

Abstract
Background
In cancer, mutations of DNA methylation modification genes have crucial roles for epigenetic modifications genome-wide, which lead to the activation or suppression of important genes including tumor suppressor genes. Mutations on the epigenetic modifiers could affect the enzyme activity, which would result in the difference in genome-wide methylation profiles and, activation of downstream genes. Therefore, we investigated the effect of mutations on DNA methylation modification genes such as DNMT1, DNMT3A, MBD1, MBD4, TET1, TET2 and TET3 through a pan-cancer analysis.
Methods
First, we investigated the effect of mutations in DNA methylation modification genes on genome-wide methylation profiles. We collected 3,644 samples that have both of mRNA and methylation data from 12 major cancer types in The Cancer Genome Atlas (TCGA). The samples were divided into two groups according to the mutational signature. Differentially methylated regions (DMR) that overlapped with the promoter region were selected using minfi and differentially expressed genes (DEG) were identified using EBSeq. By integrating the DMR and DEG results, we constructed a comprehensive DNA methylome profiles on a pan-cancer scale. Second, we investigated the effect of DNA methylations in the promoter regions on downstream genes by comparing the two groups of samples in 11 cancer types. To investigate the effects of promoter methylation on downstream gene activations, we performed clustering analysis of DEGs. Among the DEGs, we selected highly correlated gene set that had differentially methylated promoter regions using graph based sub-network clustering methods.
Results
We chose an up-regulated DEGs cluster where had hypomethylated promoter in acute myeloid leukemia (LAML) and another down-regulated DEGs cluster where had hypermethylated promoter in colon adenocarcinoma (COAD). To rule out effects of gene regulation by transcription factor (TF), if differentially expressed TFs bound to the promoter of DEGs, that DEGs did not included to the gene set that effected by DNA methylation modifiers. Consequently, we identified 54 hypomethylated promoter DMR up-regulated DEGs in LAML and 45 hypermethylated promoter DMR down-regulated DEGs in COAD.
Conclusions
Our study on DNA methylation modification genes in mutated vs. non-mutated groups could provide useful insight into the epigenetic regulation of DEGs in cancer.
Keywords: DNA methylation modifier, Sub-network clustering, DMR-DEG integration, Pan-cancer analysis, Genome-wide landscape
Background
DNA mutation is one of the major causes of many diseases, thus understanding impact of mutations in genes is an important research problem. For example, mutations in oncogenes and tumor suppressor genes have been extensively studied over the years [1–3]. Some class of genes, e.g., epigenetic genes, have roles in cancer proliferation by modifying the epigenetic status of a cell, then the epigenetic status change affects gene expression regulation then cancer phenotype. Epigenetic genes are divided into three functional groups: epigenetic modulators, modifiers, and mediators [4]. Epigenetic modulators transmit signals to epigenetic regulators. Upon receiving such signal, epigenetic modifiers modify the epigenetic status of a genome. In response to the changes in the epigenome, epigenetic mediators then could change their biological roles. In addition, abnormal mutations in the epigenetic genes can adversely affect this epigenetic system, causing tumors.
Among epigenetic genes, DNA methylation related epigenetic modifiers, DNMT1, DNMT3A, MBD1, MBD4, TET1, TET2, and TET3, have been studied related to cancer [5–16]. DNMT3A mutation was found at a high rate of 22.1 percent of acute myeloid leukemia patients [17]. In our study, mutations in DNA methylation modifier genes were found in about 13 percent (1,474/11,315) of cancer patients from The Cancer Genome Atlas (TCGA) projects [18].
In general, mutations on a gene can affect the function of a gene, even loss or gain of a function. Many DNA methylation modification genes are enzymes. Thus, mutations on the epigenetic modifiers could affect the activity of epigenetic modifiers, which would result in the difference in genome-wide methylation profiles and in turn, activation of downstream genes. However, there is no systematic study on this important topic. In this paper, we investigated the effect of mutations on DNA methylation modification genes such as DNMT1, DNMT3A, MBD1, MBD4, TET1, TET2, and TET3 through a pan-cancer analysis. First, we investigated the effect of mutations in DNA methylation modification genes on genome-wide methylation profiles in 12 major cancer types in TCGA.
As a result, we found that genome-wide methylation landscapes were significantly different between two sample groups with mutations and without mutations in the DNA methylation modifier genes. Second, we investigated the effect of DNA methylations in the promoter regions on downstream genes in 12 cancer types. To investigate the effect of mutations on gene expression further, we chose an up-regulated gene cluster where differentially expressed genes (DEGs) were mostly hypomethylated promoter regions in acute myeloid leukemia and another down-regulated gene cluster where DEGs had mostly hypermethylated promoter regions in colon adenocarcinoma.
Methods
TCGA data of DNA methylome and transcriptome
To perform pan-cancer data analysis, we downloaded data for 12 major cancer types from TCGA: bladder cancer (BLCA), breast cancer (BRCA), colon adenocarcinoma (COAD), glioblastoma (GBM), head and neck squamous carcinoma (HNSC), kidney renal carcinoma (KIRC), acute myeloid leukemia (LAML), lung adenocarcinoma (LUAD), lung squamous carcinoma (LUSC), ovarian cancer (OV), rectal adenocarcinoma (READ) and uterine corpus endometrial carcinoma (UCEC). A total of 3,644 samples that had both methylome and transcriptome data were collected. Among 3,644 samples, 580 samples had at least one or more mutation in seven DNA methylation modifier genes, and 432 mutations except for synonymous mutation samples were finally identified. Thus samples were divided into two groups, one with mutations in DNA methylation modifiers (432 samples) and the other group (3,212 samples). Among 12 cancer types, OV type had no mutation sample. Thus, we analyzed 11 cancer types (Table 1).
Table 1.
Number of samples per 12 major cancer type in TCGA
| Cancer type | Total samples | Synonymous mutation samples | Non-synonymous mutation samples |
|---|---|---|---|
| TCGA-BRCA | 784 | 14 | 47 |
| TCGA-HNSC | 521 | 19 | 41 |
| TCGA-LUAD | 455 | 14 | 66 |
| TCGA-BLCA | 408 | 24 | 70 |
| TCGA-LUSC | 369 | 16 | 55 |
| TCGA-KIRC | 319 | 3 | 17 |
| TCGA-COAD | 279 | 33 | 54 |
| TCGA-UCEC | 173 | 19 | 37 |
| TCGA-LAML | 170 | 0 | 33 |
| TCGA-READ | 93 | 3 | 8 |
| TCGA-GBM | 64 | 2 | 4 |
| TCGA-OV | 9 | 1 | 0 |
| Total | 3644 | 148 | 432 |
Each value represents the number of samples that have both methylome and transcriptome data and the number of samples that have mutations in seven DNA methylation modifier genes
DEG analysis
The mRNA-seq data labeled by “illuminahiseq rnaseqv2 RSEM genes normalized” were downloaded from the firebrowse website (http://firebrowse.org/). A Bioconductor (version 3.8) EBSeq package [19] was used for the DEG analysis of RNA data. For each cancer type, we divided the samples into two groups into mutated versus non-mutated samples and performed DEG analysis. Number of DEGs was counted with false discovery rate (FDR) less than 0.05. Fold change values of gene expression level were used in the following clustering analysis.
