

Abstract
Non-neuronal optogenetic approaches empower precise regulation of protein dynamics in live cells but often require target-specific protein engineering. To address this challenge, we developed a generalizable light-modulated protein stabilization system (GLIMPSe) to control the intracellular protein level independent of its functionality. We applied GLIMPSe to control two distinct classes of proteins: mitogen-activated protein kinase phosphatase 3 (MKP3), a negative regulator of the extracellular signal-regulated kinase (ERK) pathway, and a constitutively active form of MEK (CA MEK), a positive regulator of the same pathway. Kinetics study showed that light-induced protein stabilization could be achieved within 30 min of blue light stimulation. GLIMPSe enables target-independent optogenetic control of protein activities and therefore minimizes the systematic variation embedded within different photoactivatable proteins. Overall, GLIMPSe promises to achieve light-mediated post-translational stabilization of a wide array of target proteins in live cells.
Keywords: optogenetics, GLIMPSe, degron, protein degradation, MKP3, CA MEK
Graphical Abstract
A key factor that drives the dynamic nature of signaling pathways is spatial and temporal control of protein activities. Conventional genetic and pharmacological approaches such as gene overexpression, gene knock out, and RNA interference help delineate interaction maps of signaling components. These approaches, unfortunately, lack the flexibility to resolve signaling dynamics in live cells. Light serves as an attractive tool for the regulation of signal transduction as it can be rapidly controlled with high spatiotemporal resolution.1–3 This strategy has been widely used to control target protein activity by protein translocation,4–7 protein caging,8–11 sequestration,12,13 clustering,14,15 induced avidity,16–18 or allostery.19,20 Successful application of non-neuronal optogenetics in multicellular organisms has provided new insights into cell fate determination during embryonic development.21–26 Most of these systems, however, require target-specific protein engineering and often cannot be generalized to control a broader class of proteins.
A generalizable strategy for post-translational reduction of target protein level involved the fusion of a degradation peptide sequence, or degron, to a protein of interest. Light-mediated uncaging of the degron resulted in inducible protein degradation in Saccharomyces cerevisiae27 and mammalian cells.28 This strategy elicits a post-translational, optogenetic “knock-down” effect of a protein of interest. A comprehensive understanding of a signaling pathway would benefit from the complementary post-translational, optogenetic “knock-in” strategy, which has not been available to date.
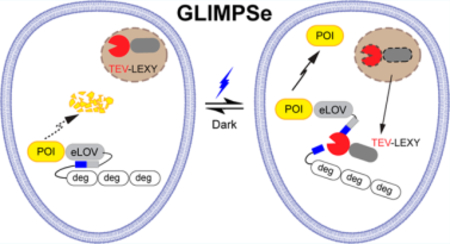
To fill in this gap, here, we developed a generalizable light-modulated protein stabilization system (GLIMPSe) that allows for optical enhancement of protein stability. GLIMPSe uses a degradation module to constitutively suppress the protein level until a photosensitive rescue module triggers protein stabilization. Compared with previous optogenetic strategies, GLIMPSe allows for optical control of different classes of proteins and stabilizes the protein level within 30 min of blue light stimulation. To demonstrate the generalizability of GLIMPSe, we achieved optical control of functionally distinct classes of proteins, including the mitogen-activated protein kinase phosphatase 3 (MKP3) and a constitutively active kinase MEK (CA MEK). Thus, GLIMPSe enables bidirectional control of the extracellular signal-regulated kinase (ERK) signaling pathway, which regulates crucial cell functions such as proliferation, differentiation, migration, and apoptosis. We expect that GLIMPSe would add a powerful capacity to the current optogenetics toolbox and promise to lower the technical barrier for achieving optical control of protein activities.
RESULTS AND DISCUSSION
Degron-Directed Constitutive Degradation Lowers Target Protein Levels
The Deadend (Dnd) protein regulates germline development in vertebrates.29–34 We have recently discovered a potent 21 amino-acid degradation sequence within Xenopus laevis Dnd protein that promotes rapid ubiquitin-independent proteasomal degradation of Dnd in Xenopus oocytes and mammalian cell lines such as HEK293T and GC-2spd.35 To determine whether this degradation sequence (here referred to as degron or deg) can be used as a general moiety to mediate degradation of intracellular proteins, we fused tandem arrays of degrons to the C-terminus of firefly luciferase (FLuc) protein (FLuc-tevS-Ndeg, N = 1, 2, and 3). A tobacco etch virus (TEV) protease recognition site (tevS) was sandwiched between FLuc and degron to render TEV-mediated rescue of protein degradation (Figure 1a). HEK293T cells were cotransfected with FLuc-tevS-Ndeg (N = 1, 2, 3) and a constitutively expressed Renilla luciferase (RLuc, loading control), recovered overnight, and harvested for a dual-luciferase analysis. The degrons significantly reduced the relative luminescence (FLuc/RLuc) signals in a dose-dependent way, with N = 3 showing the optimal contrast compared to the no-degron control (Figure 1b). Thus, we used three tandem degrons (denoted as deg) in all other constructs. When HEK293T cells were cotransfected with plasmids encoding TEV protease, a 4-fold increase of FLuc signal was observed compared with the no-TEV control (Figure 1b).
