | ✓ ✓ ✓ | ✓ | n.d. | ✓ ✓ ✓ | ✖ | ✖ |
| Trioxsalen | “ | ✓ ✓ | ✓ | n.d. | n.d. | ✖ | n.d. |
| 4′-aminomethyltrioxsalen hydrochloride | “ | ✓ ✓ ✓ | ✓ ✓ ✓ | n.d. | ✖ | ✖ | ✖ |
| Transplatin | Platinum complexes | ✓ ✓ ✓ | ✓ ✓ ✓ | ✓ ✓ | ✓ ✓ ✓ | ✓ ✓ ✓ | ✓ ✓ ✓ |
| Cisplatin | “ | ✓ ✓ ✓ | ✓ ✓ ✓ | ✓ ✓ | ✓ ✓ ✓ | ✓ ✓ ✓ | ✓ |
| trichloro(ethylene) platinate(II) | “ | n.d. | ✓ ✓ ✓ | n.d. | n.d. | ✓ ✓ ✓ | ✓ ✓ ✓ |
| Tetrachloro platin | “ | n.d. | ✓ ✓ ✓ | ✓ ✓ | n.d. | ✓ ✓ ✓ | ✓ ✓ ✓ |
| Nitrogen mustard | Mustard | n.d. | ✓ ✓ ✓ | ✓ ✓ | n.d. | ✓ ✓ ✓ | ✓ ✓ ✓ |
| Chlorambucil | “ | n.d. | ✓ ✓ ✓ | n.d. | n.d. | ✓ ✓ ✓ | ✓ ✓ |
| Carmustine | “ | n.d. | ✓ ✓ | n.d. | n.d. | ✓ ✓ | ✓ ✓ ✓ |
| Bis(2-chloroethyl)amine | Alkylamine | n.d. | ✓ ✓ | ✓ ✓ | n.d. | n.d. | ✓ ✓ |
| Bis(2-iodoethyl)amine | “ | n.d. | ✓ ✓ | ✓ ✓ | n.d. | n.d. | ✓ ✓ |
| Epibromo hydrin | epoxydes | n.d. | ✖ | ✖ | n.d. | ✖ | ✖ |
| 1,4 butanediol diglycidyl ether | “ | n.d. | ✖ | ✖ | n.d. | ✖ | ✖ |
| Busulphan | Alkyl sulphonate | n.d. | ✖ | ✖ | n.d. | n.d. | ✖ |
| Nitrous acid | Nitrous acid | n.d. | ✖ | ✖ | n.d. | n.d. | ✖ |
| Formaldehyde | aldehyde | n.d. | ✓ ✓ | ✓ ✓ | n.d. | n.d. | ✓ ✓ ✓ |
| Ethidium diazide | Photo-cross-linker | n.d. | n.d | n.d | n.d. | ✖ | ✖ |
| p-Azidophenyl isothiocyanate | “ | n.d. | n.d | ✓ ✓ ✓ | n.d. | n.d. | ✖ |
| SDA (succinimidyl 4,4-azipentanoate) | “ | n.d. | ✖ | ✓ ✓ ✓ | n.d. | ✖ | ✖ |
| succinimidyl-[4-(psoralen-8-yloxy)]-butyrate (SPB) | “ | n.d. | n.d | ✓ ✓ ✓ | n.d. | ✖ | ✖ |
| 2-iminothiolane | “ | n.d. | ✖ | ✓ ✓ | n.d. | n.d. | ✖ |
| Bis(sulfosuccinimidyl) suberate | Activated carboxylic acid | n.d. | ✖ | ✓ ✓ ✓ | n.d. | ✖ | ✖ |
A). Psoralens.
Photo-activated compounds are particularly desirable for their ability to react with a wide variety of functional groups without the release of leaving groups that may hamper subsequent characterization. Furthermore, their reaction yields can be controlled to a certain extent by varying irradiation wavelength and duration. Psoralens are characterized by relatively flat ring systems (Table 1), which must intercalate within duplex structures before engaging in photo-activated chemistry [95]. Typical reactions involve cyclo-addition to pyrimidine rings, with possible formation of both mono- and bifunctional adducts [75]. The DNA and RNA duplexes described above were employed as substrates to compare the susceptibility of different types of biopolymers. The explored reaction conditions ranged from 10:1 to 1:1 reagent to substrate ratios (see Experimental). Upon ligand binding, irradiation with 365 nm light was carried for 5 to 60 min. to evaluate the effects on overall yields. By inducing the dissociation of unreacted strands, denaturing PAGE analysis revealed the presence of inter-strand conjugates in the DNA sample treated with 8-MOP and their absence in the corresponding RNA reaction (Figure 1, compare lane 4 and 8). These results were corroborated by ESI-MS determinations, which confirmed the presence of duplex products containing up to two 8-MOP equivalents in the DNA sample versus none in the RNA counterpart treated under identical conditions (compare Figure 2A and 2B). The inter-strand nature of the DNA adducts was confirmed by in source dissociation experiments, as explained above (Figure 3). Additionally, full-fledged MS/MS analysis was also performed on the 1:1 adduct, which produced abundant fragmentation consistent with type 1 conjugates (see Figure 1S of Supporting Information).
At first sight, the results afforded by the reaction of 8-MOP with the RNA duplex seemed to contradict an earlier report that described the proficiency of 4′-aminomethyltrioxsalen (AMT) –a slightly more polar psoralen– to effectively crosslink E. coli rRNA [96]. When this compound was reacted with the DNA and RNA duplexes, denaturing PAGE provided results consistent with the formation of both intra- and inter-strand adducts (Figure 2S of Supporting Information). At the same time, however, in source dissociation during ESI-MS analysis revealed that the observed species were almost exclusively intra-strand products of type 0 and 1. Consistent with these findings, MS/MS fragmentation of the 1:1 adducts provided no evidence of conjugated type 2 products (compare for example Figure 1S and 3S of Supporting Information), thus casting doubts not only on the results of our PAGE experiments, but also on the outcome reported for E. coli rRNA [96]. The discrepancy could be explained by examining the binding process that precedes the photo-cross-linking reaction, which can be affected in different ways by the structure of the reagent. It is possible that the stacking interactions established by the psoralen aromatic system may stabilize not only intercalator binding, but also association of the contiguous base pairs. In this way, a combination of unreacted, type 0, and type 1 products could still contribute to limit strand dissociation, even in the absence of type 2 conjugates. Described as “virtual cross-linking,” this behavior has been described for other intercalators, such as anthracyclines, which are not capable of actual inter-strand conjugation, but can still limit duplex dissociation induced by denaturating PAGE [97,98]. Binding considerations could also help understand the discrepancy between the behaviors of 8-MOP and AMT. In fact, the non-covalent complexes established by the latter are much more stable than those involving the former, with dissociation constants (Kds) that are orders of magnitude apart (e.g., μM versus mM ranges) [99]. At the same time, the binding of 8-MOP to RNA duplexes tends to be at least one order of magnitude weaker than that to DNA analogs [99]. This combination of features could explain how the stronger interaction established by AMT with the RNA duplex was sufficient to induce “virtual cross-linking,” whereas that established by 8-MOP fell short. It must be noted that any attempt to enhance 8-MOP binding by increasing ligand concentration failed to produce the desired stabilizing effects on strand association, at least within the solubility limit of this molecule in water (i.e. 170 μM) [99].
The ability of psoralens to produce protein-nucleic acid conjugates was evaluated in similar fashion by using the NC•SL3 ribonucleoprotein described above. Additionally, a complex containing the viral NC protein and our duplex DNA construct (i.e., NC•17DNA(p:c)) was also prepared by capitalizing on the ability of this quintessential nucleic acid chaperone to bind both RNA and DNA structures with similar affinities [100]. No bands corresponding to protein-RNA conjugates could be observed upon denaturing PAGE of samples treated with either 8-MOP or AMT (Figure 4S of Supporting Information, lane 8 and 12). In contrast, bands corresponding to protein-DNA conjugates were observed in NC∂7DNA(p:c) samples treated with the psoralens under identical conditions (lane 6 and 10). These gels contained a positive control obtained by reacting NC•SL3 with NM cross-linker, which enabled us to determine the possible migration rate of sought-after conjugates. The fact that the results obtained from these protein-nucleic samples mirrored very closely those provided by the DNA and RNA duplexes indicates that psoralens may represent promising probes for DNA-containing substrates, but are ineffective for RNA-containing ones.
