

Abstract
OBJECTIVE
Sustained excess BMI increases the risk of type 1 diabetes (T1D) in autoantibody-positive relatives without diabetes of patients. We tested whether elevated BMI also accelerates the progression of islet autoimmunity before T1D diagnosis.
RESEARCH DESIGN AND METHODS
We studied 706 single autoantibody–positive pediatric TrialNet participants (ages 1.6–18.6 years at baseline). Cumulative excess BMI (ceBMI) was calculated for each participant based on longitudinally accumulated BMI ≥85th age- and sex-adjusted percentile. Recursive partitioning analysis and multivariable modeling defined the age cut point differentiating the risk for progression to multiple positive autoantibodies.
RESULTS
At baseline, 175 children (25%) had a BMI ≥85th percentile. ceBMI range was −9.2 to 15.6 kg/m2 (median −1.91), with ceBMI ≥0 kg/m2 corresponding to persistently elevated BMI ≥85th percentile. Younger age increased the progression to multiple autoantibodies, with age cutoff of 9 years defined by recursive partitioning analysis. Although ceBMI was not significantly associated with progression from single to multiple autoantibodies overall, there was an interaction with ceBMI ≥0 kg/m2, age, and HLA (P = 0.009). Among children ≥9 years old without HLA DR3-DQ2 and DR4-DQ8, ceBMI ≥0 kg/m2 increased the rate of progression from single to multiple positive autoantibodies (hazard ratio 7.32, P = 0.004) and conferred a risk similar to that in those with T1D-associated HLA haplotypes. In participants <9 years old, the effect of ceBMI on progression to multiple autoantibodies was not significant regardless of HLA type.
CONCLUSIONS
These data support that elevated BMI may exacerbate islet autoimmunity prior to clinical T1D, particularly in children with lower risk based on age and HLA. Interventions to maintain normal BMI may prevent or delay the progression of islet autoimmunity.
Introduction
The global rise in incidence of type 1 diabetes (T1D) has intensified efforts to identify modifiable risk factors in order to prevent or delay onset of clinical diabetes (1). Although there are several genetic loci for T1D susceptibility, heritability does not completely predict appearance of islet autoantibodies or disease development (2), highlighting the role of other influences such as the environment (3).
The parallel rise in obesity (4,5) and T1D incidence suggests a potential link between elevated body weight and T1D progression (6–9). The accelerator hypothesis proposes that obesity-induced insulin resistance triggers the autoimmune-mediated β-cell destruction that characterizes T1D (10). Additionally, obesity-induced insulin resistance may also accelerate clinical onset of T1D by increasing insulin needs in those with already compromised insulin secretory capacity, as was previously proposed by Libman et al. (11,12). The threshold hypothesis proposes that diabetes develops when the combination of genetic and environmental diabetogenic factors exceeds a threshold (13). We have recently demonstrated that longitudinal, sustained elevation in BMI, measured as cumulative excess BMI (ceBMI), increases the progression to T1D in adult (14) and pediatric (15) islet autoantibody–positive participants of the TrialNet Pathway to Prevention (PTP) cohort. These data suggest that lifestyle modifications may slow the progression to T1D. However, the mechanism is unclear.
While increased insulin resistance may lead to hyperglycemia and diagnosis of stage 3 clinical diabetes, we hypothesized that ceBMI may exacerbate the autoimmune process in preclinical stages of T1D. In order to understand the mechanism by which ceBMI increases the risk of T1D, we herein explored its influence on conversion from single to multiple autoantibody positivity in pediatric participants of the TrialNet PTP cohort. These data may reveal a therapeutically modifiable risk factor, with both genetic and environmental influences, and potential for intervention prior to the development of T1D.
Research Design and Methods
Subjects
The TrialNet PTP cohort was established in 2001 and has previously been described (16). Briefly, first-degree relatives (ages 1–45 years) and second- or third-degree relatives (ages 1–20 years) without diabetes of individuals with T1D were enrolled and screened for presence of pancreatic islet autoantibodies. Islet autoantibody status was assessed according to the Diabetes Antibody Standardization Program. Participants were tested first for the presence of GAD65 (17), insulin (18), or islet-antigen 2 (IA-2/islet cell autoantibody [ICA]512) autoantibodies (19), and if positive, they were tested for ICA (20). Measurement of zinc transporter 8 (ZnT8) autoantibodies (21) was initiated with samples stored since 2004 and was consistently measured in the PTP cohort starting in 2012. Confirmed autoantibody-positive individuals were observed longitudinally with either semiannual or annual monitoring, which included measurement of height and weight, and oral glucose tolerance testing (OGTT).
A total of 182,145 participants in the PTP study were screened from 2004 through August 2017, of whom 2,382 were determined to be single autoantibody positive and were monitored for progression to development of multiple autoantibody positivity. The current analysis focuses on the 1,224 participants who were <18 years old when identified as single autoantibody positive; 766 of these had two or more evaluations for BMI. For avoidance of confounding by weight loss that precedes T1D diagnosis, further exclusions included BMI data collected within 6 months of diabetes diagnosis as well as any subjects who were determined to be autoantibody positive >2 years prior to their first BMI evaluation. To facilitate consistent and valid calculations of BMI percentiles, and use of Centers for Disease Control and Prevention (CDC) criteria to define overweight and obesity, we restricted our analysis cohort to only include participants with at least two BMI measurements meeting the above criteria before 20 years of age. Our resulting cohort consisted of a total of 706 subjects (Supplementary Fig. 1).
