

Abstract
Rotation around a specific bond after photoexcitation is central to vision and emerging opportunities in optogenetics, super-resolution microscopy, and photoactive molecular devices. Competing roles for steric and electrostatic effects that govern bond-specific photoisomerization have been widely discussed, the latter originating from chromophore charge transfer upon excitation. We systematically altered the electrostatic properties of the green fluorescent protein chromophore in a photoswitchable variant, Dronpa2, using amber suppression to introduce electron-donating and electron-withdrawing groups to the phenolate ring. Through analysis of the absorption (color), fluorescence quantum yield, and energy barriers to ground- and excited-state isomerization, we evaluate the contributions of sterics and electrostatics quantitatively and demonstrate how electrostatic effects bias the pathway of chromophore photoisomerization, leading to a generalized framework to guide protein design.
Photoisomerizable chromophores, such as those in rhodopsins, phytochromes, photoactive yellow proteins, and fluorescent proteins (FPs), rotate around specific bonds after photoexcitation in the protein environment, which is essential to converting light energy into molecular motion (1). To investigate the role of the protein environment on tuning bound chromophore and/or ligand functionality, we chose to study FPs, a relatively simple model system consisting of an autocatalytically formed chromophore contained in a β barrel (2). The chromophore’s local environment can markedly alter its photophysical properties, leading to a wide range of colors, fluorescence quantum yields (FQYs), and photoswitching characteristics (3). The chromophore’s FQY increases by three orders of magnitude when contained in the protein scaffold compared with when it is free in solution (4). The dominant nonradiative decay process that lowers the chromophore’s FQY is isomerization about either the phenolate (P) or imidazolinone (I) bonds, resulting in a P-ring flip or cis-trans isomerization, respectively (Fig. 1A) (5). This nonradiative decay process is enhanced in photoswitching FPs that are widely used for super-resolution microscopy (3). Modulating the probability between radiative and nonradiative decay, and for the latter, the propensity for P- or I-bond isomerization, epitomizes the essential features of protein control.
Fig. 1. Model for chromophore isomerization in FPs.
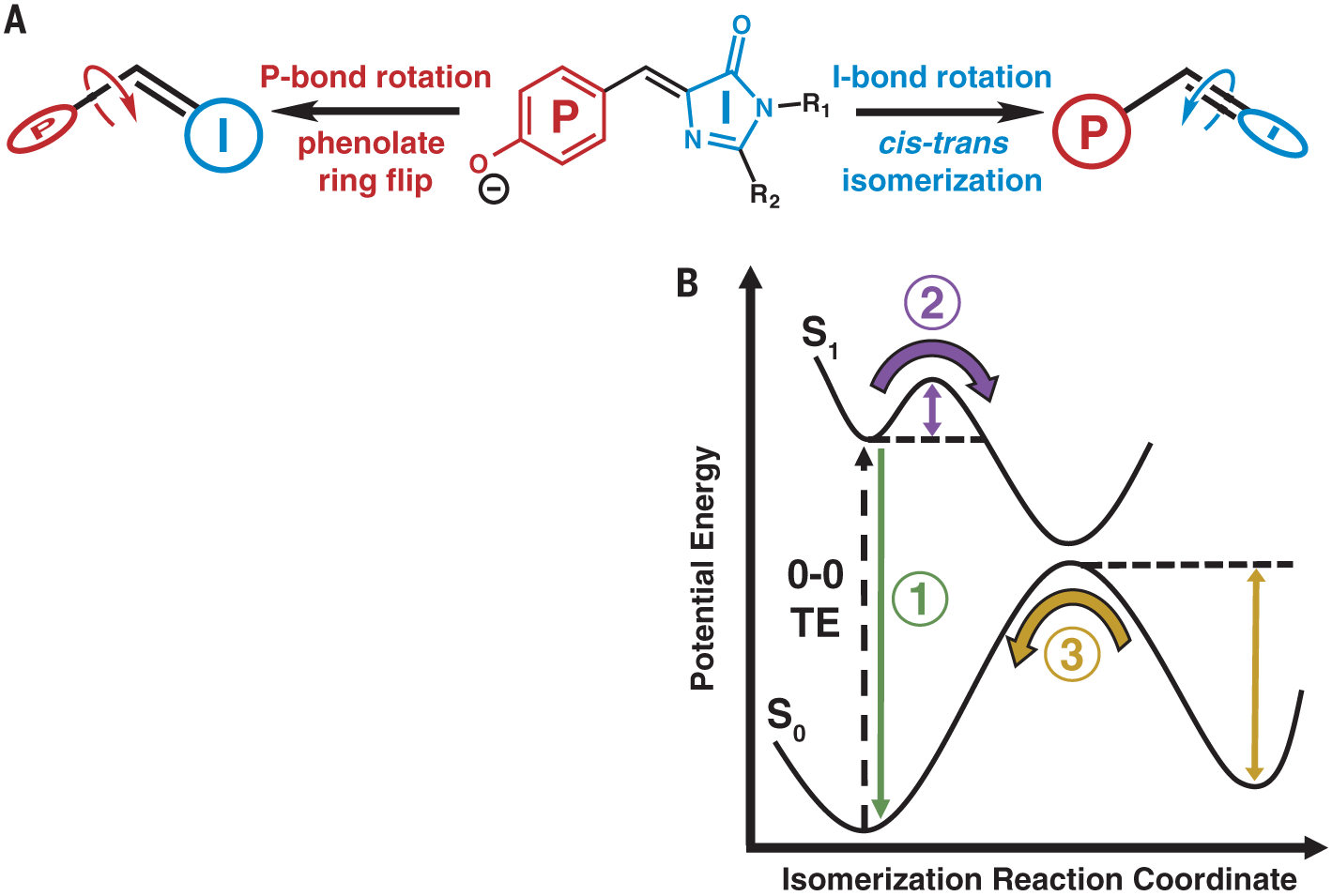
