

Abstract
Focusing on the literature progress since 2002, the present review explores the highly significant role that multicomponent reactions (MCRs) have played as a very important tool for expedite synthesis of a vast number of organic molecules, but also, highlights the fact that many of such molecules are biologically active or at least have been submitted to any biological screen. The selected papers covered in this review must meet two mandatory requirements: (1) the reported products should be obtained via a multicomponent reaction; (2) the reported products should be biologically actives or at least tested for any biological property. Given the diversity of synthetic approaches utilized in MCRs, the highly diverse nature of the biological activities evaluated for the synthesized compounds, and considering their huge structural variability, much of the reported data are organized into concise schemes and tables to facilitate comparison, and to underscore the key points of this review.
Keywords: multicomponent reactions (MCRs), medicinal chemistry, biological activity, drug discovery
1. Introduction
Multicomponent reactions (MCRs) are a type of convergent organic reactions in which three or more precursors react in only one step to form a product that incorporates substantial portions of all components (i.e., atom economy) [1]. As a result, there is very little waste or unwanted by-product formation compared to sequential synthesis. The concomitant step economy, high convergence and structural diversity of the resulting products make this sustainable approach a powerful tool for the synthesis of biologically active molecules and optimization processes in the pharmaceutical industry [2]. Even though the MCR concept for the synthesis of diverse organic structures has been well known for over a century, it has only recently started gaining more attention and there is an increasing number of research articles emphasizing on the synthesis of biologically relevant organic molecules via this approach [3]. Such an attempt is presented in this review paper with an emphasis on organic molecules synthesized via MCRs and subjected to screening for biological activity. It was found that most of such screening was focused on anti-leihsmanial, anti-inflammatory, ROCK inhibitors, bromodomain inhibitors, antifibrotic agents, human toll-like receptor 8-active, neuroprotective agents, acetylcholinesterase inhibitors, anti-HIV, antimicrobial, antioxidant, anti-mycobacterial and anticancer activities (Figure 1). It is noteworthy that 64% and 16% of the supporting literature found for this review correspond to heterocyclic structures with anticancer and antimicrobial activities, respectively.
Figure 1.

Bibliometric graphic depicting the number of published research papers vs the type of biological activity screened [Data were collected searching for the keywords “multicomponent reactions” and “biologically active molecules” by the period 2002 to date.
2. Some Aspects of the Multicomponent Reactions
Reactions like Strecker (1850) [4], Hantzsch (1890) [5], Biginelli (1893) [6], Mannich (1912) [7], Passerini (1921) [8], Asinger (1956) [9] and Ugi (1959) [10], among others, are good classical examples of MCRs. In a MCR, the product is assembled according to a cascade of elementary two-component reactions. Thus, there is a network of reaction equilibria, which all finally flow into an irreversible step to afford the expected product. The use of MCRs in all areas of the applied chemistry are very popular because they offer a wealth of products, while requiring only a minimum of effort. As opposed to the classical way to synthesize complex molecules by sequential synthesis, MCRs allow the assembly of complex molecules in a one-pot manner. Unlike the usual stepwise formation of individual bonds in the target molecule via a multi-step synthetic approach, the defining attribute of MCRs is the inherent formation of several bonds in one operation without isolating the intermediates (referred to as the bond-forming efficiency, BFE) [1,11,12], nor changing the reaction conditions or adding further reagents.
Recently, chemists have renewed their interest in MCRs. This is driven in part, by the pharmaceutical industry due to the growing need to assemble libraries of small-molecules structurally complexes for evaluation as lead scaffolds in drug discovery and development programs. New libraries of such scaffolds are becoming more and more requested after as pathogens mutate to become resistant to current medications. In addition, anti-aging agents are needed for treatment of Alzheimer’s, Parkinson’s, diabetes and cancer, among other diseases [13]. Thus, MCRs represent an excellent tool for the generation of such libraries, which are indispensable for structure–activity relationship (SAR) studies in drug discovery programs.
3. Biologically Active Compounds Obtained from Multicomponent Approaches
As multicomponent reactions represent a powerful tool in the repertoire of sustainable organic synthesis its synergistic utilization with other green chemistry principles would bring organic chemists one-step closer to the ideal synthesis [14]. The rapid and efficient access to a plethora of heterocyclic building blocks through multicomponent reactions have been recognized by the synthetic community as a preferred strategy to design and synthesize biologically active compounds [15,16].
The wide variety of MCR procedures applied to the synthesis of relevant organic molecules clearly shows that MCR-based approaches are exceptionally useful for drug discovery and optimization processes in the pharmaceutical industry due to its high atom-economy, operational simplicity, time/cost efficiency and generation of structural diversity from multifunctional substrates [17,18]. In fact, the growing number of acyclic and heterocyclic building blocks on the market and in clinical evaluation discovered and synthesized by MCR approaches manifests their growing importance in medicinal chemistry and drug discovery programs. The corresponding biological activities displayed by the diverse organic compounds synthesized via MCRs approaches during the period 2002 to date will be discussed as follow.
3.1. Anti-Leihsmanial Activity
Leishmaniasis, a parasitic disease causes a major public health problem, which is prevalent in some tropical and sub-tropical areas of the world. One of its types, visceral leishmaniasis (VL), also known as kala-azar, is highly endemic in the Indian subcontinent and in East Africa. It is transmitted by the bite of infected female phlebotomine sandflies belonging to the genus Leishmania [19]. The existing chemotherapies are not effective enough as these have various drawbacks such as significant toxicity, variable efficacy, lack of oral bioavailability, and high cost involved during the treatment [20]. Thus, for the global health programs there has been a pressing need for the discovery of new lead compounds for the treatment of leishmaniasis [21]. In that direction, a series of structurally diverse α-aminophosphonates 4 were synthesized and evaluated for in vitro anti-leishmanial activity and cytotoxicity using the MTT assay (Scheme 1). Compounds 4 were prepared through a three-component reaction involving aldehydes/ketones 1, amines 2, and phosphites 3 via a Kabachnik–Fields type reaction under catalyst- and solvent-free reaction conditions at room temperature [22]. Several of the obtained compounds exhibited anti-leishmanial potency against the L. donovani promastigote with IC50 values in the low micromolar range. The structure–activity relationships were quantitatively evaluated by a statistically reliable CoMFA model with high predictive abilities (r2pred = 0.87, r2ncv = 0.985) [23].
Scheme 1.

One-pot three-component reaction for the synthesis of diverse α-aminophosphonates 4 and their biological activities.
3.2. Anti-Inflammatory Activity
Bacterial infection and inflammation are strongly interrelated with each other. Pain and inflammation often arise due to bacterial infections [24]. Inflammation is nothing but a biological process that arises due to physical, chemical, biological and immunological stimuli to the human body [25], for that, inflammation is the significant indication in numerous pathological conditions such as Alzheimer’s disease, osteoarthritis, rheumatoid arthritis and obesity-related diseases [26,27].
In this sense, a series of curcumin 3,4-dihydropyrimidinones/thiones/imines 7 were synthesized in 90–96% yield through a one-pot multicomponent cyclocondensation reaction between curcumin (5), substituted aromatic aldehydes 1 and urea/thiourea/guanidine 6 in the presence of chitosamine hydrochloride as a biodegradable and non-toxic catalyst under solvent-free microwave irradiation (MWI) (Scheme 2). All the synthesized curcumin derivatives 7 were screened for anti-inflammatory (but also for antioxidant) activities. The biological activity data of the synthesized compounds showed that most of them exhibited greater anti-inflammatory activity than curcumin [28].
Scheme 2.

Three-component synthesis of 3,4-dihydropyrimidinones/thiones type 7 under microwave irradiation, for anti-inflammatory activity.
Patil et al., reported a one-pot pseudo-five-component synthesis of highly functionalized tetrahydropyridines 9 using Cu(OTf)2 as catalyst, Scheme 3. In vitro anti-inflammatory activity of the obtained compounds 9 was determined against matrix metalloproteinases (MMPs) as MMP-2 and MMP-9 by using gelatin zymography [29].
Scheme 3.

Multicomponent Cu(OTf)2 catalyzed synthesis of substituted tetrahydropyridines 9 for anti-inflammatory activity.
A highly diastereoselective synthesis (exclusively the cis isomer is formed), of chromeno β-lactam hybrids 13/14 was also achieved by an efficient one-pot three-component reaction between either 5,5-dimethylcyclohexane-1,3-dione (10a) or 4-hydroxycoumarin (10b), diverse benzaldehydes 11 and malononitrile (12), in the presence of DABCO under reflux conditions (Scheme 4).
Scheme 4.

Three-component synthesis of β-lactam hybrids 13/14 for anti-inflammatory activity.
The synthesized compounds (13 in 80–95% yield) and (14 in 82–95% yield) were screened for anti-inflammatory activity (as well as for anticancer activity, please see Section 3.13.13. Compound 13b (Ar = 4-ClC6H4, Ar1 = 4-MeC6H4) was the most active of all the chromeno β-lactam hybrids 13/14 tested, with a 19.8 anti-inflammatory ratio, although, it resulted less active than the reference drug dexamethasone corticosteroid used for the treatment of rheumatoid and skin inflammation [30].
3.3. ROCK Inhibitors
Rho-associated protein kinases (ROCKs) are ubiquitously expressed in most adult tissues, and are involved in modulating the cytoskeleton, protein synthesis and degradation pathways, synaptic function, and autophagy. Among the current limited number of ROCK inhibitors of clinical use, such as fasudil [31] and netarsudil [32], the synthesis and biological evaluation of a series of boronic acid-containing 3H-pyrazolo[4,3-f]quinolones 17 as potential ROCK inhibitors have recently been reported [33]. The synthetic process involved a three-component Povarov type reaction between indazol-5-amines 15, methylene active ketones 16 and aldehydes 1 in the presence of catalytic amounts of HCl, affording the expected products 17 in 70–90% yield, as depicted in Scheme 5. After a SAR analysis of the obtained products 17, the biological trials indicated that compound labeled as HSD1590 (17p), resulted more potent than the reference drug netarsudil at binding to or inhibiting ROCK enzymatic activities. This compound exhibited single digit nanomolar binding to ROCK (Kds < 2 nM) and subnanomolar enzymatic inhibition profile (i.e., ROCK2 IC50 was 0.5 nM for 17p while Netarsudil inhibited ROCK2 with IC50 = 11 nM under similar conditions) [33].
Scheme 5.
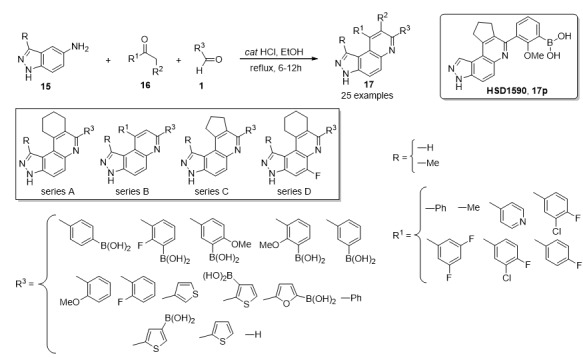
Three-component synthesis of boron containing compounds 17 for ROCK inhibition.
3.4. Bromodomain Inhibitors
The inhibition of the bromodomain and extra-terminal (BET) domain subfamily of human bromodomains from chromatin [34], has contributed new insights into gene regulation and emerged as a promising therapeutic strategy in cancer. Structural analogy of early methyltriazolo BET inhibitors has prompted a need for structurally dissimilar ligands as bromodomain function probes. Using fluorous-tagged multicomponent reactions, a focused chemical library of bromodomain inhibitors 20 with micromolar biochemical IC50 values was developed around a 3,5-dimethylisoxazole biasing element. Iterative synthesis and biochemical assessment allowed optimization of novel BET bromodomain inhibitors based on an imidazo[1,2-a]pyrazine scaffold. The synthesis of the target molecules 20 (in 11–58% yield), involved a three-component reaction between isocyanides 18, pyrazines/pyridines 2 and aldehydes 1 in the presence of Sc(OTf)3 as catalyst, followed by a Suzuki-type coupling reaction with the boronic acid derivative 19 catalyzed by Pd(dppf)Cl2 (Scheme 6). The lead compound 20c (R = tBu; R1 = R2 = H) binds BRD4 with a Kd of 550 nM and 724 nM cellular potency in BRD4-dependent lines. Additionally, compound 20c showed potency against TAF1, a bromodomain-containing transcription factor previously unapproached by discovery chemistry [35].
Scheme 6.

Elaboration of imidazopyridine scaffolds 20 as bromodomain inhibitors mediated by a three-component approach.
3.5. Antifibrotic Agents
Liver fibrosis is a critical wound healing response to chronic liver injury such as hepatitis C virus (HCV) infection. If persistent, liver fibrosis can lead to cirrhosis and hepatocellular carcinoma (HCC). The development of new therapies for preventing liver fibrosis and its progression to cancer associated with HCV infection remains a critical challenge [36]. Identification of novel anti-fibrotic compounds will provide opportunities for innovative therapeutic intervention of HCV-mediated liver fibrosis. In this sense, it was designed and synthesized a set of 5-arylthio-5H-chromenopyridines 22 as a new class of anti-fibrotic agents. Products 22 were synthesized in 16–45% yield through a pseudo-four-component reaction involving malonitrile (12, 2 mmol), thiophenols 21 (1 mmol) and 4-diethylaminosalicylaldehyde in the presence of triethylamine as catalyst (Scheme 7). Liver fibrosis assays demonstrated that compounds 22a (Ar = 4-FC6H4) and 22c (Ar = 4-BrC6H4) showed inhibitory activity towards human hepatic stellate cells (LX2) activation at 10 μM. The HCV NS3 and NS5A proteins in HCV subgenome-expressing cells were also significantly reduced in cells treated with 22a and 22c, suggesting the possible inhibitory role of the compounds in HCV translation/replication activities [37]. The reactivity of compounds 22 with medicinally-relevant metal compounds such as platinum and gold was also examined. The reactivity of these complexes with metals and during mass spectrometry suggested that C-S bond cleavage is relatively facile.
Scheme 7.

Preparation of 5-arylthio-5H-chromenopyridines 22 of anti-fibrotic interest using a multi-component reaction (MCR) approach.
3.6. Human Toll-Like Receptor 8-Active Compounds
The innate immune system utilizes germline-encoded pattern recognition receptors (PRRs) to discern pathogen-associated molecular patterns (PAMPs) that are distinct to the pathogen [38]. The transmembrane PRRs include the toll-like receptors (TLRs) [39], which are expressed either on the plasma membrane or in the endolysosomal compartments [38]. At least ten functional TLRs are encoded in the human genome, each with an extracellular domain having leucine-rich repeats and a cytosolic domain called the toll/IL-1 receptor domain [40]. The discovery of TLRs has not only served to greatly accelerate the understanding of the interplay between the innate and adaptive immune systems, but is also catalyzing novel approaches to vaccine design and development. The ligands for these receptors are highly conserved microbial molecules [40,41]. For instance, it was proposed that imidazo[1,2-a]pyridine/pyrazines 23 (Scheme 8), could work as TLR7/8 ligands. Compounds 23, were obtained (in 13–86% yield) via a Groebke-Blackburn-Bienaymé type multicomponent reaction [42], along with the unplanned furo[2,3-c]pyridine/pyrazines 25 (in 10–84% yield), when pyridoxal (24) was used as aldehyde (Scheme 8) [43]. Both libraries of structures 23 and 25 were subjected to screening for TLR7/8 agonistic activities, as potential vaccine adjuvants.
Scheme 8.

Two step one-pot three-component synthetic processes for TLR7/8 agonistic ligands 23 and 25.
The biological assays showed that most of the obtained compounds 23 were inactive in NF-κB reporter gene assays specific for human TLR-3, -7, -8, and -9; however, most compounds 25 were found to specifically activate NF-κB signaling in TLR8-transfected HEK293 cells [43].
3.7. Neuroprotective Agents
Neurodegenerative disorders constitute a significant public health problem worldwide, and among these, cerebrovascular accidents represent one of the leading causes of death, neurological disability, and cognitive impairment [44]. During cerebral ischemia, oxygen and glucose deprivation induces a metabolic cascade that leads to neuronal death. One of the most significant consequences of these changes is the dysregulation of Ca2+ homeostasis, leading to brain damage. In this context, a set of C5-unsubstituted-C6-aryl-1,4-dihydropyridines 28 were prepared by a CAN-catalyzed three-component reaction from chalcones 26, β-dicarbonyl compounds 27, and ammonium acetate in refluxing EtOH (Scheme 9). Compounds 28 were able to block Ca2+ entry after a depolarizing stimulus and showed an improved Cav1.3/Cav1.2 selectivity in comparison with nifedipine. Furthermore, they were able to protect neuroblastoma cells against Ca2+ overload and oxidative stress models. It is highlighted that the selectivity ratio of 28 makes them highly interesting for the treatment of neurological disorders where Ca2+ dyshomeostasis and high levels of oxidative stress was demonstrated. Furthermore, their low potency toward the cardiovascular channel subtype makes them safer by reducing their probable side effects, in comparison to classical 1,4-dihydropyridines. Some of the obtained compounds 28 afforded good protective profile in a postincubation model that simulates the real clinical situation of ictus patients, offering a therapeutic window of opportunity of great interest for patient recovery after a brain ischemic episode. Good activities were also found in acute ischemia/reperfusion models of oxygen and glucose deprivation [45].
Scheme 9.

Three-component synthesis of 5-unsubstituted dihydropyridines 28 with improved Cav1.3 selectivity as potential neuroprotective agents against ischemic injury.
3.8. Acetylcholinesterase Inhibitors
It is suggested that compounds that can inhibit cholinesterase enzyme may be considered as anti-Alzheimer, anti-Parkinson, and anti-autism drugs [46]. Due to the fact donepezil and other FDA approved drugs used for treatment of above diseases present some side effects [47], many efforts trying to find, develop, and explore more potent and permissive anti-Alzheimer drugs without any harmful side effects have been made so far. Among them, a borax-catalyzed protocol for the synthesis of a set of 4-aryl-substituted-4H-pyran derivatives fused to α-pyrone ring 30 in a one-pot procedure, as potential acetylcholinesterase inhibitors (AChEIs), was described. In this approach, products 30 were obtained in good to excellent yields, from a three-component reaction between aryl aldehydes 1, 4-hydroxy-6-methyl-2H-pyran-2-one (29) and malononitrile (12), in the presence of borax as catalyst and THF as solvent (Scheme 10). Subsequently, compounds 30 were evaluated in silico against acetylcholinesterase enzyme (AChE) and their Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMETox) properties were also studied, to make the results more reliable and introduce them as remarkable potential candidates for inhibition of AChE, in the treatment of Alzheimer’s, Parkinson’s and autism diseases. Among the evaluated products, compound 30f (R = p-OCH2C6H4Br) showed the best activity against AChE [48].
Scheme 10.

Three-component derivatization of compounds 30 for evaluation of their activity against AChE.
3.9. Anti-HIV Activity
Reverse transcriptase (RT) is a key enzyme which plays an essential and multifunctional role in the replication of the human immunodeficiency virus (HIV) and thus represents an attractive target for the development of new drugs useful in AIDS therapy [49,50]. In view of the increasing incidence of resistance to current drug regimens and the frequency of adverse events, the development of novel, selective, potent, safe, inexpensive antiviral agents, that are also effective against mutant HIV strains, remains a high priority for medical research. In that direction, the design, synthesis, and the structure-activity relationship studies of a series of 2,3-diaryl-1,3-thiazolidin-4-ones 31 was performed. The synthesis of products 31 (in 8–87% yield), involved a three-component procedure reacting a suiTable 2,6-dihalo-substituted benzaldehydes 1 with an equimolar amount of a (hetero)aromatic amines 2 in the presence of an excess of mercaptoacetic acid 30 in refluxing toluene (Scheme 11). Some derivatives 31 proved to be highly effective in inhibiting HIV-1 replication at nanomolar concentrations with minimal cytotoxicity, thereby acting as nonnucleoside HIV-1 RT inhibitors (NNRTIs). Computational studies were used to delineate the ligand-RT interactions and to probe the binding of the ligands 31 to HIV-1 RT [51].
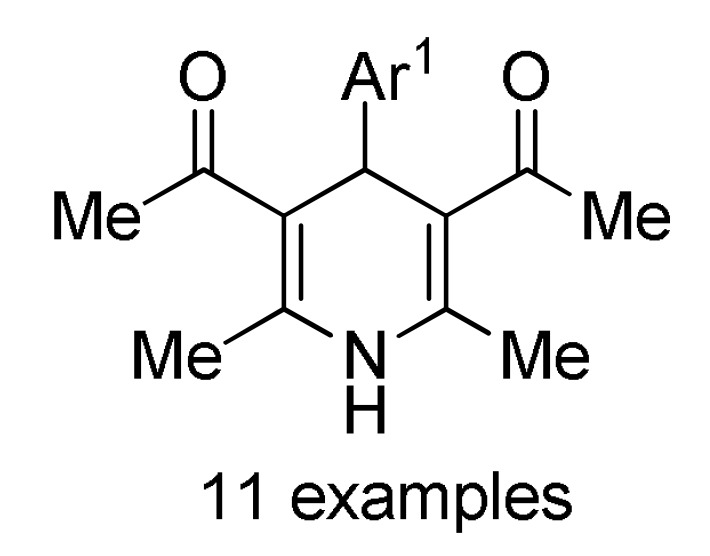
Table 2.
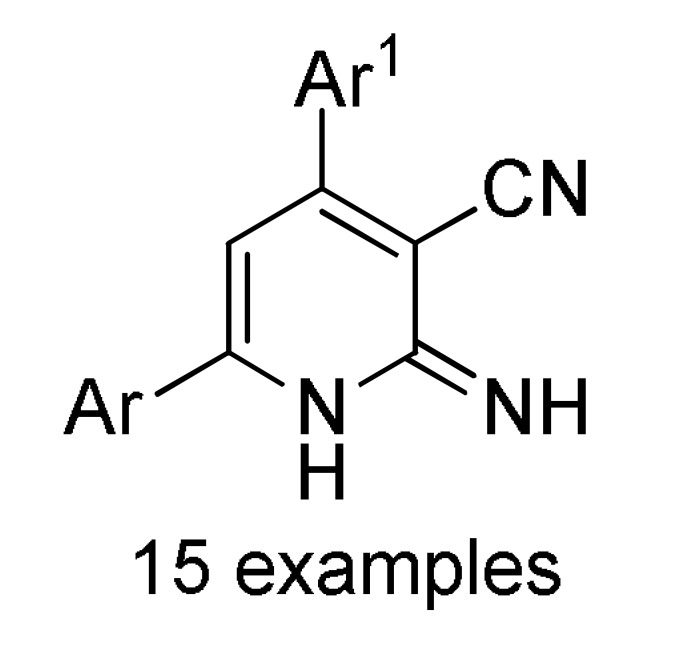
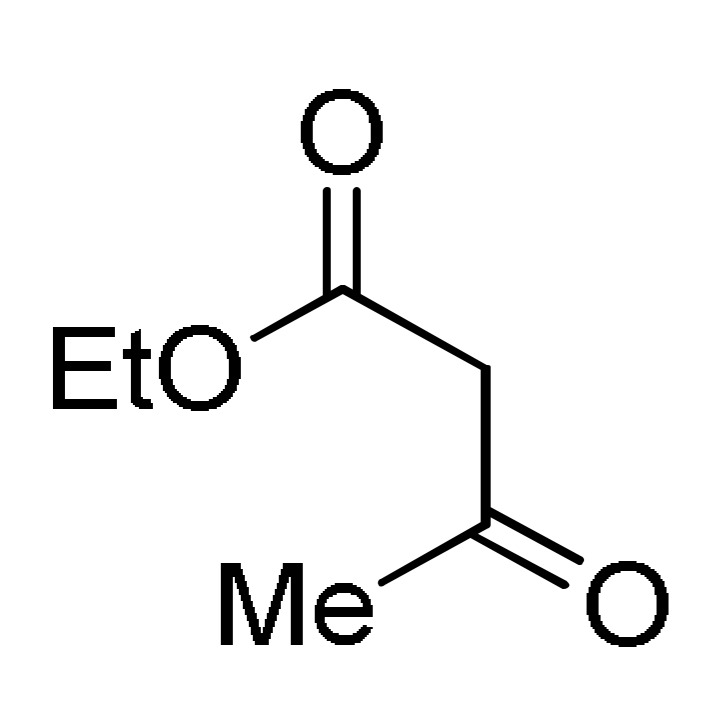
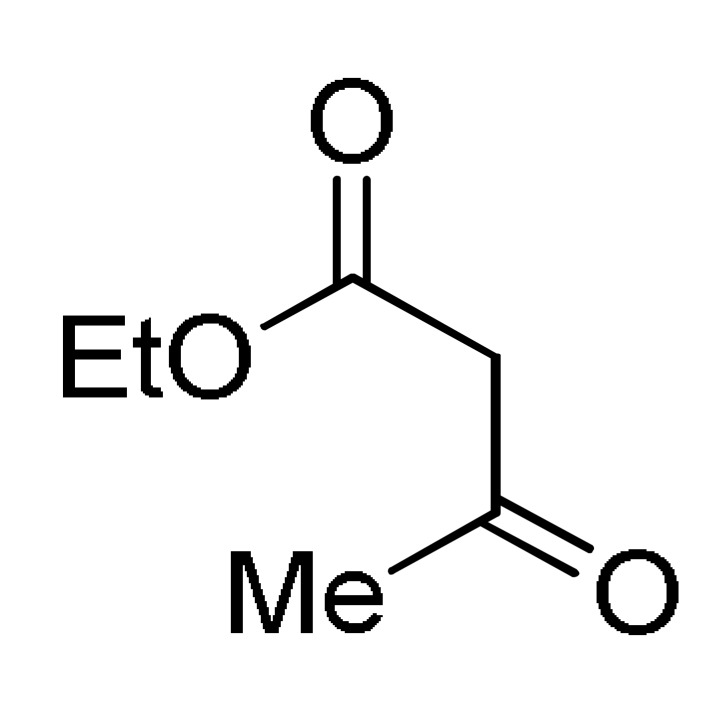
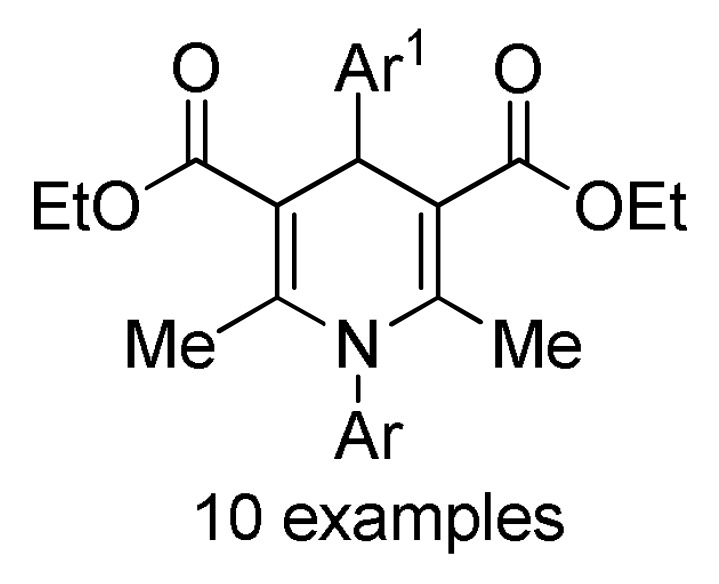
Multicomponent synthesis of dihydropyridine-based heterocyclic systems 157–161 of potential anticancer activity.
| Reagents | Conditions | Product | Reference | |||
|---|---|---|---|---|---|---|
| Ar1CHO 1 |
ArCOMe 16 |
38 |
AcONH4 | EtOH, reflux |
157 a |
[176] |
12 |
158 a |
|||||
8 |
8 |
ArNH2 2 |
PEG-400, 200 W, 80 °C |
159 b |
[177] | |
27 |
27 |
(NH4)2CO3 |
160 b |
|||
47 |
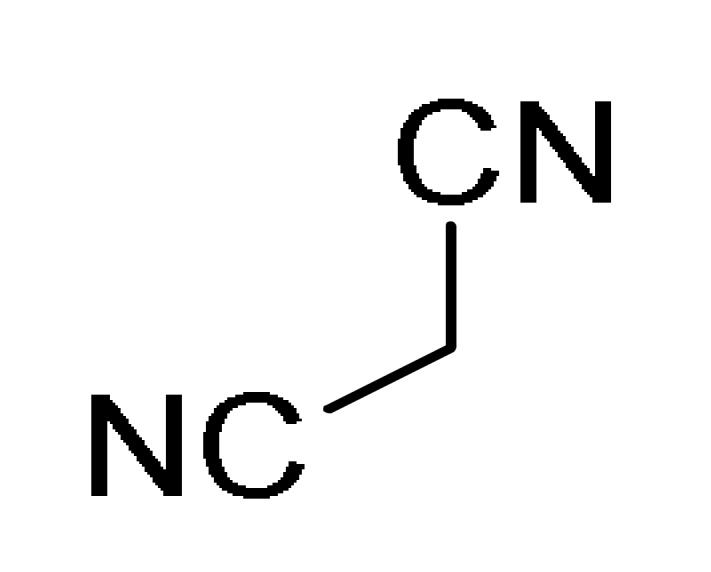
12 |
ArNH2 2 |
Montmorillonite-K10, H2O/EtOH (2:1), 60 °C |
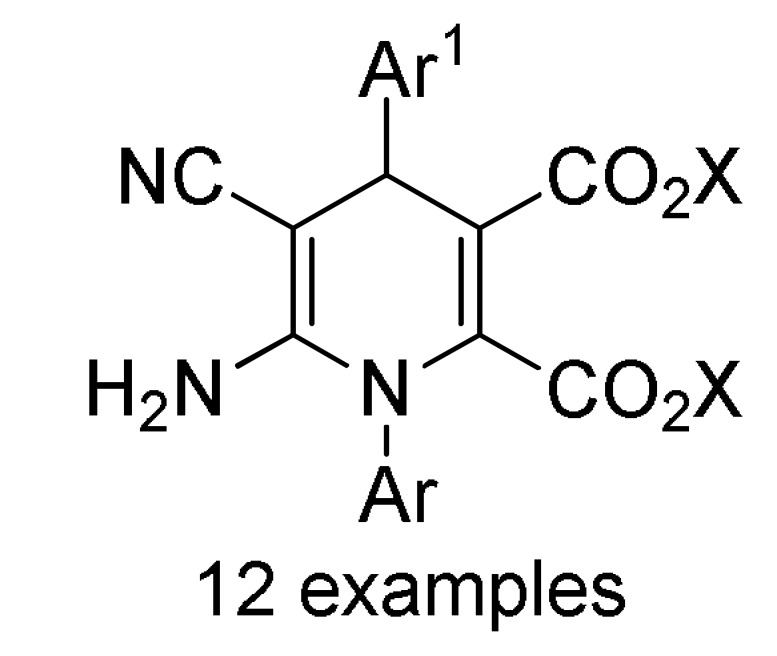
161 c |
[178] | |
a For compounds 157/158: Ar1 = 2-HOC6H4, 2-ClC6H4, 3-ClC6H4, 2,5-(MeO)2C6H3, 2,4-(MeO)2C6H3, 3-thienyl, 4-EtOC6H4, 2-EtOC6H4, 4-MeOC6H4, 2-MeOC6H4, 4-ClC6H4; Ar = 4-BrC6H4, 3-BrC6H4, 2-BrC6H4, Ph, 3-thienyl. b For compounds 159/160: Ar1 = 3-NO2C6H4, 3-PhOC6H4, 3-ClC6H4, 4-HO-3-MeOC6H3, 4-Me2NC6H4, 4-MeOC6H4, 4-ClC6H4, 4-MeSC6H4, 3-BrC6H4, 2-ClC6H4, Ph, 2-NO2C6H4, 3-MeC6H4, 2-MeC6H4; Ar = 4-MeSC6H4, 4-MeOC6H4, 3-NO2C6H4, 4-ClC6H4, 3-MeOC6H4, 4-HO-3-MeOC6H3. c For compounds 161: X = Me, Et; Ar1 = Ph, 4-BrC6H4, 3-BrC6H4, 4-MeOC6H4, 4-ClC6H4, 3-MeC6H4, 2-HOC6H4; Ar = Ph, 4-MeOC6H4, 4-BrC6H4, 4-IC6H4, 4-FC6H4, 4-NO2C6H4.
Scheme 11.

Three-component synthesis of 2,3-diaryl-1,3-thiazolidin-4-ones 31 for structure-activity relationships, molecular modeling and studies as potent anti-HIV agents.
3.10. Antimicrobial Activity
An alarming increment in pathogenic resistance to existing drugs is a serious problem with antimicrobial therapy, indicating the necessity of continuing with the research for new classes and more effective of antimicrobials [52], possibly acting through mechanisms different from those of existing drugs [53]. In this context, it is very essential to successfully develop novel and efficient antimicrobial agents with clinically unexploited mode of action. As a contribution to this topic, Lakshmi et al., reported an InCl3-catalyzed three-component reaction for the synthesis of 3-pyranyl indole derivatives 33 as antimicrobial agents. The process was mediated by a tandem Knoevenagel–Michael reaction of 3-cyanoacetyl indole (32) diverse aromatic aldehydes 1 and malononitrile (12) catalyzed by InCl3 in refluxing ethanol (Scheme 12). The antibacterial activity was screened by paper disc diffusion method against two Gram-positive bacteria (Staphylococcus aureus, Bacillus cereus), and two Gram-negative bacteria (Escherichia coli, Klebsiella pneumoniae) by using ciprofloxacin as reference compound. As shown in Scheme 12, compound 33e (R = 2-Cl) was found to exhibit the more potent in vitro antibacterial activity, with MIC values of 12.4, 16.4, 16.5, and 16.1 μM against S. aureus, B. cereus, E. coli, and K. pneumoniae, respectively. In addition, compounds 33a (R = H), 33d (R = 4-Cl), 33h (R = 4-F), and 33m (R = 4-OMe) exhibited significant antibacterial activity when compared to the standard drug ciprofloxacin [54].
Scheme 12.

InCl3-catalyzed three-component synthesis of 3-pyranyl indole derivatives 33 for assays against Gram-positive and Gram-negative bacteria.
A new class of pyrano[3,2-c]chromene derivatives 34 incorporating a validated molecular target was synthesized through a one-pot multicomponent cyclocondensation reaction between β-aryloxyquinoline-3-carbaldehydes 1, 4-hydroxycoumarins 10b and malononitrile (12) in ethanol containing a catalytic amount of piperidine, Scheme 13. Antibacterial activity was screened against three Gram-positive bacteria (Bacillus subtilis MTCC 441, Clostridium tetani, Streptococcus pneumoniae) and three Gram-negative bacteria (Escherichia coli, Salmonella typhi, Vibrio cholerae) by using ampicillin as a standard antibacterial drug. Remarkably, compounds 34f (R = Me, R1 = Cl, R2 = H), 34l (R = H, R1 = Cl, R2 = Me) and 34q (R = MeO, R1 = Me, R2 = Me) exhibited excellent in vitro antibacterial activity. The majority of compounds 34 were found to possess higher potency as compared to standard bactericidal ampicillin against Gram-positive bacteria B. subtilis [55].
Scheme 13.

Piperidine-catalyzed three-component synthesis of pyrano[3,2-c]chromene derivatives 34 as potential Gram-positive and Gram-negative bactericidal agents.
Vijesh et al., reported the synthesis of 1,4-dihydropyridine derivatives (1,4-DHPs) 35 containing substituted pyrazole moiety as potent antimicrobial, as well as, antioxidant agents [56]. The synthetic process involved a pseudo-four-component Hantzsch reaction between 3-aryl-1H-pyrazole-4-carbaldehydes 1, 1,3-dicarbonyl compounds 8 (ethyl acetoacetate and methyl acetoacetate) and ammonium acetate in ethanol under reflux conditions (Scheme 14). Antibacterial activity was screened against one Gram-positive bacteria (Staphylococcus aureus) and two Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa) by using streptomycin as standard drug. The results indicated that among the tested compounds, 35c (R = Et, Ar = 4-MeSC6H4) and 35f (R = Et, Ar = 4-ClC6H4) showed excellent activity against all the tested microbial strains E. coli, S. aureus and P. aeruginosa at concentrations of 1, 0.5 and 0.25 mg/mL compared to the standard drug streptomycin.
Scheme 14.

Synthesis of 1,4-dihydropyridine derivatives 35 through a Hantzsch multicomponent reaction for antibacterial assays.
El-borai et al., reported an efficient protocol for the microwave-assisted synthesis of pyrazolo[3,4-b]pyridine derivatives 37 in good to excellent yields [57]. In this approach, products 37 were obtained from a three-component reaction between 5-amino-1-phenyl-3-(pyridin-3-yl)-1H-pyrazole type 2, pyruvic acid (36) and diverse aromatic aldehydes 1 using acetic acid as solvent under MWI at 160 °C for 20 min (Scheme 15). Subsequently, products 37 were screened for antibacterial, as well as, antifungal and antitumor activity. In particular, antibacterial activity was screened against one Gram-positive bacteria (Bacillus cereus) and three Gram-negative bacteria (Escherichia coli, Enterobacter cloaca and Serratia marcescens). Among the tested compounds, only 37a (R = MeO, R1 = H), 37d (R = OH, R1 = H), 37e (R = Br, R1 = H) and 37f (R = R1 = MeO) exhibited antibacterial activity of high order against all strains of the bacteria yeast tested.
Scheme 15.

