

Opto-PROTAC adds a light-inducible switch on PROTAC, enabling optical control for targeted degradation of proteins of interest.
Abstract
By hijacking endogenous E3 ligase to degrade protein targets via the ubiquitin-proteasome system, PROTACs (PRoteolysis TArgeting Chimeras) provide a new strategy to inhibit protein targets that were regarded as undruggable before. However, the catalytic nature of PROTAC potentially leads to uncontrolled degradation that causes systemic toxicity issues, limiting the application of PROTAC in the clinic. Here, we introduce a light-inducible switch on PROTACs, thereafter termed as opto-PROTAC, to enable the degradation of protein targets in a spatiotemporal manner. By adding a photolabile caging group on pomalidomide as a parental compound and two additional PROTACs, dBET1 and dALK, we demonstrated light-inducible protein degradation. These opto-PROTACs display no activities in the dark, while the restricted degradation can be induced at a specific time and rate by ultraviolet A irradiation. Our approach provides a generalizable platform for the development of light-controlled PROTACs and enables PROTAC to be a precision medicine.
INTRODUCTION
The PROteolysis TArgeting Chimera (PROTAC) technique emerged as the result of identifying peptides or small-molecule chemical ligands that specifically bind with specific endogenous E3 ligases, such as F-box/WD repeat-containing protein 1A (FBXW1A, also known as b-TRCP1) (1), the von Hippel–Lindau tumor suppressor (2–5), mouse double minute 2 homolog (MDM2) (6), and cereblon (CRBN) (5, 7, 8). Structurally, PROTAC is a bifunctional small molecule that consists two functional parts, a “warhead” that displays high specificity in binding protein of interest (POI) and another part that functions as a ligand to be recognized by E3 ligase, which are connected by a linker (5, 9, 10). PROTACs provide an alternative approach for those so-called undruggable targets, such as transcriptional factors. Theoretically, any intracellular substrate can be targeted for proteolysis if there is a usable small molecule that specifically binds with those protein targets. Unlike most of the small-molecule inhibitors, which mainly block the catalytic activity of their target enzyme, PROTACs dictate the POI for proteolysis (1, 11, 12). This unique catalytic feature offers several advantages for PROTACs over the conventional small-molecule inhibitors. For example, it can target those noncatalytic enzymes, reduces drug exposure time and dosage required to suppress signaling, and eliminates both catalytic activity and scaffolding function of bifunctional proteins, such as receptor tyrosine kinases (RTKs) (10, 13). However, the powerful catalytic nature of PROTAC also raises potential disadvantages. When systemically administered, it might potentially lead to uncontrolled degradation of the POIs in any cells it can access, e.g., normal cell versus cancer cells. To this end, inhibition of BET (bromodomain and extra-terminal) bromodomains is relatively well tolerated, while complete loss of bromodomain-containing protein 2 (BRD2) and BRD4 is lethal, which might reduce the therapeutic window of dBET1 (7, 14). Therefore, it would be of critical importance to add a controllable switch on those PROTACs, enabling their spatial activation in desirable tissues and cells where cancer cells reside in, to eliminate potential nonspecific toxicity issues of PROTACs.
RESULTS
Design of opto-pomalidomide as a parent lead compound for opto-PROTAC
Given its noninvasive ability and highly spatiotemporal precision, light-induced regulation has been extensively used in biological field, such as optogenetics (15). By adopting the photolysis concept, we designed and synthesized light-inducible PROTACs, thereafter termed as opto-PROTACs, by adding a photolabile caging group on pomalidomide to block its interaction with the E3 ligase CRBN. The activity of the opto-PROTACs can be subjected to photolysis-mediated regulation. Given that the key hydrogen bond is formed between glutarimide NH of pomalidomide and the backbone carbonyl of His380 in CRBN (Fig. 1A) (8, 16, 17), we installed a nitroveratryloxycarbonyl group (18–20) on the glutarimide nitrogen as the lead parental compound, which was referred as opto-pomalidomide (fig. S1, A and B). This engineered opto-pomalidomide molecule could be induced to undergo photolysis by ultraviolet A (UVA) irradiation in vitro (Fig. 1, B and C, and fig. S2, A to D), in a time-dependent manner (Fig. 1D and fig. S2, E to I). Furthermore, in contrast to free pomalidomide, the inert opto-pomalidomide was largely ineffective to bind with CRBN in vitro (Fig. 1E), while UVA irradiation efficiently uncaged it from the caged status to be functionally activated, as demonstrated in its regained ability to bind with CRBN (Fig. 1, A and E).
Fig. 1. Design of opto-pomalidomide as a parent lead compound for opto-PROTAC.
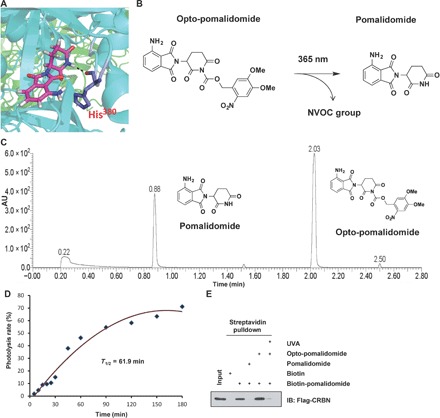
(A) The key hydrogen bond (black dashes) is formed between glutarimide NH of pomalidomide and backbone carbonyl of His380 of CRBN, based on the structure of DDB1-CRBN E3 ubiquitin ligase in complex with pomalidomide (Protein Data Bank: 4CI3). (B) Opto-pomalidomide can be uncaged by UVA irradiation (365 nm). (C) Ultraperformance liquid chromatography–mass spectrometry (UPLC-MS) analysis of opto-pomalidomide after irradiation with UVA (365 nm) for 30 min in vitro. (D) Time course uncaging of opto-pomalidomide by UVA irradiation in vitro. Opto-pomalidomide (1 mM) was irradiated with UVA for the indicated time and then subjected to the UV-Vis absorption analysis. (E) Opto-pomalidomide regains the ability to bind with CRBN after UVA irradiation (365 nm, 30 min) in a competitive binding assay. Biotin-pomalidomide was used to pull down Flag-CRBN purified from human embryonic kidney (HEK) 293T cells, with or without the indicated drug (pomalidomide or opto-pomalidomide with or without UVA irradiation). Biotin was used as a negative control. AU, arbitrary units; IB, immunoblot.
Light induces the uncaging of opto-pomalidomide to mediate degradation of Ikaros zinc finger transcription factors
Pomalidomide and its derivatives, such as lenalidomide and thalidomide, are widely used as immunomodulatory drugs for treating diseases such as multiple myeloma, by inducing the proteolysis of IKZF1/3 (Ikaros zinc finger transcription factor 1/3), Ikaros family transcriptional factors by the CRBN E3 ligase (21–23). To determine whether opto-pomalidomide functions in a light-induced controllable manner in cells, we pretreated human embryonic kidney (HEK) 293T cells with opto-pomalidomide and determined the binding between CRBN and IKZF1 (Fig. 2A). Notably, in keeping with previous reports (21, 22), we found that functioning like a “molecular glue,” pomalidomide bound both CRBN and IKZF1/3 to subsequently transfer the ubiquitin chain onto the target proteins, IKZFs (Fig. 2, B and C). In contrast, opto-pomalidomide was inert as it is ineffective in guiding the protein-protein interaction between CRBN and IKZF1 (Fig. 2B), while after activation by UVA irradiation, the uncaged opto-pomalidomide regained the ability to promote the binding between CRBN and IKZF1, leading to CRBN-mediated IKZF1 ubiquitination (Fig. 2C). Moreover, pomalidomide induced IKZF1/3 degradation in a CRBN-dependent manner, while opto-pomalidomide lost this function without UVA irradiation, even with 10-folds of excess drug concentration (Fig. 2D), excluding the possibility of undesirable off-target effects without external light stimuli.