B). Alternative photo-cross-linkers.
Additional agents known to engage in photo-activated chemistry were also evaluated as possible cross-linking reagents. In particular, we investigated the possibility to exploit the ability of the azido group to form a reactive nitrene upon UV activation, which is capable of reacting with nucleic acid and protein structures [101,102]. The test compound ethidium diazide [103], which combines a nucleic acid intercalator scaffold with two identical groups in symmetrical positions, failed to produce detectable crosslinks with any of the nucleic acid or ribonucleoprotein substrates. The fact that no bridging products could be observed upon varying the reagent to substrate ratio up to 100:1, or extending irradiation at 254 nm up to 60 min. could be likely due to the possible orientation and distance between the active functions in the reagent. Unfavorable spacing and geometric arrangement may prevent the proper completion of a bridging conjugate upon initial mono-functional reaction.
We also tested putative probes that combined a protein reactive group with photoreactive group, such p-azidophenyl isothiocyanate, a hetero-bifunctional reagent combining an azido function with an electrophilic isothiocyanate function. To overcome the unsatisfactory yields afforded by the photo-activated reaction, we evaluated a stepwise strategies in which the first step was conducted in the absence of light to identify the best possible conditions to increase the isothiocyanate activity, whereas the second was carried out by irradiating the sample at 365 nm to activate the azido group and complete the bifunctional conjugation (see Experimental). When the ribonucleoprotein substrate was considered, the initial step performed in the dark provided abundant modification of the protein in the complex, but only negligible reactivity with the nucleic acid component. In contrast, subsequent photo-activation induced negligible reactivity with either component under the explored experimental conditions. In similar fashion, succinimidyl 4,4-azipentanoate (SDA), succinimidyl-[4-(psoralen-8-yloxy)]-butyrate) (SPB), and 2-iminothiolane (Traut’s reagent) were also included in the study on the basis of their combinations of functions capable of reacting with proteins, and photoactivable groups that may be able to attack the aromatic systems of nucleic acid components. For all these reagents, the dark reaction produced abundant type 0 products, while subsequent light exposure failed to provide the sought-after bifunctional bridging. Also in this case, unfavorable structural contexts could be responsible for the unsatisfactory outcomes, if initial alkylation placed the second reactive center outside bridging range from any possible target.
C). Platinated complexes.
The realization that the cytotoxic properties of cisplatin were due to the ability to form stable adducts with nucleic acids has promoted the development of numerous platinated analogs as cancer therapeutics [104]. At the same time, this capability has spurred interest also in their application as possible structural probes for these types of biopolymers [77]. The general mechanism of action involves the formal replacement of chloride ligands in the metal’s coordination sphere with a variety of electron donors that, in the case of nucleic acids, correspond predominantly to the N7 of guanine nucleotides [105,106]. In the case of cisplatin, the geometry of the coordination sphere leads to the preferred formation of intra-strand bonds, whereas its transplatin analog can form relatively stable inter-strand bridges [107]. Over the years, the reactivity of this class of compounds with DNA substrates has been the object of intense study, whereas little is known about their activity towards RNA. For this reason, we tested the activity of platinum compounds with both DNA and RNA models, and analyzed the respective reaction mixtures according to the approaches described above. The study included also trichloro(ethylene)platinate(II) and tetrachloro-platin to test their ability to bridge more than two functional groups at a time, with the understanding that their multivalent nature may make them better suited for interactomics applications, in which the goal may be to identify components that are in direct contact with one another, than for structural investigations aimed at identifying specific points of contact as spatial constraints for modeling operations.
Analyzed by denaturing PAGE, the mixtures obtained by reacting cisplatin with our DNA and RNA duplexes displayed type 0 and/or 1 and 2 products. In contrast, equivalent samples treated with transplatin displayed bands corresponding to type 0 and/or 1 adducts, whereas putative type 2 conjugates were masked by widespread smearing caused by the possible presence of multiple platinum adducts. The results obtained by ESI-MS helped clarify these observations and provided additional insights. In the case of the RNA substrate, for example, cisplatin provided greater yields of bifunctional type 1 and 2 products (Figure 5SA of Supporting Information) than the transplatin analogue that produced a greater yield of mono-functional type 0 (panel B). This observation was consistent with the specific stereochemistry of the cisplatin coordination sphere, which places two sequential purine nucleobases on the same strand in ideal positions to replace the initial chloride ligands forming type 1 products [108–111]. The orientation of such ligands was instead unfavorable in transplatin, as reflected by the reduction of detectable bifunctional products [107,112].
The corresponding ESI-MS data contained numerous mono-functional type 0 products, in which the chloride groups were partially replaced by different electron-rich ligands present in solution, such as ammonia, acetate, and hydroxyl ions (Figure 5S of Supporting Information). Considering that replacement of chloride with hydroxide is a necessary activation step in the reaction of platinum compounds [107,112], the initial type 0 adducts could potentially retain residual reactivity towards suitable electron-rich substrates. In bottom-up approaches, these types of adducts could then lead to bridging reactions between oligonucleotide products released in solution by the digestion procedure, thus producing artifacts that do not reflect the original structure of the target substrate. For this reason, the addition of sulfhydryl reagents, such a β-mercaptoethanol, is recommended before digestion to deactivate the residual activity of initial type 0 adducts. Alternatively, longer incubation intervals in ammonium acetate environment could serve to replace hydroxide with the much less reactive ammonia.
The various products were submitted to gas-phase activation to achieve further characterization. Consistent with their structure, mono-functional type 0 products displayed greater propensity toward strand dissociation than typical backbone fragmentation under the selected conditions (see Experimental). This effect is exemplified by the data obtained from the putative [17RNA(p:c) + Pt(NH3)3 − 6H+]6− precursor ion detected at m/z 1831.75. The dissociation process produced two abundant species corresponding to the unmodified 17RNAp strand and its modified 17RNAc counterpart, whereas only weak signals were detected for modified 17RNAp and unmodified 17RNAc (panel A of both Figure 6S and 7S of Supporting Information). These observations suggested that the platinum center coordinated preferentially one strand of the duplex, whereas the rest of the coordination sphere was occupied by three equivalents of ammonia. The presence of 4 guanine nucleotides in the 17RNAp strand versus only one in the complementary 17RNAc (Scheme 1) could explain this modification pattern, which may just reflect the probability of cis/transplatin to react with either strand. The handful of backbone fragments detected under these conditions corresponded to the putative loss of short c/y ions from the intact platinated duplex (panels C and D of both Figure 6S and 7S of Supporting Information). This observation could be ascribed to the stability of the base pairing interactions in the duplex structure, which prevented strand dissociation even in the absence of inter-strand crosslinks [113]. It has been shown that top-down activation of duplex constructs can induce backbone fragmentation of the fraying ends of the oligonucleotide components, while preserving base pairing in the remaining sections [114]. As a possible alternative, the initial platinated duplex was submitted to in-source dissociation and the ensuing platinated 17RNAc was further activated to achieve backbone fragmentation (see Experimental). This atypical MS3 experiment enabled the detection of characteristic c/y ions that narrow down the positions of the platinum center to contiguous nucleotides (Figure 8S of Supporting Information). It is important to note that, unlike the outcome obtained from NM adducts, no sign of weakening of the N-glycosidic bond were observed for platinated counterparts, as substantiated by the detection of abasic oligonucleotides fragments retaining platinum complexes (i. e. [17RNAc + Pt(NH3)3 -G]3−). This effect could be explained by the fact that platinum coordination by the N7 position of purines does not tend to undermine the stability of the N-glycosidic bond [115]. At the same time, the platinum adducts were also found capable of progressively losing its amino ligands. Taken together, these fragmentation channels contributed greatly to the increased complexity of the observed fragmentation spectra.