DMR analysis
The methylation data labeled by ”humanmethylation450 within bioassay data set function” were downloaded from the firebrowse website. For the methylation data analysis, the DMR was analyzed with a FDR of 0.05 using “bumperhunter” in the minfi package [20] of Bioconductor (version 3.8). For each cancer type, we divided the samples into two groups into mutated versus non-mutated samples as same as DEG analysis. The DMRs found were annotated using “matchgene” to select the genes with DMR in the promoter.
Random sample test
Random sampling was performed to compare the seven DNA methylation modifier mutation samples of each cancer types. Random samples were selected with the same size as the seven DNA methylation modifiers mutation samples, and DEG and DMR analysis were performed 10,000 times using the selected and remaining samples.
Log ratio of average methylation levels in promoter regions
To compare the methylation levels of each promoter region between the samples of which the seven DNA methylation modifier genes were mutated and the other samples, we firstly calculated the average of methylation levels of each promoter region for the samples with mutation and the other samples, respectively. After that, the log2 ratio of the averaged methylation levels was calculated and the equation is shown below:
where j indicates each probe, i is the index of cancer, Avg_mutij is the average of the methylation levels of probe j for the samples with mutation in cancer i, Avg_nonij is the average of the methylation levels of probe j for the samples without mutation in cancer i and LRij is the log2 ratio of two average values of probe j in cancer i. Pseudo is the value of 0.001 we added to the averages to avoid the error caused by dividing by zero.
Gene expression correlation analysis
For transcriptome data, correlation values between genes were calculated using Pearson’s correlation of “pearsonr” of scipy for each cancer type. The final correlation value between the final genes was calculated using the weight value of PPI score of STRING database. These correlation values are used the following clustering analysis.
Graph-based clustering
We used igraph package [21] of R to detect multilevel community and perform sub-network clustering. For the graph-based clustering, we used the fold-change value of the gene and correlation values between genes. Before clustering, we discard genes with fold-change less than 0.2 and edge of correlation with less than 0.5. After clustering, we perform the GO enrichment test and one-sample t-test for each cluster.
Network visualization with cytoscape
Visualization of the sub-network cluster is shown using Cytoscape (version 3.7.1).
Promoter binding TF search by TRANSFAC
To search all TFs to bind the promoter sequence of DEG, we used TRANSFAC.
Workflow
The analysis of the mutation data of seven DNA methylation modifiers on the pan-cancer scale was performed in three phases and the analysis workflow is shown in a schematic diagram (Fig. 1). In this section, the analysis process is briefly explained to help understand the analysis results. Detailed analysis methods are written in the “Methods” section.
Fig. 1.

Workflow. See the “Workflow” section for more details
PART 1: impact of mutations in DNA methylation modifiers on genome-wide methylation landscape
First, we investigated the effect of mutations in DNA methylation modifiers on genome-wide methylation profiles.
1-1. statistics on mutations in seven DNA methylation modifiers
Before investigating the genome-wide effects of seven DNA methylation modifiers, it was confirmed the distribution of 7 methylation modifier mutations in the mutation samples. Mutation frequencies in DNA methylation modifiers were collected for each cancer.
1-2. genome-wide methylation landscapes
To investigate the genome-wide effects of seven DNA methylation modifiers, we analyzed the difference in DNA methylation profiles in pan-cancer. To compare the difference in methylation of samples that were divided into DNA methylation modifiers mutation, mutated and non-mutated samples (432 vs. 3,212 samples) in terms of log2 ratios (See “Methods” section for the detail).
1-3. statistics of the number of differentially methylated regions (DMRs) between two groups
To confirm the effect of unbalanced samples and to evaluate whether these differences are significant or not, we analyzed them statistically. We compared the number of DMRs in samples with mutations in the DNA methylation modifier with the number of DMRs in randomly selected unbalanced samples. The analysis of DMR counts was performed with randomly sampled the same size as the number of mutation samples and repeated 10,000 times to calculate the p-value.
PART 2: impact of mutations in DNA methylation modifiers on genome-wide gene expression landscape
Since DNA methylation can have significant effect on gene expression profiles, we compared gene expression profiles between the mutated and the non-mutated samples. In this part, we only compared gene expression profiles between two groups, without attempting to investigate the effect of DNA methylation on gene expression, which was reported in Part 3.
2-1. statistics on gene expression profiles
DEG counts was collected from randomly chosen same size samples, repeating 10,000 times to calculate p-values.
2-2. clustering analysis of transcriptome
To investigate biological functions of DEGs, we divided DEGs into smaller gene sets based on network based gene clustering analysis and then performed gene ontology (GO) term enrichment test on each set of DEGs to compare the difference in functions of genes between the mutated and non-mutated groups. Before performing sub-network clustering, correlation values between genes were calculated. Pearson’s correlation value was calculated for transcriptome data, and protein-protein interaction (PPI) score from STRING [22] database was multiplied by weight. Using the log2 fold-change value obtained from the DEG analysis, we removed genes that had opposite interaction or the small change amount. Thus, we selected a set of gene with over 0.15 of absolute value of log2 fold change of gene expression and over 0.5 positive correlated genes network. We performed graph-based sub-network clustering using iCluster (see “Methods” section) with fold change of gene expression using pre-processed gene-gene interaction score. To select meaningful clusters after clustering, we performed one sample t-test with gene expression levels and Fisher’s exact test using GO term enrichment test. Clusters with p-value under 10−9 was selected.
PART 3: integrated analysis of DMR and DEG
Now, we tried to associate DEGs and DMRs between the two groups as below.
3-1. integration of gene expression and methylation expression
To investigate the effect of DMRs on DEGs, we focused on methylation difference in the promoter regions. First, we selected gene clusters with significantly enriched DEGs and DMRs using a Fisher’s exact test for each of gene clusters. Then, gene sets were selected by considering negative correlation between promoter methylation and the corresponding gene expression.
3-2. transcription factor (TF) binding site search with TRANSFAC
In addition to negative correlation between promoter methylation and the corresponding gene expression, we considered expression levels of TFs that could bind to the promoter regions. Thus, we searched for all TF binding sequences in the DEG promoter region using TRANSFAC [23].
3-3. comparison without TF effect
Expression level of the TFs that had binding sites in the promoter regions was considered to remove cases where gene expression difference could result from TF expression difference. For example, if TF binding to the promoter of up-regulated DEG is not up-regulated, the up-regulated DEG can be determined by the effect of DMR regardless of the effect of TF. Thus, both up-regulated DEG with up-regulated TF and down-regulated DEG with down-regulated TF were removed.
Results and discussions
Part 1 - statistic analysis of mutation effect of seven DNA methylation modifier genes
To analyze the effects of seven DNA methylation modifier genes, we collected 3,644 TCGA methylome and transcriptome data. First, the number of mutation samples in DNA methylation modifier genes was found to be between 5% and 21% of the total sample for 11 major cancer types (Table 2). Excluding OV without mutation samples, 11 cancer types were analyzed.