(A) Rotation can occur about either the P or I bond, leading to a P-ring flip or cis-trans isomerization, respectively. R1 and R2 represent residues Gly64 and Cys62, respectively, which covalently link the chromophore to the rest of the FP (Fig. 2A). (B) General potential energy diagram along the isomerization reaction coordinate for a photoisomerizable chromophore. 0–0 TE represents the TE between the lowest vibrational state of the ground and excited electronic states. Three features studied in this work are emphasized: fluorescence (1, green), ES barrier crossing (2, purple), and GS barrier crossing (3, yellow).
The most well-studied and intuitively appealing hypothesis for the chromophore’s substantial increase in FQY in the protein suggests that steric confinement of the protein scaffold physically prevents the bond rotation required for nonradiative decay, as demonstrated by studies involving chemically locked or artificially confined chromophores (6). An alternative hypothesis identifies the role of electrostatics in modulating the FQY. After a perturbation to either the chromophore’s electronic state (e.g., by photon absorption) or nuclear coordinates (e.g., by isomerization), a redistribution of the chromophore’s electron density occurs, which is usually described as charge transfer between the rings. Consequently, the electric field exerted by the environment can either promote or hinder charge transfer and thus could control whether fluorescence or isomerization is more favorable after excitation (7).
In earlier work on split green fluorescent protein (GFP), we linked structure and function with energetics (8) and showed that the dominant energetic feature governing the competition between fluorescence and isomerization is the excited-state (ES) barrier for chromophore bond rotation (Fig. 1B, process 2). Here we present a systematic study investigating the contributions of sterics and electrostatics to energetic features of the chromophore’s potential energy surface in both the ground state (GS) and ES. To experimentally probe these effects, we introduced a diverse range of substituents on the chromophore’s P ring using amber suppression (9) with substituted tyrosine residues (10), taking advantage of the chromophore’s autocatalytic maturation process (Fig. 2A). The electronic perturbation to the chromophore due to the substituent can be thought of as analogous to a perturbation of the protein environment around the chromophore that alters the chromophore’s electronic properties, as suggested by past studies on polymethine dyes (11). As a model system, we chose the widely used photoswitchable FP Dronpa2 [the Met159→Thr (M159T) mutant of Dronpa] because of the balance between its moderately high FQY and photoisomerization efficiency (12). We also include results from a nonphotoswitchable FP, a superfolder GFP construct, to generalize the scope of our conclusions.
Fig. 2. Incorporation of electron-donating and electron-withdrawing substituents into the Dronpa2 chromophore.