The putative type 1 and 2 bifunctional products, which share the same elemental composition and thus mass, were analyzed in similar fashion to carry out their structural characterization. The predominant dissociation products obtained by activating these isobaric species were platinated adducts of individual strands, which could not unambiguously discriminate between the type 1 and 2 structures (Figures 9S and 10S of Supporting Information, panels A). Also in this case, the 17RNAc strand displayed a greater incidence of modification due to the presence of multiple G nucleotides. However, it was not possible to ascertain whether any Pt(NH3)2 moiety was bridging contiguous nucleobases on the same strand (indicative of a type 1 precursor), or represented a “dangling” adduct produced by the cleavage of a coordination bond with a nucleobase on the complementary strand (indicative of a type 2 species). At the same time, the typical backbone fragments from the duplex’s fraying ends could not reveal the position of the cross-linked sites (panels B and C of Figure 9S and 10S of Supporting Information). Unfortunately, the conundrum could not be solved by performing the type of MS3 determination described above, which would have involved the activation of individual strands only after they were produced by dissociation of the initial duplex (see for example Figure 11S of Supporting Information). These types of samples highlighted the limitations of top-down approaches in the characterization of putative isobaric crosslinks. In these cases, a preferable solution would consist of bottom-up approaches that employed endonuclease digestion to produce tethered species bridging hydrolytic products from opposing strands.
The platinum compounds were tested also for their ability to induce the effective cross-linking of ribonucleoprotein complexes. In this case, a fixed concentration of NC•SL3 substrate was treated with increasing amounts of individual platinum compounds (see Experimental). Initial analysis by denaturing PAGE revealed that transplatin was significantly more effective than cisplatin at generating the sought-after conjugates. A possible explanation could be found in the propensity of transplatin to form initial “dangling” mono-adducts, which could subsequently react with protein functional groups to complete inter-molecular crosslinks. In contrast, the stereochemistry of cisplatin favors the effective formation of intra-strand crosslinks within the RNA construct, which may compete with inter-molecular bridging. In light of the relatively large size of the observed conjugates, their MS characterization was not pursued by using direct gas-phase activation, but rather by implementing a bottom-up strategy that involved digestion with RNase A and trypsin (see Experimental). Mass mapping revealed the presence of characteristic tethered species, including a prominent M1:K3 + Pt(NH3)2 + G1:C4 product. Full-fledge characterization was accomplished by MS/MS determinations, as described above, which identified K3 and G2 as the cross-linked residues (panel A and B of Figure 12S of Supporting Information). As exemplified by these data, the characteristic isotopic abundances afforded by platinum provided unmistakable patterns that greatly facilitated the mass mapping of digestion mixtures (Figure 13S of Supporting Information).
D). 2-Haloethyl-derivatives.
Named after vescicating reagents developed as chemical warfare agents, nitrogen mustards can be effectively employed as structural probes by virtue of their ability to produce stable conjugates between nucleotides located within striking distance [15,77]. Capitalizing on their cytotoxic effects, several members of this family have been employed in cancer therapy and, thus, are widely available. In addition to mechloretamine (nitrogen mustard, NM), we tested also chlorambucil and carmustine, as well as the less reactive bis(2-chloroethyl)amine and bis(2-iodoethyl)amine, which were purchased/synthesized to test the possibility of increasing the production of diagnostic bifunctional crosslinks. A common feature of these compounds is the propensity of 2-chloroethyl functions to undergo hydrolysis in the aqueous environment and produce corresponding 2-hydroxyethyl groups. Additionally, our ESI-MS analysis revealed that these functional groups are capable of reacting also with ammonia in the ammonium-based medium. These types of side reactions can readily deactivate nitrogen mustards, reduce their overall half-life in solution, and negatively affect the yield of substrate modification. After initial nucleobase alkylation by the first 2-chloroethyl function, these side reactions can effectively compete with other nucleobases for the second 2-chloroethyl, thus reducing the yield of bridging conjugates. For this reason, we tested also the demethylated analog bis(2-chloroethyl)amine, which substantially exceeds the ~6 min half-life reported for NM in aqueous environment [116]. In parallel, we also tested the effects of substituting chlorine with the better leaving group iodine. An unintended effect of solvent reactivity is that, unlike for platinated reagents, ad hoc quenching reactions are not necessary to prevent the formation of artifactual conjugates upon digestion operations.
The reactivity of bis(2-haloethyl) compounds is preferentially directed towards the N7 of guanine, which makes the alkylated purine system into an excellent leaving group. For this reason, the base loss induced by gas-phase activation of alkylated products can introduce abasic lesions in the oligonucleotide sequence, which can still reveal the original position of the modification. In type 2 conjugates, base loss can lead to the formation of a “dangling” type 0 fragment that is still tethered to the cleaved nucleobase, whereas the complementary strand displays the abasic lesion left by the cleaved base. This concept can be illustrated by examining the MS/MS spectrum of the C8:G10+NM+A11:G13 conjugate generated by RNase T1 digestion of SL3 RNA treated with NM (see Experimental). Signals corresponding to nucleobase-tethered and abasic versions of both C8:G10 and A11:G13 products can be readily recognized (Figure 14S of Supporting Information, panel A and B). Upon further activation, both types of products provided characteristic fragments that allowed the immediate identification of the cross-linked positions. The fact that NM performs as a de facto cleavable cross-linker [117–120,42] should facilitate the implementation of consecutive activation steps (e.g., MSn) to resolve possible assignment ambiguities.
A close examination of digestion maps generated during bottom-up analysis revealed that the rather bulky nature of chlorambucil and carmustine could affect the hydrolytic activity of the selected ribonucleases. Similar effects were noted also on conjugated products with multiple crosslinks, even when considerably less bulky reagents, such as NM and transplatin, were employed (see for example the miscleavages produced by RNase T1 digestion of NM-treated SL3 RNA) (Figure 14S of Supporting Information, panel B). In the specific case of RNase T1, possible steric hindrance could be exacerbated by the fact that these putative probes share guanine nucleotides as their preferential targets, which happen to represent also the preferred substrates of this nucleotide-specific ribonuclease. These observations offer a note of caution on the utilization of probing and cleavage reagents with the same residue/nucleotide specificity.
E). Alternative alkylating agents.
Additional agents that are known to modify the nucleophilic positions of nucleic acid components were included in the survey. For instance, epoxyde reagents are characterized by strained 3-member rings that are very susceptible to nucleophilic attack. Their inclusion in the study was prompted by the well-known ability to produce stable adducts with proteins and nucleic acids, which was first recognized in subjects exposed to industrial chemicals [121]. The reactivity of the epoxydic function has been also employed to target specifically adenine loops in G-quadruplex DNA structures [122]. In our hands, however, the representative diglycidyl-1,4-butanediol failed to provide detectable products under the selected experimental conditions (see Experimental). Varying the reagent to substrate ratio up to 100:1 did not produce satisfactory cross-linking yields. In this case, however, the unfavorable outcome could be ascribed to possible competition with water and buffer molecules, which may rapidly reduce the amount of active reagent available in solution [123]. The fact that even the usually abundant type 0 products eluded detection raised questions about the prospects of possible probing applications.
The reactive agent 1,4-butanediol-dimethanesulphonate (busulphan) was included in the study on the basis of earlier reports regarding its ability to produce bridging adducts in DNA substrates [124,125]. This molecule contains two methylsulphonate functions linked by a four-carbon chain, which are capable of reacting with nucleophilic functional groups of nucleic acids [126,127]. Also in this case, the anemic reactivity displayed by this reagent was ascribable to possible competition with nucleophilic species in the selected buffer. The outcome did not improve when the ammonium acetate environment was replaced by bicarbonate, cacodylate, or simple deionized water, or when the reagent to substrate ratio was increased up to 100:1 (see Experimental). The explored conditions failed to produce detectable inter-molecular cross-linking also when the double-stranded RNA and NC•SL3 substrate were tested.
Nitrous acid has been described by some authors as an efficient DNA and protein cross-linker [128], and was therefore included in the study. The reactivity of this agent has been associated with initial nucleobase nitrosation. The ensuing intermediate can subsequently react with amino groups in either proteins or nucleobases [129]. However, an unsatisfactory outcome was observed in the range of conditions explored (see Experimental). Any attempt to increase reactivity by lowering the pH to 4.0 resulted in decreased RNA stability, rather than increased conjugate formation. Similar results were obtained also with double-stranded RNA and NC•SL3 substrates.
Formaldehyde is widely employed in applications requiring the formation of stable protein-nucleic acid crosslink to identify putative regulatory relationships [130–135]. These types of applications, however, rely on immuno-precipitation techniques and sequence amplification to identify the sites of protein interaction onto DNA and RNA substrates [136,137]. Our assays confirmed these capabilities and provided also some notes of caution on their utilization. A practical concern regards the somewhat reversible nature of the alkylation reaction [138], which could lead to possible loss of conjugation, and therefore information, during the enzymatic digestion step included in bottom-up approaches. Another issue concerns the complexity of typical product mixtures, which stems from the intrinsic lack of specificity characteristic of this reagent [133].