Fig. 2. Light controls the effects of opto-pomalidomide in mediating IKZF1/3 degradation.
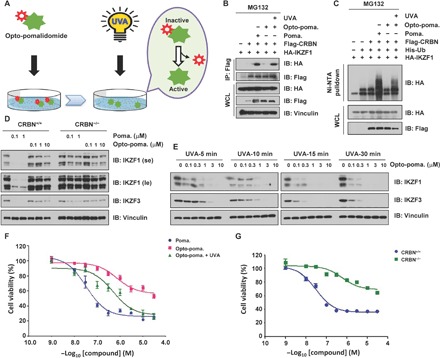
(A) A schematic diagram showing that UVA irradiation activates opto-pomalidomide in cell culture. (B) UVA irradiation activates opto-pomalidomide to mediate the interaction between CRBN and IKZF1. IB analysis of whole-cell lysis (WCL) and Flag–immunoprecipitation (IP) derived from HEK293T cells transfected with indicated plasmids in the presence of pomalidomide or opto-pomalidomide with/without UVA irradiation (365 nm) for 15 min. Cells were treated with 10 μM MG132 for 12 hours before harvest. (C) UVA irradiation activates opto-pomalidomide to mediate the ubiquitination of IKZF1 by CRBN in cells. IB analysis of WCL and Ni–nitrilotriacetic acid (NTA) pull down products derived from HEK293T cells transfected with indicated plasmids in the presence of pomalidomide or opto-pomalidomide with or without UVA irradiation (365 nm) for 15 min. Cells were treated with 10 μM MG132 for 12 hours before harvest. (D) Opto-pomalidomide does not promote the degradation of IKZF1/3 without UVA irradiation. IB analysis of WCL derived from MM.1SCRBN+/+ versus MM.1SCRBN−/− cells in the presence of pomalidomide or opto-pomalidomide for 12 hours. (E) UVA irradiation activates opto-pomalidomide to promote the degradation of IKZF1/3 in cells. IB analysis of WCL derived from MM.1S cells in the presence of opto-pomalidomide with UVA irradiation (365 nm) as indicated time. (F) UVA irradiation–activated opto-pomalidomide inhibits MM.1S cell proliferation in a dose-dependent manner. MM.1S cells were treated by pomalidomide versus opto-pomalidomide with or without UVA irradiation (365 nm) for 15 min and then subjected to CCK-8 cell viability assay. (G) Pomalidomide reduces MM.1S cell proliferation in a CRBN-dependent manner. MM.1SCRBN+/+ and MM.1SCRBN−/− cells were treated by pomalidomide for 72 hours and then subjected to CCK-8 cell viability assay.
To further determine the uncaging efficiency and in vivo controllability of opto-pomalidomide, we stimulated opto-pomalidomide pretreated cell with different durations of UVA irradiation (Fig. 2E). Notably, the observed degradation of IKZF1/3 by opto-pomalidomide occurred in a drug dose– and UVA dose–dependent manner (Fig. 2E and fig. S3A). This degradation was not induced by UVA irradiation itself when opto-pomalidomide was not present (fig. S3, B and C), which further demonstrates the specific regulation of degradation of IKZFs by the engineered opto-pomalidomide in a light-dependent manner. Moreover, as a biological consequence, we found that unlike pomalidomide, the ability of opto-pomalidomide to kill multiple myeloma cells was largely dependent on UVA irradiation, and UVA itself minimally affected cell viability in the absence of opto-pomalidomide (Fig. 2, F and G, and fig. S3, D and E).
Design of opto-dBET1 and light controls the release of dBET1
Given that pomalidomide is wildly used as a ligand of CRBN for the synthesis of various PROTACs (7, 17), we further speculated whether the lead compound opto-pomalidomide can be adapted to pomalidomide-based PROTACs to achieve similar magnitude of light-inducible control of protein degradation. To this end, we synthesized two pomalidomide-based opto-PROTACs, opto-dBET1 and opto-dALK (Figs. 3A and 4A and figs. S4, A and B, and S7, A and B) (7, 17), for further biochemical and biological validation and characterization. Notably, similar to opto-pomalidomide, opto-dBET1 could also be efficiently uncaged by UVA irradiation in vitro (Fig. 3B and fig. S5, A to J).
Fig. 3. Light controls the effects of opto-dBET1 in mediating degradation of BRDs.
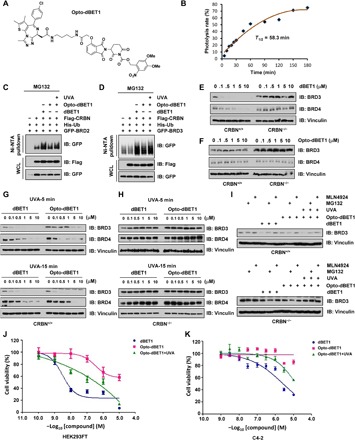
(A) A schematic illustration of the chemical structure of opto-dBET1. (B) Time course uncaging of opto-dBET1 by UVA irradiation in vitro. Opto-dBET1 (1 mM) was irradiated with UVA (365 nm) for the indicated time and then subjected to UV-Vis absorption analysis. Cells were treated with 10 μM MG132 for 12 hours before harvest. (C and D) UVA irradiation activates opto-dBET1 to mediate the ubiquitination of BRD2 (C) and BRD3 (D) by CRBN in cells. IB analysis of WCL and Ni-NTA pull down products derived from HEK293T cells transfected with indicated plasmids in the presence of dBET1 or opto-dBET1 with or without UVA irradiation (365 nm) for 15 min. Cells were treated with 10 μM MG132 for 12 hours before harvest. (E) dBET1 promotes the degradation of BRDs in a CRBN-dependent manner. IB analysis of WCL derived from HEK293FTCRBN+/+ versus HEK293FTCRBN−/− treated with dBET1 at the indicated dose for 12 hours. (F) Opto-dBET1 does not promote the degradation of BRDs in cells without UVA irradiation. IB analysis of WCL derived from HEK293FTCRBN+/+ versus HEK293FTCRBN−/− treated with opto-dBET1 at indicated dose for 12 hours. (G and H) UVA irradiation activates opto-dBET1 to promote the degradation of BRDs in cells in a CRBN-dependent manner. IB analysis of WCL derived from HEK293FTCRBN+/+ (G) versus HEK293FTCRBN−/− (H) in the presence of dBET1 versus opto-dBET1 with UVA irradiation (365 nm) for 5 or 15 min. (I) UVA irradiation–activated opto-dBET1 promotes BRD3 degradation in a ubiqutin proteosome system–dependent manner. IB analysis of WCL derived from HEK293FTCRBN+/+ (G) versus HEK293FTCRBN−/− (H) in the presence of dBET1 versus opto-dBET1 with or without UVA irradiation (365 nm). Cells were treated with either 10 μM MG132 or 1 μM MLN4924 for 12 hours. (J and K). UVA irradiation–activated opto-dBET1 inhibits HEK293FT (J) and C4-2 (K) cell proliferation in a dose-dependent manner. HEK293FT cells were treated by dBET1 versus opto-dBET1 with or without UVA irradiation (365 nm) for 15 min and then subjected to a CCK-8 cell viability assay.
Fig. 4. Light controls the effects of opto-dALK in mediating the degradation of the ALK fusion protein.