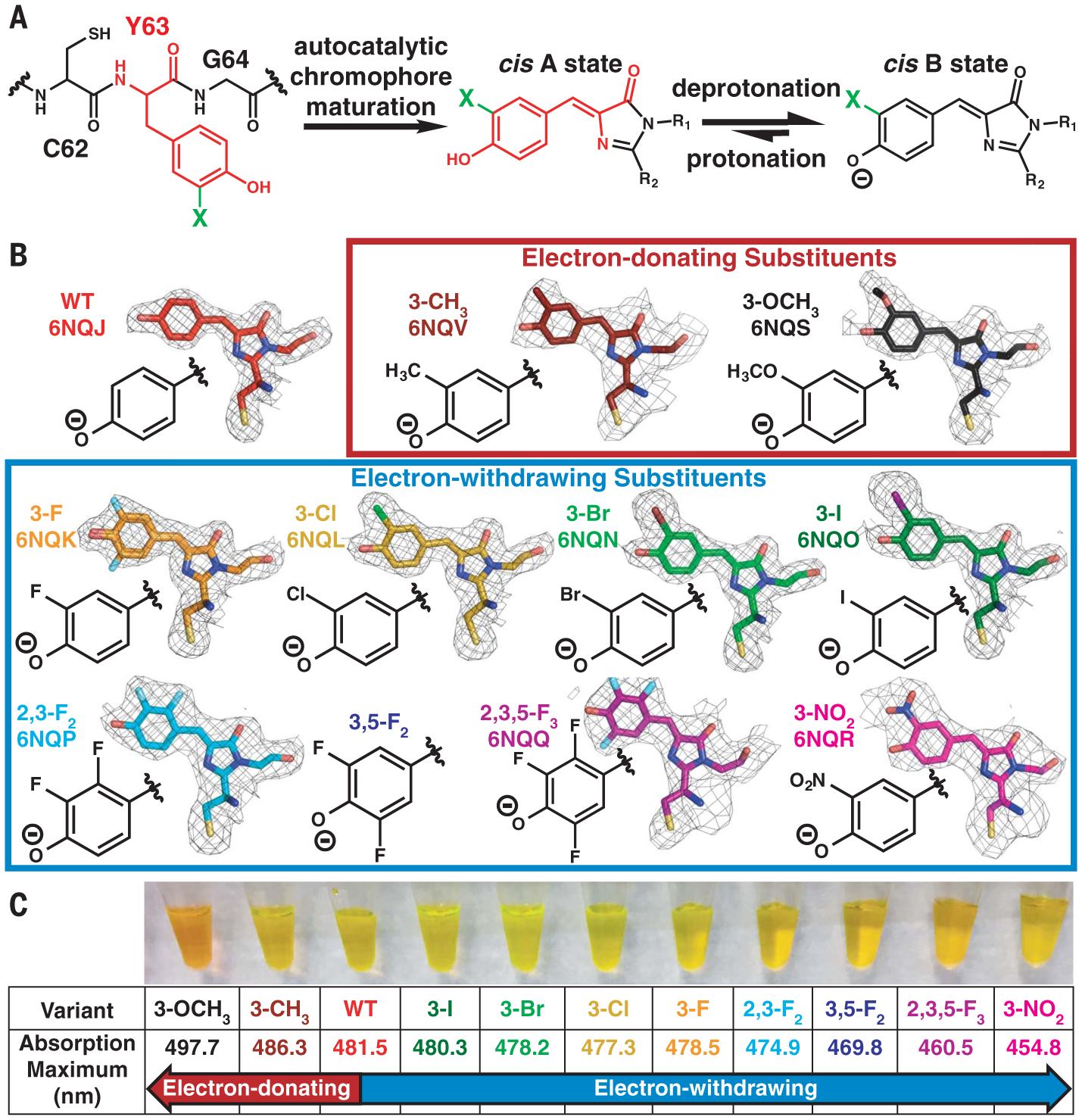
(A) Scheme depicting incorporation of substituents (represented by a green “X”) through amber suppression of Y63 and chromophore maturation in Dronpa2 variants (C, Cys; Y, Tyr; G, Gly). (B) Dronpa2 amber suppression variants grouped by electron-donating and electron-withdrawing properties. The electron density maps (2mFo – DFc, 1σ) from solved x-ray structures (except 3,5-F2, which could not be crystallized; see supplementary text S2) show substituent orientation(s) (see fig. S2 for omit maps). Two conformations were necessary for modeling the chromophore of the 3-F variant (fig. S2). The legend of fig. S2 includes the identity of the monomer displayed for each variant. WT, wild type. (C) Image of purified proteins and their corresponding 77 K absorption peak maxima.
