

SUMMARY
Transcriptional programming of the innate immune response is pivotal for host protection. However, the transcriptional mechanisms that link pathogen sensing with innate activation remain poorly under-stood. During HIV-1 infection, human dendritic cells (DCs) can detect the virus through an innate sensing pathway, leading to antiviral interferon and DC maturation. Here, we develop an iterative experimental and computational approach to map the HIV-1 innate response circuitry in monocyte-derived DCs (MDDCs). By integrating genome-wide chromatin accessibility with expression kinetics, we infer a gene regulatory network that links 542 transcription factors with 21,862 target genes. We observe that an interferon response is required, yet insufficient, to drive MDDC maturation and identify PRDM1 and RARA as essential regulators of the interferon response and MDDC maturation, respectively. Our work provides a resource for interrogation of regulators of HIV replication and innate immunity, highlighting complexity and cooperativity in the regulatory circuit controlling the response to infection.
Graphical Abstract
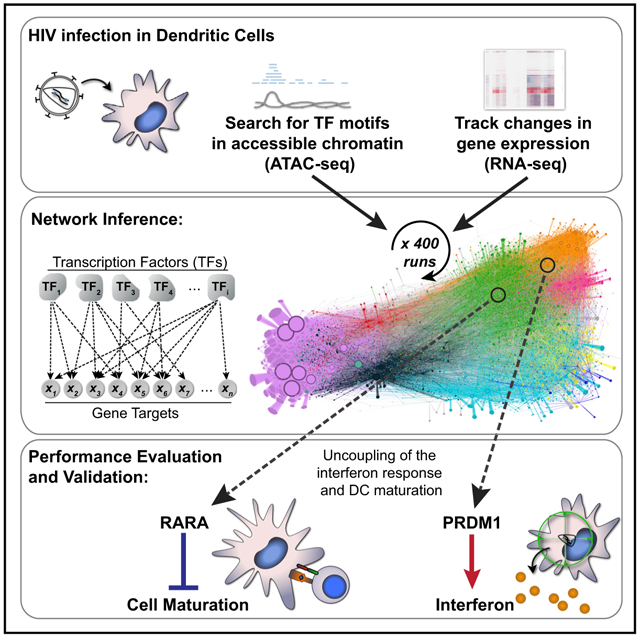
In Brief
Pathogen sensing leads to host transcriptional reprogramming to protect against infection. However, it is unclear how transcription factor activity is coordinated across the genome. Johnson et al. integrate chromatin accessibility and gene expression data to infer and validate a gene regulatory network that directs the innate immune response to HIV.
INTRODUCTION
The host’s ability to rapidly alter gene expression to defend against infection is a central element of the innate immune response. Host-encoded pattern recognition receptors (PRRs) detect components of foreign microorganisms and self-derived immunostimulatory products (Cao, 2016; Ivashkiv and Donlin, 2014; Iwasaki and Medzhitov, 2015). When a pathogen is sensed, PRRs initiate signal transduction cascades that lead to activation of multiple transcription factors (TFs), which subsequently rewire gene expression to protect the host. Considering that aberrations in innate immunity are hallmarks of many disorders, including chronic viral diseases, neurodegeneration, diabetes, and cancer (Corrales et al., 2017; Heneka et al., 2014; Wada and Makino, 2016), it is not surprising that transcriptional activation of innate immune signaling is under tight control, with the goal of maintaining a sensitive response to infectious threats while avoiding unwanted inflammation and auto-immunity. In the case of HIV-1 infection, however, innate immune responses are insufficient for host protection and become dysregulated during progression to AIDS (Fernandez et al., 2011; Sandler et al., 2014; Schoggins et al., 2011).
Dendritic cells (DCs) serve key functions in host defense and are among the first cells thought to contact HIV-1 during transmission (Iijima et al., 2008). Myeloid DCs express an arsenal of PRRs and link innate detection of microbes to activation of pathogen-specific adaptive immune responses (Banchereau et al., 2000; Théry and Amigorena, 2001). These cells express cell surface receptors for HIV-1 entry, but because of the presence of restriction factors, the virus undergoes limited productive infection in primary DCs and monocyte-derived DCs (MDDCs) and does not trigger robust immune responses (Granelli-Piperno et al., 2004; Manel et al., 2010; Silvin et al., 2017; Smed-Sörensen et al., 2005). The major restriction factor in myeloid DCs is SAMHD1, an enzyme that exhibits phosphohydrolase activity and depletes the cellular pool of deoxyribonucleotide triphosphates (dNTPs) required for HIV reverse transcription (Hrecka et al., 2011; Laguette et al., 2011). This restriction can be overcome if DCs are first exposed to virus-like particles that deliver the lentiviral accessory protein Vpx (absent from HIV-1 but encoded by simian immunodeficiency virus [SIV] and HIV-2) (Goujon et al., 2006; Mangeot et al., 2000). Vpx targets SAMHD1 for degradation, enabling productive HIV-1 infection, sensing of viral components, and activation of innate immune responses (Manel et al., 2010).
Innate immune responses against HIV-1 are triggered in primary DCs and in MDDCs by the sensor cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS), which detects reverse-transcribed HIV cDNA, in a process that requires concomitant HIV capsid protein interaction with the cellular protein NONO, and is facilitated by other proximal factors (Gao et al., 2013; Jønsson et al., 2017; Lahaye et al., 2013, 2018; Yoh et al., 2015). Downstream of innate sensing initiated by cGAS, several TFs are activated, including IRF and nuclear factor κB (NF-κB) family members, which drive induction of interferons (IFNs), IFN-stimulated genes (ISGs), and inflammatory cytokines, and promote DCs to transition from an inactive immature state to a mature, activated state. In addition to upregulating innate antiviral factors, mature DCs express at their cell surface the costimulatory factors CD80 and CD86, which are critical for programming adaptive responses (Goubau et al., 2013; Iwasaki, 2012). The DC transcriptional response circuitry involves feedback loops that engage multiple activator and repressor TFs that collectively influence thousands of gene targets during IFN signaling and DC maturation (Ivashkiv and Donlin, 2014). Therefore, it is difficult to understand the constellation of TF-target gene connections that operates during innate immune responses using traditional approaches.