Finally, the popular N-hydroxysuccinimide (NHS) ester bis(sulfosuccinimidyl)suberate (BS3), which finds extensive use in proteomics analysis to produce protein cross-linking [139], was tested here to evaluate its possible reactivity toward nucleic acid components. This reagent displayed negligible reactivity toward the nucleic acid models employed in the study, but provided abundant adducts with the protein component of the NC•SL3 substrate.
Conclusions
The purpose of this study was to evaluate the merits of a variety of bifunctional agents as possible cross-linkers for nucleic acids and protein-nucleic acid complexes, with the goals of expanding the toolkit available for the investigation of these types of biopolymers and providing a helpful practical guide to approach these types of applications. We initially focused on classes of reagents with demonstrated reactivity towards nucleic acid functional groups. We explored a broad range of experimental conditions in an effort to recognize the most consequential parameters affecting the production of inter-molecular conjugates, which are pre-eminently desirable for their ability to identify bound components and reveal spatial contiguity between susceptible groups. For this reason, we employed a combination of analytical approaches that are well versed in the detection of these types of products. In particular, denaturing PAGE proved to be a very valuable and inexpensive tool for evaluating the yield of inter-strand and protein-strand conjugation. This technique allowed us to systematically screen selected experimental parameters before performing full-fledged MS characterization, which was reserved only to samples that had already undergone substantial optimization. The latter was then employed to reveal the types of products afforded by the putative cross-linkers, corroborate the identity of conjugated components, and obtain the sequence position of the actual residues involved in bridging reactions. The characterization of possible intermediates and undesirable side-products allowed us to fine-tune the reaction conditions, as well as to identify possible factors that negatively affected the cross-linking reactions. At the same time, the concerted application of these techniques helped us identify possible problems in the analytical workflow, such as the residual reactivity of some mono-functional products, or the base loss upon gas-phase activation, which demand specific precautions to facilitate the detection of sough-after products.
The diverse outcomes produced by the test reagents clearly taught us that a general knowledge of chemical reactivity is not sufficient to inform the practical selection of cross-linking agents, which requires instead a deeper understanding of the effects of experimental conditions, analytical behavior, as well as distribution and structural context of susceptible functional groups. Consistent with these considerations, the reagents examined here did not always meet expectations, but demonstrated instead the considerable influence of the reaction environment. For example, the unsatisfactory outcomes provided by azido and diazirine compounds were attributable to the limited half-lives of their respective carbene and nitrene intermediates in aqueous solution. Similarly, the competition of water and other nucleophiles for the 2-haloethyl functions of nitrogen mustards accounted for the high yields displayed by less desirable mono-functional products. In general, sensitivity to the actual structural context is an excellent quality for structural applications, regardless of yield, but may be detrimental to interactomics work. The different reactivity manifested by psoralens with DNA and RNA substrates was traced to the different steric situation afforded by the helical topologies of these biopolymers. While uridine nucleotides are known to react at least as efficiently as thymidines, the distance between the former in complementary strands is greater in an A-type RNA helix than between the latter in a B-type DNA structure, thus preventing the formation of inter-strand cross-linking. Based on these observations, it is likely that the reported ability of AMT to produce inter-strand conjugation in RNA may have resulted from duplex stabilization induced by mere intercalation, rather than covalent bridging [99].
This survey was conceived to compare the performance of putative reagents and provide a tool to guide their selection. At the same time, the extensive information on their performance, possible factors affecting the outcome, and preferred experimental conditions will represent a valuable starting point for approaching their application to nucleic acid substrates. While proteins contain very diverse functional groups that can serve as potential targets, the choice is more limited for these types of biopolymers. Although several mono-functional reagents have been developed for nucleotide-specific footprinting applications [41 and references therein] only a handful of bifunctional cross-linkers are capable of reacting with such functional groups [53]. Developing new chemistries for phosphate groups in both DNA and RNA, or hydroxyl functions in RNA, could potentially lead to cross-linkers capable of reacting with every possible nucleotide in a given substrate, thus greatly increasing the information content provided by their application. This survey clearly indicated that the quest for a universal nucleic acid cross-linker, which may afford excellent performance with all types of nucleic acid substrates, remains very much open and will require extending the exploration beyond traditional chemistries used to chemically modify these essential biopolymers.
Supplementary Material
The report assessed a large number of putative bifunctional crosslinkers as possible tools for the structural elucidation and interactomics analysis of nucleic acids and their assemblies. The concerted utilization of denaturing PAGE and in-source activation enabled the effective assessment of inter-molecular, type 2 conjugates, which represent the most informative types of products. Consecutive steps of gas-phase activation enabled the localization of the sequence position of the crosslinked residues, which can be employed as spatial constraints for model-building operations.
Acknowledgements
Funding was provided by the National Institute of Health with award GM064328.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Giegé R, A historical perspective on protein crystallization from 1840 to the present day, FEBS Journal. 280 (2013) 6456–6497. doi: 10.1111/febs.12580. [DOI] [PubMed] [Google Scholar]
- [2].Back JW, de Jong L, Muijsers AO, de Koster CG, Chemical Cross-linking and Mass Spectrometry for Protein Structural Modeling, Journal of Molecular Biology. 331 (2003) 303–313. doi: 10.1016/S0022-2836(03)00721-6. [DOI] [PubMed] [Google Scholar]
- [3].Leitner A, Walzthoeni T, Kahraman A, Herzog F, Rinner O, Beck M, Aebersold R, Probing Native Protein Structures by Chemical Cross-linking, Mass Spectrometry, and Bioinformatics, Molecular & Cellular Proteomics. 9 (2010) 1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rappsilber J, The beginning of a beautiful friendship: Cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes, Journal of Structural Biology. 173 (2011) 530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Walzthoeni T, Leitner A, Stengel F, Aebersold R, Mass spectrometry supported determination of protein complex structure, Current Opinion in Structural Biology. 23 (2013) 252–260. doi: 10.1016/j.sbi.2013.02.008. [DOI] [PubMed] [Google Scholar]
- [6].Sinz A, The advancement of chemical cross-linking and mass spectrometry for structural proteomics: from single proteins to protein interaction networks, Expert Rev Proteomics. 11 (2014) 733–743. doi: 10.1586/14789450.2014.960852. [DOI] [PubMed] [Google Scholar]