We expressed wild-type and 10 Dronpa2 variants with chromophores containing electron-donating and electron-withdrawing substituents on the P ring (Fig. 2B). X-ray crystal structures confirm that the P-ring substituent(s) occupies a single orientation, except for the 3-F variant, which has two orientations (supplementary text S1 and fig. S2). Introduction of the substituent(s) causes little to no structural deviation compared with the wild type (supplementary text S2 and figs. S1 to S3). The absorption spectrum for each Dronpa2 variant directly reflects the electronic contribution of the substituent: Electron-donating groups red-shift, whereas electron-withdrawing groups blue-shift the absorption maximum (Fig. 2C and fig. S4). Both the trend of electrostatic color tuning and the direction of charge transfer upon excitation can be understood through either a Hammett analysis (supplementary text S3) or Olsen’s resonance color theory (13) (supplementary text S4 and fig. S8), agreeing with simulations that suggest negative charge flows from the P ring to the I ring upon photon absorption (5). We use the absorption peak maximum [an approximation of the 0–0 transition energy (TE)] (supplementary text S5) as a scale to reflect the substituents’ electron-donating and electron-withdrawing capabilities (supplementary text S4) because the initial Franck-Condon excitation is purely an electronic process.
We then sought to examine the influence of each chromophore’s steric and electronic properties on ES processes. First, we measured each variant’s FQY (Fig. 1B, process 1) and plotted the values against the corresponding TE (Fig. 3A). The trend is nonmonotonic with a peaked shape; variants with red- and blue-shifted TEs show positive and negative correlations with FQY, respectively. A variant of superfolder GFP with the same series of substituted chromophores exhibits the same trend (fig. S7), suggesting that electrostatic sensitivity is an intrinsic chromophore property.
Fig. 3. GS and ES properties of Dronpa2 amber suppression variants.
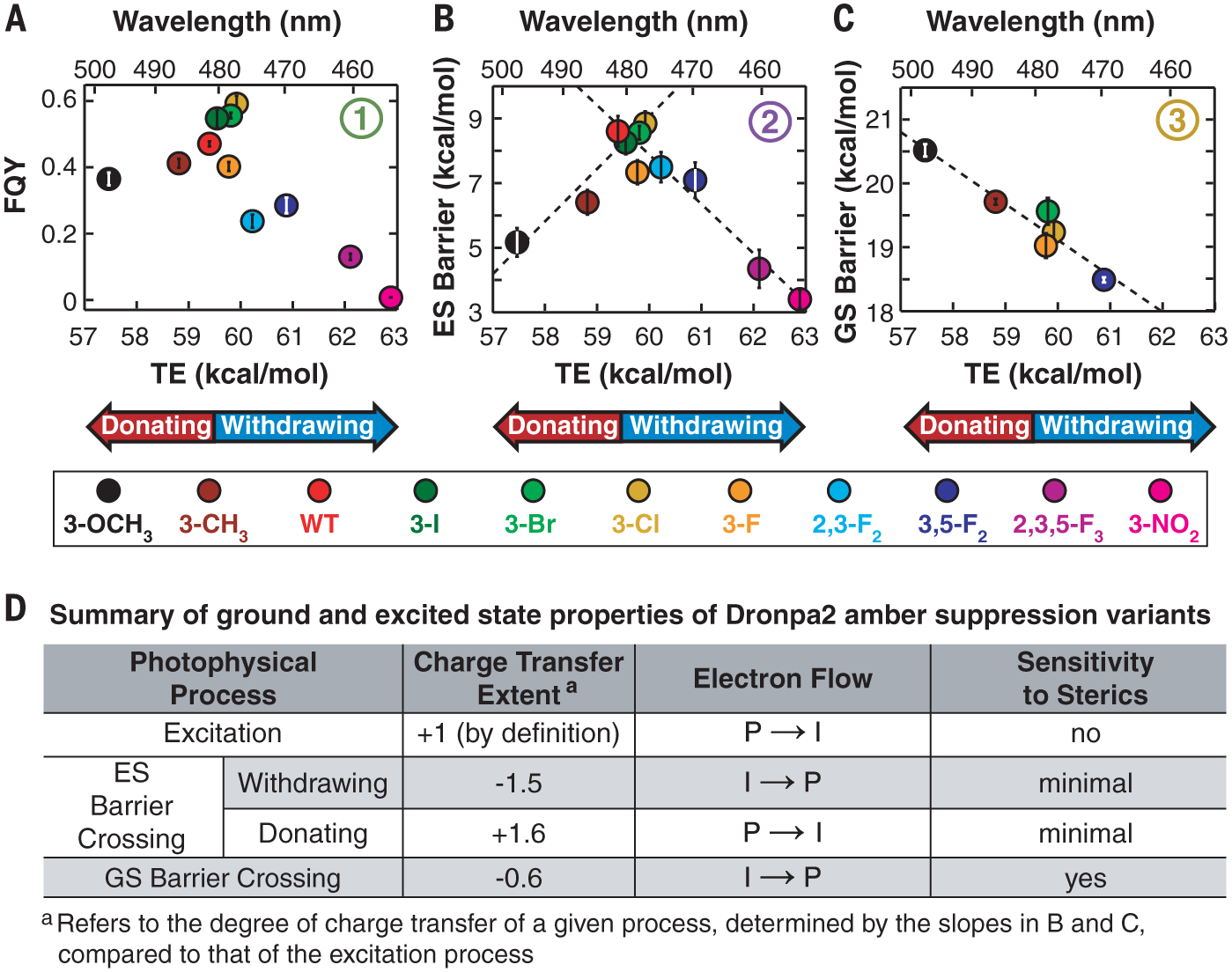
Circled numbers refer to processes depicted in Fig. 1B. (A) FQY versus TE and (B) ES energy barrier height versus TE both exhibit a peaked shape (see fig. S7 for comparable FQY results from GFP variants). Linear fits to the electron-donating and electron-withdrawing substituent data in (B) are shown as dashed lines with positive and negative slope, respectively. (C) Isosteric series of the GS energy barrier height plotted against TE, with a dashed line representing a linear fit. (D) Summary of GS and ES properties of Dronpa2 variants.