Work from our groups and others has demonstrated that gene regulatory network inference, when applied to study dynamic systems such as macrophage activation, T helper 17 (Th17) lymphocyte polarization, and innate immune response to cytosolic DNA, has predicted the functions of key transcriptional regulators whose involvement was previously unknown (Ciofani et al., 2012; Gilchrist et al., 2006; Lee et al., 2013; Ramsey et al., 2008). Our earlier computational methods pioneered the use of time series perturbations and the incorporation of structured prior information into gene regulatory network inference (Bonneau et al., 2006; Greenfield et al., 2010, 2013; Madar et al., 2010). In this report, we demonstrate that network inference is improved by ensemble learning across hundreds of individual computational runs, with each run predicated on subsampled information in the "Prior" network. We have integrated chromatin accessibility data, together with genome-wide measurements of gene expression, to infer and experimentally validate a network describing the human DC transcriptional circuitry that is engaged upon HIV sensing.
RESULTS
Perturbation of Human DCs in the Face of Diverse Innate Immune and Viral Stimuli
With the goal of better understanding how myeloid cells respond to innate immune stimuli, we exposed immature MDDCs to a battery of innate immune agonists and viral challenges, generating datasets composed of RNA sequencing (RNA-seq) and assays for transposase-accessible chromatin sequencing (ATAC-seq) (Figure 1A; Table S1). We tracked the kinetics of gene expression and MDDC maturation during HIV-1 infection compared with classic innate agonists by infecting MDDCs with a single-cycle HIV-1 reporter virus (HIV-1-GFP +Vpx), transducing with a non-replicating lentivirus (LKO-GFP +Vpx), or stimulating in parallel with the Toll-like receptor (TLR) 4 agonist lipopolysaccharide (LPS) or the double-stranded RNA mimetic polyinosinic:polycytidylic acid (poly(I:C)) for 2, 8, 24, and 48 h. LPS and poly(I:C) triggered rapid changes in gene expression in human MDDCs across several gene clusters (Figure 1B; Table S2). Similarly, infection with HIV-1-GFP led to induction of innate immune genes (Figure 1B, clusters 5 and 8) but did so with delayed kinetics compared with LPS and poly(I:C), likely because of time-dependent accumulation of reverse transcription products, integration, and virus replication progressing over the first 24 h (Gao et al., 2013; Johnson et al., 2018). Gene expression profiles were consistent with the timing of MDDC maturation as scored by flow cytometry (Figure S1A), with LPS and poly(I:C) stimulation leading to early and robust induction of CD86. MDDCs infected with HIV-1-GFP did not mature until 48 h and only minimally responded to LKO-GFP (Figure S1A), as we previously described (Johnson et al., 2018; Manel et al., 2010).
Figure 1. HIV-1 Infection Triggers a Type I IFN Response in MDDCs.

(A) Schematic of the human DC network project depicting derivation of MDDCs, time series stimulation with HIV-1-GFP (+Vpx), LKO-GFP (+Vpx), LPS, and poly(I:C), followed by network inference, network visualization, in silico quality control, and experimental validation.
(B) Heatmap of differentially expressed genes in MDDCs across time (2, 8, 24, and 48 h), selected with false discovery rate (FDR) < 0.1 and sorted by Louvain modularity cluster (see STAR Methods).
(C) GSEA plots for the hallmark IFN response in MDDCs for all described HIV-1-GFP time points compared with mock, indicating normalized enrichment score (NES) and FDR. Enrichment is considered significant with FDR < 0.25.
(D) Leading edge genes from GSEA for HIV-1-GFP infections at 24 h.
In agreement with the earlier analyses, we observed that RNA-seq samples from infected and uninfected MDDCs could be clearly distinguished when visualized by principal-component analysis (PCA) (Figure S1B). Samples at 24 and 48 h time points could be further separated when grouped specifically by time or cell sorting condition: GFP negative (HIV exposed, but not expressing GFP), GFP positive (HIV infected), HIV-CD86-low (HIV-infected, immature MDDC), and HIV-CD86-high (HIV-infected, mature MDDC) (Figure S1C). We also used gene set enrichment analysis (GSEA) (Subramanian et al., 2005) to evaluate the qualitative nature of the innate response and uncovered strong associations between HIV-infected samples and gene sets for IFN alpha/beta signaling, inflammatory signaling, and DC maturation (full GSEA results available in Supplemental Information). The most significant enrichment was found with the hallmark IFN response (Figure 1C), which peaked at 24 h post-infection. Canonical ISGs were highly upregulated during HIV-1-GFP infection (Figure 1D) and their induction correlated with activation of IFNB1 and IFNL1 (Figure S1D). Having characterized the MDDC transcriptional response to HIV-1 infection, which included known maturation and IFN response signatures, we next sought to identify regions of open chromatin that may be accessible to TF binding to define possible TF-to-gene target relationships that regulate the innate response.
ATAC-Seq Reveals Time-Dependent Chromatin Opening at Innate Immune Gene Promoters
Analyzing genome-wide chromatin accessibility represents a powerful way to assess the presence of regulatory elements such as promoters and enhancers in mammalian cells (Buenrostro et al., 2013; Johnson et al., 2018). To match the experimental conditions used for RNA-seq, we profiled changes in open chromatin by performing ATAC-seq on MDDCs that were mock treated (Vpx only) or infected with HIV-1-GFP +Vpx for 2, 8, 24, and 48 h and sorted by flow cytometry (Figures 2A and 2B). We identified 88,000 high-confidence peaks across all ATAC-seq conditions (Table S3). Similar to the RNA-seq samples, the ATAC-seq samples for mock and HIV-1-GFP could be separated by PCA through time and status of infection with minimal variation between donors (Figures S2A and S2B). By plotting genome-wide changes in gene expression, together with changes in chromatin accessibility at transcription start sites, we observed that global changes in chromatin accessibility are temporally linked with stages of HIV-1 single-round infection and MDDC maturation (Figure 2C). At 24 h post-infection, we found that the induction of IFNB1, IFNL1, and most hallmark IFN response genes in GFP-negative and GFP-positive populations was linked with increases in chromatin accessibility. By 48 h, expression intensity and chromatin accessibility of these genes began to subside, with particularly noticeable reductions in chromatin accessibility for IFNB1 and IFNL1 to levels below baseline.
Figure 2. Transient Changes in Chromatin Accessibility Correspond to Activation of the Innate Response during HIV Infection.

(A) Illustration depicting ATAC-seq sample processing.
(B) Flow cytometry plots of MDDCs sorted (red boxes) for ATAC-seq after infection with HIV-1-GFP (+Vpx) at 2, 8, 24, and 48 h (MOI = 5). Plots show CD86 versus GFP expression and are representative data from 1 of 3 donors.