Microwave-assisted three-component synthesis of pyrazolo[3,4-b]pyridine derivatives 37 of antibacterial interest.
Shah et al., reported a small library of quinoline-pyridine hybrids 40 through a three-component reaction between a series of 2-chloro-3-formylquinolines 1, active methylene compounds 12/38 and 3-(pyridine-3-ylamino)cyclohex-2-enone (39) in the presence of catalytic amount of sodium hydroxide in ethanol under refluxing conditions (Scheme 16) [58]. This protocol afforded a time-efficient synthesis of the structurally diverse quinoline-pyridine hybrids 40 in good yields for antimicrobial, as well as, for antifungal and antitubercular screening. These products were screened for their antibacterial activity against three Gram-positive bacteria (Bacillus subtilis, Clostridium tetani, Streptococcus pneumoniae), and three Gram-negative bacteria (Escherichia coli, Salmonella typhi, Vibrio cholerae). Among the products tested only 40b (R = Me, R1 = CN) and 40i (R = H, R1 = CO2Me) showed better inhibitory effects for E. coli, and 40g (R = MeO, R1 = CO2Et) showed better results for S. typhi compared to standard drugs such as ampicillin, chloramphenicol, ciprofloxacin and norfloxacin.
Scheme 16.

Three-component synthesis of quinoline-pyridine hybrids 40 as potential antibacterial agents.
Bhaskar et al., reported the synthesis of a series of spirooxindole derivatives 44 and 45 through a three-component 1,3-dipolar cycloaddition of an azomethine ylide generated in situ from sarcosine (41a) or L-proline (41b) and isatin 42 with the dipolarophile 1,4-naphthoquinone 43 followed by spontaneous air oxidation in atmospheric reflux conditions (Scheme 17) [59]. Subsequently, products 44 and 45 were screened for antibacterial (and for antifungal), activity against four Gram-positive bacteria (Staphylococcus aureus, methicillin-resistant Staphylococcus aureus, Enterobacter aerogenes, Micrococcus luteus), and four Gram-negative bacteria (Proteus vulgaris, Klebsiella pneumoniae, Salmonella typhimurium, and Salmonella paratyphi-B). Remarkably, compound 44n (R = COMe, R1 = Me) was found to be more than 1.6 times active against methicillin-resistant S. aureus bacteria than streptomycin and ciprofloxacin. Also more than 6.4 times active against M. luteus and S. typhimurium bacteria than ciprofloxacin.
Scheme 17.

Three-component synthesis of diverse spirooxindole derivatives 44 and 45 for antibacterial studies.
In the course of a synthetic study toward other spiro-oxindole, Singh et al., reported an eco-friendly strategy for the synthesis of spiro-oxindole derivatives 46 in good yields and excellent stereoselectivities by a β-cyclodextrin-catalyzed one-pot multicomponent reaction from isatins 42, cyclic 1,3-diketones 10a and (thio)urea 6 in water under mild reaction conditions (Scheme 18) [60]. In this approach β-cyclodextrin not only formed an inclusion complex with isatin, but also was involved in intermolecular hydrogen bonding with the (thio)urea to promote the reaction. These products were screened for their antibacterial activity against one Gram-positive bacterium (Staphylococcus aureus), and one Gram-negative bacterium (Escherichia coli). Notably, compounds 46c (R = H, R1 = Me, X = S), 46d (R = R1 = H, X = S), 46g (R = Br, R1 = Me, X = O), 46i (R = Br, R1 = Me, X = S) and 46j (R = Br, R1 = H, X = S) showed comparable antibacterial activity to the standard drug streptomycin. These results revealed that the presence of bromo-substituent and sulphur moiety in the synthesized compounds induced high potency, whereas the presence of methyl group decreased the effectiveness of the compounds.
Scheme 18.

β-Cyclodextrin-catalyzed three-component synthesis of spiro-oxindole derivatives 46 for their evaluation as antibacterial agents.
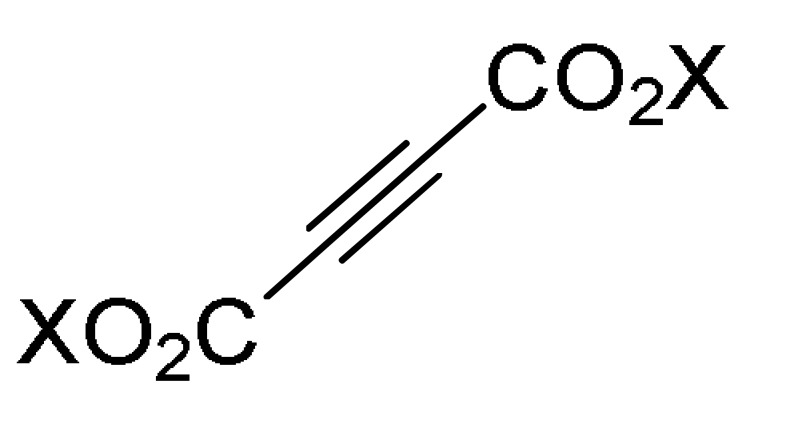
Contemporaneously, Darandale et al., proposed a green, practical and facile strategy for the synthesis of 1,2,3,6-tetrahydropyrimidine analogues as potent antimicrobial but also as antifungal agents [61]. Thus, the ZrOCl2-catalyzed pseudo-five-component reaction of substituted amines type 2, dialkyl acetylenedicarboxylates 47, and formaldehyde (1) in refluxing water furnished the target compounds 48 in good to excellent yields (Scheme 19). These compounds were screened for their antibacterial activity against two Gram-positive bacteria (Staphylococcus aureus, Bacillus subtilis), and one Gram-negative bacterium (Escherichia coli) by using ciprofloxacin and ampicillin as standard drugs. The results indicated that compounds 48a (R = H, X = C, R1 = Et), 48b (R = 4-Cl, X = C, R1 = Et), and 48e (R = H, X = N, R1 = Et), all having the diethyl but-2-ynedioate functionality, were found to be most active and potent against the tested bacterial strains. They had MIC values (15–60 µM) compatible with standard drugs, except for bacterium S. aureus which showed MIC values between 60 and 100 µM. Lastly, to develop potent antibacterial agent, diethyl but-2-ynedioate was better choice than dimethyl but-2-ynedioate.
Scheme 19.

ZrOCl2-Catalyzed multicomponent synthesis of 1,3,4,5-tetrasubstituted 1,2,3,6-tetrahydropyrimidines 48 for antibacterial assays.
Murlykina, et al., reported the synthesis of 3,6-diarylpyrazolo[3,4-b]pyridine-4-carboxylic acids 49 through a MW-assisted three-component reaction between 5-aminopyrazoles type 2, salicylic aldehydes 1 and pyruvic acid (36) in acetic acid acting both as solvent and catalyst (Scheme 20) [62]. These compounds were screened for their antibacterial activity against two Gram-positive bacteria (Bacillus subtilis, Staphylococcus aureus), and two Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa). It was found that Gram-negative bacteria (E. coli and P. aeruginosa) showed resistance to all tested compounds in the concentration range of 15–250 µM. Albeit, strains of Gram-positive bacteria (B. subtilis and S. aureus) were found more sensitive, bacteriostatic activity was fixed only in the highest concentration 250 µM during the first day of the experiment. Thus, the tested compounds displayed lower action in comparison to nitroxoline being the reference substance.
Scheme 20.

Microwave-assisted three-component synthesis of 3,6-diarylpyrazolo[3,4-b]pyridine-4-carboxylic acids 49 of antibacterial interest.
Complementarily, Trivedi’s group introduced the ortho-quinonemethide 51 (o-QM) as powerful intermediate for the generation of complex heterocyclic structures including the benzo[a]xanthenone skeleton [63]. The Ce-MCM-41-catalyzed three-component reaction between 2-naphthol (50), substituted aldehydes 1 and 1,3-diketones 10a (5,5-dimethyl-1,3-cyclohexanedione; 1,3-cyclohexanedione; indane-1,3-dione and acetyl acetone) provided a general route to synthesize the target compounds 52 in good yields under solvent-free conditions (Scheme 21). This eco-friendly protocol offered several advantages including a green and cost-effective procedure, shorter reaction times, simpler work-up, recovery and reusability of the solid acid heterogeneous catalyst. Compounds 52 were screened for their antibacterial activity against five Gram-positive bacteria (Bacillus subtilis, Micrococcus luteus, Bacillus circulans, Streptococcus mutans, Lysinibacillus sp.) and four Gram-negative bacteria (Escherichia coli, Klebsiella pneumoniae, Salmonella paratyphi, Pseudomonas putida) using streptomycin as standard drug. It was noticed that the tested compounds 52f (R1 = Me, R = iBuC6H4), 52k (R1 = Me, R = 5-Br-3-Py), 52n (R1 = Me, R = 2-HO-4-MeOC6H3), 52o (R1 = Me, R = 4-BnO-3-MeOC6H3) and 52p (R1 = Me, R = 3,4,5-(MeO)3C6H2) showed promising antibacterial activity against either Gram-positive or Gram-negative bacterial strains.
Scheme 21.

Ce-MCM-41-Catalyzed three-component synthesis of benzo[a]xanthenone derivatives 52, for antibacterial screening, under solvent-free reaction conditions.
Sable, et al., reported the synthesis of fully substituted thiophene derivatives 55 through a one-pot three-component reaction of 2-bromo/chloromethyl derivatives 53, acetyl acetone (27) and phenyl isothiocynates 54 under mild reaction conditions (Scheme 22) [64]. Synthesized compounds were tested for antibacterial activity against two Gram-positive bacteria (Bacillus subtilis, Staphylococcus aureus), and two Gram-negative bacteria (Pseudomonas aeruginosa, Escherichia coli) using azithromycin as the reference standard drug. Compounds 55 showed excellent to good activity against Gram-negative bacteria (P. aeruginosa, E. coli) having MIC values between 0.3 and 8.5 µM, and lower activity against Gram-positive bacteria (B. subtilis, S. aureus) having MIC values between 0.1 and 9.5 µM. The structure–activity relationship study (SAR) indicated that a change in the substituent might also affect the antibacterial activity. For example, compounds having R = H/Cl appeared to have more potential against Gram-positive bacteria (S. aureus) and Gram-negative bacteria (P. aeruginosa, E. coli). In addition, compounds having R = CH3/OCH3 were found to be more active against Gram-positive (B. subtilis) and Gram-negative bacteria (P. aeruginosa, E. coli).
Scheme 22.

One-pot three-component synthesis of fully substituted thiophene derivatives 55 as potential antibacterial agents.
In 2015, Sindhu et al., reported the three-component synthesis of thiazolidinedione–triazole hybrids 59 in high yields after short reaction times [65]. This copper(I)-catalyzed 1,3-dipolar cycloaddition reaction of 5-(arylidene)thiazolidine-2,4-diones 56, propargyl bromide (57) and substituted aryl azides 58 was performed in PEG-400 as green media in the presence of CuSO4.5H2O and sodium ascorbate as catalytic system (Scheme 23). The obtained compounds were screened for their antibacterial activity against two Gram-positive bacteria (Staphylococcus aureus, Bacillus subtilis) and two Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa) using ciprofloxacin as standard drug. The minimum inhibitory concentration (MIC) of compounds ranged between 32 and 256 µM against Gram-positive bacteria. Notably, compounds 59f (R = Br, R1 = Me, R2 = H), 59g (R = Br, R1 = H, R2 = Cl), 59i (R = NO2, R1 = F, R2 = Cl), and 59k (R = NO2, R1 = F, R2 = H) were found to be best as they exhibited MIC values of 64 µM against S. aureus and B. subtilis. Whereas 59h (R = Br, R1 = F, R2 = Cl) was found to be best against S. aureus and B. subtilis with lowest MIC of 32 µM. However, compounds did not exhibited any activity against Gram-negative bacteria.
Scheme 23.

Multicomponent process for the synthesis of thiazolidinedione–triazole hybrids 59 of antibacterial interest.
During the course of two decades the Groebke-Blackburn-Bienaymé (GBB-3CR) reaction has emerged as a very important multicomponent reaction (MCR), resulting in over a hundred patents and a great number of publications in various fields of interest [66]. For example, Aouali et al., described the synthesis of fully substituted imidazo[2,1-c][1,2,4]triazoles 60 by a Groebke-Blackburn-Bienaymé reaction between 5-amino-1,2,4-triazoles 2, aromatic aldehydes 1 and isocyanides 18 using scandium triflate as a Lewis acid catalyst (Scheme 24). The synthesized derivatives 60 were screened for their antibacterial but also, for antifungal and antioxidant activities [67]. Compounds 60 were screened for their antibacterial activity against two Gram-positive bacteria (Bacillus cereus, Staphylococcus aureus) and three Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa, Salmonella enteritidis). Particularly, compound 60b (R = Me, R1 = 4-Cl, R2 = cyclohexyl) exhibited excellent inhibition against the Gram-positive bacteria with inhibition zone diameter ranged from 29 to 20 mm and MIC values of 78 and 312 µM against B. cereus and S. aureus, respectively. However, these compounds were inactive against Gram-negative bacteria.
Scheme 24.

Groebke-Blackburn-Bienaymé three-component reaction for the preparation of imidazo[2,1-c][1,2,4]triazole derivatives 60 for antibacterial assays.
In 2017, an elegant microwave-assisted synthesis of pyrrolo[1,10]phenanthrolines 62 was achieved through a four-component reaction between 1,10-phenanthroline (61), aromatic aldehydes 1, malononitrile (12) and isocyanides 18, leading to the corresponding products 62 in excellent yields at 60 °C (Scheme 25) [68]. The benefits of MWI in terms of reaction times and efficiency were clearly demonstrated by a comparative study with thermal activation. Subsequently, compounds 62 were screened for antibacterial, as well as, for antitumoral and antifungal activity. These compounds were screened for their antibacterial activity against two Gram-positive bacteria (methicillin-resistant Staphylococcus aureus, Bacillus subtilis) by the agar diffusion method using erythromycin and oxacillin as standard drugs. Remarkably, compounds 62d (R = 4-Cl, R1 = 4-MeOC6H4) and 62l (R = 4-F, R1 = 4-MeOC6H4) exhibited the highest antibacterial activity with inhibition zones of 29 mm and 27 mm, respectively, against methicillin-resistant S. aureus bacteria.
Scheme 25.

Microwave-assisted multicomponent synthesis of pyrrolo[1,10]phenanthrolines 62 for antibacterial evaluation.
Alternatively, Meena et al., described a three-component synthesis of spiro[indolo-3,10′-indeno[1,2-b]quinolin]-2,4,11′-triones 65 using isatins type 42, enaminones 63 and indane-1,3-dione (64) [69]. As a result, a collection of spiro[indolo-3,10′-indeno[1,2-b]quinolin]-2,4,11′-triones 65 was synthesized in the presence of a catalytic amount of ceric ammonium nitrate (CAN) in ethanol–water (1:1) under refluxing conditions (Scheme 26). This method provided several advantages such as shorter reaction times, high yields and operational simplicity. Compounds 65 were screened for their antibacterial activity against two Gram-positive bacteria (Staphylococcus aureus, Bacillus subtilis) and two Gram-negative bacteria (Escherichia coli, Pseudomonas aeruginosa) using ciprofloxacin as standard drug. The minimum inhibitory concentration (MIC) of tested compounds ranged between 8 and 512 µM. Compounds 65c (R = H, R1 = Me, R2 = 4-ClC6H4) and 65n (R = NO2, R1 = Me, R2 = Ph) exhibited the lowest MIC of 16 µM against S. aureus. It is important to note that compound 65c exhibited lowest MIC of 8 µM and 64 µM against B. subtilis and E. coli, respectively.
Scheme 26.

CAN-mediated three-component synthesis of spiro[indolo-3,10′-indeno[1,2-b]quinolin]-2,4,11′-triones 65 of antibacterial interest.
Very recently, Ashok et al., reported an efficient synthesis of pyrano[3,2-b]xanthen-7(2H)-ones 67 in good to excellent yields [70]. This I2-catalyzed three-component reaction involved 2,2-dimethylchroman-7-ol (66), aromatic aldehydes 1 and 1,3-cyclohexanediones 10a under MWI in acetic acid (Scheme 27). This transformation featured multiple bonds breaking and forming events in a single, atom-economic operation. The synthesized pyrano[3,2-b]xanthen-7(2H)-ones were screened for antibacterial, as well as, for antifungal and antioxidant activities. Compounds 67 were screened for their antibacterial activity against two Gram-positive bacteria (Bacillus subtilis, Staphylococcus aureus) and two Gram-negative bacteria (Escherichia coli, Klebsiella pneumoniae) by the disc diffusion method using ciprofloxacin as standard antibiotic. Among all the synthesized compounds 67j (R = 4-NO2C6H4, R1 = Me) (16.6 mm) and 67n (R = 2-thienyl, R1 = Me) (15.5 mm) showed good zone of inhibition against S. aureus compared to standard ciproflaxin (13.6 mm) in both concentrations (20 µM and 40 µM).
Scheme 27.

MW-assisted three-component synthesis of pyrano[3,2-b]xanthen-7(2H)-ones 67 for antibacterial screening.
More recently, Safari et al., reported a catalyst-free four-component reaction of cyanoacetohydrazide 38, malononitrile (12), diverse cyclic CH-acids type 16 and ninhydrin (68) in refluxing ethanol to afford spiro-4H-pyran derivatives 69 in excellent yields (Scheme 28). The efficiency of this multicomponent reaction to access complex skeleton is once again remarkable [71]. Compounds 69 were screened for their antibacterial activity against one Gram-positive bacterium (Staphylococcus aureus) and one Gram-negative bacterium (Escherichia coli) by the disc diffusion method using tetracycline as standard antibiotic. Disc diffusion data showed inhibition zones of 4–15 mm for the synthesized compounds against S. aureus, except for 69a (from 16a), 69b (from 16b with R = CO2Me, R1 = Ph) and 69c (from 16c), compared to standard tetracycline (30 mm). However, E. coli was resistant against all compounds tested.
Scheme 28.

Multicomponent synthesis of spiroindenopyridazine-4H-pyran derivatives 69 of antibacterial interest.
A one-pot three-component synthesis of 2,6-bis(1-coumarin-2-yl)-4-(4-substituted phenyl) pyridine derivatives 71 by a Chichibabin reaction was designed. These compounds were synthesized in 75–91% yield by the reaction of 3-acetyl coumarins 70 with substituted aromatic aldehydes 1 and ammonium acetate under acidic conditions (Scheme 29). The synthesized compounds 71 were evaluated for antimicrobial activity, as well as for DPPH free radical scavenging activity and ferrous ion-chelating ability. The mode of action of the active compounds was established by docking with the receptor GlcN6P synthase. The antimicrobial results revealed that compounds containing halogen or electron-withdrawing substituents either on the coumarin or phenyl rings exhibited potent antimicrobial and antioxidant activities. In the antibacterial studies, compounds bearing a bromo group in addition to a chloro group exhibited greater activity than those bearing only chloro groups. This observation suggested that di-substitution in the target compounds 71 by halogens enhanced the antimicrobial and antioxidant potential [72].
Scheme 29.

Multicomponent synthesis of 2,6-bis(1-coumarin-2-yl)-4-(4-substituted phenyl)pyridine derivatives 71 for antibacterial evaluation.
A convenient, one-pot, three-component protocol for the preparation of 2-(1-(2-oxo-2H-chromen-3-yl)ethylidene)hydrazinecarbothioamide derivatives 72 was achieved. Firstly, the synthesis of 3-acetyl-2H-chromen-2-one type 70 was carried out using starch sulfuric acid and cellulose sulfuric acid as biodegradable catalysts. Subsequently, the reaction of 70 with isothiocynates type 54 and hydrazine hydrate in the presence of catalytic amount of glacial acetic acid in refluxing ethanol afforded the target products 72 in 84–94% yield (Scheme 30). All synthesized compounds 72 were screened for antimicrobial activity. All compounds were found to show good to excellent activity against Escherichia coli MTCC 443 [73].
Scheme 30.

One-pot and solvent-free three-component synthesis of coumarin-thiosemicarbazone hybrids 72 of antibacterial interest.
A library of trimethoprim-based antibiotic compounds 74 and 75 was prepared through a selective multicomponent reaction upon the known drug trimethoprim 73. A Groebke-Blackburn-Bienaymé reaction of 73 with aldehydes 1 and isocyanides 18 afforded the desired adducts 74 and 75 in 6–59% and 4–48% yields, respectively, in one-step (Scheme 31). The analogs 74 and 75 displayed meaningful structural features of the initial drug together with relevant modifications at several points, keeping antibiotic potency and showing satisfactory antimicrobial profile (good activity levels and reduced growth rates), especially against methicillin-resistant Staphylococcus aureus [74].
Scheme 31.

Three-component synthesis of mono-(74) and double-(75) Trimethoprim-based GBBR adducts for antibacterial evaluation.
3.11. Antioxidant Activity
Compounds that exhibit antioxidant activity due to their chemical structure and redox properties have an important role in the uptake of singlet and triplet oxygen species, responsible for cardiovascular diseases, inflammatory bowel syndrome, cancer, aging, atherosclerosis and Alzheimer’s disease [75]. Different areas converge on the need to synthesize antioxidant compounds as protection against these diseases. Thereby, Lakshmi et al., reported the three-component synthesis of the 3-pyranyl indoles 33 described previously in Scheme 12 [54]. The antioxidant activity of such compounds was determined using methods for antioxidant activity estimation such as DPPH and ABTS. Thus, compounds 33m (R = 4-MeO), 33n (R = 3,4-diMeO), 33o (R = 2-Br-4,5-diMeO) and 33p (R = 4-NMe2) showed the most relevant radical scavenging activity in both methods due to the presence of electron-donating groups such as –OMe and –NMe2 with an IC50 < 50 μM in DPPH method. IC50 values of 858.6 μM (33m), 867.2 μM (33n), 880.3 μM (33o) and 900.0 μM (33p) were observed using the ABTS method. Ascorbic acid was used as standard, giving an IC50 < 50 μM in the DPPH method, and 650.0 μM in the ABTS method.
Lal et al., reported a time-efficient Biginelli reaction for the synthesis of curcumin 3,4-dihydropyrimidinones/thiones/imines 7 as described previously in Scheme 2 [28]. These compounds were screened for their antioxidant activity using ferric reducing antioxidant power (FRAP), cupric reducing antioxidant capacity (CUPRAC) and DPPH radical scavenging activity (DRSA) assays. Notably, compound 7e derived from 2,4-dihydroxybenzaldehyde and urea showed maximum DRSA antioxidant activity (IC50 = 3.86 μM) compared to ascorbic acid (IC50 = 3.76 μM), while the antioxidant activity decreased in compounds derived from 2-hydroxybenzaldehyde, 3-hydroxybenzaldehyde, and 4-hydroxybenzaldehyde, confirming the importance of the location of hydroxyl groups in the structure in order to potentiate the antioxidant activity.
A combinatorial approach carried out by Dangolani, et al., allowed obtaining a wide range of heterocyclic compounds [76]. In order to obtain the pyrano[2,3-d]pyrimidines 77, diverse carbohydrates 76 such as glucose, galactose, arabinose, maltose and lactose reacted with barbituric acid (27a) and malononitrile/diethylmalonate (12)/(38) in ethanol and PTSA as catalyst, in a three-component fashion. Additionally, barbituric acid (27a) was replaced by cyclohexane-1,3-dione (27b) to give two chromene-3-carbonitriles 78 in good yields (Scheme 32).
Scheme 32.
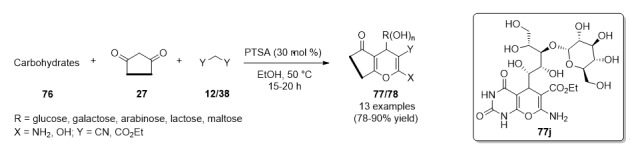
PTSA-catalyzed three-component synthesis of pyrano[2,3-d]pyrimidines 77 and chromene-3-carbonitriles 78 for antioxidant assays.
Additionally, the same authors reported a four-component reaction of barbituric acid (27a), aromatic aldehyde 1, malononitrile (12) and D-glucosamine (76a) to furnish the polyhydroxylated pyrido[2,3-d]pyrimidines 79 under the same synthetic conditions mentioned above including PTSA as Brønsted acid catalyst and ethanol as solvent (Scheme 33) [76].
Scheme 33.

Four-component synthesis of pyrido[2,3-d]pyrimidines 79 for antioxidant assays.
The above three families of synthesized compounds were evaluated using 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) antioxidant measuring system. The finding indicated that the maximum antioxidant activity occurred in the pyrano[2,3-d]pyrimidine 77j which is derived from barbituric acid, ethylcyanoacetate and maltose, Scheme 32, with a Trolox equivalent antioxidant capacity (TEAC) of 5.23 μM.
Within the heterocyclic compounds, the xanthene moiety is a key target due to the large number of biologically active molecules that contain it. Accordingly, Ashok et al., developed a microwave-assisted three-component reaction for the synthesis of pyrano[3,2-b]xanthen-7(2H)-ones 67 as previously was described in Scheme 27 [70]. The measurement of the antioxidant activity of the synthesized compounds was performed by DPPH free radical scavenging assay using ascorbate as a standard antioxidant. Interestingly, compounds 67g (R = 4-BrC6H4, R1 = H) (81.86%), 67i (R = 4-NO2C6H4, R1 = H) (83.25%) and 67k (R = 4-HOC6H4, R1 = H) (82.22%) showed a slightly lower DPPH antioxidant activity than standard ascorbate (86.50%).
The multicomponent synthesis of thiadiazolo[2,3-b]quinazolin-6-(7H)-ones 81 could be accomplished from 2-amino-5-phenyl-1,3,4-thiadiazoles 80, aromatic aldehydes 1 and dimedone (10a) in the presence of catalytic amounts of tetrabutylammonium hydrogen sulfate (Bu4NHSO4) in ethanol–water (1:1) under refluxing conditions (Scheme 34). Besides, this study showed that thiadiazolo[2,3-b]quinazolin-6-(7H)-ones 81 could be formed with high atom-economy involving the formation of one C-C and two C-N bonds in only one step [77]. The antioxidant activity of the synthesized compounds was screened by DPPH and OH radical scavenging assays using ascorbic acid as reference compound. Compounds 81a (R = H, R1 = 4-Br), 81e (R = 4-Cl, R1 = H) and 81f (R = 4-Cl, R1 = 4-Br) showed remarkable DDPH radical scavenging activity with values of 88.4, 87.6 and 85.4%, respectively, as compared with ascorbic acid (91.4%). In addition, compounds 81a, 81b (R = H, R1 = 4-Cl) and 81f showed good OH radical scavenging activity with values between 79.3 and 87.0%, as compared with ascorbic acid (89.5%).
Scheme 34.

Three-component synthesis of thiadiazolo[2,3-b]quinazolin-6-(7H)-ones 81 for antioxidant evaluation.
Very recently, a catalytic four-component synthesis of 1,3-cyclopentadiene derivatives was described by Ezzatzadeh et al. [78]. This unique and mild access to fully substituted 1,3-cyclopentadienes 84 involved terminal alkynes 82, sulfonyl azides 83, activated acetylenic compounds 47 and isocyanides 18 in the presence of zinc oxide nanoparticles (ZnO-NPs) and copper iodide as catalytic system in acetonitrile at room temperature (Scheme 35). This procedure offered several advantages such as being eco-friendly, ZnO-NPs could be re-used; the work-up was easy, non-toxic, and had a cleaner reaction profile. Antioxidant activity was investigated for the synthesized compounds 84a (R = Ph, R1 = 4-MeC6H4, R2 = Me, R3 = tBu), 84b (R = Pr, R1 = Ph, R2 = Me, R3 = tBu), 84c (R = Bu, R1 = Me, R2 = Me, R3 = tBu), and 84d (R = Pr, R1 = Me, R2 = Et, R3 = tBu) using the DPPH radical trapping and comparing results with synthetic antioxidants (TBHQ and BHT). The DPPH scavenging power was achieved in the following order: TBHQ > BHT > 84a > 84c > 84d > 84b, respectively. The free radical scavenging power was enhanced from 200 to 1000 ppm concentrations. For example, concentration 1000 ppm of 84a had 30.2% inhibition, whereas 200 ppm of 84a showed 9.6% free radical inhibition.
Scheme 35.

Four-component synthesis of 1,3-cyclopentadiene derivatives 84 for their DPPH scavenging power evaluation.
3.12. Anti-Mycobacterial Activity
Emergence of resistance against new tuberculosis (TB) drugs is an alarming issue demanding new drug profiles. For that, designing drugs for the treatment of tuberculosis has been a challenging area in medicinal chemistry in view of the multi-drug resistance [79] and high mortality rate [80] associated with this disease. Among the approved anti-tubercular drugs, benzimidazole derivatives appear as privileged and promising structures in chemotherapy of tuberculosis [81,82,83]. As a contribution to this topic, Anand et al., reported a three-component reaction for the synthesis of 2-mercaptobenzimidazole linked coumarinyl triazoles 87 as anti-tubercular agents. The process is mediated by a Click type reaction between 2-propargylthiobenzimidazole (85), 4-bromomethyl coumarins/1-aza-coumarins 86 and sodium azide, in the presence of CuSO4/sodium ascorbate as catalytic system (Scheme 36). The anti-tubercular assays of the obtained compounds 87 against M. tuberculosis (H37Rv) coupled with in silico molecular docking studies indicated that dimethyl substituted products 87c (X = O, R = 5,7-diMe) and 87d (X = O, R = 7,8-diMe) showed promising activity with higher C-score values [84].
Scheme 36.

Three-component one-pot synthesis of 1,4-substituted triazoles 87 for evaluation against M. tuberculosis (H37Rv).
Isoniazid analogues 90 were prepared by three-component condensation reactions of isoniazid (INH) 88, 3-mercaptopropionic acid 89 and various aldehydes 1 in THF as solvent and EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide) as activating agent (Scheme 37).
Scheme 37.

Three-component synthetic protocol of compounds 90 as potential antitubercular agents.
The products were evaluated against Mycobacterium tuberculosis H37Rv (MTB) and cytotoxicity. All the obtained compounds showed in vitro activity against MTB with MIC ranging from 0.12 to 41.2 μM. Thirteen compounds inhibited MTB with less than 1 μM. Six compounds were more potent compared than the standard first line drug INH (MIC of 0.36 μM) with MIC less than 0.36 μM and the compound N-(2-(4-(benzyloxy)phenyl)-4-oxo-1,3-thiazinan-3-yl)sonicotinamide inhibited MTB with MIC of 0.12 μM and was three times more potent than INH [85].
An efficient and green method was reported for the synthesis of fluorinated spiro-thiazine derivatives 92 and 93 via a three-component reaction, using 1-butyl-3-methylimidazolium hexafluorophosphate [bmim][PF6] as solvent and catalyst (Scheme 38). The synthesized compounds were subjected to antimycobacterial efficacy against Mycobacterium tuberculosis H37Rv strain and DNA cleavage activity. All compounds exhibited very poor antitubercular activities but DNA cleavage studies revealed that the tested compounds exhibited promising cleavage activity [86].
Scheme 38.

[bmim][PF6]-Mediated three-component synthesis of spiro[indole-3,2′[1,3]-thiazine]-2,4-diones 93 of antitubercular interest.
Quiroga et al., reported the synthesis and antimycobacterial activity of 4-arylbenzopyrazolo[3,4-b]quinolindiones 95 (in 70–80% yield) and 96 (in 70–85% yield). These compounds were obtained via a three-component reaction between 2-hydroxynaphthoquinone (94), benzaldehydes 1 and aminopyrazoles type 2, assisted by MWI (Scheme 39). Compounds 95 and 96 were evaluated against fifteen Mycobacterium spp strains, and six of them showed antimycobacterial activity. The highest inhibitory activity with MIC ≤ 2 μM for three of these compounds 96a (R = H, R1 = Me, R2 = Ph), 96b (R = 4-Me, R1 = Me, R2 = Ph) and 96g (R = 4-F, R1 = Me, R2 = Ph) was related with their highest lipophilicity and lesser polarity within these series [87].
Scheme 39.

Microwave-assisted three-component synthesis of benzopyrazolo[3,4-b]quinolindiones 95/96 for antimycobacterial activity evaluation.
Carbazole-tethered pyrrole derivatives 98 were synthesized via a one-pot three-component condensation of 2-nitrovinylcarbazoles 97 with aryl or alkyl amines 2 and dimethylacetylene dicarboxylate 47 (DMAD) in the presence of FeCl3 as an effective catalyst (Scheme 40). Twenty-four compounds were screened for in vitro anti-mycobacterial activity against Mycobacterium tuberculosis H37Rv. The compound dimethyl 1-(4-fluorophenyl)-4-(9-methyl-9H-carbazol-3-yl)-1H-pyrrole-2,3-dicarboxylate was the most active with MIC = 3.13 μM showing low cytotoxicity [88].
Scheme 40.

Three-component synthesis of carbazole-tethered substituted pyrrole analogs 98 for anti-mycobacterial activity against Mycobacterium tuberculosis H37Rv.
Ashok et al., reported the antitubercular and antimicrobial activity the 1,2,3-triazolyl xanthenones 99. The synthetic process involved a one-pot three-component thermal cyclization reaction of cyclic-1,3-diones type 10a, 1-aryl-1H-[1,2,3]triazole-4-carbaldehydes 1 and β-naphthol (50) in the presence of catalytic amount of iodine (Scheme 41). Xanthenone derivatives 99 were screened for in vitro antitubercular activity against M. tuberculosis H37Rv (ATCC27294) strain, as well as, for antibacterial activity against Gram-positive, Gram-negative strains and antifungal activity against a pathogenic strain of fungi. The antimycobacterial results indicated that compounds 99c (R = Me, R1 = 4-Cl), 99e (R = Me, R1 = 4-MeO) and 99k (R = H, R1 = 4-Cl) displayed good antitubercular activity, especially, compounds with substituent at the 4-position of the Ar ring (i.e., 4-ClC6H4 and 4-MeOC6H4). Moreover, derived of the 1,3-cyclohexadione type 10a containing methyl groups (i.e., R = Me) did not display activity. The molecular docking studies of compounds active against pantothenate synthetase revealed the favorable interactions with amino acid residues of such enzyme [89].
Scheme 41.

Iodine-mediated three-component synthesis of 1,2,3-triazolyl-xanthenones 99 for antitubercular activity against M. tuberculosis H37Rv (ATCC27294) strain.
3.13. Anticancer Activity
3.13.1. Quinoline Derivatives
Quinoline is one of the most important N-based heterocyclic compounds. Furthermore, quinoline derivatives are known to have a broad range of applications in medicinal, bioorganic, and industrial chemistry as well as in the field of synthetic organic chemistry [90,91]. Due to its importance, the synthesis of the quinoline scaffold has been studied for well over a century through the classical methods including the well-known Skraup, Doebner–Miller, Friedländer and Combes reactions, among others [92,93]. In 2006 Kouznetzov et al., reported the three-component cyclization of pyridinecarbaldehydes 1, anilines 2 and indene (100) in the presence of BF3∙OEt2 as catalyst [94]. The process involved an imino Diels-Alder cycloaddition affording the corresponding indeno[2,1-c]quinolines 101 in 32−98% yield (Scheme 42). Nearly all of the obtained products 101 were active against breast (MCF-7), lung (H-460) and CNS (SF-268) human cancer cell lines. Regarding the results, the activity of products 101 seems not to be related to the electronic properties of the R substituent. In contrast, the position of N-atom in the pyridine ring appears to exert some influence on the activity. Thus, compounds possessing α-pyridine were less active than the β- and γ- ones. According to the reviewed literature, this report correspond to the first example in which is combined both a synthetic approach mediated by a multicomponent reaction, as well as, the biological evaluation of the obtained products.
Scheme 42.

BF3∙OEt2–Catalyzed three-component synthesis of indeno[2,1-c]quinolines 101 of anticancer interest.
Various heterogeneous catalysts have also been encountered for the synthesis of quinoline derivatives. In 2011, Roopan et al., found that Montmorillonite K10 catalyzed a three-component cyclocondensation reaction of isatoic anhydride (102), (hetero)aromatic aldehydes 1 and AcONH4 under mild reaction conditions to afford the corresponding 2-aryl-2,3-dihydroquinazolin-4(1H)-ones 103 in 72−92% yield (Scheme 43) [95]. This methodology offered very attractive features such as reduced reaction times, higher yields and economic viability of the catalyst. Furthermore, the catalyst could be recovered and reused without change in the yield and catalytic activity. The 2-aryl-2,3-dihydroquinazolin-4(1H)-ones 103 were screened for their antitumor activity against an Ehrlich Ascites Carcinoma (EAC) tumor cell line. The result suggested that compounds 103a (R = 2-chloro-8-methylquinolin-3-yl), 103b (R = 2-chloro-7-methylquinolin-3-yl), 103c (R = 2-chloro-6-methyl-quinolin-3-yl) and 103d (R = 2-chloro-7,8-dimethylquinolin-3-yl) had the lowest IC50 values compared to the standard 5-fluorouracil (132.12 μM) and hence higher cytotoxicity effects on EAC tumor cell line than the standard drug.
Scheme 43.

