

Abstract
Rationale:
Hypertension represents a major risk factor for stroke, myocardial infarction, and heart failure and affects 30% of the adult population. Mitochondrial dysfunction contributes to hypertension, but specific mechanisms are unclear. The mitochondrial deacetylase Sirt3 is critical in the regulation of metabolic and antioxidant functions which are associated with hypertension, and cardiovascular disease risk factors diminish Sirt3 level.
Objective:
We hypothesized that reduced Sirt3 expression contributes to vascular dysfunction in hypertension but increased Sirt3 protects vascular function and decreases hypertension.
Methods And Results:
To test the therapeutic potential of targeting Sirt3 expression we developed new transgenic mice with global Sirt3 overexpression (Sirt3OX) which protects from endothelial dysfunction, vascular oxidative stress and hypertrophy, attenuates angiotensin II- and DOCA-salt induced hypertension. Global Sirt3 depletion in Sirt3−/− mice results in oxidative stress due to SOD2 hyperacetylation, increases HIF1α, reduces endothelial cadherin, stimulates vascular hypertrophy, increases vascular permeability and vascular inflammation (p65, caspase 1, VCAM, ICAM and MCP1), increases inflammatory cell infiltration in the kidney, reduces telomerase expression, and accelerates vascular senescence and age-dependent hypertension; conversely, increased Sirt3 expression in Sirt3OX mice prevents these deleterious effects. The clinical relevance of Sirt3 depletion was confirmed in arterioles from human mediastinal fat in patients with essential hypertension showing a 40% decrease in vascular Sirt3, coupled with Sirt3 dependent 3-fold increases in SOD2 acetylation, NfKB activity, VCAM, ICAM and MCP1 levels in hypertensive subjects compared with normotensive subjects.
Conclusions:
We suggest that Sirt3 depletion in hypertension promotes endothelial dysfunction, vascular hypertrophy, vascular inflammation and end-organ damage. Our data support a therapeutic potential of targeting Sirt3 expression in vascular dysfunction and hypertension.
Subject Terms: Endothelium/Vascular Type/Nitric Oxide, High Blood Pressure, Hypertension, Hypertrophy, Vascular Disease
Keywords: Hypertension, mitochondria, superoxide, superoxide dismutase, Sirtuin 3, high blood pressure, oxidative stress, endothelial dysfunction, vascular inflammation
Graphical Abstract
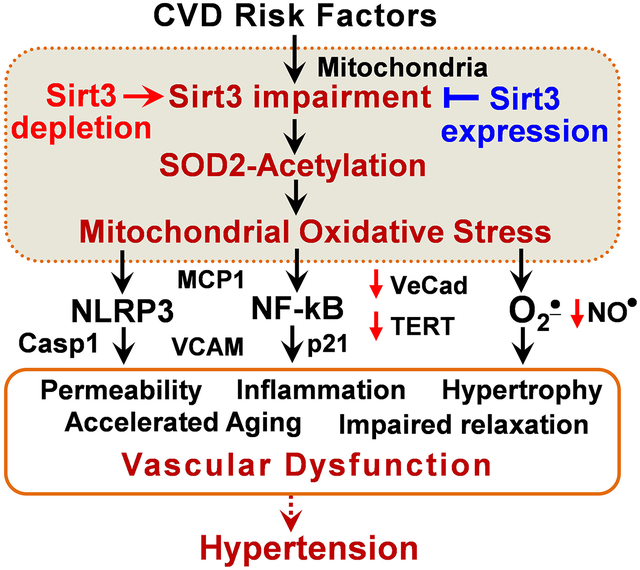
NAD+ dependent mitochondrial deacetylase Sirt3 is a key regulator of antioxidant and metabolic functions. Sirt3 level declines with age paralleling the increased incidence of hypertension. In this work we described a novel therapeutic potential of targeting Sirt3 expression using new transgenic global Sirt3 overexpressing mice. Sirt3 overexpression inhibits vascular oxidative stress and hypertrophy, preserves endothelial-dependent relaxation and vascular permeability, attenuates angiotensin II- and DOCA-salt hypertension. Sirt3 depletion induces pathophysiological metabolic and phenotypic vascular alterations by increased HIF1α, reduced VE-cadherin, elevated endothelial permeability, activation of NFkB and inflammasome pathways, vascular cell-senescence and aging, infiltration of T cells and age-dependent hypertension while increased Sirt3 expression prevents these deleterious effects. The clinical relevance of Sirt3 depletion was confirmed in arterioles from human mediastinal fat in patients with essential hypertension showing 40% decrease in vascular Sirt3, coupled with SOD2 acetylation, vascular inflammation and cell-senescence markers in hypertensive subjects compared to vessels from normotensive subjects. Our data support a therapeutic potential for targeting Sirt3 expression in treatment of vascular dysfunction and hypertension.
INTRODUCTION
Hypertension represents a major risk factor for stroke, myocardial infarction, and heart failure which causes one-third of deaths worldwide.1, 2 Hypertension is a multifactorial disorder involving perturbations of the vasculature, kidney and central nervous system.3 Despite treatment with multiple drugs, 37% of hypertensive patients remain hypertensive,4 likely due to mechanisms contributing to blood pressure elevation that are not affected by current treatments.
Vascular dysfunction is crucial in hypertension pathophysiology and exhibits a bidirectional relationship.5 Endothelial dysfunction leads to and accelerates the progression of hypertension while hypertension causes vascular dysfunction. Metabolic disorders and oxidative stress contribute to the pathogenesis of vascular dysfunction, and mitochondrial deacetylase Sirt3 is critical in the regulation of metabolic and antioxidant functions,6, 7 however, the role of Sirt3 has been largely ignored. Clinical studies show that cardiovascular disease risk factors reduce Sirt3 level and Sirt3 declines with age,8, 9 paralleling the increased incidence of cardiovascular disease and hypertension.9 We have previously shown that Sirt3 depletion in Sirt3−/− mice increases hypertension which was linked to hyperacetylation of the key mitochondrial antioxidant, superoxide dismutase 2 (SOD2), leading to SOD2 inactivation and mitochondrial oxidative stress.10, 11 Analysis of human subjects with essential hypertension showed an increase in SOD2 acetylation and decrease in Sirt3 levels in peripheral blood mononuclear cells10 supporting the association of Sirt3 depletion and hypertension. The causative role of Sirt3 depletion in vascular alterations remains unclear and the therapeutic potential of targeting Sirt3 expression in vascular dysfunction and hypertension is not known.
Mitochondria become dysfunctional in hypertension,12, 13 however, the precise role of mitochondrial dysfunction remains unclear. We have previously shown that angiotensin II and inflammatory cytokines promotes mitochondrial dysfunction.14 Activation of RAS/AngII/AT1R and inflammation reduce Sirt3 levels,15, 16 while Sirt3 expression was associated with reduced ventricular hypertrophy, attenuated cardiomyopathy and diminished inflammatory injury.17, 18 It is conceivable that mitochondria are both the target and the regulator of inflammatory pathways. Indeed, inflammation plays a critical role in the pathogenesis of endothelial dysfunction and hypertension,19 and overexpression of mitochondrial antioxidant SOD2 protects from cytokine-mediated vascular dysfunction and attenuates hypertension.12, 14 Sirt3 activates SOD2 by deacetylation of specific lysine residues.7 We hypothesized that increased Sirt3 expression prevents SOD2 hyperacetylation, attenuates mitochondrial oxidative stress and reduces vascular inflammation which can protect vascular function and reduce hypertension.
Previous studies have shown that Sirt3 overexpression protects from doxorubicin-induced cardiomyopathy in mice.20 The whole-body Sirt3-transgenic mice were generated by crossing loxP-stop-LoxP-SIRT3 transgenic mice with mice expressing Cre under the control of the human β-actin promoter.21 The whole body Sirt3 overexpressing mice showed significantly reduced expression of fibrotic markers and were found resistant to developing angiotensin-II-mediated cardiac fibrosis.22 Meanwhile, the protective potential of Sirt3 expression on vascular function and hypertension has not been studied.
Hypertension is associated with mitochondrial dysfunction23, 24 and targeting mitochondrial health is emerging as a novel approach to treat hypertension.25, 26 We have previously shown mitochondrial dysfunction in two commonly used mouse models of hypertension: (1) angiotensin II infusion; and (2) administration of deoxycorticosterone acetate (DOCA) and sodium chloride to uninephrectomised mice.12 The DOCA-salt model of hypertension differs from angiotensin II–induced hypertension because it is largely volume dependent and is associated with suppressed plasma renin activity. Interestingly, treatment of hypertensive mice with mitochondria-targeted SOD2 mimetic mitoTEMPO improved endothelial function and reduced blood pressure in both angiotensin II and DOCA-salt models.12 We hypothesized that overexpression of mitochondrial Sirt3 will prevent the SOD2 hyperacetylation linked to SOD2 inactivation in hypertension10 and, therefore, attenuate hypertension in both angiotensin II and DOCA-salt animal models.
To test the therapeutic potential of Sirt3 overexpression we developed new transgenic mice with global Sirt3 overexpression (Sirt3OX) by crossing the EIIa-cre with loxP-stop-LoxP-Sirt3 mice.22 This resulted in constitutively increased whole body Sirt3 expression on a C57Bl/6J background mice. We studied the effects of Sirt3 overexpression in mice on SOD2 acetylation, markers of vascular inflammation, oxidative stress and hypertension. The results of the animal studies were verified by analysis of Sirt3 expression, SOD2 acetylation and vascular inflammation in arterioles isolated from human mediastinal fat samples taken from patients with and without essential hypertension. These data support the hypothesis that increased Sirt3 expression reduces oxidative stress, inhibits vascular inflammation and endothelial dysfunction, reduces vascular hypertrophy, attenuates vascular aging and hypertension.
METHODS
The authors declare that all supporting data are available within the article and its online supplementary files. All methods have corresponding literature reference. Additional protocol information is available from the corresponding author upon reasonable request.
Reagents.
DHE superoxide probe was supplied by Invitrogen (Grand Island, NY). Caspase 1 (2225S), Sirt3 (54905), and p21 (S807) antibodies were from Cell Signaling. VCAM (ab174279), ICAM (ab53013), MCP1 (ab214819), p65 (ab97726), TERT (ab32020), HIF1α (187524), Vecad (ab33168) were from Abcam. The specificity of acetyl-K68-SOD2 antibodies (Abcam, ab137037) was previously validated in mice with reduced Sirt3 expression and site-directed mutagenesis of K68 in cells.27–29 SOD2 (sc30080) and β-galactosidase (sc66586) antibodies were obtained from Santa Cruz Biotechnology. The antibodies and fluorophores for flow cytometry were purchased from Biolegend (San Diego, CA) and included: 7-AAD for live/dead staining; BV510-conjugated anti-CD45 (30-F11); PE/Cy7-conjugated anti-CD4 (GK1.5); APC/Cy7-conjugated anti-CD8 (53-6.7); PE -conjugated anti-CD3 (145-2C11); FITC-conjugated CD44 (IM7); APC-conjugated anti-CD62L(MEL-14); FITC-conjugated anti-F4/80 (BM8). All other reagents were obtained from Sigma (St Louis, MO).
Animal experiments.