Figure 1.

Control intracellular protein level with degron. (a) Schematic for TEV protease-mediated rescue of firefly luciferase (FLuc) protein degradation. (b) Degradation of FLuc-tevS-deg and TEV-mediated protein stabilization demonstrated by a dual-luciferase assay in HEK293T cells. (c) Schematic for bidirectional regulation of the ERK pathway by CA MEK and MKP3, respectively. (d) Western blot analysis of pERK/pan-ERK level in PC12 cells under different conditions. The pERK/pan-ERK intensity ratio was normalized to that of CA MEK overexpression (left-most lane). (e) Schematic for regulating MKP3 stability with degron and TEV protease. (f) Quantification of PC12 cell differentiation ratio under different conditions. Values represent mean ± SD of three biological replicates (n = 3). More than 200 cells were counted per replicate. (g) Representative images of PC12 cells under different conditions. The total amount of plasmids was kept the same between each condition by including a plasmid encoding mRuby2 as a filler. Scale bar, 100 μm.
Stabilization of MKP3 via GLIMPSe Enables Optogenetic Downregulation of the ERK Signaling Pathway
This degron system inspired us to develop a post-translational, “knock-in” strategy that would allow for light-induced enhancement of the intracellular protein level. A protein of interest can be constantly degraded until light triggers a signal that removes the degron and rescues the protein. We reason that this strategy will complement current optogenetic methods and is particularly useful in controlling proteins whose activity does not depend on intracellular translocation or whose active site is difficult to cage and photouncage. One such protein is the mitogen-activated protein kinase phosphatase 3 (MKP3), a cytoplasmic phosphatase that specifically dephosphorylates the phospho-ERK1/2 (pERK1/2).36
To assess if the degron could modulate MKP3 activity, we used a neurite outgrowth assay in the rat PC12 pheochromocytoma cell line. To visualize degron-fused MKP3 in transfected PC12 cells, we inserted a gene encoding mRuby2 into the same vector with a p2A sequence (mRuby2-p2A-MKP3-tevS-deg). The p2A peptide is a consensus motif that allows for efficient, stoichiometric production of discrete proteins through a ribosomal skipping event.37 Insertion of the p2A peptide allows only MKP3 to undergo a constitutive degradation, leaving mRuby2 intact for cell visualization. Western blot analysis confirmed that degron functions normally with the fused HA-MKP3 protein, as revealed by the 15.7-fold reduction of protein level compared with the no-degron control in PC12 cells (Figure S1).
To stimulate PC12 cell differentiation, we transfected a constitutively active dual specificity mitogen-activated protein kinase (CA MEK)38 (Figure 1c) in PC12 cells. Western bot analysis showed that expression of CA MEK enhanced the pERK/pan-ERK level (Figure 1d, left-most band). CA MEK-induced ERK phosphorylation was significantly reduced by coexpressed MKP3, but not by MKP3-deg (Figure 1d, bars 2 and 3), which was poorly expressed as a result of fusing with degron. Single-cell analysis of PC12 cell differentiation showed a consistent result, where MKP3-deg did not reduce CA MEK-mediated neurite outgrowth (Figure 1e, 1f bars 1 and 2) in contrast with MKP3 (Figure 1f, bar 3). When TEV was cotransfected with MKP3-tevS-deg and CA MEK, the expression of MKP3 was successfully rescued, which in turn reduced the PC12 cell differentiation ratio to a level comparable to that of MKP3 (Figure 1f, bar 4). Representative multichannel fluorescence images under various transfection conditions were shown in Figure 1g. In these experiments, identical amounts of CA MEK- and MKP3-containing plasmids were used in each condition. A filler plasmid (mRuby2 or empty vector) was used to ensure an equal total amount of DNA in each condition.