- [7].Corgiat BA, Nordman JC, Kabbani N, Chemical crosslinkers enhance detection of receptor interactomes, Frontiers in Pharmacology. 4 (2014). doi: 10.3389/fphar.2013.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen F, Gülbakan B, Weidmann S, Fagerer SR, Ibáñez AJ, Zenobi R, Applying mass spectrometry to study non-covalent biomolecule complexes, Mass Spectrom Rev. 35 (2016) 48–70. doi: 10.1002/mas.21462. [DOI] [PubMed] [Google Scholar]
- [9].Smits AH, Vermeulen M, Characterizing Protein-Protein Interactions Using Mass Spectrometry: Challenges and Opportunities, Trends Biotechnol. (2016). doi: 10.1016/j.tibtech.2016.02.014. [DOI] [PubMed] [Google Scholar]
- [10].Yang B, Wu Y-J, Zhu M, Fan S-B, Lin J, Zhang K, Li S, Chi H, Li Y-X, Chen H-F, Luo S-K, Ding Y-H, Wang L-H, Hao Z, Xiu L-Y, Chen S, Ye K, He S-M, Dong M-Q, Identification of cross-linked peptides from complex samples, Nature Methods. 9 (2012) 904–906. doi: 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- [11].Coffman K, Yang B, Lu J, Tetlow AL, Pelliccio E, Lu S, Guo D-C, Tang C, Dong M-Q, Tamanoi F, Characterization of the Raptor/4E-BP1 Interaction by Chemical Cross-linking Coupled with Mass Spectrometry Analysis, Journal of Biological Chemistry. 289 (2014) 4723–4734. doi: 10.1074/jbc.M113.482067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Young M, High M throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry, Proc. Natl Acad. Sci. USA. 97 (2000) 5802–5806. doi: 10.1073/pnas.090099097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kluger R, Alagic A, Chemical cross-linking and protein–protein interactions—a review with illustrative protocols, Bioorganic Chemistry. 32 (2004) 451–472. doi: 10.1016/j.bioorg.2004.08.002. [DOI] [PubMed] [Google Scholar]
- [14].Sinz A, Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein–protein interactions, Mass Spectrometry Reviews. 25 (2006) 663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- [15].Yu ET, Hawkins A, Eaton J, Fabris D, MS3D structural elucidation of the HIV-1 packaging signal, Proceedings of the National Academy of Sciences. 105 (2008) 12248–12253. doi: 10.1073/pnas.0800509105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang H, Tang X, Munske GR, Tolic N, Anderson GA, Bruce JE, Identification of protein-protein interactions and topologies in living cells with chemical cross-linking and mass spectrometry, Mol. Cell Proteomics. 8 (2009) 409–420. doi: 10.1074/mcp.M800232-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J, Practically useful: what the Rosetta protein modeling suite can do for you, Biochemistry. 49 (2010) 2987–2998. doi: 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, Leitner A, Beck M, Hartl F-U, Ban N, Malmström L, Aebersold R, Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry, Science. 337 (2012) 1348–1352. doi: 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- [19].Kahraman A, Herzog F, Leitner A, Rosenberger G, Aebersold R, Malmstrom L, Cross-link guided molecular modeling with ROSETTA, PLoS ONE. 8 (2013) e73411. doi: 10.1371/journal.pone.0073411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kosinski J, von Appen A, Ori A, Karius K, Müller CW, Beck M, Xlink Analyzer: software for analysis and visualization of cross-linking data in the context of three-dimensional structures, J. Struct. Biol 189 (2015) 177–183. doi: 10.1016/j.jsb.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Leitner A, Faini M, Stengel F, Aebersold R, Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines, Trends Biochem. Sci 41 (2016) 20–32. doi: 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- [22].Lasker K, Förster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W, Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach, Proc. Natl. Acad. Sci. U.S.A 109 (2012) 1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ward AB, Sali A, Wilson IA, Integrative Structural Biology, Science. 339 (2013) 913–915. doi: 10.1126/science.1228565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Faini M, Stengel F, Aebersold R, The Evolving Contribution of Mass Spectrometry to Integrative Structural Biology, Journal of The American Society for Mass Spectrometry. 27 (2016) 966–974. doi: 10.1007/s13361-016-1382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xue J, Manigrasso M, Scalabrin M, Rai V, Reverdatto S, Burz DS, Fabris D, Schmidt AM, Shekhtman A, Change in the Molecular Dimension of a RAGE-Ligand Complex Triggers RAGE Signaling, Structure. 24 (2016) 1509–1522. doi: 10.1016/j.str.2016.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fields S, Song O, A novel genetic system to detect protein-protein interactions, Nature. 340 (1989) 245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- [27].Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y, A comprehensive two-hybrid analysis to explore the yeast protein interactome, Proc. Natl. Acad. Sci. U.S.A 98 (2001) 4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC, Green fluorescent protein as a marker for gene expression, Science. 263 (1994) 802–805. [DOI] [PubMed] [Google Scholar]
- [29].Soboleski MR, Oaks J, Halford WP, Green fluorescent protein is a quantitative reporter of gene expression in individual eukaryotic cells, FASEB J. 19 (2005) 440–442. doi: 10.1096/fj.04-3180fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG, Life with 6000 genes, Science. 274 (1996) 546, 563–567. [DOI] [PubMed] [Google Scholar]
- [31].Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwin B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM, A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae, Nature. 403 (2000) 623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- [32].Rual J-F, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, Klitgord N, Simon C, Boxem M, Milstein S, Rosenberg J, Goldberg DS, Zhang LV, Wong SL, Franklin G, Li S, Albala JS, Lim J, Fraughton C, Llamosas E, Cevik S, Bex C, Lamesch P, Sikorski RS, Vandenhaute J, Zoghbi HY, Smolyar A, Bosak S, Sequerra R, Doucette-Stamm L, Cusick ME, Hill DE, Roth FP, Vidal M, Towards a proteome-scale map of the human protein-protein interaction network, Nature. 437 (2005) 1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- [33].Bruce JE, In vivo protein complex topologies: Sights through a cross-linking lens, PROTEOMICS. 12 (2012) 1565–1575. doi: 10.1002/pmic.201100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu F, Rijkers DTS, Post H, Heck AJR, Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry, Nature Methods. 12 (2015) 1179–1184. doi: 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- [35].Schmidt C, Kramer K, Urlaub H, Investigation of protein—RNA interactions by mass spectrometry—Techniques and applications, Journal of Proteomics. 75 (2012) 3478–3494. doi: 10.1016/j.jprot.2012.04.030. [DOI] [PubMed] [Google Scholar]
- [36].Kramer K, Sachsenberg T, Beckmann BM, Qamar S, Boon K-L, Hentze MW, Kohlbacher O, Urlaub H, Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins, Nat. Methods. 11 (2014) 1064–1070. doi: 10.1038/nmeth.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Müller DR, Schindler P, Towbin H, Wirth U, Voshol H, Hoving S, Steinmetz MO, Isotope-tagged cross-linking reagents. A new tool in mass spectrometric protein interaction analysis, Anal. Chem 73 (2001) 1927–1934. [DOI] [PubMed] [Google Scholar]
- [38].Pearson KM, Pannell LK, Fales HM, Intramolecular Cross-Linking Experiments on Cytochrome C and Ribonuclease a Using an Isotope Multiplet Method, Rapid Commun. Mass Spectrom 16 (2002) 149–159. [DOI] [PubMed] [Google Scholar]