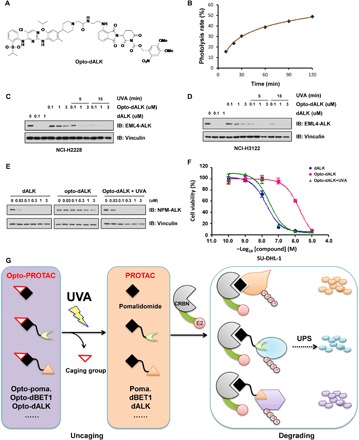
(A) A schematic illustration of the chemical structure of the engineered opto-dALK. (B) Time course uncaging of opto-dALK by UVA irradiation in vitro. Opto-dBET1 (1 mM) was irradiated with UVA (365 nm) for indicated time and then subjected to the UV-Vis absorption analysis. (C to E). UVA irradiation activates opto-dALK to promote the degradation of EML-ALK fusion proteins in cells. IB analysis of WCL derived from NCI-H2228 (C) or NCI-3122 (D) NSCLC cells or SU-DHL-1 cells (E) treated with BET1 versus opto-dBET1 at indicated dose with or without UVA irradiation (365 nm) for 5 or 15 min. (F) UVA irradiation–activated opto-dALK inhibits SU-DHL-1 cell proliferation in a dose-dependent manner. SU-DHL-1 cells were treated by dALK versus opto-dALK with or without UVA irradiation (365 nm) for 15 min and then subjected to CCK-8 cell viability assay. (G) A schematic diagram showing that working model of opto-pomalidomide in degrading POI in a UVA-dependent manner.
Light controls the uncaging of opto-dBET1 to mediate degradation of BRDs
In keeping with previous studies (7), we found that dBET1 functioned as a molecular linker that recruit bromodomain family members (BRD2/3) for CRBN-mediated ubiquitination (Fig. 3, C and D), while opto-dBET1 lost such function due to the blocking of its CRBN binding ability, thereby incapable of guiding the ubiquitination of BRD2 and BRD3 (Fig. 3, C and D). However, upon UVA irradiation that led to uncaging process (Fig. 3B), opto-dBET1 regained its ability to promote ubiquitination of BRDs in cells (Fig. 3, C and D). In support of this notion, we further showed that the dBET1 degraded BRD3/4 in a CRBN-dependent manner (Fig. 3E), while opto-dBET1 was largely inert and incapable of degrading BRD3/4 under this experimental condition (Fig. 3F).
To further explore whether UVA irradiation could reactivate opto-dBET1, we pretreated HEK293FT cells with opto-dBET1 and then subjected those to UVA irradiation. Opto-dBET1 was activated in 5 to 15 min of UVA irradiation in cells, leading to the degradation of BRD3/4 (Fig. 3G) in a CRBN (Fig. 3H) and ubiquitin proteosome system–dependent manner (Fig. 3I). High dose of dBET1 has been reported to be toxic for cells, due to its effects on completely depleting bromodomain family members that play critical role in modulating enhancer and transcription activity of many genes (14). This shortcoming limits the further application of dBET1 in the clinic. As a potential solution to this emerging concern to dBET1 and, possibly, other PROTACs in general, our results demonstrate that opto-dBET1 was an inert drug in inducing degradation of BRDs, a process that can be specifically activated by light to achieve precise degradation in a temporal and spatial manner (Fig. 3, C to I). In keeping with this notion, dBET1 inhibited cell proliferation in a dose-dependent manner (Fig. 3, J and K), while under our experimental condition, opto-dBET1 was relatively less toxic than dBET1 and could be timely activated by UVA irradiation to suppress cell proliferation (Fig. 3, J and K, and fig. S6, A to D).
Design of opto-dALK and light controls the release of dALK to mediate the degradation of anaplastic lymphoma kinase proteins
Anaplastic lymphoma kinase (ALK) is an RTK that was identified to be associated with anaplastic large cell lymphoma (ALCL) (24). ALK fusion proteins are observed in several types of cancers, such as non–small cell lung cancer (NSCLC) (25) and non-Hodgkin’s lymphoma (26). Echinoderm microtubule-associated protein-like 4 (EML4)–ALK mutations confer resistance to ALK inhibitors (27), while PROTACs targeting ALK provide a new approach for those cancers with ALK fusion proteins (13, 17). Our results demonstrated that opto-dALK could be efficiently uncaged in vitro by UVA irradiation (Fig. 4B and fig. S8, A to J). Furthermore, in accordance with our previous report (17), dALK promoted the degradation of EML4-ALK in two NSCLC cell lines, NCI-2228 and NCI-3122, in a dose-dependent and time-dependent manner (fig. S9, A to D). On the other hand, opto-dALK was largely inactive at basal level for guiding ALK degradation but could be activated by UVA irradiation in drug dose– and UVA dose–dependent manner (Fig. 4, C and D). Furthermore, after activation by UVA, opto-dALK also promoted the degradation of NPM-ALK fusion protein in the ALCL cell line, SU-DHL-1 (Fig. 4E). Previous studies by us and others have revealed that the degradation of EML-ALK fusion protein per se is not sufficient to efficiently retard the growth of NCI-2228 and NCI-3122 NSCLC cancer cell lines, whereas the cell growth of SU-DHL-1 is more sensitive to the degradation of NPM-ALK (17, 28). Hence, we demonstrated that dALK inhibited the cell proliferation of SU-DHL-1 in a dose-dependent manner (Fig. 4F), while the NSCLC cell lines are relatively more resistant to ALK degradation (fig. S9, E and F). The ability of opto-dALK to block cell proliferation of SU-DHL-1 cells, as well as, to a lesser extent, NCI-2228 and NCI-3122 cells, required prior uncaging by UVA irradiation, while UVA itself only minimally affected cell viability in the absence of opto-dALK (Fig. 4F and fig. S9, E to I). Together, our data provide experimental evidence for the development of light-controlled PROTACs and enable PROTAC to be a precision medicine approach.
DISCUSSION
By installing a light-controllable caging group on the glutarimide NH of pomalidomide to block its recruitment to the CRBN E3 ligase, we successfully established a general platform to control protein degradation in cells in a highly specific temporal and spatial manner. On the basis of the inefficiency at its inert status in the darkness and quick response to light-induced activation, opto-pomalidomide and the opto-PROTACs confer well-controllable biological characters, making opto-PROTAC an outstanding candidate for precision medicine to achieve targeting therapy, thereby rendering it more suitable for future use in the clinic. Moreover, given the dominating presence of pomalidomide in the synthesis of various PROTACs as an E3 ligase ligand, the caging-uncaging process of opto-pomalidomide could also be applied to any other pomalidomide-derived PROTACs for controllable degradation of protein targets such as cyclin-dependent kinases (29, 30), breakpoint cluster region protein (BCR)–ABL (31), Bruton’s tyrosine kinase (32), and Tau (33). This will allow the further development of photoswitchable PROTACs as a precision medicine approach for treating cancer and other diseases, such as Alzheimer’s disease, in a tissue-specific and temporal-specific manner.
On the other hand, this caging strategy may also be applied to other chemical ligands other than pomalidomide that recruit other specific E3 ligases, based on the well-characterized structure information of their interacting interface, which will largely expand the concept of opto-PROTACs. The Deiters and colleagues (34) and Pan and colleagues (35) also independently developed a similar caged method on PROTAC and achieved light-induced controlling of protein degradation. Furthermore, in addition to photolysis-induced uncaging process, other photopharmacological method such as incorporating an azobenzene photoswitch (36, 37) in the linker region of a PROTAC to achieve photochemical isomerization process can also be used to generate light-inducible version of PROTACs for the purpose of optical control of protein degradation (38, 39).
