

ABSTRACT
COTI-2 is a third-generation thiosemicarbazone, which is effective against a diverse group of human cancer cell lines at nanomolar concentrations. COTI-2 also showed superior activity against tumor cells, in vitro and in vivo. As a high efficacy and low toxicity agent, it currently candidates in a phase I clinical study of gynecological malignancies and head and neck squamous cell carcinoma (HNSCC). However, its effect in pediatric T-cell acute lymphoblastic leukemia (T-ALL) is not clear. This study investigates the effect of COTI-2 on T-ALL Jurkat cells in vitro and in vivo. Jurkat cells were exposure to COTI-2 at different concentration and time. Cell apoptosis was detected by flow cytometry to examine the sensitivity of Jurkat cell lines treated with either COTI-2 alone or in combination with MiR-203 mimic or inhibitor in vitro. An orthotopic mouse model was used to examine the sensitivity of Jurkat cells treated with COTI-2 in vivo. Western blotting and RT-qPCR were performed to dissect molecular mechanisms. The results showed that COTI-2 promotes apoptosis of Jurkat cells in dose-and time-dependent way. Enforced expression of miR-203 promotes COTI-2-mediated cell apoptosis, whereas miR-203 silencing attenuates COTI-2-mediated cell apoptosis in Jurkat cells in vitro. COTI-2 is also effective against growth of Jurkat cells in vivo. Mechanistically, COTI-2 induced miR-203 upregulation and inhibited caspase-3/9 activaty leading to inhibition of cell apoptosis. Taken together, COTI-2 inhibits tumor growth in vitro and in vivo in Jurkat cells likely through miR-203-dependent mechanisms. COTI-2 may be a potential approach for T-ALL treatment.
KEYWORDS: T-cell acute lymphoblastic leukemia, COTI-2, miR-203, caspase-3/9, apoptosis
Introduction
Acute lymphoblastic leukemia (ALL) is a malignant transformation and proliferation of lymphoid progenitor cells in the bone marrow, blood and extramedullary sites. While 80% of ALL occurs in children, it represents a devastating disease when it occurs in adults [1]. The structure of treatment of adult ALL has been adapted from pediatric protocols. Unfortunately, while long-term survival approaches 90% for standard-risk pediatric ALL, the success rate is much more modest in adults [2]. Chemotherapy consists of induction, consolidation and long-term maintenance, with CNS prophylaxis given at intervals throughout therapy. However, due to high-risk disease characteristics and significant toxicity associated with chemotherapy in adults, outcomes are far less encouraging [3,4].
COTI-2, a novel small molecule currently in a phase I clinical study of gynecological malignancies and head and neck squamous cell carcinoma (HNSCC), was designed using CHEMSAS®, a proprietary computational platform.
It has recently reported that COTI-2 is effective against a diverse group of human cancer cell lines regardless of their tissue of origin or genetic makeup. Most treated cancer cell lines were sensitive to COTI-2 at nanomolar concentrations. When compared to traditional chemotherapy or targeted-therapy agents, COTI-2 showed superior activity against tumor cells, in vitro and in vivo [5]. Lindemann et al. reported that COTI-2 decreased clonogenic survival of HNSCC cells and potentiated response to cisplatin and/or radiation in vitro and in vivo through p53-dependent and p53-independent mechanisms [6]. In addition, combining COTI-2 with multiple chemotherapeutic and targeted agents enhances the activity of these drugs in vitro and in vivo, and no overt toxicity was observed in the combination treatment groups in vivo. Furthermore, some chemo-resistant tumor cell lines only showed mild cross-resistance to COTI-2 while most remained sensitive to it [7]. However, the mechanism of action of COTI-2 is still unclear.
MicroRNA (miRNA) is a single-stranded, non-coding RNA molecule of 22–25 nucleotides, which leads to down-regulation of target protein expression [8]. Recently, hypermethylation of miR-203 has been reported in chronic myeloid leukemia (CML) and acute myeloid leukemia (AML), conferring proliferative advantage in tumor cells [9,10].
