

Here, we report a class of tryptophan trimers and tetramers that inhibit (at low micromolar range) dengue and Zika virus infection in vitro. These compounds (AL family) have three or four peripheral tryptophan moieties directly linked to a central scaffold through their amino groups; thus, their carboxylic acid groups are free and exposed to the periphery.
KEYWORDS: SPR, Zika, antivirals, dengue, envelope protein, virus attachment
ABSTRACT
Here, we report a class of tryptophan trimers and tetramers that inhibit (at low micromolar range) dengue and Zika virus infection in vitro. These compounds (AL family) have three or four peripheral tryptophan moieties directly linked to a central scaffold through their amino groups; thus, their carboxylic acid groups are free and exposed to the periphery. Structure-activity relationship (SAR) studies demonstrated that the presence of extra phenyl rings with substituents other than COOH at the N1 or C2 position of the indole side chain is a requisite for the antiviral activity against both viruses. The molecules showed potent antiviral activity, with low cytotoxicity, when evaluated on different cell lines. Moreover, they were active against laboratory and clinical strains of all four serotypes of dengue virus as well as a selected group of Zika virus strains. Additional mechanistic studies performed with the two most potent compounds (AL439 and AL440) demonstrated an interaction with the viral envelope glycoprotein (domain III) of dengue 2 virus, preventing virus attachment to the host cell membrane. Since no antiviral agent is approved at the moment against these two flaviviruses, further pharmacokinetic studies with these molecules are needed for their development as future therapeutic/prophylactic drugs.
INTRODUCTION
The Flaviviridae is a large family of viruses comprising four distinct genera, Hepaci-, Pegi-, Pesti-, and Flavivirus (1). The Flavivirus genus contains many important human pathogens, such as dengue virus (DENV) and Zika virus (ZIKV). These members are mainly transmitted by two mosquito species (Aedes aegypti and Aedes albopictus) and constitute a significant cause of morbidity and mortality worldwide (2–4). Socioeconomical events, such as population growth, uncontrolled urbanization, as well as increased international air traffic, constitute the main factors for the emergence of both DENV and ZIKV worldwide (5). Moreover, the lack or reduction of appropriate vector control programs in some geographical areas raises the possibility of their establishment, which might result in future outbreaks (6). The most recent example was in the Philippines in 2019, where an outbreak of dengue fever led to hundreds of deaths and was declared a national epidemic. Despite the long-term efforts to restrict the spread of and infection by these viruses, no antivirals are commercially available. In addition, new flaviviruses may appear in the near future, making even more urgent the need for the development of novel compounds (7, 8).
DENV is the most prevalent arbovirus, infecting approximately 400 million people per year, of which almost 100 million develop clinical symptoms (9). There are four antigenically distinct serotypes (DENV1 to -4), which cause both a mild febrile-like illness (dengue fever) and severe dengue, characterized by increased vascular permeability and plasma leakage, which may result in a shock syndrome and eventually death (10, 11).
The closely related ZIKV was first discovered in 1947 in Uganda but was neglected until 2007, after its reappearance during an outbreak on the Micronesian Yap Islands (12). In addition, two large outbreaks in French Polynesia (2013), Brazil, and other Central/South American countries (2015 to 2016) led the World Health Organization (WHO) to declare ZIKV a Public Health Emergency of International Concern in 2016 (13, 14). During that period, almost 40,000 symptomatic cases were reported in the Americas (15). In general, ZIKV causes mild symptoms, like fever, headache, and joint pain. However, the virus has gained increased interest during recent years because of its correlation with severe neurological manifestations in newborns (microcephaly) and in adults (Guillain-Barré syndrome) (16, 17). Moreover, several studies have confirmed that ZIKV is transmitted to humans not only by virus-infected mosquitoes but also through blood transfusion, through sexual contact, and vertically, lowering the viral barrier (18–20).
Over the past few years, great effort was carried out in vaccine and antiviral research against these two viruses. In particular, numerous vaccine platforms have been developed, including recombinant viral proteins, whole inactivated virus, live-attenuated virus, and DNA subunits. Among them, the live-attenuated tetravalent vaccine (Dengvaxia) was licensed against DENV in approximately 20 countries in 2016. Although the vaccine boosts a strong immune response in individuals with a history of DENV infection, the risk of severe dengue is significantly increased in those who have not experienced an infection before, resulting in large numbers of hospitalizations (21, 22). Although the underlying cause of this phenomenon is not yet identified, different reports state that the vaccine may act as a silent/primary infection, producing insufficient neutralizing antibody levels. Subsequently, a secondary infection leads to more severe disease symptoms due to antibody-dependent enhancement (ADE) (23). Despite the promising outcomes in vaccine development, important limitations need to be considered, as one of the main hurdles regarding their efficacy and safety is antibody cross-reactivity between ZIKV and DENV (24, 25).
In addition, a large number of Food and Drug Administration (FDA)-approved drugs have been reevaluated against both viruses, targeting different viral and host proteins (26, 27). However, none of these drugs is approved at the moment. Therefore, there is an imperative need for the development of novel antivirals to prevent/treat infection by these emerging flaviviruses. Among them, entry inhibitors are particularly attractive due to their ability to block de novo cell infection, averting the uptake of the virus by uninfected cells (28). In the huge family of those inhibitors, polyanionic molecules represent one of the major classes, exerting antiviral activity against several viruses, such as HIV and herpes simplex virus (29–31). However, in some cases, their mechanism of action remains unclear.
In this study, we screened a library of chemically divergent compounds (approximately 500 molecules) for their activity against DENV and ZIKV infection. Among them, a novel series of tryptophan (Trp) trimers and tetramers (belonging to the AL class, which was developed by our group) (Fig. 1) were identified as potent inhibitors against both flaviviruses in the low micromolar range. Structure-activity relationship (SAR) studies helped us to determine the structural requirements for their selective antiviral activity. These molecules were found to be active in nonhuman primate and human cell lines. Further mechanism-of-action studies revealed that these compounds inhibit viral replication during the early stages by interacting with the domain III of the envelope protein of DENV. This domain plays a pivotal role in the attachment and entry of the virus into target cells via its interaction with specific host receptors (32, 33).
FIG 1.

Structure of the tested analogues.
RESULTS
Synthesis and SAR studies.
The synthesis of trimers AL352, AL438, AL439, AL440, AL467, and AL455 (Fig. 1) has been described recently (34). To complete the structure-activity relationship (SAR) studies, some novel compounds have been synthesized: trimers AL544, AL545, AL437, and AL484, tetramer AL531, and monomers AL441 and AL442 (Fig. 1). These molecules were prepared using the synthetic strategies described in the supplemental material. As seen from Fig. 1, all the tripodal N1-substituted derivatives possess a common 4-(2-carboxyethyl)-4-nitroheptanedioic acid central core. On the other hand, the tetrapodal derivative AL531 has a pentaerythritol central core. To evaluate the antiviral activity of these analogues, the prototype strains of DENV2 (NGC) and ZIKV (MR766) were tested in African green monkey kidney cells (Vero). Cytotoxicity assessment was performed in parallel. As shown in Table 1, modifications on the structure of AL analogues lead to significant differences in their antiviral activities. In particular, the unsubstituted trimer AL352 did not show any antiviral activity up to 100 μM, and the N1-methyl trimer AL544 also proved inactive at subtoxic concentrations. However, the N1-benzyl trimers AL440 and AL439, bearing extra benzyl and 4-fluorobenzyl rings, respectively, at the N1 position of the indole side chain of Trp, showed remarkable antiviral activity against DENV2 (50% effective concentration [EC50], 3.5 and 3.6 μM, respectively) and ZIKV (EC50, 3.3 and 5.8 μM, respectively). On the contrary, trimer AL438, replaced at N1 with a 4-carboxybenzyl moiety, was not found to be active against these viruses. Displacement of the substituted phenyl ring at the C2 position of the indole side chain led to the trimers AL467 (4-CF3-phenyl at C2) and AL545 (3-CF3-phenyl rings at C2), which showed activity against both DENV and ZIKV at low micromolar ranges. On the contrary, AL455, with a 4-carboxyphenyl moiety at C2, showed no activity. Moreover, the trimer AL437, with a phenylsulfenyl moiety at the C2 position, presented no activity, while trimer AL484, with a 4-nitrophenylsulfenyl moiety at C2, was active against both DENV and ZIKV. This result confirms that phenyl rings replaced with noncarboxylated groups at the C2 position of the indole ring are beneficial for antiviral activity. In addition, the 4-carboxybenzyl group at the N1 position present in monomer AL441 abolished the antiviral activity, while that with a substituted 4-fluorobenzyl group monomer AL442 was active. On the other hand, AL442 was less potent than the trimer AL439 and tetramer AL531 despite the fact that these three analogues are altered with the same group at the N1 position (4-fluorobenzyl). Although the tetramer AL531 showed remarkable antiviral activity against DENV2 and ZIKV (EC50, 4.0 and 1.9 μM, respectively), its marked cellular cytotoxicity made it a less attractive candidate for further evaluation. These findings suggest that multivalence plays an important role in improved antiviral activity.