(C) Smooth scatter density plots of HIV-1-GFP-infected MDDCs from sorted populations as in (B), showing the genome-wide relationship between transcript levels (RNA-seq average log2 fold change) TSS chromatin accessibility (ATAC-seq average log2 fold change). Blue, density of points across the entire genome; red, hallmark IFN response genes as in Figure 1C. The positions of IFNB1, IFNL1, and select ISGs are labeled for each condition.
(D) ATAC-seq gene-associated peak values shown as counts per million (CPM) reads for the indicated time points and conditions. Lines connect samples from independent donors. Statistics were calculated on log2-transformed data using a 2-way ANOVA with multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To investigate changes in chromatin accessibility in more detail, we plotted promoter-associated ATAC-seq peak height across the time series for two housekeeping control genes (PGK1 and TBP), the IFN genes IFNB1 and IFNL1, well-defined ISGs, and IRF and NF-κB family members. Maximum chromatin accessibility for IFNB1, IFNL1, and several ISGs was detected at 24 h post-infection in cells expressing the GFP reporter (Figures 2D, S2C, and S2D). For IFN-related genes; the NF-κB family members RELA, RELB, and REL; and the related TF HIVEP1, chromatin accessibility was higher in HIV-infected, CD86-low MDDCs that were not fully mature compared with CD86-high, mature MDDCs (Figure 2D), suggesting that chromatin state is linked to MDDC maturation status.
To determine whether mapping open chromatin using ATAC-seq in HIV-infected MDDCs offered specific advantages over publicly available datasets, we compared our ATAC-seq peaks to steady-state open chromatin data from CD14+ monocytes (Figures S2C and S2D). Several ISGs displayed high chromatin accessibility at baseline (STAT1, STAT2, IRF1, IRF7, IRF9, OASL, HLA-C, and ISG20), with peaks detected in both MDDC ATAC-seq samples and CD14+ monocyte data available from ENCODE (Figure S2C). HIV-1-GFP infection led to a transient increase in chromatin accessibility at ISG promoters that corresponded with known binding sites for IRF3, STAT1, and NF-κB (RELA). In contrast, IFNB1, IFNL1, and other ISGs (CXCL10, CXCL11, ISG15, LY6E, USP18, and IFIT1) displayed open chromatin in MDDCs infected with HIV-1-GFP, but not in mock-treated cells or in CD14+ monocytes (Figure S2D), supporting the use of cell- and condition-specific ATAC-seq data as a basis for network inference.
Inferred Network of DC Transcriptomic Changes following Innate Immune Responses
To infer a predictive gene regulatory network, we adapted our previously published algorithm (the Inferelator), which was designed to learn from mixes of steady-state and dynamic data (Bonneau et al., 2006). The method can incorporate multiple data types to influence network model selection, including TF occupancy, cooperativity, and transcriptional profiling in response to innate immune stimuli (Figures 1 and 2). High-throughput methods like ATAC-seq and chromatin immunoprecipitation sequencing (ChIP-seq) can be used to guide network inference by defining prior information on network architecture, dramatically improving network model selection and predictive power (Arrieta-Ortiz et al., 2015; Miraldi et al., 2019; Siahpirani and Roy, 2017). Key to this study, we extended our previously published computational methods for learning the regulation of gene networks through the Inferelator (Arrieta-Ortiz et al., 2015; Bonneau et al., 2006; Ciofani et al., 2012; Greenfield et al., 2010; Madar et al., 2010) by integrating results obtained from RNA-seq and ATAC-seq experiments performed at the bulk level in a time course fashion (Figure 3A).
Figure 3. Repeated Subsampling of Prior Information Improves Network Inference.

(A) Pipeline for network inference. Prior information (TF accessibility) and gene expression time series data were used as inputs for the Inferelator. Output networks underwent performance evaluation and quality control as indicated.
(B) Prior matrix denotes a possible connection between TF and gene X if that TF’s binding motif is found in accessible chromatin upstream of (−1 kb) or within gene X.
(C) Schematic illustrating how TF activity is estimated by summing predicted target expression over time (red, increase; blue, decrease).
(D) TF and gene target connections in the Prior network are pruned using an EN regression to yield the final inferred network.
(E and F) Precision-recall plots (E) and area under precision-recall (AUPR) curves (F) for the indicated networks scored against the TRRUST database (see STAR Methods).
(G and H) Precision-recall plots (G) and AUPR curves (H) for the indicated networks scored against known ISGs (Schoggins et al., 2011).
Here, prior information on network architecture is derived from the 88,000 peaks of chromatin accessibility data generated by our ATAC-seq experiments that tracked MDDC responses to HIV infection (Figures 2A and 3A). Using curated TF binding motif databases, we established possible TF-gene target relationships by searching ATAC-seq peaks for TF motifs that we located within or up to 1 kb upstream of gene bodies (Figure 3B). This information was used to build a "Prior" matrix that connected each gene with its possible TF regulators (Table S4). We then estimated TF activity over the course of stimulation based on the combined expression of a TF’s predicted gene targets (Figure 3C). This step approximates the effect of unmeasured parameters, such as post-translational regulation and protein-protein interactions, on TF activity in a condition-dependent manner. We were thus able to model changes in TF activity that are semi-independent of changes in TF expression, as observed for IRF3 (Figures S3A and S3B). IRF3 activity was predicted to increase in response to LPS, poly(I:C), and HIV sensing, which are all known to drive IFN production through IRF3 phosphorylation. Our inference model also predicted that activity and expression for STAT2, IRF7, and RELA correlated with innate stimulation, as expected, given their well-defined roles in the innate response (Cao, 2016).