In this study, we aimed to study the anti-cancer role and potential mechanism of COTI-2 in acute lymphoblastic leukemia (ALL) cells in vitro and in vivo. Our results show that COTI-2 effectively induced apoptosis and inhibited growth of leukemia cells via activating miR-203.
Materials and methods
Cytotoxic drugs
COTI-2 was supplied by Critical Outcome Technologies Inc. (London, Ontario). COTI-2 was dissolved in 100% dimethyl sulfoxide stock solution and diluted in medium plus FBS such that final DMSO concentrations were 0.5–1.0% depending on the experiment
Cell culture
Jurkat cells were purchased from AmericanType Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI 1640 supplemented with 10% heat inactivated fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine (Life Technologies, Carlsbad, CA).
Transfection of cells with the miR‐203 mimics vector or the inhibitors vector
For miR‐203 overexpression, the miR‐203 mimic or corresponding negative control (miR‐NC) was purchased from GenePharma (Shanghai, China). Jurkat cells were transfected with either the miR‐203 mimic or miR‐NC at a final concentration of 100 nM using Lipofectamine® 2000 (Invitrogen) according to the manufacturer’s protocol. Cells were used for miR‐203 expression analysis or other experiments after 48 h of transfection. For miR‐203 inhibition, Jurkat cells were treated with final concentration of 100 nM miR‐203 inhibitor (Invitrogen) for 48 h, then the effect of miR‐203 on related gene expression was detected.
qPCR for miR-203
Total RNA from tissue and cells were isolated using TRIzol reagent (Invitrogen) to obtain miRs according to the manufacturer’s instructions. All RNA extractions were carried out in a sterile laminar flow hood using RNase/DNase-free laboratory ware. The single-tube TaqMan miRNA assay (Applied Biosystems, CA, US) was used to detect and quantify mature miR-203, using ViiA7 RT reader (Applied Biosystems, CA, US); the protocol was performed for 38 cycles at 95°C for 3 min, 95°C for 15 s, and 60°C for 30 s. miR-203 expression was normalized on U6, and then expressed as fold change (2ΔΔCt).
Western blotting analysis
Cells were seeded at a density of 3 × 105 per well in 6-well plates and allowed to attach for 24 h. After various treatments, the whole cell lysates were subjected to Western blotting analysis. Total proteins were extracted by using RIPA lysis buffer containing 10 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% Triton X-100 (w/v), 0.5% sodium deoxycholate, and 0.1% SDS with protease and phosphatase inhibitor mixture. The equal amounts of protein were separated in 10% SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were incubated with primary antibodies overnight at 4°C. The following primary antibodies were used: PARP, cleaved-caspase-3 and cleaved-caspase-9. The corresponding HRP-conjugated secondary IgG antibodies were used, and immunoreacted proteins were detected with chemiluminescence reagent.
Apoptosis assay
Apoptotic cells were measured by staining with FITC-conjugated Annexin V/propidium iodide (PI) (BD Pharmingen, San Diego, CA, USA) and determining with flow cytometer according to the manufacturer’s instructions. Both early apoptotic (Annexin V-positive, PI-negative) and late apoptotic (Annexin V-positive and PI-positive) cells were included in cell death determinations.
Xenograft model and miR-203 evaluation
Nude mice (5 weeks old) were purchased from Vital River Laboratories (Beijing, China). Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the university. Jurkat cells (5 x 106/0.2 ml per mouse) were suspended in sterile PBS and injected subcutaneously into the right flank of the mice (n = 10 mice per treatment group). When tumors reached a palpable volume of approximately 75–100 mm3 (3–4 weeks), treatment with COTI-2 (10 mg/kg, 5 days a week for 7 weeks) or saline intraperitoneal injection (IP) began. Tumor growth was measured every 4 days by caliper measurement. Tumor volumes were determined by a caliper and calculated according to the formula (width2 x length)/2. Absolute quantities of miR-203 in tumor tissue were obtained by qRT-PCR .
Immunohistochemical evaluation
Tumor tissue sections (4μm) were subjected to immunohistochemical staining (IHC) for cleaved-caspase-3. The percentage of positively-stained area with 40 fields of view was analyzed by Image-pro plus 6.0 (Media cybernetics).