Table 2.
Summary of mutation status of seven DNA methylation modifier genes in each cancer
| Cancer type | Total samples | Mutated samples | Non-mutated samples | Mutation sample ratio | Number of DMRs | Number of DEGs |
|---|---|---|---|---|---|---|
| TCGA-BRCA | 784 | 47 | 737 | 6% | 20,390 | 99 |
| TCGA-HNSC | 521 | 41 | 480 | 8% | 21,442 | 57 |
| TCGA-LUAD | 455 | 66 | 389 | 15% | 7,886 | 288 |
| TCGA-BLCA | 408 | 70 | 338 | 17% | 12,711 | 379 |
| TCGA-LUSC | 369 | 55 | 314 | 15% | 45,441 | 415 |
| TCGA-KIRC | 319 | 17 | 302 | 5% | 23,544 | 127 |
| TCGA-COAD | 279 | 54 | 225 | 19% | 26,491 | 535 |
| TCGA-UCEC | 173 | 37 | 136 | 21% | 44,743 | 1,494 |
| TCGA-LAML | 170 | 33 | 137 | 19% | 28,215 | 439 |
| TCGA-READ | 93 | 8 | 85 | 9% | 58,058 | 205 |
| TCGA-GBM | 64 | 4 | 60 | 6% | 77,753 | 215 |
Each value represents the number of samples that have both methylome and transcriptome data, the number of samples that have mutations in seven DNA methylation modifier genes, the number of samples that don’t have mutations, ratio of the mutation samples per non-mutation samples, the number of DMRs and the number of DEGs that were selected by 0.05 false discovery rate
The seven DNA methylation modifier genes that we studied were DNMT1, DNMT3A, MBD1, MBD4, TET1, TET2 and TET3. DNMT1 and DNMT3A function as DNA methyl-transfer and TET1, TET2 and TET3 have demethylation functions. Mutation statistics of the seven modifiers are summarized in (Fig. 2). Cancer types of BLCA, BRCA, COAD, LUAD, and LUSC were predominantly mutated in the TET genes that have demethylation functions. In the case of LAML, DNMT3A mutation samples were high, while remaining GBM, HNSC and KIRC, the ratio was similar. In the case of GBM, KIRC, and READ, the total mutation rate was less than 9%, and the number of mutations for each gene was 5 or less (Table 2). We should individually analyze to find a functional difference for each methylation modifier genes because the methylation modifier functions include methyl-transfer function and de-methylation that are opposite functions. However, since the number of samples is so small that it is very difficult to find a meaningful analysis result by each gene analysis, we first analyzed the global impact on the methylation dysfunction and then analyzed in depth. In addition, in the case of GBM and READ, the number of samples was eight or four, which makes it difficult to determine the representative characteristics of mutant cancers.
Fig. 2.

The number of samples that each of the seven DNA methylation modifier genes is mutated. A sample with mutations in multiple DNA methylation modification genes was counted redundantly as multiple genes. DNMT3A mutation is dominant in LAML samples. In COAD, mutations in TET1, TET2 and TET3 are dominant
Effect of mutations in seven DNA methylation modifier genes on genome-wide methylation landscapes
We compared genome-wide methylation landscapes between the mutated and the non-mutated groups. Since comparison of genome-wide methylation landscapes between the two groups was difficult to interpret, we compared promoter regions instead. Among the annotated 450,000 CpG sites, we selected the 140,040 sites as promoters when the sites are annotated as TSS200 or TS1500; TSS200 is the region that covers zero to 200 bases upstream of the transcription start site (TSS) and TSS1500 covers 200 to 1500 bases upstream of the TSS. For each of nine cancer types, methylation differences in 140,040 promoter regions of CpG sites were examined separately. We compared mutated and non-mutated samples of seven DNA methylation modifier genes, and the methylation values for each CpG site were expressed as log2 ratio values by comparing mean values. For the selected CpG sites, the average of DNA methylation of the mutation versus non-mutation samples was calculated as the log2 ratio and a heatmap was drawn by selecting 29,879 CpG sites with the log2 ratio value bigger than 1 or smaller than -1. Hypermethylated promoter is shown in red and hypomethylated promoter is shown in blue (Fig. 3). We measured the number of hyper-/hypo-methylated promoters in each cancer and estimated odd ratios and p-values of Fisher’s exact test. Each was calculated by applying different cutoff criteria for log2 fold changes of hyper-/hypo-methylated promoters (Table 3). In the heatmap results, COAD and UCEC have a large number of hypermethylated promoters, while LAML, LUSC, HNSC, BRCA, and BLCA have a large number of hypomethylated promoter regions. COAD showed the highest positive ratio and LAML had the most hypo-methylated promoter even when the cut-off criterion was raised. The heatmap results showed that there was a change in methylation due to the mutation of seven DNA methylation modifier genes, and detailed analysis was conducted to investigate the CpG site of promoter region with methylation changes in nine cancer types.
Fig. 3.

Genome-wide landscape of promoter methylation. Differential methylation level of gene promoter regions are profiled for 9 cancer types: bladder cancer (BLCA), breast cancer (BRCA), colon adenocarcinoma (COAD), head and neck squamous carcinoma (HNSC), kidney renal carcinoma (KIRC), acute myeloid leukemia (LAML), lung adenocarcinoma (LUAD), lung squamous carcinoma (LUSC), and uterine corpus endometrial carcinoma (UCEC). 9,580 genes showed hyper-methylation (red) or hypo-methylation (blue) in the promoter regions for at least one cancer type. In the lower panel, genes (i.e., column of the figure) are ordered according to the chromosomal position, and cancer types (i.e., row of the figures) are sorted by lexicographic order. In the upper panel, genes and cancer types are clustered in terms of methylation profile similarity
Table 3.
Number of hyper-/hypo- methylated promoter in each cancer
| Cancer | Hypermethylated promoter | Hypormethylated promoter | log2 odd ratio | p-value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.05 | 0.075 | 0.1 | 0.05 | 0.075 | 0.1 | 0.05 | 0.075 | 0.1 | 0.05 | 0.075 | 0.1 | |
| COAD | 2,923 | 921 | 246 | 348 | 41 | 3 | 3.0702 | 4.4895 | 6.3575 | 0 | 1.15E-201 | 1.E-67 |
| UCEC | 2,886 | 1,329 | 565 | 1,343 | 441 | 129 | 1.1036 | 1.5914 | 2.1308 | 1.20E-89 | 4.52E-88 | 5.E-62 |
| KIRC | 1,747 | 458 | 100 | 1,235 | 226 | 38 | 0.5003 | 1.0190 | 1.3959 | 2.52E-16 | 5.03E-18 | 1.E-07 |
| LAML | 407 | 73 | 15 | 3,274 | 1,285 | 446 | -3.0079 | -4.1377 | -4.8940 | 0 | 5.86E-258 | 2.E-107 |
| LUSC | 297 | 27 | 1 | 3,882 | 886 | 94 | -3.7082 | -5.0362 | -6.5545 | 0 | 2.33E-208 | 4.E-27 |
| HNSC | 63 | 2 | 0 | 2,500 | 284 | 15 | -5.3104 | -7.1497 | -infinity | 0 | 1.95E-80 | 6.E-05 |
| BRCA | 310 | 18 | 1 | 1,496 | 201 | 27 | -2.2707 | -3.4811 | -4.7548 | 3.10E-159 | 8.28E-40 | 2.E-07 |
| BLCA | 87 | 3 | 0 | 1,031 | 60 | 3 | -3.5668 | -4.3219 | -infinity | 1.21E-187 | 8.09E-15 | 2.E-01 |
| LUAD | 183 | 8 | 2 | 521 | 17 | 0 | -1.5094 | -1.0874 | +infinity | 3.97E-36 | 0.107299 | 5.E-01 |
DMR analysis to investigate mutation effects of seven DNA methylation modifiers.