Once TF activity was estimated, the network structure Prior was used to bias model selection of TF-to-gene target regulatory interactions toward edges with Prior information during network inference (Figure 3D). To improve the overall performance and stability of network inference, several aspects of the method (and key inputs) were tested, such as different sources of Priors (publicly available ENCODE data versus our ATAC-seq data), different TF binding motif databases (HOCOMOCO [Kulakovskiy et al., 2013] versus CisBp 2.0 [Weirauch et al., 2014]), and different model selection methods (Bayesian best subset regression [BBSR] versus elastic net [EN]). In addition, because every computational run is subject to stochasticity in the inference procedure, we evaluated whether network performance could be improved by combining hundreds of individual computation runs (selecting model components such as TF activity estimates that were stable across random subsamples of the structure Prior; see STAR Methods). To evaluate the performance of networks inferred using the parameters described earlier, we used area under precision-recall curves (Madar et al., 2010) to compare the prediction and ranking of our TF-to-gene target regulatory edges against TRRUST v.2, a gold-standard reference database of human transcriptional regulatory events (Han et al., 2018), and well-known lists of ISGs (Kane et al., 2016; Schoggins et al., 2011; Shaw et al., 2017). We found that the EN regression model outperformed BBSR when ATAC-seq-based networks were benchmarked against TRRUST v.2 (Figures 3E and 3F). We improved precision-recall on TRRUST and known ISGs by bootstrapping 400 individual EN-ATAC inference runs into a converged network (EN-ATAC x400) (Figures 3E-3H, S3C, and S3D). This final EN-ATAC network correctly predicted 90 of 97 core mammalian ISGs (Shaw et al., 2017) to be downstream of IRF and NF-κB family members (Figure S3E) and emphasized a role for IRF3 (Figure S3F). Thus, we were able to integrate gene expression and chromatin accessibility data to estimate a network with 542 TFs and 21,862 target genes, explaining >2/3 of the variance in our expression dataset (Table S5).
Network Clustering, Differential Gene Expression Analysis, and TF Enrichment Tests Define Key Subregulatory Groups
We used this MDDC transcriptional response network to group the expression changes observed into sets of coregulated genes with high-confidence regulatory subnetworks. We first applied a cutoff to visualize the top 75,000 regulatory edges that displayed the highest-confidence beta scores (Figure 4A) and then partitioned the network using Louvain modularity clustering into 10 major neighborhoods of TF and target gene communities, with each cluster encompassing at least 1% of the total number of genes in the network (MDDC network topology can be freely explored using Gephi software by downloading the Gephi-formatted supplemental file, available in the STAR Methods section). We were able to assign putative biological functions to 7 of 10 clusters based on pathway enrichment scores (see STAR Methods). The most striking enrichment scores were observed in the upper-right shoulder of the network, demarcating clusters that are predicted to function in the IFN response (cluster 5, IRFs & Interferon), inflammation and cytokine production (cluster 8, NF-κB & Inflammation), and in response to xenobiotic stress (cluster 2, Regulation of Cell Activation) (Figures 4B-4D).
Figure 4. Modularity Clustering of the Network Defines TF and Gene Target Clusters Predicted to Control Distinct Biological Functions.

(A) EN-ATAC network topology in MDDCs visualized through Gephi software. Clusters were named according to pathway and motif enrichment (see STAR Methods). Arrows indicate transcriptional regulatory events between TFs and targets and do not specify positive or negative effects on target expression. Node size denotes the relative number of edges. Clusters are color coded based on Louvain clustering and ranked based on their decreasing size (from 1 to 10) (based on the percentage of genes in each cluster versus total number of genes).
(B–E) Enrichment of pathways from Reactome (B), Genome Browser TF position weight matrices (PWMs) (C), TF perturbations (D), and JASPAR and TRANSFAC PWMs (E) for clusters 2, 5, and 8 (color coded as in A). Ranking is by Enrichr combined score (−log (p) × Z score).
Activation of IRF, STAT, and NF-κB family members is centrally linked to IFN signaling during the innate immune response (Cao, 2016; Ivashkiv and Donlin, 2014). Fittingly, we found that several IRF (IRFs 1, 2, 3, 5, 7, 8, and 9), STAT1 and STAT2, and all five NF-κB (RELA, RELB, REL, NFKB1, and NFKB2) populated the upper-right shoulder of the network (Figure S4A), consistent with their overlap in motif preferences (Figure S4B). Interestingly, IRF4 and IRF5, which also have GAA- and GAAA-rich motif features similar to those of other IRFs (Figure S4C), are positioned in different areas of the network. In particular, IRF4 is localized to the extreme lower left (Figure S4A, cluster 1, Chromatin Modifiers), close to pioneer factors and repressors that target large numbers of genes (Jankowski et al., 2016).
We next sought to establish the relative impact of each TF in the network during an innate response. Toward this end, we performed hypergeometric tests to assess TF enrichment and found high enrichment for IRF, STAT, and NF-κB family members during stimulation with HIV-1-GFP, LPS, and poly(I:C) (Figure S4D; Table S6), with kinetics closely matching what was observed for IFN production and MDDC maturation (Figures S1A and S1D). We also found mild enrichment of these TFs during LKO-GFP infection, supporting the concept that non-replicating lentiviral vectors are partially, if inefficiently, sensed by the innate immune system (Figure S4D) (Johnson et al., 2018). By highlighting network topology in which genes are differentially expressed during treatment with HIV-1-GFP, LKO-GFP, LPS, and poly(I:C), we determined that most of the transcriptional response is concentrated in clusters 2, 5, and 8 (Regulation of Cell Activation, IRFs & Interferon, and NF-κB & Inflammation, respectively) (Figure S4E).
Exploration and Validation of Modulators of the IFN Response
The first wave of the antiviral response is driven by production of type I and type III IFNs (Levy et al., 2011). In MDDCs, the major expressed type I and type III IFNs are IFNB1 and IFNL1, respectively (Figure S1D). To visualize all high-confidence edges for IFNB1, IFNL1, and their regulators that were predicted by the network, we generated a subnetwork visualization tool (Figure 5A; see STAR Methods for instructions on how to access the Jupyter subnetwork widget). Upstream of both IFNB1 and IFNL1, we identified IRF3 and additional IRFs that have been reported to contribute to their induction under various conditions (such as IRF1, IRF5, IRF7, and IRF8) (Ivashkiv and Donlin, 2014). Several unexpected factors were also predicted to be upstream regulators of IFNB1 and IFNL1 (Figure 5A), and of these TFs, HIVEP1, CBFB, STAT2, PRDM1, and KLF13 were expressed at relatively high levels in MDDCs and in some cases exhibited dynamic expression changes during the innate response (Figure 5B).
Figure 5. Subnetwork Exploration Uncovers Positive and Negative Regulators of IFN and ISGs.

(A) Jupyter widget subnetwork view (see STAR Methods) of predicted upstream regulators of IFNB1 and IFNL1. Arrows indicate transcriptional regulatory events between TFs and targets and do not specify positive or negative effects on target expression. Node size denotes the relative number of edges. Nodes are color coded by network cluster (see Figure 4).