To elucidate the underlying energetics, we estimated the ES energy barrier (Fig. 1B, process 2) for each Dronpa2 variant using temperature-dependent fluorescence lifetime measurements (supplementary texts S6 and S7 and figs. S10 to S15), which capture the combined decay rate of all relaxation processes from the S1 minimum. As with FQY, the ES barrier heights show a peaked trend when plotted against TE (Fig. 3B). Linear fits to the electron-donating and electron-withdrawing variants’ data exhibit slopes with similar magnitude but opposite sign (Fig. 3D), which describe the extent and direction of charge transfer during the ES barrier crossing. A change of 1 kcal/mol in TE in either direction corresponds to a change of ~1.5 kcal/mol in ES barrier height, implying that ES barrier crossing, a nonradiative process, is more sensitive to electronic effects than Franck-Condon excitation.
To investigate the role of steric and electronic effects on GS isomerization barrier height (Fig. 1B, process 3), we determined the isomerization rate constant through pH-dependent thermal relaxation kinetics measurements after photoexcitation to a cis-trans photostationary state, assuming the validity of transition state theory (supplementary text S8 and figs. S17 to S18). A plot of the GS barrier height versus TE (fig. S19) appears to show a lack of correlation between the GS barrier and the substituent’s electronic effects. Close examination of fig. S19 reveals that the substituent’s steric properties may also contribute to the observed trends. For example, among the data points for the 3-F, 3-Cl, 3-Br, and 3-I substituents, as highlighted by the gray box in fig. S19, the barrier height increases as a function of halogen size despite similar TEs, indicating that substituent size influences GS barrier height. To isolate the electrostatic contribution to GS barrier height, we created an isosteric substituent series (defined in supplementary text S9) and plotted the corresponding data for this subgroup, which monotonically decrease as a function of TE (Fig. 3C). The extent of charge transfer during GS barrier crossing reflected by the slope in Fig. 3C is approximately one-third of that in the ES (Fig. 3B and charge-transfer extent in Fig. 3D), suggesting that changes in the electronic properties of the chromophore have a smaller, but still evident, impact on thermal relaxation. In contrast to the GS barrier, the influence of sterics on the ES barrier is minimal (Fig. 3D and fig. S20). If sterics were the dominant factor, large substituents would be expected to increase the barrier to chromophore twisting in the ES and, consequently, FQY. However, electrostatics is clearly the dominant factor for ES isomerization in a constant protein environment (Fig. 3, A and B, and fig. S20).