TUNEL evaluation
Apoptosis in tumor tissue sections was determined using In Situ Cell Death Detection kit (Roche, Mannheim, Germany). Briefly, tumor tissue sections of formalin-fixed, paraffin-embedded specimens were dewaxed in xylene
and rehydrated in a graded series of ethanol. The tumor samples were incubated with proteinase K (2 mg/ml), and the TUNEL staining was performed according to the manufacturer’s instructions.
Densitometry and statistical analysis
Statistical analysis was performed using SPSS.22 Software. One-way analysis of variance (ANOVA) test was used for comparisons when more than two groups were analyzed and Student’s t-test when two groups were analyzed. ANOVA was performed using the Bonferroni post-hoc test. Values of P < 0.05 were considered significant.
Results
COTI-2 induces apoptosis in dose- and time-dependent manners
A dose-dependent study in Jurkat cells revealed a moderate increase in apoptosis 48 h after exposure to 100 nM COTI-2 and very extensive apoptosis at concentrations of 200 nM (Figure 1(a)). Time-course analysis of cells exposed to 200 nM COTI-2 demonstrated a significant increase in apoptosis as early as 6 h. These events became apparent after 24 h of drug exposure, and reached maximal levels after 48 h (Figure 1(b)). Consistent with these findings, the same COTI-2 concentrations and exposure intervals resulted in cleavage/activation of caspase-9 and −3, and degradation of poly-ADP-ribose polymerase (PARP) (Figure 1(c,d)).
Figure 1.
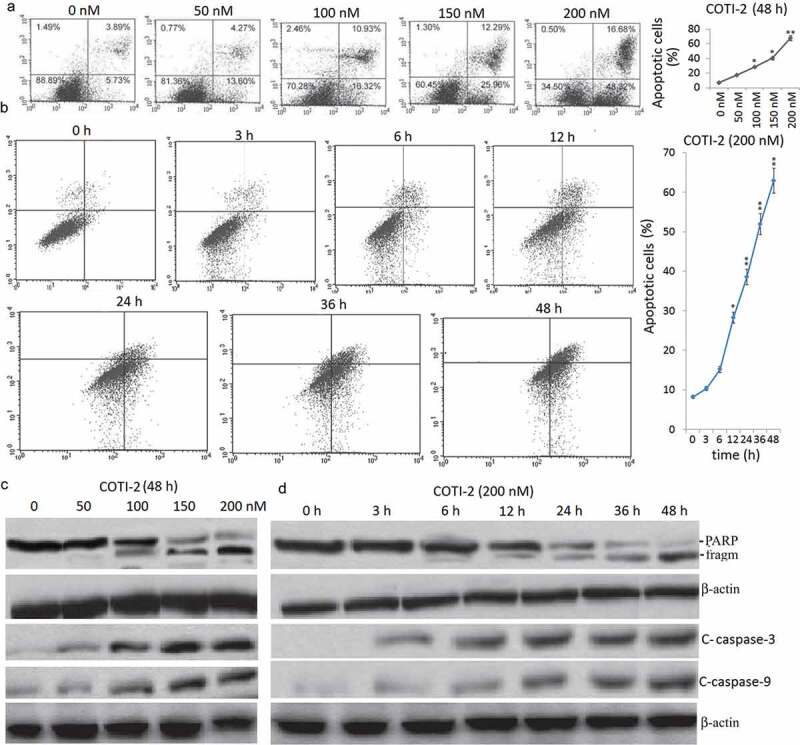
Treatment with COTI-2 results in caspase activation and apoptosis in dose- and in time-dependent manners in Jurkat cells. (a), Jurkat cells were treated with 0, 50, 100, 150, and 200 nM COTI-2 for 48 h. (b) The cells were treated with 200 nM BITC for 0–48 h. In (a) and (b), cells were stained with Annexin V/PI, and the percentage of apoptotic cells was determined using flow cytometry. (c,d), After treatment with COTI-2, total cellular extracts, nuclear extracts, and cytosolic fractions were prepared and subjected to western blot analysis using antibodies against PARP, cleaved-caspase (C-Caspase)-9, cleaved-caspase-3. Each lane was loaded with 30 mg of protein. Two additional studies yielded equivalent results.