(B) Heatmap of gene expression for the indicated TFs during treatment with LPS, poly(I:C), HIV-1-GFP (+Vpx), and LKO-GFP (+Vpx) at 2, 8, 24, and 48 h.
(C) Immunoblots of MDDC lysates after treatment with the indicated shRNAs.
(D) Flow cytometry of CD86 versus GFP expression in shRNA-modified MDDCs 48 h after mock treatment (+Vpx) or infection with HIV-1-GFP (+Vpx).
(E–G) Plots showing %GFP+ (E), %CD86+ (F), and %SIGLEC1+ (G) from MDDCs treated as in (D).
(H–I) ELISA of IFNL1 (H) and IP-10 (I) expression in supernatants from shRNA-modified MDDCs that were mock treated or infected with HIV-1-GFP for 48 h.
For (E)–(I), plots represent pooled data from 5–8 donors. **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figure S5.
We next sought to empirically test the function of several of these TFs in MDDCs, noting that PRDM1 and HIVEP1 were among the highest-ranking TFs according to hypergeometric tests (Figure S4D). Using short hairpin RNA (shRNA) vectors, we targeted these TFs for knockdown in MDDCs alongside key sensors of HIV (cGAS and NONO) and the essential TF IRF3. Among other potential TF candidates, we additionally tested KLF13, based on its relatively high expression and known role in recruiting coactivators p300/CBP and PCAF to drive expression of CCL5 (Ahn et al., 2007). We confirmed knockdown of these targets either by immunoblot ((Figure 5C) or qPCR (Figure S5A), and as expected, knockdown of cGAS and NONO potently inhibited innate immune activation in MDDCs during HIV-1-GFP infection (Johnson et al., 2018; Lahaye et al., 2013, 2018; Figures 5D-5I and S5B-S5E). The most effective shRNA clones for HIVEP1, KLF13, and PRDM1 had no effect on viral infection. Under these conditions, we could not conclude whether HIVEP1 knockdown affected MDDC maturation (Figure S5D). Knockdown of KLF13 partially inhibited CD86 induction, which was significant for two shRNAs at high HIV-1-GFP MOI (Figure S5E). Moreover, we found that greater KLF13 knockdown correlated with lower levels of CD86 and inhibition of IFNL1 expression during HIV-1-GFP infection (Figure S5F). In a more dramatic way, knockdown of PRDM1 significantly inhibited innate immune activation of MDDCs, as scored by induction of CD86, SIGLEC1, IFNL1, and IP-10 (Figures 5D-5I), and these effects were observed with three independent shRNAs. Altogether, these data suggest that although cellular factors such as cGAS, NONO, and IRF3 function as critical toggle switches during the innate response, additional TFs such as KLF13 and, to a greater extent, PRDM1 may operate to fine-tune the magnitude of the innate response. Other TFs with high enrichment scores identified from experiments with wild-type HIV-1, HIV-2, recombinant IFN, and discrete innate immune stimuli (Figure S5G; Table S6) warrant further exploration.
Mature and Immature MDDC Populations Cannot Be Distinguished Based on IRF/STAT Enrichment Alone
IRF and STAT activation is critical for inducing an IFN gene signature, but neutralization of IFN signaling does not block upregulation of inflammatory cell markers during HIV infection (Lahaye et al., 2013; Manel et al., 2010), suggesting that factors in addition to these TFs contribute to MDDC maturation. Gene set enrichment analysis on CD86-low- and CD86-high-sorted MDDCs (Figures S1A and S2B) identified DC maturation genes as significantly enriched in CD86-high compared with CD86-low populations (Figures 6A-6C). However, we found no statistically significant difference in hallmark IFN response genes, with ISG expression levels and chromatin accessibility elevated in both conditions (Figures 6B, S6A, and S6B). Thus, an IFN response is not sufficient to explain the differences in MDDC maturation status following HIV infection.
Figure 6. Mature MDDCs Cannot Be Identified Based on an IFN Signature Alone.

(A) GSEA plots comparing gene expression data for CD86-high (mature) versus CD86-low (immature) MDDCs. Enrichment is considered significant with FDR < 0.25.
(B) Smooth scatter density plots of transcript levels (RNA-seq) versus TSS chromatin accessibility (ATAC-seq) in HIV-1-GFP-infected MDDCs for CD86-low and CD86-high populations. Blue density marks the entire genome. Genes in the DC maturation gene set (pink) and the hallmark IFN response gene set (orange) are highlighted in the top and bottom plots, respectively.
(C) log2 normalized gene expression plotted for the CD86-low population (y axis) and CD86-high population sorted at 48 h. DC maturation genes (pink) and hallmark IFN response genes (orange) are shown as in (B).
(D) Heatmap of hypergeometric scores for TF enrichment displaying HIV-1-GFP compared with mock at 2, 8, and 24 h (GFP−); 24 h (GFP+); 48 h (CD86-low); and 48 h (CD86-high). TFs were ranked in descending order according to the 24 h GFP+ condition. Heatmap is color coded according to the −log p value of TF enrichment.
(E) Differential gene expression contrast between CD86-high and CD86-low populations as visualized by Gephi software, highlighting edges in clusters 2 and 8. Ranking is by Enrichr combined score (−log (p) * Z score).
(F) Enrichment of differentially expressed genes separated by network cluster for the CD86-high versus CD86-low contrast as in (E).
(G–I) Potential protein-protein interactions (G), Reactome pathway enrichment (H), and single gene perturbations (I) predicted from differentially expressed genes in the CD86-high versus CD86-low contrast.
(J) Network connections for RARA and STAT2 visualized by Gephi software, color coded by contrast, and superimposed onto a gray network backdrop.
See also Figure S6.
Consistent with our DC maturation gene set results, the NF-κB family members (RELB, REL, NFKB1, RELA, and NFKB2) showed stronger enrichment in CD86-high than in CD86-low populations (Figures 6D-6F), as did additional, unexpected TFs (CLOCK, THRB, SRF, and HIVEP1) found in cluster 2 (Regulation of Cell Activation) and cluster 8 (NF-κB & Inflammation). Interestingly, pathway analysis of differentially accessible chromatin in CD86-high versus CD86-low conditions suggested that the retinoic acid receptor (RAR) RAR alpha (RARA), PPARD, NCOR1, and the canonical NF-κB family member RELA (among other TFs) influenced the transition between immature and mature MDDCs (Figures S6C-S6E). When differentially expressed genes were assessed in CD86-high versus CD86-low conditions in a similar fashion, pathway analysis predicted strong enrichment of protein-protein interactions with the RARA partner molecule RXRA, pathways associated with nuclear receptor transcription, MyD88 signaling, and gene expression changes resembling published datasets from RARA perturbation (highest among other inflammatory activators) (Figures 6G-6I). These analyses implicated RARA/RXRA nuclear hormone receptor signaling to be involved in regulating MDDC maturation.