To develop an optogenetic system for light-induced stabilization of MKP3, we first tried to cage the tevS with a recently developed evolved LOV (eLOV) protein from Avena sativa39 (MKP3-eLOVtevS-deg) and coexpressed a cytosolic TEV. We hypothesized that blue light stimulation would uncage and expose tevS, which could then be cleaved by TEV and restore the MKP3 protein level in cells. Thus, we expected to observe a reduction of PC12 cell differentiation upon blue light stimulation. Indeed, blue light reduced the PC12 cell differentiation ratio (Figure S2, bar 1) in cotransfected cells. However, a significant decrease in differentiation ratio was also observed in the dark (Figure S2, bar 2).
The low differentiation ratio in the dark indicates a high basal level of TEV cleavage. To address this issue, we modified the recently developed light-inducible nuclear export system (LEXY).9 LEXY consists of a C-terminal caged nuclear export signal (NES) and an N-terminal nuclear localization signal (NLS) to accumulate the fusion protein into the nucleus in the dark. Blue light uncages NES and mediates active nuclear export of the fusion protein through the nuclear pores. We fused TEV into the LEXY system (TEV-LEXY) so that the TEV protease would be sequestered into the nucleus in the dark and exported into the cytoplasm in response to blue light (Figure 2a). We hypothesized that LEXY-mediated TEV compartmentalization could sequester the majority of TEV in the nucleus (in the dark) and reduce its basal activity. As expected, the addition of the TEV-LEXY successfully reduced the basal TEV cleavage activity and fully recovered the PC12 cell differentiation ratio in the dark (Figure 2b, bar 2). Upon blue light stimulation, PC12 cell differentiation was suppressed to the baseline (Figure 2b, bar 1). Light inactivation of ERK pathway in PC12 cells was also validated by the 1.9-fold reduction of the pERK level (Figure 2c) assayed by Western blot. Note that both eLOV and TEV-LEXY are required to achieve the optimal light–dark contrast because PC12 cells cotransfected with MKP3-tevS-deg (uncaged form) and TEV-LEXY show no significant difference in their differentiation ratio (0.23 vs 0.24) (Figure S3, bars 1 and 2).
Figure 2.

Generation of GLIMPSe for light-induced MKP3 stabilization. (a) Schematic for optogenetic control of MKP3 stability with GLIMPSe. (b) Quantification of differentiation ratio for PC12 cells expressing GLIMPSe-MKP3 optogenetic system. Differentiation ratio = no. of differentiated cells/no. of transfected cells. The bar graph is presented with mean ± SD averaged over three independent experiments (n = 3). (c) Western blot analysis of PC12 cells expressing CA MEK, MKP3-eLOVtevS-degron, and TEV-LEXY. Band intensities are normalized to the average intensity of lane 3 and 4. The bar graph is presented with mean ± SD averaged over four replicates (n = 4).
GLIMPSe Functions within 30 min of Blue Light Stimulation
To determine the kinetics of optical control of MKP3 stabilization, we varied the duration of blue light stimulation. We first determined the response time of TEV-LEXY with optical microscopy and found cytosolic export of TEV-LEXY occurred within 5 min of blue light exposure (Figure 3a). We then performed a kinetic analysis by controlling the light exposure time from 0.5 to 24 h (Figure 3b). Because the cleaved protein loses three degrons in response to light, its molecular weight reduces approximately 10 kDa compared with the uncleaved form (e.g., the size of HA-MKP3-eLOVtevS-deg is 72 kDa, whereas the size of HA-MKP3-eLOV is 62 kDa). Compared with the dark control (Figure 3b, left-most lane), blue light stimulation significantly increased the band intensity at 62 kDa across all illumination conditions, indicating light-mediated degron removal and protein stabilization.
Figure 3.

Kinetics of blue light-induced protein stabilization. (a) Kinetics of TEV-LEXY nuclear-cytoplasmic shuttling upon 5 min of blue light stimulation in HEK293T cells. Scale bar: 10 μm. (b) Kinetics of light-induced protein stabilization in PC12 cells with 0.5 mW/cm2 blue light stimulation probed by a gel-shift assay. Cells kept in the dark showed minimum size shift from 72 kDa to 62 kDa, indicating that most of the HA-MKP3-eLOVtevS fusion protein was still fused to degron. (c) The efficiency of protein stabilization in response to blue light stimulation. Protein stabilization efficiency in each time point was calculated by dividing the band intensity at 62 kDa with the sum of 72-kDa and 62 kDa band intensity in the corresponding lane. The efficiency vs time curve was then fit in MATLAB. Data are presented with mean ± SD averaged over two replicates (n = 2).