TABLE 1.
Cytotoxicity and antiviral activity of a series of AL compounds against DENV2 and ZIKV prototype strains in Vero cellsc
| Compound | Mol wt (Da) | CC50a (μM) | EC50b (μM) |
|
|---|---|---|---|---|
| DENV2 | ZIKV | |||
| AL352 | 835.9 | >100 | No activity | No activity |
| AL544 | 877.9 | >100 | No activity | No activity |
| AL440 | 1106.2 | 98 ± 3 | 3.5 ± 2.9 | 3.3 ± 1.4 |
| AL439 | 1160.2 | 80 ± 30 | 3.6 ± 1.1 | 5.8 ± 1.1 |
| AL438 | 1238.3 | >100 | No activity | No activity |
| AL467 | 1268.1 | >100 | 8.0 ± 1.4 | 10.5 ± 5.0 |
| AL545 | 1310.2 | 43 ± 8 | 0.7 ± 0.3 | 1.3 ± 0.5 |
| AL455 | 1196.2 | >100 | No activity | No activity |
| AL437 | 1160.3 | 83.5 ± 6.5 | No activity | No activity |
| AL484 | 1337.4 | >100 | 3.6 ± 0.4 | 15.6 ± 13.6 |
| AL531 | 1601.7 | 42.5 ± 1.3 | 4.0 ± 1.6 | 1.9 ± 0.1 |
| AL441 | 472.5 | >100 | No activity | No activity |
| AL442 | 446.5 | 45.6 ± 5 | 10.8 ± 4.3 | 6.9 ± 2.2 |
CC50, concentration required to reduce cell viability by 50%.
EC50, effective concentration of each compound able to inhibit viral replication by 50%.
The laboratory-adapted strains DENV2 NGC and ZIKV MR766 were used.
In order to gain more information on the mechanism of action of this series of molecules, compounds AL439 and AL440, with the best selectivity index (cellular cytotoxicity/antiviral activity), were selected for further studies.
Cytotoxicity and antiviral activity of AL439 and AL440 against DENV and ZIKV infection in different cell lines.
Table 2 shows the cellular cytotoxicity of each molecule, evaluated in different cell lines. AL439 and AL440 showed particularly higher cytotoxic levels in placenta choriocarcinoma (Jeg3; concentration required to reduce cell viability by 50% [CC50], 35 μM to 45 μM) and human umbilical vein endothelial cells (HUVEC; CC50, 42.4 μM to 43.7 μM) than in the other human cells. In addition, AL439 was marginally toxic in green monkey (Vero) and baby hamster kidney (BHK) cells (80 μM and 88 μM, respectively). The inhibitory effects of AL439 and AL440 against the prototype strains of DENV2 (Table 3) and ZIKV (Table 4) next were evaluated in several susceptible cell lines. Although these derivatives were active against both flaviviruses, minor differences were observed among the selected cell lines. More specifically, AL439 and AL440 demonstrated insignificantly higher activity in human hepatocarcinoma (Huh; EC50, 0.6 μM to 1.4 μM), choriocarcinoma (Jeg3; EC50, 0.3 μM to 0.9 μM), and lung adenocarcinoma (A549; EC50, 0.8 μM to 2.7 μM) cells than in hamster (BHK; EC50, 1.0 μM to 4.0 μM) and green monkey kidney (Vero; EC50, 3.3 μM to 5.8 μM) cells against both viral strains. Furthermore, the activity of AL439 and AL440 in HUVEC (EC50, 2.1 μM to 5.5 μM) was quite comparable to those observed in nonhuman cells (Vero and BHK). Finally, Table 5 exhibits the antiviral activity of both AL derivatives in monocyte-derived dendritic cells (MDDC). This cell line represents one of the primary target cell types during a flavivirus infection. Notably, both molecules were active against the prototype strains of DENV2 (EC50, 3.6 μM to 7.6 μM) and ZIKV (EC50, 1.5 μM to 3.9 μM), with no cytotoxic effects at the highest tested concentration (CC50 of >50 μM).
TABLE 2.
Cellular cytotoxicity of AL439 and AL440 in different cell lines
| Compound | CC50a (μM) |
|||||
|---|---|---|---|---|---|---|
| Vero | BHK | Huh | Jeg3 | A549 | HUVEC | |
| AL439 | 80 ± 30 | 88 ± 17 | >100 | 35 ± 10 | >100 | 42.4 ± 1.7 |
| AL440 | 98 ± 3 | >100 | >100 | 45 ± 5 | >100 | 43.7 ± 5.1 |
Data represent average ± SD values from at least three independent experiments.
TABLE 3.
Antiviral activity of AL439 and AL440 against DENV2 NGC prototype strain in different cell linesa
| Compound | EC50 (μM) |
|||||
|---|---|---|---|---|---|---|
| Vero | BHK | Huh | Jeg3 | A549 | HUVEC | |
| AL439 | 3.6 ± 1.1 | 1.4 ± 0.1 | 1.2 ± 0.1 | 0.5 ± 0.1 | 0.8 ± 0.1 | 3.3 ± 0.8 |
| AL440 | 3.5 ± 2.9 | 3.8 ± 1.8 | 1.1 ± 0.1 | 0.9 ± 0.2 | 1.0 ± 0.1 | 4.8 ± 1.6 |
Data represent average ± SD values from at least three independent experiments.
TABLE 4.
Antiviral activity of AL439 and AL440 against ZIKV prototype strain in different cell linesa
| Compound | EC50 (μM) |
|||||
|---|---|---|---|---|---|---|
| Vero | BHK | Huh | Jeg3 | A549 | HUVEC | |
| AL439 | 5.8 ± 1.1 | 4.0 ± 0.1 | 0.6 ± 0.2 | 0.6 ± 0.1 | 2.7 ± 0.6 | 2.1 ± 0.7 |
| AL440 | 3.3 ± 1.4 | 1.0 ± 0.3 | 1.4 ± 0.3 | 0.3 ± 0.1 | 2.2 ± 0.2 | 5.1 ± 2.2 |
Data represent average ± SD values from at least three independent experiments. The strain used was the ZIKV MR766 strain.
TABLE 5.
Anti-DENV and anti-ZIKV activities and cytotoxicity of AL439 and AL440 in MDDC
| Compound | CC50 (μM) | Values fora: |
|||
|---|---|---|---|---|---|
| DENV2 NGC |
ZIKV MR766 |
||||
| EC50 (μM) | SI | EC50 (μM) | SI | ||
| AL439 | >50 | 7.6 ± 0.8 | >6.5 | 1.5 ± 0.2 | >33 |
| AL440 | >50 | 3.6 ± 0.8 | >13.9 | 3.9 ± 0.6 | >12.8 |
All EC50 values were calculated as averages ± SD from four different donors. Selectivity index (SI; CC50/EC50) for each molecule is also included.
Evaluation of antiviral activity against ZIKV by immunofluorescence staining.
We further assessed the antiviral activity of AL440 against the prototype ZIKV MR766 strain using immunofluorescence staining for viral envelope glycoprotein. As shown in Fig. 2, incubation of Vero cells with 20 μM (Fig. 2C) and 10 μM (Fig. 2D) AL440 led to inhibition of virus infection. However, treatment of cells with less than 2 μM AL440 (Fig. 2E and F) was not able to hamper the infection. Negative (dimethyl sulfoxide [DMSO]-treated cells in the absence of AL440) (Fig. 2A) and positive (virus-infected cells in the absence of AL440) (Fig. 2B) controls were also included.