Exposure of jurkat cells to COTI-2 results in the upregulation of miR-203
The effects of COTI-2 on the expression of miR-203 was examined in Jurkat cells. A marked dose-dependent increase of miR-203 expression was noted in COTI-2-treated cells in 24 h (Figure 2(a)). At 48 h, the miR-203 levels reached the normal level (Figure 2(a)).
Figure 2.
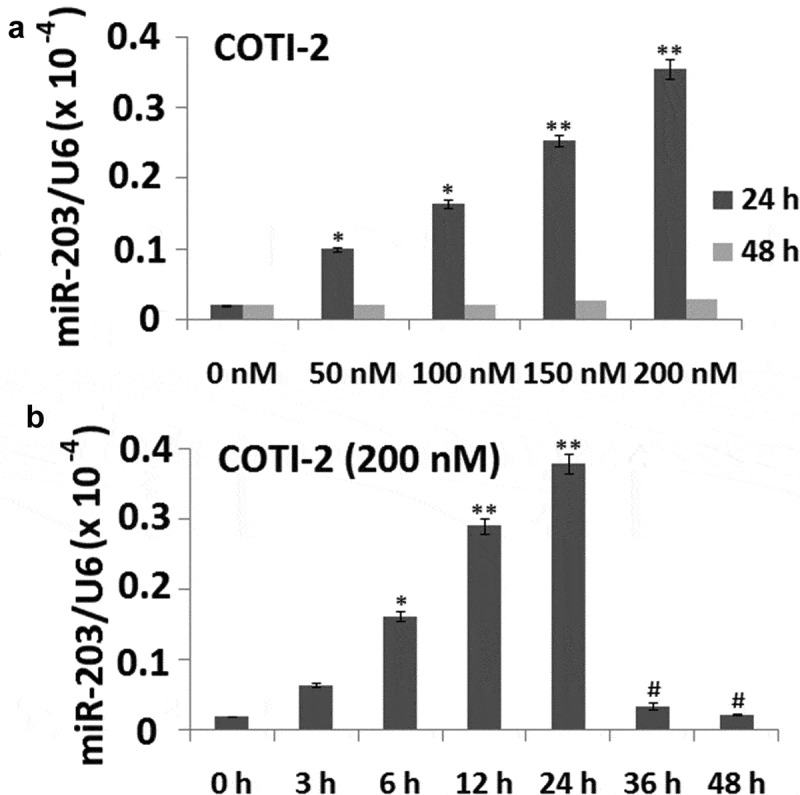
Treatment with COTI-2 results in increased miR-203 expression in dose- and in time-dependent manners in Jurkat cells. (a), Jurkat cells were treated with 0, 50, 100, 150, and 200 nM COTI-2 for 48 h. (b) The cells were treated with 200 nM BITC for 0–48 h. In (a) and (b), miR-203 expression was detected by qRT-PCR assay.
Exposure of cells to 200 nM COTI-2 for 6 h resulted in a modest increase in levels of miR-203. These events became apparent after 12 h. Then miR-203 expression was increased and reached the highest level after 24 h of drug exposure, and reached the normal level at 48 h (Figure 2(b)).
Enforced expression of miR-203 enhances coti-2-mediated apoptosis
To further confirm the functional role of miR-203 in COTI-2-mediated lethality in leukemia cells, Jurkat cells expressing miR-203 mimics were employed. Exposure of Jurkat cells to miR-203 mimics resulted in a significant increase of miR-203 expression compared with miR-NC transfected Jurkat cells (Figure 3(a)). In addition, miR-203 overexpression promotes the activation of caspase-9 and −3 and degradation of PARP (Figure 3(b)). Furthermore, miR-203 overexpression in Jurkat cells increased COTI-2-mediated apoptosis compared with control cells in a dose- and time- dependent manner (Figure 3(c,d)). Taken together, these findings indicate that miR-203 upregulation has a significant functional role in COTI-2-mediated lethality.