We noted that the band intensity for 62 kDa at 0.5 h is denser than that of 72 kDa at 0 h. Given a translation rate of 5.6 amino acids/sec in mammalian cells,40 the synthesis of a 72 kDa protein takes 117 s. Thus, the increase of 62 kDa band intensity at 0.5 h should result from the stabilization of newly synthesized proteins via light-mediated removal of degrons. To demonstrate this point, we treated cells with 100 ng/μL cycloheximide, a translational inhibitor, for 18 h to block new protein synthesis during light treatment (Figure S4). As expected, the amount of total protein remained consistent between light-stimulated samples and dark control.
One way to define the protein stabilization efficiency is the ratio of cleaved protein over the total amount of protein. Such normalization avoids batch-to-batch variation of the absolute protein level. The efficiency vs time curve was then fit in MATLAB by the following eq (Figure 3c)
where I is the band intensity at corresponding molecular weight, b represents the basal activity, a represents the maximum enhancement of cleavage activity from the basal level, and τ0 represents the time to reach half the maximum of a. The fit result suggests that optical control of protein stabilization occurs on the order of 2 min (τ0 = 1.79 min) of blue light stimulation. At this time scale, the newly stabilized protein should arise from the equilibrated pool of degron-containing, undegraded MKP3. Thus, GLIMPSe functions through a different mechanism from chemical-inducible transcription approaches. Interestingly, there was a transient increase of the uncleaved (72 kDa) protein 0.5 and 1 h after blue light illumination, which could imply that a transient decrease of protein concentration triggers either an increase of protein synthesis, a reduction of degron-mediated protein degradation, or both. The slight decrease of the 62 kDa protein level after 1 h (Figure 3b, second and third lanes from the right) could arise from spontaneous degradation of cleaved MKP3.
Stabilization of CA MEK via GLIMPSe Enables Optogenetic Upregulation of the ERK Signaling Pathway
To demonstrate the generalizability of GLIMPSe, we set out to use the same strategy to control kinases, a different class of protein from MKP3. We chose the constitutively active dual specificity mitogen-activated protein kinase (CA MEK) that activates the ERK pathway. We first verified that CA MEK could be degraded by degron, and its degradation could be rescued by coexpression of TEV protease, which was assayed by PC12 cell differentiation and Western blot (Figure 4). As expected, PC12 cells transfected with CA MEK-tevS-deg showed a significant reduction of differentiation ratio compared to cells transfected with CA MEK (Figure 4a,b), as well as a 2.2-fold reduction of the pERK activity (Figure 4c). Coexpression of a TEV protease with CA MEK-tevS-deg rescued CA MEK degradation and resulted in a 2.7-fold increase of the PC12 differentiation ratio (Figure 4d,e) as well as a 1.4-fold elevation of pERK level compared to cells without TEV-EGFP (Figure 4f).
Figure 4.

Control the intracellular level of CA MEK with degron and TEV. (a) Representative images of PC12 cells transfected with CA MEK-mRuby2 (top) or mRuby2-p2A-CA MEK-tevS-degron (bottom). Scale bar: 100 μm. (b) Cells transfected with degron-fused CA MEK showed significantly less neurite outgrowth than those transfected with CA MEK. (c) Western blot analysis of the pERK/pan-ERK level in PC12 cells transfected with CA MEK or CA MEK-deg. Values at each condition were normalized to the intensity of CA MEK-degron. The bar graph is presented with mean ± SD averaged over three independent sets of experiments (n = 3). (d) Schematic for controlling CA MEK protein stability with C-terminal fused degron and TEV protease. (e) Quantification of PC12 cell differentiation ratio under different conditions. PC12 cells transfected with mRuby2-p2A-CA MEK-tevS-degron showed significantly less differentiation, which was rescued by cotransfection of TEV-EGFP. (f) Western blot analysis for TEV-mediated rescue of CA MEK stability and elevation of pERK level in PC12 cells. pERK/pan-ERK intensity was normalized to the intensity of CA MEK-tevS-degron in the absence of TEV protease. The bar graph is presented with mean ± SD averaged over three replicates (n = 3).