Identification of RARA as a Critical TF for Controlling MDDC Maturation
At steady state, RARs bind retinoic acid response elements (RAREs), together with retinoid X receptors (RXRs) in a complex with nuclear corepressors (NCOR1/2) and suppress transcription of RARE-dependent genes (Cunningham and Duester, 2015). Ligand binding leads to the dissociation of NCOR1/2 from RARs and the recruitment of coactivators, which then drive gene expression. Given that RARA occupies a central region of cluster 2 (Regulation of Cell Activation) (Figures 4D and 6J) and was predicted to influence MDDC maturation, we sought to assess its network connections and function compared with STAT2 and cGAS. Subnetwork visualization of RARA and close network edges displayed many connections between cluster 2 (Regulation of Cell Activation) and Clusters 5 and 8 (IRF & Interferon and NF-κB & Inflammation, respectively) (Figure 7A). Many of these connections were detected using the Search Tool for the Retrieval of Interacting Genes (STRING) database (Szklarczyk et al., 2015), highlighting the position of RARA close to TFs involved in controlling cell fate, lipid and sterol metabolism, IFN signaling, and inflammation (Figure 7B).
Figure 7. RARA Is a Negative Regulator of MDDC Maturation.

(A) Jupyter widget subnetwork view (see STAR Methods) showing edges between RARA, its predicted targets, STAT2, and the remaining top 30 TFs ranked by hypergeometric p value. Arrows point from TFs to targets but do not suggest positive or negative activity.
(B) STRING database connections for nodes shown in (A), including NCOR1 and NCOR2. Brackets indicate groups and biological pathways. Nodes connected in the EN-ATAC network with no direct STRING connections are shown to the left. In (A) and (B), nodes are color coded by network cluster.
(C) Flow cytometry plots of MDDCs modified by shRNA that were mock infected (+Vpx) or challenged with HIV-GFP (+Vpx) at day 4 for 48 h. Plots show CD86 versus GFP expression and represent 1 of 4 donors (MOI = 0.5, 1.5, and 5).
(D) qPCR validation of target knockdown in MDDCs modified by the indicated shRNAs.
(E) qPCR of ISG15, CXCL10, and GFP expression in shRNA-modified MDDCs that were either mock treated or infected with HIV-1-GFP (MOI = 5) for 24 h.
(F) Flow cytometry of MDDCs treated with vehicle or the RARA agonist Am80 (10 μM) ± infection with HIV-1-GFP for 48 h.
(G) Pooled data of CD86 MFI in MDDCs from 4 donors treated as in (F). Am80 concentrations = 10 nM, 100 nM, 1 μM, and 10 μM.
(H) Flow cytometry of MDDCs treated with vehicle or the RARA antagonist BMS 195614 (10 μM) ± infection with HIV-1-GFP for 48 h.
(I) Pooled data of CD86 MFI in MDDCs from 4 donors treated as in (H). BMS 195614 concentrations = 1 nM, 100 nM, and 10 μM.
For (D), (E), (G), and (I), n = 4 donors. *p < 0.05, **p < 0.01, ***p < 0.001. Data represent mean ± SEM. See also Figure S7.
Knockdown of cGAS or STAT2 led to a pronounced loss of innate immune activation in MDDC during infection with HIV-1-GFP (Figures 7C and 7D). In contrast, knockdown of RARA upregulated CD86 at baseline and potentiated MDDC maturation during infection, suggesting RARA acts as a negative regulator of inflammation. Consistent with this notion, expression levels of many predicted targets of RARA increased in response to LPS and poly(I:C) treatment and to a lesser degree in response to HIV-1-GFP (Figure S7A). Moreover, we found an inverse relationship between gene expression of RARA and costimulatory molecules CD80 and CD86 (Figure S7B), supported by the negative correlation between the estimated transcriptional activity of RARA and its measured gene expression (Figure S7C). Knockdown of RARA was associated with a slight but statistically significant decrease in production of IFN and ISGs ISG15 and CXCL10 (Figures 7E and S7E-S7H), leading us to consider that loss of RARA might affect the balance between antiviral IFN responses and inflammatory responses that benefit virus replication. In agreement with this idea, we observed heightened virus transcription in RARA knockdown conditions (measured by GFP reporter expression) (Figures 7C and 7E), because increased inflammation is a correlate of increased HIV replication (Deeks et al., 2013).
Direct modulation of RARA using the pharmacological agonist Am80 (Delescluse et al., 1991) triggered increased CD86 surface expression both at baseline and following HIV-1-GFP infection (Figures 7F and 7G). We also observed that treatment with Am80 or the classic RARA ligand all-trans retinoic acid (ATRA), increased gene expression of CD86 (Figure S7D). Conversely, use of the RARA antagonist BMS 195614 reduced upregulation of CD86 during virus infection (Figures 7H and 7I). Although our findings from RARA agonists and antagonists may appear inconsistent with those of RARA knockdown, they are in line with the dual functions of nuclear receptors in recruiting either coactivators or corepressors of transcription, which will be discussed later. Altogether, these data support our network predictions that spotlight a role for RARA in the regulation of MDDC maturation.
DISCUSSION
Multiple TFs are known to drive the innate response downstream of pathogen sensing in myeloid DCs (Ivashkiv and Donlin, 2014), but it remains unclear how production of IFN, differential expression of thousands of host genes, and DC maturation are coordinately regulated across the genome in response to HIV detection. To address this issue, we employed a systems biology approach to understand how MDDCs respond to stimulation, generating steady-state and time series RNA-seq under a battery of viral and innate immune perturbations combined with ATAC-seq measurements of chromatin accessibility (Figure 1A). In this study, we inferred a transcriptomic network linking 542 TFs to 21,862 gene targets, revealed unexpected roles for TFs in the innate response through validation experiments (PRDM1 and RARA), and provided tools for further network exploration that accompany this manuscript (see STAR Methods).