Mutated samples of seven DNA methylation genes were compared with non-mutated samples using bumperhunter of minfi package for DMR analysis. The significance of the number of DMRs potentially caused by the mutation of seven DNA methylation modifiers was compared with the number of DMRs in random samples. Random sampling DMR analysis was performed by repeatedly choosing samples of the same size for 10,000 times. P-value of the mutant sample was calculated from the distribution of DEG and DMR values obtained from 10,000 repeated tests. In the result of DMR test, 8 cancer types of 11, as BRCA, HNSC, LUAD, BLCA, LUSC, COAD, UCEC and LAML, showed significantly low p-value (Aditional file 1: Figure S2). The other cancer type, KIRC, READ and GBM, were not significant due to have few mutation samples (See Fig. 2). Overall, it seemed that mutations of seven DNA methylation modifier genes affected genome-wide promoter methylation differences.
Part2 - genome-wide association analysis of mutation effect of seven DNA methylation modifier genes
Sub-network clustering result in pan-cancer scale
We performed graph-based clustering of DEGs. First, we used the network topology of STRING database and chose edges between two genes only when expression values of the two genes were highly correlated. Edges were weighted by the STRING database confidence scores. After that, the clustering was performed and the clusters were filtered using t-test.
The selected clusters were visualized using Cytoscape [24] (Fig. 4). Up-regulated DEG is displayed in a gradual red color and down-regulated DEG is displayed in a gradual blue color by the fold change value of gene expressions. Promoter DMR information was integrated into the DEG clusters and the case of DMR in the promoter of the up- and down-regulated DEG was marked in the cluster. DEGs with methylated promoter regions were colored in pink for hypermethylation and sky blue for hypomethylation.
Fig. 4.

Graph-based clustering results. Up-regulated DEGs are colored in red, and down-regulated DEGs are colored in blue. The diamond borders of the genes are colored in pink or sky blue when the promoters of the genes are either hypermethylated or hypomethylated, respectively. The red circles indicate the selected clusters in LAML and COAD
Cluster selection for in-depth analysis
We performed Fisher’s exact test with the number of DMR-DEGs (differentially expressed gene with differentially methylated promoter region) in each cluster to select statistically significant clusters.
A cluster in LAML was selected in which mutated samples of DNMT3A were abundant and DEGs were up-regulated. There were four clusters with up-regulated genes with hypo-methylated promoter, and one cluster containing genes with large log2 fold change of expression level was selected. In COAD clusters, TET1/2/3 genes were mutated with promoter hypermethylated, so we selected a cluster that contained the largest number of down-regulated DEGs. In the case of COAD, the most significant cluster with the highest number of DMR-DEG was selected. For the functional analysis of DEGs in the clusters, we selected a cluster of up-regulated DEGs in LAML and a cluster of down-regulated DEGs in COAD (Fig. 5).
Fig. 5.
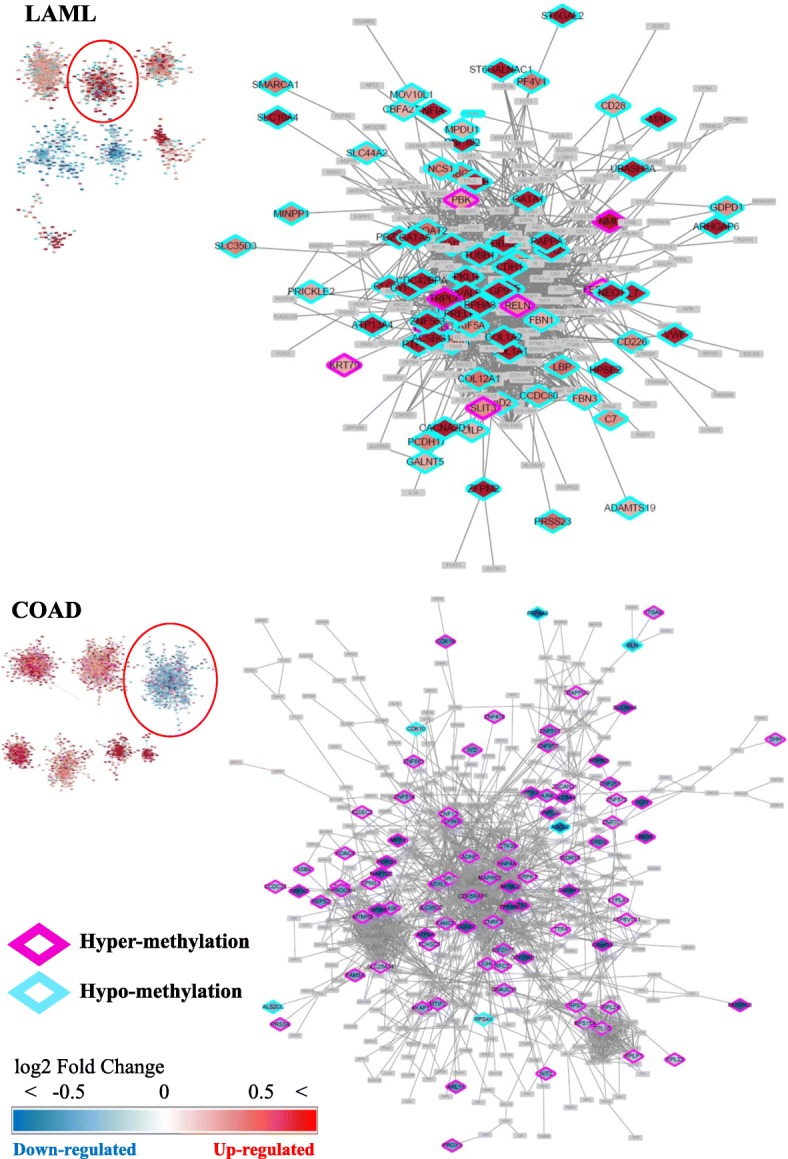
Selected sub-network clusters in LAML and COAD. Up-regulated genes were colored in red, and down-regulated genes were colored in blue according to the expression fold change level. DEGs without a differentially methylated promoter is shown in translucent gray. The borders of the genes are colored in pink or sky blue when the promoters of the genes are either hypermethylated or hypomethylated, respectively
TF selection related with DMR-DEGs
Among the genes in the clusters of COAD and LAML, we selected DEGs that the expression changes were not associated with TFs. To investigate TF-DNA-methylation interaction, we searched for all TF binding sties in the promoter regions using TRANSFAC [23] database. In COAD, there were 86 DMR-DEGs and we detected 170 TFs. In LAML, 75 DMR-DEGs were selected, and 179 TFs were detected by TRANSFAC using a promoter sequence of DEGs.