Whole-body Sirt3-transgenic Sirt3 overexpressing mice (Sirt3OX) were generated by crossing loxP-stop-LoxP-SIRT3 transgenic mice22 with EIIa-cre (C57Bl/6J background, Jackson Labs) resulting in constitutively increased Sirt3 (~4-fold) in the whole body (Sirt3OX). In this work we used both male and female Sirt3OX mice at the ages three to twelve months.22 Mice lacking the Sirt3 protein (Sirt3−/−)30 and their wild-type littermates on a C57BL/6J background were provided by Dr. David H. Wasserman and Louise Lantier.31 We used both male and female Sirt3−/− mice at the ages three to fifteen months. Hypertension was induced by angiotensin II (0.7 mg/kg/min) or DOCA-salt as described previously.32 To test the therapeutic potential of Sirt3 overexpression, wild-type and Sirt3OX mice received saline or angiotensin II minipump placement, Sham or DOCA-salt. Blood pressure was monitored by the telemetry as previously described.33, 34 The Vanderbilt Institutional Animal Care and Use Committee approved the procedures. Simple randomization was used to select animals for sham, angiotensin II or DOCA-salt groups for equal chance of being allocated to treatment groups. Animal samples were assigned a code and post-study unblinding released the masked data upon completion of the study.
Superoxide measurements using HPLC.
Mouse aortic segments were loaded with DHE (50 μM) in KHB buffer by 30-minute incubation in a tissue culture incubator at 37°C. Next, aortic segments were placed in methanol (300 μl) and homogenized with a glass pestle. The tissue homogenate was passed through a 0.22 μm syringe filter and methanol filtrates were analyzed by HPLC according to previously published protocols.35 DHE-superoxide specific product 2-hydroxyethidium was detected using a C-18 reverse-phase column (Nucleosil 250 to 4.5 mm) and a mobile phase containing 0.1% trifluoroacetic acid and an acetonitrile gradient (from 37% to 47%) at a flow rate of 0.5 ml/min. 2-Hydroxyethidium was quantified by fluorescence detector using an emission wavelength of 580 nm and an excitation of 480 nm as described previously.12
Nitric oxide measurements by Electron Spin Resonance.
Nitric oxide production in endothelial cells and vessels was quantified by ESR and colloid Fe(DETC)2 as we have described previously.36 All ESR samples were placed in quartz Dewar (Corning, New York, NY) filled with liquid nitrogen. ESR spectra were recorded using a EMX ESR spectrometer (Bruker Biospin Corp., Billerica, MA) and a super high Q microwave cavity. The ESR settings were as follows: field sweep, 160 Gauss; microwave frequency, 9.42 GHz; microwave power, 10 milliwatts; modulation amplitude, 3 Gauss; scan time, 150 msec; time constant, 5.2 sec; and receiver gain, 60 dB (n = 4 scans).
Vasodilatation study.
Isometric tension studies were performed on 2 mm mouse aortic rings dissected free of perivascular fat from C57B//6J and Sirt3−/− mice. Studies were performed in a horizontal wire myograph (DMT, Aarhus, Denmark, models 610M and 620M) containing physiological salt solution with the composition of 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM NaHCO3, 11 mM glucose, and 1.8 mM CaCl2. The isometric tone of each vessel was recorded using LabChart Pro v7.3.7 (AD Instruments, Australia). The aortic rings were equilibrated over a 2-hour period by heating and stretching the vessels to an optimal baseline tension of 36 mNewtons before contracting them with three cycles of 60 mM KCl physiological saline solution. Endothelial dependent and independent vascular relaxation was tested after pre-constriction with 1μM phenylephrine. Once the vessels reach a steady state contraction, increasing concentrations of acetylcholine were administered, and the response to each concentration of drug was recorded.
Flow cytometry.
To examine renal inflammatory cell infiltration, single cell suspensions were prepared and analyzed by flow cytometry as previously described.37
Human studies.
Arterioles were harversted from human mediastinal fat samples obtained from patients during cardiac surgery enrolled in a Risk of Oxygen during Cardiac Surgery (ROCS) randomized clinical trial with essential hypertension (BP>140/90 mmHg) and normotensive subjects as previously described38 for Western blot analysis of Sirt3, SOD2, SOD2 acetylation, NFkB activity marker p65, cell-senescence marker p21, antiaging marker telomerase, inflammation markers VCAM, ICAM and MCP1. Full informed consent was obtained for all tissue samples.
Statistics.
Data are expressed as mean ± SEM. To compare the effect of Sirt3 knockout or overexpression in response to angiotensin II infusion, two-way analysis of variance (ANOVA) was used followed with Bonferroni post hoc test. For data involve more than two groups, one-way ANOVA followed with Bonferroni post hoc test was used. For telemetry blood pressure measurements over time, two-way ANOVA with repeated-measures was employed using GraphPad Prizm 7. P levels < 0.05 were considered significant.
RESULTS
Sirt3 overexpression attenuates vascular oxidative stress and diminishes hypertension.
We tested whether global Sirt3 overexpression in mice improves endothelial function and attenuates angiotensin II-induced hypertension. A new mice strain was produced by crossing the EIIa-cre with loxP-stop-LoxP-Sirt322 resulting in constitutively increased Sirt3 expression in the whole body (Sirt3OX) on C57Bl/6J background. Sirt3OX and wild-type mice underwent telemetry placement for subsequent blood pressure monitoring. Ten days later, osmotic pumps with angiotensin II (0.7 mg/kg/day) were implanted. Sirt3OX mice had slightly reduced basal blood pressure compared to wild-type mice. Infusion of wild-type mice with angiotensin II led to severe hypertension with night systolic maximum blood pressure of 180 mm Hg. Sirt3 overexpression attenuated angiotensin II-induced hypertension by 25 mm Hg (Figure 1A). Angiotensin II-induced hypertension was linked to impaired endothelial-dependent relaxation in wild-type mice but Sirt3 overexpressing mice had completely protected endothelial-dependent relaxation (Figure 1B). Hypertension is linked to vascular oxidative stress but Sirt3 overexpression prevented vascular superoxide overproduction (Figure 1C) and significantly attenuated the loss of endothelial nitric oxide in angiotensin II-infused mice (Figure 1D).39 These data support an important role of Sirt3 in vascular function and hypertension.
Figure 1: Blood pressure, vascular relaxation, aortic superoxide and nitric oxide production in wild-type and Sirt3OX mice in angiotensin II model.
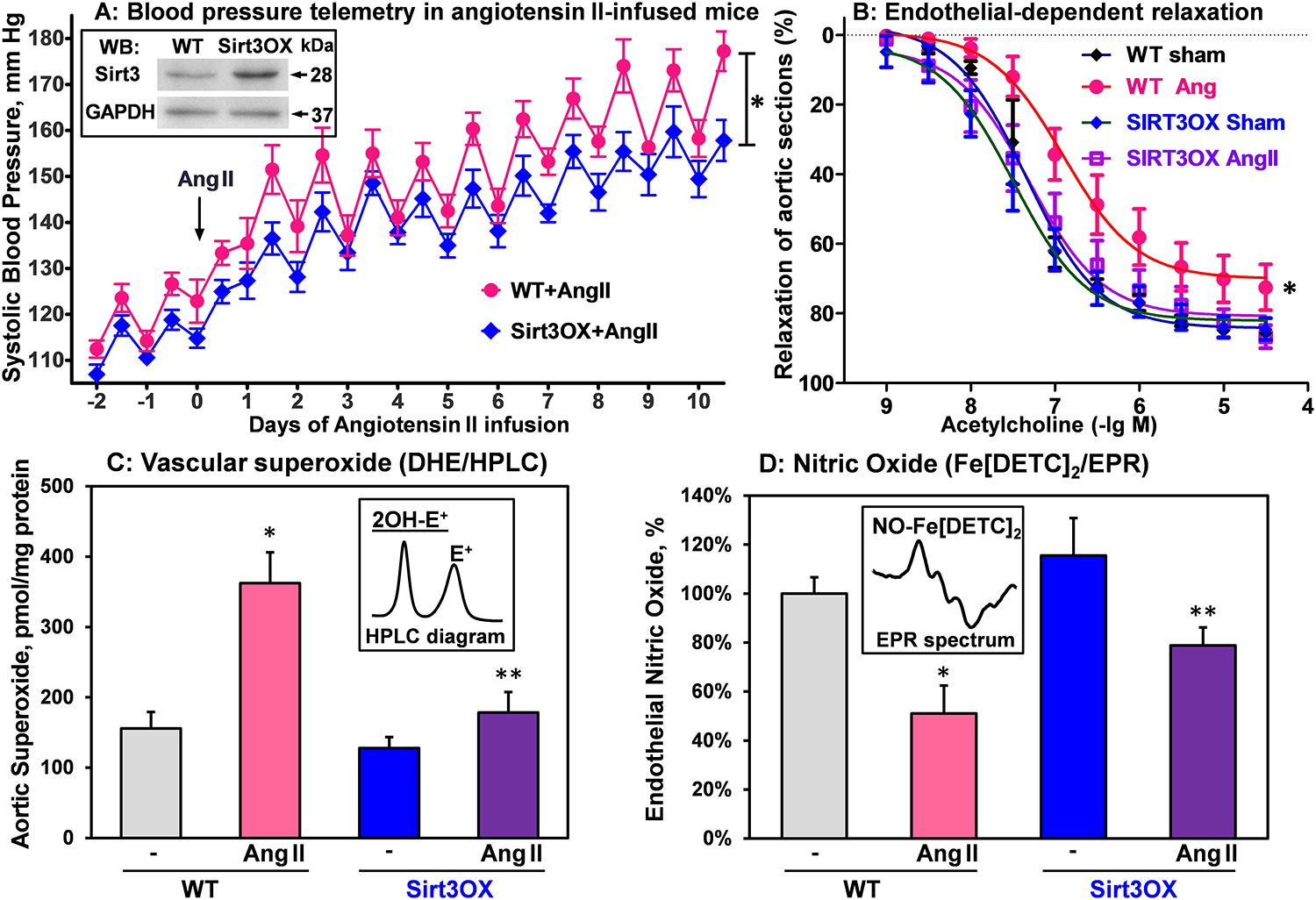
(A) Systolic blood pressure was measured by telemetry before and after Ang II-infusion (0.7 mg/kg/day). Data were analyzed using 2-way ANOVA with repeated measurements. *P=0.02, n=8 in each group. Insert shows representative Western blot of Sirt3 aortas isolated from Sirt3OX mice and wild-type C57Bl/6J littermates. (B) Endothelial-dependent relaxation to acetylcholine. Vasculare relaxation data were analyzed using 2-way ANOVA with repeated measurements. *P=0.01 Sirt3OX+Ang II vs WT+Ang II, n=8 in each group. (C) Aortic superoxide (O2•) was measured by DHE-HPLC assay of O2• specific product, 2-hydroxyethidium (2-OH-E+).12 (D) Endothelial nitric oxide (NO) measured by NO spin trap FeDETC2 and Electron Paramagnetic Resonance (EPR).12 For (C) and (D), data were analyzed using 2-way ANOVA and Bonferroni post-hoc multiple comparisons. Results are mean ± SEM (n=5). *P<0.01 vs WT, **P<0.01 Sirt3OX+Ang II vs WT+Ang II.