Figure 3.
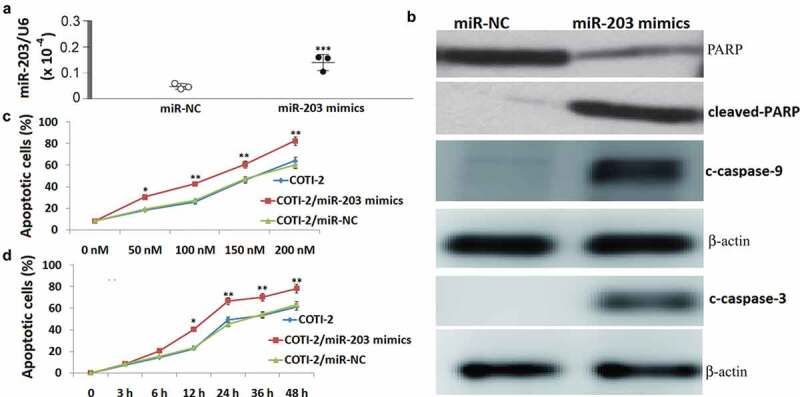
Enforced expression of miR-203 enhanced BITC-mediated apoptosis in Jurkat cells. Total cellular extracts were prepared from Jurkat cells transfected with miR-NC (control) and miR-203 mimic, and then subjected to qRT-PCR assay for miR-203 (a), and western blot assay for cleaved caspase-9 and −3 and PARP (b). (c), Jurkat cells transfected with miR-NC or miR-203 mimic were treated with 0, 50, 100, 150, and 200 nM COTI-2 for 48 h. After treatment, cells were stained with Annexin V/PI, and apoptosis was determined using flow cytometry. (d), Jurkat cells transfected with miR-NC or miR-203 mimic were treated with 200 nM COTI-2 for 3–48 h. After treatment, cells were stained with Annexin V/PI, and apoptosis was determined using flow cytometry. Significant difference from controls, Student’s t-test, *p < 0.05; **p < 0.01;***P < 0.001.
Knockdown of miR-203 substantially attenuates COTI-2-mediated apoptosis
To further confirm the functional role of miR-203 in COTI-2-mediated lethality in leukemia cells, Jurkat cells expressing miR-203 inhibitors were employed. The results showed that miR-203 inhibitor transfection significantly decreased COTI-2-induced miR-203 expression in Jurkat cells (Figure 4(a)). Significantly, miR-203 downregulation attenuated COTI-2-mediated apoptosis (Figure 4(b)), and completely inhibited the activation of caspase-9 and −3 and degradation of PARP. These data indicate that miR-203 downgulation has a significant functional role in COTI-2-mediated apoptosis.
Figure 4.
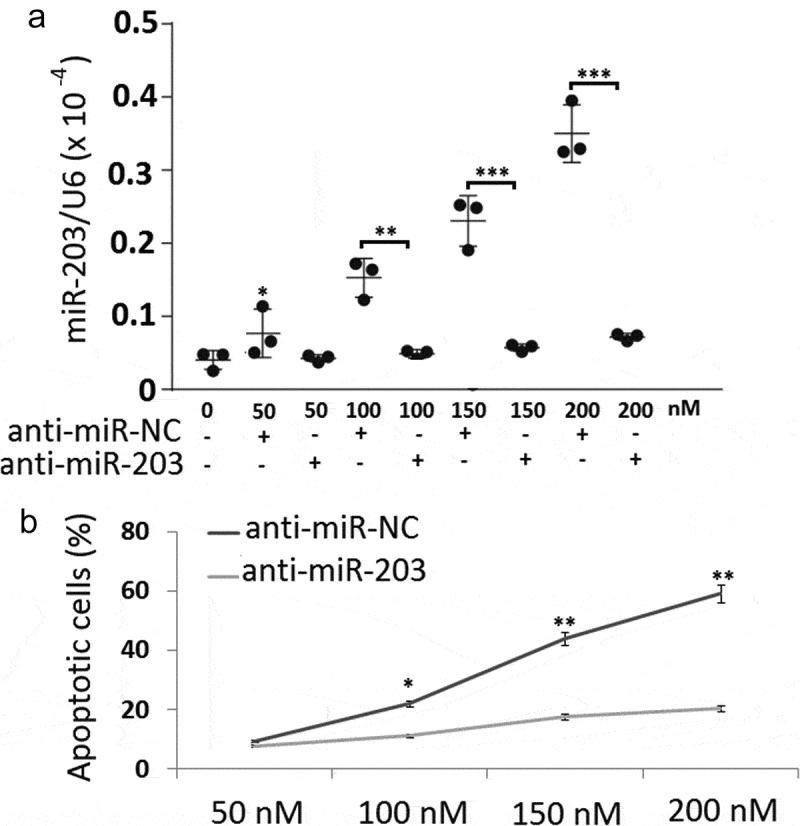
Downregulation of miR-203 inhibited BITC-mediated apoptosis in Jurkat cells. (a), Total cellular extracts were prepared from Jurkat cells transfected with NC inhibitor (control) and miR-203 inhibitor, and then subjected to qRT-PCR assay for miR-203; (b), Jurkat cells transfected with miR-NC or miR-203 mimic were treated with 0, 50, 100, 150, and 200 nM COTI-2 for 48 h. After treatment, cells were stained with Annexin V/PI, and apoptosis was determined using flow cytometry. Significant difference from controls, Student’s t-test, *p < 0.05; **p < 0.01;***P < 0.001.