Computational strategies that combine ATAC-seq and gene expression data have recently been employed to identify key regulators of tissue-resident memory CD8+ T cells (Milner et al., 2017), define TF specificity across cell types (Cusanovich et al., 2018), and chart the landscape of human cancers (Corces et al., 2018). Here, we improved the stability of network inference by performing ensembles of analyses, with each run subsampling information from a structured network Prior that was generated from our ATAC-seq data. By modeling changes in gene expression to be a function of combined TF activities, we reduced the complexity of multiple factors known to influence gene expression into a single parameter (such as changes in TF expression, post-translational modifications, and epigenetic marks on target genes). In doing so, our network revealed strong enrichment of TFs known to regulate the innate response, notably including IRF3, a key factor that is activated by phosphorylation and, because it is constitutively expressed, is rarely identified in pathway analyses that emphasize changes in TF expression (Amit et al., 2009). Our network Prior only considered chromatin accessibility from 1 kb upstream through target gene bodies, which may have biased the network toward proximal factors, and it did not cleanly distinguish between TFs with overlapping motif preferences. Future network inference models will likely incorporate distal enhancer elements and improved TF motif databases.
To date, this study reports one of the largest gene regulatory networks that has been charted in mammalian cells. Because our full reported network (500,000 edges) is potentially promiscuous, we have provided beta values (Table S5) that can be used to truncate edges at any specified cutoff to yield smaller networks composed of high-confidence connections. We anticipate further refinements to our network rendered possible through the use of the tools we have included as companion applications to this manuscript (i.e., network visualization and exploration via Gephi and our Jupyter widget; see STAR Methods) and through subsequent experimental validation. The final gene regulatory network reported here (75,000 edges) exhibited a high level of precision-recall against TFs targeting known ISGs and correctly predicted prominent roles for IRF, STAT, and NF-κB family members in the IFN response. We recovered 90 of 97 core mammalian ISGs as targets of IRFs; the remaining 7 core ISGs were predicted to be downstream of STAT and AP-1 family TFs (Table S5). Of the 22 IFN species measured by RNA-seq, we found IFNL1 to be upregulated more than any type I IFN, including IFNB1, during stimulation with LPS, poly(I:C), or HIV-1. We know IFN signaling can provide both beneficial and detrimental effects during progression to AIDS (Sandler et al., 2014); the contribution of type I versus type III IFN in an in vivo response to HIV remains to be determined.
Among several TFs predicted to be upstream of IFN genes in our network, we found that PRDM1 (also known as positive regulatory domain-I, PR/SET domain-1, and BLIMP1) is an essential positive regulator of innate immune activation in response to HIV-1 infection in MDDCs. For almost 30 years, PRDM1 has been known to bind IFN promoter sequences (Keller and Maniatis, 1991). Since then, reports have suggested that PRDM1 is important for differentiation of plasma cells, is a risk factor for autoimmune disease, and can act to either positively or negatively regulate cell-specific cytokine production (Bönelt et al., 2019; Kim et al., 2013; Ko et al., 2018; Nutt et al., 2007). PRDM1 is known to be rapidly induced by aryl hydrocarbon receptor (AHR) ligands, and its expression is required for AHR-induced differentiation of MDDCs (Goudot et al., 2017). Here, we revealed that PRDM1 promotes innate immune activation during HIV infection, in accordance with its high hypergeometric enrichment and cluster association with key IRFs in the network. In our system, expression levels of PRDM1 are not dramatically affected by innate stimulation, but nonetheless, it plays an important role in regulating the IFN response during HIV infection. Further exploration of our network could help uncover whether factors associated with PRDM1 determine how PRDM1 operates to promote or inhibit IFN production in a cell-specific manner.
By analyzing RNA-seq data from sorted cell populations, we determined that induction of a robust ISG signature is insufficient for MDDC maturation, suggesting that factors in addition to PRDM1, IRF, and STAT family TF members influence the transition from immature to mature MDDCs (Figure 6). In support of this notion, we demonstrated that RARA acts as a negative regulator of this notion, we demonstrated that RARA acts as a negative regulator of MDDC maturation at basal state (Figures 7 and S7). Based on our network analysis predicting that RARA and NF-κB activities govern the transition from immature to mature MDDC, together with our data indicating that RARA modulates innate immune responses, we propose a model in which, at basal state, RXR/RARA heterodimers bind to RAREs and inhibit MDDC maturation driven by NF-κB family members (Figure S7I). Perturbation of RARA expression, either by shRNA or by unknown factors during the innate response, relieves this inhibition and permits MDDC maturation (Figure S7J). Binding of ligands/retinoids to RARA results in the exchange of bound nuclear corepressors with nuclear coactivators that will also permit MDDC maturation. Thus, RARA knockdown and administration of RARA agonists can achieve a similar biological end, albeit through different means (Figure S7K). In agreement with this model, using an RARA antagonist to block the release of nuclear corepressors inhibits MDDC maturation in response to HIV infection (Figure S7L). These results reinforce the reported involvement of retinoids in DC maturation (Geissmann et al., 2003; Szatmari and Nagy, 2008). Given that retinoic acid is known to activate mitogen-activated protein kinase (MAPK) phosphorylation and abrogate tolerance in a stressed intestinal environment (DePaolo et al., 2011), it is not unreasonable to envision that RARA activation might influence derepression of inflammatory gene expression during HIV infection.
Our model thus suggests that RARA function could provide another layer of control to govern DC maturation through sensing changes in metabolism. It is known that DCs derived from monocytes in vitro resemble inflammatory DCs in vivo, with the derivation process requiring NCOR2, an established regulator of RAR/RXR signaling (Cunningham and Duester, 2015; Sander et al., 2017). In a similar vein, differentiation of DCs in vitro and in vivo is known to depend on fatty acid synthesis (Rehman et al., 2013). In addition, in plasmacytoid DCs, lipid metabolism, RXR, and PPAR-related pathways can modulate type I IFN responses, suggesting that environmental cues such as retinoids help shape immune responses (Wu et al., 2016). It is tempting to draw parallels with how changes in fatty acid oxidation and cholesterol biosynthesis influence innate responses and determine cell fate (tolerogenic versus immunogenic DC) (Maldonado and von Andrian, 2010; York et al., 2015). Retinoic acid is an established modulator of DC differentiation and intestinal immune cell trafficking (Czarnewski et al., 2017), and we speculate that metabolism and RAR/RXR signaling in gut-associated tissues can affect innate responses during HIV infection.