Laboratory and Anthropometric Measurements
At each study visit, a standard protocol OGTT was performed and HbA1c was obtained. Glucose was measured using the glucose oxidase method (22). Diabetes was diagnosed according to American Diabetes Association criteria (fasting glucose ≥126 mg/dL, random glucose ≥200 mg/dL, 2-h OGTT ≥200 mg/dL) (23), which must have been met on two occasions. An HbA1c ≥6.5% could be used as part of confirmatory testing (16).
BMI was calculated as weight in kilograms divided by the square of height in meters. The CDC 2000 growth charts (www.cdc.gov/nccdphp/dnpao/growthcharts/resources/sas.htm) were used to obtain the BMI value corresponding to the 85th percentile for age- and sex- adjusted BMI for each subject at the time of each study visit. ceBMI has been used previously as a measure of persistent elevation of BMI beyond the overweight threshold (24,25). The weighted sums of the differences between the actual BMI and BMI corresponding to the 85th percentile for the sex and age at that evaluation were calculated using the method described by Lee et al. (25) and Bouchard et al. (24). Briefly, a ceBMI score was calculated by summing the difference calculated at each BMI assessment while accounting for the irregular timing between evaluations (Eq. 1):
 |
where ceBMIyrsj is ceBMI-years for subject j in units kg/m2 * years and m is the number of BMI evaluations for subject j. ti is the time i (i.e., some future time point after baseline), and ti+1 is the time i+1, which is the next time point after ti. We further annualized ceBMIyrs to accommodate the irregular timing of BMI assessment in relation to the time at multiple autoantibody positivity, or censoring (Eq. 2):
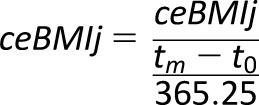 |
where ceBMIj is a value representing the annual average ceBMI in kg/m2 for subject j over the number of years subject j had m BMI evaluations, tm is time in days at the last BMI measurement, and t0 is the time of first BMI evaluation.
Statistical Considerations
Categorical variables were compared among groups by Pearson χ2 tests or Fisher exact tests when cell sizes were insufficient. The majority of continuous variables summarized had skewed distributions and were compared between groups using nonparametric tests (Wilcoxon rank sum tests or Kruskal-Wallis tests, depending on the number of groups). Nonparametric Spearman rank correlation tests were used to assess correlation between continuous measures at baseline. Analyses of BMI were based on age- and sex-adjusted BMI percentiles. As such, baseline underweight status was defined as <5th percentile, normal weight between the 5th and 85th percentile, overweight ≥85th percentile, and obesity ≥95th percentile. ceBMI was analyzed both as a continuous measure and a dichotomized measure. ceBMI ≥0 kg/m2 indicated an individual’s BMI on average ≥85th percentile for their sex and age during the observation period.
The main clinical outcome for analysis was time to conversion from single to multiple autoantibody positivity, defined as the time from the first BMI evaluation with single autoantibody positivity to the date of appearance of multiple positive autoantibodies. Those who did not convert from single to multiple autoantibody positivity were censored at their last date of follow-up. Kaplan-Meier methods were used to assess differences in the time to conversion to multiple autoantibody positivity between groups of interest, and Cox proportional hazards models were used to assess the influence and significance of continuous and categorical variables. Assumptions for proportionality of hazards were tested for in these models. Given the known existence of risk factors and their potentially confounding effects, all time-to-event analyses were adjusted for age and sex as well as other factors as appropriate. To assess possible cut points for age at autoantibody positivity presentation in terms of their influence and stratification of risk on time to the appearance of multiple autoantibodies, we utilized recursive partitioning analyses (26) (rpart package in R) as previously described (14,15). A model-based and iterative approach, specifically, recursive partitioning analyses, was used to identify the “optimal” cut point of the marker that best discriminated the outcome of interest, i.e., time to progression to multiple positive autoantibodies.
Overall, inferential tests were two sided, with P values <0.05 considered to be statistically significant. For interaction terms, P values <0.1 were considered sufficient for further exploration and evaluation of relationships given the sample size and number of events. All analyses were conducted in the statistical program R (version 3.5.1 for Windows).
Results
Demographics
A total of 706 single-positive-autoantibody pediatric subjects between the ages of 2 and 18 years at the first BMI evaluation from the TrialNet PTP study were included in these analyses (Table 1). Of these, 144 subjects (20%) progressed to multiple autoantibody positivity during the observation time. The median age at the first visit with BMI data available was 10.2 years (range 1.6–18.6), and the median BMI percentile at their first evaluation was 61.9% (interquartile range 84.6–35.1). At baseline, 12% of participants were overweight (BMI ≥85th to <95th percentile) and 13% were obese (≥95th percentile). Supplementary Table 2 summarizes BMI percentile by subgroups.
Table 1.