FIG 2.
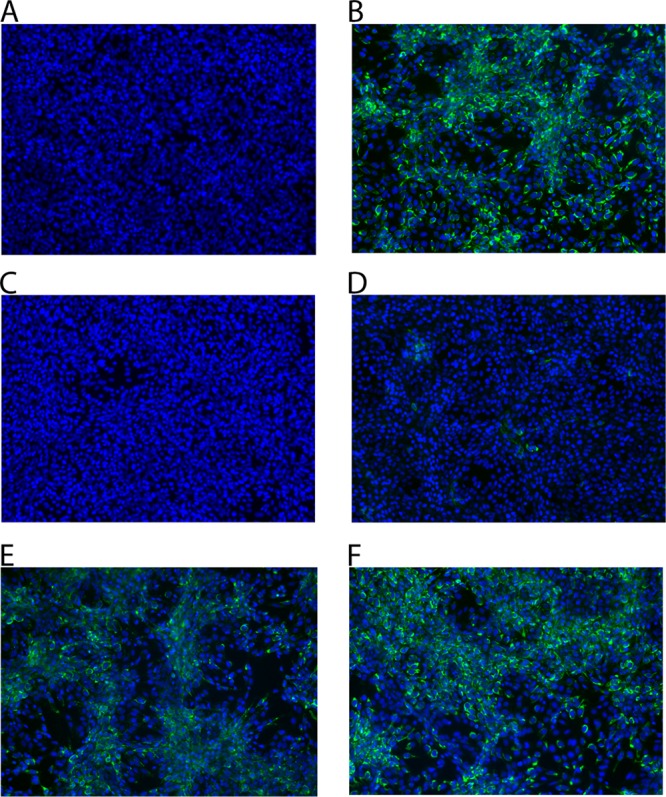
Detection of ZIKV infection using immunofluorescence staining for envelope glycoprotein in the absence and presence of AL440. Exposure of Vero cells to the prototype ZIKV MR766 strain (MOI, 0.2) along with AL440 at different concentrations. Fluorescence images were acquired 72 h postinfection. (A) Cell control. (B) Virus control. Strains were exposed to 20 μM (C), 10 μM (D), 2 μM (E), and 0.4 μM (F) AL440.
Antiviral activity of AL439 and AL440 against different DENV and ZIKV laboratory and clinical strains.
In order to assess broad-spectrum activity, AL439 and AL440 were tested against a panel of different laboratory and clinical strains of DENV and ZIKV in Vero cells. Tables 6 and 7 summarize the results of these experiments. In general, comparable antiviral activity was noticed against the laboratory-adapted and clinical strains of DENV for both compounds. However, an improvement in antiviral activity was observed for AL439 against the DENV4 clinical strain (EC50, 1.2 ± 0.1 μM) compared to the one against the DENV4 Dak laboratory strain (EC50, 7.3 ± 2.8 μM) (Table 6).
TABLE 6.
Antiviral activity of AL439 and AL440 against several DENV laboratory and clinical strains in Vero cells
| Compound | EC50 (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Laboratory |
Clinical |
|||||||
| DENV1 Djibouti | DENV2 NGC | DENV3 H87 | DENV4 Dak | DENV1 | DENV2 | DENV3 | DENV4 | |
| AL439 | 1.7 ± 0.4 | 3.6 ± 1.1 | 3.9 ± 3.0 | 7.3 ± 2.8 | 1.5 ± 0.2 | 2.4 ± 0.9 | 1.1 ± 0.1 | 1.2 ± 0.1 |
| AL440 | 2.5 ± 0.8 | 3.5 ± 2.9 | 5.1 ± 4.5 | 5.7 ± 1.1 | 2.1 ± 0.3 | 2.5 ± 0.7 | 5.5 ± 2.5 | 4.4 ± 0.2 |
TABLE 7.
Antiviral activity of AL439 and AL440 against several ZIKV laboratory and clinical strains in Vero cells
| Compound | EC50 (μM) |
||
|---|---|---|---|
| Laboratory strain MR766 | Clinical strain |
||
| PRVAVC59 | FLR | ||
| AL439 | 5.8 ± 1.1 | 5.2 ± 0.6 | 4.5 ± 1.0 |
| AL440 | 3.3 ± 1.4 | 4.0 ± 0.7 | 0.7 ± 0.2 |
Regarding their activity against ZIKV, no differences were observed between the laboratory (MR766) and the clinical (PRVAVC59 and FLR) isolates in the presence of AL439. On the contrary, the activity of AL440 was slightly higher against the ZIKV FLR strain (EC50, 0.7 ± 0.2 μM) than MR766 (EC50, 3.3 ± 1.4 μM) and PRVAVC59 (EC50, 4.0 ± 0.7 μM) (Table 7).
AL compounds inhibit DENV infection during the early stages.
To pinpoint at which stage of the viral replicative cycle AL439 and AL440 act, a time-of-drug-addition assay was performed. As shown in Fig. 3A, pretreatment of cells with 10 μM AL compounds for 2 h before virus addition (time point 0) resulted in a complete inhibition of viral infection. A similar inhibition profile was observed when the compounds and the virus were added to the cells at the same time. On the other hand, viral infection was only inhibited by approximately 25% when AL439 and AL440 were added 2 h postinfection (p.i.). Addition of compounds at later time points did not affect viral infection. Results obtained using dextran sulfate (DS; entry inhibitor) and 7-deaza-2′-C-methyladenosine (7DMA; viral polymerase inhibitor) are also represented in Fig. 3. Specifically, DS showed a suppression of viral infection during the early stages (−2 h and 0 h p.i.), while 7DMA was able to limit the infection even at 12 h p.i. Based on these data, we suggest that AL439 and AL440 interfere with the early stages of viral infection.
FIG 3.

AL compounds inhibit infection during the early stages without any effect on the replication complex. (A, upper) Schematic representation of the time-of-drug-addition assay. (Lower) Pretreatment of cells with the compounds during the first hours (−2 h and 0 h) inhibits DENV infection. No inhibition was noticed after addition of the molecules at later time points (2 h to 18 h). The AL analogues (green-red) show a profile similar to that of the established entry inhibitor DS (blue). On the other hand, viral polymerase inhibitor 7DMA (black) is able to suppress the infection even at later stages (12 h p.i.). Dashed line represents virus control. (B) Stably transfected BHK cells with a DENV subgenomic replicon were incubated with serial dilutions of AL molecules (0.8 to 100 μM). A decrease in the luciferase activity was only observed for the replication inhibitor 7DMA but not for AL439, AL440, and DS (blue). Incubation of AL439 and AL440 at the highest concentration led to the reduction of cellular metabolism (red). All data are averages from two independent experiments.
To further confirm these findings, a subgenomic replicon assay was carried out. In particular, different concentrations of AL439 and AL440 were added to BHK/DENV cells. Based on the data shown in Fig. 3B, we observed that neither compound inhibited replication (no change in luciferase activity), indicating an interaction with the structural proteins of the virus. However, we noticed that treatment of cells with 100 μM molecules led to a decrease in luciferase activity. This observation can be explained by the increased cellular cytotoxicity. A comparable result was obtained for the reference entry inhibitor (DS). On the other hand, 7DMA presented a strong inhibition of viral replication (approximately 100% luciferase reduction was observed at 20 μM).
In addition, we examined the effect of AL439 and AL440 on DENV attachment. Specifically, we incubated Vero cells in the presence of several concentrations of AL molecules along with a DENV2 strain for 2 h at 4°C, allowing viral attachment to the cells. Cells then were washed and incubated with medium for an additional 72 h. Interestingly, we observed that virus infection can be effectively inhibited even if the compounds were removed 2 h p.i. (EC50 for AL439 and AL440, 3.6 ± 0.04 μM and 3.8 ± 0.1 μM, respectively). These findings are quite comparable to our observations from the initial screening and indicate that our compounds interfere with the attachment of the virus to the cells, corroborating the results that we obtained from the aforementioned assays.
AL compounds interact with DENV particles in a direct way.