In additional experiments, we investigated DOCA-salt induced hypertension in wild-type and Sirt3OX mice. Both angiotensin II and DOCA-salt induced hypertension depends on inflammation and endothelial dysfunction.40 However, the DOCA-salt model of hypertension differs from angiotensin II-induced hypertension because the former is largely volume dependent and is associated with suppressed plasma renin activity. Wild-type and Sirt3OX mice had DOCA-salt surgery as we described previously.12 Briefly, the left kidney was removed through a left-flank incision, a DOCA pellet (50 mg) was inserted subcutaneously through a midscapular incision and drinking water was changed to 1% saline. Control mice were sham operated. Blood pressure was monitored by telemetry. Twenty-one days after surgery, the animals were sacrificed and we isolated aortas for analysis of vascular superoxide using DHE probe and HPLC analysis of the superoxide specific product, 2-hydroxyethydium.12 Studies in the DOCA-salt model of hypertension showed significant antihypertensive effect of Sirt3 overexpression (Figure 2B). Interestingly, Sirt3 overexpression completely inhibited the DOCA-salt induced superoxide overproduction (Figure 2B).39 These data support a therapeutic potential of increased Sirt3 expression.
Figure 2: Blood pressure and vascular superoxide in wild-type and Sirt3OX mice in DOCA-salt model.
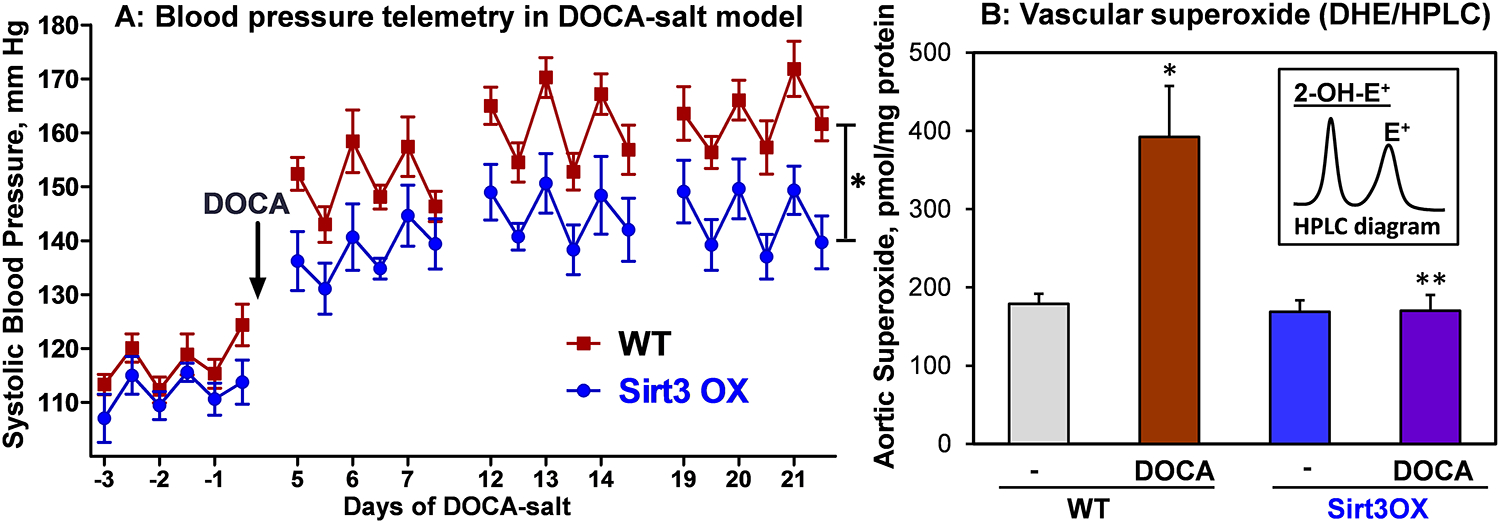
(A) Systolic blood pressure was measured by telemetry before and after DOCA-salt treatment. Results are mean ± SEM. Blood pressure data were analyzed using 2-way ANOVA with repeated measurements. *P=0.03 vs WT+DOCA, n=7 in each group. (B) Aortic superoxide was measured by HPLC analysis of DHE-O2• specific product, 2-hydroxyethidium (2-OH-E+).12 Results are mean ± SEM (n=5). Data were analyzed using 2-way ANOVA and Bonferroni post-hoc multiple comparisons. *P<0.001 vs WT, **P<0.001 Sirt3OX+DOCA vs WT+DOCA.
Sirt3 overexpression attenuates angiotensin II-induced vascular hypertrophy.
We had previously shown that increased vascular oxidative stress promotes vascular hypertrophy while antioxidant treatment with superoxide scavengers reduces vascular hypertrophy.32 The pathophysiological significance of Sirt3 in vascular hypertrophy has not been tested. We hypothesized that Sirt3 overexpression reduces vascular hypertrophy since Sirt3 prevents vascular oxidative stress. To test this hypothesis, we quantified aortic hypertrophy following two weeks of angiotensin II infusion as we have described previously.32 As expected, angiotensin II infusion increased vascular hypertrophy in wild-type mice; however, Sirt3 overexpression significantly diminished smooth muscle hypertrophy in angiotensin II-infused Sirt3OX mice (Figure 3). Furthermore, Sirt3 depletion in sham Sirt3−/− mice increased vascular hypertrophy compared to wild-type littermates while Sirt3 overexpression significantly reduced aortic wall thickness in sham Sirt3OX mice (Figure 3). These data support the pathophysiological role of Sirt3 depletion in vascular hypertrophy.
Figure 3: Vascular hypertrophy in sham and Ang II-infused wild-type, Sirt3−/− and Sirt3OX mice.
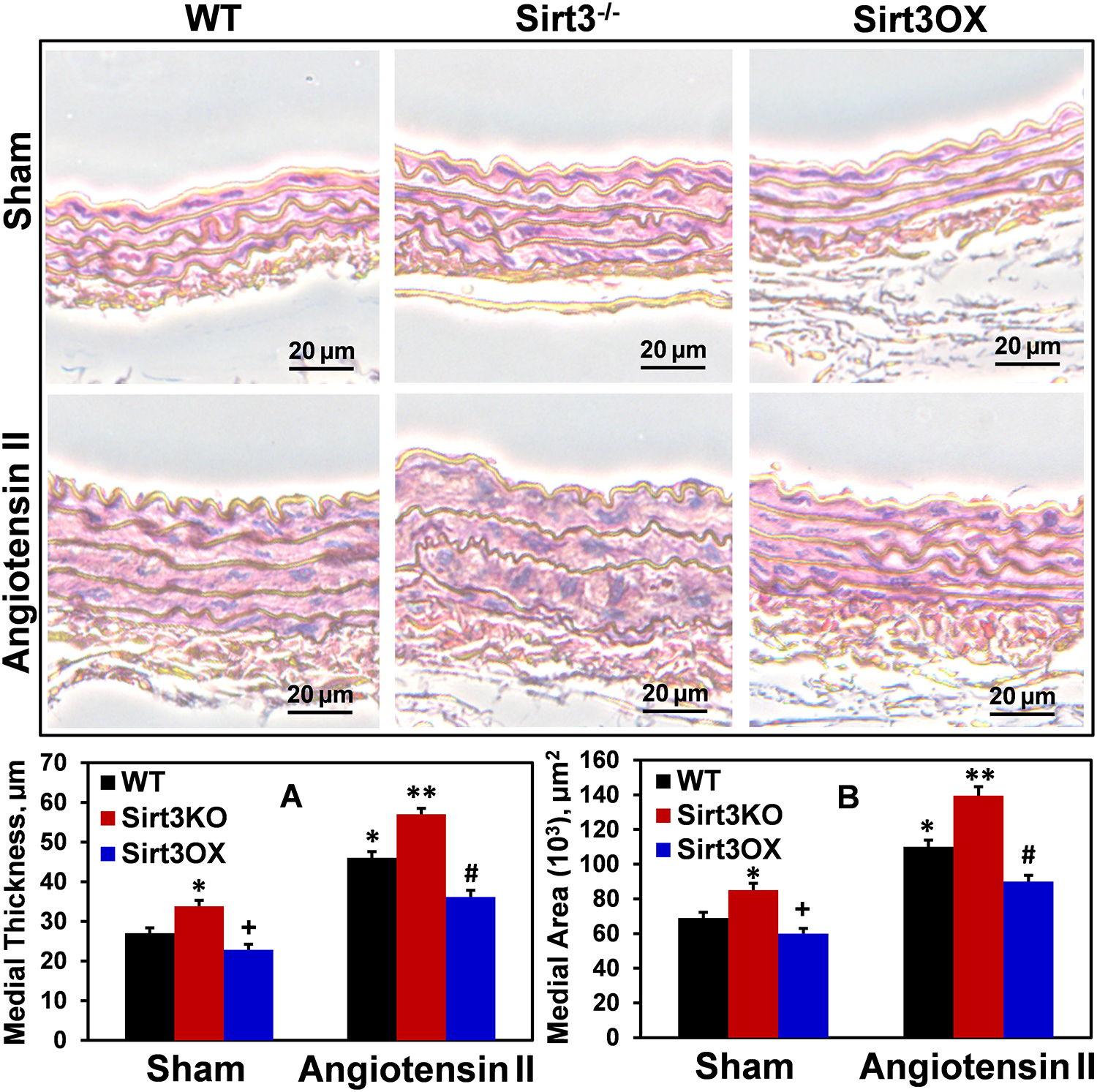
Representative images of hematoxylin and eosin-stained aortic cross sections are shown. Aortic thickness (A) and medial area (B) were quantified as described previously.32 Supplemental Online Figure I displays typical low magnification images with the intact vascular lumen to show the range of vessel wall thickness. Aortic hypertrophy was increased in Sirt3−/− mice, and Sirt3 overexpression reduced vascular hypertrophy in Sirt3OX mice compared to wild-type littermates. Data were analyzed using 2-way ANOVA and Bonferroni post-hoc multiple comparisons. Values are mean ± SEM. (A) *P=0.007 Sirt3−/− Sham vs WT sham, *P=0.00006 WT+Ang II vs WT sham, +P=0.003 vs WT sham, **P=0.0035 vs WT+Ang II, #P=0.0038 vs WT+Ang II (n=5). (B) *P=0.0012 Sirt3−/− Sham vs WT sham, *P=0.00005 WT+Ang II vs WT sham, +P=0.04 vs WT sham, **P=0.001 vs WT+Ang II, #P=0.0007 vs WT+Ang II (n=5).
Sirt3 depletion rises HIF1α and vascular permeability which is normalized by Sirt3 overexpression.
Metabolism is critical in the regulation of endothelial cells, and inhibition of fatty acid oxidation and glycolysis leads to phenotypic alterations, endothelial permeability and inflammation.41, 42 Sirt3 is critical in fatty acid metabolism and antioxidant regulation.6, 25 We hypothesized that Sirt3 depletion promotes a metabolic switch to glycolysis via upregulation of redox-dependent HIF1α, leading to diminished expression of VE-cadherin42 and dysfunction of the endothelial barrier.43 To test this hypothesis, we measured HIF1α and VE-cadherin in aorta isolated from sham and angiotensin II-infused Sirt3−/−, Sirt3OX and wild-type mice. Angiotensin II infusion significantly increased SOD2 acetylation in wild-type mice and this was further exacerbated in Sirt3−/− mice but Sirt3 overexpression substantially diminished SOD2 acetylation while SOD2 protein levels were similar in wild-type, Sirt3−/− and Sirt3OX mice. This data support the SOD2 inactivation due to SOD2 hyperacetylation in response to angiotensin II infusion and Sirt3 depletion (Figure 4A). Angiotensin II significantly increased vascular HIF1α by 2-fold and reduced VE-cadherin which was associated with increased vascular permeability in wild-type mice (Figure 4). Sirt3 depletion in Sirt3−/− mice significantly increased both basal and angiotensin II-induced HIF1α, and reduced VE-cadherin compared with wild-type mice. As endothelial permeability increases, the access of cytokines and vasoactive substances to tissue is greater contributing to inflammation, hypertrophy, hypertension, and end-organ-damage. Interestingly, Sirt3 overexpression completely prevents upregulation of HIF1α, preserves VE-cadherin level and normalizes the endothelial permeability (Figure 4).