Demographics and baseline characteristics of PTP subjects
| All subjects | ceBMI <0 kg/m2 | ceBMI ≥0 kg/m2 | ||
|---|---|---|---|---|
| Characteristic | N = 706 | N = 517 | N = 189 | P |
| Age at single Ab+ (years), median (range) | 9.8 (1.2–17.9) | 9.9 (1.2–17.9) | 9.4 (1.6–17.6) | 0.97 |
| Sex | 0.78 | |||
| Male | 347 (49.2) | 252 (48.7) | 95 (50.3) | |
| Female | 359 (50.8) | 265 (51.3) | 94 (49.7) | |
| Ab type | 0.85 | |||
| GADA+ | 460 (65.1) | 340 (65.8) | 120 (63.5) | |
| IA-2A+ | 33 (4.7) | 24 (4.6) | 9 (4.8) | |
| IAA+ | 213 (30.2) | 153 (29.6) | 60 (31.7) | |
| Race | 0.67 | |||
| White | 577 (81.7) | 425 (82.2) | 152 (80.4) | |
| Nonwhite/unknown | 129 (18.3) | 92 (17.8) | 37 (19.6) | |
| Ethnicity | 0.0001 | |||
| Hispanic/Latino | 118 (16.7) | 69 (13.3) | 49 (25.9) | |
| Non-Hispanic/Latino | 564 (79.9) | 430 (83.2) | 134 (70.9) | |
| Unknown/missing | 24 (3.4) | 18 (3.5) | 6 (3.2) | |
| HLA haplotype | ||||
| DR3 and/or DR4-DQ8 | 0.92 | |||
| No | 205 (29.1) | 151 (29.3) | 54 (28.6) | |
| Yes | 499 (70.9) | 364 (70.7) | 135 (71.4) | |
| DR3-DQ2 | 0.99 | |||
| No | 408 (58.0) | 298 (57.9) | 110 (58.2) | |
| Yes | 296 (42.0) | 217 (42.1) | 79 (41.8) | |
| DR4-DQ8 | 0.80 | |||
| No | 432 (61.4) | 318 (61.7) | 114 (60.3) | |
| Yes | 272 (38.6) | 197 (38.3) | 75 (39.7) | |
| Missing | 2 | 2 | 0 | |
| Baseline BMI percentile | <0.0001 | |||
| Underweight (<5%) | 26 (3.7) | 24 (4.6) | 2 (1.0) | |
| Normal (≥5%, <85%) | 505 (71.5) | 468 (90.5) | 37 (19.6) | |
| Overweight (≥85%, <95%) | 84 (11.9) | 20 (3.9) | 64 (33.9 | |
| Obese (≥95%) | 91 (12.9) | 5 (1.0) | 86 (45.5) | |
| Baseline ceBMI (kg/m2) | <0.0001 | |||
| Median | −1.92 | −2.6 | 2.54 | |
| Range | −9.2 to 15.7 | −9.2 to 15.7 | −5.7 to 14.2 | |
| Baseline BMI category | <0.0001 | |||
| Normal (<0 kg/m2) | 531 (75.2) | 492 (95.2) | 39 (20.6) | |
| Overweight (≥0 kg/m2) | 175 (24.8) | 25 (4.8) | 150 (79.4) | <0.0001 |
Data are n (%) unless otherwise indicated. Ab, autoantibody; GADA, GAD autoantibody; IA-2A, IA-2 autoantibody; IAA, insulin autoantibody.
There was a spectrum of ceBMI from −9.2 to 15.6 kg/m2 (median −1.91 kg/m2 [interquartile range −3.6 to 0.23]). In this pediatric cohort, 26.8% (189 of 706) had ceBMI values ≥0 kg/m2 representing sustained excess BMI above the CDC threshold defining elevated BMI (overweight or obesity). There were no significant differences in age at first visit between those who were persistently overweight or obese compared with those of normal ceBMI (median ages 9.4 and 9.9 years, respectively, P = 0.97). Similarly, no significant differences in distribution of males to females based on ceBMI ≥0 kg/m2 vs. <0 kg/m2 (P = 0.78) and islet autoantibody type (P = 0.85) or differences in HLA haplotype distribution (i.e., having DR3-DQ2 and/or DR4-DQ8, DR3-DQ2, or DR4-DQ8; all P ≥0.80). We did find a higher proportion of Hispanic/Latino individuals among those who had a ceBMI ≥0 kg/m2 compared with subjects with a ceBMI <0 kg/m2 (26% vs. 13%, respectively, P = 0.0001).
Age and HLA Type Are Inherent Characteristics That Influence the Progression From Single to Multiple Autoantibody Positivity
Older age at first autoantibody presentation was a significant protective risk factor for the development of multiple autoantibody positivity. For every year of age, the associated rate of progression to multiple autoantibody positivity was reduced by 7% (hazard ratio [HR] 0.93, 95% C: 0.89–0.97; P = 0.0007), after adjustment for ceBMI ≥0 kg/m2, sex, autoantibody type, and T1D-associated HLA haplotype. As expected, carrying at least one of the high-risk HLA haplotypes (i.e., DR3-DQ2 and DR4-DQ8) also had a significant impact on the progression from single to multiple positive autoantibodies. Based on that same multivariable model, individuals with HLA DR3-DQ2 and/or DR4-DQ8 had a nearly twofold increased risk of developing multiple autoantibodies compared with those with neither DR3-DQ2 nor DR4-DQ8 (HR 1.86, 95% CI 1.23–2.82; P = 0.003), persistent after adjustments for sex, autoantibody type, and ceBMI ≥0 kg/m2.