To find out whether AL440 directly interacts with the virus, an ultrafiltration assay was performed. Specifically, viral stock was incubated with AL440 at different concentrations (50 μM, 10 μM, and 2 μM) for 1 h. The samples then were passed through a concentrator filter unit (10-kDa cutoff) to remove any unbound material (due to smaller size), and the retained fraction on the filter was evaluated in a plaque assay for inhibition of viral infectivity. As shown in Fig. 4A, cells were completely infected after their treatment with 2 μM AL440. On the other hand, incubation of cells with 10 μM resulted in a reduction of the number of viral plaques. Finally, the presence of AL440 at the highest concentration (50 μM) protected the cells from viral infection (less than 10 viral plaques were observed). As expected, incubation with epigallocatechin gallate (EGCG; 0.1 mg/ml, positive control) demonstrated a decrease in the number of viral plaques. Furthermore, these findings were confirmed by the virus inactivation assay. More specifically, viral particles were allowed to incubate with 10 μM AL440 and the sample was subsequently diluted to a suboptimal concentration (conditions where the compound is no longer active while viral particles are still present and infectious, unless they are inactivated by the molecule) before infecting the cells. As shown in Fig. 4B, the presence of AL440 led to protection of the cells. On the other hand, absence of AL440 (virus control) resulted in the complete destruction of the cell monolayer. The data described above clearly showed that AL440 had a direct impact on free viral particles as such.
FIG 4.
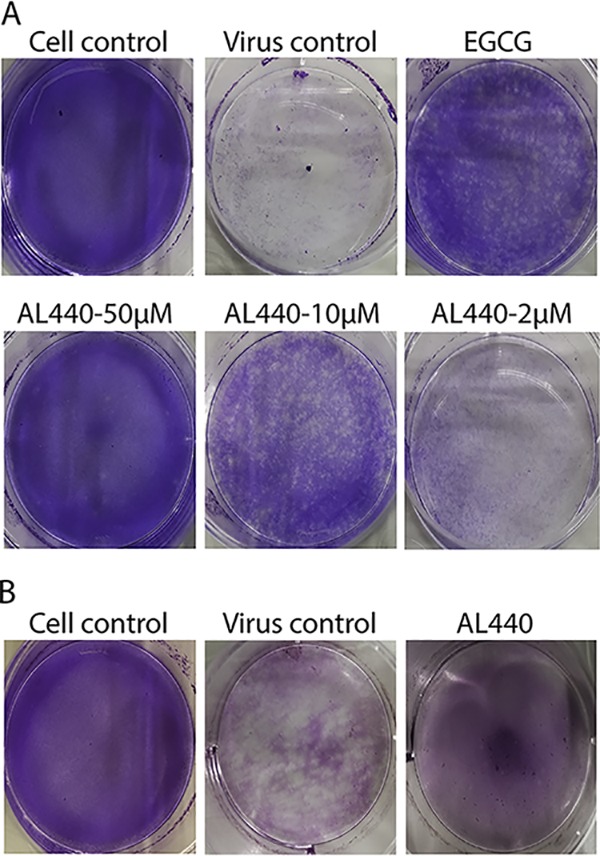
AL compounds interact with the viral particles. (A) Ultrafiltration. EGCG (0.1 mg/ml) and different concentrations of AL440 were mixed with DENV2 stock and further passed through a filter unit. Incubation of each sample (retained at the filter) on BHK cells showed that increasing concentrations of the compound reduce the number of viral plaques compared to that of virus control. (B) Virus inactivation. DENV2 stock treated with 10 μM AL440 was subsequently diluted 100 times, and the mixture was used to infect BHK cells. The diluted sample demonstrated a strong inhibition of viral infection compared to that of the virus control (no treatment with AL440).
AL compounds inhibit DENV infection by interacting with domain III of the virus envelope protein.
To investigate the potential binding of AL439 and AL440 to envelope (E) glycoprotein of DENV, a surface plasmon resonance (SPR) analysis was implemented. As shown in Fig. 5, AL compounds were able to bind to E protein (domain III) in a dose-dependent manner. More specifically, the binding capacity (association phase corresponding to response units) of AL440 was slightly stronger than that of AL439 (Fig. 5A and B). However, both molecules presented a similar binding profile, with an association phase from 0 to 125 s, followed by an immediate dissociation phase, where the compounds leave from the target protein. In addition, DS (positive control) showed a strong interaction with the E protein of DENV. In contrast, the negative-control AL438 (a compound from our series displaying no antiviral activity against DENV; Table 1) showed no interaction with the E protein (Fig. 5C and D). Comparison between AL molecules and DS revealed distinct differences, as the binding efficiency of DS to E protein was more pronounced (approximately 2× stronger) and its full dissociation from the target appeared at later time points than those for AL439 and AL440 (Fig. 5A to C).
FIG 5.

Surface plasmon resonance technology shows the interaction of AL compounds with domain III of the DENV envelope protein. Sensograms show the binding signal between the envelope protein of DENV (domain III) and the AL compounds. Twofold serial dilutions of AL439 (A) and AL440 (B) were added to the chip (the concentration of each analogue is shown in nanomolar range). The binding capacity is measured from 0 s to 125 s, followed by the quick dissociation phase. The binding profiles of DS (C) and AL438 (D) are also depicted. The y axis indicates the resonance signal in resonance units (RU), whereas the x axis represents the time of experiment (seconds).
DISCUSSION
In this study, a library of several classes of molecules was tested against DENV and ZIKV to evaluate their activity. Among these, a new generation of Trp trimers and tetramers was identified to have broad-spectrum activity against several laboratory and clinical strains of both flaviviruses.
To define the structural requirements, which are crucial for the antiviral activity of the molecules, SAR studies were performed. Most modifications took place at N1 and C2 positions of the indole side chain of tryptophan and clearly showed the importance of extra phenyl rings bearing substituents different from -COOH at these positions for the antiviral activity. However, no substituent leads to loss of antiviral activity. Introduction of -COOH on the indole side chain results in an increase not only in the size of the molecule but also the density (charge distribution), leading to the conclusion that different parameters might have an effect on the potency of molecules. We also showed that the increasing number of Trp residues on the central scaffold of molecules slightly increases their inhibitory activity, as indicated from the comparison of AL439 (three groups) and AL442 (one group), allowing us to conclude that there is a certain degree of multivalence that contributes to the enhancement of antiviral activity. Regarding the tetramer derivative, elevated cytotoxic levels could be explained by its physiochemical properties and, more specifically, the increased lipophilicity that determines parameters, such as the absorption and cytotoxicity of a molecule (35, 36). To ascertain the mode of action of this series of molecules, the most potent compounds (AL439 and AL440) were chosen for further analysis.
Screening studies showed that AL439 and AL440 are active against several laboratory and clinical strains of both flaviviruses. However, slight differences in antiviral activity against these strains can be explained by several conformational changes on the target protein (37). In addition, we noticed that these molecules are active in all tested cell lines (EC50 for DENV2 and ZIKV, 0.5 to 7.6 μM and 0.3 to 5.8 μM, respectively); however, they also presented different cytotoxic profiles. More specifically, AL439 and AL440 displayed higher cytotoxic levels in HUVEC and Jeg3 cells (CC50, <50 μM) than the other cell lines. A reason for this observation is that the molecules follow a distinct pattern for their activity and, possibly, they are able to influence several metabolic processes, affecting cell viability (38).
Time-of-drug-addition and subgenomic replicon experiments showed that these analogues act during the early stages of viral replication, possibly on the entry process. To further determine which step of virus entry is targeted, an attachment assay was performed. In this experiment, inhibition of viral infection was observed even after the removal of compounds 2 h p.i. We confirmed these data by SPR, where both analogues displayed an interaction with domain III of E glycoprotein (located at the C terminus). This particular domain participates in cell receptor recognition and contains antigenic epitopes that interact with potent neutralizing antibodies (39–42). Blocking this part of the protein has an unfavorable impact on viral entry into host cells (43). Interestingly, we observed a difference in binding capacity between the DS and AL analogues with E protein. We hypothesize that the binding site on viral E glycoprotein has positively charged residues to form interactions with the negative charges of the Trp residues and the extra phenyl rings on the N1 or C2 indole side chains in a weaker manner (faster dissociation rate) than the DS-E interaction (44). Unfortunately, we were not able to determine the apparent equilibrium dissociation constant (KD) values for the binding of AL439 and AL440 to domain III of the viral E glycoprotein. This could be due to self-aggregation problems that give a prominent and constant signal, which results in nonsaturation of the whole complex (45). In addition, the fact that the dissociation phase of our molecules is too fast (Fig. 5A and B) can be explained not only by the nature of the analogues (typical SPR profile of small molecules) but also due to the presence of an additional binding site(s) on other parts of the envelope protein.