Figure 4: Analysis of SOD2 acetylation, HIF1α, VE-cadherin and vascular permeability in aortas isolated from Sham and Ang II-infused Sirt3−/−, Sirt3OX and wild-type mice.
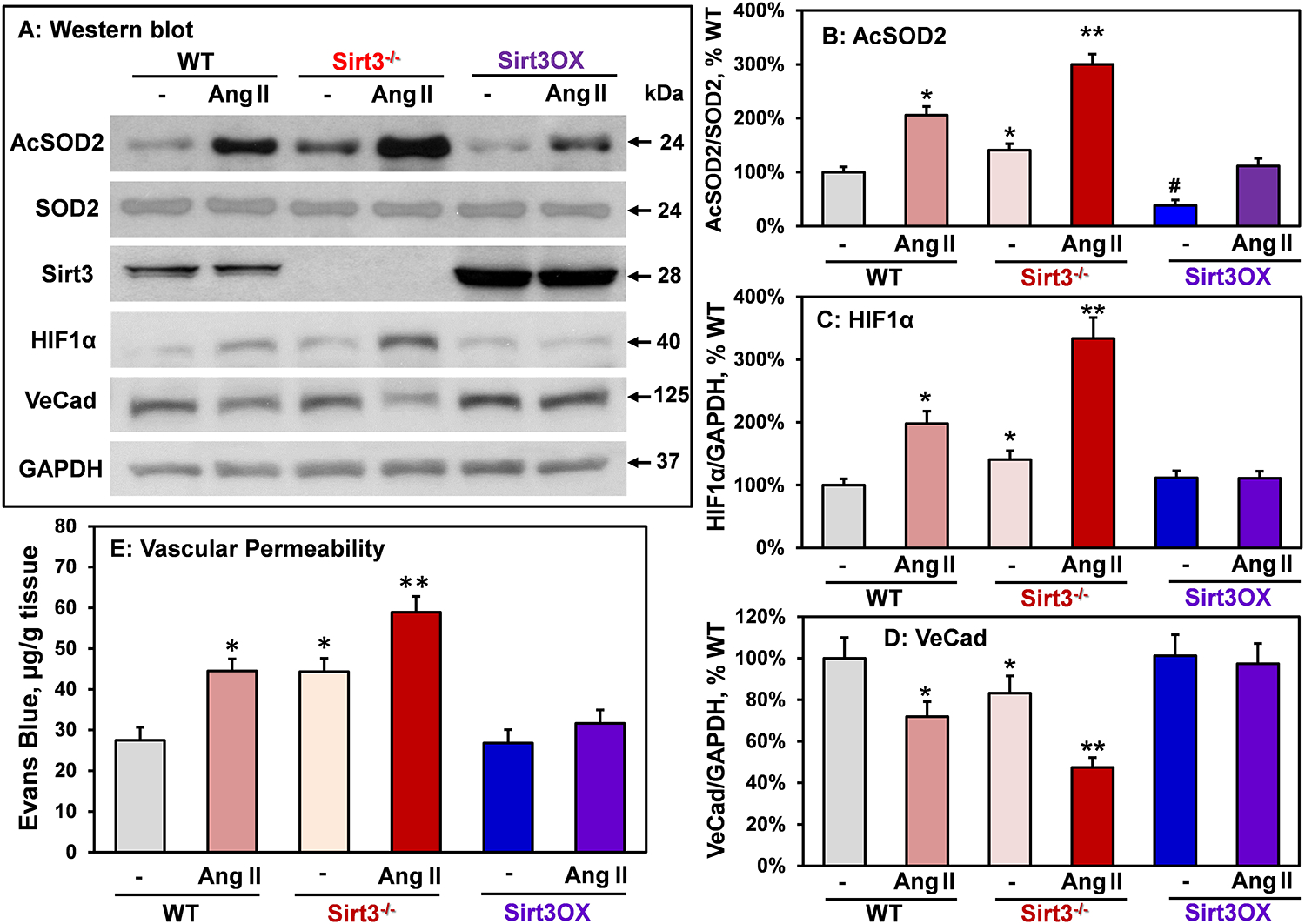
(A) Typical Western blots of Acetyl-K68-SOD2, total SOD2, HIF1α and VE-cadherin normalized for GAPDH in aortas. (B) SOD2-K68 acetylation, (C) HIF1α expression and (D) VE-cadherin levels normalized by GAPDH compared to Sham wild-type mice (100%). *P<0.05 vs WT sham, #P<0.05 vs WT sham **P<0.01 vs WT+Ang II (n=4). (E) Vascular permeability was measured by accumulation of Evans Blue dye in aortas. Male mice were infused with Ang II (0.7 mg/kg/day) or saline for two weeks prior to Miles assay.78 Data were analyzed using 2-way ANOVA and Bonferroni post-hoc multiple comparisons. *P<0.01 vs WT, **P<0.01 vs WT+Ang II (n=6).
Sirt3 depletion induces vascular inflammation and inflammatory cell infiltration which are prevented by Sirt3 overexpression.
We hypothesized that global Sirt3 depletion raises vascular inflammation via redox activated NFkB44 and NLRP3 inflammasome45 which induce vascular cell adhesion molecule-1 (VCAM), ICAM and monocyte chemoattractant protein-1 (MCP1)46 to augment vascular inflammation and dysfunction. Indeed, Western blot of aortas isolated from Sham and angiotensin II-infused Sirt3−/− mice showed significant increase in p65 (NFkB subunit)47 and caspase-1 (Casp1)45 indicating NFkB and inflammasome activation (Figure 5). Vascular cell inflammation was supported by increased levels of VCAM and ICAM (surrogate markers of NFkB activity) and MCP1 in Sirt3−/− aortas. Interestingly, Sirt3 overexpression reduced basal vascular p65, VCAM, ICAM and MCP1 levels and attenuated angiotensin II-induced vascular inflammation (Figure 5A–D).
Figure 5: Analysis of inflammasome activation, vascular inflammation and of inflammatory cells infiltration in Sirt3−/−, Sirt3OX and wild-type mice.
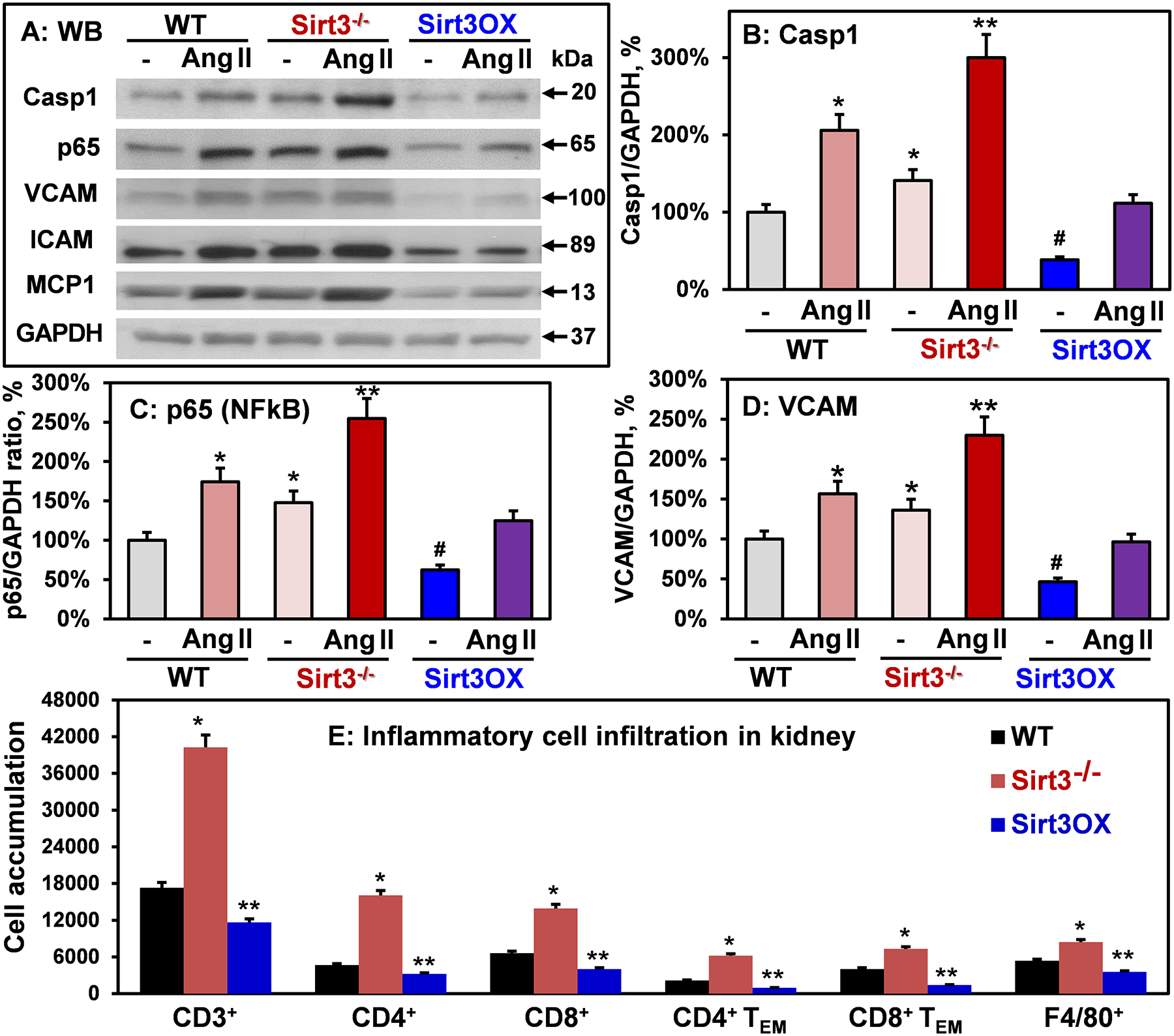
(A) Typical Western blots of caspase 1, p65, VCAM, ICAM and MCP1 normalized for GAPDH in aortas. (B) caspase 1, (C) p65 (NFkB subunit) and (D) VCAM levels normalized by GAPDH compared to Sham wild-type mice (100%). Results are mean ± SEM (n=6). *P<0.05 vs WT Sham, **P<0.01 vs WT+Ang II, #P<0.01 vs WT Sham. (E) Accumulation of T cells and macrophages in kidneys of six month old Sirt3−/−, Sirt3OX and wild-type mice measured by flow cytometry as described previously.53 Online Figure II shows the representative images of flow cytometry and gating strategy. Results are mean ± SEM (n=4). Data were analyzed using 2-way ANOVA and Bonferroni post-hoc multiple comparisons. *P<0.01 vs WT, **P<0.05 vs WT.