ceBMI Influences Risk of Developing Multiple Islet Autoantibodies in an Age-Specific Manner
Using recursive partitioning cut point analyses, we identified the age cut point of ≥9 years at autoantibody presentation as a threshold that best discriminated the rate of progression from single to multiple positive autoantibodies, with those ≥9 years old having a reduced rate compared with those younger than 9 years of age even with adjustments for other factors listed above (HR 0.51, P < 0.0001). Within each of the age subgroups (i.e., ≥9 and <9 years), age as a continuous variable was no longer a significant influence on the likelihood of progression to multiple positive autoantibodies. The presence of HLA DR3-DQ2 and/or DR4-DQ8 corresponded to a significantly increased rate of progression to multiple autoantibody positivity in individuals ≥9 years of age (HR 2.27; P = 0.019) and, although not reaching statistical significance, it showed a similar directional impact in children <9 years of age at autoantibody presentation (HR 1.65; P = 0.059) even after adjustment for additional risk factors.
In the overall cohort, ceBMI was not independently associated with progression from single to multiple autoantibodies as a continuous measure (HR 1.03, 95% CI 0.99–1.07; P = 0.19) or dichotomized at the overweight threshold (ceBMI ≥0 kg/m2) after adjustment for age, HLA type, autoantibody subtype, and sex (HR 1.18, 95% CI 0.82–1.68; P = 0.37). Race or ethnicity were not significant in the analysis with the overall cohort or in the age subgroups, and thus they were not included in the final models. We observed evidence of a significant interaction effect between ceBMI (≥0 kg/m2 vs. not) and age (≥9 years vs. not) (P = 0.02); specifically, ceBMI increased the rate of progression to multiple positive autoantibodies for individuals ≥9 years of age. For each 1 kg/m2 increase in continuous ceBMI measure, the risk increased by 7% (HR 1.07, 95% CI 1.01–1.13; P = 0.02). When we dichotomized at ceBMI ≥0 kg/m2, we again found that participants who were 9 years old or older at single autoantibody presentation were at greater risk if they were persistently overweight or obese, with a near twofold increased rate of developing multiple positive autoantibodies compared with those who on average remained below the overweight threshold over time (HR 1.92, 95% CI 1.12–3.29; P = 0.018) (Fig. 1 and Supplementary Table 3). In children <9 years of age at autoantibody presentation, the influence of ceBMI was not significant either as continuous (HR 0.98, 95% CI 0.92–1.05; P = 0.57) or as dichotomized (HR 0.81, 95% CI 0.5–1.32; P = 0.40) measure (Fig. 1 and Supplementary Table 3). Of note, although an age of 9 years was identified through the recursive partitioning analysis, the same patterns persisted with other age cut points in the peri-pubertal period (e.g., 8, 10, 12, 14 years). At each of these ages, the effect of ceBMI on the risk of progression was minimal in younger children but more pronounced in older youth.
Figure 1.

ceBMI increased progression to multiple autoantibody positivity in an age-specific manner. ceBMI above the age- and sex-adjusted 85th percentile (i.e., ceBMI ≥0 kg/m2) did not increase risk in subjects <9 years of age (HR 0.81, 95% CI 0.5–1.31; P = 0.39) (A) but was influential in those ≥9 years of age at first autoantibody presentation, where ceBMI ≥0 kg/m2 (i.e., being persistently obese or overweight) almost doubled the risk of progression from single to multiple autoantibody positivity (HR 1.93, 95% CI 1.13–3.31; P = 0.017) (B).
ceBMI Is Important for Rate of Multiple Autoantibody Progression in Participants With Less Aggressive Inherent Risk Factors
We additionally found a significant three-way interaction between age, ceBMI, and HLA type (P = 0.009), leading to further stratification to determine the effect of ceBMI in each of the four subgroups based on HLA status (carrying DR3-DQ2 and/or DR4-DQ8 vs. none) and age (≥9 vs. <9 years old). As shown in Table 2, having HLA DR3-DQ2 and/or DR4-DQ8 was significantly associated with an increased rate of progression to multiple autoantibody positivity in participants ≥9 years of age compared with the absence of DR3-DQ2 or DR4-DQ8 haplotypes. Importantly, in individuals ≥9 years old without either of the high-risk DR3-DQ2 or DR4-DQ8 HLA haplotypes, having ceBMI ≥0 kg/m2 was a significant factor in differentiating risk of progression to multiple autoantibodies (HR 7.32, 95% CI 1.88–28.5; P = 0.004) (Table 2). In fact, ceBMI ≥0 kg/m2 appeared to best differentiate the likelihood of progression in participants without the T1D-associated HLA haplotypes, and their risk of progression became similar to that of the participants who did have T1D-associated HLA haplotypes (Fig. 2). In contrast, ceBMI ≥0 kg/m2 was not a significant factor for risk of progression to multiple autoantibody positivity in those older children (≥9 years) with DR3-DQ2 and/or DR4-DQ8 (HR 1.38, 95% CI 0.32–2.59, P = 0.31). In children <9 years of age, ceBMI did not influence the progression to multiple autoantibodies, regardless of HLA type (Table 2). Supplementary Table 3 illustrates additional comparisons.