Furthermore, several AL derivatives included in this study were also evaluated against HIV-1 (strain NL4.3) replication. Interestingly, we observed that AL439 and AL440 were not active at concentrations of up to 40 μM, while AL437 and AL455 (not active against DENV and ZIKV) showed activity in the low micromolar range (EC50, 10 μM and 2.4 μM, respectively). These findings clearly suggest that the activity of AL439 and AL440 is specific against DENV and ZIKV.
In the literature, there are several reports about the role of polyanions as entry inhibitors, especially against HIV infections (46, 47). One of the main advantages of this interesting class over small-molecule drugs is their ability to bind to the target in a multivalent way, making them promising antiviral candidates (48). Comparison between our molecules and the established entry inhibitor DS (the control in this study) revealed numerous advantages, as AL439 and AL440 have a precise structure, well-defined composition, and moderate molecular size. These properties provide higher diffusion rates of these compounds than the sulfated polysaccharide DS (49). In addition, the sulfated groups (-OSO3H) present on DS can be exposed to changes in the human microenvironment that lead to the loss of its activity (50). However, some problems associated with polyanionic substances should also be noted, as poor absorption, anticoagulant properties, and reduced in vivo efficacy have been considered major drawbacks for their use as antiviral agents (51). Based on the synthetic accessibility of our molecules and the aforementioned reasons, defined molecular structure, relatively moderate molecular weight, and the presence of COOH instead of OSO3H can increase the therapeutic properties of these compounds to levels that might be broader than those of DS.
To summarize, a novel class of Trp trimers and tetramers has been synthesized and evaluated for its antiviral activity against two emerging flaviviruses, DENV and ZIKV. These compounds were active in the low micromolar range against several strains of different susceptible cell lines. The mechanism of action was also determined, suggesting their role as entry inhibitors through interaction with the DENV E protein. Finally, taking into account the high degree of conservation in nucleotide/amino acid sequences among different flaviviruses, the assessment of antiviral activity of these compounds should be performed against other members of this family, such as yellow fever and Japanese encephalitis virus, to determine their efficacy and qualify them as potential antiviral agents with broad-spectrum activity.
MATERIALS AND METHODS
Cell lines and viruses.
African green monkey kidney cells (Vero; ATCC) and human lung epithelial adenocarcinoma cells (A549; ATCC) were grown in Eagle minimum essential medium (1× MEM-Rega3; Gibco, Belgium) supplemented with 8% fetal bovine serum (FBS; HyClone), 1 mM sodium bicarbonate (Gibco), 2 mM l-glutamine (Gibco), and 1× nonessential amino acids (100×; Gibco). Baby hamster kidney cells (BHK; kindly provided by M. Diamond, Washington University, USA) and hepatocarcinoma cells (Huh) were grown in Dulbecco’s modified Eagle medium (1× DMEM; Gibco) supplemented with 10% FBS, 1 mM sodium pyruvate (Gibco), 0.01 M HEPES (Gibco), and 1× nonessential amino acids. Placenta choriocarcinoma cells (Jeg3; ATCC) were grown in 10% FBS, 1 mM sodium pyruvate, and 1× nonessential amino acids. Finally, low-passage-number human umbilical vein endothelial cells (HUVEC; Lonza) were grown in endothelial cell growth basal medium 2 (EBM-2; Lonza) supplemented with an EGM-2 SingleQuots kit (Lonza). All cell lines were maintained at 37°C in a humidified 5% CO2 incubator.
Several laboratory and clinical strains of DENV and ZIKV were used in this study. DENV serotype 1 to 4 clinical isolates were obtained by Kevin Ariën from the Institute of Tropical Medicine (ITM; Antwerp, Belgium). All samples for routine diagnostics from patients presenting at the ITM polyclinic are stored after completion of the routine tests. The ITM has a policy that sample leftovers of patients presenting at the ITM polyclinic can be used for research unless the patients explicitly state their objection. The Institutional Review Board of ITM approved the policy of this presumed consent, as long as patients’ identity is not disclosed to third parties. DENV was isolated from stored serum samples from previously diagnosed acute dengue infections. Laboratory-adapted DENV serotype 1 Djibouti strain D1/H/IMTSSA/98/606 (GenBank accession number AF298808.1) (52), DENV serotype 3 H87 prototype strain (M93130.1) (53), and DENV serotype 4 Dak strain HD 34460 (MF004387.1; unpublished data) were kindly provided by X. de Lamballerie (Université de la Méditerranée, Marseille, France). Laboratory-adapted DENV serotype 2 New Guinea-C strain (NGC; M29095.1) (54) was a kind gift from V. Deubel (formerly at Institute Pasteur, Paris, France). Finally, the prototype ZIKV strain MR766 (MK105975.1; ATCC) and two clinical strains, PRVAVC59 (MH916806; ATCC) and FLR (KX087102.2; ATCC), were also tested. All viral strains were propagated in C6/36 mosquito cells from Aedes albopictus (ATCC). The viruses present in the supernatant of these cells were collected 5 to 6 days postinfection (p.i.) and stored at −80°C. Titration of the virus was performed by plaque assay using BHK cells. More specifically, serial 10-fold dilutions of each viral strain were inoculated for 2 h at 37°C on BHK cells (8 × 105 cells/well) grown in 6-well plates (Corning, USA). Cells were washed twice with infection medium (DMEM–2% FBS) and incubated with an overlay containing 2% Avicel (IMCD Benelux) and DMEM with 2% FBS at a 1:1 ratio for 30 min at room temperature. The cells were incubated for 5 days at 37°C and then washed with cold phosphate-buffered saline (PBS), fixed with 70% ethanol, and finally stained with 0.1% crystal violet. The viral titer was calculated by counting the number of plaques and expressed as PFU per milliliter.
Cell viability and antiviral activity.
All tested compounds were dissolved in dimethyl sulfoxide (DMSO). A final concentration of DMSO was maintained at 0.01% to evaluate its potential effect in all performed cellular assays.
The viability of cells in the presence of the compounds was assessed by the MTS method [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; Promega, Leiden, The Netherlands]. More specifically, every cell line was seeded at 104 cells/well in a 96-well plate (Corning, USA). After 24 h, cells were incubated with 5-fold serial dilutions of each compound (range, 0.8 μM to 100 μM) for 72 h. The medium then was replaced by 50 μl of 0.2% MTS in PBS and incubated for 3 to 4 h at 37°C. The CC50 values (compound concentration reducing the number of cells by 50%) were determined by measuring the optical density at 490 nm (SpectraMax Plus 384; Molecular Devices).
For antiviral testing, cells were seeded at 104 cells/well (100 μl of infection medium) in 96-well plates. After an overnight incubation, compounds were added in 5-fold serial dilutions as previously described. Cells were infected at different multiplicities of infection (MOI of 1 for DENV and 0.2 for ZIKV). After 2 h, cells were washed to remove unbound viral particles and further incubated with new medium in the presence of the compounds. Supernatant was collected 72 h p.i., and the virus yield was measured by the Cells Direct one-step quantitative reverse transcription-PCR (qRT-PCR) kit (Thermo Fisher) according to the manufacturer’s instructions. RT-PCR was performed using the ABI 7500 Fast real-time PCR system (Applied Biosystems, NJ, USA), and all data were analyzed with ABI PRISM 7000 software (v2.0.5; Applied Biosystems). The primer and probe sequences used for qRT-PCR were 5′-GGA TAG ACC AGA GAT CCT GCT GT-3′ (DENV 1–4 forward), 5′-CAT TCC ATT TTC TGG GGT TC-3′ (DENV 1–3 reverse), 5′-CAA TCC ATC TTG CGG CGC TC-3′ (DENV 4 reverse), 5′-FAM-CAG CAT CAT TCC AGG CAC AG-MGB-3′ (DENV 1–3 probe), 5′-FAM-CAA CAT CAA TCC AGG CAC AG-MGB-3′ (DENV 4 probe), 5′-TGA CTC CCC TCG TAG ACT G-3′ (ZIKV forward), 5′-CAC CTT TAG TCA CCT TCC TCT C-3′ (ZIKV reverse), and 5′-6-FAM/AGA TCC CAC/ZEN/AAA TCC CCT CTT CCC/3IABkFQ-3′ (ZIKV probe). Finally, the amplification conditions were 50°C for 15 min, 95°C for 2 min, and then 45 cycles of 95°C for 15 s and 60°C for 1 min. The infection efficiency was calculated using a curve of serial dilutions of standard samples for each virus. All data were analyzed in GraphPad Prism 7 software.