Sirt3 depletion accelerates vascular aging and senescence promoting age dependent hypertension.
Sirt3 is associated with human longevity,48, 49 and cardiovascular disease risk factors and aging are linked to reduced Sirt3,8, 9 paralleling the increased incidence of hypertension. We hypothesized that Sirt3 depletion accelerates vascular aging and development of age-associated hypertension. To test this hypothesis we analyzed the aortic expression of genes linked to aging and cell-senescence such as telomerase reverse transcriptase (TERT),50 cyclin-dependent kinase inhibitor 1 (p21),51 senescence-associated β-galactosidase (SA-β-gal)52 and age-dependent systolic blood pressure in wild-type, Sirt3−/− and Sirt3 overexpressing six-month old male and female mice. Sirt3 depletion reduced TERT level by 50% while Sirt3 overexpression doubled the TERT level compared to the wild-type mice. Markers of cell-senescence p21 and SA-β-gal were significantly increased in the Sirt3−/− aortas but Sirt3 overexpression reduced cell-senescence markers compared to wild type mice (Figure 6A–C). The increase in vascular aging and cell senescence markers in Sirt3−/− mice was associated with the age dependent increase of systolic blood pressure (Figure 6D). These data support the role of Sirt3 depletion in accelerated vascular aging and age-related hypertension.
Figure 6: Western blot analysis of aging and cell senescence markers and age dependent hypertension in Sirt3−/− mice.
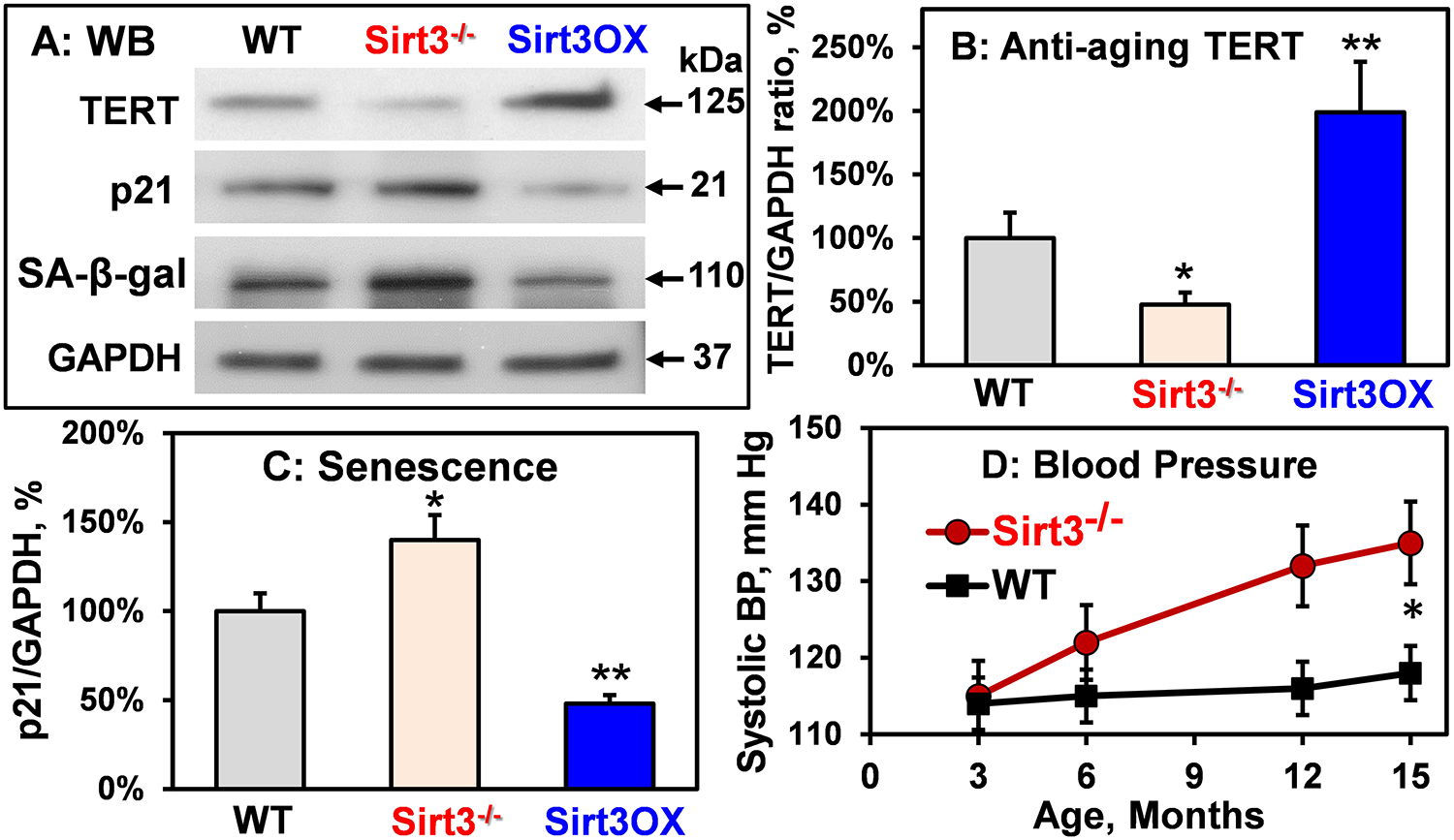
(A) Typical Western blots of aortic TERT, p21, and SA-β-gal in 6-month-old wild-type, Sirt3−/− and Sirt3OX mice. (B) TERT and (C) p21 levels normalized by GAPDH compared to Sham wild-type mice (100%). Results are mean ± SEM (n=5). Data were analyzed using 1-way ANOVA and Bonferroni post-hoc multiple comparisons. *P<0.01 vs WT, **P<0.01 vs WT. (D) Systolic blood pressure in Sirt3−/− and WT mice. Data were analyzed with 2-way ANOVA with repeated measurements. *P=0.008 vs WT (n=6).
As described above, Sirt3 depletion increases vascular permeability, vascular inflammation, and vascular aging. We tested if this is associated with increased end-organ inflammation. T cells and macrophages are important in end-organ damage and hypertension.53 As determined by flow cytometry, at six months of age, kidneys from Sirt3−/− mice have 2 to 3-fold increase in CD3+ total T lymphocytes, both CD4+ and CD8+ T cells, as well as a marked increase in F4/80+ monocytes/macrophages. Of note, as the predominant sources of interferon-γ and interleukin-17A in the kidney, known to contribute to hypertension,37 CD4+ and CD8+ effector memory T cells were markedly elevated in Sirt3−/− mice. In contrast, Sirt3 overexpression significantly reduced renal inflammatory cells compared with wild-type littermates, including CD3+ total T cells, CD4+ and CD8+ T cells, respective memory T cell populations, and F4/80+ monocytes/macrophages (Figure 5E). These findings are in line with increased vascular permeability, vascular inflammation and hypertension in Sirt3−/− mice; however, Sirt3 overexpression in Sirt3OX mice significantly reduced kidney inflammation compared with wild-type littermates (Figure 5E).
Essential hypertension is linked to reduced Sirt3, SOD2 acetylation and vascular inflammation.
To define the clinical relevance of Sirt3 depletion, SOD2 hyperacetylation and Sirt3-dependent vascular alterations described above, we studied Sirt3 levels, SOD2 acetylation and markers of vascular inflammation in human subjects with and without essential hypertension by Western blot analysis of arterioles from human mediastinal fat. We observed a 40% decrease in vascular Sirt3 level and 3-fold increase in SOD2 acetylation in hypertensive subjects while SOD2 levels were not affected (Figure 7). To test the potential role of the Sirt3-dependent vascular metabolic, inflammatory and cell-senescence pathways described above, we performed Western blot analyses of HIF1α, VE-cadherin, p65, VCAM, ICAM, MCP1, p21 and TERT levels in arterioles from hypertensive and normotensive subjects. It was found that essential hypertension was associated with a 3- to 4-fold increases in HIF1α, inflammatory and cell-senescence markers compared with normotensive subjects. These data support the potential role of Sirt3 depletion and SOD2 hyperacetylation in human hypertension.
Figure 7: Western blot of SOD2 acetylation, SOD2, Sirt3, inflammation and cell-senescence markers in arterioles isolated from human mediastinal fat in patients with essential hypertension compared with normotensive subjects.
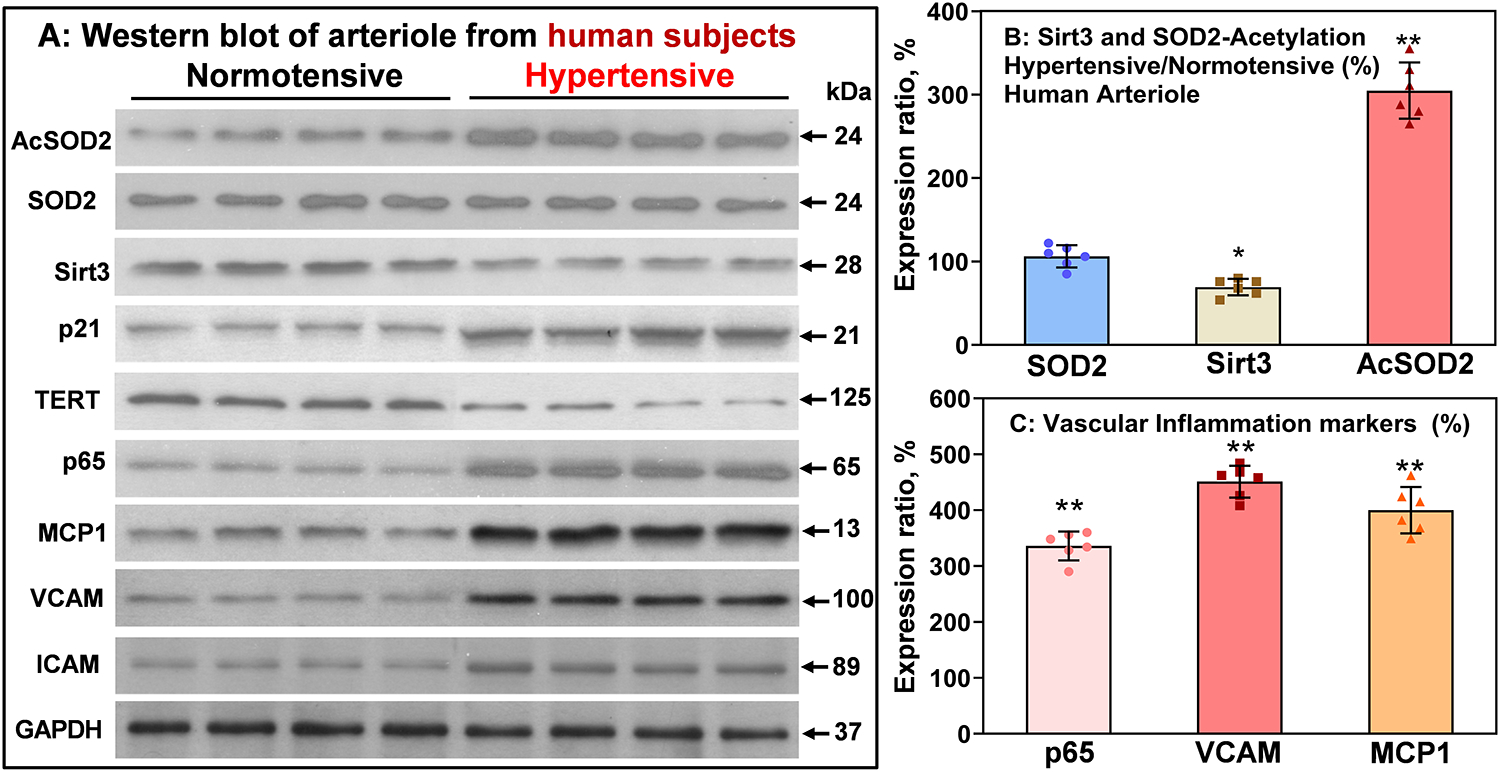
Data were normalized by GAPDH. Results are mean ± SEM. Data were analyzed using 1-way ANOVA and Bonferroni post-hoc multiple comparisons. (A) *P=0.01, *P=0.0001, n=6. (B) **P=0.000001 vs Normotensive, n=6.