Table 2.
ceBMI increased risk of progression to multiple autoantibody positivity in older children with lower-risk HLA haplotype
| Age | HLA haplotype | n | HR (95% CI) | P | ceBMI (kg/m2) | n | HR (95% CI) | P |
|---|---|---|---|---|---|---|---|---|
| <9 years | Neither DR3 nor DR4-DQ8 | 87 | Ref | 0.068 | <0 | 65 | Ref | 0.15 |
| ≥0 | 22 | 0.34 (0.08–1.49) | ||||||
| DR3 and/or DR4-DQ8 | 226 | 1.62 (0.96–2.73) | <0 | 161 | Ref | 0.87 | ||
| ≥0 | 65 | 0.96 (0.57–1.61) | ||||||
| ≥9 years | Neither DR3 nor DR4-DQ8 | 118 | Ref | 0.018 | <0 | 86 | Ref | 0.004 |
| ≥0 | 32 | 7.32 (1.88–28.5) | ||||||
| DR3 and/or DR4-DQ8 | 273 | 2.28 (1.15–4.53) | <0 | 203 | Ref | 0.31 | ||
| ≥0 | 70 | 1.38 (0.32–2.59) |
Ref, reference.
Figure 2.

ceBMI influenced progression to multiple autoantibody positivity in subjects ≥9 years of age at first autoantibody progression with a lower-risk HLA haplotype (i.e., neither DR3 nor DR4-DQ8): In this subset of children, ceBMI ≥0 kg/m2 (i.e., being persistently obese or overweight) increased by more than sevenfold the risk of progression to multiple autoantibody positivity (HR 7.32, 95% CI 1.88–28.5; P = 0.004), and their risk was similar to that in the children who had the high-risk HLA haplotypes. In contrast, ceBMI did not further increase the progression to multiple autoantibodies in children with high-risk HLA haplotypes.
Conclusions
T1D is now recognized as a spectrum of diseases with heterogenous preclinical stages prior to the onset of symptomatic hyperglycemia. While T1D prevention trials focus on halting the development of clinical onset of T1D (stage 3), there is also great interest in arresting the development of multiple autoantibody positivity, which marks entry into stage 1 of T1D. In the TrialNet PTP cohort, the presence of HLA DR3-DQ2 and/or DR4-DQ8 haplotypes and younger age are inherent, independent risk factors for progressing from single to multiple positive autoantibodies (27–29). We have demonstrated that ceBMI above the threshold for overweight/obese increases the risk for multiple autoantibody development in youth >9 years old who do not have HLA DR3-DQ2 or DR4-DQ8, thus diminishing the protective influence of older age and absence of high-risk HLA haplotypes. Our findings that ceBMI had influence in some subgroups but not in others (causing lack of detectable effect on the overall cohort) highlight the recognized heterogeneity of T1D and the importance of stratified analysis.
Investigations into the role of BMI in T1D pathophysiology have not been consistent. Many of these prior prospective studies limited analyses to one measurement of BMI (e.g., birth weight, weight at cohort enrollment) without considering the interval values (30–32). Our previous analysis of longitudinal BMI measurements demonstrated that sustained elevation in BMI increased the risk of T1D onset in both pediatric and adult subjects followed in the TrialNet PTP cohort (14,15). The mechanism of this influence may be related to insulin resistance in the setting of compromised β-cell function (11,12). This current study proposes that obesity-induced exacerbation of autoimmunity may be an additional mechanism by which longitudinal elevation in BMI contributes to the preclinical stages of T1D. The accelerator hypothesis first promoted by Wilkin and colleagues (6,10) proposed that both type 1 and type 2 diabetes have a common origin in obesity-induced insulin resistance, while our data suggest that elevated BMI is associated with an acceleration in the progression of islet autoimmunity in certain subgroups of individuals.
The appearance of multiple islet autoantibodies has been shown to be a significant milestone for T1D development, with 70% of individuals progressing to clinical diabetes (stage 3) within 10 years of seroconversion (33). Of note, older age is a protective factor from progression to clinical T1D (2,26). Given that only a minority of single antibody subjects develop T1D (27), identifying ceBMI as modifiable risk factor, with both genetic and environmental causes, that influences the conversion from single to multiple autoantibodies in the PTP cohort has clinical implications, as it could be targeted to prevent or delay progression to clinical T1D. Importantly, subjects ≥9 years old without HLA DR3 or DR4, who should have less aggressive autoimmunity and lower incidence of multiple autoantibody conversion, may acquire high risk of entering stage 1 of T1D if BMI is sustained above the overweight/obese threshold. Thus, in this subgroup with lower intrinsic risk (i.e., older and lacking high-risk HLA haplotypes), risk factors such as elevated BMI may play a more critical role in initiating the first preclinical phase of T1D.