Antiviral activity in MDDC.
Peripheral blood mononuclear cells (PBMCs) obtained from four different healthy donors (provided from Rode Kruis Vlaanderen, Leuven, Belgium) were resuspended in Roswell Park Memorial Institute medium (RPMI 1640) supplemented with 10% FBS and 2 mM l-glutamine. The next day, cells were stimulated with 50 ng/ml of granulocyte-macrophage colony-stimulating factor and 25 ng/ml of interleukin-4 and seeded in a 6-well plate (1.5 × 106 cells/well) for 5 days at 37°C to obtain MDDCs. The cells were seeded at 3 × 104 cells/well in 24-well plates (Corning, USA) and incubated with the compounds in 5-fold serial dilutions for 30 min. DENV-2 NGC or ZIKV MR766 then was added to the cells. After 2 h, MDDCs were centrifuged and washed two times with fresh medium. Finally, cells were incubated in infection medium (RPMI, 2% FBS) for 48 h, collected, and then subjected to one-step qRT-PCR analysis.
Immunofluorescence assay.
Vero cells (104 cells/well) were seeded in a black 96-well plate (Falcon, Germany). After 24 h, cells were infected with ZIKV MR766 at an MOI of 0.2 in the presence of AL440 (concentrations of 20 μM, 10 μM, 2 μM, and 0.4 μM) at 37°C for 2 h. Cells were washed twice with PBS to remove the unbound viral particles and incubated at 37°C with infection medium for 72 h. Cells next were fixed for 5 min with 2% paraformaldehyde (PFA; Sigma-Aldrich, USA) at room temperature and permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, USA) for 10 more minutes. In addition, 1% bovine serum albumin (BSA; Cell Signaling Technology, The Netherlands) diluted in PBS was used to block the fixed cells for 1 h. After two washing steps, cells were incubated with the primary 4G2 pan-flavivirus antibody (Merck Millipore, Belgium) overnight at 4°C. Goat anti-mouse IgG Alexa Fluor 488 (Invitrogen) was used as a secondary antibody. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). Fluorescence microscopy was performed on a Zeiss Axiovert 200M inverted microscope (Germany) using a 10× enhanced green fluorescent protein objective.
Time-of-drug-addition assay.
Vero cells (104 cells/well) were seeded in a 96-well plate and infected with DENV-2 NGC strain at an MOI of 1 for 2 h. Cells were washed and treated with an active concentration of each compound (3-fold higher than EC50 values) at different time points before (2 h) and after (0, 2, 4, 6, 8, 12 and 18 h) infection. After 20 h (time point corresponding to one complete viral replication cycle), cells were collected and the viral RNA levels were measured by real-time RT-PCR. In addition, dextran sulfate (DS), with an average molecular weight of 8,000 Da, and 7-deaza-2’-C-methyladenosine (7DMA) were included as reference compounds. DS is a negatively charged inhibitor of the entry/fusion process, while 7DMA is a nucleosidic viral polymerase inhibitor. Both compounds were added at a 10 μM final concentration.
Subgenomic replicon assay.
To evaluate whether the compounds interact with the nonstructural (NS) proteins or the replication complex (indirectly), BHK cells stably transfected with a DENV subgenomic replicon (BHK/DENV; obtained from M. Diamond) were used. The replicon system encodes all NS proteins, as well as a Firefly luciferase expression cassette, instead of prM and E proteins of a DENV2 viral strain. Briefly, BHK/DENV (6 × 103 cells/well) was seeded in DMEM–2% FBS supplemented with 3 μg/ml puromycin for antibiotic selection (Sigma-Aldrich, USA) in a white-bottom 96-well plate (Nunclon Delta Surface; ThermoFischer Scientific). The next day, serial dilutions of AL439 and AL440 were added to the cells. After 3 days, supernatant was removed, cells were washed with PBS, the Renilla-Glo luciferase assay reagent (ThermoFischer Scientific) was added to each well, and the plate was incubated for 15 min. Finally, luminescence was measured at 490 nm (Tecan Spark 10M multimode microplate reader; BioExpress). DS and 7DMA were included as control compounds, while the cytotoxicity of the AL compounds in BHK/DENV cells was determined in parallel using the MTS method described above.
Viral attachment assay.
Vero cells were exposed to DENV2 NGC (MOI of 1) in the presence of different concentrations of compounds (40 μM, 20 μM, 10 μM, 5 μM, and 2.5 μM) for 2 h at 4°C (condition that allows virus attachment but no internalization). Cells were washed twice with infection medium to remove nonspecific material and incubated at 37°C (facilitates virus entry/fusion) without the compounds for 72 h. Supernatant was collected and virus yield was measured by one-step real-time RT-PCR.
Ultrafiltration and virus inactivation assay.
Direct interactions between AL440 and the DENV2 NGC strain were evaluated using two different methodologies.
First, in the ultrafiltration assay, viral stock (104 PFU) was incubated with different concentrations of AL440 (2 μM, 10 μM, and 50 μM) at 37°C for 1 h. Each sample was further passed through a 10-kDa-cutoff concentrator filter unit (Vivaspin 500; Vivaproducts), followed by three centrifugation steps, in order to allow nonbinding, free compound (<10 kDa) to infiltrate. The fraction retained by the filter (virus-compound complex) was added into BHK cells for a plaque assay. Epigallocatechin gallate (EGCG; 0.1 mg/ml) was used as a positive control to assess the specificity of the assay, as it has been shown to interact with the viral particles. Viral stock was also incubated without the presence of the compounds (virus control) and treated the same way. Finally, an equal volume of normal medium was added to control plates. The presence/number of plaques was evaluated per condition and compared with the virus control.
Second, during the virus inactivation assay, aliquots of DENV2 stock (104 PFU) were incubated with a concentration of AL440 (10 μM) able to inhibit viral infection at 37°C for 1 h. Sample was next diluted 100 times with fresh medium to reach a concentration of the compound below the EC50 values and used to infect susceptible Vero cells. Virus infectivity was further assessed by the plaque reduction assay (titration) using BHK cells. Virus and cell controls were also included.
SPR technology.
To study interactions of the compounds with the envelope protein of DENV, SPR analysis was performed on a Biacore T200 instrument (GE Healthcare, Uppsala, Sweden). Briefly, domain III of the recombinant envelope protein (GenWay Biotech Inc., San Diego, CA) of DENV2 was immobilized on a CM5 sensor chip in 10 mM sodium acetate buffer, pH 5, using standard amine coupling chemistry, resulting in a chip density of 250 RU. A reference flow cell was used to evaluate nonspecific binding to the chip. AL439 and AL440 were diluted in PBS supplemented with 0.005% surfactant P20 and 5% DMSO (pH 7.4) to obtain different concentrations between 1.25 μM and 20 μM (in 2-fold dilution steps). AL438 was included as a negative control in this study. Samples were injected for 2 min at a flow rate of 30 μl/min, and the dissociation was monitored for 4 min. Several buffer blanks were used for double referencing. The chip was finally regenerated with 1 M NaCl in 10 mM NaOH followed by 10 mM glycine-HCl, pH 1.5. All responses were adjusted for DMSO effects using a solvent correction procedure.
Data availability.
Accession numbers (GenBank) for each viral strain are AF298808.1 (DENV serotype 1 Djibouti D1/H/IMTSSA/98/606 strain), M29095.1 (DENV serotype 2 NGC strain), M93130.1 (DENV serotype 3 H87 strain), MF004387.1 (DENV serotype 4 Dak strain), MK105975.1 (ZIKV MR766 strain), MH916806.1 (ZIKV PRVAVC59 strain), and KX087102.2 (ZIKV FLR strain).