Notably, photodynamic therapy (PDT) has been widely used to treat kinds of diseases, including cancer (40–42). To this end, UVA light has been used as PDT light for treating disease for years (43). Recently, UVA light has already been used to activate riboflavin for corneal collagen cross-linking, which has been approved by the U.S. Food and Drug Administration in 2016 (44). Although our results showed that UVA illumination alone has minimal nonspecific effect on degrading the POI (fig. S3, B and C) and short term of UVA irradiation has minimal effect on the cell viability (figs. S3E; S6, C and D; and S9, G to I), previous studies have showed that UVA exposure might cause DNA damages (45–47). Thus, it will be important to develop uncaging method other than UVA illumination to achieve better anticancer therapeutic effects. Furthermore, given that the prototype UVA switch could be currently applied to only limited cancer types such as blood and skin cancers, due to its inefficiency in penetrating tissues, future efforts are aimed to develop better caging groups with absorption within the near-infrared region (48, 49) for enhanced tissue penetration for better clinical outputs.
MATERIALS AND METHODS
General chemistry methods
High-performance liquid chromatography (HPLC) spectra were acquired using an Agilent 1200 Series system with diode-array detection (DAD) detector for all the intermediates and Opto-PROTACs below. Chromatography was performed on a 2.1 mm by 150 mm Zorbax 300SB-C18 5-μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 ml/min. The gradient program was as follows: 1% B (0 to 1 min), 1 to 99% B (1 to 4 min), and 99% B (4 to 8 min). High-resolution mass spectra (HRMS) data were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Nuclear magnetic resonance (NMR) spectra were acquired on either a Bruker DRX-600 spectrometer with 600 MHz for proton (1H NMR) and 151 MHz for carbon (13C NMR) or a Bruker Avance III 800 MHz spectrometer (201 MHz 13C); chemical shifts were reported in (δ). Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to 254 nm. Samples were injected onto a Phenomenex Luna 250 mm by 30 mm, 5 μm, C18 column at room temperature. The flow rate was 40 ml/min. A linear gradient was used with 10% of acetonitrile (A) in H2O [with 0.1% trifluoroacetic acid (TFA)] (B) to 100% of acetonitrile (A). HPLC was used to establish the purity of target compounds. All final compounds had >95% purity using the HPLC methods described above.
4,5-Dimethoxy-2-nitrobenzyl 3-(4-amino-1,3-dioxoisoindolin-2-yl)-2,6-dioxopiperidine-1-carboxylate (Opto-pomalidomide)
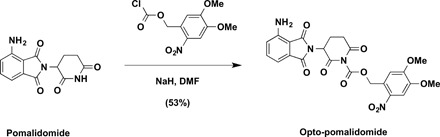
To a solution of pomalidomide (27 mg, 0.1 mmol, 1.0 equivalent) in N,N′-dimethylformamide (DMF; 1 ml) was added NaH (4.8 mg, 60% in mineral oil, 0.12 mmol, 1.2 equivalent) at 0°C. After stirring for 10 min, 4,5-dimethoxy-2-nitrobenzyl carbonochloridate (33 mg, 0.12 mmol, 1.2 equivalent) was added to the mixture at 0°C. The reaction mixture was then warmed up to room temperature and stirred for additional 3 hours. The resulting mixture was purified by preparative HPLC (10 to 100% acetonitrile/0.1% TFA in H2O) to afford Opto-pomalidomide as yellow solid (27.0 mg, 53%). 1H NMR [600 MHz, dimethyl sulfoxide (DMSO)–d6]: δ 7.74 (s, 1H), 7.49 (dd, J = 8.5, 7.0 Hz, 1H), 7.26 (s, 1H), 7.04 (d, J = 8.6 Hz, 1H), 7.02 (d, J = 7.1 Hz, 1H), 6.58 (s, 2H), 5.81 (d, J = 14.5 Hz, 1H), 5.77 (d, J = 14.4 Hz, 1H), 5.40 (dd, J = 12.9, 5.5 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.14 (ddd, J = 17.4, 13.8, 5.5 Hz, 1H), 2.86 (ddd, J = 17.4, 4.4, 2.6 Hz, 1H), 2.63 (qd, J = 13.3, 4.4 Hz, 1H), and 2.18 to 2.11 (m, 1H). 13C NMR (151 MHz, DMSO-d6): δ 170.8, 168.6, 168.6, 167.6, 153.8, 150.8, 148.5, 147.3, 139.6, 136.0, 132.2, 125.3, 122.3, 111.5, 110.9, 108.6, 67.5, 56.7, 56.5, 48.7, 31.2, and 21.8. ESI mass/charge ratio (m/z) = 535.2 [M + Na+]. HRMS calcd for C23H24N5O10+ [M + NH4+], 530.1518; found, 530.1536.
4,5-Dimethoxy-2-nitrobenzyl 3-(4-(2-((4-aminobutyl)amino)-2-oxoethoxy)-1,3-dioxoisoindolin-2-yl)-2,6-dioxopiperidine-1-carboxylate (Opto-dBET1-L)
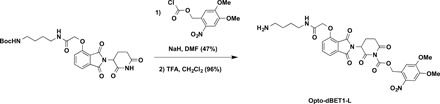
To a solution of tert-butyl (4-(2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetamido)butyl)carbamate [see (7)for the details of synthesis] (50 mg, 0.1 mmol, 1.0 equivalent) in DMF (1 ml) was added NaH (4.8 mg, 60% in mineral oil, 0.12 mmol, 1.2 equivalent) at 0°C. After stirring for 10 min, 4,5-dimethoxy-2-nitrobenzyl carbonochloridate (33 mg, 0.12 mmol, 1.2 equivalent) was added to the mixture at 0°C. The reaction mixture was then warmed up to room temperature and stirred for additional 3 hours. The resulting mixture was purified by preparative HPLC (10 to 100% acetonitrile/0.1% TFA in H2O) to afford desired product as white solid (34.6 mg, 47%). ESI m/z = 642.3 [M − Boc + H+]. HRMS calcd for C34H39N5O14Na+ [M + Na+], 764.2386; found, 764.2400.
To a solution of obtained above compound (34.6 mg, 0.047 mmol, 1.0 equivalent) in CH2Cl2 (2 ml) was added TFA (1 ml) at room temperature. After stirring for 1 hour, the resulting mixture was concentrated under reduced pressure. The resulting residue was purified by preparative HPLC (10 to 100% acetonitrile/0.1% TFA in H2O) to afford Opto-dBET1-L as white solid in TFA salt form (34.0 mg, 96%). 1H NMR (600 MHz, DMSO-d6): δ 8.08 (t, J = 5.9 Hz, 1H), 7.85 (dd, J = 8.5, 7.3 Hz, 1H), 7.75 (s, 1H), 7.72 (s, 3H), 7.53 (d, J = 7.2 Hz, 1H), 7.41 (d, J = 8.6 Hz, 1H), 7.27 (s, 1H), 5.81 (d, J = 14.4 Hz, 1H), 5.77 (d, J = 14.4 Hz, 1H), 5.47 (dd, J = 12.8, 5.5 Hz, 1H), 4.80 (s, 2H), 3.89 (s, 3H), 3.87 (s, 3H), 3.19 to 3.12 (m, 3H), 2.88 (ddd, J = 17.5, 4.4, 2.7 Hz, 1H), 2.80 (h, J = 6.0 Hz, 2H), 2.63 (qd, J = 13.2, 4.4 Hz, 1H), 2.19 to 2.12 (m, 1H), and 1.57 to 1.45 (m, 4H). 13C NMR (201 MHz, DMSO): δ 170.8, 168.4, 167.3, 167.0, 165.7, 155.7, 153.9, 150.8, 148.6, 139.8, 137.6, 133.4, 125.3, 121.0, 117.1, 116.7, 111.1, 108.8, 68.1, 67.6, 56.8, 56.6, 49.1, 39.0, 38.2, 31.2, 26.5, 24.9, and 21.6. ESI m/z = 642.3 [M + H+]. HRMS calcd for C29H32N5O12+ [M + H+], 642.2042; found, 642.2036.