There is growing literature that suggests that obesity-induced inflammation may be a contributing factor leading to the development of autoimmune disease (34,35). Elevated levels of the proinflammatory adipokine leptin have been implicated in several autoimmune diseases, such as rheumatoid arthritis, lupus, and inflammatory bowel disease, among others. In a study by Marzullo et al. (36), positivity for thyroid peroxidase antibodies was associated with elevated leptin levels independent of BMI status. The levels of the anti-inflammatory adipokine, adiponectin, were found to be similar in patients with T1D compared with healthy subjects, but monocytes from T1D subjects had reduced adiponectin receptors, blunting adiponectin inhibition of T-cell proliferation (37). Moreover, adiponectin has been shown to have direct effects on the β-cells, promoting function and survival (38), further highlighting the adipose tissue as an active participant in T1D pathophysiology, although the nature of the associations is still unclear.
The potential relationship between obesity-induced inflammation and autoimmune disease, including islet autoimmunity, could also underlie our previous observations that the type 2 diabetes–associated TCF7L2 genetic variant, which may be involved in fatty acid metabolism (39), accelerates antigen spreading in individuals with a single positive ICA512/IA-2 or insulin autoantibody who are obese or overweight (40). In this smaller study of pediatric and adult TrialNet participants who had available ImmunoChip data, the effect of ceBMI or age strata was not evaluated. In addition, this genetic variant is associated with single autoantibody expression at the onset of clinical T1D (41). In these individuals with only mild islet autoimmunity, TCF7L2 variants, via β-cell dysfunction, insulin resistance, and diabetogenic interaction with obesity (39) could precipitate the progression to clinical T1D, reflecting a complex interplay between metabolic risk variants and autoimmunity in the setting of T1D (41). Taken together, these findings suggest that type 2 diabetes–related mechanisms (e.g., genetics, obesity/overweight) not only can add to the risk of clinical diabetes in individuals with evidence of islet autoimmunity but also interact with the autoimmune process (reviewed in ref. 42). When the combination (and interaction) of diabetogenic factors exceeds a certain threshold, clinical diabetes develops, as proposed by the threshold hypothesis (13). This concept could justify therapeutic strategies that address type 2 diabetes–related mechanisms, when present, as adjuvants to immunomodulatory therapies aiming to prevent or delay T1D progression.
A major strength of this study is the size of the study cohort followed longitudinally at regular intervals using a standard protocol, which allows for sophisticated analysis including recursive partitioning to identify specific subgroups who are susceptible to the effects of ceBMI. The inclusion of subjects carrying low-, intermediate-, and high-risk HLA class genotypes in the PTP cohort allowed us to identify ceBMI as a modifiable risk factor for autoantibody progression in older individuals who would be considered to have a less aggressive autoimmune profile based on HLA subtype. Our study does, however, have some limitations. The TrialNet PTP cohort is not followed from birth, and therefore the duration of a single positive autoantibody is unknown. Additionally, some individuals are followed for a short period of time, which may have influenced these results. We do note an increased proportion of Hispanic/Latino subjects among those with a ceBMI ≥0 kg/m2, which is consistent with higher prevalence of elevated BMI among Hispanic children from the general population (5), and while we did accommodate HLA subtype in our analysis, there may be other genetic contributors. Per TrialNet study protocol, ZnT8 autoantibodies were not measured in one-third of the participants, with ∼20–25% (i.e., 8% of the entire cohort) expected to be positive. Some of the subgroups analyzed in this unique cohort of participants were relatively small in size; thus, results should be confirmed in the future. Finally, the optimal cut points identified for age that influence progression to multiple positive autoantibodies are data derived using recursive partitioning analysis and may not be broadly applicable to other populations.
We provide evidence that sustained elevation in BMI may at least in part contribute to T1D pathophysiology through an acceleration of the autoimmune process, particularly in those without high-risk factors, in addition to the known effect of increasing β-cell demand. Future studies are required to identify signaling molecules responsible for obesity-induced autoimmunity specifically in T1D, but our data demonstrate the important interaction of diverse risk factors in T1D, e.g., age, HLA subtype, and BMI. Targeted lifestyle interventions to maintain BMI <85th percentile may help prevent or delay the progression to multiple autoantibody positivity and could halt the growing incidence of clinical stage 3 T1D.
Supplementary Material
Article Information
Funding. The Type 1 Diabetes TrialNet Study Group is a clinical trials network currently funded by the National Institutes of Health (NIH) through the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of Allergy and Infectious Diseases, and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, through the cooperative agreements U01 DK061010, U01 DK061034, U01 DK061042, U01 DK061058, U01 DK085461, U01 DK085465, U01 DK085466, U01 DK085476, U01 DK085499, U01 DK085509, U01 DK103180, U01 DK103153, U01 DK103266, U01 DK103282, U01 DK106984, U01 DK106994, U01 DK107013, U01 DK107014, UC4 DK106993, UC4DK117009, and JDRF. This work was also partially supported by the Pediatric Endocrine Society Clinical Scholars, award number A128964, and the National Center for Advancing Translational Sciences, National Institutes of Health, through UCSF-CTSI grant UL1 TR001872 (to C.F.-C.) and U01-DK-103180-01 (to M.J.R.).