To construct GLIMPSe-CA MEK (Figure 5a), we replaced MKP3 in GLIMPSe-MKP3 with CA MEK. PC12 cells transfected with GLIMPSe-CA MEK and exposed to 45 h of blue light stimulation showed a 2.5-fold increase in their differentiation ratio (Figure 5b, bar 3) and 2-fold elevation of pERK level compared to cells kept in the dark (Figure 5c). Reducing the exposure time to 30 min was sufficient to induce significant neurite outgrowth (Figure S5). Negative controls without TEV-LEXY showed a base-level differentiation ratio (Figure 5b, bars 1 and 2) and pERK level (Figure 5c, bars 1 and 2). Similar to the case of MKP3, both eLOV and TEV-LEXY are required to achieve the optimal light-dark contrast because PC12 cells cotransfected with CA MEK-tevS-deg (uncaged form) and TEV-LEXY show no significant difference in their differentiation ratio (0.4 vs 0.3) (Figure S6, bars 1 and 2).
Figure 5.

Generation of GLIMPSe for light-induced CA MEK stabilization. (a) Schematic for optogenetic control of CA MEK stability with GLIMPSe. (b) Quantification of differentiation ratio for PC12 cells expressing GLIMPSe-CA MEK optogenetic system. Cells were illuminated with 0.5 mW/cm2 blue light or kept in the dark for 45 h before imaging. The bar graph is presented with mean ± SD averaged over three biological replicates (n = 3) with more than 200 cells counted per replicate. (c) Western blot analysis of PC12 cells expressing GLIMPSe-CA MEK under dark and light. Band intensities are normalized to the average intensity of lanes 7 and 8. The bar graph is presented with mean ± SD averaged over four biological replicates (n = 4).
Development of a generalizable optogenetic platform would significantly lower the technical barrier for the utilization of non-neuronal optogenetics. For example, a recent computational framework has been developed to facilitate the design of split proteins for optogenetic control of protein activities.41 Here, we developed GLIMPSe optogenetic system by integrating a degron derived from the Xenopus laevis Dnd protein into two optogenetic modules (LEXY and eLOV).
Several features make GLIMPSe an attractive optogenetic system. First, the GLIMPSe system takes advantage of the Dnd degron, a very potent degron with only 21 amino acid residues, which functions by promoting ubiquitin-independent proteasomal turnover. Inside the cell, nonubiquitinated proteins are destructed by proteasomes containing three proteasome activators, including the PSME1/2 heteroheptamer, PSME3 homoheptamer, or PSME4 monomer.42–45 We previously reported that degradation mediated by the Dnd degron was blocked only when all three types of the ubiquitin-independent proteasome were inhibited simultaneously.35 Ubiquitin-independent proteasomes are widely present in the majority of cell types. Thus, different from the majority of degrons, which are often recognized by tissue-specific E3 ubiquitin ligases, the Dnd degron can keep the protein of interest at a very low expression level in a wide variety of cells. Upon TEV-dependent removal of the degron, the protein of interest can accumulate rapidly within minutes.
In addition to the degron, the GLIMPSe system relies on the TEV-LEXY, which exhibits a low background activity, and is highly sensitive to blue light. In our experiments, degron-mediated protein degradation can be prevented by a short exposure to blue light (30 min) at an amenable power dosage (0.5 mW/cm2), which produces negligible phototoxicity with 45-h exposure time (Figure S7), consistent with our previous studies.7,15
The above features make the GLIMPSe system well-suited for optogenetic expression of a wide range of intracellular proteins in the majority of cell types. To demonstrate a prototypical system, we used GLIMPSe to achieve efficient optogenetic control of two different classes of proteins: a phosphatase (MKP3) that acts as a negative regulator of ERK pathway and a kinase (CA MEK), an activator of the same pathway. The full DNA and protein sequences of GLIMPSe have been shown in Figure S8.
Empowered by the high spatiotemporal resolution as well as its capacity to delineate signaling subcircuits, optogenetic technology promises to offer new insights into signal transduction in live cells. It has been increasingly realized, however, that quantitative analysis of signaling outcomes suffers from systematic variations embedded within different photoactivatable proteins such as binding affinity, dynamic range, as well as the expression level. GLIMPSe offers a platform that allows for target-independent optogenetic control of protein activities and therefore minimizes the systematic variation. Note that the basal activity should always be characterized if a different protein of interest were to be used. Although GLIMPSe requires blue light that has a limited penetration depth in biological tissues, we believe that the recent development of wireless optogenetics based on emerging nanomaterials such as upconversion nanoparticles would address this challenge.46–48
METHODS
Dual-Luciferase Assay
HEK293T cells were grown in 24-well plates and transiently transfected at 60–80% confluency using Turbofect (Thermo Fisher Scientific) transfection reagent for 3 h. After 3 h, cells were replenished with the growth medium (DMEM + 10% FBS) and recovered overnight. Cells were harvested 20–24 h later by the addition of 1× passive lysis buffer (Promega). Transfection for luciferase assays included 25 ng of Firefly luciferase construct and 425 ng of TEV protease or a mRuby2 filler plasmid. A 50 ng aliquot of Renilla luciferase (pRL-TK) plasmid was added to each transfection reaction to normalize for cell number and transfection efficiency. Firefly and Renilla luciferase activities were measured in lysates using the dual luciferase assay kit (Promega). Firefly luciferase activity was normalized to the Renilla luciferase activity and reported as relative luciferase activity units.