The observation of two approximately equal but opposite slopes between ES barrier height and TE (Fig. 3B) suggests a mechanism change for barrier crossing that depends on the electronic properties of the chromophore (supplementary text S3). Charge transfer is coupled with bond twisting in the ES of monomethine dyes (5, 14). In the GFP chromophore, this twisting can proceed about either the P or I bond (Fig. 1A). Both P and I twisting in the isolated anionic chromophore have comparable barrier heights but opposite charge-transfer directions, suggesting that small perturbations to the chromophore or its environment could influence which twisting pathway is more energetically favorable (14). Our data reveal a decrease in FQY and ES barrier height compared with those from wild-type Dronpa2 regardless of the substituent’s electronic effect (Fig. 3, A and B), indicating that the electronic properties of the chromophore can tune the relative ES barrier heights for P and I twisting, thus biasing toward the pathway with the lower barrier. By contrast, the GS chromophore follows the same isomerization mechanism regardless of the substituent’s electronic effect (Fig. 3C).
The observed trends in energetics can be linked to structural intuition through a valence-bond model depicting the deprotonated chromophore as an allylic anion (Fig. 4, A and B) (5, 14, 15). In Fig. 4C, we present an energetic model for chromophore isomerization guided by the adiabatic states in Fig. 4B. The electronic properties of the chromophore, governed in this work by the substituent on the P ring, determine the ES barrier height difference between P and I twisting. For GS relaxation from trans to cis, we are limited by what we can measure spectroscopically, namely the photochromic cis-trans isomerization. We can therefore observe only one GS twisting mechanism (I twist), which explains the monotonic trend in GS barrier height as a function of TE (Fig. 3C). In the potential energy diagram (Fig. 4C), the transition state for either the P- or I-twist pathway in S1 (ES) lies closer to planarity than the corresponding transition state in S0 (GS). As a result, reaching the transition state on the GS surface requires greater bond rotation, explaining the enhanced steric sensitivity observed for GS isomerization.
Fig. 4. Allylic anion model of isomerization for a chromophore containing an electron-withdrawing substituent.
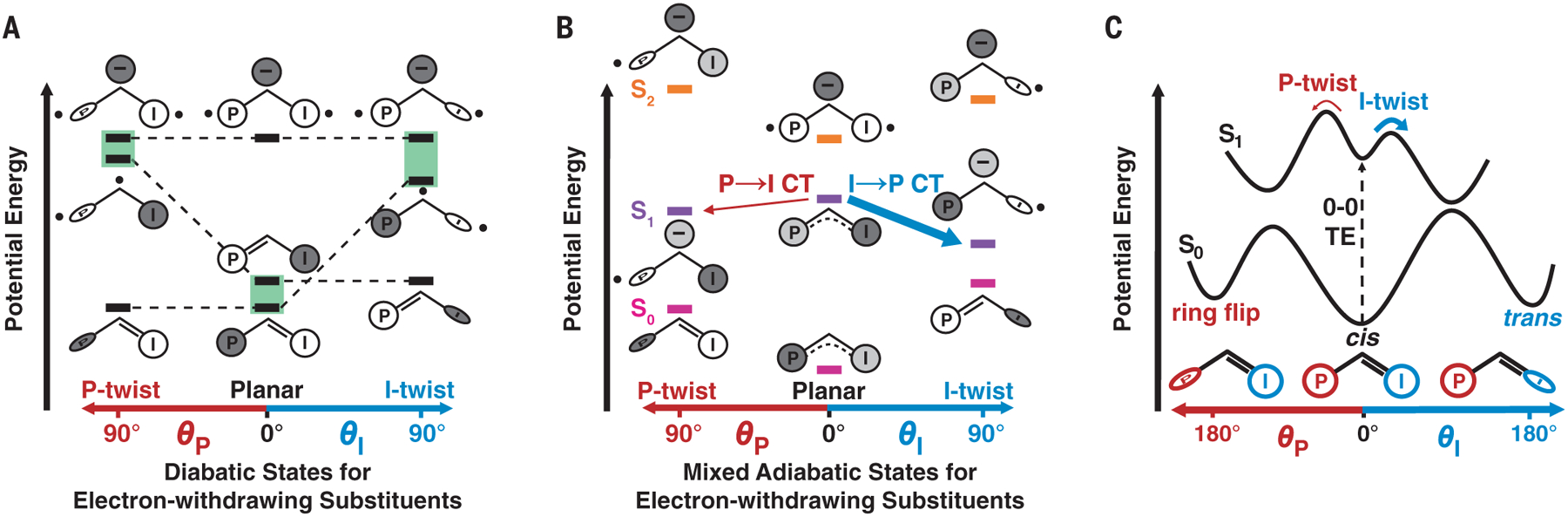
Shades of gray represent relative magnitude of negative charge localized to the methine bridge, P ring, or I ring, and dots represent unpaired electrons. The color scheme is equivalent to that in Fig. 1A. (A) Three diabatic states of the chromophore in planar, I-twisted, and P-twisted geometries, with energetic penalties required for breaking double bonds for rotation. Mixing of the coupled states (highlighted in green) leads to the adiabatic states shown in (B). For variants with electron-withdrawing substituents, the I-twist pathway is more energetically downhill, and thus preferred, compared with the P-twist pathway. Electron-donating substituents would have the opposite energetic effect and favor the P-twist pathway (not shown for