4,5-Dimethoxy-2-nitrobenzyl 3-(4-(2-((4-(2-((S)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetamido)butyl)amino)-2-oxoethoxy)-1,3-dioxoisoindolin-2-yl)-2,6-dioxopiperidine-1-carboxylate (Opto-dBET1)
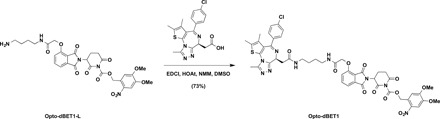
To a solution of Opto-dBET1-L (23.0 mg, 0.03 mmol, 1.1 equivalent) in DMSO (1 ml) were added (S)-2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetic acid [see (7) for the details of synthesis] (14.4 mg, 0.028 mmol, 1 equivalent), EDCI [1-ethyl-3-(3-dimethylaminopropyl)carbodiimide] (8.1 mg, 0.042 mmol, 1.5 equivalent), HOAt (1-hydroxy-7-azabenzo-triazole) (5.7 mg, 0.042 mmol, 1.5 equivalent), and NMM (N-methylmorpholine) (14.2 mg, 0.14 mmol, 5.0 equivalent). After being stirred overnight at room temperature, the resulting mixture was purified by preparative HPLC (10 to 100% acetonitrile/0.1% TFA in H2O) to afford Opto-dBET1 as light yellow solid in TFA salt form (23.2 mg, 73%). 1H NMR (600 MHz, DMSO-d6): δ 8.21 (t, J = 5.7 Hz, 1H), 8.02 (t, J = 5.8 Hz, 1H), 7.83 (dd, J = 8.5, 7.3 Hz, 1H), 7.74 (s, 1H), 7.53 to 7.46 (m, 3H), 7.45 to 7.39 (m, 3H), 7.26 (s, 1H), 5.81 (d, J = 14.4 Hz, 1H), 5.77 (d, J = 14.4 Hz, 1H), 5.47 (dd, J = 12.9, 5.4 Hz, 1H), 4.80 (s, 2H), 4.52 (dd, J = 8.1, 6.1 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 3.29 to 3.05 (m, 7H), 2.92 to 2.82 (m, 1H), 2.71 to 2.57 (m, 4H), 2.41 (s, 3H), 2.20 to 2.11 (m, 1H), 1.62 (s, 3H), and 1.54 to 1.43 (m, 4H). 13C NMR (201 MHz, DMSO): δ 170.8, 169.9, 168.4, 167.1, 167.0, 165.7, 163.5, 155.7, 155.6, 153.9, 150.8, 150.3, 148.6, 139.7, 137.6, 137.2, 135.7, 133.4, 132.7, 131.2, 130.6, 130.3, 130.1, 128.9, 125.3, 121.0, 117.1, 116.6, 111.0, 108.8, 68.1, 67.6, 56.8, 56.6, 54.3, 49.1, 38.6, 38.6, 38.1, 31.2, 27.1, 27.0, 21.7, 14.5, 13.1, and 11.8. ESI m/z = 1024.3 [M + H+]. HRMS calcd for C48H47N9O13SCl+ [M + H+], 1024.2697; found, 1024.2734.
3,4-Dimethoxy-2-nitrobenzyl 3-(4-((2-aminoethyl)amino)-1,3-dioxoisoindolin-2-yl)-2,6-dioxopiperidine-1-carboxylate (Opto-dALK-L)
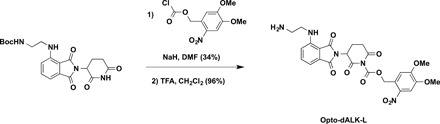
To a solution of tert-butyl (2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethyl)carbamate [see (17) for the details of synthesis] (20.0 mg, 0.048 mmol, 1.0 equivalent) in DMF (1 ml) was added NaH (2.9 mg, 60% in mineral oil, 0.072 mmol, 1.5 equivalent) at 0°C. After stirring for 10 min, 4,5-dimethoxy-2-nitrobenzyl carbonochloridate (16.0 mg, 0.058 mmol, 1.2 equivalent) was added to the mixture at 0°C. The reaction mixture was warmed up to room temperature and stirred for additional 3 hours. The resulting mixture was purified by preparative HPLC (10 to 100% acetonitrile/0.1% TFA in H2O) to afford desired product as yellow solid (10.7 mg, 34%). ESI m/z = 556.2 [M − Boc + H+]. HRMS calcd for C25H26N5O10+ [M − Boc + H+], 556.1674; found, 556.1690.
To a solution of obtained above compound (10.7 mg, 0.016 mmol, 1.0 equivalent) in CH2Cl2 (2 ml) was added TFA (1 ml) at room temperature. After stirring for 1 hour, the resulting mixture was concentrated under reduced pressure. The resulting residue was purified by preparative HPLC (10 to 100% acetonitrile/0.1% TFA in H2O) to afford Opto-dALK-L as yellow solid in TFA salt form (10.6 mg, 96%). 1H NMR (600 MHz, methanol-d4): δ 7.78 (s, 1H), 7.66 (dd, J = 8.6, 7.1 Hz, 1H), 7.28 (s, 1H), 7.21 to 7.16 (m, 2H), 5.85 (d, J = 15.2 Hz, 1H), 5.76 (d, J = 15.3 Hz, 1H), 5.32 (dd, J = 12.8, 5.5 Hz, 1H), 3.92 (s, 3H), 3.90 (s, 3H), 3.71 (t, J = 6.1 Hz, 2H), 3.22 (t, J = 6.1 Hz, 2H), 3.08 (ddd, J = 17.6, 13.7, 5.3 Hz, 1H), 2.98 (ddd, J = 17.6, 4.5, 2.8 Hz, 1H), 2.83 (qd, J = 13.2, 4.4 Hz, 1H), and 2.24 to 2.17 (m, 1H). 13C NMR (151 MHz, methanol-d4): δ 170.0, 168.8, 167.9, 167.4, 154.1, 150.3, 148.3, 146.0, 139.0, 136.2, 132.5, 125.6, 116.5, 111.6, 110.9, 109.4, 107.8, 67.1, 55.8, 55.3, 48.8, 39.5, 38.2, 30.8, and 21.6. ESI m/z = 556.2 [M + H+]. HRMS calcd for C25H26N5O10+ [M + H+], 556.1674; found, 556.1712.