COTI-2 exhibits antitumor activity in xenografts of leukemia Jurkat cells by induction of apoptosis and upregulation of miR-203
We assessed the effectiveness of COTI-2 in inhibiting the growth of Jurkat xenografts in immunocompromised mice when administered intraperitoneally (IP). COTI-2 significantly inhibited tumor growth in the Jurkat xenografts at a dose of 10 mg/kg (Figure 5(a)). We further determined apoptosis in tumor tissue of leukemia xenograft using TdT-mediated dUTP-biotin nick-end labeling (TUNEL) assay. TUNEL-positive apoptotic cells of tumor sections significantly increased in COTI-2-treated Jurkat xenograft mice compared with the control groups (Figure 5(b)). Exposure of mice to COTI-2 also caused a rapid increase in immunoreactivity for cleaved-caspase-3 in tumor sections (Figure 5(c)), indicative of apoptosis. Furthermore, miR-203 expression in tumor sections of Jurkat xenograft mice increased upon COTI-2 treatment (Figure 5(d)). Such findings suggest that COTI-2-mediated antileukemic activity in vivo is associated with the upregulation of miR-203.
Figure 5.
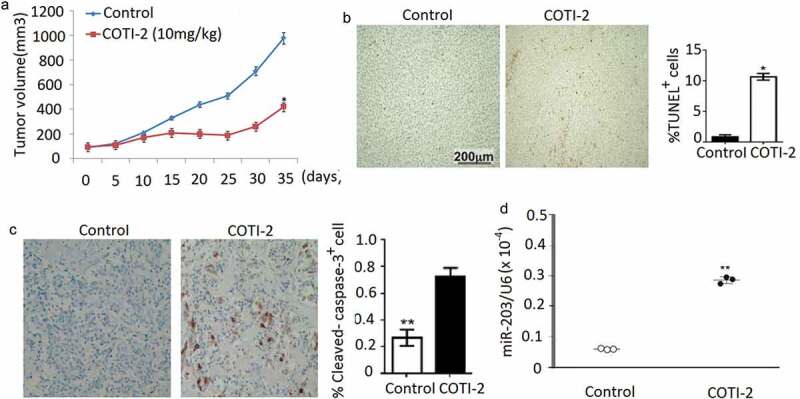
COTI-2 treatment inhibits Jurkat xenograft growth (a). Jurkat cells (5 × 106) were injected into the flanks of mice (n = 5 mice per group). Xenografts were allowed to reach 75 ~ 100 mm3 before IP treatment initiation with COTI-2 (10 mg/kg, 5 days a week for 7 weeks) or saline alone. Tumor growth was measured every 5 days by caliper measurement. (b), Tumors were obtained from animals 35 days after drug exposure. Tumors were fixed and stained to examine apopptosis using TUNEL assay. (c), The levels of cleaved-caspase-3 was detected by immunohistochemistry; (d), The levels of miR-203 was detected by qRT-PCR. Significant difference from controls, Student’s t-test, *p < 0.05. **p < 0.01.