- [39].Schmidt A, Kalkhof S, Ihling C, Cooper DMF, Sinz A, Mapping protein interfaces by chemical cross-linking and Fourier transform ion cyclotron resonance mass spectrometry: application to a calmodulin / adenylyl cyclase 8 peptide complex, Eur J Mass Spectrom (Chichester). 11 (2005) 525–534. doi: 10.1255/ejms.748. [DOI] [PubMed] [Google Scholar]
- [40].Trester-Zedlitz M, Kamada K, Burley SK, Fenyö D, Chait BT, Muir TW, A modular cross-linking approach for exploring protein interactions., J. Am. Chem. Soc (2003) 2416–2425. [DOI] [PubMed] [Google Scholar]
- [41].Fujii N, Jacobsen RB, Wood NL, Schoeniger JS, Guy RK, A novel protein crosslinking reagent for the determination of moderate resolution protein structures by mass spectrometry (MS3-D), Bioorg. Med. Chem. Lett 14 (2004) 427–429. [DOI] [PubMed] [Google Scholar]
- [42].Petrotchenko EV, Serpa JJ, Borchers CH, An Isotopically Coded CID-cleavable Biotinylated Crosslinker for Structural Proteomics, Molecular & Cellular Proteomics. 10 (2011) M110.001420. doi: 10.1074/mcp.M110.001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fabris D, Yu ET, Elucidating the higher-order structure of biopolymers by structural probing and mass spectrometry: MS3D, Journal of Mass Spectrometry. 45 (2010) 841–860. doi: 10.1002/jms.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Arora B, Tandon R, Attri P, Bhatia R, Chemical Crosslinking: Role in Protein and Peptide Science, Curr. Protein Pept. Sci 18 (2017) 946–955. doi: 10.2174/1389203717666160724202806. [DOI] [PubMed] [Google Scholar]
- [45].Schilling B, Row RH, Gibson BW, Guo X, Young MM, MS2Assign, automated assignment and nomenclature of tandem mass spectra of chemically crosslinked peptides, J Am Soc Mass Spectrom. 14 (2003)834–50. [DOI] [PubMed] [Google Scholar]
- [46].Bragg PD, Hou C, Subunit composition, function, and spatial arrangement in the Ca2+- and Mg2+-activated adenosine triphosphatases of Escherichia coli and Salmonella typhimurium, Archives of Biochemistry and Biophysics. 167 (1975) 311–321. doi: 10.1016/0003-9861(75)90467-1. [DOI] [PubMed] [Google Scholar]
- [47].Lomant AJ, Fairbanks G, Chemical probes of extended biological structures: synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate), J. Mol. Biol 104 (1976) 243–261. [DOI] [PubMed] [Google Scholar]
- [48].Hermanson GT, Bioconjugate techniques, 3. ed, Acad. Press, Amsterdam, 2013. [Google Scholar]
- [49].Gilchrist TL, Rees CW, Carbenes, nitrenes and arynes, Nelson, London, 1969. [Google Scholar]
- [50].McKenzie JA, Raison RL, Rivett DE, Development of a bifunctional crosslinking agent with potential for the preparation of immunotoxins, Journal of Protein Chemistry. 7 (1988) 581–592. doi: 10.1007/BF01024876. [DOI] [PubMed] [Google Scholar]
- [51].Chen Z, Stokes DL, Rice WJ, Jones LR, Spatial and Dynamic Interactions between Phospholamban and the Canine Cardiac Ca 2+ Pump Revealed with Use of Heterobifunctional Cross-linking Agents, Journal of Biological Chemistry. 278 (2003) 48348–48356. doi: 10.1074/jbc.M309545200. [DOI] [PubMed] [Google Scholar]
- [52].Novak P, Kruppa GH, Young MM, Schoeniger J, A top down method for the determination of residue specific solvent accessibility in proteins., J. Mass Spectrom 39 (2004) 322–8. [DOI] [PubMed] [Google Scholar]
- [53].Zhang Q, Crosland E, Fabris D, Nested Arg-specific bifunctional crosslinkers for MS-based structural analysis of proteins and protein assemblies., Anal. Chim. Acta 627 (2008) 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hofmann T, Fischer AW, Meiler J, Kalkhof S, Protein structure prediction guided by crosslinking restraints--A systematic evaluation of the impact of the crosslinking spacer length, Methods. 89 (2015) 79–90. doi: 10.1016/j.ymeth.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zybailov BL, Glazko GV, Jaiswal M, Raney KD, Large Scale Chemical Cross-linking Mass Spectrometry Perspectives, J Proteomics Bioinform. 6 (2013) 001. doi: 10.4172/jpb.S2-001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Agou F, Ye F, Véron M, In vivo protein cross-linking, Methods Mol. Biol 261 (2004) 427–442. doi: 10.1385/1-59259-762-9:427. [DOI] [PubMed] [Google Scholar]
- [57].Petrotchenko EV, Borchers CH, Crosslinking combined with mass spectrometry for structural proteomics, Mass Spectrom Rev. 29 (2010) 862–876. doi: 10.1002/mas.20293. [DOI] [PubMed] [Google Scholar]
- [58].Pappalardo L, Kerwood DJ, Pelczer I, Borer PN, Three-dimensional folding of an RNA hairpin required for packaging HIV-1, J. Mol. Biol 282 (1998) 801–18. [DOI] [PubMed] [Google Scholar]
- [59].Morellet N, Jullian N, De Roquigny H, Maigret B, Darlix JL, Roques BP, Determination of the structure of the nucleocapsid protein NCp7 from the human immunodeficiency virus type 1 by 1H NMR, Embo J. 11 (1992) 3059–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hagan N, Fabris D, Direct mass spectrometric determination of the stoichiometry and binding affinity of the complexes between nucleocapsid protein and RNA stem-loop hairpins of the HIV-1 Psi-recognition element, Biochemistry. 42 (2003) 10736–10745. doi: 10.1021/bi0348922. [DOI] [PubMed] [Google Scholar]
- [61].Woolley P, Dohrmann S, Crosslinking of double-helical nucleic acids with a photoreactive analog of ethidium, Biochemistry. 22 (1983) 3226–3231. doi: 10.1021/bi00282a029. [DOI] [PubMed] [Google Scholar]
- [62].Graves DE, Yielding LW, Watkins CL, Yielding KL, Synthesis, separation and characterization of the mono- and diazide analogs of ethidium bromide, Biochim. Biophys. Acta 479 (1977) 98–104. [DOI] [PubMed] [Google Scholar]
- [63].Finkelstein H, Darstellung organischer Jodide aus den entsprechenden Bromiden und Chloriden, Berichte der deutschen chemischen Gesellschaft. 43 (1910) 1528–1532. doi: 10.1002/cber.19100430257. [DOI] [Google Scholar]
- [64].Richter S, Fabris D, Binaschi M, Gatto B, Capranico G, Palumbo M, Effects of common buffer systems on drug activity: the case of clerocidin., Chem. Res. Toxicol 17 (2004) 492–501. [DOI] [PubMed] [Google Scholar]
- [65].Richter SN, Fabris D, Moro S, Palumbo M, Dissecting reactivity of clerocidin toward common buffer systems by means of selected drug analogues., Chem Res Toxicol. 18 (2005) 35–40. [DOI] [PubMed] [Google Scholar]
- [66].Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW, Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry., J. Am. Chem. Soc 121 (1999) 806–812. [Google Scholar]
- [67].Kellersberger KA, Yu E, Kruppa GH, Young MM, Fabris D, Top-down characterization of nucleic acids modified by structural probes using high-resolution tandem mass spectrometry and automated data interpretation, Anal. Chem 76 (2004) 2438–2445. doi: 10.1021/ac0355045. [DOI] [PubMed] [Google Scholar]
- [68].Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR, Direct analysis of protein complexes using mass spectrometry, Nature Biotechnology. 17 (1999) 676–82. [DOI] [PubMed] [Google Scholar]
- [69].Yu E, Fabris D, Direct probing of RNA structures and RNA-protein interactions in the HIV-1 packaging signal by chemical modification and electrospray ionization Fourier transform mass spectrometry., J. Mol. Biol 330 (2003) 211–223. [DOI] [PubMed] [Google Scholar]
- [70].Fabris D, Turner KB, Hagan NA, Electrospray Ionization-Mass Spectrometry for the Investigation of Protein-Nucleic Acids Interactions, in: Mass Spectrometry of Nucleosides and Nucleic Acids, Banoub J and Limbach P eds., CRC Press, Taylor and Francis Group, LLC, London, U.K., 2010: pp. 303–327. [Google Scholar]
- [71].Fabris D, MS analysis of nucleic acids in the post-genomic era, Analytical Chemistry. 83 (2011) 5810–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Fabris D, Turner KB, Hagan NA, Electrospray Ionization - Mass Spectrometry for the Investigation of Protein-Nucleic Acid Interactions, in: Mass Spectrometry of Nucleosides and Nucleic Acids, J. H. Banoub and Limbach PA, Ed.s, CRC Press, 2009: pp. 303–327. [Google Scholar]
- [73].Rozenski J: Mongo Oligo Mass Calculator, v2.06, (1999).