Long-Term Light Stimulation for PC12 Cells Neurite Outgrowth Assay
After 3 h of transfection, cells were switched to growth medium (F12K + 15% horse serum + 2.5% FBS) and exposed to 0.5 mW/cm2 blue light stimulation. Both the LED device and the cell culture plate were placed into a CO2 incubator. After overnight recovery in the complete growth medium, cell culture was exchanged to low-serum starvation medium (F12K + 0.15% horse serum + 0.025% FBS), followed by another 24 h of continuous blue light stimulation. Cells that were transfected but kept in the dark were used as a negative control. Neurite outgrowth was quantified by the end of light stimulation.
Kinetic Analysis of GLIMPSe-Mediated Protein Stabilization
PC12 cells were plated and transfected in a 12-well tissue culture plate with 100 ng EGFP-p2A-HA-MKP3-eLOVtevS-deg and 50 ng of NLS-mCherry-TEV-LEXY plasmid. After 3 h of transfection, cells were recovered overnight in growth medium (F12K + 15% horse serum + 2.5% FBS). The next day, cells were exposed to 0.5 mW/cm2 blue light stimulation for different timespans ranging from 30 min to 24 h. After light exposure, cells were harvested for Western blot analysis with HA tag and GAPDH primary antibody.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the School of Molecular and Cellular Biology at UIUC and the National Institute of General Medical Sciences of the National Institutes of Health under Award No. R01GM132438 (K.Z.) and NIH R35 GM131810 (J.Y.). P.M. is grateful to the Dissertation Completion Fellowship at UIUC for support. S.R.S. is grateful to the Westcott Bioscience Fellowship from the Biochemistry Department at UIUC for support. We thank Dr. Tobias Meyer from Stanford University for the gift of PC12 NS1 cells and Dr. Lin-Feng Chen (UIUC) for the gift of HEK293T cells. We also thank Ranajay Mandal from Purdue University for technical support in the construction of the LED lightbox.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.9b00285.
Description of molecular cloning, cell assays, construction of programmable LED device, and statistical analysis; Supplementary Figures S1–S8 describing characterization of degron, PC12 cell differentiation under various treatment conditions, and DNA and amino acid sequences of GLIMPSe (PDF)
REFERENCES
- (1).Knopfel T, Lin MZ, Levskaya A, Tian L, Lin JY, and Boyden ES (2010) Toward the second generation of optogenetic tools. J. Neurosci 30 (45), 14998–15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Dagliyan O, and Hahn KM (2019) Controlling protein conformation with light. Curr. Opin. Struct. Biol 57, 17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang K, and Cui B (2015) Optogenetic control of intracellular signaling pathways. Trends Biotechnol 33 (2), 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Levskaya A, Weiner OD, Lim WA, and Voigt CA (2009) Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461 (7266), 997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, and Tucker CL (2010) Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 7 (12), 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Toettcher JE, Weiner OD, and Lim WA (2013) Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155 (6), 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang K, Duan L, Ong Q, Lin Z, Varman PM, Sung K, and Cui B (2014) Light-mediated kinetic control reveals the temporal effect of the Raf/MEK/ERK pathway in PC12 cell neurite outgrowth. PLoS One 9 (3), e92917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wu YI, Frey D, Lungu OI, Jaehrig A, Schlichting I, Kuhlman B, and Hahn KM (2009) A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 461 (7260), 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Niopek D, Wehler P, Roensch J, Eils R, and Di Ventura B (2016) Optogenetic control of nuclear protein export. Nat. Commun 7, 10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhou XX, Chung HK, Lam AJ, and Lin MZ (2012) Optical control of protein activity by fluorescent protein domains. Science 338 (6108), 810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhou XX, Fan LZ, Li P, Shen K, and Lin MZ (2017) Optical control of cell signaling by single-chain photoswitchable kinases. Science 355 (6327), 836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chen D, Gibson ES, and Kennedy MJ (2013) A light-triggered protein secretion system. J. Cell Biol. 201 (4), 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lee S, Park H, Kyung T, Kim NY, Kim S, Kim J, and Heo WD (2014) Reversible protein inactivation by optogenetic trapping in cells. Nat. Methods 11 (6), 633–636. [DOI] [PubMed] [Google Scholar]