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or JDRF.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. C.F.-C., S.M.G., C.E.-M., I.M.L., D.J.B., S.E.G., and M.J.R. are members of the Type 1 Diabetes TrialNet Study Group. C.F.-C. designed the study and analysis plan and wrote the first draft of the manuscript. S.M.G. analyzed the data and contributed statistical support and editing of the manuscript. C.E.-M., I.M.L., D.J.B., and S.E.G. contributed to interpretation of results and revised the manuscript. M.J.R. contributed to study and data analysis design and interpretation of results and critically revised the manuscript. C.F.-C. and M.J.R. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 2018 Pediatric Endocrine Society Annual Meeting, 5–8 May 2018, Toronto, Canada; at the 2019 Pediatric Endocrine Society Annual Meeting, 24 April–1 May 2019, Baltimore, MD; and at the 79th Scientific Sessions of the American Diabetes Association, San Francisco, CA, 7–11 June 2019.
Footnotes
A complete list of the members of the Type 1 Diabetes TrialNet Study Group can be found in Supplementary Data.
This article contains Supplementary Data online at https://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc19-1167/-/DC1.
References
- 1.Lawrence JM, Imperatore G, Dabelea D, et al.; SEARCH for Diabetes in Youth Study Group . Trends in incidence of type 1 diabetes among non-Hispanic white youth in the U.S., 2002-2009. Diabetes 2014;63:3938–3945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Redondo MJ, Yu L, Hawa M, et al. . Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia 2001;44:354–362 [DOI] [PubMed] [Google Scholar]
- 3.Pierce BG, Eberwine R, Noble JA, et al. . The missing heritability in T1D and potential new targets for prevention. J Diabetes Res 2013;2013:737485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA 2012;307:491–497 [DOI] [PubMed] [Google Scholar]
- 5.Ogden CL, Carroll MD, Lawman HG, et al. . Trends in obesity prevalence among children and adolescents in the United States, 1988-1994 through 2013-2014. JAMA 2016;315:2292–2299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betts P, Mulligan J, Ward P, Smith B, Wilkin T. Increasing body weight predicts the earlier onset of insulin-dependant diabetes in childhood: testing the ‘accelerator hypothesis’ (2). Diabet Med 2005;22:144–151 [DOI] [PubMed] [Google Scholar]
- 7.Lauria A, Barker A, Schloot N, et al. . BMI is an important driver of β-cell loss in type 1 diabetes upon diagnosis in 10 to 18-year-old children. Eur J Endocrinol 2015;172:107–113 [DOI] [PubMed] [Google Scholar]
- 8.Fourlanos S, Narendran P, Byrnes GB, Colman PG, Harrison LC. Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia 2004;47:1661–1667 [DOI] [PubMed] [Google Scholar]
- 9.Viner RM, Hindmarsh PC, Taylor B, Cole TJ. Childhood body mass index (BMI), breastfeeding and risk of type 1 diabetes: findings from a longitudinal national birth cohort. Diabet Med 2008;25:1056–1061 [DOI] [PubMed] [Google Scholar]
- 10.Wilkin TJ. The accelerator hypothesis: weight gain as the missing link between type I and type II diabetes. Diabetologia 2001;44:914–922 [DOI] [PubMed] [Google Scholar]
- 11.Libman IM, Becker DJ. Coexistence of type 1 and type 2 diabetes mellitus: “double” diabetes? Pediatr Diabetes 2003;4:110–113 [DOI] [PubMed] [Google Scholar]
- 12.Libman IM, Pietropaolo M, Arslanian SA, LaPorte RE, Becker DJ. Changing prevalence of overweight children and adolescents at onset of insulin-treated diabetes. Diabetes Care 2003;26:2871–2875 [DOI] [PubMed] [Google Scholar]
- 13.Wasserfall C, Nead K, Mathews C, Atkinson MA. The threshold hypothesis: solving the equation of nurture vs nature in type 1 diabetes. Diabetologia 2011;54:2232–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrara CT, Geyer S, Evans-Molina C, et al.; Type 1 Diabetes TrialNet Study Group . The role of age and excess body mass index in progression to type 1 diabetes in at-risk adults. J Clin Endocrinol Metab 2017;102:4596–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrara CT, Geyer SM, Liu YF, et al.; Type 1 Diabetes TrialNet Study Group . Excess BMI in childhood: a modifiable risk factor for type 1 diabetes development. Diabetes Care 2017;40:698–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skyler JS, Greenbaum CJ, Lachin JM, et al.; Type 1 Diabetes TrialNet Study Group . Type 1 Diabetes TrialNet--an international collaborative clinical trials network. Ann N Y Acad Sci 2008;1150:14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baekkeskov S, Aanstoot HJ, Christgau S, et al. . Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase [published correction appears in Nature 1990;347:782]. Nature 1990;347:151–156 [DOI] [PubMed] [Google Scholar]
- 18.Palmer JP, Asplin CM, Clemons P, et al. . Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 1983;222:1337–1339 [DOI] [PubMed] [Google Scholar]
- 19.Solimena M, Dirkx R Jr., Hermel JM, et al. . ICA 512, an autoantigen of type I diabetes, is an intrinsic membrane protein of neurosecretory granules. EMBO J 1996;15:2102–2114 [PMC free article] [PubMed] [Google Scholar]
- 20.Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974;2:1279–1283 [DOI] [PubMed] [Google Scholar]