Part 3 - DMR-DEGs in-depth analysis
Selection of cancers for in-depth analysis.
For the in-depth analysis to investigate the effect of mutations in DNA methylation modifiers, we first selected cancers based on the mutation profiles Fig. 2. In COAD, the number of the samples of which the demethylation-related genes, TET1, TET2 and TET3, were mutated was bigger than that of the samples with mutations in the methylation-related genes. On the contrary, in LAML, mutations in the methylation-related genes, e.g., DNMT3A, were dominant. We also looked genome-wide promoter methylation landscape to see relations between the mutations in the methylation-related genes and the methylation status of the promoters of the genes. As shown in Fig. 3, we were able to observe that there was a distinct signature of promoter hypermethylation in COAD (Fig. 5). On the contrary, in LAML, the promoters were hypomethylated rather than hypermethylated. GBM also showed the promoter hypomethylation but the number of samples with mutations was too small to analyze the effect of mutations (Fig. 2). Thus, we selected COAD and LAML for further analyses.
Selection of DMR-DEG possibly without TF-mediated regulation.
Before associating DMR-DEG, we excluded the DMR-DEGs that the expression changes were possibly affected by TFs. Among selected TFs that had binding sites in the promoter regions (see cluster selection in PART 2), if expression levels of TFs were different significantly between the mutated and non-mutated sample groups, TF expression difference could affect expression levels of downstream genes, thus we remove genes whose promoter regions had binding sites of such TFs. We set 0.2 and -0.2 as cutoff values for log2 fold change to determine if a gene or a TF is up-regulated or down-regulated. When a gene is up-regulated and a TF targeting the gene is up-regulated, the DEG was removed. Likewise, when a gene is down-regulated and a TF targeting the gene is also down-regulated, the DEG was removed. Finally, 54 DMR-DEGs in LAML and 45 DMR-DEGs in COAD were selected and studied for functional effects (Table 4).
Table 4.
List of 54 DMR-DEGs in LAML and 45 DMR-DEGs in COAD
| Selected cluster | DMR-DEGs |
|---|---|
| Cluster of LAML | ACSBG1, ARHGAP6, ATP13A4, C7, CACNA2D1, CBFA2T3, CCDC80, CD226, CD28, CILP, COL12A1, COL1A1, COL1A2, DBX2, EPHA3, FBN1, FBN3, FERMT2, FN3K, GATA1, GATA5, GDPD1, GFI1B, GP9, HBG2, HPSE2, IL7, KIF5A, LBP, LRRC8E, MAL, MBOAT2, MINPP1, NCS1, NEO1, NID2, NLGN1, NMU, PAPPA, PF4V1, PKLR, PRELP, PRICKLE2, PTPRR, RNF182, SLC35D3, SLC44A2, SLIT3, ST6GAL2, ST6GALNAC1, TUBB1, VWF, ZFPM2, ZNF563 |
| Cluster of COAD | ADNP, AKAP11, ARL11, ASB9, ASXL1, ATP9A, BBS5, C8orf33, CASK, CDK18, DNAJC15, FCHSD1, GTF2IRD1, HDAC8, HUNK, IFT52, IQSEC2, IYD, LYPLA1, MOSPD3, MPP1, MRPS33, MTIF3, NIT2, PIPOX, PRSS8, REPS2, RPL24, RPLP1, RPS7, SHH, SLC25A14, SPIN3, SPNS1, TARBP1, TP53RK, TTPA, UCHL3, WDR35, ZNF12, ZNF251, ZNF514, ZNF517, ZNRF2, ZXDB |
up-regulated DEGs related with hypo-DMR in LAML
54 up-regulated DEGs with hypomethylated promoters were selected in LAML. To investigate the biological function of these genes, we searched the literature to find relevance of these genes to LAML. For 54 DEGs in LAML, we searched with the terms “methylation” or “acute myeloid leukemia”. CACNA2D1, CBFA2T3, CD226, EPHA3, GATA1, GFI1B, IL7, NMU, PTPRR, SLIT3 and ST6GAL2 genes are related with disorder of methylation in LAML. CACNA2D1 (Voltage-dependent calcium channel subunit alpha-2/delta-1) encodes a member of the alpha-2/delta subunit family, a protein in the voltage-dependent calcium channel complex. CACNA2D1 has DMR in oxytocin signaling pathway in LAML [25].
CBFA2T3 is known to operate via a fusion gene mechanism with INADL and TM2D1 in AML [26].
CD226 (Cluster of Differentiation 226, DNAM-1 (DNAX Accessory Molecule-1)) is a 65 kDa glycoprotein expressed on the surface of natural killer cells, platelets, monocytes and a subset of T cells. TIGIT binding with CD226 is up-regulated on CD8(+) T cells in LAML [27]. EPHA3 (ephrin type-A receptor 3) has been implicated in mediating developmental events, particularly in the nervous system. Receptors in the EPH subfamily typically have a single kinase domain and an extracellular region containing a Cys-rich domain and 2 fibronectin type III repeats. EphA3 was methylated in leukemia patients [28]. GATA1 (GATA-binding factor 1) regulates the expression of an ensemble of genes that mediate the development of red blood cells and platelets. Its critical roles in red blood cell formation include promoting the maturation of precursor cells. GATA-1 binds to the PU.1 gene and inhibits expression in LAML [29]. IL7 (Interleukin 7) stimulates proliferation of all cells in the lymphoid lineage (B cells, T cells and NK cells). IL-7 has abnormal methylation in peripheral blood of LAML patients [30]. GFI1B (Growth factor independent 1b, Zinc finger protein Gfi-1b) are highly expressed in LAML [31].
NMU induced specifically acute promyelocytic leukemia in Sprague-Dawley rats [32]. PTPRR has been recently identified as a fusion partner of the ETV6 gene in AML patients bearing an inv(12)(p13q13) and leads to GM-CFS-independent STAT3 activation [33].
SLIT3 (Slit homolog 3 protein) is a ligand-receptor SLIT-ROBO family. Low expression of SLIT and high expression of ROBO1 and ROBO2 suggests their participation in LAML pathogenesis [34].
ST6GAL2 was detected with unique DMR gene for AML subtype [35].
SLC44A2 is related with LAML. SLC44A2 (Choline transporter-like protein 2) is located in a pathway controlling DNA damage and repair, and affects the survival in LAML [36].
In GO-term enrichment test with “blood coagulation”, “cell adhesion”, “platelet activation”, “extracellular matrix organization”, “cellular response to transforming growth factor beta stimulus”, “response to stimulus”, “collagen fibril organization”, “multicellular organismal process”, “response to endogenous stimulus”, “skin morphogenesis” and “cell activation” (Table 5).