Supplementary Material
ACKNOWLEDGMENTS
We thank Leentje Persoons, Daisy Ceusters, Sandra Claes, Eric Fonteyn, and Els Vanstreels for their excellent technical assistance.
This work was supported by KU Leuven and the Fonds Wetenschappelijk Onderzoek (FWO grant PF/10/018), the Spanish MINECO (projects SAF2015-64629-C2-1-R and SAF2015-64629-C2-2-R [MINECO/FEDER]), and the Spanish Agencia Estatal Consejo Superior de Investigaciones Científicas (CSIC; projects CSIC201680E079 and 201980E028). The Spanish MEC/MINECO is also acknowledged for grants to B.M.-G. and O.M.-M.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Simmonds P, Becher P, Bukh J, Gould EA, Meyers G, Monath T, Muerhoff S, Pletnev A, Rico-Hesse R, Smith DB, Stapleton JT, ICTV Consortium. 2017. ICTV virus taxonomy profile: Flaviviridae. J Gen Virol 98:2–3. doi: 10.1099/jgv.0.000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suwanmanee S, Luplertlop N. 2017. Dengue and Zika viruses: lessons learned from the similarities between these Aedes mosquito-vectored arboviruses. J Microbiol 55:81–89. doi: 10.1007/s12275-017-6494-4. [DOI] [PubMed] [Google Scholar]
- 3.Dissanayake HA, Seneviratne SL. 2018. Liver involvement in dengue viral infections. Rev Med Virol 28:e1971-11. doi: 10.1002/rmv.1971. [DOI] [PubMed] [Google Scholar]
- 4.Weaver SC, Costa F, Garcia-Blanco MA, Ko A, Ribeiro GS, Saade G, Shi PY, Vasilakis N. 2016. Zika virus: history, emergence, biology, and prospects for control. Antiviral Res 130:69–80. doi: 10.1016/j.antiviral.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paixão ES, Teixeira MG, Rodrigues LC. 2018. Zika, chikungunya and dengue: the causes and threats of new and reemerging arboviral diseases. BMJ Glob Heal 3:e000530. doi: 10.1136/bmjgh-2017-000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gubler DJ. 2002. Epidemic dengue. Trends Microbiol 10:100–103. doi: 10.1016/s0966-842x(01)02288-0. [DOI] [PubMed] [Google Scholar]
- 7.Gould E, Pettersson J, Higgs S, Charrel R, de Lamballerie X. 2017. Emerging arboviruses: why today? One Health 4:1–13. doi: 10.1016/j.onehlt.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dallmeier K, Neyts J. 2016. Zika and other emerging viruses: aiming at the right target. Cell Host Microbe 20:420–422. doi: 10.1016/j.chom.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raekiansyah M, Mori M, Nonaka K, Agoh M, Shiomi K, Matsumoto A, Morita K. 2017. Identification of novel antiviral of fungus-derived brefeldin A against dengue viruses. Trop Med Health 45:1–7. doi: 10.1186/s41182-017-0072-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yung CF, Lee KS, Thein TL, Tan LK, Gan VC, Wong JGX, Lye DC, Ng LC, Leo YS. 2015. Dengue serotype-specific differences in clinical manifestation, laboratory parameters and risk of severe disease in adults, Singapore. Am J Trop Med Hyg 92:999–1005. doi: 10.4269/ajtmh.14-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vervaeke P, Vermeire K, Liekens S. 2015. Endothelial dysfunction in dengue virus pathology. Rev Med Virol 25:50–67. doi: 10.1002/rmv.1818. [DOI] [PubMed] [Google Scholar]
- 12.Wikan N, Smith DR. 2016. Zika virus: history of a newly emerging arbovirus. Lancet Infect Dis 16:e119–e126. doi: 10.1016/S1473-3099(16)30010-X. [DOI] [PubMed] [Google Scholar]
- 13.Chan JFW, Choi GKY, Yip CCY, Cheng VCC, Yuen KY. 2016. Zika fever and congenital Zika syndrome: an unexpected emerging arboviral disease. J Infect 72:507–524. doi: 10.1016/j.jinf.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roos RP. 2016. Zika virus–a public health emergency of international concern. JAMA Neurol 73:1395–1396. doi: 10.1001/jamaneurol.2016.3677. [DOI] [PubMed] [Google Scholar]
- 15.Lozier M, Adams L, Febo MF, Torres-Aponte J, Bello-Pagan M, Ryff KR, Munoz-Jordan J, Garcia M, Rivera A, Read JS, Waterman SH, Sharp TM, Rivera-Garcia B. 2016. Incidence of Zika virus disease by age and sex–Puerto Rico, November 1, 2015-October 20, 2016. MMWR Morb Mortal Wkly Rep 65:1219–1223. doi: 10.15585/mmwr.mm6544a4. [DOI] [PubMed] [Google Scholar]
- 16.Ming G, Song H, Tang H. 2017. Racing to uncover the link between Zika virus and microcephaly. Cell Stem Cell 20:749–753. doi: 10.1016/j.stem.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Pinto-Díaz CA, Rodríguez Y, Monsalve DM, Acosta-Ampudia Y, Molano González N, Anaya JM, Ramírez-Santana C. 2017. Autoimmunity in Guillain Barré syndrome associated with Zika virus infection and beyond. Autoimmun Rev 16:327–334. doi: 10.1016/j.autrev.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Magnus MM, Espósito DLA, da Costa VA, de Melo PS, Costa-Lima C, da Fonseca BAL, Addas-Carvalho M. 2018. Risk of Zika virus transmission by blood donations in Brazil. Hematol Transfus Cell Ther 40:250–254. doi: 10.1016/j.htct.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hastings AK, Fikrig E. 2017. Zika virus and sexual transmission: a new route of transmission for mosquito-borne flaviviruses. Yale J Biol Med 90:325–330. [PMC free article] [PubMed] [Google Scholar]
- 20.Cao B, Diamond MS, Mysorekar IU. 2017. Maternal-fetal transmission of Zika virus: routes and signals for infection. J Interferon Cytokine Res 37:287–294. doi: 10.1089/jir.2017.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aguiar M, Stollenwerk N, Halstead SB. 2016. The impact of the newly licensed dengue vaccine in endemic countries. PLoS Negl Trop Dis 10:e0005179. doi: 10.1371/journal.pntd.0005179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halstead SB. 2018. Safety issues from a phase 3 clinical trial of a live-attenuated chimeric yellow fever tetravalent dengue vaccine. Hum Vaccin Immunother 14:2158–2162. doi: 10.1080/21645515.2018.1445448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martínez-Vega RA, Carrasquila G, Luna E, Ramos-Castañeda J. 2017. ADE and dengue vaccination. Vaccine 35:3910–3912. doi: 10.1016/j.vaccine.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Diamond MS, Ledgerwood JE, Pierson TC. 2019. Zika virus vaccine development: progress in the face of new challenges. Annu Rev Med 70:121–135. doi: 10.1146/annurev-med-040717-051127. [DOI] [PubMed] [Google Scholar]
- 25.Blackman MA, Kim IJ, Lin JS, Thomas SJ. 2018. Challenges of vaccine development for Zika virus. Viral Immunol 31:117–123. doi: 10.1089/vim.2017.0145. [DOI] [PubMed] [Google Scholar]
- 26.Low JGH, Ooi EE, Vasudevan SG. 2017. Current status of dengue therapeutics research and development. J Infect Dis 215(Suppl 2):S96–S102. doi: 10.1093/infdis/jiw423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saiz JC, Martín-Acebes MA. 2017. The race to find antivirals for Zika virus. Antimicrob Agents Chemother 61:e00411-17. doi: 10.1128/AAC.00411-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Simmons G. 2012. Development of novel entry inhibitors targeting emerging viruses. Expert Rev Anti Infect Ther 10:1129–1138. doi: 10.1586/eri.12.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Witvrouw M, De Clercq E. 1997. Sulfated polysaccharides extracted from sea algae as potential antiviral drugs. Gen Pharmacol 29:497–511. doi: 10.1016/s0306-3623(96)00563-0. [DOI] [PubMed] [Google Scholar]