- 21.Wenzlau JM, Juhl K, Yu L, et al. . The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A 2007;104:17040–17045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sosenko JM, Mahon J, Rafkin L, et al.; Diabetes Prevention Trial-Type 1 and TrialNet Study Groups . A comparison of the baseline metabolic profiles between Diabetes Prevention Trial-Type 1 and TrialNet Natural History Study participants. Pediatr Diabetes 2011;12:85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.American Diabetes Association Standards of medical care in diabetes--2014. In Clinical Care Recommendations, 2014. Diabetes Care 2014;37(Suppl. 1):S14–S80 [DOI] [PubMed] [Google Scholar]
- 24.Bouchard DR, Porneala B, Janssen I, et al. . Risk of type 2 diabetes and cumulative excess weight exposure in the Framingham Offspring Study. J Diabetes Complications 2013;27:214–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JM, Gebremariam A, Vijan S, Gurney JG. Excess body mass index-years, a measure of degree and duration of excess weight, and risk for incident diabetes. Arch Pediatr Adolesc Med 2012;166:42–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu P, Krischer JP; Type 1 Diabetes TrialNet Study Group . Prognostic classification factors associated with development of multiple autoantibodies, dysglycemia, and type 1 diabetes-a recursive partitioning analysis. Diabetes Care 2016;39:1036–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bingley PJ, Boulware DC, Krischer JP; Type 1 Diabetes TrialNet Study Group . The implications of autoantibodies to a single islet antigen in relatives with normal glucose tolerance: development of other autoantibodies and progression to type 1 diabetes. Diabetologia 2016;59:542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Redondo MJ, Babu S, Zeidler A, et al.; Diabetes Prevention Trial Type 1 Study Group . Specific human leukocyte antigen DQ influence on expression of antiislet autoantibodies and progression to type 1 diabetes. J Clin Endocrinol Metab 2006;91:1705–1713 [DOI] [PubMed] [Google Scholar]
- 29.Barker JM, Barriga KJ, Yu L, et al.; Diabetes Autoimmunity Study in the Young . Prediction of autoantibody positivity and progression to type 1 diabetes: Diabetes Autoimmunity Study in the Young (DAISY). J Clin Endocrinol Metab 2004;89:3896–3902 [DOI] [PubMed] [Google Scholar]
- 30.Meah FA, DiMeglio LA, Greenbaum CJ, et al.; Type 1 Diabetes TrialNet Study Group . The relationship between BMI and insulin resistance and progression from single to multiple autoantibody positivity and type 1 diabetes among TrialNet Pathway to Prevention participants. Diabetologia 2016;59:1186–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dabelea D, D’Agostino RB Jr., Mayer-Davis EJ, et al.; SEARCH for Diabetes in Youth Study Group . Testing the accelerator hypothesis: body size, β-cell function, and age at onset of type 1 (autoimmune) diabetes. Diabetes Care 2006;29:290–294 [DOI] [PubMed] [Google Scholar]
- 32.Kuchlbauer V, Vogel M, Gausche R, et al. . High birth weights but not excessive weight gain prior to manifestation are related to earlier onset of diabetes in childhood: ‘accelerator hypothesis’ revisited. Pediatr Diabetes 2014;15:428–435 [DOI] [PubMed] [Google Scholar]
- 33.Ziegler AG, Rewers M, Simell O, et al. . Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013;309:2473–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Versini M, Aljadeff G, Jeandel PY, Shoenfeld Y. Obesity: an additional piece in the mosaic of autoimmunity. Isr Med Assoc J 2014;16:619–621 [PubMed] [Google Scholar]
- 35.Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev 2014;13:981–1000 [DOI] [PubMed] [Google Scholar]
- 36.Marzullo P, Minocci A, Tagliaferri MA, et al. . Investigations of thyroid hormones and antibodies in obesity: leptin levels are associated with thyroid autoimmunity independent of bioanthropometric, hormonal, and weight-related determinants. J Clin Endocrinol Metab 2010;95:3965–3972 [DOI] [PubMed] [Google Scholar]
- 37.Pang TT, Chimen M, Goble E, et al. . Inhibition of islet immunoreactivity by adiponectin is attenuated in human type 1 diabetes. J Clin Endocrinol Metab 2013;98:E418–E428 [DOI] [PubMed] [Google Scholar]
- 38.Wijesekara N, Krishnamurthy M, Bhattacharjee A, Suhail A, Sweeney G, Wheeler MB. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J Biol Chem 2010;285:33623–33631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grant SFA. The TCF7L2 locus: a genetic window into the pathogenesis of type 1 and type 2 diabetes. Diabetes Care 2019;42:1624–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Redondo MJ, Steck AK, Sosenko J, et al.; Type 1 Diabetes TrialNet Study Group . Transcription Factor 7-Like 2 (TCF7L2) gene polymorphism and progression from single to multiple autoantibody positivity in individuals at risk for type 1 diabetes. Diabetes Care 2018;41:2480–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Redondo MJ, Geyer S, Steck AK, et al.; Type 1 Diabetes TrialNet Study Group . TCF7L2 genetic variants contribute to phenotypic heterogeneity of type 1 diabetes. Diabetes Care 2018;41:311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Redondo MJ, Evans-Molina C, Steck AK, Atkinson MA, Sosenko J. The influence of type 2 diabetes–associated factors on type 1 diabetes. Diabetes Care 2019;42:1357–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.