DISCUSSION
This study provides the first evidence that genetic Sirt3 overexpression reduces vascular oxidative stress, inhibits vascular hypertrophy, protects endothelial-dependent relaxation, normalizes vascular permeability, and attenuates angiotensin II- and DOCA-salt hypertension. We found that whole body Sirt3 depletion in Sirt3−/− mice induces pathophysiological metabolic and phenotypic vascular alterations, including increased HIF1α, reduced VE-cadherin, elevated endothelial permeability, activation of NFkB and NLRP3 inflammasome pathways, increased vascular inflammation, increased markers of cell-senescence p21 and SA-β-gal, reduced telomerase expression, increased infiltration of T cells and macrophages and development of age-dependent hypertension. Interestingly, genetic Sirt3 overexpression significantly prevents these deleterious effects both in sham mice and hypertension models. For the first time we report diminished Sirt3 levels and SOD2 hyperacetylation in arterioles from human subjects with essential hypertension compared to normotensive subjects which was accompanied by alterations of vascular metabolic, inflammatory and cell-senescence pathways identical to Sirt3 depleted mice. These data support the pathophysiological role of Sirt3 depletion in vascular dysfunction and suggest a therapeutic potential of targeting Sirt3 expression for treatment of vascular dysfunction and hypertension.
Cardiovascular disease risk factors reduce Sirt3 levels8 and activity,54 and Sirt3 declines with age9, 15, 55 paralleling the increased incidence of hypertension. Meanwhile, the specific Sirt3-dependent pathways contributing to these pathological conditions remain elusive. This work shows that Sirt3 depletion has a causative role in vascular dysfunction and hypertension. NAD+ dependent mitochondrial deacetylase Sirt3 activates a key mitochondrial antioxidant, superoxide dismutase 2 (SOD2), by deacetylation of specific lysine residues.28, 29, 56 We have previously shown by mass spectrometry an increased abundance of SOD2-K68 acetylation and reduced SOD2 activity in hypertension.10 Furthermore, treatment of Sirt3−/− mice with mitochondria targeted SOD2 mimetic mitoTEMPO rescued endothelial-dependent relaxation and diminished hypertension.10 In this work we showed that SOD2-K68 acetylation is strongly associated with increased HIF1α, reduced VE-cadherin, increased endothelial permeability, activation of NFkB and NLRP3 inflammasome pathways, vascular inflammation, markers of cell-senescence and increased infiltration of inflammatory cells in Sirt3−/− mice which were prevented in Sirt3OX mice. Analysis of vascular tissue from human subjects with essential hypertension support the potential role of Sirt3 impairment and SOD2 acetylation in vascular inflammation and dysfunction.
Hypertension is a multifactorial disorder associated with oxidative stress and inflammation.57 We have previously shown an increased production of mitochondrial superoxide12 and reduced activity of mitochondrial superoxide dismutase10 in animal models of hyperytension. The imbalance between the increased mitochondrial superoxide and reduced antioxidant SOD2 activity leads to mitochondrial oxidative stress.11 We have previously reported a feed-forward cross-talk between mitochondrial ROS and cellular NADPH oxidases.13, 58, 59 In this work we did not measure specific superoxide production in mitochondria but analyzed the whole cellular superoxide production using DHE probe and HPLC.35 Dispite this apparent limitation we found that Sirt3 overexpression completely prevents the vascular superoxide overproduction both in angiotensin II- and DOCA-salt model of hypertension. The Sirt3 effect on whole cell superoxide production was associated with reduced SOD2 acetylation and inhibition of vascular inflammation. These data support the role of Sirt3 impairment in both oxidative stress and vascular inflammation. Meanwhile, additional studies are required to establish the specific role of mitochondrial superoxide and cellular NADPH oxidases in response to Sirt3 impaiment.
In this work we used new transgenic mice with global Sirt3 overexpression. The cell-specific effects of Sirt3 expression, however, remain unclear. Our data show that global Sirt3 depletion promotes vascular hypertrophy and endothelial dysfunction while increased Sirt3 expression attenuates vascular hypertrophy and protects endothelial function suggesting the important role of Sirt3 in vascular homeostasis. Further studies with cell-specific Sirt3 overexpression are warranted to define the specific role of endothelial Sirt3 in the regulation of endothelial metabolism, endothelial barrier function, and endothelial-dependent relaxation. Recent studies implicate reduced Sirt3 and SOD2 activity in endothelial progenitor cell dysfunction in hypertension which impairs endothelial repair capacity.60 Knockdown of Sirt3 in endothelial progenitor cells suppressed the reendothelialization capacity while SOD2 mimetic mitoTEMPO reduced mitochondrial damage and rescued the endothelial progenitor function60 supporting the role of Sirt3-SOD2 impairment in endothelial injury.
Our data suggest a potential role of Sirt3 in the regulation of smooth muscle cells. This could be further tested in smooth muscle specific Sirt3 overexpressing mice. Sirt3 also contributes to regulation of inflammatory cells. Increased Sirt3 expression in macrophages by viniferin attenuates LPS-induced lung injury.18 Sirt3 activators protect from doxorubicin-induced cardiotoxicity and inhibit tumor growth.61, 62 In keep with these findings, we also observed that Sirt3 depletion exacerbates renal inflammatory cell infiltration, and Sirt3 overexpression effectively reduced inflammatory cell numbers in the kidney. Therefore, development of cell-specific models and specific Sirt3 agonists are of particular importance for future clinical translation.
The precise mechanisms of vascular alterations in response to Sirt3 depletion are not clear. Sirt3 depletion can induce mitochondrial damage in response to mitochondrial oxidative stress due to SOD2 acetylation and SOD2 inactivation. Indeed, Sirt3 depletion activates the NLRP3 inflammasome which can be induced by a broad range of stimuli including mitochondrial DNA45 and cardiolipin.63 Sirt3 depletion causes mitochondrial damage accompanied by release of mitochondrial DNA which directly activates the NLRP3 inflammasome leading to caspase-1 activation, IL-1β release and MCP1 formation.45 An additional mechanisms of NLRP3 inflammasome activation include mitochondrial cardiolipin. It normally resides on the matrix side of the inner mitochondrial membrane and it can be found in the membranes of most bacteria. Mitochondrial oxidative stress leads to cardiolipin translocation to the outer mitochondrial membrane and induces cardiolipin oxidation. Bacteria-like cardiolipin is required for assembly and activation of the NLRP3 inflammasome.63, 64 Interestingly, we have recently shown increased cardiolipin oxidation in hypertension associated with Sirt3 depletion and SOD2 acetylation.11 Sirt3 depletion can also activate the redox dependent proinflammatory transcriptional factor NFkB.44 Indeed, Sirt3 depletion increases endothelial and vascular production of reactive oxygen species10 and mitochondrial H2O2 activates NFkB pathway.65
Our data show that Sirt3 depletion accelerates vascular aging and promotes age-dependent hypertension while increased Sirt3 expression raises TERT levels, reduces markers of cell-senescence and attenuates hypertension. Telomere uncapping and p21-induced senescence are two-fold greater in hypertensive patients compared with nonhypertensive individuals66 and TERT protects microvascular endothelial function.67 Vascular aging has been linked to the pathogenesis of vascular dysfunction and hypertension.5 Interestingly, global Sirt3 depletion reduces lifespan68 and increases blood pressure.22 The precise retrograde signaling responsible for the “antiaging” and “antihypertensive” effects of Sirt3 are not clear. We suggest that Sirt3 depletion leads to SOD2 inactivation due to SOD2 hyperacetylation. These changes promote mitochondrial damage and vascular inflammation, accelerate vascular aging and hypertension. Indeed, SOD2 depletion which mimics SOD2 inactivation by acetylation promotes age-dependent and salt-sensitive hypertension.69 Our data show that increased Sirt3 expression attenuates SOD2 acetylation and, therefore, protects vascular function and reduces hypertension.
It is possible that both Sirt3 activity and Sirt3 expression is diminished in pathological conditions. Indeed, our data show a 40% decrease in Sirt3 level and 3-fold increase in SOD2 acetylation in hypertensive subjects implicating Sirt3 inactivation (Figure 7). The specific mechanisms of Sirt3 inactivation, however, remain elusive. Sirt3 is a NAD+ dependent enzyme, therefore, NAD+ decline may contribute to Sirt3 inactivation and boosting NAD + level can be beneficial in pathological conditions.70 Indeed, supplementation with NAD+ donor nicotinamide riboside, a pan-sirtuin agonist, modestly reduces blood pressure71, 72 and age-related vascular dysfunction.73 Alternatively, increased Sirt3 expression can be protective.74 Indeed, calorie restriction and exercise inreases Sirt3 levels, reduce inflammation45 and risk of cardiovascular disease.75 We previously suggested a potential role Sirt3 redox inactivation and scavenging of mitochondrial H2O2 reduces SOD2 aceylation and improves blood pressure.10 Therefore, multiple metabolic and redox pathways can contribute to Sirt3 impairment,25 and further studies are needed to define the most effective targeting Sirt3 function in human pathological conditions.
Our work suggests the potential role of Sirt3 depletion in the pathogenesis of vascular dysfunction and hypertension. Interestingly, multiple factors such as a sedentary lifestyle, smoking, aging, metabolic conditions, and inflammation are associated with Sirt3 depletion11, 15, 16 and increased hypertension. These factors can be partially mediated by SOD2 hyperacetylation.76 Furthermore, in humans, a variable number tandem repeat48, 49 and two non-synonymous human Sirt3 SNPs77 impact the Sirt3 expression, longevity and pathological conditions. We suggest that genetic and cardiovascular risk factors may reduce Sirt3 expression which can contribute to progression of multiple pathologic conditions associated with mitochondrial dysfunctions such as neurodegeneration, inflammation, hypertension and cardiovascular disease. It is conceivable that strategies directed to increase Sirt3 expression may have therapeutic potential in these conditions.
Supplementary Material
Figure 8: Proposed role of Sirt3 depletion in vascular dysfunction and hypertension. Sirt3 depletion results in SOD2 hyperacetylation and inactivation leading to mitochondrial oxidative stress.