clarity). Although the relative energy levels of this allylic anion model are qualitative, they are consistent with high-level calculations on the free chromophore at different bond rotation geometries (15). Negative charge transfer (CT) occurs from I to P for the I-twist pathway and from P to I for the P-twist pathway, which agrees with a Hammett analysis (supplementary text S3) and simulations of the free chromophore (5, 14). (C) Potential energy diagram for FP chromophore isomerization with two competing bond rotation pathways inspired by the mixed adiabatic states in (B). The GS cis chromophore is excited from S0 to S1 and relaxes to an S1 local minimum (relaxation coordinate not shown) (13). From the S1 minimum, the chromophore rotates about either the P or the I bond, depending on the relative ES barrier heights of the competing processes. The diagram represents Dronpa2 variants with electron-withdrawing substituents; variants with electron-donating substituents would have an inverted barrier height ratio between the two competing twisting pathways.
By engineering chromophore variants using amber suppression, we have systematically elucidated the role of electrostatics on chromophore color and isomerization in an FP environment. The electrostatic sensitivities of the chromophore stem from the intrinsic direction of charge transfer during electronic transitions and photoisomerizable bond rotations, which is ubiquitous in other photoisomerizable systems (8, 16–24). By tuning the environment of the chromophore in these protein systems, with an emphasis on the often-overlooked electrostatic component, it may be possible to finely control properties of interest, such as regioselective isomerization, because of distinctive charge redistributions as different bonds are rotated. On the basis of our results in FPs, introducing hydrogen-bond–donating residues around the P ring of the chromophore would bias toward the I-twist photoisomerization pathway (13). In the photoisomerizable retinal chromophore in rhodopsins, theoretical studies have suggested that different bond-specific photoisomerization intermediates have different electronic distributions (21), allowing for similar targeted environmental modifications to bias bond rotation pathways. As such, our conclusions provide an initial, generalizable framework to incorporate electrostatic and steric effects into the design of other photoisomerizable systems to help develop improved variants and new functionalities in optogenetics and imaging (1).
Supplementary Material
ACKNOWLEDGMENTS
We dedicate this manuscript to the memory of Seth Olsen, whose theoretical studies of the GFP chromophore motivated much of the analysis of this work. We thank S. Lynch of the Stanford NMR Facility for assistance with NMR data collection and interpretation.R. A. Mehl of the Unnatural Protein Facility was instrumental in providing an aminoacyl-tRNA synthetase for 3-methyltyrosine incorporation. S. H. Schneider helped develop the MATLAB code for statistical analysis of the fluorescence lifetime data. We thank J. I. Brauman, T. J. Martínez, N. H. List, S. D. Fried, and L. M. Oltrogge for discussions regarding this work.