3,4-Dimethoxy-2-nitrobenzyl 3-(4-((2-(2-(4-(4-((5-chloro-4-((2-(isopropylsulfonyl)phenyl)amino)pyrimidin-2-yl)amino)-5-isopropoxy-2-methylphenyl)piperidin-1-yl)acetamido)ethyl)amino)-1,3-dioxoisoindolin-2-yl)-2,6-dioxopiperidine-1-carboxylate (Opto-dALK)
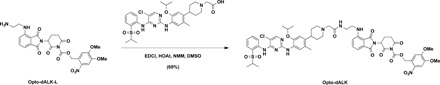
To a solution of Opto-dALK-L (8.1 mg, 0.012 mmol, 1.1 equivalent) in DMSO (1 ml) were added 2-(4-(4-((5-chloro-4-((2-(isopropylsulfonyl)phenyl)amino)pyrimidin-2-yl)amino)-5-isopropoxy-2-methylphenyl)piperidin-1-yl)acetic acid [see (17) for the details of synthesis] (8.3 mg, 0.011 mmol, 1.0 equivalent), EDCI (3.2 mg, 0.017 mmol, 1.5 equivalent), HOAt (2.3 mg, 0.017 mmol, 1.5 equivalent), and NMM (5.6 mg, 0.055 mmol, 5.0 equivalent). After being stirred overnight at room temperature, the resulting mixture was purified by preparative HPLC (10 to 100% acetonitrile/0.1% TFA in H2O) to afford Opto-dALK as yellow solid in TFA salt form (10.0 mg, 69%). 1H NMR (600 MHz, methanol-d4): δ 8.34 (d, J = 8.3 Hz, 1H), 8.23 (s, 1H), 7.98 (d, J = 7.9 Hz, 1H), 7.72 (d, J = 6.0 Hz, 1H), 7.66 to 7.57 (m, 3H), 7.46 (t, J = 7.7 Hz, 1H), 7.19 (d, J = 8.6 Hz, 1H), 7.11 (d, J = 7.1 Hz, 1H), 7.07 (s, 1H), 6.79 (s, 1H), 5.61 (d, J = 15.3 Hz, 1H), 5.49 (d, J = 15.3 Hz, 1H), 5.28 (dd, J = 12.9, 5.4 Hz, 1H), 4.59 (p, J = 6.0 Hz, 1H), 3.94 (s, 2H), 3.87 (s, 3H), 3.82 (s, 3H), 3.69 to 3.51 (m, 6H), 3.42 (p, J = 6.8 Hz, 1H), 3.23 to 3.13 (m, 2H), 3.08 to 3.01 (m, 2H), 2.93 (ddd, J = 17.5, 4.4, 2.8 Hz, 1H), 2.83 to 2.74 (m, 1H), 2.22 to 2.14 (m, 1H), 2.12 (s, 3H), 2.07 to 1.93 (m, 4H), 1.33 (d, J = 6.0 Hz, 6H), and 1.28 (d, J = 6.8 Hz, 6H). 13C NMR (201 MHz, MeOD): δ 170.0, 169.0, 168.0, 167.5, 164.6, 160.9, 160.7, 156.4, 155.2, 154.1, 150.3, 148.2, 146.7, 146.1, 138.6, 137.2, 137.1, 136.2, 134.8, 132.3, 131.2, 127.3, 126.4, 126.2, 125.7, 124.6, 122.5, 117.0, 111.0, 110.6, 109.8, 109.0, 107.7, 105.4, 71.4, 67.2, 57.2, 55.9, 55.4, 53.8, 48.8, 48.5, 48.1, 41.6, 38.0, 34.8, 30.8, 29.6, 29.5, 21.6, 21.0, 17.7, and 14.1. ESI m/z = 1153.5 [M + H+]. HRMS calcd for C55H62N10O14SCl+ [M + H+], 1153.3851; found, 1153.3848.
Plasmids and chemicals
Flag-CRBN and hemagglutinin (HA)–IKZF1 were gifts from W. G. Kaelin (Dana-Farber Cancer Institute). Green fluorescent protein (GFP)–BRD2 and GFP-BRD3 were purchased from Addgene. dBET1 was obtained from J. E. Bradner’s group at the Dana-Farber Cancer Institute. Pomalidomide was purchased from Sigma-Aldrich. dALK was synthesized, as described previously (17).
Cell culture
HEK293T cells and HEK293FT were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS), 100 U of penicillin, and streptomycin (100 μg/ml). MM.1S, MT2, C4-2, SU-DHL-1, NCI-H2228, and NCI-H3122 cells were cultured in RPMI 1640 containing 10% FBS, 100 U of penicillin, and streptomycin (100 μg/ml). HEK293FTCREB+/+, HEK293FTCREB−/−, MM.1SCRBN+/+, and MM.1SCRBN−/− cells were gifts from W. G. Kaelin (Dana-Farber Cancer Institute). For UVA irradiation, cells were pretreated with opto-PROTACs for 2 to 4 hours and then subjected to UVA irradiation for indicated durations.
Antibodies
Anti-IKZF1 (ab191394) antibody was purchased from Abcam. Anti-IKZF3 (NBP22449) antibody was purchased from Novus Biologicals. Anti-BRD3 (11859-1-AP) antibody was purchased from Proteintech. Anti-BRD4 (A301-985A-M) antibody was purchased from Bethyl Laboratories. Anti-ALK (3633) was purchased from Cell Signaling Technologies. Monoclonal anti-HA antibody (MMS-101P) was purchased from BioLegend. Polyclonal anti-HA (sc-805) antibody was purchased from Santa Cruz Biotechnologies. Anti-GFP antibody (632380) was purchased from Invitrogen. Polyclonal anti-Flag antibody (F-2425), monoclonal anti-Flag antibody (F-3165, clone M2), anti-tubulin antibody (T-5168), anti-vinculin antibody (V-4505), anti-Flag agarose beads (A-2220), anti-HA agarose beads (A-2095), peroxidase-conjugated anti-mouse secondary antibody (A-4416), and peroxidase-conjugated anti-rabbit secondary antibody (A-4914) were purchased from Sigma-Aldrich. All antibodies were used at a 1:1000 dilution in 5% bovine serum albumin in tris-buffered saline and tween 20 (TBST) buffer for Western blots.
UV-Visible absorption spectrum
UV-visible (UV-Vis) spectrometry was performed on a NanoDrop-2000 UV-Vis Spectrophotometer. Pomalidomide, opto-pomalidomide, dBET1, opto-dBET1, dALK, and opto-dALK were dissolved in DMSO and diluted to indicate concentration, followed by UVA irradiation for indicated duration of time. Then, the samples were subjected to UV-Vis spectrometry analysis. Pomalidomide and opto-pomalidomide mixture solution was made with indicated ratio, and the absorption value at 364 nm was measured to draw standard curve. dBET1 and opto-dBET1 mixture solution was made with indicated ratio, and the absorption value at 340 nm was measured to draw standard curve. dALK and opto-dALK mixture solution was made with indicated ratio, and the absorption value at 370 nm was measured to draw standard curve.
Immunoblots and immunoprecipitation
Cells were lysed in EBC buffer [50 mM tris (pH 7.5), 120 mM NaCl, and 0.5% NP-40] supplemented with protease inhibitors (cOmplete Mini, Roche) and phosphatase inhibitors (phosphatase inhibitor cocktail sets I and II, Calbiochem). The protein concentrations of the lysates were measured using the Bio-Rad protein assay reagent on a Beckman Coulter DU-800 spectrophotometer. The lysates were then resolved by SDS–polyacrylamide gel electrophoresis (PAGE) and immunoblotted with indicated antibodies. For immunoprecipitation (IP), 1 mg of lysates were incubated with the appropriate sepharose beads for 4 hours at 4°C. Immunocomplexes were washed four times with NETN buffer [20 mM tris (pH 8.0), 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40] before being resolved by SDS-PAGE and immunoblotted with indicated antibodies.
In vitro pulldown assays
Flag-CRBN was expressed in HEK293T cells lysed in PROTAC buffer B [50 mM tris (pH 7.5), 150 mM NaCl, and 0.5% NP-40] supplemented with protease inhibitors (cOmplete Mini, Roche) and phosphatase inhibitors (phosphatase inhibitor cocktail sets I and II, Calbiochem). A total of 3 mg of cell lysate was incubated with 10 μl of 10 mM biotin-pomalidomide and 8 μl of streptavidin beads for 1 hour at 4°C in the absence or presence of pomalidomide or opto-pomalidomide. Then, the beads were washed four times with NETN buffer [20 mM tris (pH 8.0), 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40] before being resolved by SDS-PAGE and immunoblotted with indicated antibodies.