- [74].Wilkins MR, Lindskog I, Gasteiger E, Bairoch A, Sanchez JC, Hochstrasser DF, Appel RD, Detailed peptide characterization using PEPTIDEMASS--a World-Wide-Web-accessible tool, Electrophoresis. 18 (1997) 403–408. doi: 10.1002/elps.1150180314. [DOI] [PubMed] [Google Scholar]
- [75].Kitamura N, Kohtani S, Nakagaki R, Molecular aspects of furocoumarin reactions: Photophysics, photochemistry, photobiology, and structural analysis, Journal of Photochemistry and Photobiology C: Photochemistry Reviews. 6 (2005) 168–185. doi: 10.1016/j.jphotochemrev.2005.08.002. [DOI] [Google Scholar]
- [76].De Guzman RN, Wu ZR, Stalling CC, Pappalardo L, Borer PN, Summers MF, Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element, Science. 279 (1998) 384–388. [DOI] [PubMed] [Google Scholar]
- [77].Zhang Q, Yu ET, Kellersberger KA, Crosland E, Fabris D, Toward building a database of bifunctional probes for the MS3D investigation of nucleic acids structures, J. Am. Soc. Mass Spectrom 17 (2006) 1570–1581. doi: 10.1016/j.jasms.2006.06.002. [DOI] [PubMed] [Google Scholar]
- [78].Thomas CB, Kohn KW, Bonner WM, Characterization of DNA-protein cross-links formed by treatment of L1210 cells and nuclei with bis(2-chloroethyl)methylamine (nitrogen mustard), Biochemistry. 17 (1978)3954–3958. [DOI] [PubMed] [Google Scholar]
- [79].Loeber R, Michaelson E, Fang Q, Campbell C, Pegg AE, Tretyakova N, Cross-linking of the DNA repair protein Omicron6-alkylguanine DNA alkyltransferase to DNA in the presence of antitumor nitrogen mustards, Chem. Res. Toxicol 21 (2008) 787–795. doi: 10.1021/tx7004508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Cimino GD, Gamper HB, Isaacs ST, Hearst JE, Psoralens as photoactive probes of nucleic acid structure and function: organic chemistry, photochemistry, and biochemistry, Annu. Rev. Biochem 54 (1985) 1151–1193. doi: 10.1146/annurev.bi.54.070185.005443. [DOI] [PubMed] [Google Scholar]
- [81].Scalabrin M, Siu Y, Asare-Okai PN, Fabris D, Structure-Specific Ribonucleases for MS-Based Elucidation of Higher-Order RNA Structure, J. Am. Soc. Mass Spectrom (2014). doi: 10.1007/s13361-014-0911-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Smith SI, Brodbelt JS, Rapid characterization of cross-links, mono-adducts, and non-covalent binding of psoralens to deoxyoligonucleotides by LC-UV/ESI-MS and IRMPD mass spectrometry, Analyst. 135 (2010) 943–952. doi: 10.1039/b924023c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Biemann K, Papayannopoulos IA, Amino acid sequencing of proteins, Accounts of Chemical Research. 27 (1994) 370–378. doi: 10.1021/ar00047a008. [DOI] [Google Scholar]
- [84].Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW, Top Down versus Bottom Up Protein Characterization by Tandem High-Resolution Mass Spectrometry, Journal of the American Chemical Society. 121 (1999) 806–812. doi: 10.1021/ja973655h. [DOI] [Google Scholar]
- [85].Schmidt C, Kramer K, Urlaub H, Investigation of protein–RNA interactions by mass spectrometry—Techniques and applications, Journal of Proteomics. 75 (2012) 3478–3494. doi: 10.1016/j.jprot.2012.04.030. [DOI] [PubMed] [Google Scholar]
- [86].Mcluckey SA, Berkel GJ, Glish GL, Tandem Mass Spectrometry of Small, Multiply Charged Oligonucleotides, Journal of the American Society for Mass Spectrometry. 3 (1992) 60–70. doi: 10.1016/1044-0305(92)85019-G. [DOI] [PubMed] [Google Scholar]
- [87].Wan KX, Gross J, Hillenkamp F, Gross ML, Fragmentation mechanisms of oligodeoxynucleotides studied by H/D exchange and electrospray ionization tandem mass spectrometry, Journal of the American Society for Mass Spectrometry. 12 (2001) 193–205. doi: 10.1016/S1044-0305(00)00208-7. [DOI] [PubMed] [Google Scholar]
- [88].Kirpekar F, Krogh TN, RNA fragmentation studied in a matrix-assisted laser desorption/ionisation tandem quadrupole/orthogonal time-of-flight mass spectrometer, Rapid Communications in Mass Spectrometry. 15 (2001) 8–14. doi:. [DOI] [PubMed] [Google Scholar]
- [89].Hathout Y, Fabris D, Han MS, Sowder RC, Henderson LE, Fenselau C, Characterization of intermediates in the oxidation of zinc fingers in human immunodeficiency virus type 1 nucleocapsid protein P7, Drug Metab. Dispos 24 (1996) 1395–1400. [PubMed] [Google Scholar]
- [90].Antoine MD, Fabris D, Fenselau C, Covalent sequestration of the nitrogen mustard mechloretamine by metallothionein., Drug Metab. Dispos 26 (1998) 921–926. [PubMed] [Google Scholar]
- [91].Fabris D, Hathout Y, Fenselau C, Investigation of zinc chelation in zinc-finger arrays by electrospray mass spectrometry, Inorg. Chem 38 (1999) 1322–1325. [DOI] [PubMed] [Google Scholar]
- [92].Leitner A, Reischl R, Walzthoeni T, Herzog F, Bohn S, Förster F, Aebersold R, Expanding the Chemical Cross-Linking Toolbox by the Use of Multiple Proteases and Enrichment by Size Exclusion Chromatography, Mol Cell Proteomics. 11 (2012). doi: 10.1074/mcp.M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kang S, Mou L, Lanman J, Velu S, Brouillette WJ, Prevelige PE, Synthesis of biotin-tagged chemical cross-linkers and their applications for mass spectrometry, Rapid Communications in Mass Spectrometry. 23 (2009) 1719–1726. doi: 10.1002/rcm.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Richter FM, Hsiao H-H, Plessmann U, Urlaub H, Enrichment of protein-RNA crosslinks from crude UV-irradiated mixtures for MS analysis by on-line chromatography using titanium dioxide columns, Biopolymers. 91 (2009) 297–309. doi: 10.1002/bip.21139. [DOI] [PubMed] [Google Scholar]
- [95].Demaret JP, Brunie S, Ballini JP, Vigny P, Geometry of intercalation of psoralens in DNA approached by molecular mechanics, Photochem. Photobiol 50 (1989) 7–21. [DOI] [PubMed] [Google Scholar]
- [96].Rabin D, Crothers DM, Analysis of RNA secondary structure by photochemical reversal of psoralen crosslinks, Nucleic Acids Res. 7 (1979) 689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L, Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity, Pharmacol. Rev 56 (2004) 185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- [98].Scalabrin M, Quintieri L, Palumbo M, Riccardi Sirtori F, Gatto B, Virtual Cross-Linking of the Active Nemorubicin Metabolite PNU-159682 to Double-Stranded DNA, Chem. Res. Toxicol 30 (2017) 614–624. doi: 10.1021/acs.chemrestox.6b00362. [DOI] [PubMed] [Google Scholar]
- [99].Isaacs ST, Shen CK, Hearst JE, Rapoport H, Synthesis and characterization of new psoralen derivatives with superior photoreactivity with DNA and RNA, Biochemistry. 16 (1977) 1058–1064. [DOI] [PubMed] [Google Scholar]
- [100].Fisher RJ, Fivash MJ, Stephen AG, Hagan NA, Shenoy SR, Medaglia MV, Smith LR, Worthy KM, Simpson JT, Shoemaker R, McNitt KL, Johnson DG, Hixson CV, Gorelick RJ, Fabris D, Henderson LE, Rein A, Complex interactions of HIV-1 nucleocapsid protein with oligonucleotides, Nucleic Acids Res. 34 (2006) 472–484. doi: 10.1093/nar/gkj442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Chen JL, Nolan JM, Harris ME, Pace NR, Comparative photocross-linking analysis of the tertiary structures of Escherichia coli and Bacillus subtilis RNase P RNAs, EMBO J. 17 (1998) 1515–1525. doi: 10.1093/emboj/17.5.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Buchmueller KL, Hill BT, Platz MS, Weeks KM, RNA-tethered phenyl azide photocrosslinking via a short-lived indiscriminant electrophile, J. Am. Chem. Soc 125 (2003) 10850–10861. doi: 10.1021/ja035743+. [DOI] [PubMed] [Google Scholar]
- [103].Graves DE, Watkins CL, Yielding LW, Ethidium bromide and its photoreactive analogues: spectroscopic analysis of deoxyribonucleic acid binding properties, Biochemistry. 20 (1981) 1887–1892. [DOI] [PubMed] [Google Scholar]
- [104].Cohen SM, Lippard SJ, Cisplatin: from DNA damage to cancer chemotherapy, Prog. Nucleic Acid Res. Mol. Biol 67 (2001) 93–130. [DOI] [PubMed] [Google Scholar]
- [105].Pinto AL, Lippard SJ, Binding of the antitumor drug cis-diamminedichloroplatinum(II) (cisplatin) to DNA, Biochim. Biophys. Acta 780 (1985) 167–180. [DOI] [PubMed] [Google Scholar]