Three-component synthesis of 2-aryl-2,3-dihydroquinazolin-4(1H)-ones 103 for anticancer activity analysis.
The synthesis of quinoline derivatives has also been carried out with homogeneous catalysts such as, piperidine (20 mol%) under mild reaction conditions. In 2014, Sangani et al., applied this chemistry to the synthesis of biquinoline-pyridine hybrids 104 [96]. The piperidine-catalyzed three-component reaction of tetrazolo[1,5-a]quinoline-4-carbaldehydes type 1, β-pyridinyl enaminone 39 and malononitrile/methylcyanoacetate/ethylcyanoacetate (12)/38 led to the corresponding biquinoline–pyridine hybrids 104 in ethanol under reflux conditions (Scheme 44). All compounds 104 were tested for in vitro anticancer activities against two cancer cell lines A549 (adenocarcinomic human alveolar basal epithelial) and Hep G2 (liver cancer). Enzyme inhibitory activities of all compounds were carried out against EGFR and HER-2 kinase. Remarkably, compound 104i (R = H, R1 = CO2Et) displayed the most potent inhibitory activity with IC50 values of 0.09 µM and 0.2 µM against EGFR and HER-2 kinase, respectively.
Scheme 44.

Piperidine-catalyzed three-component synthesis of biquinoline-pyridine hybrids 104 for in vitro anticancer activity against A549 and Hep G2 cancer cell lines.
Alternatively, the use of an asymmetric Povarov reaction allowed Alonso et al., to synthesize 1,2,3,4-tetrahydroquinolinylphosphanes 106 through a regio- and stereoselective aza-Diels-Alder reaction from aldehydes 1, styrenes 105 and phosphine oxide aniline 2a in the presence of BF3∙OEt2 as Lewis acid and 4 Å molecular sieves in chloroform under reflux conditions (Scheme 45). The amount of BF3∙OEt2 (3.0 equiv) and reaction times (48 h) are relatively high without any specific explanation as to why by the authors [97]. Furthermore, the multicomponent reaction also proceeded with an aniline containing the phosphine sulfide group type 2b to form 1,2,3,4-tetrahydro-quinolinylphosphine sulfides 107 in good yields using homogeneous acidic catalysis. The synthesized compounds were screened against three human cancer cell lines such as lung adenocarcinoma (A549), ovarian carcinoma (SKOV03) and embryonic kidney (HEK293). Notably, compound 106c (R = 4-FC6H4, R1 = 4-F) with an IC50 value of 0.08 µM showed excellent activity against the A549 cell line, while 1,2,3,4-tetrahydroquinolinylphosphine sulfide 107f (R = 4-FC6H4, R1 = 4-F) with an IC50 value of 0.03 µM was the most active against the A549 cell line.
Scheme 45.

Povarov three-component reaction for the synthesis of 1,2,3,4-tetrahydroquinolinylphosphanes 106 and 1,2,3,4-tetrahydroquinolinylphosphine sulfides 107 for antitumor screening.
In 2018, Castillo et al., described a catalyst-free method to construct diversely substituted 1,2,3,4-tetrahydroquinolines 110 through a Domino Mannich/Friedel-Crafts alkylation reaction of N-arylamines 108, paraformaldehyde (109) and electron-rich olefins 105 in ACN at room temperature [98]. This work showed that the choice of solvent was crucial and could greatly influence the reaction course: for example, γ-aminoethers were observed when methanol, ethanol, n-propanol or n-butanol were used as solvent [99]. These conditions enabled the construction of a broad library of 1,2,3,4-tetrahydroquinolines 110 in good to excellent yields via the formation of N-aryl-N-alkylmethyleneiminium ions as the key intermediates (Scheme 46). Interestingly, nine of the synthesized compounds were evaluated by the U.S. National Cancer Institute (NCI), where compound 110f (R = 6-MeO, R1 = 4-ClC6H4, X = pyrrolidin-2-onyl) presented a remarkable activity against 57 cancer cell lines, with the most important GI50 values ranging from 1.46 to 8.28 µM.
Scheme 46.

Three-component Domino-Mannich/Friedel-Crafts alkylation reaction for the synthesis of 1,2,3,4-tetrahydroquinolines 110 of anticancer interest.
An elegant synthesis of spiro-indeno[1,2-b]quinoxaline pyrrolothiazoles 113 in a highly regio- and stereoselective manner was reported in 2018 by Mani et al. [100]. The use of chalcones type 26 containing quinoline moiety in combination with ninhydrin (68), o-phenylenediamine (111) and thiazolidine-2-carboxylic acid (112) led to pyrrolothiazoles 113 in good yields and short reaction times (Scheme 47). This four-component reaction proceeded through cyclocondensation of ninhydrin (68) and o-phenylenediamine (111) to form an indeno[1,2-b]quinoxalin-11-one, which further condensed with thiazolidine-2-carboxylic acid (112) to produce an azomethine ylide derivative. This 1,3-dipole subsequently underwent cycloaddition reaction with chalcones type 26 to obtain a small library of pyrrolothiazole derivatives 113. The latter compounds showed interesting in vitro antitumor activity against two human cancer cell lines such as breast cancer (MCF-7) and lung adenocarcinoma (A549). Noteworthily, compound 113e (R = Me, R1 = H, R2 = Me) showed excellent activity with IC50 values of 11.0 µM and 15.0 µM against MCF-7 and A-549 cell lines, respectively.
Scheme 47.

Regioselective multicomponent synthesis of spiro-indeno[1,2-b]quinoxalin-pyrrolothiazoles 113 for antitumor screening.
Very recently, an Ugi four-component synthesis of quinoline-coumarin hybrids 115 was described by Taheri et al. [101]. This efficient and simple access to quinoline-coumarin derivatives involved coumarin-3-carboxylic acid (114), diverse 2-chloroquinoline-3-carbaldehydes 1, aniline derivatives 2 and aliphatic isocyanides 18 in methanol at room temperature (Scheme 48).
Scheme 48.

Ugi multicomponent reaction for the synthesis of quinoline-coumarin derivatives 115 to be investigated against A2780 human Ovarian cancer cell line.
Cytotoxic effects of fourteen products were investigated in A2780 human ovarian cancer cells. Interestingly, compound 115k (R = 5,8-diMe, R1 = H, R2 = cyclohexyl) showed excellent anticancer activity with IC50 values of 0.042 µM. Furthermore, the treatment of A2780 cells with compound 115k significantly (p-value ≤ 0.05) induced apoptosis by down-regulation of Bcl-2 and survivin both in mRNA and protein level via a single dose (0.042 µM), as well as, activation of caspase 9 and 3, loss of mitochondrial membrane potential (MMP), and high reactive oxygen species (ROS).
3.13.2. Pyrazole Derivatives
Pyrazole is a heteroaromatic compound of 5-membered containing two adjacent nitrogen atoms. The wide range of biological and synthetic applications displayed by pyrazoles, as well as, by their fused heterocyclic systems has been well documented in several comprehensive reviews [102,103,104,105,106]. In particular, formylpyrazoles occupy a noticeable place in the field of organic and medicinal chemistry despite somewhat being less popular than the amino derivatives, since such heteroaryl aldehydes are key intermediates for obtaining a wide range of biologically active compounds. In 2017, Kamble et al., employed a series of 3-(substituted)-1-phenyl-1H-pyrazole-4-carbaldehydes 1 in combination with isatoic anhydride (102) and benzylamine (2) in a K2CO3-mediated process conducted under reflux conditions in methanol (Scheme 49). This three-component reaction resulted in the synthesis of pyrazol-4-yl-2,3-dihydroquinazolin-4(1H)-ones 116 in high yields and short reaction times [107]. Compounds 116a (R = Ph), 116d (R = 4-FC6H4), 116f (R = 3-Py) and 116g (R = 2-thienyl) were in vitro screened by the NCI against 60 human tumor cell lines at a single dose of 10 µM. Results for each tested compound were reported as a mean graph of the percentage growth inhibition (%GI) of the treated cells, and deliberated by comparing with untreated control cells as percentage of growth inhibition (%GI) over all the tested cell lines. Remarkably, compound 116d exhibited the highest activity being breast cancer MCF7 (%GI 66.73), leukemia MOLT-4 (%GI 60.28), prostate cancer PC-3 (%GI 59.04), renal cancer UO-31 (%GI 57.83), non-small cell lung cancer EKVX (%GI 54.20) and leukemia HL-60 (TB) (%GI 54.03) the most sensitive strains. In addition, compound 116a exhibited good activity being renal cancer UO-31 (%GI 51.80), breast cancer T47D (%GI 50.85), prostate cancer PC-3 (%GI 45.38) and ovarian cancer IGROV1 (%GI 44.57) the most sensitive strains.
Scheme 49.

Three-component synthesis of pyrazol-4-yl-2,3-dihydroquinazolin-4(1H)-ones 116 for in vitro screening by the US National Cancer Institute (NCI) against 60 human tumor cell lines.
Nikalje’s group developed an efficient and green synthetic protocol to prepare 6-amino-4-substituted-3-methyl-2,4-dihydropyrano[2,3-c]pyrazole-5-carbonitriles 117 by four-component condensation reactions of various aromatic aldehydes 1, malononitrile (12), hydrazine hydrate and ethyl acetoacetate (8) using the ionic liquid triethylammonium hydrogen sulphate [Et3NH][HSO4] as a green reaction medium and also as a catalyst (Scheme 50). The fused heterocyclic products were isolated in high yields after short reaction times [108]. It is noteworthy that the catalyst was reused up to 4 times without much loss of catalytic activity. The in vitro anticancer activity of the synthesized compounds was carried out by the sulforhodamine B (SRB) assay against four human cancer cell lines including melanoma (SK-MEL-2), breast (MDA-MB-231), leukemia (K-562) and cervix (HeLa). The results indicated that compounds 117b (Ar = 4-ClC6H4), 117d (Ar = 4-MeOC6H4), 117g (Ar = 3,4-diMeOC6H3) and 117h (Ar = 3-NO2C6H4) exhibited significant cancer cell growth inhibition against the SK-MEL-2, MDA-MB-231 and K-562 cancer cell lines. Notably, compound 117b containing one chlorine atom in the para position of the phenyl ring exhibited excellent in vitro anticancer activity against the SK-MEL-2, MDA-MB-231 and K-562 cell lines with GI50 concentrations of 0.1 µM, 0.74 µM and 11.20 µM, respectively.
Scheme 50.

Ionic liquid-catalyzed four-component synthesis of dihydropyrano[2,3-c]pyrazoles 117 for anticancer evaluation.
3.13.3. Curcumin Derivatives
Curcumin (diferuloylmethane or 1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) is an edible natural pigment extracted from Curcuma longa [109]. The therapeutic activity of curcumin has been widely investigated over the last few decades and reports suggest the role of curcumin in innumerable biological activities, particularly its prominent in vitro and in vivo anticancer activity [109,110]. However, multiple structural-pharmacokinetic challenges such as low solubility and poor bioavailability during oral administration greatly limit its clinical application. Albeit, several strategies have been developed to overcome these disadvantages, one of the most common is the synthesis of novel curcumin derivatives that have better therapeutic properties and bioavailability [111,112]. From the viewpoint of the organic chemistry, curcumin exists in solution as a tautomeric keto form in acidic and neutral solutions while enol form in the alkaline medium. Furthermore, it has both an electrophilic Michael acceptor and an active methylene Michael donor units. In that direction, a series of structurally diverse 4-dihydropyrimidin-2(H)-one/thione derivatives of curcumin 7, as shown in Scheme 2, were synthesized through a three-component reaction involving curcumin (5), substituted aromatic aldehydes 1, and urea/thiourea 6 via a Biginelli type reaction in ethanol and concentrated sulphuric acid under reflux conditions [113]. Compounds 7b (Ar = 4-MeOC6H4, X = O), 7d (Ar = 4-ClC6H4, X = O) and 7g (Ar = 3-MeO-4-OH C6H3, X = O) were in vitro screened by the NCI against 60 human tumor cell lines at a single dose of 10 µM. Remarkably, compound 7d showed the highest activity, being leukemia CCRF-CEM (%GI 78.55), renal cancer UO-31 (%GI 75.92), prostate cancer PC-3 (%GI 75.87), central nervous system (CNS) SNB-75 (%GI 75.49), breast cancer MDA-MB-231 (%GI 73.63) and leukemia MOLT-4 (%GI 73.57) the most sensitive strains. In addition, compound 7g exhibited good activity being prostate cancer PC-3 (%GI 76.71), colon cancer HCT-116 (%GI 73.27), leukemia MOLT-4 (%GI 70.26), breast cancer MDA-MB-231 (%GI 67.70) and central nervous system (CNS) SNB-75 (%GI 65.81) the most sensitive strains.
Bhuvaneswari et al., reported that 1,4-diazabicyclo[2.2.2]octane (DABCO) catalyzed a three-component reaction of curcumin (5), substituted aromatic aldehydes 1 and malononitrile/ethyl 2-cyanoacetate (12)/(38) in ethanol at room temperature to afford functionalized curcumin derivatives 118 in 80−92% yield, Scheme 51. The construction of the cyclohexene ring proceeded through a Knoevenagel/Michael/cyclization sequence catalyzed by DABCO (10 mol%) [114]. Compounds 118j (R = 4-Me, R1 = CO2Et) and 118k (R = 2-NO2, R1 = CO2Et) were screened for their in vitro antitumor activity against human breast cancer cells (MCF-7) using the MTT assay. Compound 118j showed excellent activity with IC50 value of 10.0 µM against human breast cancer cells (MCF-7). In addition, molecular docking studies allowed rationalizing the anti-apoptotic Bcl-2 binding of all synthesized compounds and revealed that the docking of compounds 118j and 118k with Bcl-2 was more potent compared to curcumin 5.
Scheme 51.

DABCO-catalyzed three-component synthesis of functionalized curcumin derivatives 118 of anticancer interest.
3.13.4. Pyrrole Derivatives
Pyrrole is one of the most relevant N-heterocyclic unit because of its presence in diverse natural and synthetic compounds with a broad range of applications in medicinal chemistry, drug discovery and materials science [115,116,117]. Although, several synthetic approaches for this scaffold have been developed, there remains a great need to find simpler and atom-economical approaches for the construction of functionalized pyrrole-fused derivatives, which have been considered of interest in view of their pharmacological importance [118,119,120]. In this sense, Magedov et al., employed a series of N-(aryl- and alkylsulfonamido)acetophenones 119 in combination with diverse aromatic aldehydes 1 and malonitrile (12) in a TEA-catalyzed three-component procedure in ethanol under reflux conditions to afford highly functionalized 2-pyrrolines 120 in good yields, high regioselectivity and short reaction times (Scheme 52) [121]. However, the poor diastereoselectivity generated a mixture of cis- and trans-2-pyrrolines in 1:1.2 to 1:2 ratio. Rationalization of the low diastereoselectivity was not discussed. The synthesized compounds 120 were screened against two human cancer cell lines such as cervical adenocarcinoma (HeLa) and breast cancer (MCF-7). Because of the labor-intensive separation of the stereoisomeric pyrroline mixtures, the tests were performed with these mixtures. Remarkably, compound 120m (R = Ph, R1 = 4-MeOC6H5, R2 = Ph) showed the most potent inhibitory activity with GI50 values of 36.0 µM and 50.0 µM against the HeLa and MCF-7 cell lines, respectively.
Scheme 52.

Three-component synthesis of highly functionalized 2-pyrrolines 120 for anticancer evaluation.
As originally described by Pagadala [122], highly functionalized pyrroles 122 are also available by means of a catalyst-free four-component reaction in aqueous medium. This green transformation involved an aromatic aldehyde 1, malononitrile (12), an isocyanide 18 and a cyclic secondary amine 121, allowing an efficient access to polysubstituted pyrroles 122 in good yields (Scheme 53). It is noteworthy that aromatic aldehydes 1 were successfully employed while aliphatic aldehydes such as acetaldehyde and n-butyraldehyde failed to provide the corresponding polysubstituted pyrrole. Easy accessibility of starting materials, short reaction times and catalyst-free reaction, as well as, the use of water as solvent are claimed as the key advantages of this procedure. The synthesized compounds 122 were screened for their antitumoral activity against breast (MCF-7) and colon (HT-29) human cancer cell lines using doxorubicin and cisplatin as reference standards. Notably, compound 122g (R = 2,4-diMeOC6H3, R1 = 3,4-diClC6H3, X = O) with an IC50 value of 1.24 µM showed the highest activity against the MCF-7 cell line, while compound 122h (R = 2-BrC6H4, R1 = 3,4-diClC6H3, X = O) with an IC50 value of 1.47 µM was the most active against the HT-29 cell line.
Scheme 53.

Multicomponent synthesis of fully substituted pyrrole-3-carbonitriles 122 in aqueous medium to be screened for their antitumoral activity against breast (MCF-7) and colon (HT-29) human cancer cell lines.
3.13.5. Chromone and Chromene Derivatives
The oxygen-containing heterocycles are an important class of compounds in organic chemistry. The fusion of an aromatic ring to an oxygen-containing heterocycle will alter the electron density; thereby, the physical, chemical and biological properties will change. In particular, chromone is an oxygen-containing heterocyclic system with a benzoannelated γ-pyrone ring being chromone (4H-chromen-4-one, 4H-1-benzopyran-4-one) the parent compound [123,124]. Furthermore, bicyclic oxygen-containing heterocycles resulting from fusion of benzene ring with 5,6-positions of either 2H- or 4H-pyran ring system are designated as 2H-chromene and 4H-chromene, respectively. Hence, the structural importance of the chromone and chromene moiety has elicited a great deal of interest in the field of organic synthesis, medicinal chemistry and drug discovery to develop novel and improved synthesis of these molecular skeletons [125,126,127]. In this sense, Huang et al., described a catalyst-free access to dithiocarbamate substituted chromones 124 by means of a three-component reaction (Scheme 54) [128]. A mixture of 3-chloromethyl chromone derivatives 123, cyclic secondary amines 121 and an excess of carbon disulfide was stirred at room temperature in DMF affording compounds 124 in good yields. The synthesized compounds were screened for their in vitro antiproliferative activity against six cancer cell lines, including HCCLM-7 (hepatocellular carcinoma cell), He-La (cervical carcinoma cell), MDA-MB-435S (mammary adenocarcinoma cell), SW-480 (colon carcinoma cell), Hep-2 (laryngocarcinoma cell) and MCF-7 (mammary adenocarcinoma cell) by using the MTT method. It should be noted that compound 124u (R = 6-Cl, R1 = piperidinyl) was identified as the most promising candidate due to their high potency against all cancer cell lines with IC50 values ranging from 0.24 to 0.85 µM. Further flow-activated cell sorting analysis revealed that compound 124u arrest the cell cycle of MDA-MB-435S and SW-480 both in G2/M phase with dose-dependent effect and might display apoptosis-inducing effect on these tumor cell lines.
Scheme 54.

Three-component synthesis of dithiocarbamate substituted chromones 124 to be screened for their in vitro antiproliferative activity by mean of the MTT method.
In 2010, Kumar et al., reported an intriguing three-component synthesis of benzochromenes 125 in good yields by mixing 2-naphthol (50), an aldehyde 1 and malononitrile/ethyl cyanoacetate (12)/(38) in the presence of a catalytic amount of ceric ammonium nitrate (CAN) under solvent-free conditions (Scheme 55) [129]. Great functional diversity was achieved with this thoughtful strategy that accommodated aromatic, heteroaromatic, and aliphatic aldehydes. The advantages of this method include the use of a recyclable catalyst, short reaction times, simple work-up procedure and easy isolation. From a mechanistic standpoint, a sequence initiated by the formation of an ortho-quinonemethide through the nucleophilic addition of 2-naphthol to the aldehyde catalyzed by CAN. Then, Michael addition of malononitrile or ethyl cyanoacetate onto the ortho-quinonemethide, followed by intramolecular 6-exo-dig cyclization/imine-enamine tautomerization sequence generated the benzochromene ring. The synthesized compounds 125 were screened for their antiproliferative activity in prostate cancer (DU-145), breast cancer (MCF-7), cervical carcinoma (C-33A) and lung carcinoma (A-549) human cell lines. It is noteworthy that compound 125b (R = indol-3-yl, R1 = CN) exhibited the highest activity against all cancer cell lines with IC50 values ranging from 5.4 to 12.2 µM.
Scheme 55.

CAN-Catalyzed three-component synthesis of 3-amino-1-substituted-1H-benzo[f]chromenes 125 under solvent-free conditions for evaluation of their antiproliferative activity.
Shortly after, the Paliwal´s group reported the three-component reaction between dimedone (10a) an heteroaryl aldehyde e 1 and malononitrile (12) at room temperature in the ionic liquid (IL) [TEA][OAc] (TEAA), gave rise to a series of 5,6,7,8-tetrahydro-4H-chromene-3-carbonitriles 126 in good yields (Scheme 56) [130]. According to the authors, [TEA][OAc] acted as a green catalyst as well as a reusable solvent. Use of IL media for this transformation resulted in improved yields within shorter reaction times. A complementary study on these latter conditions suggested that IL could be reused for six times without apparent loss of catalytic activity. The synthesized compounds 126 were screened for their in vitro antitumor activity in breast cancer (MDA-MB-435), prostate cancer (PC-3) and ovarian cancer (Ovkar-3) human cell lines. However, the synthesized compounds did not give the satisfactory results between 10−7 and 10−4 molar concentrations in comparison with doxorubicin.
Scheme 56.

TEAA-promoted three-component synthesis of 5,6,7,8-tetrahydro-4H-chromene-3-carbonitriles 126 of antitumor interest.
An alternative access to chromenyl phosphonates from dialkylphosphites was reported in 2014 [131]. Thus, an ethanolic solution of a substituted salicylaldehyde type 1, a dialkylphosphite type 3 and malononitrile (12) was stirred at room temperature in the presence of a catalytic amount of dibutylamine, afforded the 2-amino-3-cyano-4H-chromen-4-ylphosphonates 127 in very good yields, (Scheme 57).
Scheme 57.

Three-component synthesis of 2-amino-3-cyano-4H-chromen-4-ylphosphonates 127 to be screened against two cancer cell lines using MTT assay.
The ready availability of this non-toxic catalyst, high yields, and simple work-up make this approach an eco-friendly alternative to the currently existing protocols. The synthesized compounds 127 were screened against two cancer cell lines, including adenocarcinomic alveolar basal epithelial (A-549) and epidermoid cancer (KB) using MTT assay. Albeit all synthesized compounds showed moderate activity at 20 and 40 µM concentrations, compounds 127a (R = H, R1 = Et) and 127d (R = 6-Br, R1 = Et) showed remarkable activity against the two tested cell lines.
Perumal et al., reported an ultrasound-assisted three-component reaction of 2-hydroxy-1,4-naphthoquinone 94, an aromatic aldehyde 1 and malononitrile 12 in the presence of Cu(OTf)2 catalyst and eco-friendly polyethylene glycol solvent, thus giving access to highly functionalized benzo[g]chromene derivatives 128 in good yields and short reaction times (Scheme 58) [132]. The synthesized compounds 128 were tested for their in vitro anticancer activity against cervical cancer cell line (HeLa). Most of the compounds showed higher antitumor activity that doxorubicin. Particularly, compounds 128c (R = 4-Br), 128g (R = 3-benzyloxy-4-MeO), 128h (R = 2-hydroxy-3-MeO) and 128j (R = 3-Cl) displayed the highest activity with IC50 values ranging from 1.2 to 4.1 µM.
Scheme 58.

Cu(OTf)2-Catalyzed three-component synthesis of benzo[g]chromenes 128 under ultrasound irradiation for testing of their in vitro anticancer activity against cervical cancer cell line (HeLa).
3.13.6. Pyridone Derivatives
2-Pyridone, the tautomer of 2-hydroxypyridine, is one of the privileged heteroaromatic rings in natural products, bioactive molecules and pharmaceutical agents [133,134]. Furthermore, pyridone fused with a benzene or heterocyclic ring gives rise to diverse heterocyclic systems with innumerable pharmacological properties. Thus, the selective synthesis of functionalized 2-pyridone derivatives and fused-pyridone heterocycles has been one of the important longstanding subjects in organic synthetic chemistry [135,136,137]. In that direction, a series of highly functionalized pyrano[3,2-c]pyridones 130 were synthesized through a TEA-catalyzed three-component reaction of 4-hydroxy-1,6-dimethylpyridin-2(1H)-one (129), an aromatic aldehyde 1 and malononitrile (12) in ethanol under reflux conditions (Scheme 59) [138]. Annexin-V staining and DNA laddering assays.
Scheme 59.

Three-component synthesis of pyrano[3,2-c]pyridone derivatives 130 for antiproliferative assays.
The biheterocyclic products 129 were isolated in high yields after short reaction times. The obtained compounds 129 were screened for their in vitro antitumor activity against cervical cancer cell line (HeLa). Notably, compounds exhibited submicromolar or low micromolar potencies for the inhibition of proliferation of HeLa cells with GI50 values ranging from 0.33 to 43.3 µM. Furthermore, the antiproliferative effect from the potent apoptosis inducing ability of these heterocycles, was confirmed by the flow cytometric
Within their research efforts, the Hajela group reported the regioselective synthesis of densely substituted 2-pyridones 132 through a three-component reaction of aromatic aldehydes 1, substituted acetophenones 16 and phenyl acetamides 131 in the presence of sodium hydride in DMSO to afford the corresponding 3,4,6-triaryl-2-pyridones 132 in good yields as a single regioisomer (Scheme 60) [139]. The obtained compounds 132 were evaluated for their antiproliferative activity against human breast carcinoma cell line MCF-7 and MDA-MB-231. Thus, compound 132h (R = 3,4-diMeO, R1 = 4-MeO, R2 = H) displayed the highest anti-breast cancer activity with IC50 values of 90.5 µM and 15.1 µM against MCF-7 and MDA-MB-231 cell line, respectively. In addition, cell cycle analysis showed that compound 132h induced statistically significant arrest of cells in G1 phase and reduction in S-phase cells in a dose-dependent manner.
Scheme 60.

Three-component synthesis of 3,4,6-triaryl-2-pyridones 132 for antiproliferative activity against human Breast carcinoma cell line.
Alternatively, Kumar et al., described a three-component synthesis of quinolinone derivatives 134 using diverse aromatic aldehydes 1, 2,3,6,7-tetramethoxyphenanthren-9-amine (2) and Meldrum’s acid (133) (Scheme 61) [140]. As a result, a collection of 3,4-dihydrodibenzo[f,h]quinolin-2(1H)-ones 134 was elaborated in the presence of a catalytic amount of sulfamic acid in refluxing ethanol. The authors demonstrated that the mechanism consisted of the preliminary Knoevenagel condensation of Meldrum’s acid with the aldehyde, affording the arylidene intermediate. Then, Michael addition of amine and subsequent dehydration, cyclization by loss of CO2 and acetone gave quinolinone derivatives 134. The obtained compounds were evaluated for their in vitro cytotoxic potential against human lung (A549), prostate (PC-3 and DU145), breast (MCF-7) and colon (HT-29 and HCT-116) cancer cell lines. Notably, compound 134p (R = 3,4-OCH2O) showed excellent antiproliferative activity against A549 lung cancer cell line with an IC50 of 3.17 µM. Flow cytometric analyses revealed that compound 134p arrested both Sub G1 and G2/M phases of cell cycle in a dose dependent manner. Additionally, compound 134p also displayed significant inhibition of tubulin polymerization and disruption of microtubule network with an IC50 of 5.15 µM.
Scheme 61.

Sulfamic acid-catalyzed three-component synthesis of 3,4-dihydrodibenzo[f,h]quinolin-2(1H)-ones 134 for evaluation of their in vitro cytotoxic potential.
3.13.7. Thiazole Derivatives
S-Heterocycles have maintained their status as an important nucleus with high medicinal value and low toxicity profile, in comparison to previous N-heterocycles. Particularly, thiazole has been widely found in diverse pharmacologically active substances and some naturally-occurring compounds [141]. For that reason, thiazole is a versatile building-block for the preparation of thiazole-based compounds with innumerable biological properties [142]. Thus, thiazole derivatives possessing anticancer activity will reignite the interest of the scientific community in the usefulness of these S-heterocycles in the medicinal chemistry and drug discovery [143,144]. In this context, tetronic acid type 27 was used as the 1,3-dicarbonyl parent in a straightforward construction of the 4-aza-podophyllotoxin skeleton embedded with a thiazole unit [145]. Thus, tetronic acid (27) reacted with aromatic aldehydes 1 and 2-methylbenzo[d]thiazol-5-amine (2) at 120 °C in acetic acid under MWI, leading to a series of novel 4-aza-podophyllotoxin analogs 135 (Scheme 62). The triheterocyclic products 135 were isolated in high yields after short reaction times. Then, some selected compounds were subject to the test of in vitro cytotoxicity to malignant melanin carcinoma cell line M14, mammary carcinoma cell line MCF-7, and colon carcinoma cell line SW1116. Interestingly, the 4-aza-podophyllotoxin analog 135j (Ar = 4-OH-3-NO2C6H3) displayed the most potent inhibitory activity with IC50 values of 33.9, 56.0 and 69.4 µM against M14, MCF-7 and SW1116 human cell lines, respectively.
Scheme 62.

Microwave-assisted three-component synthesis of 4-aza-podophyllotoxin analogs 135 for testing of their in vitro cytotoxicity.
Combining two potentially bioactive moieties to form heterocyclic scaffolds is a known process in drug discovery. In 2012, Gu et al., reported an impressive three-component reaction of 3-nitro-2-bromopyridine 136, primary 1-aminophosphonates 3 and carbon disulfide in the presence of copper(II) chloride (1 equiv.), tin(II) chloride (4 equiv.) and potassium carbonate (3 equiv.) at 100 °C in DMF, leading to α-aminophosphonates 137 containing thiazole[5,4-b]pyridine moiety with yields up to 85% (Scheme 63) [146]. According to the authors, SnCl2∙2H2O enabled reduction of the nitro group to amine, while CuCl2∙2H2O promoted C–S bond cross-coupling reaction between dithiocarbamate salts and 3-amino-2-bromopyridine. This unique transformation featured multiple bonds breaking and forming events in a single, atom-economic process. The antitumor activity of the obtained compounds 137 was determined by the MTT assay against three human cancer cell lines such as PC-3, Bcap-37 and H460. Notably, compound 137f (R = Et, Ar = 4-FC6H4) showed the most potent inhibitory activity with IC50 values of 1.04, 0.81 and 2.23 µM against PC-3, Bcap-37 and H460 cell lines, respectively.
Scheme 63.

Three-component synthesis of α-aminophosphonate derivatives 137 to be assayed against PC-3, Bcap-37 and H460 human cancer cell lines by the MTT method.
In order to develop new drugs, an elegant and efficient synthesis of 5-arylidenethiazolidinones 139 was discovered in 2016. This three-component reaction involved thiazolidine-2,4-dione 138, substituted aromatic aldehydes 1 and an excess of piperidine (121) in refluxing ethanol (Scheme 64) [147]. A reasonable mechanism was suggested to rationalize the formation of these 5-arylidenethiazolidinones 139. Initially, piperidine-promoted Knoevenagel condensation between thiazolidine-2,4-dione 138 and aromatic aldehyde 1 afforded a Knoevenagel adduct. Then, nucleophilic addition of piperidine 121 onto one of the carbonyl functionalities followed by dehydration would give the observed 5-arylidenethiazolidinone 139. These compounds were screened for their in vitro antitumor activity against human breast cancer cells such as MCF-7 and MDA-MB-453. The result suggested that compound 139i (Ar = 4-PrOC6H4) displayed the highest inhibitory activity against MCF-7 (%GI 66.4) and MDA-MB-453 (%GI 60.0) cell lines. Furthermore, cell cycle analysis showed that compound 139i arrest the progression of MCF-7 cells at G0/G1 phase.
Scheme 64.

Three-component synthesis of 5-arylidenethiazolidinones 139 for screening of their in vitro antitumor activity against human Breast cancer cells.
Alternatively, an eco-friendly access to thiadiazolo[3,2-a]pyrimidine-6-carbonitriles from a 1,3,4-thiadiazol-2-amine was reported in 2016 [148]. An ethanolic solution of 5-(4-chlorophenyl)-1,3,4-thiadiazol-2-amine 2, an aromatic aldehyde 1 and malononitrile (12) was sonicated at 80 °C in the presence of catalytic amounts of NaOH, affording the thiadiazolo[3,2-a]pyrimidine-6-carbonitriles 140 in good yields (Scheme 65). A comparative study shed light on the benefits of ultrasound irradiation in terms of yields and reaction times in comparison to the same transformation under conventional heating. The obtained compounds were evaluated for their in vitro anticancer activity against MCF-7, K562, HeLa and PC-3 cancer cell lines using fluorouracil as positive control. From the anticancer activity results, compound 140i (Ar = 3-HO-4-MeOC6H3) showed the highest GI50 values of 32.7, 34.3, 55.3, and 28.9 µM for the MCF-7, HeLa, K562 and PC-3 cancer cell lines, respectively. A docking study of the synthesized compounds showed good binding mode in the active site of thymidylate synthase enzyme.
Scheme 65.

Ultrasound-assisted three-component synthesis of thiadiazolo[3,2-a]pyrimidine-6-carbonitriles 140 to be evaluated for their in vitro anticancer activity against MCF-7, K562, HeLa and PC-3 cancer cell lines using fluorouracil as positive control.
In 2017, Semenov’s group developed similar reactions using Meldrum’s acid (133) as the 1,3-dicarbonyl partner [149]. Since 3-arylisothiazol-4-aminium chloride 2∙HCl has been rarely exploited as a nucleophile in multicomponent processes, this substrate was reacted by refluxing glacial acetic acid with an aromatic aldehyde 1 and Meldrum’s acid (133) in the presence of sodium acetate, affording bicyclic derivatives 141 with modest yields (Scheme 66). Selected products were screened for their in vitro antitumor activity using a panel of human cancer cell lines, including CAOV-3 and TOV-112D from ovarian tumors, MDA-MB231, MDA-361/DYT2 and MDA-MB-468 from breast adenocarcinoma, MDA-MB-435 from melanoma, and NCI-H1975 from non-small cell lung cancer. Notably, compound 141b (R = 4-MeO, Ar = 3-thienyl) exhibited the highest activity against all cancer cell lines with IC50 values ranging from 80.72 to 309.17 nM. Furthermore, compound 141b blocked cell cycle in mitosis and disintegrated interphase microtubule network, suggesting its tubulin-targeting microtubule destabilizing effect.
Scheme 66.

Three-component synthesis of isothiazolo[4,5-b]pyridin-5-one derivatives 141 for screening of their in vitro antitumor activity against a panel of human cancer cell lines.
In 2018, a time-efficient three-component synthesis of a series of pyrazolo-oxothiazolidine derivatives 142 was achieved through the reaction between 1-(benzofuran-2-yl)-3-(substituted-arylprop-2-en-1-ones type 26, thiosemicarbazide 38 and dialkyl acetylenedicarboxylates 47 in the presence of sodium hydroxide in refluxing ethanol (Scheme 67) [150]. Synthesized compounds were evaluated for their antiproliferative activity against the A549 lung cancer cell line. Particularly, compounds 142a (R = 4-F, R1 = Et), 142f (R = 4-F, R1 = Me) and 142h (R = 3,4-diMeO, R1 = Me) showed much better activity than the standard drug sorafenib (IC50 = 3.77 µM), with IC50 values of 0.93 µM, 0.80 µM and 0.96 µM, respectively. Molecular docking studies indicated that compound 142f had the greatest affinity for catalytic site of receptors EGFR and VEGFR2.
Scheme 67.

Three-component synthesis of pyrazolo-oxothiazolidine derivatives 142 for evaluation of their antiproliferative activity against A549 Lung cancer cell line.
Indoleamine 2,3-dioxygenase 1 (IDO1) has emerged as an attractive target for cancer immunotherapy. In this context, the Passerini reaction was employed to assemble a small library of imidazothiazoles 143 that target IDO1 [151]. The reaction of isocyanides 18 with aqueous formaldehyde (1) and functionalized phenylacetic acids 114 led to imidazothiazoles 143 containing a α-acyloxyamide moiety in the side chain, in moderate to good yields (Scheme 68). Notably, compound 143d (R = 4-hydroxybenzyl) showed an IC50 value of 0.20 µM in the IDO1-based assay, a full biocompatibility at 10 µM, together with a modest inhibitory activity in A375 cells. Furthermore, molecular docking studies showed that 143d displayed a unique binding mode in the indoleamine 2,3-dioxygenase 1 (IDO1) active site, with the side-chain protruding in an additional pocket C, where a crucial hydrogen bond is formed with Lys238.
Scheme 68.