Funding: M.G.R. was supported by a Center for Molecular Analysis and Design graduate fellowship. C.-Y.L. was supported by a Kenneth and Nina Tai Stanford Graduate Fellowship and the Taiwanese Ministry of Education. This work was supported, in part, by the NIH (grant GM118044 to S.G.B.). Part of this work was performed at the Stanford Nano Shared Facilities (SNSF) and supported by the National Science Foundation (award ECCS-1542152). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (contract no. DE-AC02-76SF00515). The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health (NIH), National Institute of General Medical Sciences (NIGMS) (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIGMS or NIH.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: All x-ray density maps and atomic models for Dronpa2 variants have been deposited in the Protein Data Bank (wild type: 6NQJ; 3-F: 6NQK; 3-Cl: 6NQL; 3-Br: 6NQN; 3-I: 6NQO; 2,3-F2: 6NQP; 2,3,5-F3: 6NQQ; 3-NO2: 6NQR; 3-CH3: 6NQV; 3-OCH3: 6NQS). All other data are presented in the main text or supplementary materials.
REFERENCES AND NOTES
- 1.O’Banion CP, Lawrence DS, ChemBioChem 19, 1201–1216 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Tsien RY, Annu. Rev. Biochem 67, 509–544 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Bourgeois D, Adam V, IUBMB Life 64, 482–491 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Acharya A et al. , Chem. Rev 117, 758–795 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Martin ME, Negri F, Olivucci M, J. Am. Chem. Soc 126, 5452–5464 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Walker CL et al. , Curr. Opin. Chem. Biol 27, 64–74 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Park JW, Rhee YM, J. Am. Chem. Soc 138, 13619–13629 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Lin C-Y, Both J, Do K, Boxer SG, Proc. Natl. Acad. Sci. U.S.A 114, E2146–E2155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amber suppression allows for the site-specific incorporation of noncanonical amino acids into recombinant proteins. The amino acid site of interest on the relevant gene is mutated to the amber stop codon, TAG. Non-native tRNA containing an anticodon recognizing the amber stop codon is encoded on a separate plasmid. A non-native aminoacyl tRNA synthetase is also introduced into the cell that is capable of charging the non-native tRNA with the noncanonical amino acid. The tRNA then recognizes the amber stop codon on the mRNA within the translation machinery of the cell, allowing for the incorporation of the noncanonical amino acid into the growing polypeptide chain.
- 10.Dumas A, Lercher L, Spicer CD, Davis BG, Chem. Sci 6, 50–69 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marder SR et al. , Science 265, 632–635 (1994). [DOI] [PubMed] [Google Scholar]
- 12.Stiel AC et al. , Biochem. J 402, 35–42 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin C-Y, Romei MG, Oltrogge LM, Mathews II, Boxer SG,J. Am. Chem. Soc 141, 15250–15265 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olsen S, Lamothe K, Martínez TJ, J. Am. Chem. Soc 132, 1192–1193 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Olsen S, McKenzie RH, J. Chem. Phys 130, 184302 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Schenkl S, van Mourik F, van der Zwan G, Haacke S,Chergui M, Science 309, 917–920 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Ko C, Virshup AM, Martínez TJ, Chem. Phys. Lett 460, 272–277 (2008). [Google Scholar]
- 18.Wolfe LS et al. , Proc. Natl. Acad. Sci. U.S.A 107, 16863–16868 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gozem S, Schapiro I, Ferré N, Olivucci M, Science 337, 1225–1228 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Bassolino G et al. , J. Am. Chem. Soc 136, 2650–2658 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Gozem S, Luk HL, Schapiro I, Olivucci M, Chem. Rev 117, 13502–13565 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Smyrnova D, Marín M. d. C., Olivucci M, Ceulemans A,J. Chem. Theory Comput 14, 3163–3172 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Marín M. d. C. et al. , J. Am. Chem. Soc 141, 262–271 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Punwong C, Hannongbua S, Martínez TJ, J. Phys. Chem. B 123, 4850–4857 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.