In vivo ubiquitination assays
Denatured in vivo ubiquitination assays were performed, as previously described (14). Briefly, HEK293T cells were transfected with Flag-CRBN, His-ubiquitin, and HA-IKZF1 or GFP-BRD2 or GFP-BRD3. Twenty-four hours after transfection, pomalidomide, opto-pomalidomide, dBET1, or opto-dBET1 were added to the cell culture together with 10 μM MG132 for 12 hours, and cells were harvested in denatured buffer [6 M guanidine-HCl (pH 8.0), 0.1 M Na2HPO4/NaH2PO4, and 10 mM imidazole]. After sonication, the ubiquitinated proteins were purified by incubation with Ni–nitrilotriacetic acid (NTA) matrices for 3 hours at room temperature. The pulldown products were washed sequentially twice in buffer A, twice in buffer A/TI mixture (buffer A, buffer TI = 1:3), and once in buffer TI [25 mM tris-HCl (pH 6.8) and 20 mM imidazole]. The polyubiquitinated proteins were separated by SDS-PAGE for immunoblot (IB) analysis.
CCK-8 cell proliferation assay
Cell in 96-well plates were incubated with 10 μl per well of CCK-8 (cell counting Kit-8) solution and incubated for 2 hours, followed by the measurement of optical density at 450 nm.
Statistical analysis
The quantitative data from multiple repeat experiments were analyzed by a two-tailed unpaired Student’s t test or one-way analysis of variance (ANOVA) and presented as means ± SEM. When P < 0.05, the data were considered as statistically significant.
Supplementary Material
Acknowledgments
We thank Wei laboratory members for critical reading of the manuscript and members of the Wei, Jin, and Gray laboratories for helpful discussions. Funding: This work was supported in part by the NIH grants (GM089763 and CA200573 to W.W.). Author contribution: W.W., J.J., H.Ü.K., and H.C. designed the synthesis strategy. H.C. synthesized the compounds and characterized the compounds using HPLC, LCMS, and NMR. J.L. and Z.H. performed the HPLC assay. T.Z. and N.G guided the HPLC assay. W.W. and J.L. designed and performed the in vitro and in vivo release assay. J.L., L.M., D.W., Y.L., and Q.L. performed the cell experiments. W.W. led the whole study including design of the study, interpretation of the results, and writing and editing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay5154/DC1
Fig. S1. The schematic illustration of the synthesis of opto-pomalidomide and the working model for opto-pomalidomide to achieve light-induced degradation of IKZF1/3.
Fig. S2. UPLC-MS characterization and the time-dependent uncaging of pomalidomide and opto-pomalidomide.
Fig. S3. Uncaging of opto-pomalidomide by UVA irradiation leads to active degradation of IKZFs in multiple myeloma cancer cell lines.
Fig. S4. The schematic illustration of the synthesis of opto-dBET1 and the working model for opto-dBET1 to achieve light-induced degradation of BRDs.
Fig. S5. UPLC-MS characterization and the time-dependent uncaging of opto-dBET1 by UVA irradiation in vitro.
Fig. S6. dBET1 inhibits cell proliferation in a CRBN-dependent manner.
Fig. S7. The schematic illustration of the synthesis of opto-dALK and the working model for opto-dALK to achieve light-induced degradation of ALK fusion proteins.
Fig. S8. UPLC-MS characterization and the time-dependent uncaging process of opto-dALK.
Fig. S9. Dose-dependent degradation of ALK fusion proteins by dALK.
REFERENCES AND NOTES
- 1.Sakamoto K. M., Kim K. B., Kumagai A., Mercurio F., Crews C. M., Deshaies R. J., Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U.S.A. 98, 8554–8559 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakamoto K. M., Kim K. B., Verma R., Ransick A., Stein B., Crews C. M., Deshaies R. J., Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteomics 2, 1350–1358 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Schneekloth J. S. Jr., Fonseca F. N., Koldobskiy M., Mandal A., Deshaies R., Sakamoto K., Crews C. M., Chemical genetic control of protein levels: Selective in vivo targeted degradation. J. Am. Chem. Soc. 126, 3748–3754 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Bondeson D. P., Mares A., Smith I. E., Ko E., Campos S., Miah A. H., Mulholland K. E., Routly N., Buckley D. L., Gustafson J. L., Zinn N., Grandi P., Shimamura S., Bergamini G., Faelth-Savitski M., Bantscheff M., Cox C., Gordon D. A., Willard R. R., Flanagan J. J., Casillas L. N., Votta B. J., den Besten W., Famm K., Kruidenier L., Carter P. S., Harling J. D., Churcher I., Crews C. M., Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bondeson D. P., Smith B. E., Burslem G. M., Buhimschi A. D., Hines J., Jaime-Figueroa S., Wang J., Hamman B. D., Ishchenko A., Crews C. M., Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 25, 78–87.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schneekloth A. R., Pucheault M., Tae H. S., Crews C. M., Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 18, 5904–5908 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winter G. E., Buckley D. L., Paulk J., Roberts J. M., Souza A., Dhe-Paganon S., Bradner J. E., Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K., Hines J., Winkler J. D., Crew A. P., Coleman K., Crews C. M., Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22, 755–763 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng J., Guo J., North B. J., Tao K., Zhou P., Wei W., The emerging role for Cullin 4 family of E3 ligases in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 1871, 138–159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pettersson M., Crews C. M., PROteolysis TArgeting Chimeras (PROTACs) — Past, present and future. Drug Discov. Today Technol. 31, 15–27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo J., Liu J., Wei W., Degrading proteins in animals: “PROTAC”tion goes in vivo. Cell Res. 29, 179–180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paiva S. L., Crews C. M., Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 50, 111–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burslem G. M., Smith B. E., Lai A. C., Jaime-Figueroa S., McQuaid D. C., Bondeson D. P., Toure M., Dong H., Qian Y., Wang J., Crew A. P., Hines J., Crews C. M., The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem Biol 25, 67–77.e33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai X., Gan W., Li X., Wang S., Zhang W., Huang L., Liu S., Zhong Q., Guo J., Zhang J., Chen T., Shimizu K., Beca F., Blattner M., Vasudevan D., Buckley D. L., Qi J., Buser L., Liu P., Inuzuka H., Beck A. H., Wang L., Wild P. J., Garraway L. A., Rubin M. A., Barbieri C. E., Wong K.-K., Muthuswamy S. K., Huang J., Chen Y., Bradner J. E., Wei W., Prostate cancer–associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 23, 1063–1071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deisseroth K., Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci. 18, 1213–1225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer E. S., Böhm K., Lydeard J. R., Yang H., Stadler M. B., Cavadini S., Nagel J., Serluca F., Acker V., Lingaraju G. M., Tichkule R. B., Schebesta M., Forrester W. C., Schirle M., Hassiepen U., Ottl J., Hild M., Beckwith R. E., Harper J. W., Jenkins J. L., Thomä N. H., Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C., Han X.-R., Yang X., Jiang B., Liu J., Xiong Y., Jin J., Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). Eur. J. Med. Chem. 151, 304–314 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klán P., Šolomek T., Bochet C. G., Blanc A., Givens R., Rubina M., Popik V., Kostikov A., Wirz J., Photoremovable protecting groups in chemistry and biology: Reaction mechanisms and efficacy. Chem. Rev. 113, 119–191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.San Miguel V., Bochet C. G., del Campo A., Wavelength-selective caged surfaces: How many functional levels are possible? J. Am. Chem. Soc. 133, 5380–5388 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Hansen M. J., Velema W. A., Lerch M. M., Szymanski W., Feringa B. L., Wavelength-selective cleavage of photoprotecting groups: Strategies and applications in dynamic systems. Chem. Soc. Rev. 44, 3358–3377 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Lu G., Middleton R. E., Sun H., Naniong M., Ott C. J., Mitsiades C. S., Wong K.