Passerini three-component reaction for the synthesis of imidazothiazoles 143 for targeting IDO1 active site.
3.13.8. Indole-Based Anticancer Heterocyclic Systems
Interest in synthesizing new indole derivatives continue due to their biological properties displayed [152]. In particular, substituted indole derivatives play a key role in the synthesis of biologically active compounds especially with anticancer, antitumor, and anti-inflammatory activities [153,154,155]. They are also reported as potent inducers of apoptosis through a cell-based HTS caspase assay [156].
Indole-Substituted Heterocyclic Derivatives
A series of 4-aryl-6-indolyl-nicotinonitrile-2-ones 144 were synthesized in 77–87% yield, through a one-pot four-component reaction of 3-acetylindole type 32 (X = H), aromatic aldehydes 1, ethyl cyanoacetate (38) and ammonium acetate in the presence of piperidine as a catalyst under microwave irradiation (Scheme 69). Products 144 were evaluated for antiproliferative activity against human ovarian adenocarcinoma (SK-OV-3), breast adenocarcinoma (MCF-7), and cervix adenocarcinoma (HeLa) cells to establish a structure–activity relationship [157]. In the same direction, Lakshmi et al., reported a one-pot three-component method for the synthesis of 3-pyranylindoles 33 in 68–87% yield. The procedure was accomplished by a tandem Knoevenagel–Michael reaction of 3-cyanoacetyl indole type 32 (X = CN), various aromatic aldehydes 1 and malononitrile (12) catalyzed by InCl3 in ethanol under refluxing conditions (Scheme 69). The synthesized compounds 33 were evaluated for anticancer activity, in addition to anti-microbial and antioxidant activity, some of them showing good anticancer activity against MCF-7 breast cancer cell lines in comparison with the standard drug used [54].
Scheme 69.

One-pot three-component methods for the synthesis of 3-pyranylindoles 33 and 4-aryl-6-indolyl-nicotinonitrile-2-ones 144, mediated by 3-acetylindole type 32 as common precursor, for evaluation of their anticancer activity.
Based on previous molecular hybridization techniques [158], Gupta et al., designed a series of indole-chalcone based benzopyran hybrid compounds 145 in order to identify molecules that can inhibit DNA ligation and cell proliferation, and may serve as drug-like molecules. Products 145 were obtained in 70–78% yield, via a three-component reaction between substituted 2-methylindoles 32, chalcones 26 and malononitrile (12) in the presence of L-proline as catalyst and acetonitrile as solvent, Scheme 70. The synthetic molecules 145 were tested for their antiligase and antiproliferative activities in cancer cells. A detailed study of the most active compound 145a (R = H, R1 = tBu, R2 = C4H3S) was carried out in order to verify the mode of action and cytotoxicity in in vitro and 3D tumor models [159].
Scheme 70.

l-Proline-catalyzed three-component synthesis of indole-chalcone based benzopyran hybrids 145 for testing of their antiligase and antiproliferative activities in cancer cells.
Carbazole (Benzo[b]Indole)-Substituted Heterocyclic Derivatives
Carbazole is a benzo[b]indole in which the benzene ring is fused with the 2,3-position of the indole ring. This motif is a privileged pharmacophore scaffold found in many biologically active compounds of diverse origins, from natural products to synthetic sources. Widespread interests of chemists have been attracted to these structures due to their biological activities and potential applications as pharmacological agents [160].
Thus, Indumathi et al., reported the synthesis of pyrido[2,3-a]carbazoles 147, in 81–85% yield, by a four-component reaction of 6-methyl-2,3,4,9-tetrahydro-1H-carbazol-1-one (146), aromatic aldehydes 1, malononitrile/ethylcyanoacetate (12)/(38), ammonium acetate and L-proline as catalyst in ethanol (Scheme 71). The obtained compounds 147 were evaluated for their in vitro cytotoxicity against five cancer cell lines, as well as, antimicrobial and antioxidant assays. Additionally, their primary structure-activity relationships were established [161].
Scheme 71.

L-Proline catalyzed multicomponent synthesis of pyrido[2,3-a]carbazoles 147 for evaluation of their in vitro cytotoxicity against five cancer cell lines.
A library of carbazole Mannich bases 149 was synthesized in 85–94% yield from a three-component reaction of 4-hydroxycarbazole (148), aromatic aldehydes 1 and cyclic amines type 121 in toluene under reflux in the presence of TEA as catalyst (Scheme 72). Further, the synthesized Mannich bases 149 were tested for their in vitro antiproliferative activity against three cancer cell lines (i.e., Hela, MDA-MB-231, and HepG2). The results indicated that compounds 149 showed selective cytotoxicity against Hela cells. Additionally, in silico molecular docking study of carbazole Mannich bases 149 against colchicine binding site of the tubulin polymer was investigated [162].
Scheme 72.

Three-component synthesis of a library of carbazole Mannich bases 149 to be tested for their in vitro antiproliferative activity against three cancer cell lines.
Synthesis of highly functionalized pyrrolo[3,2-a]carbazoles 151 via ring contraction through an aerobic metal free rearrangement and intramolecular Michael addition reaction using one-pot three-component reaction was reported. Thus, a mixture of (tetrahydro-carbazol-1’-ylidene)-propanedinitriles type 146, ninhydrin (150) and o-phenylenediamines 111 were subjected to reflux in methanol as the solvent and catalytic amount of TEA to afford the target products 151 in 67–90% yield (Scheme 73). The obtained compounds were studied for colorectal cancer activity, as well as, free radical scavenging in vitro via MTT and DPPH assays, respectively. Further, the structure-activity relationships were also carried out [163].
Scheme 73.