Discussion
The results of this study indicate that treatment with COTI-2 results in apoptosis in human leukemia cells. Notably, this study demonstrates for the first time that upregulation of miR-203 has an important role in COTI-2-mediated lethality.
COTI-2 appears to be a cytotoxic agent since it inhibited cellular proliferation by inducing apoptosis in cancer cells [11]. In the present study, COTI-2 demonstrated effective anti-proliferative and pro-apoptotic activity against leukemia cell lines in vitro.
Extensive evidence is accumulating that miR-203 expression has a critical role in inducing cell apoptosis and inhibiting the survival of transformed cells [12,13], particularly those of hematopoietic origin [9]. The development of anticancer agents that enhanced miR-203 levels has been the focus of intense interest. Indeed, a number of studies have documented miR-203 upregulation during apoptosis by a variety of agents, including lncRNA MALAT1 [14], Sevoflurane [15], Berberine, the active compound of traditional Chinese medicine Huang Lian [16]. Evidence revealed that miR-203 levels are regulated through several different mechanisms [17–19].
In the present study, enforced expression of miR-203 markedly enhanced COTI-2-mediated lethality in Jurkat cells, arguing that upregulation of miR-203 has a critical role in COTI-2-related lethality. Consistent with this notion, targeting miR-203 largely inhibited caspase activation. These findings are consistent with previous study, which demonstrated that miR-203 operates upstream of caspases signals [20]. In addition, the miR-203 overexpression in Jurkat cells led to a significant increase in COTI-2-mediated caspase activation and apoptosis. Such findings are accordant with the studies that indicated that upregulation of miR-203 through miR-203 mimic transfection is sufficient to induce apoptosis in multiple myeloma cells [21].
Previous studies have shown that COTI-2 markedly inhibits tumor growth of human HNSCC cell xenografts [6]. However, little is known about inhibitory effects of COTI-2 on tumor growth of human leukemia xenograft model.
In our studies using a nude mice Jurkat xenograft model, tumor volumes were reduced compared with controls after COTI-2 treatment, indicating an antileukemia activity of this compound. To further validate the apoptotic mechanism found in vitro, we next examined the TUNEL staining, cleaved-caspase-3, and miR-203 expression in tumor specimens obtained from control- and COTI-2-treated animals. The increase in TUNEL-positive cells, caspase-3 activation, and the increase in miR-203 expression were detected in the COTI-2-treated xenografts compared with the control group. To the best of our knowledge, this is the first report that describes an effective extrapolation of the in vitro apoptosis-inducing effects of COTI-2 on the leukemia cells to the in vivo situation.