- 30.Vacas-Córdoba E, Maly M, De la Mata FJ, Gómez R, Pion M, Muñoz-Fernández MÁ. 2016. Antiviral mechanism of polyanionic carbosilane dendrimers against HIV-I. Int J Nanomedicine 11:1281–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Witvrouw M, Fikkert V, Pluymers W, Matthews B, Mardel K, Schols D, Raff J, Debyser Z, De Clercq E, Holan G, Pannecouque C. 2000. Polyanionic (i.e., polysulfonate) dendrimers can inhibit the replication of human immunodeficiency virus by interfering with both virus adsorption and later steps (reverse transcriptase/integrase) in the virus replicative cycle. Mol Pharmacol 58:1100–1108. doi: 10.1124/mol.58.5.1100. [DOI] [PubMed] [Google Scholar]
- 32.Alhoot MA, Rathinam AK, Wang SM, Manikam R, Sekaran SD. 2013. Inhibition of dengue virus entry into target cells using synthetic antiviral peptides. Int J Med Sci 10:719–729. doi: 10.7150/ijms.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watterson D, Kobe B, Young PR. 2012. Residues in domain III of the dengue virus envelope glycoprotein involved in cell-surface glycosaminoglycan binding. J Gen Virol 93:72–82. doi: 10.1099/vir.0.037317-0. [DOI] [PubMed] [Google Scholar]
- 34.Martínez-Gualda B, Sun L, Martí-Marí O, Noppen S, Abdelnabi R, Bator CM, Quesada E, Delang L, Mirabelli C, Lee H, Schols D, Neyts J, Hafenstein S, Camarasa MJ, Gago F, San-Felix A. 20 December 2019. A scaffold simplification strategy leads to a novel generation of dual human immunodeficiency virus and enterovirus A71 entry inhibitors. J Med Chem doi: 10.1021/acs.jmedchem.9b01737. [DOI] [PubMed] [Google Scholar]
- 35.Gao Y, Gesenberg C, Zheng W. 2017. Oral formulations for preclinical studies: principle, design, and development considerations, p 455–495. In Qiu Y, Chen Y, Zhang G, Yu L, Mantri RV (ed), Developing solid oral dosage forms: pharmaceutical theory and practice, 2nd ed Elsevier, Inc, San Diego, CA. doi: 10.1016/B978-0-12-802447-8.00017-0. [DOI] [Google Scholar]
- 36.Arnott JA, Planey SL. 2012. The influence of lipophilicity in drug discovery and design. Expert Opin Drug Discov 7:863–875. doi: 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- 37.Witvrouw M, Weigold H, Pannecouque C, Schols D, De Clercq E, Holan G. 2000. Potent anti-HIV (type 1 and type 2) activity of polyoxometalates: structure-activity relationship and mechanism of action. J Med Chem 43:778–783. doi: 10.1021/jm980263s. [DOI] [PubMed] [Google Scholar]
- 38.Olsen DB, Eldrup AB, Bartholomew L, Bhat B, Bosserman MR, Ceccacci A, Colwell LF, Fay JF, Flores OA, Getty KL, Grobler JA, LaFemina RL, Markel EJ, Migliaccio G, Prhavc M, Stahlhut MW, Tomassini JE, MacCoss M, Hazuda DJ, Carroll SS. 2004. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob Agents Chemother 48:3944–3953. doi: 10.1128/AAC.48.10.3944-3953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Jia R, Shen H, Wang M, Yin Z, Cheng A. 2017. Structures and functions of the envelope glycoprotein in flavivirus infections. Viruses 9:338–314. doi: 10.3390/v9110338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marzinek JK, Lakshminarayanan R, Goh E, Huber RG, Panzade S, Verma C, Bond PJ. 2016. Characterizing the conformational landscape of flavivirus fusion peptides via simulation and experiment. Sci Rep 6:19160–19115. doi: 10.1038/srep19160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu KP, Wu CW, Tsao YP, Kuo TW, Lou YC, Lin CW, Wu SC, Cheng JW. 2003. Structural basis of a flavivirus recognized by its neutralizing antibody: solution structure of the domain III of the Japanese encephalitis virus envelope protein. J Biol Chem 278:46007–46013. doi: 10.1074/jbc.M307776200. [DOI] [PubMed] [Google Scholar]
- 42.Chávez JH, Silva JR, Amarilla AA, Moraes Figueiredo LT. 2010. Domain III peptides from flavivirus envelope protein are useful antigens for serologic diagnosis and targets for immunization. Biologicals 38:613–618. doi: 10.1016/j.biologicals.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Modis Y, Ogata S, Clements D, Harrison SC. 2005. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J Virol 79:1223–1123. doi: 10.1128/JVI.79.2.1223-1231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martínez-Gualda B, Sun L, Martí-Marí O, Mirabelli C, Delang L, Neyts J, Schols D, Camarasa M-J, San-Félix A. 2019. Modifications in the branched arms of a class of dual inhibitors of HIV and EV71 replication expand their antiviral spectrum. Antiviral Res 168:210–214. doi: 10.1016/j.antiviral.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giannetti AM, Koch BD, Browner MF. 2008. Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J Med Chem 51:574–580. doi: 10.1021/jm700952v. [DOI] [PubMed] [Google Scholar]
- 46.Lüscher-Mattli M. 2000. Polyanions–a lost chance in the fight against HIV and other virus diseases? Antivir Chem Chemother 11:249–259. doi: 10.1177/095632020001100401. [DOI] [PubMed] [Google Scholar]
- 47.Leydet A, Jeantet-Segonds C, Bouchitté C, Moullet C, Boyer B, Roque JP, Witvrouw M, Este J, Snoeck R, Andrei G, De Clercq E. 1997. Polyanion inhibitors of human immunodeficiency virus and other viruses. 6. Micelle-like anti-HIV polyanionic compounds based on a carbohydrate core. J Med Chem 40:350–356. doi: 10.1021/jm960348y. [DOI] [PubMed] [Google Scholar]
- 48.MacIel D, Guerrero-Beltrán C, Ceña-Diez R, Tomás H, Muñoz-Fernández MÁ, Rodrigues J. 2019. New anionic poly(alkylideneamine) dendrimers as microbicide agents against HIV-1 infection. Nanoscale 11:9679–9690. doi: 10.1039/c9nr00303g. [DOI] [PubMed] [Google Scholar]
- 49.Keeney M, Jiang XY, Yamane M, Lee M, Goodman S, Yang F. 2015. Nanocoating for biomolecule delivery using layer-by-layer self-assembly. J Mater Chem B 3:8757–8770. doi: 10.1039/c5tb00450k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balzarini J, Van Damme L. 2007. Microbicide drug candidates to prevent HIV infection. Lancet 369:787–797. doi: 10.1016/S0140-6736(07)60202-5. [DOI] [PubMed] [Google Scholar]
- 51.Pirrone V, Wigdahl B, Krebs FC. 2011. The rise and fall of polyanionic inhibitors of the human immunodeficiency virus type 1. Antiviral Res 90:168–182. doi: 10.1016/j.antiviral.2011.03.176. [DOI] [PubMed] [Google Scholar]
- 52.Tolou HJ, Couissinier-Paris P, Durand JP, Mercier V, de Pina JJ, de Micco P, Billoir F, Charrel RN, de Lamballerie X. 2001. Evidence for recombination in natural populations of dengue virus type 1 based on the analysis of complete genome sequences. J Gen Virol 82:1283–1290. doi: 10.1099/0022-1317-82-6-1283. [DOI] [PubMed] [Google Scholar]
- 53.Osatomi K, Sumiyoshi H. 1990. Complete nucleotide sequence of dengue type 3 virus genome RNA. Virology 176:643–647. doi: 10.1016/0042-6822(90)90037-R. [DOI] [PubMed] [Google Scholar]
- 54.Irie K, Mohan PM, Sasaguri Y, Putnak R, Padmanabhan R. 1989. Sequence analysis of cloned dengue virus type 2 genome (New Guinea-C strain). Gene 75:197–211. doi: 10.1016/0378-1119(89)90266-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession numbers (GenBank) for each viral strain are AF298808.1 (DENV serotype 1 Djibouti D1/H/IMTSSA/98/606 strain), M29095.1 (DENV serotype 2 NGC strain), M93130.1 (DENV serotype 3 H87 strain), MF004387.1 (DENV serotype 4 Dak strain), MK105975.1 (ZIKV MR766 strain), MH916806.1 (ZIKV PRVAVC59 strain), and KX087102.2 (ZIKV FLR strain).