Three-component synthesis of highly poly functionalized pyrrolo[3,2-a]carbazoles 151, via a TEA-catalyzed ring contraction process, for their study against Colorectal cancer cells.
3.13.9. Pyrimidine-Based Anticancer Heterocyclic Systems
The great interest in pyrimidine and dihydropyrimidine (DHPM) derivatives lies in the fact that these classes of compounds has, in principle, pronounced biological activity [164,165]. Consequently, diverse methodologies have been developed to improve the synthesis of this attractive family of compounds [166].
Biginelli-Mediated Synthesis of Dihydropyrimidines
The Biginelli reaction, is an MCR that involves the cyclocondensation of acetoacetic esters, aromatic aldehydes and (thio)urea [167]. The products of this three-component synthesis are identified as 3,4-dihydropyrimidin-2(1H)-ones (DHPMs). Thus, a library of Biginelli adducts 152 was synthesized in 31–92% yield, as described in Table 1, and evaluated as potential inhibitors of in vitro cancer cells proliferation, but also, as scavengers of reactive nitrogen and oxygen species (RNS and ROS, respectively). The capacity of all compounds 152 to inhibit cancer cells growth was dependent on the histological origin of cells, except for 152p (X = S, R = MeSC6H4), which was highly active against all cell lines. Compounds 152k (X = O, R = 4-HO-3-MeOC6H3), and 152x (X = S, R = C6H11), were as potent as the reference drug doxorubicin against adriamycin-resistant ovarian and prostate cancer cells, respectively [168].
Table 1.
Synthesis of dihydropyrimidone(thione)s 152–156 of antiproliferative interest mediated by Biginelli type three-component reactions.
| Reagents | Conditions | Product | Reference | ||
|---|---|---|---|---|---|
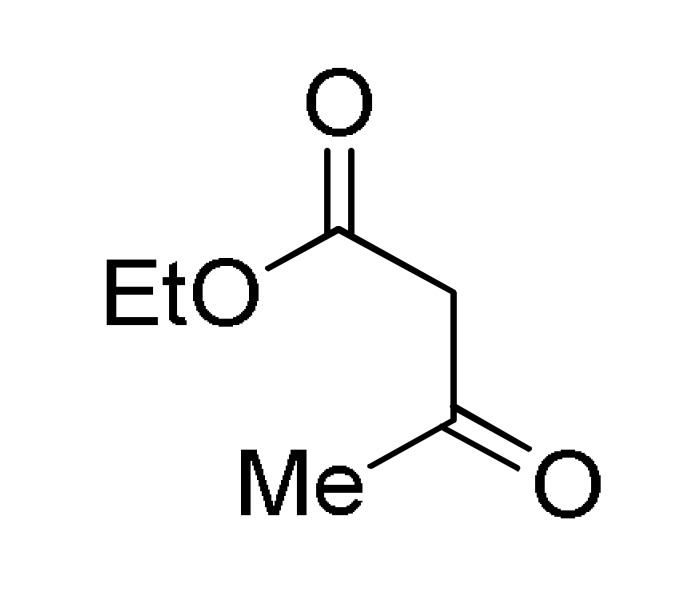 type 8 |
RCHO type 1 |
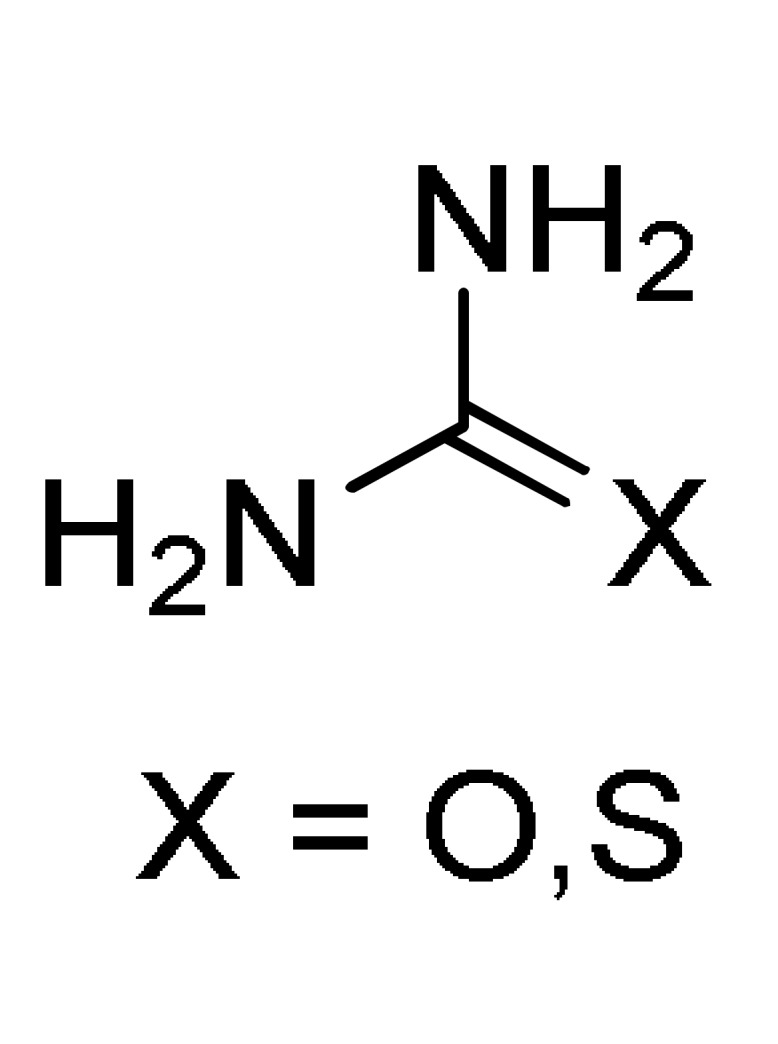
6 |
p-sulfonic acid calix[4]arene (0.5% mol), EtOH, reflux |
152 a |
[169] |
 type 27 |
 type 1 |
p-sulfonic acid, EtOH, reflux |
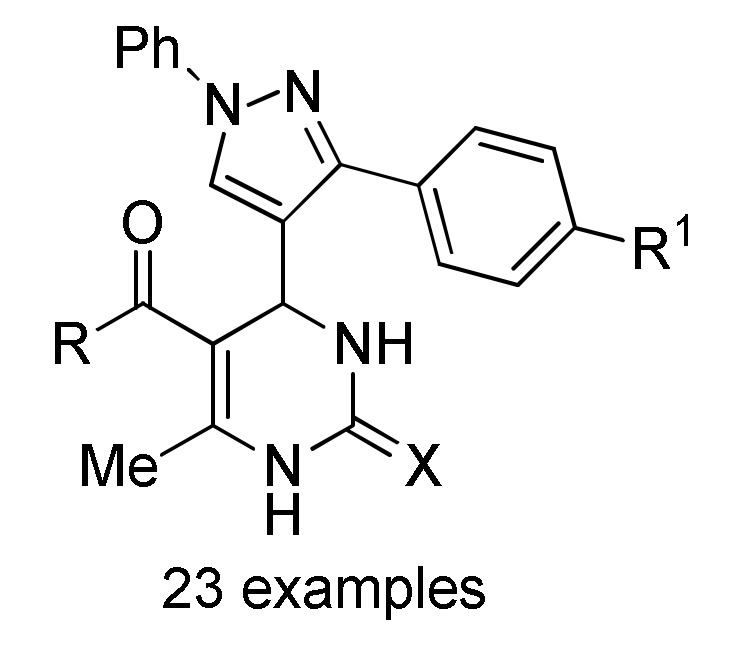
153 b |
[169] | |
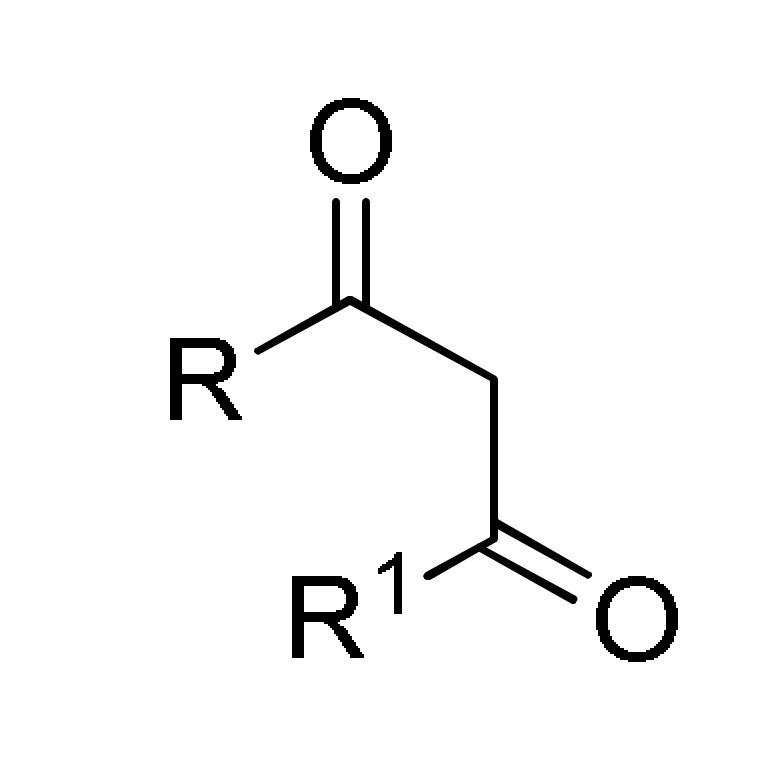 type 27 |
ArCHO/R2CHO 1 |
MAI·Fe2Cl7 (5 mol%), BMI·BF4 IL (1 mL), 80 °C |
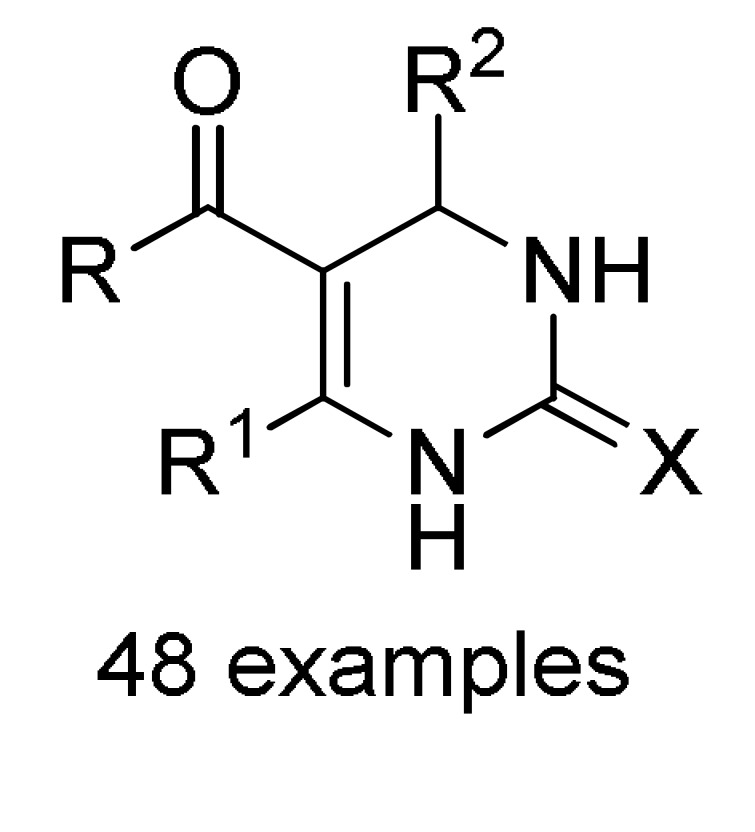
154 c |
[170] | |
| Heterogeneous Zn- and Cd-based CPs catalysts (5 mol%), 70–100 °C, Continuous flow |
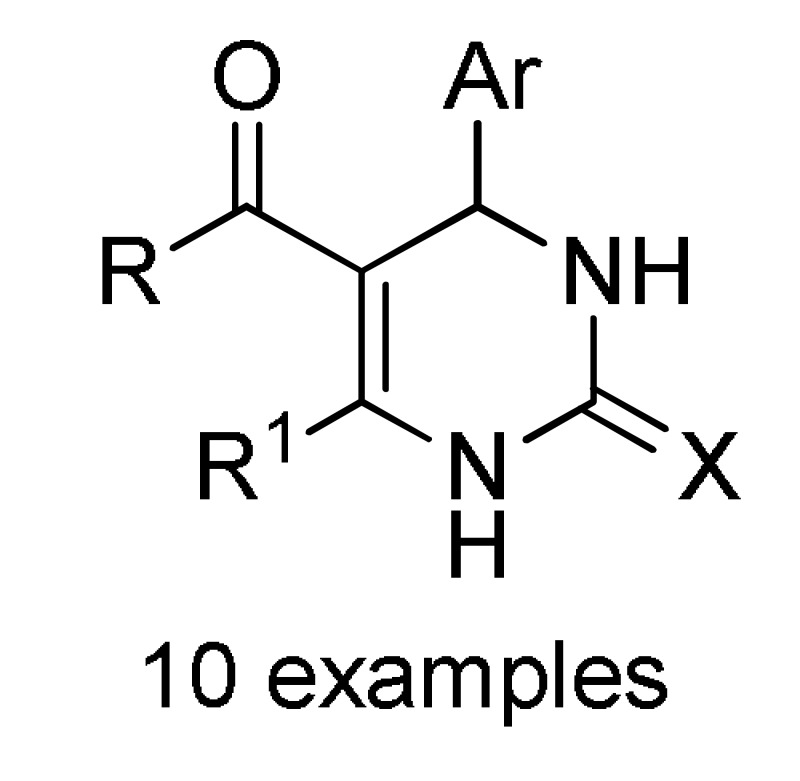
155 d |
[171] | |||
 type 27 |
 type 6 |
Bi(NO3)3.5H2O, 70 °C, solvent free |
156 e |
[172] | |
a For compounds 152: X = O, S; R = 3-HOC6H4, Ph, 4-HOC6H4, 3,4-(HO)2C6H3, 4-MeOC6H4, 3-MeOC6H4, 4-HO-3-MeOC6H3, 4-HO-3,5-(MeO)2C6H2, 4-MeSC6H4, 3,4-(OCH2O)C6H3, 4-FC6H4, Pr, cyclohexyl. b For compounds 153: X = O, S; R = OEt, PhNH, 4-MeC6H4NH, 4-MeOC6H4NH, 4-ClC6H4NH, 2-ClC6H4NH, 4-NO2C6H4NH; R1 = H, Cl. c For compounds 154: X = O, S; R = OEt, Me; R1 = Me; R2 = Ph, 4-ClC6H4, 3-HOC6H4, 2-HOC6H4, 3-NO2C6H4, 2-NO2C6H4, 4-HO-3-MeOC6H3, H, Me, 2-furyl, 3,4-(OCH2O)C6H3. d For compounds 155: X = O, S; R = OEt; R1 = Me; Ar = Ph, 3-HOC6H4, 3-NO2C6H4, 4-HO-3-MeOC6H3, 3,4-(OCH2O)C6H3. e For compounds 156: R = OMe, OEt, Me; Ar = Ph, 4-MeOC6H4, 4-MeC6H4, 4-NO2C6H4, 2,4-(Cl)2C6H3, 2,3-(Cl)2C6H3, 2,3-(F)2C6H3, 2-furyl.
Yadlapalli et al., reported the synthesis of a series of dihydropyrimidine derivatives 153 in 46–93% yield via a Biginelli reaction, as described in Table 1. The obtained compounds were evaluated for their in vitro anticancer activity against MCF-7 human breast cancer (HBC) cell line using sulforhodamine B (SRB) assay, but also, for their antitubercular activity against Mycobacterium tuberculosis (MTB) H37Rv using the Microplate Alamar Blue Assay (MABA). Interestingly, compounds 153p (X = S, R = OEt, R1 = Cl) and 153t (X = S, R = 4-MeC6H4NH, R1 = Cl) exhibited 70.6% and 63.7% of HBC cell growth inhibition, respectively, at 10 μM concentration. Compound 153p was also found to be the most potent in the series against MTB H37Rv with MIC value of 0.125 μM [169].
The synthesis, characterization, and application of a reusable ion-tagged iron catalyst was described. The catalyst was employed in the Biginelli reaction with impressive performance. High yields (42–99%) of DHPMs 154 were achieved when the reaction was carried out in imidazolium-based ionic liquids (ILs) (BMI·PF6, BMI·NTf2, and BMI·BF4) (Table 1), thus showing that the IL effects play a role in the reaction. Further, the cytotoxicity of the obtained compounds 154 was evaluated against MCF-7 cancer cell linages with encouraging results of some derivatives, which were virtually non-toxic against healthy cell linages (fibroblasts) [170].
On the other hand, two coordination polymers (CPs) were synthesized, characterized and successfully applied as robust heterogeneous catalysts for the Biginelli multicomponent reaction to obtain 3,4-dihydropyrimidin-2(1H)-one/thione (DHPMs) derivatives 155 in 80–99% yield (Table 1). The reaction was initially developed using both CPs and the Zn-based material showed much better catalytic activity. After the reaction optimization under batch conditions, a continuous flow protocol was developed and applied with impressive results. The mechanism of the transformation was also investigated by electrospray (tandem) mass spectrometry (ESI-MS(/MS)) analyses. Nine of the obtained DHPMs 155 had their antitumoral activities evaluated against MCF-7 (human breast cancer cells), A549 (human alveolar basal epithelial cells) and Caco-2 (human epithelial colorectal cells) cancer cell linages. Fibroblasts (healthy cells) were not affected by the tested DHPMs showing an excellent selectivity for tumor cells. Three DHPMs returned impressive results, being capable of inhibiting tumor cell proliferation in 72 h [171].
An additional series of DHPMs 156 was synthesized in 32–93% yield through a bismuth(III) catalyzed Biginelli reaction, Table 1, and their in vitro antiproliferative activity was evaluated in different human cell lines. A quantitative structure-activity relationship (QSAR) analysis was performed using Bayesian regularized artificial neural networks to model the relationships between in silico molecular descriptors and the observed antiproliferative activity of molecules across the tested cell lines. Among the compounds prepared, the molecules containing chloro atoms in their structure demonstrated a relevant potency and a selective antiproliferative activity against a Hepatic cancer cell line (HepaRG) without exhibiting noticeable cytotoxicity in normal dermal cells (NHDF). In prostatic (LNCaP), colon (Caco-2) and Breast (T47D and MCF-7) cancer cell lines generally compounds 156 did not exhibit relevant cytoxicity. A statistically valid QSAR model was obtained (internal validation Q2 = 0.663, RMSECV = 0.071, 10-fold cross-validation procedure, and external validation R2pred = 0.740, RMSE = 0.077), which allowed the analysis of the involved relationships between molecular descriptors and the reliable prediction of the antiproliferative activity for hypothetical related compounds in the studied cell lines. Flow cytometry analysis showed that in HepaRG and MCF-7 cell lines, compound 156p (R = MeO, Ar = 2,4-diCl2C6H3) did not decrease cell viability but, led to an accumulation of cells in G0/G1 phase of the cell cycle [172].
3.13.10. Dihydropyridine-Based Anticancer Heterocyclic Systems
Dihydropyridine derivatives (DHPs) are recognized because of their potential antioxidant, antituberculosis, analgesic, antimicrobial, and antitumor activity [173,174]. Particularly, there is much interest in the anticancer activity of these compounds owing to different types of biological targets they might interfere with for this effect to occur (e.g., PDE3, PIM1 Kinase, and Survivin protein) [175].
In this rigard, two series of 2-oxo-1,2-dihydropyridine-3-carbonitriles 157 and their isosteric 2-imino-1,2-dihydropyridine-3-carbonitriles 158 were synthesized in 62–92% and 60–85%, respectively, through one-pot four-component reaction of the appropriate acetophenones 16, aldehydes 1, and ammonium acetate with ethyl cyanoacetate (38) or malononitrile (12), respectively (Table 2). The synthesized compounds were evaluated for their tumor cell growth inhibitory activity against the human HT-29 colon tumor cell line, as well as their PDE3 inhibitory activity. Compound 157p (Ar1 = 3-thienyl, Ar = 2-EtOC6H4) showed tumor cell growth inhibitory activity with an IC50 value of 1.25 µM. Meanwhile, compound 158m (Ar1 = 3-thienyl, Ar = 4-EtOC6H4) showed inhibitory effect upon PDE3 using cAMP or cGMP as substrate. No correlation was found between PDE3 inhibition and the tumor cell growth inhibitory activity. Docking compound 157p to other possible molecular targets showed the potential to bind PIM1 Kinase [176].
On the other hand, a series of symmetrical DHPs 159 and 160 were synthesized in 87–95% and 38–55% yield, respectively, through a rapid, four-component MWI-based protocol (Table 2). Compounds 159/160 were evaluated for their tumor cell cytotoxicity against HL-60 tumor cells. A 3D-QSAR study using CoMFA and CoMSIA was carried out to decipher the factors governing MDR reversing ability in cancer. The resulting contour maps derived by the best 3D-QSAR models provided a good insight into the molecular features relevant to the biological activity in this series of analogs. 3D contour maps as a result of 3D-QSAR were utilized to identify some novel features that can be incorporated into the DHP framework to enhance the activity [177].
A sequence of DHP analogues 161 was also synthesized in 79–93% yield by Kumari et al., through a tetracomponent green synthetic method mediated by Montmorillonite-K10 (Table 2). Besides, promoter reusability, easy handling of the chemical reagent, simple reaction process, time minimization, ethanol–water solvent compatibility, and cost reduction reagent were key tools for this fruitful path. In addition, all compounds 161 were evaluated for their cytotoxic activities against three human cancer cell lines and mouse melanoma and figured out the most active compounds. Thus, these examinations recommended that DHPs and their derivatives are motivating moieties for the discovery of new anticancer drugs [178].
3.13.11. Fused Dihydroquinoline-Based Anticancer Heterocyclic Systems
Dihydroquinolines (DHQs) and their fused analogues are heterocyclic scaffolds that are ubiquitous in natural products, therapeutics, fluorophores and dyes [179]. They are structures of great versatility, and their physical and chemical properties can be finely tuned using synthetic chemistry [180].
Thus, Shi et al., reported the synthesis of a series of 4-aza-podophyllotoxin analogs 162 in 70–82% yield, containing the DHQ unit in their structures. Products 162 were obtained via a three-component reaction of tetronic acid type 27, aldehydes 1 and 2-methylbenzo[d]thiazol-5-amine 2 under MWI (Table 3). The method not only provided a valuable tool in design and synthesis of the fused systems 162 but also had the advantages of atom-economy, environmental-friendliness, good yields and operational simplicity. Additionally, the preliminary evaluation on the cytotoxic activity of this type of compounds resulted in the finding of several structures with potent and efficacious cytotoxicity to three carcinoma cell lines M14, MCF7 and SW1116 [145].
Table 3.
Multicomponent synthesis of fused dihydroquinolines 162–165 for evaluation of their potential anticancer properties.
| Reagents | Conditions | Product | Reference | ||
|---|---|---|---|---|---|
27 |
ArCHO 1 |
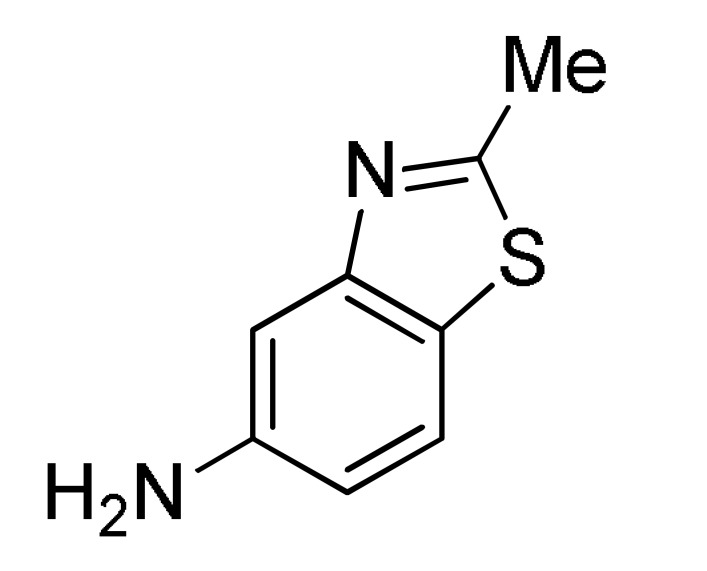
2 |
AcOH, MWI, 120 °C |
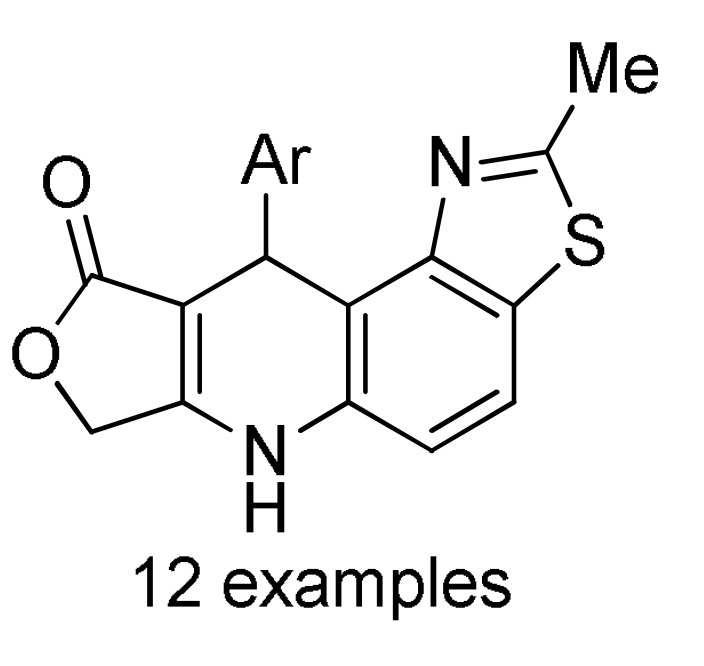
162 a |
[145] |
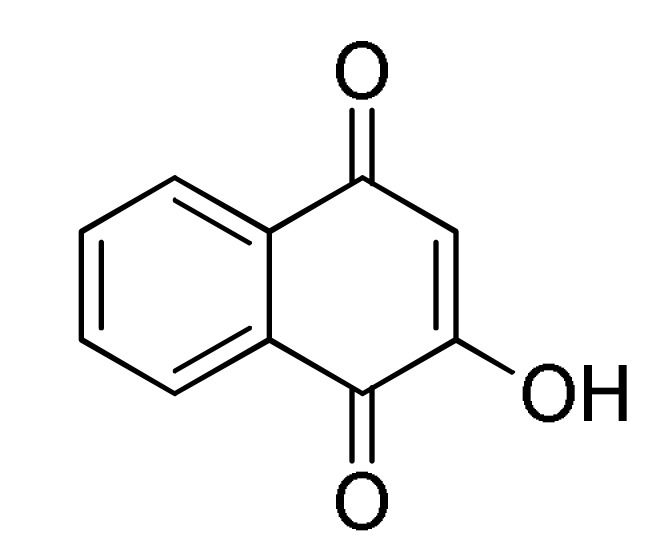
94 |
RCHO 1 |
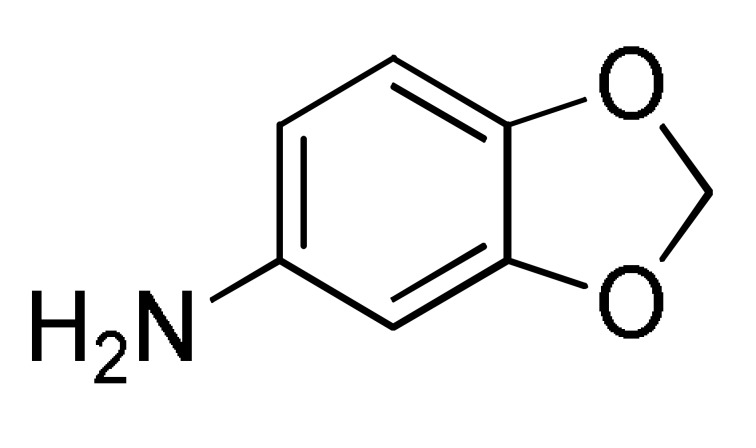
2 |
L-Proline, EtOH, reflux |
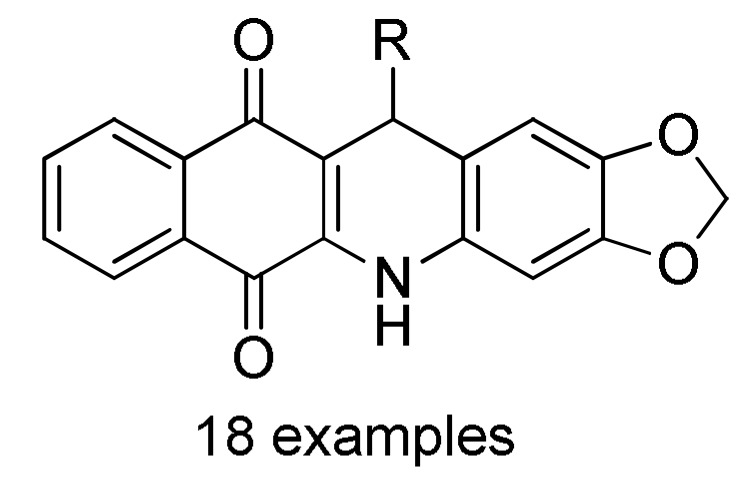
163 b |
[181] |
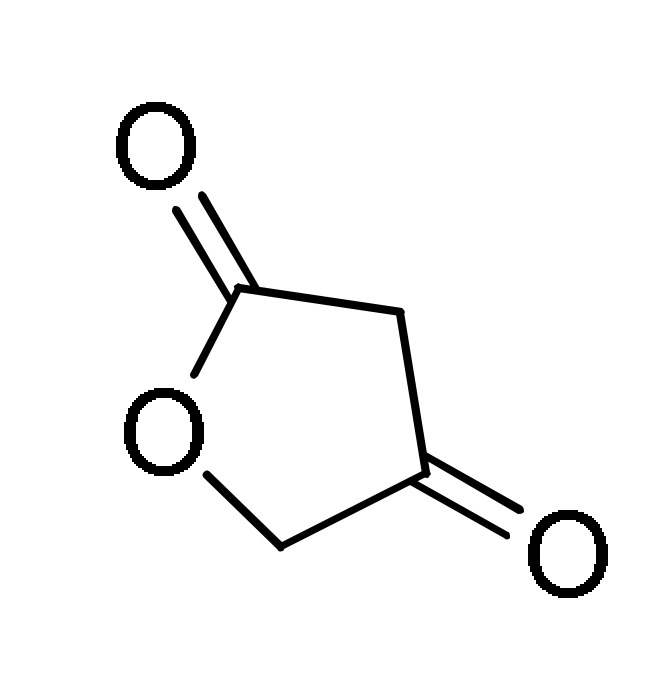
27 |
ArCHO 1 |
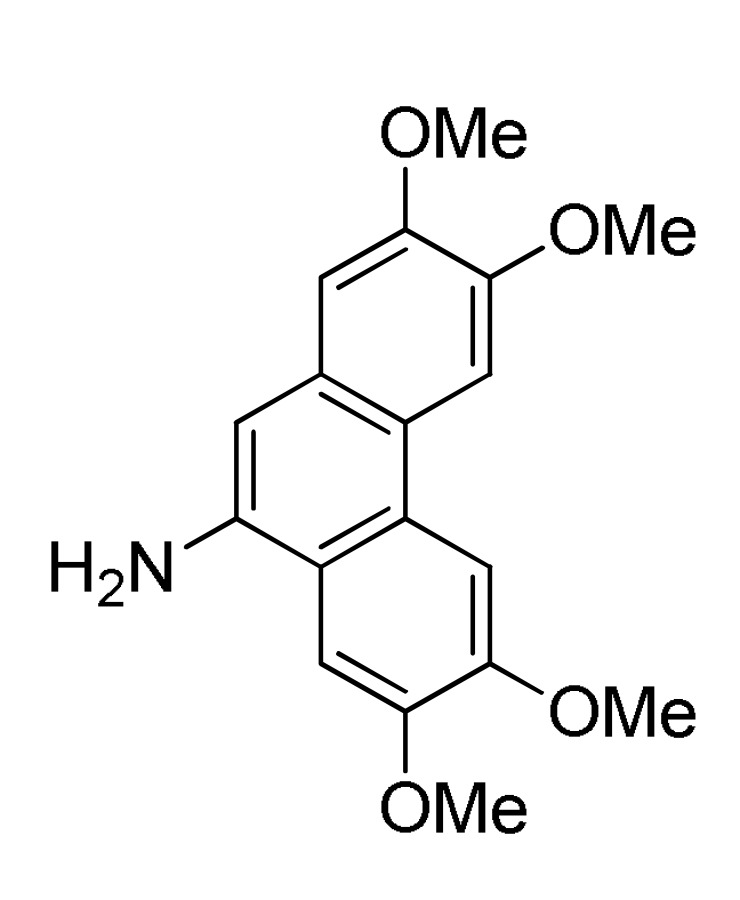
2 |
EtOH, reflux |
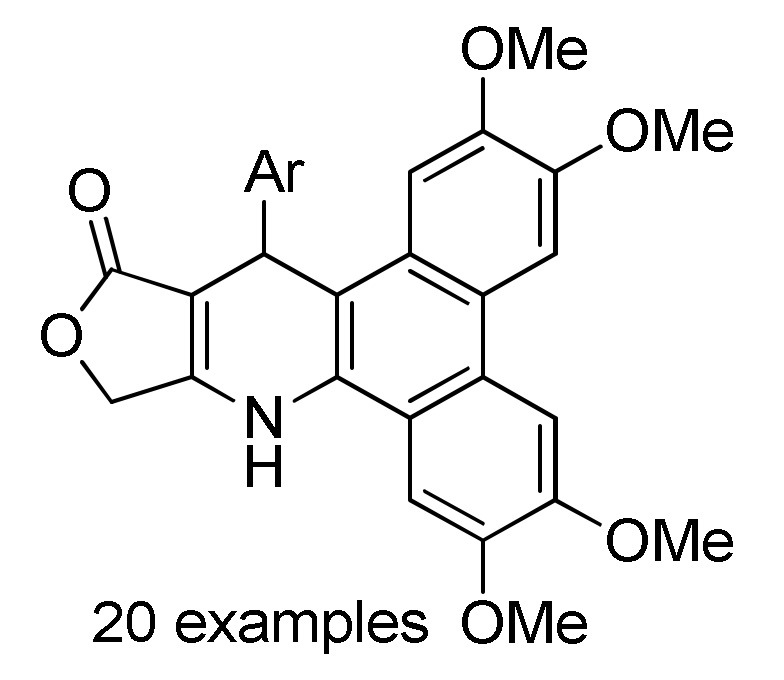
164 c |
[182] |
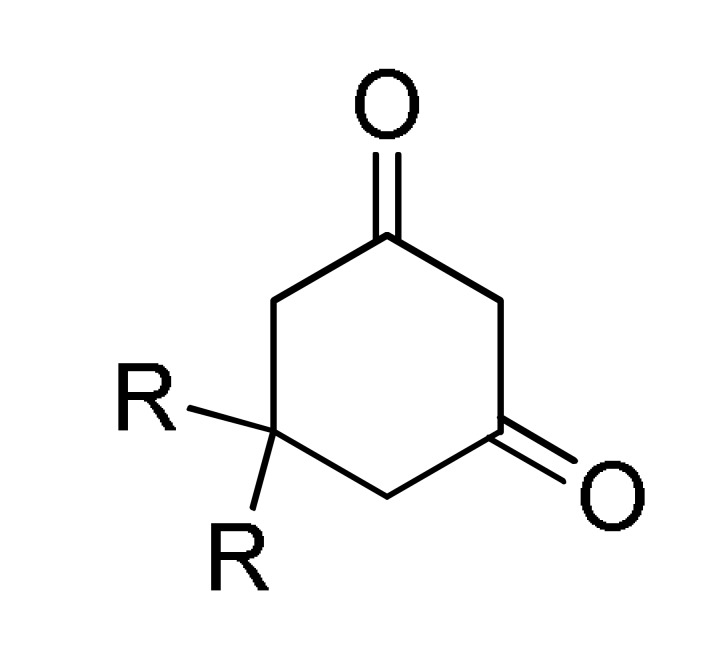
10a |
EtOH, MWI, 150 °C |
165 d |
[183] | ||
a For compounds 162: Ar = 4-FC6H4, 4-ClC6H4, 4-BrC6H4, 3-NO2C6H4, 4-NO2C6H4, Ph, 4-MeC6H4, 4-MeOC6H4, 2,4-(Cl)2C6H3, 4-HO-3-NO2C6H3, 3,4,5-(MeO)3C6H2, 2-thienyl. b For compounds 163: R = Ph, 4-MeC6H4, 4-MeOC6H4, 3-MeOC6H4, 4-ClC6H4, 2-ClC6H4, 4-FC6H4, 2-FC6H4, 3-NO2C6H4, 4-NO2C6H4, 3,4-(Cl)2C6H3, 2,4-(Cl)2C6H3, 2,5-(MeO)2C6H3, 3,5-(MeO)2C6H3, 3,4,5-(MeO)3C6H2, 2-furyl, 2-thienyl, Me. c For compounds 164: Ar = 3,4-(MeO)2C6H3, 3,4,5-(MeO)3C6H2, 2,4-(F)2C6H3, 3,4-(HO)2C6H3, 4-ClC6H4, 4-HO-3,5-(MeO)2C6H2, 4-CNC6H4, 3-BrC6H4, 4-FC6H4, 4-MeOC6H4, 3-CF3C6H4, 4-iPrC6H4, 4-MeC6H4, Ph, 2-naphthyl, 3-MeOC6H4, 2-thienyl, 3-HOC6H4, 2-FC6H4, 4-BrC6H4. d For compounds 165: R = H, Me; Ar = 4-ClC6H4, 3,4-(MeO)2C6H3, 4-CF3C6H4, 4-iPrC6H4, 3,4,5-(MeO)3C6H2, 4-BrC6H4, 4-HO-3-MeOC6H3, 4-HO-3,5-(MeO)2C6H2, 3,4-(OCH2O)C6H3, 4-MeC6H4, 4-MeOC6H4, 3-CF3C6H4, 1-naphthyl, 2-thienyl, 5-Me-2-thienyl.
Another series of para-naphthoquinone 163 embodied 4-aza-podophyllotoxin hybrids, designed via molecular hybridization approach, were synthesized in 7–99% yield using a one-pot three-component condensation of 2-hydroxy-1,4-naphthoquinone (94), aldehydes 1 and 3,4-methylene-dioxyaniline (2) in the presence of L-proline as catalyst (Table 3). The synthetic derivatives 163 were evaluated for their antitumor activity on human hepatoma cells (HepG2) and Henrietta Lacks strain of cancer cells (Hela). Among the eighteen compounds screened, 163o (R = 3,4,5-triMeOC6H2) has pronounced activity. The results demonstrated potential importance of molecular hybridization in the development of 163o as potential antitumor agent [181].
An alternative route for the synthesis of a series of 4-aza-2,3-dihydropyridophenanthrene derivatives 164 were obtained in 74–90% yield via a three-component reaction of tetronic acid type 27, substituted aldehydes 1 and 2,3,6,7-tetramethoxy phenanthrene amine 2 (Table 3). These compounds were evaluated for their cytotoxic potential against human lung (A549), prostate (PC-3 and DU145), breast (MDA-MB-231 and 4T1), gastric (HGC-27), colon (Caco-2) and cervical (HeLa) cancer cell lines. Compound 164l (Ar = 4-iPrC6H4) showed significant anticancer profile against DU145 cell line with an IC50 value of 2.6 μM.
Disruption of F-actin cytoskeleton structure and cell migration inhibition in DU145 cells indicate that the tumor progression and metastasis are affected by compound 164l. Cell cycle analysis revealed that it arrests the cells in G2/M phase. Acridine orange/ethidium bromide (AO/EB) staining, Hoechst staining and annexin-V binding assays showed that cell proliferation is inhibited through induction of apoptosis [182].
Additionally, an expeditious microwave-assisted one-pot three-component synthesis of new cytotoxic phenanthrene fused-tetrahydrodibenzo-acridinones 165 was successfully accomplished in 89–95% yield. This protocol offers wide substrate scope, catalyst-free synthesis, atom-economy, simple recrystallization, high yields, and ethanol was used as green solvent (Table 3). The obtained compounds 165 were tested for their in vitro cytotoxicity against cervical (HeLa), prostate (PC-3), fibrosarcoma (HT-1080), ovarian (SKOV-3) cancer cells, and were safer to normal (Hek-293T) kidney cell line. All the compounds displayed significant cytotoxicity profile, among them 165m (R = H, Ar = 3-CF3C6H4) being the most potent compound with an IC50 0.24 μM against SKOV-3 ovarian cancer cells. Flow cytometry analysis revealed that cells were blocked at the G2/M phase of the cell cycle. The effect of 165m on F-actin polymerisation was also studied. Hoechst staining showed the decreased number of viable cells and indicated apoptosis progression. Compound 165m caused the collapse of mitochondrial membrane potential as observed via JC-1 staining and also enhanced the generation of reactive oxygen species. The increase of caspase-3 activation by 3.7 folds supported the strong apoptosis induction. In addition, an in vitro 3D-spheroid progression assay was performed with 165m that significantly suppressed the tumor cells [183].
3.13.12. Purine-Like Pyrrolo[2,3-d]Pyrimidine Systems of Anticancer Interest
Several purine-derivatives have been considered as important and effective drugs used in cancer chemotherapy, for immunosuppression in kidney or heart transplantation and autoimmune diseases [184,185]. Various pharmacological effects of such compounds including antiviral, antibacterial, antitumor, and antifungal activity were developed for treatment of patients suffering different illness [186]. For this reason, many thiopurine derivatives and analogs have been synthesized for evaluation of their biological activities and reduced toxicity.
In that direction a synthetic approach to access the marine alkaloid rigidins and over forty synthetic analogues based on the 7-deazaadenine (166), 7-deazapurine (167) and 7-deaza-hypoxanthine skeletons 168 and 169 was developed (Table 4). Analogues based on the 7-deaza-hypoxanthine skeleton 169 exhibited nanomolar potencies against cell lines representing cancers with dismal prognoses, tumor metastases and multidrug resistant cells. Studies aimed at elucidating the modes of action of compounds 169 in cancer cells revealed that they inhibited in vitro tubulin polymerization and disorganized microtubules in live HeLa cells. Experiments evaluating the effects of compounds 169 on the binding of [3H]colchicine to tubulin identified the colchicine site on tubulin as the most likely target for these compounds in cancer cells [187].
Table 4.
Multicomponent approaches for the synthesis of 7-deazaadenine (166) 7-deazapurine (167) and 7-deazahypoxanthine skeletons 168 and 169 of potential antitumor activity.
| Reagents | Conditions | Product | Reference | |||
|---|---|---|---|---|---|---|
| ArCHO 1 |
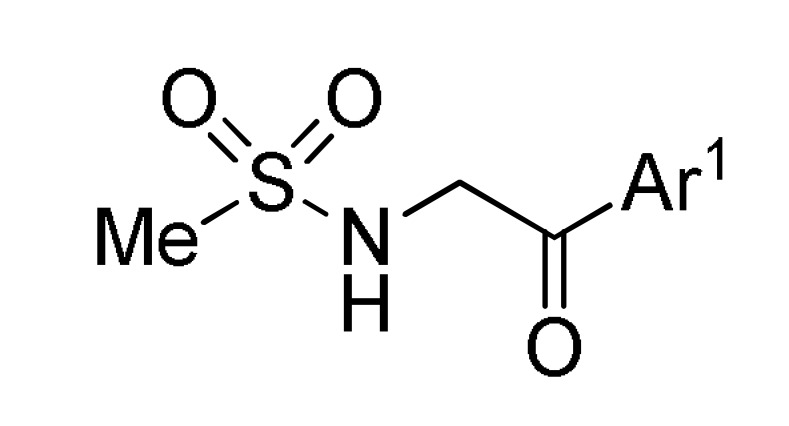
119 |
12 |
HCONH2 | K2CO3, 90 °C → 150 °C |
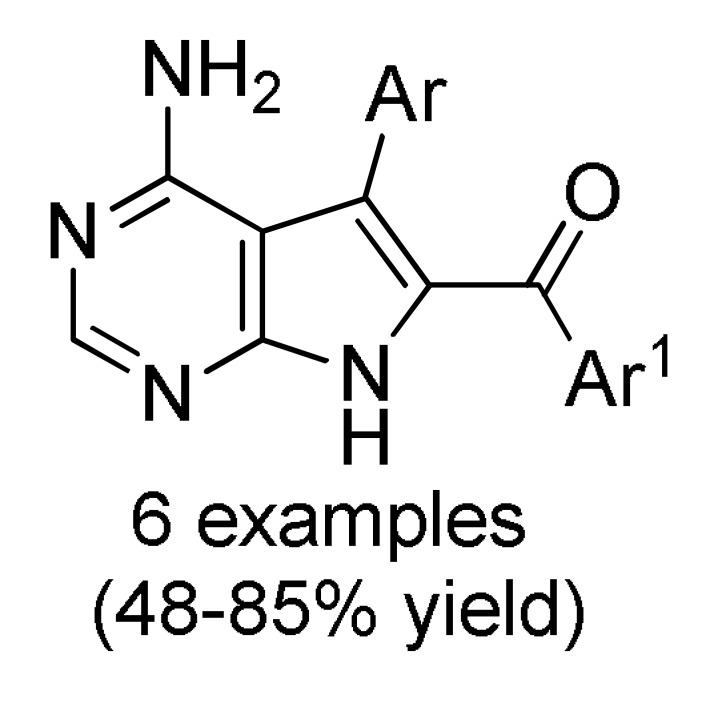
166 a |
[187] |
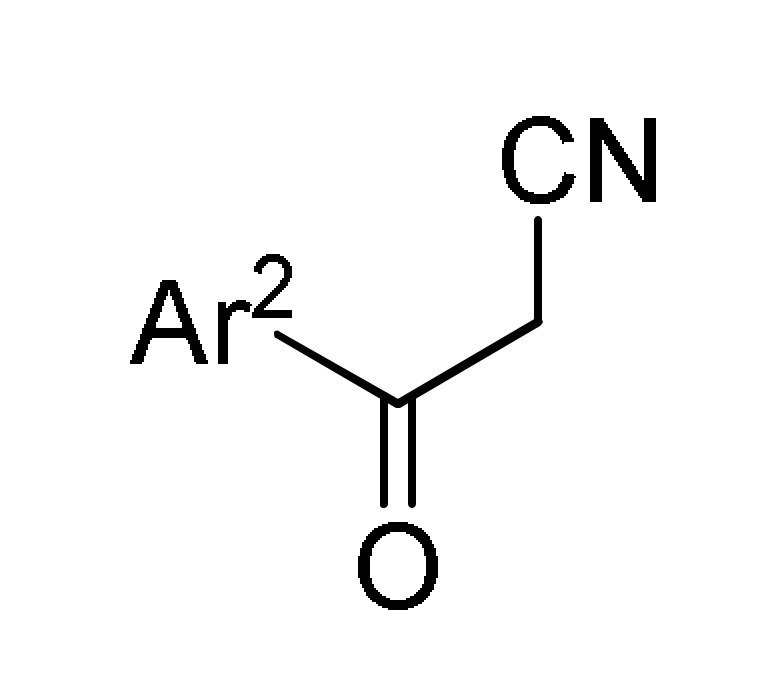
38 |
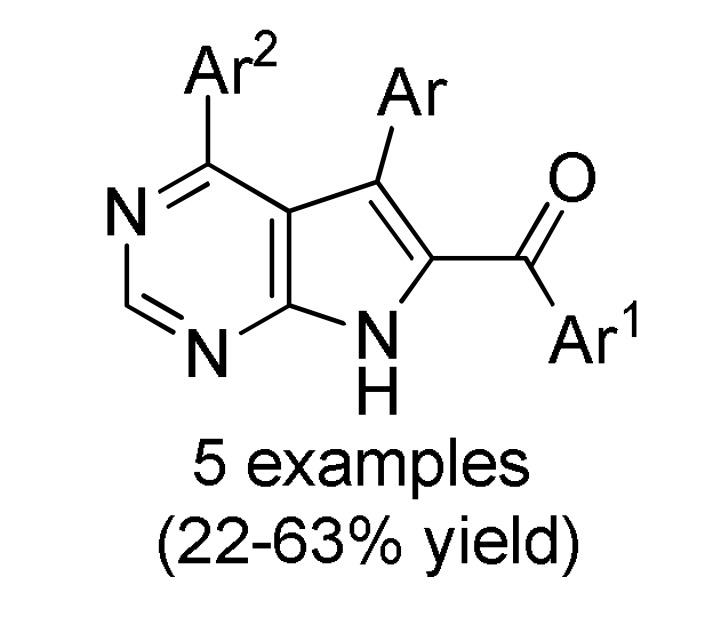
167 b |
|||||
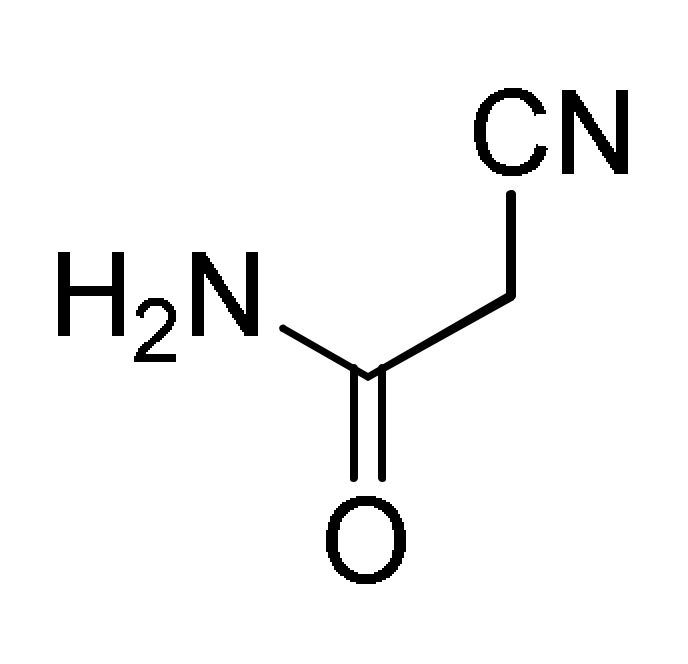
38 |
HC(OEt)3 | EtOH, K2CO3, 90 °C → 150 °C |
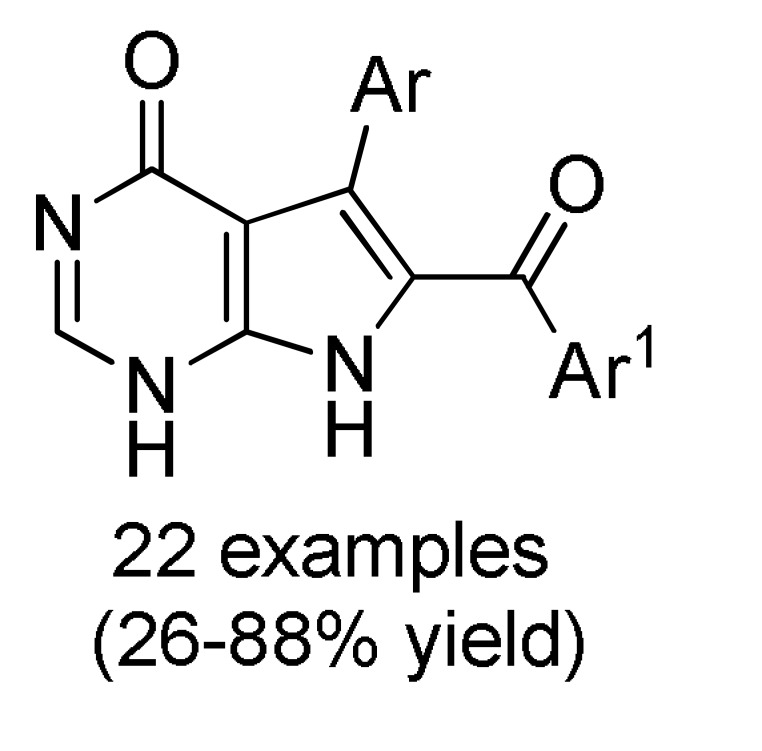
168 c |
|||
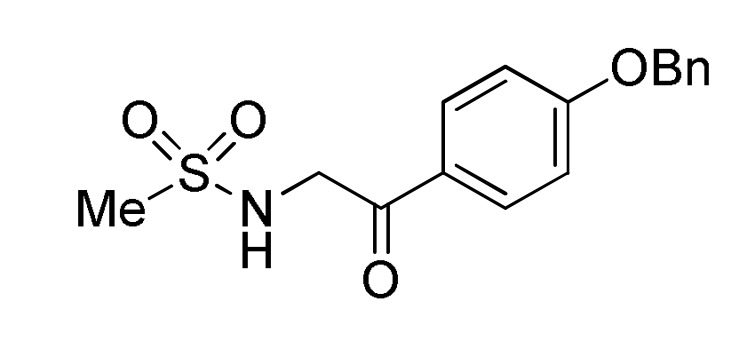
119 |
HCO2H | EtOH, K2CO3, 90 °C → 100 °C |
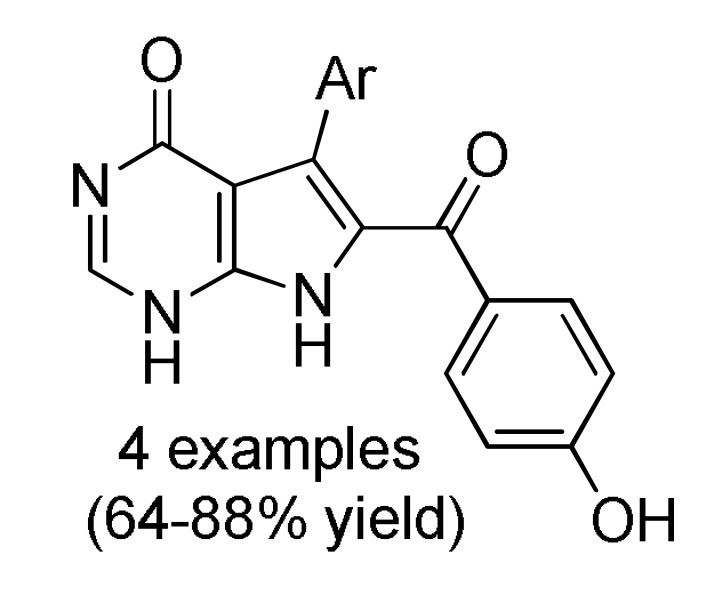
169 d |
|||
a For compounds 166: Ar = 3,5-(Br)2C6H3, 5-bromo-3-Py, Ph, 2-thienyl, 3-CNC6H4, 2,6-(Cl)2C6H3; Ar1 = Ph, 4-BnOC6H4. b For compounds 167: Ar = 4-MeOC6H4, 3,5-(Br)2C6H3, 3-Py; Ar1 = Ph, 4-MeOC6H4; Ar2 = Ph, 4-MeOC6H4, 4-MeC6H4. c For compounds 168: Ar = Ph, 3-IC6H4, 3-BrC6H4, 3-ClC6H4, 2,6-(Cl)2C6H3, 3,5-(Br)2C6H3, 3-HOC6H4, 3-CNC6H4, 3-FC6H4, 6-Br-2-Py, 5-Br-3-Py, 3-Py, 3,4,5-(MeO)3C6H2, 3,4-(MeO)2-5-IC6H2, 5-Br-3,4-(MeO)2C6H2, 4-Br-2-thienyl; Ar1 = Ph, 4-MeOC6H4, 4-FC6H4, 4-BnOC6H4, 4-BrC6H4. d For compounds 169: Ar = Ph, 3,5-(Br)2C6H3, 4-HOC6H4, 4-HO-3-MeOC6H3.
3.13.13. Coumarin Derivatives
Coumarins are one of the largest classes of naturally occurring compounds and are an essential component in the pharmaceutical, cosmetics and perfumes industry. Moreover, coumarins are considered as a privileged scaffold in the design of compounds with several biological targets. Coumarin derivatives displayed a wide range of pharmacological activities like anti-cancer [188], among others [189,190,191,192,193].
The coumarin nucleus has been fused with different classes of heterocyclic compounds with the aim of increasing the biological activity, especially the anticancer activity. For example, the 7,12-dihydro-6H-chromeno[4,3-b]quinoline 170 showed good anticancer activity against human cancer cell lines A-549 and MCF-7 with IC50 values from 0.05 μM. The derivatives 170 were synthesized via a three-component condensation of 4-hydroxycoumarin (10b) aromatic amines 2 and aldehydes 1 in water, catalyzed by sulfonic acid functionalized ionic liquid L-2-(hydroxymethyl)-1-(4-sulfobutyl)pyrrolidinium hydrogen sulfate ([HYSBPI]∙HSO4) (Scheme 74) [194]. Furthermore, the hibrids 171 and 13/14 containing dihydropyrano[c]chromene substituted quinazolines and β-lactams showed good anti-cancer activity against breast and colon cancer cell lines (MDA-MB 231, MDA-MB 453 and SW1116). Compounds 13/14 were previously depicted in Scheme 4 [30], while 171 were obtained by a DABCO-catalyzed three-component reaction using various substituted coumarins type 10b, 4-(benzyloxy)quinazoline-2-carbaldehyde 1 and ethyl cyanoacetate (38) (Scheme 74) [195].
Scheme 74.
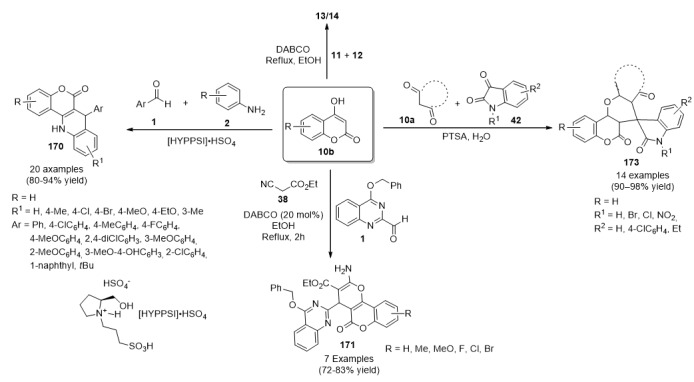
Multicomponent approaches for the synthesis of fused coumarin derivatives 13/14, 170, 171 and 173 of potential antitumor activity.
In another study, Kumar et al., developed a green, efficient and straightforward three-component procedure for the one-pot regioselective synthesis of spiro-chromeno indoline-triones 173 using cyclic diketones type 10a, isatin type 42 and 4-hydroxycoumarins type 10b in the presence of PTSA as catalyst and water as green solvent (Scheme 74). The main advantages of this protocol were short reaction time, good yield, easy work-up, high regioselectivity and reduced pollutant. Compounds 173 were effective as alkaline phosphatase (ALP) inhibitors and prostate cancer medication capabilities [196].
A simple and efficient one-pot three-component method for the synthesis of a series thiazolyl-coumarin hybrids 175 was reported by Kavitha et al. [197]. The hybrids 175 were synthesized by treating equimolar amounts of 3-(2-bromoacetyl)coumarins type 70 with various aryl/heteryl aldehydes 1 and 2-cyanothioacetamide 174 and catalyzed by L-proline in MeOH (Scheme 75). The synthesized compounds were screened for the anti-hepatocarcinoma activity. Remarkably, compounds 175 showed good to excellent anti-hepatocarcinoma activity with the support of molecular docking studies. Similarly, Vaarla et al., reported the synthesis of a series of coumarin substituted thiazolyl-3-arylpyrazole-4-carbaldehydes 176 via an efficient, one-pot multicomponent approach involving 3-(2-bromoacetyl)coumarins type 70, thiosemicarbazide and substituted acetophenones type 16 via a Vielsmeier-Haack reaction (Scheme 75).
Scheme 75.
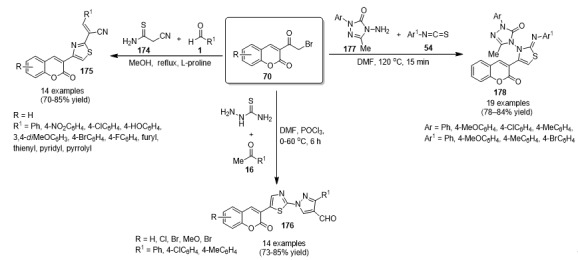
Multicomponent approaches for the synthesis of non-fused coumarin derivatives 175, 176 and 178 for evaluation of their antitumor activity.
These coumarin derivatives 176 showed moderate to appreciable cytotoxic activities against MCF-7, DU-145 and Hela cell lines. Compounds 176m (R = 6,8-diCl, R1 = 4-MeC6H4) and 176n (R = 6,8-diBr, R1 = 4-MeC6H4) exhibited significant cytotoxic activity with IC50 values of 5.75 and 6.25 μM, respectively, against Hela cell line [198]. In 2015, Shaikh et al., described a microwave-expedite green synthesis of coumarin-3-yl-thiazol-3-yl-1,2,4-triazolin-3-ones 178 by a one-pot multicomponent method from 3-(2-bromoacetyl)coumarins type 70, 1,2,4-triazolone 177 and aryl isothiocyanate type 54 at 120 0C in DMF without catalyst, Scheme 75. The obtained compounds were evaluated for their anticancer activity against MDA-MBA-231(breast cancer), A549 (lung cancer), K562 (leukaemia) and HeLa (human cervical cancer) cell lines by MTT assay. The synthesized compounds 178 showed promising inhibition with IC50 values in the range of 0.16-1.12 μM and the docking studies revealed that the compounds displayed polar and hydrophobic interactions with the active site of EGFR-TKD with amino acid residues like Met769, Lys721 and THR830 [199].
Chougala et al., designed and synthesized a series of coumarin-pyridine hybrids 179. These hibrids were prepared via a one-pot four-component condensation reaction using MWI-assisted solvent free conditions from 4-formylcoumarins 1, ammonium acetate, different ketones 16 and malononitrile (12) (Scheme 76). All synthesized compounds presented a remarkable anticancer activity against HT29, HepG2 and KB cell lines in the range of 4.22 to 27.58 μM. Compound 179f (6-methyl on coumarin and 6-coumarin on pyridine) and 179k (6-methyl on coumarin and 6-bromocoumarin on pyridine) presented prominent antitumor activity with IC50 values of 4.22 and 5.62 μM for HT-29, 5.02 and 6.94 μM for HepG2 and 6.82 and 9.64 μM for KB, respectively [200].
Scheme 76.

MW-assisted multicomponent synthesis of a series of coumarin-pyridine hybrids 179 for anticancer activity against HT29, HepG2 and KB cell lines.
In the continuous search of new coumarin derivatives for anticancer activity, Sashidhara et al., synthetized, a series of biologically active coumarin-5-imidazo[1,2-a]pyridine hybrids 180 by employing the silver(I)-catalyzed Groebke-Blackburn-Bienayme multicomponent reaction (Scheme 77). Some of the synthetized compounds were evaluated for cancer cell inhibition using MTT assay against breast cancer (MCF-7, MDAMB-231) and cervical cancer (Ishikawa) cell line. Compound 180o (R = Me, R1 = iPr, R2 = 4-Me) was the most active against MDA-MB-231 cells with IC50 of 14.12 μΜ. In addition, it was suggested that induced apoptosis in MDA-MB-231 cells was associated with mitochondrial depolarization and could induce cell cycle arrest in MDA-MB-231 cells at G0/G1 phase with a concomitant reduction in S-phase and G2/M phase [201].
Scheme 77.

Silver(I)-catalyzed three-component synthesis of a series of coumarin-5-imidazo[1,2-a]pyridine hybrids 180 for evaluation of their cancer cell growth inhibition using MTT assay.
3.13.14. Spiro Derivatives
Spiro compounds are considered privileged structures and often show interesting biological activity. They are found in a number of natural and synthetic compounds exhibiting wide range of activities against a variety of disease areas, such as anticancer [202], among others [203,204,205,206,207]. Multicomponent reaction (MCR) is a powerful tool for get new spiro-derivates with potential biological activity. In this sense, Arun et al., synthesized a series of dispirooxindole-pyrrolidine derivatives 182 and 184 through a one-pot tandem/domino approach via a 1,3-dipolar cycloaddition reaction. This reaction was performed by heating an equimolar mixture of substituted isatins 42, sarcosine (41a), 3-(1H-indol-3-yl)-3-oxo-2-(2-oxoindolin-3-ylidene)propanenitrile (181) and 3-(1H-imidazol-2-yl)-2-(1H-indole-3-carbonyl)acrylonitrile (183), respectively. The reaction proceeded in ethanol under reflux for 90–120 min, affording the spirooxindole derivatives 182/184 in 80–92% yields (Scheme 78). These compounds were evaluated against A549 human lung adenocarcinoma cancer cell lines, where the spirooxindoles 182/184 exhibited very good anticancer activity with percentage of inhibition in the range of 60.22–67.67%. Compound 182i (R = CH≡CCH2-, R1 = H) showed IC50 value of 50 μM [208,209]. In a similar study, it was reported a simple, mild and efficient one-pot multicomponent reaction for the synthesis of pyrazolopyridine- and benzodiazepine-based spirooxindoles 185 and 186, respectively. This one-pot protocol proceeded via a three-component reaction of isatins type 42, tetronic acid 27, 5-phenyl-1H-pyrazol-3-amine type 2 (for spirooxindoles 185) and o-phenylenediamines type 111 (for spirooxindoles 186), using sulfamic acid (H2NSO3H) as a green catalyst and water as solvent (Scheme 78). Compounds 185/186 were evaluated for their in vitro cytotoxic activities against a selected human cancer cell lines, such as Breast cancer (MCF-7 and MDA-MB-231), prostate cancer (DU-145), cervical cancer (HeLa) and lung cancer (A549). The spirooxindoles 185/186 showed moderate to very good cytotoxicity with IC50 values in the range of 0.35–75.20 μM. Compounds 185o (R = pyperonyl, R1 = 5-Cl) and 186h (R = R1 = R2 = H) exhibited significant cytotoxicity with IC50 0.35 μM and 1.14 μM values against MDA-MB-231 and DU-145 cell lines, respectively [210,211].
Scheme 78.
Multicomponent approaches for the synthesis of spirooxindole derivatives 182, 184, 185, 186, and 188 for evaluation of their antitumor activity.
Hui et al., published a direct route to prepare biologically relevant spirooxindole-pyrrolidine, pyrrolizidines and pyrrolothiazoles 188 by a one-pot, multicomponent 1,3-dipolar cycloaddition reaction via azomethine ylides from isatin type 42 and an amino acid derivative 114 (Scheme 78). The advantages of this protocol included, high yields, simple work-up procedure and regio- and diastereo-selectivities. Compounds 188 were evaluated for their antiproliferative activity against various cancer cell lines. Although all compounds showed good cytotoxicity activity against the tested cell lines, studies showed that compound 188j (R = R1 = H, R3 = Ph) displayed the highest inhibitory activity against HCT116 (colon cancer) and HepG2 (hepatocellular carcinoma) at a concentration of 10 μM [212].
Similarly, a class of diastereoselective spiropyrrolidine-oxindole derivatives 189, 191 and 192 were synthesized from isatins type 42, α,β-unsaturated ketones 26 and 190 and amino acids 41a and 112 in a one-pot multicomponent reaction via 1,3-dipolar cycloaddition (Scheme 79). Most of the spiro-oxindoles 189, 191 and 192 exhibited potent antitumor properties against different cancer cell lines. For example, compounds 192 inhibited the growth of MCF-7, T47D and MDA-MB-231 cells with an IC50 less than 20.0 μM. Compound 191 (R2 = Me, R3 = 4-MeC6H4) showed bio-potency against the HepG2 (hepatocellular cancer) cell line, comparable to that for doxorubicin hydrochloride (standard reference), and the spiropyrrolidine-oxindole 189k (R = R1 = H, R2 = 4-FC6H4) presented a high cytotoxic activity and selectivity against colon cancer cells HCT-116 (IC50 = 7 μM, SI: 3.7), and HepG2 (IC50 = 5.5 μM, SI: 4.7) [213,214,215].
Scheme 79.
Multicomponent approaches for the synthesis of spirooxindole derivatives 189, 191, 192, 194, and 195 for evaluation of their tumor cell growth inhibitory activity.
Parthasarathy et al., reported a catalytic efficiency of Cu(OTf)2 for the synthesis of spiropyrano[3,2-b]pyran-4(8H)-ones 194 through a three-component reaction between isatins 42, nitriles 38 and kojic acid (193) (Scheme 79). The synthesized compounds were evaluated for their tumor cell growth inhibitory activity against the human lung cancer cell line (A549). Compounds exhibited moderate to good cytotoxicity, out of which, two derivatives 194e (R = Me, R1 = H, Z = CN) and 194c (R = H, R1 = Cl, Z = CN) exhibited good anticancer potency with IC50 values of 51.1 and 51.4 μM, respectively [216].
A library of spirooxindole-O-naphthoquinone-tetrazolo[1,5-a]pyrimidine hybrids 195 was designed, synthesized and evaluated as potent antitumor agents. Hybrids 195 were obtained in 31–69% yield via a one-pot three-component reaction of isatins type 42, 5-aminotetrazole type 2 and 2-hydroxy-1,4-naphthoquinone 94 in refluxing AcOH (Scheme 79). These hybrids exhibited relatively high cytotoxic activity against cancer cell line HepG2 (IC50 = 2.86–36.34 μM), while normal cell line LO2 was less sensitive to these hybrids (IC50 = 36.37–248.39 μM). Among all the tested compounds, structure 195e (R = 1-Me, R1 = 7-F) was the most active derivative with a mean IC50 value of 2.86 μM [217].
In other study, a series of spirochromenocarbazoles 197 was synthesized via a click chemistry-based one-pot, five-component reaction between N-propargyl-isatins 42, alkyl/arylalkyl halides 196, 4-hydroxycarbazole (148), malononitrile (12) and sodium azide in a mixture DMF/H2O at 70 0C using cellulose supported CuI nanoparticles (Cell-CuI NPs) as the heterogeneous catalyst, Scheme 80. The synthesized compounds were screened against a panel of six human cancer cells namely MCF-7 and MDA-MB-231 (breast cancer), HeLa (cervical cancer), PANC-1 (pancreatic cancer), A-549 (lung cancer) and THP-1 (acute monocytic leukemia). Some compounds presented excellent activity against MCF-7, MDA-MB-231 and HeLa cancer cell lines. Particularly, compound 197j (R = H, R1 = Bu) displayed the highest activity with IC50 values 4.8, 8.4 and 9.2 µM, respectively [218].
Scheme 80.