Table 5.
Enriched GO terms of 54 DMR-DEGs in LAML
| GO-term ID | term description | gene count | FDR |
|---|---|---|---|
| GO:0007596 | blood coagulation | 9 of 288 | 0.0002 |
| GO:0007155 | cell adhesion | 13 of 843 | 0.0002 |
| GO:0030168 | platelet activation | 6 of 120 | 0.00039 |
| GO:0030198 | extracellular matrix organization | 8 of 296 | 0.00045 |
| GO:0071560 | cellular response to transforming growth factor beta stimulus | 5 of 140 | 0.0075 |
| GO:0050896 | response to stimulus | 35 of 7824 | 0.0269 |
| GO:0030199 | collagen fibril organization | 3 of 39 | 0.0269 |
| GO:0032501 | multicellular organismal process | 31 of 6507 | 0.0272 |
| GO:0009719 | response to endogenous stimulus | 12 of 1353 | 0.0298 |
| GO:0043589 | skin morphogenesis | 2 of 9 | 0.0416 |
| GO:0001775 | cell activation | 10 of 1024 | 0.0446 |
Down-regulated DEGs related with hyper-DMR in COAD
45 down-regulated DEGs with hypermethylated promoters were selected in COAD. To investigate the biological function of these genes, we searched the literature to find relevance of these genes to COAD. For 45 DEGs selected in the cluster of COAD, we searched the literature with the terms “methylation” or “Colon adenocarcinoma”. HDAC8, HUNK, PRSS8, RPS7 and UCHL3 genes are related with disorder of methylation in COAD.
HDAC8, one of the histone deacetylase (HDAC) family of transcriptional co-repressors, has emerged as important regulators of colon cell maturation and transformation [37]. Abnormal changes in DNA methylation level of HUNK were found in tumor tissues of patients [38]. PRSS8 acts as a tumor suppressor by inhibiting Sphk1/S1P/Stat3/Akt signaling pathway [39].
RPS7 (40S ribosomal protein S7) is a component of the 40S subunit. In eukaryotes, ribosomes, the organelles that catalyze protein synthesis, consist of a small 40S subunit and a large 60S subunit. Aberrant promoter hypermethylation of RPS7 inhibits colorectal cancer growth [40].
UCHL3, a member of the ubiquitin C-terminal hydrolase family, has a similar activity to UCHL1 and is ubiquitously expressed in various tissues. Methylation of the UCHL3 promoter CpG island was completely unmethylated in the colorectal cancer [41].
ADNP, ASB9 and NIT2 genes are related with COAD. ADNP is a repressor of WNT signaling in colon cancer [42]. Low ASB9 expression have higher malignant potential, such as cell invasiveness and liver metastasis resulting in a poor prognosis for human colorectal cancer [43].
NIT2 (Nitrilase Family Member 2) has a omega-amidase activity to remove potentially toxic intermediates by converting alpha-ketoglutaramate and alpha-ketosuccinamate to biologically useful alpha-ketoglutarate and oxaloacetate. Downregulation of NIT2 inhibits COAD cell proliferation and induces cell cycle arrest [44].
SHH and WDR35 genes are related with abnormal methylation in cancer. The increased and constitutive SHH expression is implicated in gastric carcinogenesis, and that promoter methylation may be an important regulatory mechanism of SHH expression [45]. WDR35 has functions in cell signaling and apoptosis. The methylation levels of WDR35 was consistent with an inverse relationship with the mRNA expression levels in a large number of ALL cells [46].
In GO-term enrichment test with “Biological Process” category, the 45 genes in COAD were found to be related with “cytoplasmic translation”, “ peptide biosynthetic process”, “SRP-dependent cotranslational protein targeting to membrane”, “cotranslational protein targeting to membrane”, “protein targeting to ER”, “translation”, “viral gene expression”, “nuclear-transcribed mRNA catabolic process, nonsense-mediated decay” and “viral transcription” (Table 6).
Table 6.
Enriched GO terms of 45 DMR-DEGs in COAD
| GO-term ID | term description | gene count | FDR |
|---|---|---|---|
| GO:0002181 | cytoplasmic translation | 3 of 55 | 0.0002 |
| GO:0043043 | peptide biosynthetic process | 4 of 175 | 0.0006 |
| GO:0006614 | SRP-dependent cotranslational protein targeting to membrane | 3 of 90 | 0.001 |
| GO:0006613 | cotranslational protein targeting to membrane | 3 of 94 | 0.0012 |
| GO:0045047 | protein targeting to ER | 3 of 98 | 0.0013 |
| GO:0006412 | translation | 4 of 233 | 0.0018 |
| GO:0019080 | viral gene expression | 3 of 111 | 0.0019 |
| GO:0000184 | nuclear-transcribed mRNA catabolic process, nonsense-mediated decay | 3 of 113 | 0.002 |
| GO:0019083 | viral transcription | 3 of 114 | 0.0021 |
Conclusions
High-dimensional feature space data analysis using sub-network clustering
Determining DEGs affected by methylation changes is a research problem of dealing with high dimensional feature spaces that need s to combine gene expression levels and methylation expression levels. Our approach to dealing with this challenging problem was to use a network based approach.
Clustering genes by combining protein-protein interaction scores and gene correlation values were effective in identifying DEGs clusters that could be affected by DNA methylation modifiers. In addition, we considered TF-DNA interference by DNA methylations in the promoter regions to focus more on the effect of mutations in DNA methylation modifiers only. Many of the genes that were identified by our approach have been shown to be related to cancer development in the literature regarding the effects of methylation. Some genes that were determined in this study are also likely to be related to cancer expression by methylation, which could be good testable hypotheses for additional biological experiments.
Biological meaning and functions of the identified DMR-DEGs
Recently, the effects of epigenetic changes in phenotypic changes including disease developments have been investigated extensively. However, the effects of mutations in epigenetic modifiers have not been well studied so far. To investigate how epigenetic changes are acquired, it is very important to investigate biological mechanisms that could cause epigenetic changes. In this context, we investigated the effects of mutations in DNA methylation modifier genes on the transcriptomic profiles in samples of mutated vs. non-mutated DNA methylation modifiers in the pan-cancer scale. We identified 54 DEGs affected by seven DNA methylation gene mutations in LAML patient samples and 61 DEGs in COAD patients. Gene expression levels of these genes increased (DEGs of LAML) or decreased (DEGs of COAD) without potential effects of TFs that could bind to the promoter regions of the genes. In other words, differences in methylation status in the promoter regions of the genes could be the main reason why these genes were expressed differentially. 28 of 33 mutant samples in LAML had mutations in DNMT3A, and 34 of the 54 samples in COAD had mutations in TET2. Mutations in DNMT3A could result in hypomethylation in the promoter regions due to abnormal methyl transfer, resulting in increased gene expression. Mutations in TET2 could result in hypermethylation in the promoter regions of genes due to abnormalities in demethylation function. In the clusters of LAML, 10 of 54 DMR-DEGs, that has highest number of DMR, were known to be associated with LAML in the literature. 7 of the 10 genes were associated with abnormal methylation with LAML in the literature. In case of COAD, 8 of 45 DMR-DEGs were associated with COAD and 4 genes among the 8 genes were associated with abnormal methylation in COAD in the literature. In this study, we reported that these genes are likely to be related to the development of cancer due to changes in DNA methylation. However, functional impact and biological interpretation of our findings are yet to be confirmed although we provided GO term enrichment analysis and related papers in the literature. As we have more samples available, our approach could contribute to elucidating testable hypotheses on the roles of mutations in DNA methylation modifiers.