- (14).Shin Y, Berry J, Pannucci N, Haataja MP, Toettcher JE, and Brangwynne CP (2017) Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 168 (1–2), 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Khamo JS, Krishnamurthy VV, Chen Q, Diao J, and Zhang K (2019) Optogenetic Delineation of Receptor Tyrosine Kinase Subcircuits in PC12 Cell Differentiation. Cell Chem. Biol 26 (3), 400–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang X, Chen XJ, and Yang Y (2012) Spatiotemporal control of gene expression by a light-switchable transgene system. Nat. Methods 9 (3), 266–269. [DOI] [PubMed] [Google Scholar]
- (17).Bugaj LJ, Choksi AT, Mesuda CK, Kane RS, and Schaffer DV (2013) Optogenetic protein clustering and signaling activation in mammalian cells. Nat. Methods 10 (3), 249–252. [DOI] [PubMed] [Google Scholar]
- (18).Motta-Mena LB, Reade A, Mallory MJ, Glantz S, Weiner OD, Lynch KW, and Gardner KH (2014) An optogenetic gene expression system with rapid activation and deactivation kinetics. Nat. Chem. Biol 10 (3), 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dagliyan O, Tarnawski M, Chu PH, Shirvanyants D, Schlichting I, Dokholyan NV, and Hahn KM (2016) Engineering extrinsic disorder to control protein activity in living cells. Science 354 (6318), 1441–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Strickland D, Moffat K, and Sosnick TR (2008) Light-activated DNA binding in a designed allosteric protein. Proc. Natl. Acad. Sci. U. S. A 105 (31), 10709–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Krishnamurthy VV, Khamo JS, Mei W, Turgeon AJ, Ashraf HM, Mondal P, Patel DB, Risner N, Cho EE, Yang J, and Zhang K (2016) Reversible optogenetic control of kinase activity during differentiation and embryonic development. Development 143 (21), 4085–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Buckley CE, Moore RE, Reade A, Goldberg AR, Weiner OD, and Clarke JD (2016) Reversible Optogenetic Control of Subcellular Protein Localization in a Live Vertebrate Embryo. Dev. Cell 36 (1), 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Johnson HE, Goyal Y, Pannucci NL, Schupbach T, Shvartsman SY, and Toettcher JE (2017) The Spatiotemporal Limits of Developmental Erk Signaling. Dev. Cell 40 (2), 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Johnson HE, and Toettcher JE (2019) Signaling Dynamics Control Cell Fate in the Early Drosophila Embryo. Dev. Cell 48 (3), 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Adams DS, Lemire JM, Kramer RH, and Levin M (2014) Optogenetics in Developmental Biology: using light to control ion flux-dependent signals in Xenopus embryos. Int. J. Dev. Biol 58 (10–12), 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Khamo JS, Krishnamurthy VV, Sharum SR, Mondal P, and Zhang K (2017) Applications of Optobiology in Intact Cells and Multicellular Organisms. J. Mol. Biol 429 (20), 2999–3017. [DOI] [PubMed] [Google Scholar]
- (27).Renicke C, Schuster D, Usherenko S, Essen LO, and Taxis C (2013) A LOV2 Domain-Based Optogenetic Tool to Control Protein Degradation and Cellular Function. Chem. Biol 20 (4), 619–626. [DOI] [PubMed] [Google Scholar]
- (28).Bonger KM, Rakhit R, Payumo AY, Chen JK, and Wandless TJ (2014) General Method for Regulating Protein Stability with Light. ACS Chem. Biol 9 (1), 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yamaji M, Jishage M, Meyer C, Suryawanshi H, Der E, Yamaji M, Garzia A, Morozov P, Manickavel S, McFarland HL, Roeder RG, Hafner M, and Tuschl T (2017) DND1 maintains germline stem cells via recruitment of the CCR4-NOT complex to target mRNAs. Nature 543 (7646), 568–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gross-Thebing T, Yigit S, Pfeiffer J, Reichman-Fried M, Bandemer J, Ruckert C, Rathmer C, Goudarzi M, Stehling M, Tarbashevich K, Seggewiss J, and Raz E (2017) The Vertebrate Protein Dead End Maintains Primordial Germ Cell Fate by Inhibiting Somatic Differentiation. Dev. Cell 43 (6), 704–715. [DOI] [PubMed] [Google Scholar]