Cell-CuI NPs-catalyzed multicomponent synthesis of spirochromenocarbazoles 197 for evaluation of their antitumor activity.
In 2016, a three-component 1,3-dipolar cycloaddition reaction of pyrimidine-fused 3-alkenyloxindole 198 with azomethine ylides (thermally generated in situ from sarcosine (41a) and polyformaldehyde) for the synthesis of a series of pyrimidine-fused spiropyrrolidine oxindoles 199 was investigated by Liu et al. The products were obtained in high yields (up to 90% yield) with good diastereoselectivity (up to > 20:1) (Scheme 81). The anticancer activity was screened by MTT assays against lung cancer cells A549, human prostate cancer cells PC-3, and human leukemia cells K562 by using cisplatin as a positive control. Some spiropyrrolidine oxindoles 199 showed GI50 ranging from 8.9 µM to 23.2 µM in vitro inhibitory activity against human leukemia cells K562, and exhibited equipotent or more potent activity than the positive control cisplatin (up to 3.0 times) [219].
Scheme 81.

Three-component synthesis of pyrimidine-fused 3-alkenyloxindole 199 for evaluation of their tumor cell growth inhibitory activity.
Sudhapriya et al., reported the synthesis of spirocarbocycle derivaties 201 by a multicomponent-domino reaction that involved cyclic nucleophiles 16/10a, vinyl malononitriles 200 and different aldehydes 1 using L-proline as catalyst in methanol as solvent (Scheme 82). The reaction proceeded smoothly in good yields with good diastereoselectivities. The synthesized spirocarbocycles 201c (R = 4-MeC6H4, n = 3), 201i (R = 4-ClC6H4, n = 3) and 201h (R = thiophenyl, n = 2) showed good anticancer activity against A549 cancer cell line with IC50 values of 50, 30 and 20 μM, respectively. The docking scores showed that the spirocarbocyclic molecules 201 had good potential against the human lung cancer cells [220].
Scheme 82.

L-Proline-catalyzed three-component synthesis of spirocarbocycle derivaties 201 for evaluation of their anticancer activity.
3.13.15. Miscellaneous of Diverse Heterocyclic Systems with Potential Antitumor Activity
Pirali et al., reported the synthesis of highly functionalized structures 205 and 208 in 60–62% and 60–68% yields, respectively, as intermediates of macrocyclic peptide mimetics by exploiting a three-component and an azide–alkyne [3+2] cycloaddition reaction catalyzed by ammonium chloride (Scheme 83). The obtained compounds were screened as HDAC inhibitors allowing to identify some of these compounds with promising biological activity. Moreover, in order to rationalize the biological results, computational studies were also performed [221].
Scheme 83.

NH4Cl-Catalyzed three-component synthesis of highly functionalized oxazoles 205 and 208 for evaluation of their anticancer activity.
On the basis of structures of known topoisomerase II catalytic inhibitors and initial molecular docking studies, bicyclic N-fused aminoimidazoles 210–214 were predicted by Baviskar et al., as potential topoisomerase II inhibitors. These compounds were synthesized in high yields by a three-component reaction, as described in Scheme 84, and evaluated against human topoisomerase IIα (hTopoIIα) in decatenation, relaxation, cleavage complex, and DNA intercalation in vitro assays. Several of the obtained compounds exhibited potent inhibition of catalytic activity of hTopoIIα while not showing DNA intercalation. Molecular docking studies and molecular dynamics (MD) simulation analysis, ATPase-kinetics and ATP-dependent plasmid relaxation assay revealed the catalytic mode of inhibition of the obtained compounds plausibly by blocking the ATP-binding site. Some of the obtained compounds also showed potent anticancer activities in kidney and breast cancer cell lines, low toxicity to normal cells, relatively higher potency compared to etoposide and 5-fluorouracil in kidney cancer cell lines, and potent inhibition in cell migration. These compounds were found to exert apoptotic effect in G1/S phase [222].
Scheme 84.

ZrCl4-Catalyzed three-component synthesis of bicyclic N-fused aminoimidazoles 210–214 for evaluation of their anticancer activity.
A series of 2,3-tri- and tetrasubstituted γ-butyrolactone 217 analogous to paraconic acids were synthesized in 14–99% yield, through a one-step and straightforward three-component reaction between carbonyl compounds type 1/16, dimethyl itaconate (215) and aryl bromides 216 (Scheme 85). The in vitro cytotoxic activity of representative compounds was evaluated against a panel of human cancer cell lines (KB, HCT-116, MCF-7, HL60). While most molecules 217 exhibited a low to moderate background activity on both KB and HL60 cancer cell lines, compound 217m (R = Ph, R1 = Me, R2 = 4-OMe), showed increased antiproliferative activity against both cell lines with IC50 values in the 0.1 to 1.0 μM range. An extended evaluation indicated that this compound also inhibited PC3, SK-OV3, MCF-7R and HL60R cell growth in the same fashion [223].
Scheme 85.

Zn-Mediated three-component synthesis of polysubstituted γ-butyrolactone 217 for evaluation of their antiproliferative activity.
In a study Shi et al., achieved the design and diversity-oriented synthesis of libraries of 1,4-thiazepine derivatives 218 (88–93% yield), 219 (73–93% yield), 220 (90–94% yield) and 221 (90–93% yield) embedded with carbazole, pyrazole or isoxazole motifs via MWI-assisted three-component reactions under solvent-free condition (Scheme 86).
Scheme 86.

MWI-Assisted three-component synthesis of 1,4-thiazepine-embedded with carbazole, pyrazole or isoxazole derivatives 218–221 for evaluation of their in vitro cytotoxic activity.
This strategy led to a green and facile access to the above compounds with prominent features of high structural diversity, short reaction times, high yields and environmental friendliness. Further, the obtained compounds were subjected to the test of their in vitro cytotoxic, as well as, antioxidant activities, resulting in the finding that these compounds not only displayed significant antioxidant activity, but also exhibited remarkably selective cytotoxicity to carcinoma cell line HCT-116 [224].
Due to its antitumor interest, two series of third generation of spin-labeled podophyllotoxin analogs 223 and 224 were synthesized via isocyanide-mediated four- and three-component reactions. Both processes were inspired by Ugi (Ugi-4CR) and Passerini (P-3CR) multicomponent reactions, respectively, through the isocyanopodophyllotoxin (222) as the key starting material (Scheme 87). Subsequently, the obtained products 223 (39–84% yield) and 224 (36–85% yield) were subjected to evaluation of their cytotoxicity against four human cancer cell lines (A-549, DU-145, KB and KBvin). Most of the evaluated compounds exhibited potent cytotoxic activity against all four cell lines, mainly against the drug resistant KBvin cancer cell line. Compounds 224e (R = H, R1 = 4-MeOC6H4, R2 = 2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl-N-oxide) (IC50 = 0.60-0.75 μM) and 224h (R = H, R1 = 3,4-methylenedioxy-C6H3, R2 = 2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl-N-oxide) (IC50 = 1.12-2.03 μM) showed superior potency to etoposide (IC50 = 2.03 to >20 μM), a clinically available anticancer drug. According to these results, compounds 224e and 224h were claimed for further developments as a new generation of epipodophyllotoxin-derived antitumor clinical trial candidates [225].
Scheme 87.

Ugi- and Passerini-mediated multicomponent synthesis of spin-labeled podophyllotoxin analogs 223 and 224 for evaluation of their in vitro cytotoxicity against four human cancer cell lines.
Synthesis and structure elucidation of two series of polyacetylated fused 1,2,4-triazine derivatives 226 (68–92% yield) and 227 (65–94% yield) was established by heating the starting Schiff’s bases 225 under an excess of acetic anhydride (Scheme 88). The inhibitory effect of compounds 226 and 227 toward the CPY1A1 activity was screened to determine their potential as promising anticancer drugs. Data showed that compound 226e (R = Me, Ar = Ph, Ar1 = 4-ClC6H4) displayed the highest inhibitory effects among all tested 1,2,4-triazine derivatives. Furthermore, docking analysis showed that these compounds bind only at the interface of substrate recognition site 2 (SRS2) and (SRS6) at the outer surface of the protein, and that amino-acids ASN214, SER216 and ILE462 participate in the binding of these compounds through H-bonds [226].
Scheme 88.

Multicomponent synthesis of polyacetylated fused 1,2,4-triazine derivatives 226/227 to be screened their potential toward CPY1A1 inhibition.
A one-pot four-component approach was established for site selective synthesis of 1,3,6-trisubstituted 3,6-diunsaturated (3Z,6Z)-2,5-diketopiperazine derivatives 229 (38–60% yield) and 230 (31–64% yield), mediated by double-aldolic condensations between pyrazine 228 and two equivalents of aldehydes type 1A/1B, involving also a mono-alkylation reaction in the same reaction pot, Scheme 89. The computational studies revealed that the steric hindrances between the 2-hydrogen atoms on the aromatic rings and the carbonyl, as well as, the steric repulsions between the hydrogen atoms of the CH group in the benzylidene and the CH2 group in the N-alkylative part might be responsible for the Z/E selectivity. The obtained compounds were tested in vitro against five cancer cell lines (i.e., U937, HL60, DU145, HT29 and K562). Results reveled that compound 230h (R = Me, Ar = 3-MeOC6H4) (IC50 = 11 nM) had a close activity to the positive compound plinabulin (IC50 = 15 nM) against the cancer cell line HL60 [227].
Scheme 89.

Cs2CO3-Catalyzed multicomponent synthesis of 1,3,6-trisubstituted 3,6-diunsaturated (3Z,6Z)-2,5-diketopiperazine derivatives 229/230 to be tested in vitro against five cancer cell lines.
A one-pot three-component synthesis of some 2-(2-naphthoyl)-6,6-dimethyl-3-aryl-2,3,6,7 tetrahydrobenzofuran-4(5H)-ones 231 in 84–92% yield is described in Scheme 90.
Scheme 90.

Three-component synthesis of 2-(2-naphthoyl)-6,6-dimethyl-3-aryl-2,3,6,7 tetrahydrobenzofuran-4(5H)-ones 231 for evaluation of their potential as inhibitors of cathepsins B, H and L.
The synthesized compounds 231 were found to be potential inhibitors to cathepsins B, H and L. The extent of inhibition varied with the substitution. Among the synthesized compounds, derivative 231d (Ar = 4-NO2C6H4) was selected as most inhibitory to cathepsin H, however compound 231f (Ar = 4-FC6H4) was the best inhibitor of cathepsin B and cathepsin L. In vitro inhibition studies correlated well when tested using MTT assay on HepG2 cells, a hepatocellular carcinoma cell line. The results validated by in silico studies performed with iGemDock predicted that among the synthesized compounds, 231d experienced the highest affinity for cathepsin B and H sites, whereas 231f had the highest affinity to cathepsin L [228].
In the current research scenario, an efficient synthesis of tetrazole scaffolds 232 in 93–96% yield was developed by a single step four-component reaction. The synthesis of the target compounds 232 was undertaken by the Ugi multicomponent approach with the condensation of various aryl amines 2, TMS-N3, cyclohexyl isocyanide 18 and aromatic aldehyde containing active pharmacophore 1, under catalyst-free reaction condition at room temperature (Scheme 91). The potency of the obtained tetrazoles 232 was checked at the NIH using sixty different cell-lines with respect to nine cancer panels, among which compounds 232a (Ar = 2,5-diMeC6H3) and 232b (Ar = 4-FC6H4) displayed the higher activity against different cell lines [229].
Scheme 91.

Ugi-Mediated multicomponent synthesis of tetrazole scaffolds 232 to be evaluated by the NCI for their in vitro cytotoxicity against sixty different cancer cell lines.
A series of benzimidazole-isoquinolinone derivatives (BIDs) 233 was synthesized in 52–82% yield. The process was conducted by an Ugi four-component reaction (U-4CR) using an amine 2, carboxylic acid 114, isonitrile 18, and the methyl 2-formylbenzoate type 1 in methanol at room temperature (Scheme 92). Subsequently, the solvent was removed under a stream of nitrogen, and the intermediate crude Ugi product obtained was deprotected and cyclized in situ under MWI using 10% TFA/DCE at 150 °C. Further, the target products 233 were screened to identify novel scaffolds for Colorectal cancer CRC. Among the compounds evaluated, 233g (R = iBu, R1 = 3-Py) exhibited the most promising anti-cancer properties. Employing two CRC cell lines, SW620 and HT29, 233g was found to suppress growth and proliferation of the cell lines at a concentration of ∼20 μM. Treatment followed an increase in G2/M cell cycle arrest, which was attributed to cyclin B1 and cyclin-dependent kinase 1 (CDK1) signaling deficiencies with simultaneous enhancement in p21 and p53 activity. In addition, mitochondrial mediated apoptosis was induced in CRC cells. Interestingly, 233g decreased phosphorylated AKT, mTOR and 4E-BP1 levels, while promoting the expression/stability of PTEN [230].
Scheme 92.

Ugi-Mediated multicomponent synthesis of benzimidazole-isoquinolinone derivatives (BIDs) 233 for screening to identify novel scaffolds for Colorectal cancer CRC.
Series of diverse thiophene, pyran, azole and azine derivatives 234–242 in 63–89% yield were synthesized from cyclopentanone type 16 as common starting material, which reacted with malononitrile and ethyl cyanoacetate (12)/(38), among other reagents (Table 5 and Table 6). The biological evaluation of the obtained compounds 234–242 was tested on three different tumor cell lines such as breast adenocarcinoma, CNS cancer, and non-small lung cancer and was compared to the inhibitory effect of doxorubicin. The results revealed that, among the heterocyclic products, furan derivatives 234e (Ar = 4-MeOC6H4, Y = CONH2, X = NH2) and 234f (Ar = 2-furyl, Y = CN, X = OH) had the highest inhibitory effect [231].
Table 5.
Multicomponent synthesis of cyclopentanone-based dihydropyran/dihydropyridine heterocyclic systems 234–238 of potential anticancer activity.
| Reagents | Conditions | Product | Reference | |||
|---|---|---|---|---|---|---|
 12/38 X = CN, CO2Et, CONH2 |
16 |
ArCHO 1 |
TEA | EtOH, reflux |
234 a |
[231] |
1 |
235 b |
|||||
| ArCHO 1 |
AcONH4 |
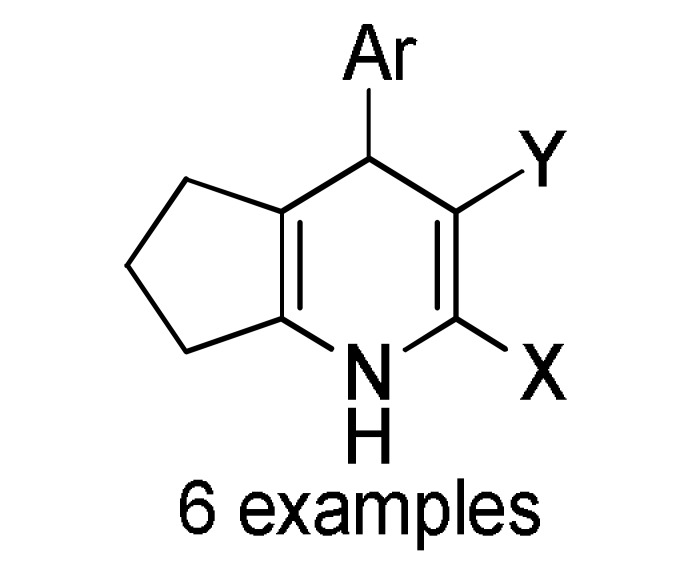
236 a |
||||
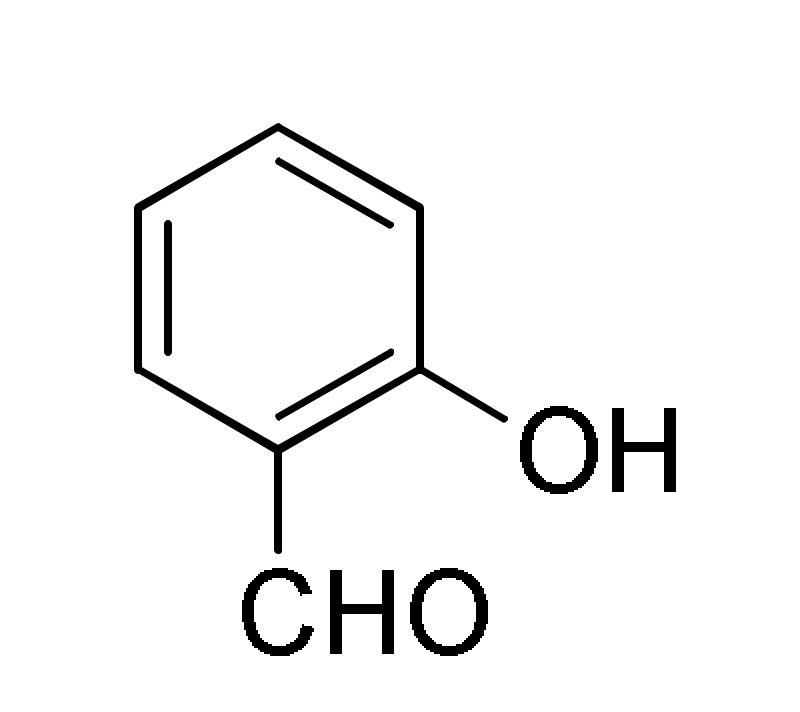
1 |
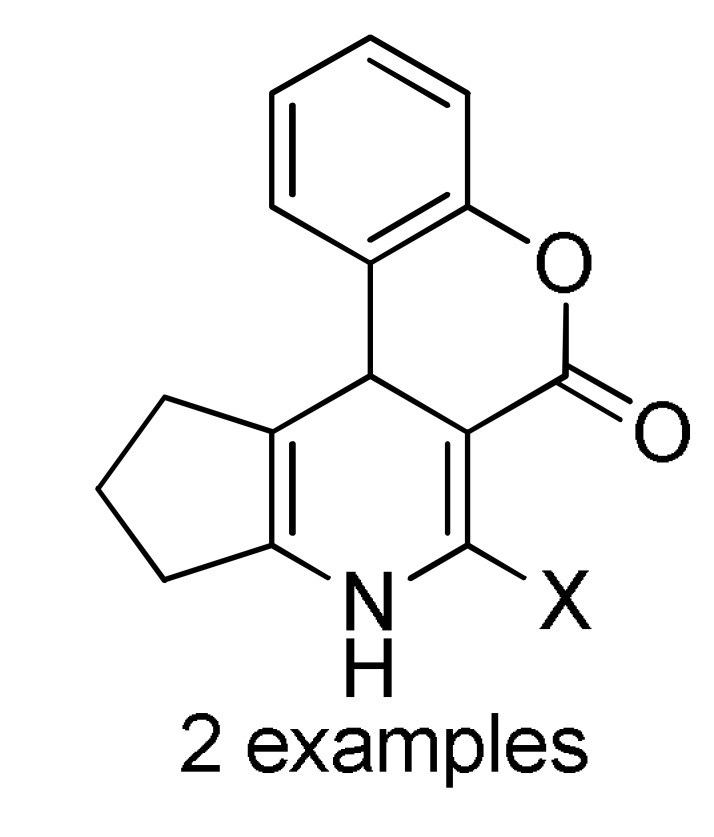
237 b |
|||||
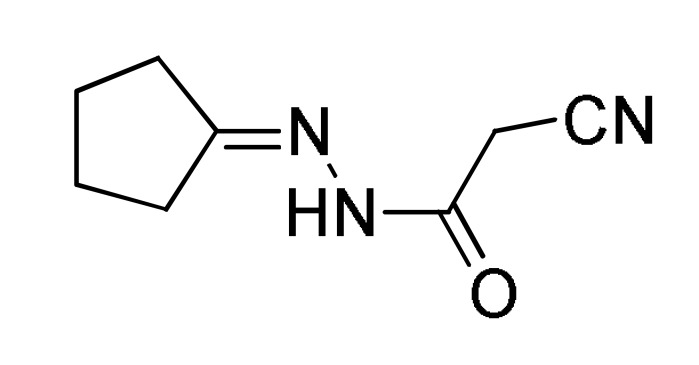
119 |
PhCHO 1 |
CH2(CN)2 12 |
----- | 1,4-dioxane, TEA, reflux |
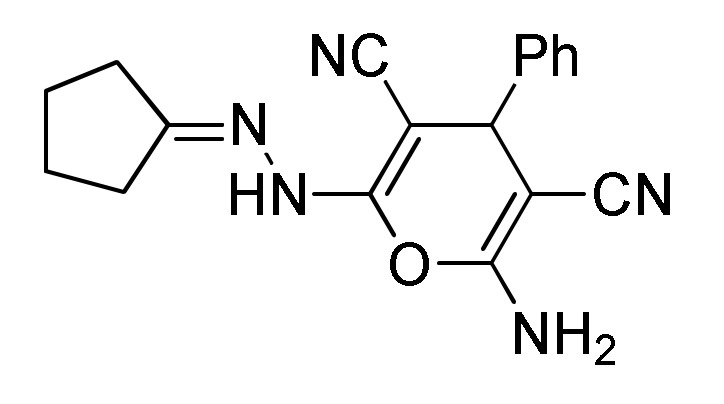
238 |
|
a For compounds 234/236: X = NH2, OH; Y = CN, CONH2; Ar = Ph, 2-furyl, 4-MeOC6H4. b For compounds 235/237: X = NH2, OH.
Table 6.
Multicomponent synthesis of cyclopentanone-based thiophene/thiazol heterocyclic systems 239–242 of potential anticancer activity.
| Reagents | Conditions | Product | Reference | ||
|---|---|---|---|---|---|
| S8 |
 type 16 |
PhNCS type 54 |
EtOH, TEA, reflux |
239 |
[231] |
 type 119 |
1,4-dioxane, TEA, reflux |
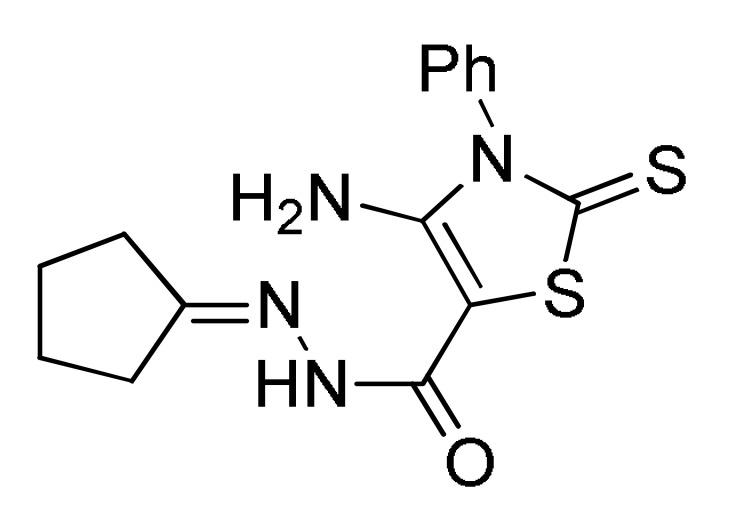
240 |
|||
| CH2(CN)2 12 |
241 | ||||
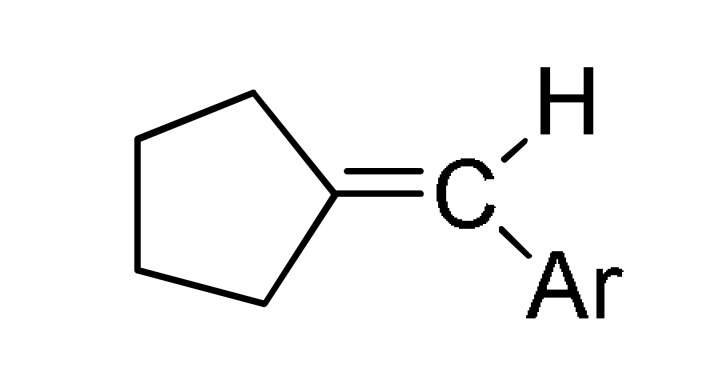 type 200 |
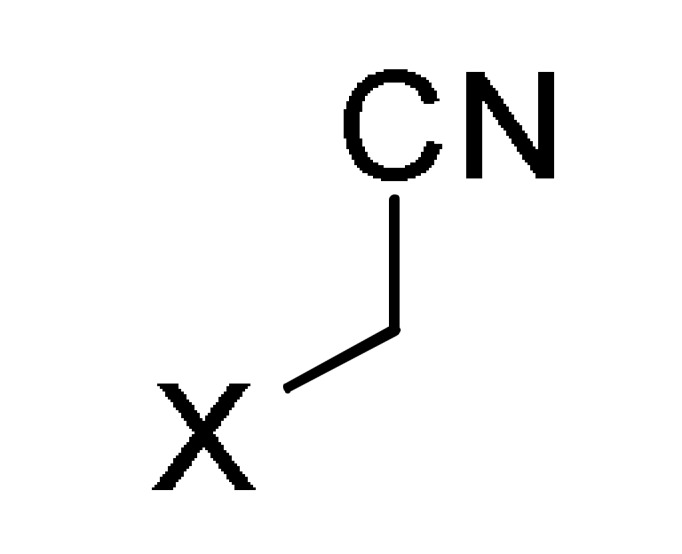 type 12/38 X = CN, CO2Et |
EtOH, TEA, reflux |
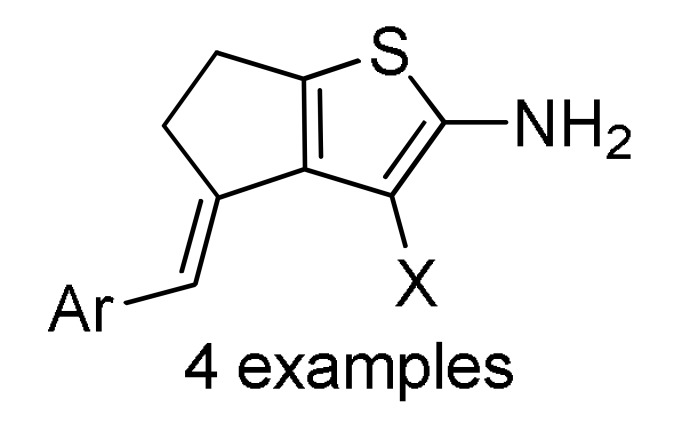
242 a |
||
a For compounds 242: X = CN, CO2Et; Ar = 4-MeOC6H4, 4-ClC6H4.
Finally, in the search for new drug-like selective G-quadruplex binders, a bioinspired design focused on the use of nucleobases as synthons in a multicomponent reaction was provided by Pelliccia et al. [232]. Thus a series of multifunctionalized imidazo[2,1-i]purine derivatives 246 (57% yield), 247 (28% yield) and 248 (22–84% yield) were synthesized via a convergent Groebke-Blackburn-Bienaymé three-component reaction (GBB-3CR) of amino-aza-heterocycles 243–245, benzaldehydes 1, and isocyanide 18, followed by a SN2 with aminoalkyl chlorides type 216 (Scheme 93).
Scheme 93.