Supplementary information
Additional file 1 Figure S1 Case ratio of 7 DNA methylation modifiers mutations in each TCGA 33 projects. Figure S2 Violin plot result of DMR analysis.
Acknowledgements
Not applicable
About this supplement
This article has been published as part of BMC Medical Genomics Volume 13 Supplement 3, 2020: Proceedings of the Joint International GIW & ABACBS-2019 Conference: medical genomics (part 2). The full contents of the supplement are available online at https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-13-supplement-3.
Abbreviations
- DEG
Differentially expressed gene
- DMR
Differentially methylated region
- DNA
Deoxyribonucleic acid
- GO
Gene ontology
- RNA-seq
Whole transcriptome sequencing
- RNA
Ribonucleic acid
- STRING
Search tool for the retrieval of interacting genes/proteins
- TCGA
The cancer genome atlas
- TF
Transcription factor
Authors’ contributions
SK designed the project. SK and CL directed the development of the algorithm. CL collected and processed the data. CL, HA, DJ, MP and JM implemented the program of the algorithm and conducted experiments of network construction. CL interpreted the network analysis results biologically. CL, JM and SK wrote the paper. All authors read and approved the final manuscript.
Funding
This research is supported by National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT (No. NRF-2017M3C4A7065887), the Collaborative Genome Program for Fostering New Post-Genome Industry of the National Research Foundation (NRF) funded by the Ministry of Science and ICT (MSIT) (No. NRF-2014M3C9A3063541), and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C3224). The funding bodies provided financial support but had no other role in the design of the study, data collection, analysis, and interpretation of data, decision to publish, or preparation of the manuscript.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chai-Jin Lee, Email: chaijin.lee@snu.ac.kr.
Hongryul Ahn, Email: hrahn@snu.ac.kr.
Dabin Jeong, Email: dabin1816@snu.ac.kr.
Minwoo Pak, Email: mwpak@snu.ac.kr.
Ji Hwan Moon, Email: jihmoon@snu.ac.kr.
Sun Kim, Email: sunkim.bioinfo@snu.ac.kr.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12920-020-0659-4.
References
- 1.Wee Y, Liu Y, Bhyan SB, Lu J, Zhao M. The pan-cancer analysis of gain-of-functional mutations to identify the common oncogenic signatures in multiple cancers. Gene. 2019;697:57–66. doi: 10.1016/j.gene.2019.02.039. [DOI] [PubMed] [Google Scholar]
- 2.Kim H, Kim Y-M. Pan-cancer analysis of somatic mutations and transcriptomes reveals common functional gene clusters shared by multiple cancer types. Sci Rep. 2018;8(1):6041. doi: 10.1038/s41598-018-24379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173(2):371–85. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17(5):284. doi: 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan X-J, Xu J, Gu Z-H, Pan C-M, Lu G, Shen Y, Shi J-Y, Zhu Y-M, Tang L, Zhang X-W, et al. Exome sequencing identifies somatic mutations of dna methyltransferase gene dnmt3a in acute monocytic leukemia. Nat Genet. 2011;43(4):309. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 6.Couronné L, Bastard C, Bernard OA. Tet2 and dnmt3a mutations in human t-cell lymphoma. N Engl J Med. 2012;366(1):95–6. doi: 10.1056/NEJMc1111708. [DOI] [PubMed] [Google Scholar]
- 7.Grossmann V, Haferlach C, Weissmann S, Roller A, Schindela S, Poetzinger F, Stadler K, Bellos F, Kern W, Haferlach T, et al. The molecular profile of adult t-cell acute lymphoblastic leukemia: mutations in runx1 and dnmt3a are associated with poor prognosis in t-all. Genes Chromosome Cancer. 2013;52(4):410–22. doi: 10.1002/gcc.22039. [DOI] [PubMed] [Google Scholar]
- 8.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, et al. Genetic characterization of tet1, tet2, and tet3 alterations in myeloid malignancies. Blood. 2009;114(1):144–7. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, et al. Acquired mutations in tet2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 10.Network CGA, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumann M, Heesch S, Schlee C, Schwartz S, Gökbuget N, Hoelzer D, Konstandin NP, Ksienzyk B, Vosberg S, Graf A, et al. Whole-exome sequencing in adult etp-all reveals a high rate of dnmt3a mutations. Blood. 2013;121(23):4749–52. doi: 10.1182/blood-2012-11-465138. [DOI] [PubMed] [Google Scholar]
- 14.Delhommeau F, Dupont S, Valle VD, James C, Trannoy S, Masse A, Kosmider O, Le Couedic J-P, Robert F, Alberdi A, et al. Mutation in tet2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 15.Scourzic L, Mouly E, Bernard OA. Tet proteins and the control of cytosine demethylation in cancer. Genome Med. 2015;7(1):9. doi: 10.1186/s13073-015-0134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. Exome sequencing identifies recurrent somatic rac1 mutations in melanoma. Nat Genet. 2012;44(9):1006. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. Dnmt3a mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tomczak K, Czerwińska P, Wiznerowicz M. The cancer genome atlas (tcga): an immeasurable source of knowledge. Contemp Oncol. 2015;19(1A):68. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BM, Haag JD, Gould MN, Stewart RM, Kendziorski C. Ebseq: an empirical bayes hierarchical model for inference in rna-seq experiments. Bioinformatics. 2013;29(8):1035–43. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium dna methylation microarrays. Bioinformatics. 2014;30(10):1363–9. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Csardi G, Nepusz T, et al. The igraph software package for complex network research. InterJournal Complex Syst. 2006;1695(5):1–9. [Google Scholar]
- 22.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al.The string database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016:937. [DOI] [PMC free article] [PubMed]
- 23.Matys V, Fricke E, Geffers R, Gößling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. Transfac®: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31(1):374–8. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao C, Zhuang J, Zhou C, Liu L, Liu C, Li H, Zhao M, Liu G, Sun C. Developing dna methylation-based prognostic biomarkers of acute myeloid leukemia. J Cell Biochem. 2018;119(12):10041–50. doi: 10.1002/jcb.27336. [DOI] [PubMed] [Google Scholar]