- (31).Weidinger G, Stebler J, Slanchev K, Dumstrei K, Wise C, Lovell-Badge R, Thisse C, Thisse B, and Raz E (2003) dead end, a novel vertebrate germ plasm component, is required for zebrafish primordial germ cell migration and survival. Curr. Biol 13 (16), 1429–1434. [DOI] [PubMed] [Google Scholar]
- (32).Youngren KK, Coveney D, Peng X, Bhattacharya C, Schmidt LS, Nickerson ML, Lamb BT, Deng JM, Behringer RR, Capel B, Rubin EM, Nadeau JH, and Matin A (2005) The Ter mutation in the dead end gene causes germ cell loss and testicular germ cell tumours. Nature 435 (7040), 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Horvay K, Claussen M, Katzer M, Landgrebe J, and Pieler T (2006) Xenopus Dead end mRNA is a localized maternal determinant that serves a conserved function in germ cell development. Dev. Biol 291 (1), 1–11. [DOI] [PubMed] [Google Scholar]
- (34).Cook MS, Coveney D, Batchvarov I, Nadeau JH, and Capel B (2009) BAX-mediated cell death affects early germ cell loss and incidence of testicular teratomas in Dnd1(Ter/Ter) mice. Dev. Biol 328 (2), 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hwang H, Jin Z, Krishnamurthy VV, Saha A, Klein PS, Garcia B, Mei W, King ML, Zhang K, and Yang J (2019) Novel functions of the ubiquitin-independent proteasome system in regulating Xenopus germline development. Development 146 (8), dev172700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bermudez O, Jouandin P, Rottier J, Bourcier C, Pages G, and Gimond C (2011) Post-transcriptional regulation of the DUSP6/MKP-3 phosphatase by MEK/ERK signaling and hypoxia. J. Cell. Physiol 226 (1), 276–284. [DOI] [PubMed] [Google Scholar]
- (37).Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, Kim MK, Shin BA, and Choi SY (2011) High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 6 (4), e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Schramek H, Feifel E, Healy E, and Pollack V (1997) Constitutively active mutant of the mitogen-activated protein kinase kinase MEK1 induces epithelial dedifferentiation and growth inhibition in madin-darby canine kidney-C7 cells. J. Biol. Chem 272 (17), 11426–11433. [DOI] [PubMed] [Google Scholar]
- (39).Wang W, Wildes CP, Pattarabanjird T, Sanchez MI, Glober GF, Matthews GA, Tye KM, and Ting AY (2017) A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nat. Biotechnol 35 (9), 864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ingolia NT, Lareau LF, and Weissman JS (2011) Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147 (4), 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Dagliyan O, Krokhotin A, Ozkan-Dagliyan I, Deiters A, Der CJ, Hahn KM, and Dokholyan NV (2018) Computational design of chemogenetic and optogenetic split proteins. Nat. Commun 9 (1), 4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Hwang J, Winkler L, and Kalejta RF (2011) Ubiquitin-independent proteasomal degradation during oncogenic viral infections. Biochim. Biophys. Acta, Rev. Cancer 1816 (2), 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rechsteiner M, and Hill CP (2005) Mobilizing the proteolytic machine: cell biological roles of proteasome activators and inhibitors. Trends Cell Biol. 15 (1), 27–33. [DOI] [PubMed] [Google Scholar]
- (44).Kish-Trier E, and Hill CP (2013) Structural biology of the proteasome. Annu. Rev. Biophys 42, 29–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ben-Nissan G, and Sharon M (2014) Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 4 (3), 862–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yu N, Huang L, Zhou Y, Xue T, Chen Z, and Han G (2019) Near-Infrared-Light Activatable Nanoparticles for Deep-Tissue-Penetrating Wireless Optogenetics. Adv. Healthcare Mater 8 (6), e1801132. [DOI] [PubMed] [Google Scholar]
- (47).He L, Zhang YW, Ma GL, Tan P, Li ZJ, Zang SB, Wu X, Jing J, Fang SH, Zhou LJ, Wang YJ, Huang Y, Hogan PG, Han G, and Zhou YB (2015) Near-infrared photoactivatable control of Ca2+ signaling and optogenetic immunomodulation. eLife 4, e10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chen S, Weitemier AZ, Zeng X, He L, Wang X, Tao Y, Huang AJY, Hashimotodani Y, Kano M, Iwasaki H, Parajuli LK, Okabe S, Teh DBL, All AH, Tsutsui-Kimura I, Tanaka KF, Liu X, and McHugh TJ (2018) Near-infrared deep brain stimulation via upconversion nanoparticle-mediated optogenetics. Science 359 (6376), 679–684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.