Groebke-Blackburn-Bienaymé-mediated three-component synthesis of imidazo[2,1-i]purine derivatives 246–248 for the identification of dual BCL2/c-MYC gene promoter G-quadruplex ligands.
Biophysical studies over products 246–248 allowed for the identification of the first dual BCL2/c-MYC gene promoter G-quadruplex ligand, which involved circular dichroism melting experiments, microscale thermophoresis measurements, NMR titrations, and computational docking calculations, as well as biological investigations including cytotoxicity and apoptotic assays, and quantitative polymerase chain reaction and Western blot analyses. Resulst permitted to assess the potency and to characterize the binding mode of the newly identified lead compound. The absence of toxicity toward normal cells, together with the small molecular weight (≅500 Da), the water solubility, the ease of functionalization, and the selectivity profile, showed to be promising and desirable features to develop G-quadruplex binders as safe and effective anticancer agents.
4. Conclusions
Through this review it has been underscored the tremendous potential of MCRs as a very important tool for the synthesis of a vast number of organic acyclic and heterocyclic molecules, coupled to the prospect that many of them are biologically active or at least have been submitted to any biological screen. After a trip through the most relevant literature dealt with libraries of organic molecules synthesized via MCRs and subjected to screening for biological activity, it was found that most of such screening were addressed to anticancer, antimicrobial, anti-leihsmanial, anti-inflammatory, ROCK inhibitor, antioxidant, antimycobacterial, bromodomain inhibitor, antifibrotic agents, human receptor 8-active, neuroprotective agents, acetylcholinesterase inhibitor and anti-HIV activities. Interestingly, more than 60% of the found literature was oriented to anticancer activity evaluation. Thus, after the searching for appropriated reports to support the subject of this review we could establish that MCRs still represent an excellent synthetic strategy for the generation of a vast number of organic scaffolds, suitable for structure–activity relationship (SAR) studies in Medicinal Chemistry and drug discovery programs.
Acknowledgments
D.I. thanks Universidad del Norte for partial financial support of this work. J.C., D.B. and H.R. thank Universidad Pedagógica y Tecnológica de Colombia for partial financial support of this work. R.A. thanks COLCIENCIAS and Universidad del Valle for partial financial support of this work.
Author Contributions
Conceptualization; Writing–Original Draft Preparation; Writing–Review and Editing, all the authors were involved; Supervision, R.A.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Dömling A., Ugi I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 2000;39:3168–3210. doi: 10.1002/1521-3773(20000915)39:183168::AID-ANIE31683.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 2.Rotstein B.H., Zaretsky S., Rai V., Yudin A.K. Small heterocycles in multicomponent reactions. Chem. Rev. 2014;114:8323–8359. doi: 10.1021/cr400615v. [DOI] [PubMed] [Google Scholar]
- 3.Dömling A., Wang W., Wang K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012;12:3083–3135. doi: 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jarusiewicz J., Choe Y., Yoo K.S., Park C.P., Jung K.W. Efficient three-component Strecker reaction of aldehydes/ketones via NHC-amidate palladium (II) complex catalysis. J. Org. Chem. 2009;74:2873–2876. doi: 10.1021/jo900163w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debache A., Boulcina R., Belfaitah A., Rhouati S., Carboni B. One-pot synthesis of 1,4-dihydropyridines via a phenylboronic acid catalyzed Hantzsch three-component reaction. Synlett. 2008:509–512. doi: 10.1055/s-2008-1032093. [DOI] [Google Scholar]
- 6.Ryabukhin S.V., Plaskon A.S., Ostapchuk E.N., Volochnyuk D.M., Tolmachev A.A. N-substituted ureas and thioureas in Biginelli reaction promoted by chlorotrimethylsilane: Convenient synthesis of N1-alkyl-, N1-aryl-, and N1,N3-dialkyl-3,4-dihydropyrimidin-2(1H)-(thi)ones. Synthesis. 2007:417–427. doi: 10.1002/chin.200723152. [DOI] [Google Scholar]
- 7.Cordoba A., Watanabe S., Tanaka F., Notz W., Barbas C.F. A highly enantioselective route to either enantiomer of both α- and β-amino acid derivatives. J. Am. Chem. Soc. 2002;124:1866–1867. doi: 10.1021/ja017833p. [DOI] [PubMed] [Google Scholar]
- 8.Andrade C.K.Z., Takada S.C.S., Suarez P.A.Z., Alves M.B. Revisiting the Passerini reaction under eco-friendly reaction conditions. Synlett. 2006:1539–1541. doi: 10.1055/s-2006-941606. [DOI] [Google Scholar]
- 9.Asinger F. Über die gemeinsame Einwirkung von Schwefel und Ammoniak auf Ketone. Angew. Chem. 1956;68:413. doi: 10.1002/ange.19560681209. [DOI] [Google Scholar]
- 10.Akritopoulou-Zanze I., Gracias V., Djuric S.W. A versatile synthesis of fused triazolo derivatives by sequential Ugi/alkyne-azide cycloaddition reactions. Tetrahedron Lett. 2004;45:8439–8441. doi: 10.1016/j.tetlet.2004.09.117. [DOI] [Google Scholar]
- 11.Tietze L.F. Domino reactions in organic synthesis. Chem. Rev. 1996;96:115–136. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]
- 12.Coquerel Y., Boddaert T., Presset M., Mailhol D., Rodriguez J. Multiple bond-forming transformations: The key concept toward eco-compatible synthetic organic chemistry. In: Pignataro B., editor. Ideas in Chemistry and Molecular Sciences: Advances in Synthetic Chemistry. Wiley-VCH; Weinheim, Germany: 2010. pp. 187–202. [DOI] [Google Scholar]
- 13.Ganem B. Strategies for innovation in multicomponent reaction design. Acc. Chem. Res. 2009;42:463–472. doi: 10.1021/ar800214s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Răzvan C.C., Ruijter E., Orru R.V.A. Multicomponent reactions: Advanced tools for sustainable organic synthesis. Green Chem. 2014;16:2958–2975. doi: 10.1039/C4GC00013G. [DOI] [Google Scholar]
- 15.Zhi S., Ma X., Zhang W. Consecutive multicomponent reactions for the synthesis of complex molecules. Org. Biomol. Chem. 2019;17:7632–7650. doi: 10.1039/C9OB00772E. [DOI] [PubMed] [Google Scholar]
- 16.Saranya S., Rohit K.R., Radhika S., Anilkumar G. Palladium-catalyzed multicomponent reactions: An overview. Org. Biomol. Chem. 2019;17:8048–8061. doi: 10.1039/C9OB01538H. [DOI] [PubMed] [Google Scholar]
- 17.Weber L. The application of multicomponent reactions in drug discovery. Curr. Med. Chem. 2002;9:2085–2093. doi: 10.2174/0929867023368719. [DOI] [PubMed] [Google Scholar]
- 18.Hulme C., Gore V. Multicomponent reactions: Emerging chemistry in drug discovery” ‘from xylocain to crixivan’. Curr. Med. Chem. 2003;10:51–80. doi: 10.2174/0929867033368600. [DOI] [PubMed] [Google Scholar]
- 19.Chappuis F., Sundar S., Hailu A., Ghalib H., Rijal S., Peeling R.W., Alvar J., Boelaert M. Visceral leishmaniasis: What are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007;5:873–882. doi: 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- 20.Desjeux P. Leishmaniasis: Current situation and new perspectives. Comp. Immunol. Microbiol. Infect. Dis. 2004;27:305–318. doi: 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 21.Kobes T., Grekov I., Lipoldová M. Leishmaniasis: Prevention, parasite detection and treatment. Curr. Med. Chem. 2012;19:1443–1474. doi: 10.2174/092986712799828300. [DOI] [PubMed] [Google Scholar]
- 22.Fields E.K. The synthesis of esters of substituted amino phosphonic acids. J. Am. Chem. Soc. 1952;74:1528–1531. doi: 10.1021/ja01126a054. [DOI] [Google Scholar]
- 23.Bhagat S., Shah P., Garg S.K., Mishra S., Kaur P.K., Singh S., Chakraborti A.K. α-Aminophosphonates as novel anti-leishmanial chemotypes: Synthesis, biological evaluation, and CoMFA studies. Med. Chem. Commun. 2014;5:665–670. doi: 10.1039/C3MD00388D. [DOI] [Google Scholar]
- 24.Zayed M.F., Hassan M.H. Synthesis and biological evaluation studies of novel quinazolinone derivatives as antibacterial and anti-inflammatory agents. Saudi Pharm. J. 2014;22:157–162. doi: 10.1016/j.jsps.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bansal Y., Sethi P., Bansal G. Coumarin: A potential nucleus for anti-inflammatory molecules. Med. Chem. Res. 2013;22:3049–3060. doi: 10.1007/s00044-012-0321-6. [DOI] [Google Scholar]
- 26.Arora R.K., Kaur N., Bansal Y., Bansa G. Novel coumarin-benzimidazole derivatives as antioxidants and safer anti-inflammatory agents. Acta Pharm. Sin. B. 2014;4:368–375. doi: 10.1016/j.apsb.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madar J.M., Shastri L.A., Shastri S.L., Holiyachi M., Naik N., Kulkarni R., Shaikh F., Sungar V. Design, synthesis, characterization, and biological evaluation of pyrido[1,2-a] pyrimidinone coumarins as promising anti-inflammatory agents. Synth. Commun. 2018;48:375–386. doi: 10.1080/00397911.2017.1397698. [DOI] [Google Scholar]
- 28.Lal J., Gupta S.K., Thavaselvam D., Agarwal D.D. Synthesis and pharmacological activity evaluation of curcumin derivatives. Chin. Chem. Lett. 2016;27:1067–1072. doi: 10.1016/j.cclet.2016.03.032. [DOI] [Google Scholar]
- 29.Patil K.N., Mane R.A., Jadhav S.B., Mane M.M., Helavi V.B. One pot multicomponent synthesis of highly functionalized tetrahydropyridine using copper (II) triflate as catalyst and their anti-inflammatory activity. Chem. Data Collect. 2019;21:100233. doi: 10.1016/j.cdc.2019.100233. [DOI] [Google Scholar]
- 30.Borazjani N., Sepehri S., Behzadi M., Jarrahpour A.M., Rad J.A., Sasanipour M., Mohkam M., Ghasemi J., Akbarizadeh A.R., Digiorgio C., et al. Three-component synthesis of chromeno β-lactam hybrids for inflammation and cancer screening. Eur. J. Med. Chem. 2019;179:389–403. doi: 10.1016/j.ejmech.2019.06.036. [DOI] [PubMed] [Google Scholar]
- 31.Olson M.F. Applications for ROCK kinase inhibition. Curr. Opin. Cell Biol. 2008;20:242–248. doi: 10.1016/j.ceb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoy S.M. Netarsudil ophthalmic solution 0.02%: First global approval. Drugs. 2018;78:389–396. doi: 10.1007/s40265-018-0877-7. [DOI] [PubMed] [Google Scholar]
- 33.Dayal N., Mikek C.G., Hernandez D., Naclerio G.A., Yin Chu E.F., Carter-Cooper B.A., Lapidus R.G., Sintim H.O. Potently inhibiting cancer cell migration with novel 3H-pyrazolo[4,3-f]quinoline boronic acid ROCK inhibitors. Eur. J. Med. Chem. 2019;180:449–456. doi: 10.1016/j.ejmech.2019.06.089. [DOI] [PubMed] [Google Scholar]
- 34.Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I., et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKeown M.R., Shaw D.L., Fu H., Liu S., Xu X., Marineau J.J., Huang Y., Zhang X., Buckley D.L., Kadam A., et al. Biased multicomponent reactions to develop novel bromodomain inhibitors. J. Med. Chem. 2014;57:9019–9027. doi: 10.1021/jm501120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J.D., Roberts L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010;7:448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patil R., Ghosh A., Cao P.S., Sommer R.D., Grice K.A., Waris G., Patil S. Novel 5-arylthio-5H-chromenopyridines as a new class of anti-fibrotic agents. Bioorg. Med. Chem. Lett. 2017;27:1129–1135. doi: 10.1016/j.bmcl.2017.01.089. [DOI] [PubMed] [Google Scholar]
- 38.Iwasaki A., Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawai T., Akira S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 40.Kumagai Y., Takeuchi O., Akira S. Pathogen recognition by innate receptors. J. Infect. Chemother. 2008;14:86–92. doi: 10.1007/s10156-008-0596-1. [DOI] [PubMed] [Google Scholar]
- 41.Takeda K., Akira S. Toll-like receptors. Curr. Protoc. Immunol. 2015;109:1–10. doi: 10.1002/0471142735.im1412s109. [DOI] [PubMed] [Google Scholar]
- 42.Shaaban S., Abdel-Wahab B.F. Groebke-Blackburn-Bienaymé multicomponent reaction: Emerging chemistry for drug discovery. Mol. Divers. 2016;20:233–254. doi: 10.1007/s11030-015-9602-6. [DOI] [PubMed] [Google Scholar]
- 43.Salunke D.B., Yoo E., Shukla N.M., Balakrishna R., Malladi S.S., Serafin K.J., Day V.W., Wang X., David S.A. Structure-activity relationships in human Toll-like receptor 8-active 2,3-diamino-furo[2,3-c]pyridines. J. Med. Chem. 2012;55:8137–8151. doi: 10.1021/jm301066h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaffer H., Morris V.B., Stewart D., Labhasetwar V. Advances in stroke therapy. Drug Deliv. Transl. Res. 2011;1:409–419. doi: 10.1007/s13346-011-0046-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tenti G., Parada P., León R., Egea J., Martínez-Revelles S., Briones A.M., Sridharan V., López M.G., Ramos M.T., Menéndez J.C. New 5-unsubstituted dihydropyridines with improved CaV1.3 selectivity as potential neuroprotective agents against ischemic injury. J. Med. Chem. 2014;57:4313–4323. doi: 10.1021/jm500263v. [DOI] [PubMed] [Google Scholar]
- 46.Colovic M.B., Krstic D.Z., Lazarevic-Pasti T.D., Bondzic A.M., Vasic V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013;11:315–335. doi: 10.2174/1570159X11311030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitt J., Irons R., Al-Obaidi M., Missouris C. A case of donepezil-related torsades de pointes. BMJ Case Rep. 2015:1–3. doi: 10.1136/bcr-2015-211900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pourshojaei Y., Abiri A., Eskandari R., Dourandish F., Eskandari K., Asadipour A. Synthesis, biological evaluation, and computational studies of novel fused six-membered O-containing heterocycles as potential acetylcholinesterase inhibitors. Comp. Biol. Chem. 2019;80:249–258. doi: 10.1016/j.compbiolchem.2019.04.004. [DOI] [PubMed] [Google Scholar]
- 49.Jonckheere H., Anné J., De Clercq E. The HIV-1 reverse transcription (RT) process as target for RT inhibitors. Med. Res. Rev. 2000;20:129–154. doi: 10.1002/(SICI)1098-1128(200003)20:2<129::AID-MED2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 50.De Clercq E. Novel compounds in preclinical/early clinical development for the treatment of HIV infections. Rev. Med. Virol. 2000;10:255–277. doi: 10.1002/1099-1654(200007/08)10:4<255::AID-RMV282>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 51.Barreca M.L., Balzarini J., Chimirri A., De Clercq E., De Luca L., Höltje H.D., Höltje M., Monforte A.M., Monforte P., Pannecouque C., et al. Design, synthesis, structure-activity relationships, and molecular modeling studies of 2,3-diaryl-1,3-thiazolidin-4-ones as potent anti-HIV agents. J. Med. Chem. 2002;45:5410–5413. doi: 10.1021/jm020977+. [DOI] [PubMed] [Google Scholar]
- 52.Woodford N. Novel agents for the treatment of resistant Gram-positive infections. Expert Opin. Investig. Drugs. 2003;12:117–137. doi: 10.1517/13543784.12.2.117. [DOI] [PubMed] [Google Scholar]
- 53.Ma Z., Lynch A.S. Development of a dual-acting antibacterial agent (TNP-2092) for the treatment of persistent bacterial infections. J. Med. Chem. 2016;59:6645–6657. doi: 10.1021/acs.jmedchem.6b00485. [DOI] [PubMed] [Google Scholar]
- 54.Lakshmi N.V., Thirumurugan P., Noorulla K.M., Perumal P.T. InCl3 mediated one-pot multicomponent synthesis, anti-microbial, antioxidant and anticancer evaluation of 3-pyranyl indole derivatives. Bioorg. Med. Chem. Lett. 2010;20:5054–5061. doi: 10.1016/j.bmcl.2010.07.039. [DOI] [PubMed] [Google Scholar]
- 55.Mungra D.C., Patel M.P., Rajani D.P., Patel R.G. Synthesis and identification of β-aryloxyquinolines and their pyrano[3,2-c]chromene derivatives as a new class of antimicrobial and antituberculosis agents. Eur. J. Med. Chem. 2011;46:4192–4200. doi: 10.1016/j.ejmech.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 56.Vijesh A.M., Isloor A.M., Peethambar S.K., Shivananda K.N., Arulmoli T., Isloor N.A. Hantzsch reaction: Synthesis and characterization of some new 1,4-dihydropyridine derivatives as potent antimicrobial and antioxidant agents. Eur. J. Med. Chem. 2011;46:5591–5597. doi: 10.1016/j.ejmech.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 57.El-borai M.A., Rizk H., Abd-Aal M.F., El-Deeb I.Y. Synthesis of pyrazolo[3,4-b] pyridines under microwave irradiation in multicomponent reactions and their antitumor and antimicrobial activities. Eur. J. Med. Chem. 2012;48:92–96. doi: 10.1016/j.ejmech.2011.11.038. [DOI] [PubMed] [Google Scholar]
- 58.Shah N.M., Patel M.P., Patel R.G. Synthesis of a novel class of some biquinoline pyridine hybrids via one-pot, three-component reaction and their antimicrobial activity. J. Chem. Sci. 2012;124:669–677. doi: 10.1007/s12039-012-0254-0. [DOI] [Google Scholar]
- 59.Bhaskar G., Arun Y., Balachandran C., Saikumar C., Perumal P.T. Synthesis of novel spirooxindole derivatives by one pot multicomponent reaction and their antimicrobial activity. Eur. J. Med. Chem. 2012;51:79–91. doi: 10.1016/j.ejmech.2012.02.024. [DOI] [PubMed] [Google Scholar]
- 60.Singh S.B., Tiwari K., Verma P.K., Srivastava M., Tiwari K.P., Singh J. A new eco-friendly strategy for the synthesis of novel antimicrobial spiro-oxindole derivatives via supramolecular catalysis. Supramol. Chem. 2013;25:255–262. doi: 10.1080/10610278.2012.761341. [DOI] [Google Scholar]
- 61.Darandale S.N., Pansare D.N., Mulla N.A., Shinde D.B. Green synthesis of tetrahydropyrimidine analogues and evaluation of their antimicrobial activity. Bioorg. Med. Chem. Lett. 2013;23:2632–2635. doi: 10.1016/j.bmcl.2013.02.099. [DOI] [PubMed] [Google Scholar]
- 62.Murlykina M.V., Sakhno Y.I., Desenko S.M., Konovalova I.S., Shishkin O.V., Sysoiev D.A., Kornet M.N., Chebanov V.A. Features of switchable multicomponent heterocyclizations of salicylic aldehydes and 5-aminopyrazoles with pyruvic acids and antimicrobial activity of the reaction products. Tetrahedron. 2013;69:9261–9269. doi: 10.1016/j.tet.2013.08.055. [DOI] [Google Scholar]
- 63.Akondi A.M., Kantam M.L., Trivedi R., Sreedhar B., Buddana S.K., Prakasham R.S., Bhargava S. Formation of benzoxanthenones and benzochromenones viacerium-impregnated-MCM-41 catalyzed, solvent-free, three-component reaction and their biological evaluation as antimicrobial agents. J. Mol. Catal. A Chem. 2014;386:49–60. doi: 10.1016/j.molcata.2014.02.012. [DOI] [Google Scholar]
- 64.Sable P.N., Ganguly S., Chaudhari P.D. An efficient one-pot three-component synthesis and antimicrobial evaluation of tetra substituted thiophene derivatives. Chin. Chem. Lett. 2014;25:1099–1103. doi: 10.1016/j.cclet.2014.03.044. [DOI] [Google Scholar]
- 65.Sindhu J., Singh H., Khurana J.M., Sharma C., Aneja K.R. Multicomponent domino process for the synthesis of some novel 5-(arylidene)-3-((1-aryl-1H-1,2,3-triazol-4-yl)methyl)-thiazolidine-2,4-diones using PEG-400 as an efficient reaction medium and their antimicrobial evaluation. Chin. Chem. Lett. 2015;26:50–54. doi: 10.1016/j.cclet.2014.09.006. [DOI] [Google Scholar]
- 66.Boltjes A., Dömling A. The Groebke-Blackburn-Bienaymé reaction. Eur. J. Org. Chem. 2019;2019:7007–7049. doi: 10.1002/ejoc.201901124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aouali M., Mhalla D., Allouche F., El Kaim L., Tounsi S., Trigui M., Chabchoub F. Synthesis, antimicrobial and antioxidant activities of imidazotriazoles and new multicomponent reaction toward 5-amino-1-phenyl [1,2,4] triazole derivatives. Med. Chem. Res. 2015;24:2732–2741. doi: 10.1007/s00044-015-1322-z. [DOI] [Google Scholar]
- 68.Dhinakaran I., Padmini V., Ganesan K., Selvarasu K. A one-pot four-component and microwave-assisted synthesis of pyrrolo [1,10] phenanthrolines. ChemistrySelect. 2017;2:6154–6158. doi: 10.1002/slct.201700819. [DOI] [Google Scholar]
- 69.Meena K., Kumari S., Khurana J.M., Malik A., Sharma C., Panwar H. One-pot three-component synthesis of spiro[indolo-3,10’-indeno[1,2-b]quinolin]-2,4,11’-triones as a new class of antifungal and antimicrobial agents. Chin. Chem. Lett. 2017;28:136–142. doi: 10.1016/j.cclet.2016.06.025. [DOI] [Google Scholar]
- 70.Ashok D., Devulapally M.G., Aamate V.K., Gundu S., Adam S., Murthy S.D.S., Balasubramanian S., Naveen B., Parthasarathy T. Novel pyrano [3,2-b] xanthen-7(2H)-ones: Synthesis, antimicrobial, antioxidant and molecular docking studies. J. Mol. Struct. 2019;1177:215–228. doi: 10.1016/j.molstruc.2018.08.091. [DOI] [Google Scholar]
- 71.Safari F., Hosseini H., Bayat M., Ranjbar A. Synthesis and evaluation of antimicrobial activity, cytotoxic and pro-apoptotic effects of novel spiro-4H-pyran derivatives. RSC Adv. 2019;9:24843–24851. doi: 10.1039/C9RA03196K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kenchappa R., Bodke Y.D., Chandrashekar A., Telkar S., Manjunatha K.S., Sindhe M.A. Synthesis of some 2,6-bis(1-coumarin-2-yl)-4-(4-substituted phenyl) pyridine derivatives as potent biological agents. Arab. J. Chem. 2017;10:S1336–S1344. doi: 10.1016/j.arabjc.2013.03.020. [DOI] [Google Scholar]
- 73.Vekariya R.V., Patel K.D., Rajani D.P., Rajani S.D., Patel H.D. A one pot, three component synthesis of coumarin hybrid thiosemicarbazone derivatives and their antimicrobial evolution. J. Assoc. Arab. Univ. Basic Appl. Sci. 2017;23:10–19. doi: 10.1016/j.jaubas.2016.04.002. [DOI] [Google Scholar]
- 74.Pedrola M., Jorba M., Jardas E., Jardi F., Ghashghaei O., Viñas M., Lavilla R. Multicomponent reactions upon the known drug trimethoprim as a source of novel antimicrobial agents. Front. Chem. 2019;7:1–9. doi: 10.3389/fchem.2019.00475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsolaki E., Nobelos P., Geronikaki A., Rekka E.A. Selected heterocyclic compounds as antioxidants. Synthesis and biological evaluation. Curr. Top. Med. Chem. 2014;14:2462–2477. doi: 10.2174/1568026614666141203120425. [DOI] [PubMed] [Google Scholar]
- 76.Dangolani S.K., Panahi F., Tavaf Z., Nourisefat M., Yousefi R., Khalafi-Nezhad A. Synthesis and antioxidant activity evaluation of some novel aminocarbonitrile derivatives incorporating carbohydrate moieties. ACS Omega. 2018;3:10341–10350. doi: 10.1021/acsomega.8b01124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khansole G.S., Prasad D., Angulwar J.A., Atar A.B., Nagaraja B.M., Jadhav A.H., Bhosalee V.N. Tetrabutylammonium hydrogen sulfate mediated three-component reaction for the synthesis of thiadiazolo[2,3-b]quinazolin-6-(7H)-ones and antioxidant activity. Mater. Today Proc. 2019;9:653–660. doi: 10.1016/j.matpr.2018.10.389. [DOI] [Google Scholar]
- 78.Ezzatzadeh E., Hossaini Z., Moradi A.V., Salimifard M., Abad S.A.S. Copper iodide and ZnO nanoparticles catalyzed multicomponent synthesis of 1,3-cyclopentadiene: Study of antioxidant activity. Can. J. Chem. 2019;97:270–276. doi: 10.1139/cjc-2018-0154. [DOI] [Google Scholar]
- 79.Cohen J. New TB drug promises shorter, simpler treatment. Science. 2004;306:1872. doi: 10.1126/science.306.5703.1872. [DOI] [PubMed] [Google Scholar]
- 80.Dye C., Watt C.J., Bleed D.M., Hosseini S.M., Raviglione M.C. Evolution of tuberculosis control and prospects for reducing tuberculosis incidence, prevalence, and deaths globally. J. Am. Med. Assoc. 2005;293:2767–2775. doi: 10.1001/jama.293.22.2767. [DOI] [PubMed] [Google Scholar]
- 81.Ramprasad J., Nayak N., Dalimba U., Yogeeswari P., Sriram D., Peethambar S.K., Achur R., Kumar H.S.S. Synthesis and biological evaluation of new imidazo [2,1-b] [1,3,4] thiadiazole-benzimidazole derivatives. Eur. J. Med. Chem. 2015;95:49–63. doi: 10.1016/j.ejmech.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 82.Chandrasekera N.S., Alling T., Bailey M.A., Files M., Early J.V., Ollinger J., Ovechkina Y., Masquelin T., Desai P.V., Cramer J.W., et al. Identification of phenoxyalkylbenzimidazoles with antitubercular activity. J. Med. Chem. 2015;58:7273–7285. doi: 10.1021/acs.jmedchem.5b00546. [DOI] [PubMed] [Google Scholar]
- 83.Kalalbandi V.K.A., Seetharamappa J., Katrahalli U., Bhat K.G. Synthesis, crystal studies, anti-tuberculosis and cytotoxic studies of 1-[(2E)-3-phenylprop-2-enoyl]-1H-benzimidazole derivative. Eur. J. Med. Chem. 2014;79:194–202. doi: 10.1016/j.ejmech.2014.04.017. [DOI] [PubMed] [Google Scholar]
- 84.Anand A., Kulkarni M.V., Joshi S.D., Dixit S.R. One pot Click chemistry: A three component reaction for the synthesis of 2-mercaptobenzimidazole linked coumarinyl triazoles as anti-tubercular agents. Bioorg. Med. Chem. Lett. 2016;26:4709–4713. doi: 10.1016/j.bmcl.2016.08.045. [DOI] [PubMed] [Google Scholar]
- 85.Ramani A.V., Monika A., Indira V.L., Karyavardhi G., Venkatesh J., Jeankumar V.U., Manjashetty T.H., Yogeeswari P., Sriram D. Synthesis of highly potent novel anti-tubercular isoniazid analogues with preliminary pharmacokinetic evaluation. Bioorg. Med. Chem. Lett. 2012;22:2764–2767. doi: 10.1016/j.bmcl.2012.02.091. [DOI] [PubMed] [Google Scholar]
- 86.Dandia A., Singh R., Saini D. Ionic liquid-mediated three-component synthesis of fluorinated spiro-thiazine derivatives and their antimycobacterial and DNA cleavage activities. J. Chem. Sci. 2013;125:1045–1053. doi: 10.1007/s12039-013-0493-8. [DOI] [Google Scholar]
- 87.Quiroga J., Diaz Y., Bueno J., Insuasty B., Abonia R., Ortiz A., Nogueras M., Cobo J. Microwave induced three-component synthesis and antimycobacterial activity of benzopyrazolo [3,4-b] quinolindiones. Eur. J. Med. Chem. 2014;74:216–224. doi: 10.1016/j.ejmech.2013.12.037. [DOI] [PubMed] [Google Scholar]
- 88.Surineni G., Yogeeswari P., Sriram D., Kantevari S. Design and synthesis of novel carbazole tethered pyrrole derivatives as potent inhibitors of Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2015;25:485–491. doi: 10.1016/j.bmcl.2014.12.040. [DOI] [PubMed] [Google Scholar]
- 89.Goud G.L., Ramesh S., Ashok D., Reddy V.P., Yogeeswari P., Sriram D., Saikrishna B., Manga V. Design, synthesis, molecular-docking and antimycobacterial evaluation of some novel 1,2,3-triazolyl xanthenones. Med. Chem. Commun. 2017;8:559–570. doi: 10.1039/C6MD00593D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abonia R., Insuasty D., Castillo J., Insuasty B., Quiroga J., Nogueras M., Cobo J. Synthesis of novel quinoline-2-one based chalcones of potential anti-tumor activity. Eur. J. Med. Chem. 2012;57:29–40. doi: 10.1016/j.ejmech.2012.08.039. [DOI] [PubMed] [Google Scholar]
- 91.Insuasty B., Montoya A., Becerra D., Quiroga J., Abonia R., Robledo S., Vélez I.V., Upegui Y., Nogueras M., Cobo J. Synthesis of novel analogs of 2-pyrazoline obtained from [(7-chloroquinolin-4-yl)amino]chalcones and hydrazine as potential antitumor and antimalarial agents. Eur. J. Med. Chem. 2013;67:252–262. doi: 10.1016/j.ejmech.2013.06.049. [DOI] [PubMed] [Google Scholar]
- 92.Bianchini G., Ribelles P., Becerra D., Ramos M.T., Menéndez J.C. Efficient synthesis of 2-acylquinolines based on an aza-vinylogous Povarov reaction. Org. Chem. Front. 2016;3:412–422. doi: 10.1039/C6QO00037A. [DOI] [Google Scholar]
- 93.Nainwal L.M., Tasneem S., Akhtar W., Verma G., Khan M.F., Parvez S., Shaquiquzzaman M., Akhter M., Alam M.M. Green recipes to quinoline: A review. Eur. J. Med. Chem. 2019;164:121–170. doi: 10.1016/j.ejmech.2018.11.026. [DOI] [PubMed] [Google Scholar]
- 94.Kouznetsov V.V., Ochoa C., Romero A.R., Zacchino S.A., Sortino M., Gupta M., Vázquez Y., Bahsas A., Amaro-Luis J. A straightforward synthetic approach to antitumoral pyridinyl substituted 7H-indeno [2,1-c] quinoline derivatives via three-component imino Diels-Alder reaction. Lett. Org. Chem. 2006;3:300–304. doi: 10.1002/chin.200639175. [DOI] [Google Scholar]
- 95.Roopan S.M., Khan F.N., Jin J.S., Kumar R.S. An efficient one pot–three component cyclocondensation in the synthesis of 2-(2-chloroquinolin-3-yl)-2,3-dihydroquinazolin-4(1H)-ones: Potential antitumor agents. Res. Chem. Intermed. 2011;37:919–927. doi: 10.1007/s11164-011-0301-3. [DOI] [Google Scholar]
- 96.Sangani C.B., Makawana J.A., Duan Y.-T., Yin Y., Teraiya S.B., Thumar N.J., Zhu H.-L. Design, synthesis and molecular modeling of biquinoline–pyridine hybrids as a new class of potential EGFR and HER-2 kinase inhibitors. Bioorg. Med. Chem. Lett. 2014;24:4472–4476. doi: 10.1016/j.bmcl.2014.07.094. [DOI] [PubMed] [Google Scholar]
- 97.Alonso C., Fuertes M., Martín-Encinas E., Selas A., Rubiales G., Tesauro C., Knudssen B.K., Palacios F. Novel topoisomerase I inhibitors. Syntheses and biological evaluation of phosphorus substituted quinoline derivates with antiproliferative activity. Eur. J. Med. Chem. 2018;149:225–237. doi: 10.1016/j.ejmech.2018.02.058. [DOI] [PubMed] [Google Scholar]
- 98.Castillo J.-C., Jimenez E., Portilla J., Insuasty B., Quiroga J., Moreno-Fuquen R., Kennedy A.R., Abonia R. Application of a catalyst-free Domino Mannich/Friedel-Crafts alkylation reaction for the synthesis of novel tetrahydroquinolines of potential antitumor activity. Tetrahedron. 2018;74:932–947. doi: 10.1016/j.tet.2017.12.049. [DOI] [Google Scholar]
- 99.Abonia R., Castillo J., Insuasty B., Quiroga J., Nogueras M., Cobo J. Efficient catalyst-free four-component synthesis of novel γ-aminoethers mediated by a Mannich type reaction. ACS Comb. Sci. 2013;15:2–9. doi: 10.1021/co300105t. [DOI] [PubMed] [Google Scholar]
- 100.Mani K.S., Murugesapandian B., Kaminsky W., Rajendrana S.P. Enantioselective approach towards the synthesis of spiro-indeno [1,2-b] quinoxaline pyrrolothiazoles as antioxidant and antiproliferative. Tetrahedron Lett. 2018;59:2921–2929. doi: 10.1016/j.tetlet.2018.06.035. [DOI] [Google Scholar]
- 101.Taheri S., Nazifi M., Mansourian M., Hosseinzadeh L., Shokoohiniad Y. Ugi efficient synthesis, biological evaluation and molecular docking of coumarin-quinoline hybrids as apoptotic agents through mitochondria-related pathways. Bioorg. Chem. 2019;91:103147. doi: 10.1016/j.bioorg.2019.103147. [DOI] [PubMed] [Google Scholar]
- 102.Castillo J.-C., Portilla J. Recent advances in the synthesis of new pyrazole derivatives. Targets Heterocycl. Syst. 2018;22:194–223. doi: 10.17374/targets.2019.22.194. [DOI] [Google Scholar]
- 103.Karrouchi K., Radi S., Ramli Y., Taoufik J., Mabkhot Y.N., Al-Aizari F.A., Ansar M. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules. 2018;23:134. doi: 10.3390/molecules23010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Castillo J.C., Quiroga J., Abonia R., Rodriguez J., Coquerel Y. The aryne aza-Diels–Alder reaction: Flexible syntheses of isoquinolines. Org. Lett. 2015;17:3374–3377. doi: 10.1021/acs.orglett.5b01704. [DOI] [PubMed] [Google Scholar]
- 105.Galvez J., Castillo J.C., Quiroga J., Rajzmann M., Rodriguez J., Coquerel Y. Divergent chemo-, regio-, and diastereoselective normal electron-demand Povarov-type reactions with α-oxo-ketene dienophiles. Org. Lett. 2014;16:4126–4129. doi: 10.1021/ol5018245. [DOI] [PubMed] [Google Scholar]
- 106.Abonia R., Castillo J., Insuasty B., Quiroga J., Nogueras M., Cobo J. An efficient synthesis of 7-(arylmethyl)-3-tert-butyl-1-phenyl-6,7-dihydro-1H,4H-pyrazolo[3,4-d][1,3]oxazines. Eur. J. Org. Chem. 2010;33:6454–6463. doi: 10.1002/ejoc.201000678. [DOI] [Google Scholar]
- 107.Kamble A.A., Kamble R.R., Chougala L.S., Kadadevarmath J.S., Maidur S.R., Patil P.S., Kumbar M.N., Marganakop S.B. Photophysical, electrochemical studies of novel pyrazol-4-yl-2,3-dihydroquinazolin-4(1H)-ones and their anticancer activity. ChemistrySelect. 2017;2:6882–6890. doi: 10.1002/slct.201700498. [DOI] [Google Scholar]
- 108.Nimbalkar U.D., Seijas J.A., Vazquez-Tato M.P., Damale M.G., Sangshetti J.N., Nikalje A.P.G. Ionic liquid-catalyzed green protocol for multi-component synthesis of dihydropyrano[2,3-c]pyrazoles as potential anticancer scaffolds. Molecules. 2017;22:1628. doi: 10.3390/molecules22101628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao S., Pi C., Ye Y., Zhao L., Wei Y. Recent advances of analogues of curcumin for treatment of cancer. Eur. J. Med. Chem. 2019;180:524–535. doi: 10.1016/j.ejmech.2019.07.034. [DOI] [PubMed] [Google Scholar]
- 110.Noureddin S.A., El-Shishtawy R.M., Al-Footy K.O. Curcumin analogues and their hybrid molecules as multifunctional drugs. Eur. J. Med. Chem. 2019;182:111631. doi: 10.1016/j.ejmech.2019.111631. [DOI] [PubMed] [Google Scholar]
- 111.Rodrigues F.C., Kumar N.V.A., Thakur G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J. Med. Chem. 2019;177:76–104. doi: 10.1016/j.ejmech.2019.04.058. [DOI] [PubMed] [Google Scholar]
- 112.Khor P.Y., Moh-Aluwi M.F.F., Rullah K., Lam K.W. Insights on the synthesis of asymmetric curcumin derivatives and their biological activities. Eur. J. Med. Chem. 2019;183:111704. doi: 10.1016/j.ejmech.2019.111704. [DOI] [PubMed] [Google Scholar]
- 113.Sharma R., Jadav S.S., Yasmin S., Bhatia S., Khalilullah H., Ahsan M.J. Simple, efficient, and improved synthesis of Biginelli-type compounds of curcumin as anticancer agents. Med. Chem. Res. 2015;24:636–644. doi: 10.1007/s00044-014-1146-2. [DOI] [Google Scholar]
- 114.Bhuvaneswari K., Sivaguru P., Lalitha A. Synthesis, biological evaluation and molecular docking of novel curcumin derivatives as Bcl-2 inhibitors targeting human breast cancer MCF-7 cells. ChemistrySelect. 2017;2:11552–11560. doi: 10.1002/slct.201702406. [DOI] [Google Scholar]
- 115.Estévez V., Villacampa M., Menéndez J.C. Recent advances in the synthesis of pyrroles by multicomponent reactions. Chem. Soc. Rev. 2014;43:4633–4657. doi: 10.1039/C3CS60015G. [DOI] [PubMed] [Google Scholar]
- 116.Ahmad S., Alam O., Naim M.J., Shaquiquzzaman M., Alam M.M., Iqbal M. Pyrrole: An insight into recent pharmacological advances with structure activity relationship. Eur. J. Med. Chem. 2018;157:527–561. doi: 10.1016/j.ejmech.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 117.Abonia R., Insuasty B., Quiroga J., Kolshorn H., Meier H. A versatile synthesis of 4,5-dihydropyrrolo [1,2-a] quinoxalines. J. Heterocycl. Chem. 2001;38:671–674. doi: 10.1002/jhet.5570380321. [DOI] [Google Scholar]
- 118.Abonia R., Cuervo P., Insuasty B., Quiroga J., Nogueras M., Cobo J. A simple two-step sequence for the synthesis of novel 4-aryl-4,5-dihydro-6H-[1,3] dioxolo [4,5-h] pyrrolo[1,2-a][1]benzazepin-6-ones from 6-amino-3,4-methylenedioxyacetophenone. Eur. J. Org. Chem. 2008;27:4684–4689. doi: 10.1002/ejoc.200800493. [DOI] [Google Scholar]