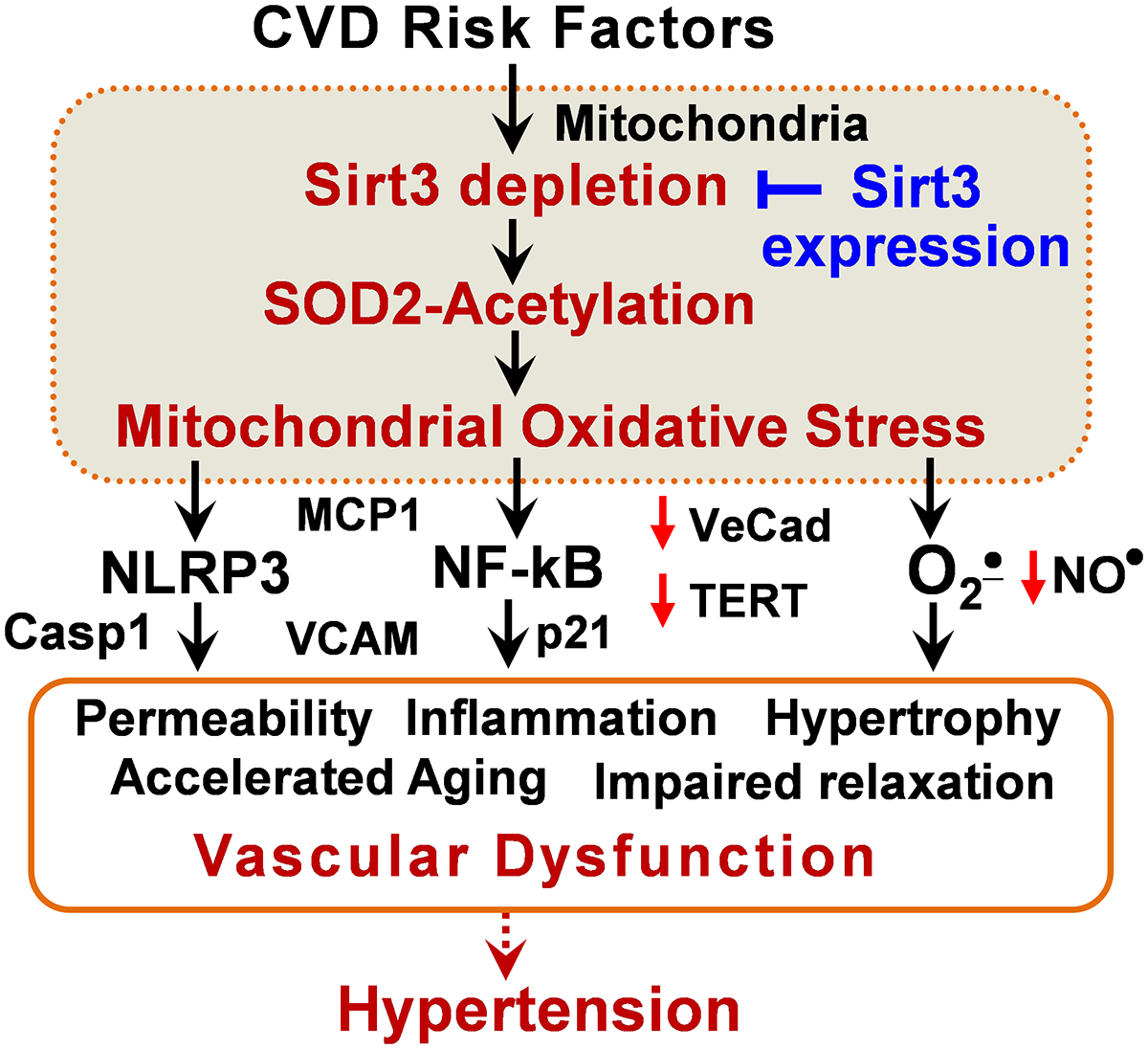
This in turn inactivates endothelial nitric oxide (NO), induces redox-dependent NF-kB activation and mitochondrial damage, and activates the NLRP3 inflammasome. These pathways disrupt the endothelial barrier, impair vasorelaxation, promote smooth muscle hypertrophy and vascular inflammation, accelerate vascular aging and increase hypertension. These deleterious effects are ameliorated by Sirt3 expression.
NOVELTY AND SIGNIFICANCE.
What Is Known?
Depletion of mitochondrial deacetylase Sirtuin 3 (Sirt3) inactivates SOD2 due to acetylation of specific lysine residues.
Sirt3 depletion in Sirt3−/− mice promotes oxidative stress and hypertension.
Mitochondria-targeted SOD2 mimetic mitoTEMPO rescues Sirt3−/− mice from hypertension.
What New Information Does This Article Contribute?
Sirt3 overexpression prevents vascular oxidative stress and attenuates hypertension in angiotensin II and DOCA-salt mouse models while Sirt3 depletion accelerates vascular inflammation, vascular aging and age-dependent hypertension.
Sirt3 overexpression inhibits vascular hypertrophy, protects endothelial barrier function, prevents end-organ inflammation and preserves endothelial-dependent relaxation.
Sirt3 level is decreased in arterioles from human subjects with essential hypertension compared to normotensive subjects, and this is accompanied by alterations in vascular metabolic, inflammatory and cell-senescence pathways similar to Sirt3 depleted mice.
ACKNOWLEDGMENTS
We thank Dr. David H. Wasserman and Louise Lantier for providing the mice lacking the Sirt3 protein (Sirt3−/−) for these studies, which were generated in the Auwerx laboratory.
SOURCES OF FUNDING
This work was supported by funding from National Institute of Health (R01HL124116, P01HL129941 and R01GM112871), Vanderbilt University (VR7040 and UL1 RR024975), Swiss National Science Foundation (SNSF 310030B-160318), and from the Ecole Polytechnique Fédérale de Lausanne (EPFL). Dr. Dikalova was supported by American Heart Association Grant-In-Aid (16GRNT31230017) and Transformational Project Award (19TPA34910157).
Nonstandard Abbreviations and Acronyms:
- Ang II
angiotensin II
- AcSOD2
acetylated (inactive) mitochondrial superoxide dismutase
- Casp1
caspase 1, NLRP3 inflammasome activation marker
- DHE
fluorescent O2• probe dihydroethidium
- DOCA
deoxycorticosterone acetate
- HIF1α
hypoxia-inducible factor-1
- HPLC
high-performance liquid chromatography
- H2O2
hydrogen peroxide
- NfKB
nuclear factor kappa-light-chain-enhancer of activated B cells
- p65
one of the five components of NFκB transcription factor
- ICAM
intercellular adhesion molecule 1
- MCP1
monocyte chemoattractant protein 1
- O2•
superoxide radicals
- p21
cyclin-dependent kinase inhibitor 1, marker of cell-senescence
- ROS
reactive oxygen species
- SA-β-gal
senescence-associated β-galactosidase
- Sirt3
Sirtuin 3
- Sirt3−/−
mice with global Sirt3 depletion
- Sirt3OX
transgenic mice with global Sirt3 overexpression
- SOD2
mitochondrial isoform of superoxide dismutase
- TERT
telomerase reverse transcriptase
- VCAM
vascular cell adhesion molecule-1
- WT
wild-type C57Bl/6J mice
Footnotes
DISCLOSURES
J.A. is a consultant for Mitobridge and MetroBiotech, companies that develop NAD boosting agents.
REFERENCES
- 1.Sacco RL, Benjamin EJ, Broderick JP, Dyken M, Easton JD, Feinberg WM, Goldstein LB, Gorelick PB, Howard G, Kittner SJ, Manolio TA, Whisnant JP, Wolf PA. American heart association prevention conference. Iv. Prevention and rehabilitation of stroke. Risk factors. Stroke. 1997;28:1507–1517 [DOI] [PubMed] [Google Scholar]
- 2.Roth GA, Johnson C, Abajobir A. et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70:1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Front Physiol. 2012;3:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrd JB, Zeng C, Tavel HM, Magid DJ, O’Connor PJ, Margolis KL, Selby JV, Ho PM. Combination therapy as initial treatment for newly diagnosed hypertension. Am Heart J. 2011;162:340–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guzik TJ, Touyz RM. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension. 2017;70:660–667 [DOI] [PubMed] [Google Scholar]
- 6.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr., Alt FW, Kahn CR, Verdin E. Sirt3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D. Regulation of mnsod enzymatic activity by sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal. 2014;20:1646–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freitas M, Rodrigues AR, Tomada N, Fonseca J, Magalhaes A, Gouveia AM, Neves D. Effects of aging and cardiovascular disease risk factors on the expression of sirtuins in the human corpus cavernosum. J Sex Med. 2015;12:2141–2152 [DOI] [PubMed] [Google Scholar]
- 9.Chaudhry KN, Chavez P, Gasowski J, Grodzicki T, Messerli FH. Hypertension in the elderly: Some practical considerations. Cleve Clin J Med. 2012;79:694–704 [DOI] [PubMed] [Google Scholar]
- 10.Dikalova AE, Itani HA, Nazarewicz RR, McMaster WG, Fessel JP, Flynn CR, Gamboa JL, Harrison DG, Dikalov SI. Sirt3 impairment and sod2 hyperacetylation in vascular oxidative stress and hypertension. Circ Res. 2017;121:664–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dikalov S, Itani HA, Richmond B, Arslanbaeva L, Vergeade A, Rahman SMJ, Boutaud O, Blackwell T, Massion PP, Harrison DG, Dikalova AE. Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction and enhances hypertension. Am J Physiol Heart Circ Physiol. 2019;316:H639–H646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res. 2010;107:106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassegue B, Griendling K, Harrison DG, Dikalova A. Nox2-induced production of mitochondrial superoxide in angiotensin ii - mediated endothelial oxidative stress and hypertension. Antioxid Redox Signal. 2014;20:281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itani HA, Dikalova AE, McMaster WG, Nazarewicz RR, Bikineyeva AT, Harrison DG, Dikalov SI. Mitochondrial cyclophilin d in vascular oxidative stress and hypertension. Hypertension. 2016;67:1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capettini LS, Montecucco F, Mach F, Stergiopulos N, Santos RA, da Silva RF. Role of renin-angiotensin system in inflammation, immunity and aging. Curr Pharm Des. 2012;18:963–970 [DOI] [PubMed] [Google Scholar]
- 16.Storka A, Fuhrlinger G, Seper M, Wang L, Jew M, Leisser A, Wolzt M. E. Coli endotoxin modulates the expression of sirtuin proteins in pbmc in humans. Mediators Inflamm. 2013;2013:876943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winnik S, Auwerx J, Sinclair DA, Matter CM. Protective effects of sirtuins in cardiovascular diseases: From bench to bedside. Eur Heart J. 2015;36:3404–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurundkar D, Kurundkar AR, Bone NB, Becker EJ Jr., Liu W, Chacko B, Darley-Usmar V, Zmijewski JW, Thannickal VJ. Sirt3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight. 2019;4:pii: 120722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. 2015;116:1022–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pillai VB, Bindu S, Sharp W, Fang YH, Kim G, Gupta M, Samant S, Gupta MP. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am J Physiol Heart Circ Physiol. 2016;310:H962–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown KD, Maqsood S, Huang JY, Pan Y, Harkcom W, Li W, Sauve A, Verdin E, Jaffrey SR. Activation of sirt3 by the nad(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014;20:1059–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sundaresan NR, Bindu S, Pillai VB, Samant S, Pan Y, Huang JY, Gupta M, Nagalingam RS, Wolfgeher D, Verdin E, Gupta MP. Sirt3 blocks aging-associated tissue fibrosis in mice by deacetylating and activating gsk3beta. Mol Cell Biol. 2015;36:678–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eirin A, Lerman A, Lerman LO. Mitochondrial injury and dysfunction in hypertension-induced cardiac damage. Eur Heart J. 2014;35:3258–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eirin A, Lerman A, Lerman LO. Mitochondria: A pathogenic paradigm in hypertensive renal disease. Hypertension. 2015;65:264–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dikalov SI, Dikalova A. Crosstalk between mitochondrial hyperacetylation and oxidative stress in vascular dysfunction and hypertension. Antioxid Redox Signal. 2019;31:710–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eirin A, Lerman A, Lerman LO. Enhancing mitochondrial health to treat hypertension. Curr Hypertens Rep. 2018;20:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurt I, Artsi H, Cohen-Kfir E, Hamdani G, Ben-Shalom G, Feinstein B, El-Haj M, Dresner-Pollak R. The sirt1 activators srt2183 and srt3025 inhibit rankl-induced osteoclastogenesis in bone marrow-derived macrophages and down-regulate sirt3 in sirt1 null cells. PLoS One. 2015;10:e0134391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Han L, Ma R, Hou X, Yu Y, Sun S, Xu Y, Schedl T, Moley KH, Wang Q. Sirt3 prevents maternal obesity-associated oxidative stress and meiotic defects in mouse oocytes. Cell Cycle. 2015;14:2959–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, Xiong Y. Tumour suppressor sirt3 deacetylates and activates manganese superoxide dismutase to scavenge ros. EMBO Rep. 2011;12:534–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–14452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M, James FD, Gius D, Wasserman DH. Sirt3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat-fed mice. Diabetes. 2015;64:3081–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676 [DOI] [PubMed] [Google Scholar]
- 33.Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension. 1995;25:1111–1115 [DOI] [PubMed] [Google Scholar]