- 26.Micci F, Thorsen J, Haugom L, Zeller B, Tierens A, Heim S. Translocation t (1; 16)(p31; q24) rearranging cbfa2t3 is specific for acute erythroid leukemia. Leukemia. 2011;25(9):1510. doi: 10.1038/leu.2011.100. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez-Correa B, Gayoso I, Bergua JM, Casado JG, Morgado S, Solana R, Tarazona R. Decreased expression of dnam-1 on nk cells from acute myeloid leukemia patients. Immunol Cell Biol. 2012;90(1):109–15. doi: 10.1038/icb.2011.15. [DOI] [PubMed] [Google Scholar]
- 28.Rush LJ, Raval A, Funchain P, Johnson AJ, Smith L, Lucas DM, Bembea M, Liu T-H, Heerema NA, Rassenti L, et al. Epigenetic profiling in chronic lymphocytic leukemia reveals novel methylation targets. Cancer Res. 2004;64(7):2424–33. doi: 10.1158/0008-5472.CAN-03-2870. [DOI] [PubMed] [Google Scholar]
- 29.Burda P, Vargova J, Curik N, Salek C, Papadopoulos GL, Strouboulis J, Stopka T. Gata-1 inhibits pu. 1 gene via dna and histone h3k9 methylation of its distal enhancer in erythroleukemia. PloS ONE. 2016;11(3):0152234. doi: 10.1371/journal.pone.0152234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Liu Y, Gao S. Correlation between il-7 genomic protein methylation level and acute myeloid leukemia. Eur Rev Med Pharmacol Sci. 2019;23(3):1196–202. doi: 10.26355/eurrev_201902_17012. [DOI] [PubMed] [Google Scholar]
- 31.Vassen L, Khandanpour C, Ebeling P, van der Reijden BA, Jansen JH, Mahlmann S, Dührsen U, Möröy T. Growth factor independent 1b (gfi1b) and a new splice variant of gfi1b are highly expressed in patients with acute and chronic leukemia. Int J Hematol. 2009;89(4):422–30. doi: 10.1007/s12185-009-0286-5. [DOI] [PubMed] [Google Scholar]
- 32.Chang Y-C, Hsu J-D, Lin W-L, Lee Y-J, Wang C-J. High incidence of acute promyelocytic leukemia specifically induced by n-nitroso-n-methylurea (nmu) in sprague–dawley rats. Arch Toxicol. 2012;86(2):315–27. doi: 10.1007/s00204-011-0753-7. [DOI] [PubMed] [Google Scholar]
- 33.Nakamura F, Nakamura Y, Maki K, Sato Y, Mitani K. Cloning and characterization of the novel chimeric gene tel/ptprr in acute myelogenous leukemia with inv (12)(p13q13) Cancer Res. 2005;65(15):6612–21. doi: 10.1158/0008-5472.CAN-04-4631. [DOI] [PubMed] [Google Scholar]
- 34.Gołos Aleksandra, Jesionek-Kupnicka Dorota, Gil Lidia, Braun Marcin, Komarnicki Mieczyslaw, Robak Tadeusz, Wierzbowska Agnieszka. The Expression of the SLIT–ROBO Family in Adult Patients with Acute Myeloid Leukemia. Archivum Immunologiae et Therapiae Experimentalis. 2019;67(2):109–123. doi: 10.1007/s00005-019-00535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saied MH, Marzec J, Khalid S, Smith P, Down TA, Rakyan VK, Molloy G, Raghavan M, Debernardi S, Young BD. Genome wide analysis of acute myeloid leukemia reveal leukemia specific methylome and subtype specific hypomethylation of repeats. PLoS ONE. 2012;7(3):33213. doi: 10.1371/journal.pone.0033213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruedigam C, Bagger FO, Heidel FH, Kuhn CP, Guignes S, Song A, Austin R, Vu T, Lee E, Riyat S, et al. Telomerase inhibition effectively targets mouse and human aml stem cells and delays relapse following chemotherapy. Cell Stem Cell. 2014;15(6):775–90. doi: 10.1016/j.stem.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mariadason JM. Hdacs and hdac inhibitors in colon cancer. Epigenetics. 2008;3(1):28–37. doi: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- 38.Xue G, Lu C-J, Pan S-J, Zhang Y-L, Miao H, Shan S, Zhu X-T, Zhang Y. Dna hypomethylation of cbs promoter induced by folate deficiency is a potential noninvasive circulating biomarker for colorectal adenocarcinomas. Oncotarget. 2017;8(31):51387. doi: 10.18632/oncotarget.17988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bao Y, Li K, Guo Y, Wang Q, Li Z, Yang Y, Chen Z, Wang J, Zhao W, Zhang H, et al. Tumor suppressor prss8 targets sphk1/s1p/stat3/akt signaling in colorectal cancer. Oncotarget. 2016;7(18):26780. doi: 10.18632/oncotarget.8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, Tong D, Liu F, Li D, Li J, Cheng X, Wang Z. Rps7 inhibits colorectal cancer growth via decreasing hif-1 α-mediated glycolysis. Oncotarget. 2016;7(5):5800. doi: 10.18632/oncotarget.6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okochi-Takada E, Nakazawa K, Wakabayashi M, Mori A, Ichimura S, Yasugi T, Ushijima T. Silencing of the uchl1 gene in human colorectal and ovarian cancers. Int J Cancer. 2006;119(6):1338–44. doi: 10.1002/ijc.22025. [DOI] [PubMed] [Google Scholar]
- 42.Blaj C, Bringmann A, Urbischek M, Krebs S, Blum H, Fröhlich T, Arnold G, Jung A, Kirchner T, Horst D. Adnp is a repressor of wnt signaling in colon cancer that can be therapeutically induced. Eur J Cancer. 2016;61:172. doi: 10.1016/S0959-8049(16)61611-8. [DOI] [Google Scholar]
- 43.Tokuoka M, Miyoshi N, Hitora T, Mimori K, Tanaka F, Shibata K, Ishii H, Sekimoto M, Doki Y, Mori M. Clinical significance of asb9 in human colorectal cancer. Int J Oncol. 2010;37(5):1105–11. doi: 10.3892/ijo_00000762. [DOI] [PubMed] [Google Scholar]
- 44.Zheng B, Chai R, Yu X. Downregulation of nit2 inhibits colon cancer cell proliferation and induces cell cycle arrest through the caspase-3 and parp pathways. Int J Mol Med. 2015;35(5):1317–22. doi: 10.3892/ijmm.2015.2125. [DOI] [PubMed] [Google Scholar]
- 45.Wang L-H, Choi Y-L, Hua X-Y, Shin Y-K, Song Y-J, Youn S-J, Yun H-Y, Park S-M, Kim W-J, Kim H-J, et al. Increased expression of sonic hedgehog and altered methylation of its promoter region in gastric cancer and its related lesions. Modern Pathol. 2006;19(5):675. doi: 10.1038/modpathol.3800573. [DOI] [PubMed] [Google Scholar]
- 46.Nordlund J, Milani L, Lundmark A, Lönnerholm G, Syvänen A-C. Dna methylation analysis of bone marrow cells at diagnosis of acute lymphoblastic leukemia and at remission. PloS ONE. 2012;7(4):34513. doi: 10.1371/journal.pone.0034513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Figure S1 Case ratio of 7 DNA methylation modifiers mutations in each TCGA 33 projects. Figure S2 Violin plot result of DMR analysis.