- 119.Castillo J.C., Abonia R., Cobo J., Glidewell C. A chain of [π]-stacked molecules in 4-(2-chloro-phenyl)pyrrolo[1,2-a]quinoxaline and a hydrogen-bonded sheet in (4RS)-4-(1,3-benzodioxol-6-yl)-4,5-dihydropyrrolo[1,2-a]quinoxaline. Acta Cryst. 2013;69:544–548. doi: 10.1107/S0108270113009098. [DOI] [PubMed] [Google Scholar]
- 120.Castillo J.C., Tigreros A., Coquerel Y., Rodríguez J., Macías M.A., Portilla J. Synthesis of pyrrolo[2,3-c]isoquinolines via the cycloaddition of benzyne with arylideneaminopyrroles: Photophysical and crystallographic Study. ACS Omega. 2019;4:17326–17339. doi: 10.1021/acsomega.9b02043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Magedov I.V., Luchetti G., Evdokimov N.M., Manpadi M., Steelant W.F.A., Van Slambrouck S., Tongwa P., Antipin M.Y., Kornienko A. Novel three-component synthesis and antiproliferative properties of diversely functionalized pyrrolines. Bioorg. Med. Chem. Lett. 2008;18:1392–1396. doi: 10.1016/j.bmcl.2008.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pagadala R., Kommidi D.R., Kankala S., Maddila S., Singh P., Moodley B., Koorbanally N.A., Jonnalagadda S.B. Multicomponent one-pot synthesis of highly functionalized pyrrole-3-carbonitriles in aqueous medium and computational study. Org. Biomol. Chem. 2015;13:1800–1806. doi: 10.1039/C4OB02229G. [DOI] [PubMed] [Google Scholar]
- 123.Sharma S.K., Kumar S., Chand K., Kathuria A., Gupta A., Jain R. An update on natural occurrence and biological activity of chromones. Curr. Med. Chem. 2011;18:3825–3852. doi: 10.2174/092986711803414359. [DOI] [PubMed] [Google Scholar]
- 124.Keri R.S., Budagumpi S., Pai R.K., Balakrishna R.G. Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 2014;78:340–374. doi: 10.1016/j.ejmech.2014.03.047. [DOI] [PubMed] [Google Scholar]
- 125.Pratap R., Ram V.J. Natural and synthetic chromenes, fused chromenes, and versatility of dihydrobenzo[h]chromenes in organic synthesis. Chem. Rev. 2014;114:10476–10526. doi: 10.1021/cr500075s. [DOI] [PubMed] [Google Scholar]
- 126.Majumdar N., Paul N.D., Mandal S., de Bruin B., Wulff W.D. Catalytic synthesis of 2H-chromenes. ACS Catal. 2015;5:2329–2366. doi: 10.1021/acscatal.5b00026. [DOI] [Google Scholar]
- 127.Costa M., Dias T.A., Brito A., Proença F. Biological importance of structurally diversified chromenes. Eur. J. Med. Chem. 2016;123:487–507. doi: 10.1016/j.ejmech.2016.07.057. [DOI] [PubMed] [Google Scholar]
- 128.Huang W., Ding Y., Miao Y., Liu M.-Z., Li Y., Yang G.-F. Synthesis and antitumor activity of novel dithiocarbamate substituted chromones. Eur. J. Med. Chem. 2009;44:3687–3696. doi: 10.1016/j.ejmech.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 129.Kumar A., Sharma S., Maurya R.A., Sarkar J. Diversity oriented synthesis of benzoxanthene and benzochromene libraries via one-pot, tricomponent reactions and their anti-proliferative activity. J. Comb. Chem. 2010;12:20–24. doi: 10.1021/cc900143h. [DOI] [PubMed] [Google Scholar]
- 130.Paliwal P.K., Jetti S.R., Jain S. Green approach towards the facile synthesis of dihydropyrano(c)chromene and pyrano[2,3-d]pyrimidine derivatives and their biological evaluation. Med. Chem. Res. 2013;22:2984–2990. doi: 10.1007/s00044-012-0288-3. [DOI] [Google Scholar]
- 131.Kalla R.M.N., Choi J.-S., Yoo J.-W., Byeon S.J., Heo M.S., Kim I. Synthesis of 2-amino-3-cyano-4H-chromen-4-ylphosphonates and their anticancer properties. Eur. J. Med. Chem. 2014;76:61–66. doi: 10.1016/j.ejmech.2014.02.025. [DOI] [PubMed] [Google Scholar]
- 132.Perumal M., Sengodu P., Venkatesan S., Srinivasan R., Paramsivam M. Environmentally benign copper triflate-mediated multicomponent one-pot synthesis of novel benzo[g]chromenes possess potent anticancer activity. ChemistrySelect. 2017;2:5068–5072. doi: 10.1002/slct.201700170. [DOI] [Google Scholar]
- 133.Torres M., Gil S., Parra M. New synthetic methods to 2-pyridone rings. Curr. Org. Chem. 2005;9:1757–1779. doi: 10.2174/138527205774610886. [DOI] [Google Scholar]
- 134.Hamama W.S., Waly M., El-Hawary I., Zoorob H.H. Developments in the chemistry of 2-pyridone. Synth. Commun. 2014;44:1730–1759. doi: 10.1080/00397911.2013.862836. [DOI] [Google Scholar]
- 135.Hirano K., Miura M. A lesson for site-selective C–H functionalization on 2-pyridones: Radical, organometallic, directing group and steric controls. Chem. Sci. 2018;9:22–32. doi: 10.1039/C7SC04509C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Prendergast A.M., McGlacken G.P. Transition metal mediated C–H activation of 2-pyrones, 2-pyridones, 2-coumarins and 2-quinolones. Eur. J. Org. Chem. 2018;44:6068–6082. doi: 10.1002/ejoc.201800299. [DOI] [Google Scholar]
- 137.Abonia R., Castillo J., Cuervo P., Insuasty B., Quiroga J., Ortíz A., Nogueras M., Cobo J. A simple one-pot synthesis of new imidazol-2-yl-1H-quinolin-2-ones from the direct reaction of 2-chloroquinolin-3-carbaldehyde with aromatic o-diamines. Eur. J. Org. Chem. 2010;2:317–325. doi: 10.1002/ejoc.200901014. [DOI] [Google Scholar]
- 138.Magedov I.V., Manpadi M., Ogasawara M.A., Dhawan A.S., Rogelj S., Van Slambrouck S., Steelant W.F.A., Evdokimov N.M., Uglinskii P.Y., Elias E.M., et al. Structural simplification of bioactive natural products with multicomponent synthesis. 2. Antiproliferative and antitubulin activities of pyrano[3,2-c]pyridones and pyrano[3,2-c]quinolones. J. Med. Chem. 2008;51:2561–2570. doi: 10.1021/jm701499n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ansari M.I., Arun A., Hussain M.K., Konwar R., Hajela K. Discovery of 3,4,6-triaryl-2-pyridones as potential anticancer agents that promote ROS-independent mitochondrial-mediated apoptosis in human breast carcinoma cells. ChemistrySelect. 2016;1:4255–4264. doi: 10.1002/slct.201600893. [DOI] [Google Scholar]
- 140.Kumar N.P., Thatikonda S., Tokala R., Kumari S.S., Lakshmi U.J., Godugu C., Shankaraiah N., Kamal A. Sulfamic acid promoted one-pot synthesis of phenanthrene fused-dihydrodibenzo-quinolinones: Anticancer activity, tubulin polymerization inhibition and apoptosis inducing studies. Bioorg. Med. Chem. 2018;26:1996–2008. doi: 10.1016/j.bmc.2018.02.050. [DOI] [PubMed] [Google Scholar]
- 141.Ayati A., Emami S., Asadipour A., Shafiee A., Foroumadi A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015;97:699–718. doi: 10.1016/j.ejmech.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 142.Jain S., Pattnaik S., Pathak K., Kumar S., Pathak D., Jain S., Vaidya A. Anticancer potential of thiazole derivatives: A retrospective review. Mini Rev. Med. Chem. 2018;18:640–655. doi: 10.2174/1389557517666171123211321. [DOI] [PubMed] [Google Scholar]
- 143.Pathania S., Narang R.K., Rawal R.K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019;180:486–508. doi: 10.1016/j.ejmech.2019.07.043. [DOI] [PubMed] [Google Scholar]
- 144.Abonia R., Castillo J., Insuasty B., Quiroga J., Sortino M., Nogueras M., Cobo J. Catalyst-, solvent- and desiccant-free three-component synthesis of novel C-2,N-3 disubstituted thiazolidin-4-ones. Arab. J. Chem. 2019;12:122–133. doi: 10.1016/j.arabjc.2016.11.016. [DOI] [Google Scholar]
- 145.Shi F., Zeng X.-N., Zhang G., Ma N., Jiang B., Tu S. Facile synthesis of new 4-aza-podophyllotoxin analogs via microwave-assisted multi-component reactions and evaluation of their cytotoxic activity. Bioorg. Med. Chem. Lett. 2011;21:7119–7123. doi: 10.1016/j.bmcl.2011.09.082. [DOI] [PubMed] [Google Scholar]
- 146.Gu L., Jin C. Synthesis and antitumor activity of α-aminophosphonates containing thiazole[5,4-b]pyridine moiety. Org. Biomol. Chem. 2012;10:7098–7102. doi: 10.1039/c2ob25875g. [DOI] [PubMed] [Google Scholar]
- 147.Bathula C., Tripathi S., Srinivasan R., Jha K.K., Ganguly A., Chakraborty G., Singh S., Munshi P., Sen S. Synthesis of novel 5-arylidenethiazolidinones with apoptotic properties via a three-component reaction using piperidine as a bifunctional reagent. Org. Biomol. Chem. 2016;14:8053–8063. doi: 10.1039/C6OB01257D. [DOI] [PubMed] [Google Scholar]
- 148.Tiwari S.V., Seijas J.A., Vazquez-Tato M.P., Sarkate A.P., Lokwani D.K., Nikalje A.P.G. Ultrasound mediated one-pot, three-component synthesis, docking and ADME prediction of novel 5-amino-2-(4-chlorophenyl)-7-substituted phenyl-8,8a-dihydro-7H-(1,3,4)thiadiazolo(3,2-α)pyrimidine-6-carbonitrile derivatives as anticancer agents. Molecules. 2016;21:894. doi: 10.3390/molecules21080894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Semenov V.V., Lichitsky B.V., Komogortsev A.N., Dudinov A.A., Krayushkin M.M., Konyushkin L.D., Atamanenko O.P., Karmanova I.B., Strelenko Y.A., Shor B., et al. Synthesis and anti-mitotic activity of 6,7-dihydro-4H-isothiazolo[4,5-b]pyridin-5-ones: In vivo and cell-based studies. Eur. J. Med. Chem. 2017;125:573–585. doi: 10.1016/j.ejmech.2016.09.075. [DOI] [PubMed] [Google Scholar]
- 150.Yakaiah S., Kumar P.S.V., Rani P.B., Prasad K.D., Aparna P. Design, synthesis and biological evaluation of novel pyrazolooxothiazolidine derivatives as antiproliferative agents against human lung cancer cell line A549. Bioorg. Med. Chem. Lett. 2018;28:630–636. doi: 10.1016/j.bmcl.2018.01.027. [DOI] [PubMed] [Google Scholar]
- 151.Griglio A., Torre E., Serafini M., Bianchi A., Schmid R., Zabetta G.C., Massarotti A., Sorba G., Pirali T., Fallarini S. A multicomponent approach in the discovery of indoleamine 2,3-dioxygenase 1 inhibitors: Synthesis, biological investigation and docking studies. Bioorg. Med. Chem. Lett. 2018;28:651–657. doi: 10.1016/j.bmcl.2018.01.032. [DOI] [PubMed] [Google Scholar]
- 152.Wu Y.-J. New indole-containing medicinal compounds. In: Gribble G., editor. Heterocyclic Scaffolds II: Reactions and Applications of Indoles. Volume 26. Springer; Berlin/Heidelberg, Germany: 2010. pp. 1–29. Topics in Heterocyclic Chemistry. [DOI] [Google Scholar]
- 153.Sakemi S., Sun H.H. Nortopsentins, A, B, and C. Cytotoxic and antifungal imidazolediylbis [indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem. 1991;56:4304–4307. doi: 10.1021/jo00013a044. [DOI] [Google Scholar]
- 154.Wijeratne E.M.K., Turbyville T.J., Zhang Z., Bigelow D., Pierson L.S., VanEtten H.D., Whitesell L., Canfield L.M., Gunatilaka A.A.L. Cytotoxic constituents of Aspergillus terreus from the rhizosphere of Opuntia versicolor of the Sonoran desert. J. Nat. Prod. 2003;66:1567–1573. doi: 10.1021/np030266u. [DOI] [PubMed] [Google Scholar]
- 155.Radwan M.A.A., Ragab E.A., Sabry N.M., El-Shenawy S.M. Synthesis and biological evaluation of new 3-substituted indole derivatives as potential anti-inflammatory and analgesic agents. Bioorg. Med. Chem. 2007;15:3832–3841. doi: 10.1016/j.bmc.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 156.Zhang H.Z., Drewe J., Tseng B., Kasibhatla S., Cai S.X. Discovery and SAR of indole-2-carboxylic acid benzylidene-hydrazides as a new series of potent apoptosis inducers using a cell-based HTS assay. Bioorg. Med. Chem. 2004;12:3649–3655. doi: 10.1016/j.bmc.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 157.El-Sayed N.S., Shirazi A.N., El-Meligy M.G., El-Ziaty A.K., Rowley D., Sun J., Nagib Z.A., Parang K. Synthesis of 4-aryl-6-indolylpyridine-3-carbonitriles and evaluation of their antiproliferative activity. Tetrahedron Lett. 2014;55:1154–1158. doi: 10.1016/j.tetlet.2013.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sashidhara K.V., Modukuri R.K., Singh S., Bhaskara Rao K., Aruna Teja G., Gupta S., Shukla S. Design and synthesis of new series of coumarin-aminopyran derivatives possessing potential anti-depressant-like activity. Bioorg. Med. Chem. Lett. 2015;25:337–341. doi: 10.1016/j.bmcl.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 159.Gupta S., Maurya P., Upadhyay A., Kushwaha P., Krishna S., Siddiqi M.I., Sashidhara K.V., Banerjee D. Synthesis and bio-evaluation of indole-chalcone based benzopyrans as promising antiligase and antiproliferative agents. Eur. J. Med. Chem. 2018;143:1981–1996. doi: 10.1016/j.ejmech.2017.11.015. [DOI] [PubMed] [Google Scholar]
- 160.Knölker H.J., Reddy K.R. Isolation and synthesis of biologically active carbazole alkaloids. Chem. Rev. 2002;102:4303–4427. doi: 10.1021/cr020059j. [DOI] [PubMed] [Google Scholar]
- 161.Indumathi T., Ahamed V.S.J., Moon S.S., Fronczek F.R., Prasad K.J.R. L-Proline anchored multicomponent synthesis of novel pyrido[2,3-a]carbazoles; investigation of in vitro antimicrobial, antioxidant, cytotoxicity and structure activity relationship studies. Eur. J. Med. Chem. 2011;46:5580–5590. doi: 10.1016/j.ejmech.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 162.Reddy P.N., Padmaja P., Reddy B.R., Jadav S.S. Synthesis, in vitro antiproliferative activity, antioxidant activity and molecular modeling studies of new carbazole Mannich bases. Med. Chem. Res. 2017;26:2243–2259. doi: 10.1007/s00044-017-1927-5. [DOI] [Google Scholar]
- 163.Sathiyachandran P., Manogaran P., Nesterov V.N., Padma V.V., Rajendra Prasad K.J. Design and synthesis of novel pyrrolo[2,3-a]carbazoles: 7-Chloro-2-oxo-3a-(2′oxo-2′,3′-dihydro-1′H-indol-3′-yl)-2,3,3a,4,5,10-hexahydro-pyrrolo[3,2-a]carbazole-1-carbonitrile as an efficient anticancer agent. Eur. J. Med. Chem. 2018;150:851–863. doi: 10.1016/j.ejmech.2018.03.056. [DOI] [PubMed] [Google Scholar]
- 164.Canto R.F.S., Bernardi A., Battastini A.M.O., Russowsky D., Eifler-Lima V.L. Synthesis of dihydropyrimidin-2-one/thione library and cytotoxic activity against the human U138-MG and Rat C6 glioma cell lines. J. Braz. Chem. Soc. 2011;22:1379–1388. doi: 10.1590/S0103-50532011000700025. [DOI] [Google Scholar]
- 165.Kappe C.O. Recent advances in the Biginelli dihydropyrimidine synthesis. New tricks from an old dog. Acc. Chem. Res. 2000;33:879–888. doi: 10.1021/ar000048h. [DOI] [PubMed] [Google Scholar]
- 166.Climent M.J., Corma A., Iborra S. Homogeneous and heterogeneous catalysts for multicomponent reactions. RSC Adv. 2012;2:16–58. doi: 10.1039/C1RA00807B. [DOI] [Google Scholar]
- 167.Biginelli P. Synthesis of 3,4-dihydropyrimidin-2(1H)-ones. Gazz. Chim. Ital. 1893;23:360–416. [Google Scholar]
- 168.Da Silva D.L., Reis F.S., Muniz D.R., Ruiz A.L.T.G., de Carvalho J.E., Sabino A.A., Modolo L.V., de Fátima A. Free radical scavenging and antiproliferative properties of Biginelli adducts. Bioorg. Med. Chem. 2012;20:2645–2650. doi: 10.1016/j.bmc.2012.02.036. [DOI] [PubMed] [Google Scholar]
- 169.Yadlapalli R.K., Chourasia O.P., Vemuri K., Sritharan M., Perali R.S. Synthesis and in vitro anticancer and antitubercular activity of diarylpyrazole ligated dihydropyrimidines possessing lipophilic carbamoyl group. Bioorg. Med. Chem. Lett. 2012;22:2708–2711. doi: 10.1016/j.bmcl.2012.02.101. [DOI] [PubMed] [Google Scholar]
- 170.Ramos L.M., Guido B.C., Nobrega C.C., Corrêa J.R., Silva R.G., de Oliveira H.C.B., Gomes A.F., Gozzo F.C., Neto B.A.D. The Biginelli reaction with an imidazolium–tagged recyclable iron catalyst: Kinetics, mechanism, and antitumoral activity. Chem. Eur. J. 2013;19:4156–4168. doi: 10.1002/chem.201204314. [DOI] [PubMed] [Google Scholar]
- 171.Silva G.C.O., Correa J.R., Rodrigues M.O., Alvim H.G.O., Guido B.C., Gatto C.C., Wanderley K.A., Fioramonte M., Gozzo F.C., de Souza R.O.M.A., et al. The Biginelli reaction under batch and continuous flow conditions: Catalysis, mechanism and antitumoral activity. RSC Adv. 2015;5:48506–48515. doi: 10.1039/C5RA07677C. [DOI] [Google Scholar]
- 172.Matiasa M., Camposa G., Santosa A.O., Falcao A., Silvestre S., Alves G. Potential antitumoral 3,4-dihydropyrimidin-2-(1H)-ones: Synthesis, in vitro biological evaluation and QSAR studies. RSC Adv. 2016;6:84943–84958. doi: 10.1039/C6RA14596E. [DOI] [Google Scholar]
- 173.Carosati E., Ioan P., Micucci M., Broccatelli F., Cruciani G., Zhorov B.S., Chiarini A., Budriesi R. 1,4-Dihydropyridine scaffold in medicinal chemistry, the story so far and perspectives (part 2): Action in other targets and antitargets. Curr. Med. Chem. 2012;19:4306–4323. doi: 10.2174/092986712802884204. [DOI] [PubMed] [Google Scholar]
- 174.Gadotti V.M., Bladen C., Zhang F.X., Chen L., Gündüz M.G., Şimşek R., Şafak C., Zamponi G.W. Analgesic effect of a broad-spectrum dihydropyridine inhibitor of voltage-gated calcium channels. Pflug. Arch. 2015;467:2485–2493. doi: 10.1007/s00424-015-1725-1. [DOI] [PubMed] [Google Scholar]
- 175.Thompson P.E., Manganiello V., Degerman E. Re-discovering PDE3 inhibitors--new opportunities for a long neglected target. Curr. Top. Med. Chem. 2007;7:421–436. doi: 10.2174/156802607779941224. [DOI] [PubMed] [Google Scholar]
- 176.Abadi A.H., Abouel-Ella D.A., Lehmann J., Tinsley H.N., Gary B.D., Piazza G.A., Abdel-Fattah M.A.O. Discovery of colon tumor cell growth inhibitory agents through a combinatorial approach. Eur. J. Med. Chem. 2010;45:90–97. doi: 10.1016/j.ejmech.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Radadiya A., Khedkar V., Bavishi A., Vala H., Thakrar S., Bhavsar D., Shah A., Coutinho E. Synthesis and 3D-QSAR study of 1,4-dihydropyridine derivatives as MDR cancer reverters. Eur. J. Med. Chem. 2014;74:375–387. doi: 10.1016/j.ejmech.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 178.Kumari A.K., Reddy V.H., Reddy G.M., Reddy Y.V.R., Leelavathi S. Synthesis of dihydropyridine derivatives under eco-friendly approach and investigation of cytotoxic activity. J. Heterocycl. Chem. 2019;56:1661–1666. doi: 10.1002/jhet.3561. [DOI] [Google Scholar]
- 179.Safak C., Simsek R. Fused 1,4-dihydropyridines as potential calcium modulatory compounds. Mini Rev. Med. Chem. 2006;6:747–755. doi: 10.2174/138955706777698606. [DOI] [PubMed] [Google Scholar]
- 180.Sharma V.K., Singh S.K. Synthesis, utility and medicinal importance of 1,2- and 1,4-dihydropyridines. RSC Adv. 2017;7:2682–2732. doi: 10.1039/C6RA24823C. [DOI] [Google Scholar]
- 181.Yang X., Zhang C., Wu L. L-Proline catalyzed three-component synthesis of para-naphthoquinone-4-aza-podophyllotoxin hybrids as potent antitumor agents. RSC Adv. 2015;5:18945–18951. doi: 10.1039/C4RA16372A. [DOI] [Google Scholar]
- 182.Kumar N.P., Sharma P., Reddy T.S., Nekkanti S., Shankaraiah N., Lalita G., Sujanakumari S., Bhargava S.K., Naidu V.G.M., Kamal A. Synthesis of 2,3,6,7-tetramethoxyphenanthren-9-amine: An efficient precursor to access new 4-aza-2,3-dihydropyridophenanthrenes as apoptosis inducing agents. Eur. J. Med. Chem. 2017;127:305–317. doi: 10.1016/j.ejmech.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 183.Kumar N.P., Sharma P., Reddy T.S., Shankaraiah N., Bhargava S.K., Kamal A. Microwave-assisted one-pot synthesis of new phenanthrene fused-tetrahydrodibenzoacridinones as potential cytotoxic and apoptosis inducing agents. Eur. J. Med. Chem. 2018;151:173–185. doi: 10.1016/j.ejmech.2018.03.069. [DOI] [PubMed] [Google Scholar]
- 184.Relling M.V., Gardner E.E., Sandborn W.J., Schmiegelow K., Pui C.H., Yee S.W., Stein C.M., Carrillo M., Evans W.E., Hicks J.K., et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharm. 2013;93:324–325. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.D´Souza P.F., Ashoka S., Rajan M.S., Shabaraya A.R. Antitumor activity of mercaptopurine in combination with trikatu and gomutra on 20-methylcholantrene induced carcinogenesis. J. Appl. Pharm. Sci. 2013;3:20–24. doi: 10.7324/JAPS.2013.3804. [DOI] [Google Scholar]
- 186.Kowalska A., Latocha M., Pluta K. Synthesis and anticancer activity of thiosubstituted purines. Med. Chem. Res. 2015;24:3107–3116. doi: 10.1007/s00044-015-1364-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Frolova L.V., Magedov I.V., Romero A.E., Karki M., Otero I., Hayden K., Evdokimov N.M., Banuls L.M., Rastogi S.K., Smith W.R., et al. Exploring natural product chemistry and biology with multicomponent reactions. 5. Discovery of a novel tubulin-targeting scaffold derived from the rigidin family of marine alkaloids. J. Med. Chem. 2013;56:6886–6900. doi: 10.1021/jm400711t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Thakur A., Singla R., Jaitak V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015;101:476–495. doi: 10.1016/j.ejmech.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 189.Bubols G.B., Da Rocha Vianna D., Medina-Remon A., von Poser G., Lamuela-Raventos R.M., Eifler-Lima V.L., Garcia S.C. The antioxidant activity of coumarins and flavonoids. Mini Rev. Med. Chem. 2013;13:318–334. doi: 10.2174/138955713804999775. [DOI] [PubMed] [Google Scholar]
- 190.Hu Y.Q., Xu Z., Zhang S., Wu X., Ding J.W., Lv Z.S., Feng L.S. Recent developments of coumarin-containing derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017;136:122–130. doi: 10.1016/j.ejmech.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 191.Hadjipavlou-Litina D., Litinas K., Kontogiorgis C. The anti-inflammatory effect of coumarin and its derivatives. Anti Inflamm. Anti Allergy Agents Med. Chem. 2007;6:293–306. doi: 10.2174/187152307783219989. [DOI] [Google Scholar]
- 192.Tan N., Yazıcı-Tütüniş S., Bilgin M., Tan E., Miski M. Antibacterial activities of pyrenylated coumarins from the roots of Prangos hulusii. Molecules. 2017;22:1098. doi: 10.3390/molecules22071098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Al-Amiery A.A., Kadhum A.A.H., Mohamad A.B. Antifungal activities of new coumarins. Molecules. 2012;17:5713–5723. doi: 10.3390/molecules17055713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Chen Z., Bi J., Su W. Synthesis and antitumor activity of novel coumarin derivatives via a three-component reaction in water. Chin. J. Chem. 2013;31:507–514. doi: 10.1002/cjoc.201201130. [DOI] [Google Scholar]
- 195.Vodnala S., Bhavani A.K.D., Kamutam R., Naidu V.G.M., Prabhakar C. DABCO-catalyzed one-pot three component synthesis of dihydropyrano[3,2-c]chromene substituted quinazolines and their evaluation towards anticancer activity. Bioorg. Med. Chem. Lett. 2016;26:3973–3977. doi: 10.1016/j.bmcl.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 196.Kumar M.R., Manikandan A., Sivakumar A., Dhayabaran V.V. An eco-friendly catalytic system for multicomponent, one-pot synthesis of novel spiro-chromeno indoline-triones and their anti-prostate cancer potentials evaluated via alkaline phosphatase inhibition mechanism. Bioorg. Chem. 2018;81:44–54. doi: 10.1016/j.bioorg.2018.07.037. [DOI] [PubMed] [Google Scholar]
- 197.Kavitha K., Srikrishna D., Aparna P. An efficient one-pot three-component synthesis of 2-(4-(2-oxo-2H-chromen-3-yl)thiazol-2-yl)-3-arylacrylonitriles and their cytotoxic activity evaluation with molecular docking. J. Saudi Chem. Soc. 2019;23:325–337. doi: 10.1016/j.jscs.2018.08.007. [DOI] [Google Scholar]
- 198.Vaarla K., Kesharwani R.K., Santosh K., Vedula R.R., Kotamraju S., Toopurani M.K. Synthesis, biological activity evaluation and molecular docking studies of novel coumarin substituted thiazolyl-3-aryl-pyrazole-4-carbaldehydes. Bioorg. Med. Chem. Lett. 2015;25:5797–5803. doi: 10.1016/j.bmcl.2015.10.042. [DOI] [PubMed] [Google Scholar]
- 199.Shaikh S.K.J., Sannaikar M.S., Kumbar M.N., Bayannavar P.K., Kamble R.R., Inamdar S.R., Joshi S.D. Microwave-expedited green synthesis, photophysical, computational studies of coumarin-3-yl-thiazol-3-yl-1,2,4-triazolin-3-ones and their anticancer activity. ChemistrySelect. 2018;3:4448–4462. doi: 10.1002/slct.201702596. [DOI] [Google Scholar]
- 200.Chougala B.M., Shastri S.L., Holiyachi M., Naik N.S., Shastri L.A., Dodamani S.S., Jalalpure S.S., Dixit S.R., Joshi S.D., Sunagar V.A. Microwave synthesis of coumarinyl substituted pyridine derivatives as potent anticancer agents and molecular docking studies. ChemistrySelect. 2017;2:5234–5242. doi: 10.1002/slct.201700358. [DOI] [Google Scholar]
- 201.Sashidhara K.V., Singh L.R., Choudhary D., Arun A., Gupta S., Adhikary S., Palnati G.R., Konwar R., Trivedi R. Design, synthesis and in vitro evaluation of coumarin–imidazo[1,2-a]pyridine derivatives against cancer induced osteoporosis. RSC Adv. 2016;6:80037–80048. doi: 10.1039/C6RA15674F. [DOI] [Google Scholar]
- 202.Saxena R., Gupta G., Manohar M., Debnath U., Popli P., Prabhakar Y.S., Konwar R., Kumar S., Kumar A., Dwivedi A. Spiro-oxindole derivative 5-chloro-4′,5′-diphenyl-3′-(4-(2-(piperidin-1-yl) ethoxy) benzoyl) spiro[indoline-3,2′-pyrrolidin]-2-one triggers apoptosis in breast cancer cells via restoration of p53 function. Int. J. Biochem. Cell Biol. 2016;70:105–117. doi: 10.1016/j.biocel.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 203.Barmak A., Niknam K., Mohebbi G., Pournabi H. Antibacterial studies of hydroxyspiro[indoline-3,9-xanthene]trione against spiro[indoline3,9-xanthene]trione and their use as acetyl and butyrylcholinesterase inhibitors. Microb. Pathog. 2019;130:95–99. doi: 10.1016/j.micpath.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 204.Guardia A., Baiget J., Cacho M., Pérez A., Ortega-Guerra M., Nxumalo W., Khanye S.D., Rullas J., Ortega F., Jiménez E. Easy-to-synthesize spirocyclic compounds possess remarkable in vivo activity against Mycobacterium tuberculosis. J. Med. Chem. 2018;61:11327–11340. doi: 10.1021/acs.jmedchem.8b01533. [DOI] [PubMed] [Google Scholar]
- 205.Maurya R., Soni A., Anand D., Ravi M., Raju K.S.R., Taneja I., Naikade N.K., Puri S.K., Wahajuddin Kanojiya S., Yadav P.P. Synthesis and antimalarial activity of 3,3-spiroanellated 5,6-disubstituted 1,2,4-trioxanes. ACS Med. Chem. Lett. 2013;4:165–169. doi: 10.1021/ml300188t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Rajanarendar E., Ramakrishna S., Govardhan Reddy K., Nagaraju D., Reddy Y.N. A facile synthesis, anti-inflammatory and analgesic activity of isoxazolyl-2,3-dihydrospiro[benzo[f]isoindole-1,3′-indoline]-2′,4,9-triones. Bioorg. Med. Chem. Lett. 2013;23:3954–3958. doi: 10.1016/j.bmcl.2013.04.053. [DOI] [PubMed] [Google Scholar]
- 207.Ali M.A., Ismail R., Choon T.S., Yoon Y.K., Wei A.C., Pandian S., Kumar R.S., Osman H., Manogaran E. Substituted spiro[2.3′]oxindolespiro[3.2′′]-5,6-dimethoxy-indane-1′′-one-pyrrolidine analogue as inhibitors of acetylcholinesterase. Bioorg. Med. Chem. Lett. 2010;20:7064–7066. doi: 10.1016/j.bmcl.2010.09.108. [DOI] [PubMed] [Google Scholar]
- 208.Arun Y., Bhaskar G., Balachandran C., Ignacimuthu S., Perumal P.T. Facile one-pot synthesis of novel dispirooxindole-pyrrolidine derivatives and their antimicrobial and anticancer activity against A549 human lung adenocarcinoma cancer cell line. Bioorg. Med. Chem. Lett. 2013;23:1839–1845. doi: 10.1016/j.bmcl.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 209.Arun Y., Saranraj K., Balachandran C., Perumal P.T. Novel spirooxindole–pyrrolidine compounds: Synthesis, anticancer and molecular docking studies. Eur. J. Med. Chem. 2014;74:50–64. doi: 10.1016/j.ejmech.2013.12.027. [DOI] [PubMed] [Google Scholar]
- 210.Kamal A., Babu K.S., Vishnu Vardhan M.V.P.S., Hussaini S.M.A., Mahesh R., Shaik S.P., Alarifi A. Sulfamic acid promoted one-pot three-component synthesis and cytotoxic evaluation of spirooxindoles. Bioorg. Med. Chem. Lett. 2015;25:2199–2202. doi: 10.1016/j.bmcl.2015.03.054. [DOI] [PubMed] [Google Scholar]
- 211.Nagaraju B., Kovvuri J., Babu K.S., Adiyala P.R., Nayak V.L., Alarifi A., Kamal A. A facile one-pot C-C and C-N bond formation for the synthesis of spiro-benzodiazepines and their cytotoxicity. Tetrahedron. 2017;73:6969–6976. doi: 10.1016/j.tet.2017.10.060. [DOI] [Google Scholar]
- 212.Dong H., Song S., Li J., Xu C., Zhang H., Ouyang L. The discovery of oxazolones-grafted spirooxindoles via three-component diversity oriented synthesis and their preliminary biological evaluation. Bioorg. Med. Chem. Lett. 2015;25:3585–3591. doi: 10.1016/j.bmcl.2015.06.076. [DOI] [PubMed] [Google Scholar]
- 213.Kumar A., Gupta G., Srivastava S., Bishnoi A.K., Saxena R., Kant R., Khanna R.S., Maulik P.R., Dwivedi A. Novel diastereoselective synthesis of spiropyrrolidine-oxindole derivatives as anti-breast cancer agents. RSC Adv. 2013;3:4731–4735. doi: 10.1039/c3ra21595d. [DOI] [Google Scholar]
- 214.George R.F., Panda S.S., Shalaby E.-S.M., Srour A.M., Farag I.S.A., Girgis A.S. Synthesis and molecular modeling studies of indole-based antitumor agents. RSC Adv. 2016;6:45434–45451. doi: 10.1039/C6RA07061B. [DOI] [Google Scholar]
- 215.Islam M.S., Ghawas H.M., El-Senduny F.F., Al-Majid A.M., Elshaier Y.A.M.M., Badria F.A., Barakat A. Synthesis of new thiazolo-pyrrolidine–(spirooxindole) tethered to 3-acylindole as anticancer agents. Bioorg. Chem. 2019;82:423–430. doi: 10.1016/j.bioorg.2018.10.036. [DOI] [PubMed] [Google Scholar]
- 216.Parthasarathy K., Praveen C., Balachandran C., Senthil Kumar P., Ignacimuthu S., Perumal P.T. Cu(OTf)2 catalyzed three component reaction: Efficient synthesis of spiro[indoline-3,4′-pyrano[3,2-b]pyran derivatives and their anticancer potency towards A549 human Lung cancer cell lines. Bioorg. Med. Chem. Lett. 2013;23:2708–2713. doi: 10.1016/j.bmcl.2013.02.086. [DOI] [PubMed] [Google Scholar]
- 217.Wu L., Liu Y., Li Y. Synthesis of spirooxindole-O-naphthoquinone-tetrazolo[1,5-a]pyrimidine hybrids as potential anticancer agents. Molecules. 2018;23:2330. doi: 10.3390/molecules23092330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chavan P.V., Desai U.V., Wadgaonkar P.P., Tapase S.R., Kodam K.M., Choudhari A., Sarkar D. Click chemistry based multicomponent approach in the synthesis of spirochromenocarbazole tethered 1,2,3-triazoles as potential anticancer agents. Bioorg. Chem. 2019;85:475–486. doi: 10.1016/j.bioorg.2019.01.070. [DOI] [PubMed] [Google Scholar]
- 219.Liu X.-L., Feng T.-T., Jiang W.-D., Yang C., Tian M.-Y., Jiang Y., Lin B., Zhao Z., Zhou Y. Molecular hybridization-guided 1,3-dipolar cycloaddition reaction enabled pyrimidine-fused spiropyrrolidine oxindoles synthesis as potential anticancer agents. Tetrahedron Lett. 2016;57:4411–4416. doi: 10.1016/j.tetlet.2016.08.063. [DOI] [Google Scholar]
- 220.Sudhapriya N., Perumal P.T., Balachandran C., Ignacimuthu S., Sangeetha M., Doble M. Synthesis of new class of spirocarbocycle derivatives by multicomponent domino reaction and their evaluation for antimicrobial, anticancer activity and molecular docking studies. Eur. J. Med. Chem. 2014;83:190–207. doi: 10.1016/j.ejmech.2014.05.065. [DOI] [PubMed] [Google Scholar]
- 221.Pirali T., Faccio V., Mossetti R., Grolla A.A., Di Micco S., Bifulco G., Genazzani A.A., Tron G.C. Synthesis, molecular docking and biological evaluation as HDAC inhibitors of cyclopeptide mimetics by a tandem three-component reaction and intramolecular [3+2] cycloaddition. Mol. Divers. 2010;14:109–121. doi: 10.1007/s11030-009-9153-9. [DOI] [PubMed] [Google Scholar]
- 222.Baviskar A.T., Madaan C., Preet R., Mohapatra P., Jain V., Agarwal A., Guchhait S.K., Kundu C.N., Banerjee U.C., Bharatam P.V. N-Fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase IIr and induce apoptosis in G1/S phase. J. Med. Chem. 2011;54:5013–5030. doi: 10.1021/jm200235u. [DOI] [PubMed] [Google Scholar]
- 223.Le Floch C., Le Gall E., Léonel E., Martens T., Cresteil T. Synthesis and cytotoxic evaluation of novel paraconic acid analogs. Bioorg. Med. Chem. Lett. 2011;21:7054–7058. doi: 10.1016/j.bmcl.2011.09.092. [DOI] [PubMed] [Google Scholar]
- 224.Shi F., Zeng X.-N., Cao X.-D., Zhang S., Jiang B., Zheng W.-F., Tu S.-J. Design and diversity-oriented synthesis of novel 1,4-thiazepan-3-ones fused with bioactive heterocyclic skeletons and evaluation of their antioxidant and cytotoxic activities. Bioorg. Med. Chem. Lett. 2012;22:743–746. doi: 10.1016/j.bmcl.2011.09.081. [DOI] [PubMed] [Google Scholar]
- 225.Kou L., Wang M.-J., Wang L.-T., Zhao X.-B., Nan X., Yang L., Liu Y.-Q., Morris-Natschke S.L., Lee K.-H. Toward synthesis of third-generation spin-labeled podophyllotoxin derivatives using isocyanide multicomponent reactions. Eur. J. Med. Chem. 2014;75:282–288. doi: 10.1016/j.ejmech.2014.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.EL Massry A.M., Asal A.M., Khattab S.N., Haiba N.S., Awney H.A., Helmy M., Langer V., Amer A. Synthesis and structure elucidation of novel fused 1,2,4-triazine derivatives as potent inhibitors targeting CYP1A1 activity. Bioorg. Med. Chem. 2012;20:2624–2637. doi: 10.1016/j.bmc.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 227.Liao S.-R., Du L.-J., Qin X.-C., Xu L., Wang J.-F., Zhou X.-F., Tu Z.-F., Li L., Liu Y.-H. Site selective synthesis of cytotoxic 1,3,6-trisubstituted 3,6-diunsaturated (3Z,6Z)-2,5-diketopiperazines via a one-pot multicomponent method. Tetrahedron. 2016;72:1051–1057. doi: 10.1016/j.tet.2015.12.073. [DOI] [Google Scholar]
- 228.Raghav N., Jangra S., Kumar A., Bhattacharyya S., Wadhwa D., Sindhu J. Cathepsin B, H and L inhibitors as cell proliferating agents: Design, synthesis, computational and pharmacological studies of some novel 2-(2-naphthoyl)-6,6-dimethyl-3-aryl-2,3,6,7-tetrahydrobenzofuran-4(5H)-ones. RSC Adv. 2016;6:34588–34599. doi: 10.1039/C6RA06480A. [DOI] [Google Scholar]
- 229.Gohel J.N., Lunagariya K.S., Kapadiya K.M., Khunt R.C. An efficient protocol for the synthesis of 1, 5-disubstituted tetrazole derivatives via a TMS-N3 based Ugi reaction and their anti-cancer activity. ChemistrySelect. 2018;3:11657–11662. doi: 10.1002/slct.201802638. [DOI] [Google Scholar]
- 230.He L.-J., Yang D.-L., Li S.-K., Zhang Y.-J., Tang Y., Lei J., Frett B., Lin H.-K., Li H.-Y., Chen Z.Z., et al. Facile construction of fused benzimidazole-isoquinolinones that induce cell-cycle arrest and apoptosis in colorectal cancer cells. Bioorg. Med. Chem. 2018;26:3899–3908. doi: 10.1016/j.bmc.2018.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mohareb R.M., Hassaneen H.M., Ahmed N.A.M. New approaches for the synthesis of thiophenes, thiazole, pyran derivatives, and their antitumor evaluation. Jacobs J. Org. Chem. 2016;1:1–13. [Google Scholar]
- 232.Pelliccia S., Amato J., Capasso D., Di Gaetano S., Massarotti A., Piccolo M., Irace C., Tron G.C., Pagano B., Randazzo A., et al. Bio-inspired dual-selective BCL-2/c-MYC G-quadruplex binders: Design, synthesis, and anticancer activity of drug-like imidazo[2,1-i]purine derivatives. J. Med. Chem. 2019 doi: 10.1021/acs.jmedchem.9b00262. [DOI] [PubMed] [Google Scholar]