Conclusion
In conclusion, these findings indicate that COTI-2 effectively induces apoptosis in human leukemia cell lines and leukemia xenograft. This effect occurs in association with the upregulation of miR-203, leading to caspase activation. This study could provide a better understanding of how COTI-2 exerts its antileukemic activity in vivo and aid in developing this compound to treat leukemia and possibly other hematologic malignancies.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Hrabovský Š, Folber F, Doubek M.. Therapy of relapsed/refractory acute lymphoblastic leukemia today and tomorrow. Klin Onkol. 2019;32:90–96. [DOI] [PubMed] [Google Scholar]
- [2].Garas A, McLean LA, De Luca CR, et al. Health-related quality of life in paediatric patients up to five years post-treatment completion for acute lymphoblastic leukaemia: a systematic review. Support Care Cancer. 2019;27:4341–4351. [DOI] [PubMed] [Google Scholar]
- [3].Winestone LE, Aplenc R. Disparities in survival and health outcomes in childhood leukemia. Curr Hematol Malig Rep. 2019;14:179–186. [DOI] [PubMed] [Google Scholar]
- [4].Rafei H, Kantarjian HM, Jabbour EJ. Recent advances in the treatment of acute lymphoblastic leukemia. Leuk Lymphoma. 2019;60:2606–2621. [DOI] [PubMed] [Google Scholar]
- [5].Salim KY, Maleki Vareki S, Danter WR, et al. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget. 2016;7:41363–41379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lindemann A, Patel AA, Silver NL, et al. COTI-2, A novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. Clin Cancer Res. 2019;25:5650–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Maleki Vareki S, Salim KY, Danter WR, et al. Novel anti-cancer drug COTI-2 synergizes with therapeutic agents and does not induce resistance or exhibit cross-resistance in human cancer cell lines. PLoS One. 2018;13:e0191766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. [DOI] [PubMed] [Google Scholar]
- [9].Bueno MJ, Pérez de Castro I, Gómez de Cedrón M, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13:496–506. [DOI] [PubMed] [Google Scholar]
- [10].Chim CS, Wong KY, Leung CY, et al. Epigenetic inactivation of the hsa-miR-203 in haematological malignancies. J Cell Mol Med. 2011;15:2760–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kummar S, Gutierrez M, Doroshow JH, et al. Drug development in oncology: classical cytotoxics and molecularly targeted agents. Br J Clin Pharmacol. 2006;62:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang J, Zhou Y, Wu YJ, et al. Hyper-methylated miR-203 dysregulates ABL1 and contributes to the nickel-induced tumorigenesis. Toxicol Lett. 2013;223:42–51. [DOI] [PubMed] [Google Scholar]
- [13].Gasque Schoof CR, Izzotti A, Jasiulionis MG, et al. The roles of miR-26, miR-29, and miR-203 in the silencing of the epigenetic machinery during melanocyte transformation. Biomed Res Int. 2015;2015:634749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen W, Xu XK, Li JL, et al. MALAT1 is a prognostic factor in glioblastoma multiforme and induces chemoresistance to temozolomide through suppressing miR-203 and promoting thymidylate synthase expression. Oncotarget. 2017;8:22783–22799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen M, Zhou L, Liao Z, et al. Sevoflurane inhibited osteosarcoma cell proliferation and invasion via targeting miR-203/WNT2B/Wnt/β-Catenin Axis. Cancer Manag Res. 2019;11:9505–9515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].You HY, Xie XM, Zhang WJ, et al. Berberine modulates cisplatin sensitivity of human gastric cancer cells by upregulation ofmiR-203. In Vitro Cell Dev Biol Anim. 2016;52:857–863. [DOI] [PubMed] [Google Scholar]
- [17].Hamano R, Miyata H, Yamasaki M, et al. Overexpression of miR-200c induces chemoresistance in esophageal cancers mediated through activation of the Akt signaling pathway. Clin Cancer Res. 2011;17:3029–3038. [DOI] [PubMed] [Google Scholar]
- [18].Bian K, Fan J, Zhang X, et al. MicroRNA-203 leads to G1 phase cell cycle arrest in laryngeal carcinoma cells by directly targeting survivin. FEBS Lett. 2012;586:804–809. [DOI] [PubMed] [Google Scholar]
- [19].Zhang F, Yang Z, Cao M, et al. MiR-203 suppresses tumor growth and invasion and down-regulates MiR-21 expression through repressing Ran in esophageal cancer. Cancer Lett. 2014;342:121–129. [DOI] [PubMed] [Google Scholar]
- [20].Li J, Chen Y, Zhao J, et al. miR-203 reverses chemoresistance in p53-mutated colon cancer cells through downregulation of Akt2 expression. Cancer Lett. 2011;304:52–59. [DOI] [PubMed] [Google Scholar]
- [21].Wu SQ, Niu WY, Li YP, et al. miR-203 inhibits cell growth and regulates G1/S transition by targeting Bmi-1 in myelomacells. Mol Med Rep. 2016;14:4795–4801. [DOI] [PubMed] [Google Scholar]