-K., Bradner J. E., Kaelin W. G. Jr., The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krönke J., Udeshi N. D., Narla A., Grauman P., Hurst S. N., McConkey M., Svinkina T., Heckl D., Comer E., Li X., Ciarlo C., Hartman E., Munshi N., Schenone M., Schreiber S. L., Carr S. A., Ebert B. L., Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H., Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Chiarle R., Voena C., Ambrogio C., Piva R., Inghirami G., The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 8, 11–23 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Soda M., Choi Y. L., Enomoto M., Takada S., Yamashita Y., Ishikawa S., Fujiwara S.-i., Watanabe H., Kurashina K., Hatanaka H., Bando M., Ohno S., Ishikawa Y., Aburatani H., Niki T., Sohara Y., Sugiyama Y., Mano H., Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Morris S. W., Kirstein M. N., Valentine M. B., Dittmer K. G., Shapiro D. N., Saltman D. L., Look A. T., Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 263, 1281–1284 (1994). [DOI] [PubMed] [Google Scholar]
- 27.Choi Y. L., Soda M., Yamashita Y., Ueno T., Takashima J., Nakajima T., Yatabe Y., Takeuchi K., Hamada T., Haruta H., Ishikawa Y., Kimura H., Mitsudomi T., Tanio Y., Mano H.; ALK Lung Cancer Study Group , EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 363, 1734–1739 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Kang C. H., Lee D. H., Lee C. O., Du Ha J., Park C. H., Hwang J. Y., Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem. Biophys. Res. Commun. 505, 542–547 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Robb C. M., Contreras J. I., Kour S., Taylor M. A., Abid M., Sonawane Y. A., Zahid M., Murry D. J., Natarajan A., Rana S., Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem. Commun. 53, 7577–7580 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olson C. M., Jiang B., Erb M. A., Liang Y., Doctor Z. M., Zhang Z., Zhang T., Kwiatkowski N., Boukhali M., Green J. L., Haas W., Nomanbhoy T., Fischer E. S., Young R. A., Bradner J. E., Winter G. E., Gray N. S., Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 14, 163–170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai A. C., Toure M., Hellerschmied D., Salami J., Jaime-Figueroa S., Ko E., Hines J., Crews C. M., Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 55, 807–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang H. T., Dobrovolsky D., Paulk J., Yang G., Weisberg E. L., Doctor Z. M., Buckley D. L., Cho J. H., Ko E., Jang J., Shi K., Choi H. G., Griffin J. D., Li Y., Treon S. P., Fischer E. S., Bradner J. E., Tan L., Gray N. S., A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem Biol. 25, 88–99.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kargbo R. B., Treatment of Alzheimer’s by PROTAC-Tau protein degradation. ACS Med. Chem. Lett. 10, 699–700 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Y. Naro, K. Darrah, A. Deiters, Optical control of small molecule-induced protein degradation. ChemRxiv 8216714 [Preprint], 3 June 2019. 10.26434/chemrxiv.8216714.v1. [DOI] [PMC free article] [PubMed]
- 35.Xue G., Wang K., Zhou D., Zhong H., Pan Z., Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc. 141, 18370–18374 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Beharry A. A., Woolley G. A., Azobenzene photoswitches for biomolecules. Chem. Soc. Rev. 40, 4422–4437 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Hüll K., Morstein J., Trauner D., In vivo photopharmacology. Chem. Rev. 118, 10710–10747 (2018). [DOI] [PubMed] [Google Scholar]
- 38.R. Martin, M. Bryan, M. Bérouti, D. Simoneschi, A. Marzio, M. Pagano, D. Trauner, PHOTACs enable optical control of protein degradation. ChemRxiv 8206688 [Preprint]. 31 May 2019. 10.26434/chemrxiv.8206688.v2. [DOI]
- 39.Pfaff P., Samarasinghe K. T. G., Crews C. M., Carreira E. M., Reversible spatiotemporal control of induced protein degradation by bistable photoPROTACs. ACS Cent. Sci. 5, 1682–1690 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shafirstein G., Battoo A., Harris K., Baumann H., Gollnick S. O., Lindenmann J., Nwogu C. E., Photodynamic therapy of non–small cell lung cancer. Narrative review and future directions. Ann. Am. Thorac. Soc. 13, 265–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agostinis P., Berg K., Cengel K. A., Foster T. H., Girotti A. W., Gollnick S. O., Hahn S. M., Hamblin M. R., Juzeniene A., Kessel D., Korbelik M., Moan J., Mroz P., Nowis D., Piette J., Wilson B. C., Golab J., Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 61, 250–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore C. M., Pendse D., Emberton M., Photodynamic therapy for prostate cancer—A review of current status and future promise. Nat. Clin. Pract. Urol. 6, 18–30 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Bethea D., Fullmer B., Syed S., Seltzer G., Tiano J., Rischko C., Gillespie L., Brown D., Gasparro F. P., Psoralen photobiology and photochemotherapy: 50 years of science and medicine. J. Dermatol. Sci. 19, 78–88 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Randleman J. B., Santhiago M. R., Kymionis G. D., Hafezi F., Corneal cross-linking (CXL): Standardizing terminology and protocol nomenclature. J. Refract. Surg. 33, 727–729 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Oikawa S., Tada-Oikawa S., Kawanishi S., Site-specific DNA damage at the GGG sequence by UVA involves acceleration of telomere shortening. Biochemistry 40, 4763–4768 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Mouret S., Baudouin C., Charveron M., Favier A., Cadet J., Douki T., Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. U.S.A. 103, 13765–13770 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cadet J., Douki T., Oxidatively generated damage to DNA by UVA radiation in cells and human skin. J. Invest. Dermatol. 131, 1005–1007 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Gorka A. P., Nani R. R., Zhu J., Mackem S., Schnermann M. J., A near-IR uncaging strategy based on cyanine photochemistry. J. Am. Chem. Soc. 136, 14153–14159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen S., Weitemier A. Z., Zeng X., He L., Wang X., Tao Y., Huang A. J. Y., Hashimotodani Y., Kano M., Iwasaki H., Parajuli L. K., Okabe S., Teh D. B. L., All A. H., Tsutsui-Kimura I., Tanaka K. F., Liu X., McHugh T. J., Near-infrared deep brain stimulation via upconversion nanoparticle–mediated optogenetics. Science 359, 679–684 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay5154/DC1
Fig. S1. The schematic illustration of the synthesis of opto-pomalidomide and the working model for opto-pomalidomide to achieve light-induced degradation of IKZF1/3.
Fig. S2. UPLC-MS characterization and the time-dependent uncaging of pomalidomide and opto-pomalidomide.
Fig. S3. Uncaging of opto-pomalidomide by UVA irradiation leads to active degradation of IKZFs in multiple myeloma cancer cell lines.
Fig. S4. The schematic illustration of the synthesis of opto-dBET1 and the working model for opto-dBET1 to achieve light-induced degradation of BRDs.
Fig. S5. UPLC-MS characterization and the time-dependent uncaging of opto-dBET1 by UVA irradiation in vitro.
Fig. S6. dBET1 inhibits cell proliferation in a CRBN-dependent manner.
Fig. S7. The schematic illustration of the synthesis of opto-dALK and the working model for opto-dALK to achieve light-induced degradation of ALK fusion proteins.
Fig. S8. UPLC-MS characterization and the time-dependent uncaging process of opto-dALK.
Fig. S9. Dose-dependent degradation of ALK fusion proteins by dALK.