- 34.Widder JD, Guzik TJ, Mueller CF, Clempus RE, Schmidt HH, Dikalov SI, Griendling KK, Jones DP, Harrison DG. Role of the multidrug resistance protein-1 in hypertension and vascular dysfunction caused by angiotensin ii. Arterioscler Thromb Vasc Biol. 2007;27:762–768 [DOI] [PubMed] [Google Scholar]
- 35.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dikalov S, Fink B. Esr techniques for the detection of nitric oxide in vivo and in tissues. Methods Enzymol. 2005;396:597–610 [DOI] [PubMed] [Google Scholar]
- 37.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. Cd70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118:1233–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gamboa JL, Billings FTt, Bojanowski MT, Gilliam LA, Yu C, Roshanravan B, Roberts LJ 2nd, Himmelfarb J, Ikizler TA, Brown NJ. Mitochondrial dysfunction and oxidative stress in patients with chronic kidney disease. Physiol Rep. 2016;4:pii: e12780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dikalova AE, Itani HA, Pandey AK, Harrison DG, Dikalov SI. Overexpression of mitochondrial deacetylase sirt3 protects endothelial function and attenuates hypertension while sirt3 depletion increases oxidative stress and endothelial dysfunction due to sod2 hyperacetylation. Hypertension. 2017;70:AP345 [Google Scholar]
- 40.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiong J, Kawagishi H, Yan Y, Liu J, Wells QS, Edmunds LR, Fergusson MM, Yu ZX, Rovira, II, Brittain EL, Wolfgang MJ, Jurczak MJ, Fessel JP, Finkel T. A metabolic basis for endothelial-to-mesenchymal transition. Mol Cell. 2018;69:689–698 e687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bierhansl L, Conradi LC, Treps L, Dewerchin M, Carmeliet P. Central role of metabolism in endothelial cell function and vascular disease. Physiology (Bethesda). 2017;32:126–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harris ES, Nelson WJ. Ve-cadherin: At the front, center, and sides of endothelial cell organization and function. Curr Opin Cell Biol. 2010;22:651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of nf-kappab activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403 [DOI] [PubMed] [Google Scholar]
- 45.Traba J, Geiger SS, Kwarteng-Siaw M, Han K, Ra OH, Siegel RM, Gius D, Sack MN. Prolonged fasting suppresses mitochondrial nlrp3 inflammasome assembly and activation via sirt3-mediated activation of superoxide dismutase 2. J Biol Chem. 2017;292:12153–12164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. Potential role of mcp-1 in endothelial cell tight junction ‘opening’: Signaling via rho and rho kinase. J Cell Sci. 2003;116:4615–4628 [DOI] [PubMed] [Google Scholar]
- 47.Maguire O, Collins C, O’Loughlin K, Miecznikowski J, Minderman H. Quantifying nuclear p65 as a parameter for nf-kappab activation: Correlation between imagestream cytometry, microscopy, and western blot. Cytometry A. 2011;79:461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giblin W, Skinner ME, Lombard DB. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014;30:271–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bellizzi D, Rose G, Cavalcante P, Covello G, Dato S, De Rango F, Greco V, Maggiolini M, Feraco E, Mari V, Franceschi C, Passarino G, De Benedictis G. A novel vntr enhancer within the sirt3 gene, a human homologue of sir2, is associated with survival at oldest ages. Genomics. 2005;85:258–263 [DOI] [PubMed] [Google Scholar]
- 50.Fuster JJ, Diez J, Andres V. Telomere dysfunction in hypertension. J Hypertens. 2007;25:2185–2192 [DOI] [PubMed] [Google Scholar]
- 51.Najjar SS, Scuteri A, Lakatta EG. Arterial aging: Is it an immutable cardiovascular risk factor? Hypertension. 2005;46:454–462 [DOI] [PubMed] [Google Scholar]
- 52.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: Role of telomere in endothelial dysfunction. Circulation. 2002;105:1541–1544 [DOI] [PubMed] [Google Scholar]
- 53.Itani HA, McMaster WG Jr., Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, Konior A, Prejbisz A, Januszewicz A, Norlander AE, Chen W, Bonami RH, Marshall AF, Poffenberger G, Weyand CM, Madhur MS, Moore DJ, Harrison DG, Guzik TJ. Activation of human t cells in hypertension: Studies of humanized mice and hypertensive humans. Hypertension. 2016;68:123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown K, Xie S, Qiu X, Mohrin M, Shin J, Liu Y, Zhang D, Scadden DT, Chen D. Sirt3 reverses aging-associated degeneration. Cell Rep. 2013;3:319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, Yu C, Shen G, Li G, Shen J, Xu Y, Gong J. Sirt3-mnsod axis represses nicotine-induced mitochondrial oxidative stress and mtdna damage in osteoblasts. Acta Biochim Biophys Sin (Shanghai). 2015;47:306–312 [DOI] [PubMed] [Google Scholar]
- 56.Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates mnsod activity in response to stress. Mol Cell. 2010;40:893–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrison DG. The mosaic theory revisited: Common molecular mechanisms coordinating diverse organ and cellular events in hypertension. J Am Soc Hypertens. 2013;7:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nazarewicz RR, Dikalova AE, Bikineyeva A, Dikalov SI. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am J Physiol Heart Circ Physiol. 2013;305:H1131–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He J, Liu X, Su C, Wu F, Sun J, Zhang J, Yang X, Zhang C, Zhou Z, Zhang X, Lin X, Tao J. Inhibition of mitochondrial oxidative damage improves reendothelialization capacity of endothelial progenitor cells via sirt3 (sirtuin 3)-enhanced sod2 (superoxide dismutase 2) deacetylation in hypertension. Arterioscler Thromb Vasc Biol. 2019;39:1682–1698 [DOI] [PubMed] [Google Scholar]
- 61.Akamata K, Wei J, Bhattacharyya M, Cheresh P, Bonner MY, Arbiser JL, Raparia K, Gupta MP, Kamp DW, Varga J. Sirt3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget. 2016;7:69321–69336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pillai VB, Kanwal A, Fang YH, Sharp WW, Samant S, Arbiser J, Gupta MP. Honokiol, an activator of sirtuin-3 (sirt3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget. 2017;8:34082–34098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS. Mitochondrial cardiolipin is required for nlrp3 inflammasome activation. Immunity. 2013;39:311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Elliott EI, Miller AN, Banoth B, Iyer SS, Stotland A, Weiss JP, Gottlieb RA, Sutterwala FS, Cassel SL. Cutting edge: Mitochondrial assembly of the nlrp3 inflammasome complex is initiated at priming. J Immunol. 2018;200:3047–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee S, Tak E, Lee J, Rashid MA, Murphy MP, Ha J, Kim SS. Mitochondrial h2o2 generated from electron transport chain complex i stimulates muscle differentiation. Cell Res. 2011;21:817–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morgan RG, Ives SJ, Walker AE, Cawthon RM, Andtbacka RH, Noyes D, Lesniewski LA, Richardson RS, Donato AJ. Role of arterial telomere dysfunction in hypertension: Relative contributions of telomere shortening and telomere uncapping. J Hypertens. 2014;32:1293–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ait-Aissa K, Kadlec AO, Hockenberry J, Gutterman DD, Beyer AM. Telomerase reverse transcriptase protects against angiotensin ii-induced microvascular endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2018;314:H1053–H1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Benigni A, Cassis P, Conti S, Perico L, Corna D, Cerullo D, Zentilin L, Zoja C, Perna A, Lionetti V, Giacca M, Trionfini P, Tomasoni S, Remuzzi G. Sirt3 deficiency shortens lifespan and impairs cardiac mitochondrial function rescued by opa1 gene transfer. Antioxid Redox Signal. 2019;31. [DOI] [PubMed] [Google Scholar]
- 69.Rodriguez-Iturbe B, Sepassi L, Quiroz Y, Ni Z, Wallace DC, Vaziri ND. Association of mitochondrial sod deficiency with salt-sensitive hypertension and accelerated renal senescence. J Appl Physiol. 2007;102:255–260 [DOI] [PubMed] [Google Scholar]
- 70.Katsyuba E, Auwerx J. Modulating nad(+) metabolism, from bench to bedside. Embo J. 2017;36:2670–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, Chonchol M, Seals DR. Chronic nicotinamide riboside supplementation is well-tolerated and elevates nad(+) in healthy middle-aged and older adults. Nat Commun. 2018;9:1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kane AE, Sinclair DA. Sirtuins and nad(+) in the development and treatment of metabolic and cardiovascular diseases. Circ Res. 2018;123:868–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Csiszar A, Tarantini S, Yabluchanskiy A, Balasubramanian P, Kiss T, Farkas E, Baur JA, Ungvari Z. Role of endothelial nad(+) deficiency in age-related vascular dysfunction. Am J Physiol Heart Circ Physiol. 2019;316:H1253–H1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP, Gius D, Gupta MP. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial sirt3. Nat Commun. 2015;6:6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57:2933–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dikalov SI, Dikalova AE. Contribution of mitochondrial oxidative stress to hypertension. Curr Opin Nephrol Hypertens. 2016;25:73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dransfeld CL, Alborzinia H, Wolfl S, Mahlknecht U. Sirt3 snps validation in 640 individuals, functional analyses and new insights into sirt3 stability. Int J Oncol. 2010;36:955–960 [DOI] [PubMed] [Google Scholar]
- 78.Han J, Weisbrod RM, Shao D, Watanabe Y, Yin X, Bachschmid MM, Seta F, Janssen-Heininger YMW, Matsui R, Zang M, Hamburg NM, Cohen RA. The redox mechanism for vascular barrier dysfunction associated with metabolic disorders: Glutathionylation of rac1 in endothelial cells. Redox Biol. 2016;9:306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.