- [106].Reedijk J, Lohman PH, Cisplatin: synthesis, antitumour activity and mechanism of action, Pharm Weekbl Sci. 7 (1985) 173–180. [DOI] [PubMed] [Google Scholar]
- [107].Sherman SE, Lippard SJ, Structural aspects of platinum anticancer drug interactions with DNA, Chemical Reviews. 87 (1987) 1153–1181. doi: 10.1021/cr00081a013. [DOI] [Google Scholar]
- [108].Fichtinger-Schepman AMJ, Van der Veer JL, Den Hartog JHJ, Lohman PHM, Reedijk J, Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation, Biochemistry. 24 (1985) 707–713. doi: 10.1021/bi00324a025. [DOI] [PubMed] [Google Scholar]
- [109].Takahara PM, Rosenzweig AC, Frederick CA, Lippard SJ, Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin, Nature. 377 (1995) 649–652. doi: 10.1038/377649a0. [DOI] [PubMed] [Google Scholar]
- [110].Coste F, Malinge J-M, Serre L, Leng M, Zelwer C, Shepard W, Roth M, Crystal structure of a double-stranded DNA containing a cisplatin interstrand cross-link at 1.63 A resolution: Hydration at the platinated site, Nucleic Acids Research. 27 (1999) 1837–1846. doi: 10.1093/nar/27.8.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Papsai P, Aldag J, Persson T, Elmroth SKC, Kinetic preference for interaction of cisplatin with the G–C-rich wobble basepair region in both tRNA Ala and Mh Ala, Dalton Trans. (2006) 3515–3517. doi: 10.1039/B603833F. [DOI] [PubMed] [Google Scholar]
- [112].Jamieson ER, Lippard SJ, Structure, Recognition, and Processing of Cisplatin-DNA Adducts, Chem. Rev 99 (1999) 2467–2498. [DOI] [PubMed] [Google Scholar]
- [113].Fabris D, Kellersberger KA, Wilhide JA, Higher-order structure of nucleic acids in the gas phase: Top-down analysis of base-pairing interactions, International Journal of Mass Spectrometry. 312 (2012) 155–162. doi: 10.1016/j.ijms.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Gabelica V, Pauw E, Comparison of the collision-induced dissociation of duplex DNA at different collision regimes: Evidence for a multistep dissociation mechanism, Journal of the American Society for Mass Spectrometry. 13 (2002) 91–98. doi: 10.1016/S1044-0305(01)00335-X. [DOI] [PubMed] [Google Scholar]
- [115].Baik M-H, Friesner RA, Lippard SJ, Theoretical Study on the Stability of N-Glycosyl Bonds: Why Does N7-Platination Not Promote Depurination?, J. Am. Chem. Soc 124 (2002) 4495–4503. [DOI] [PubMed] [Google Scholar]
- [116].Gamcsik MP, Hamill TG, Colvin M, NMR studies of the conjugation of mechlorethamine with glutathione, J. Med. Chem 33 (1990) 1009–1014. [DOI] [PubMed] [Google Scholar]
- [117].Bennett KL, Kussmann M, Björk P, Godzwon M, Mikkelsen M, Sørensen P, Roepstorff P, Chemical cross-linking with thiol-cleavable reagents combined with differential mass spectrometric peptide mapping--a novel approach to assess intermolecular protein contacts, Protein Sci. 9 (2000) 1503–1518. doi: 10.1110/ps.9.8.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Soderblom EJ, Goshe MB, Collision-Induced Dissociative Chemical Cross-Linking Reagents and Methodology: Applications to Protein Structural Characterization Using Tandem Mass Spectrometry Analysis, Analytical Chemistry. 78 (2006) 8059–8068. doi: 10.1021/ac0613840. [DOI] [PubMed] [Google Scholar]
- [119].Müller MQ, Dreiocker F, Ihling CH, Schäfer M, Sinz A, Cleavable Cross-Linker for Protein Structure Analysis: Reliable Identification of Cross-Linking Products by Tandem MS, Analytical Chemistry. 82 (2010) 6958–6968. doi: 10.1021/ac101241t. [DOI] [PubMed] [Google Scholar]
- [120].Petrotchenko EV, Borchers CH, ICC-CLASS: isotopically-coded cleavable crosslinking analysis software suite, BMC Bioinformatics. 11 (2010) 64. doi: 10.1186/1471-2105-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Kotin P, Falk HL, Organic peroxides, hydrogen peroxide, epoxides, and neoplasia, Radiat. Res. Suppl 3 (1963)193–211. [PubMed] [Google Scholar]
- [122].Doria F, Nadai M, Folini M, Scalabrin M, Germani L, Sattin G, Mella M, Palumbo M, Zaffaroni N, Fabris D, Freccero M, Richter SN, Targeting loop adenines in G-quadruplex by a selective oxirane, Chemistry. 19 (2013) 78–81. doi: 10.1002/chem.201203097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Richter S, Fabris D, Binaschi M, Gatto B, Capranico G, Palumbo M, Effects of common buffer systems on drug activity: the case of clerocidin, Chem. Res. Toxicol 17 (2004) 492–501. doi: 10.1021/tx034210b. [DOI] [PubMed] [Google Scholar]
- [124].Tong WP, Ludlum DB, Crosslinking of DNA by busulfan Formation of diguanyl derivatives, Biochimica et Biophysica Acta (BBA) - Nucleic Acids and Protein Synthesis. 608 (1980) 174–181. doi: 10.1016/0005-2787(80)90145-8. [DOI] [PubMed] [Google Scholar]
- [125].Iwamoto T, Hiraku Y, Oikawa S, Mizutani H, Kojima M, Kawanishi S, DNA intrastrand cross-link at the 5′-GA-3′ sequence formed by busulfan and its role in the cytotoxic effect, Cancer Sci. 95 (2004) 454–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Beranek DT, Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents, Mutat. Res 231 (1990) 11–30. [DOI] [PubMed] [Google Scholar]
- [127].Wyatt MD, Pittman DL, Methylating Agents and DNA Repair Responses: Methylated Bases and Sources of Strand Breaks, Chemical Research in Toxicology. 19 (2006) 1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Suzuki T, Yamada M, Nakamura T, Ide H, Kanaori K, Tajima K, Morii T, Makino K, Products of the reaction between a diazoate derivative of 2′-deoxycytidine and L-lysine and its implication for DNA-nucleoprotein cross-linking by NO or HNO(2), Chem. Res. Toxicol 13 (2000) 1223–1227. [DOI] [PubMed] [Google Scholar]
- [129].Nakano T, Terato H, Asagoshi K, Masaoka A, Mukuta M, Ohyama Y, Suzuki T, Makino K, Ide H, DNA-protein cross-link formation mediated by oxanine. A novel genotoxic mechanism of nitric oxide-induced DNA damage, J. Biol. Chem 278 (2003) 25264–25272. doi: 10.1074/jbc.M212847200. [DOI] [PubMed] [Google Scholar]
- [130].Solomon MJ, Varshavsky A, Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures, Proc. Natl. Acad. Sci. U.S.A 82 (1985) 6470–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Gilmour DS, Lis JT, In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster, Mol. Cell. Biol 5 (1985) 2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Solomon MJ, Larsen PL, Varshavsky A, Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene, Cell. 53 (1988) 937–947. [DOI] [PubMed] [Google Scholar]
- [133].Sutherland BW, Toews J, Kast J, Utility of formaldehyde cross-linking and mass spectrometry in the study of protein–protein interactions, Journal of Mass Spectrometry. 43 (2008) 699–715. doi: 10.1002/jms.1415. [DOI] [PubMed] [Google Scholar]
- [134].Klockenbusch C, O’Hara JE, Kast J, Advancing formaldehyde cross-linking towards quantitative proteomic applications, Anal Bioanal Chem. 404 (2012) 1057–1067. doi: 10.1007/s00216-012-6065-9. [DOI] [PubMed] [Google Scholar]
- [135].Hoffman EA, Frey BL, Smith LM, Auble DT, Formaldehyde crosslinking: a tool for the study of chromatin complexes, J. Biol. Chem 290 (2015) 26404–26411. doi: 10.1074/jbc.R115.651679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hendrickson DG, Kelley DR, Tenen D, Bernstein B, Rinn JL, Widespread RNA binding by chromatin-associated proteins, Genome Biology. 17 (2016). doi: 10.1186/s13059-016-0878-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Mundade R, Ozer HG, Wei H, Prabhu L, Lu T, Role of ChIP-seq in the discovery of transcription factor binding sites, differential gene regulation mechanism, epigenetic marks and beyond, Cell Cycle. 13 (2014) 2847–2852. doi: 10.4161/15384101.2014.949201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Kennedy-Darling J, Smith LM, Measuring the Formaldehyde Protein–DNA Cross-Link Reversal Rate, Analytical Chemistry. 86 (2014) 5678–5681. doi: 10.1021/ac501354y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Staros JV, N-hydroxysulfosuccinimide active esters: bis(N-hydroxysulfosuccinimide) esters of two dicarboxylic acids are hydrophilic, membrane-impermeant, protein cross-linkers, Biochemistry. 21 (1982) 3950–3955. doi: 10.1021/bi00260a008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.