In addition to profiling how human MDDCs respond to HIV, we profiled responses to a battery of classic innate immune stimuli, including LPS, poly(I:C), cGAMP, R848, and recombinant IFN. The transcriptomic responses to TLR stimulation we observed in human MDDCs resembles what has been reported for mouse bone marrow-derived DCs (Amit et al., 2009; Chevrier et al., 2011). This work represents a source of information that one can explore to compare similarities and differences among transcriptomic pathways bridging innate immune detection and production of inflammatory cytokines. Much of the transcriptomic activity observed for HIV infection in our network overlaps with that in response to LPS and poly(I:C) exposure; however, we found that HIV drives unique changes in fatty acid oxidation pathways (Johnson et al., 2018). These findings support the existence of an interplay among HIV infection, lipid metabolism, and retinoids that control IFN production and DC maturation, which will require further investigation.
In addition, HIV-1 infection drives transient changes in chromatin accessibility that likely precede or perhaps coincide with the induction and resolution phases of the innate response. Interestingly, we observed global changes in chromatin accessibility that were not always associated with changes in host gene expression. We speculate that this could either reflect the nature of the innate response or correspond with events in the virus life cycle, such as opening of gene-dense regions known to be hotspots for virus integration (Table S3) (Wang et al., 2007).
Collective analyses of these data will help to further decipher the mechanisms of innate immune regulation that span initiation, feedforward amplification, and resolution of aberrant hyperactivation. Understanding the transcriptional networks that underpin IFN production and innate immune activation in MDDCs could also shed light on regulatory mechanisms in primary DCs. We know that several aspects of HIV infection and innate immune responses are conserved between MDDCs and select primary DC subsets (e.g., SAMHD1-mediated restriction of reverse transcription, the enhancement of virus infection and innate sensing by Vpx, and the requirement for cGAS and NONO in HIV-driven IFN responses are all shared between MDDCs and CD1c+ cDC2s) (Lahaye et al., 2018; Silvin et al., 2017). However, caution should be exercised when extrapolating from our network to primary DCs. Monocyte-derived cells are not always appropriate models of primary DC behavior (Sander et al., 2017), and considering the recent identification of numerous specialized DC subtypes (Alcántara-Hernández et al., 2017; Villani et al., 2017), perhaps this is not too unexpected, because each DC subtype has a unique lineage ontogeny and largely distinct functionality (Eisenbarth, 2019).
Nevertheless, our network may inform future antiviral strategies, because the robust activation of T cells, which is facilitated by mature, activated DCs, is crucial for immunological control of HIV (Walker and McMichael, 2012). We anticipate that the quality of network inference will further improve as motif databases are refined and large-scale genetic perturbations become tractable in primary human DCs. The network presented herein highlights how the innate response depends on the coordinated action of multiple TFs and serves as a resource for further exploration of pathways that govern HIV replication and innate immunity.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mickaël M. Ménager (mickael.menager@institutimagine.org). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
To generate MDDCs, we acquired leukocytes from de-identified normal human donors from a variety of sources (Bloodworks Northwest, Renton, WA, USA; New York Blood Center, New York, NY, USA; ARUP Blood Services, Sandy, UT, USA; and from venipunctures (approved by the Institut National de la Santé et de la Recherche Médicale ethics committee, Paris, France). The authors cannot report on the sex, gender, or age of the donors since the samples were de-identified and donors remain anonymous. Peripheral blood mononuclear cells (PBMCs) were layered over Ficoll-Paque Plus (GE Healthcare). CD14+ monocytes from PBMC buffy coats were isolated with anti-human CD14 magnetic beads (Miltenyi) and cultured in RPMI (Thermo Fisher) containing 10% heat-inactivated fetal bovine serum (FBS, Peak Serum, Inc), 50 U/ml penicillin, 50 μg/ml streptomycin (P/S, Thermo Fisher), 10mM HEPES (Sigma), 2-Mercaptoethanol (Thermo Fisher), and 2 mM L-glutamine (Thermo Fisher), in the presence of recombinant human GM-CSF at 10 ng/ml and IL-4 at 50 ng/ml (Peprotech). Multiple lots of FBS were tested to identify batches leading to minimal baseline induction of CD86 over the course of MDDC differentiation. Fresh media and cytokines were added to cells (40% by volume) one day after CD14+ cell isolation. On day 4, cells were collected, resuspended in fresh media with cytokines used for infection or stimulation. Immature MDDCs on day 6 were routinely assessed by flow cytometry surface marker staining to be CD11c+ (Thermo Fisher Cat# 17-0116-42, RRID:AB_1659668), HLA-DR+ (BioLegend Cat# 307607, RRID:AB_314685), DC-SIGN+ (R and D Systems Cat# FAB161P, RRID:AB_357064), and CD86- (eBioscience Cat# 15-0869-42 RRID:AB_11042003). MDDC experiments were performed using biological replicates from blood-derived cells from multiple individual donors as indicated in the figure legends.
293FT female cells (Life Technologies Cat# R70007, RRID:CVCL_6911) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher) that was supplemented with 10% FBS, P/S, 10mM HEPES, and supplemented with 0.1 mM MEM non-essential amino acids (Thermo Fisher), 6 mM glutamine, and 1 mM sodium pyruvate (Thermo Fisher).
HL116 male cells were cultured in DMEM supplemented with 10% FBS, P/S, 10mM HEPES, 0.1 mM MEM non-essential amino acids, 6 mM glutamine, 1 mM sodium pyruvate, and HAT supplement (Thermo Fisher; hypoxanthine (5 mM), aminopterin (20 μM) and thymidine (0.8 mM)) diluted 1:50 as recommended.
All cell lines were thawed from early passages, kept in culture no longer than 4 weeks, and were regularly tested for mycoplasma contamination (every 6 months). All cells were maintained at 37°C and 5% CO2.
METHOD DETAILS
Plasmids
HIV-1-GFP has been used previously to study immune responses in human DCs (Manel et al., 2010) and is env- vpu- vpr- vif- nef-, with the GFP open reading frame in place of nef. We generated virus like particles packaging Vpx from the plasmid pSIV3+ (based on SIVmac251, GenBank acc. no. M19499), which has been described elsewhere (Mangeot et al., 2000). Target sequences for shRNA vectors are listed in Table S7. All lentiviral constructs were transformed into Stbl3 bacteria (ThermoFisher) for propagation of plasmid DNA. All plasmids were prepped and purified using Nucleobond Xtra Maxi Kit (Takara) or similar maxi prep reagents.
Virus and virus-like particle production
To produce recombinant lentiviral vectors, reporter viruses, and Vpx-containing virus-like particles, we transfected plasmids into 293FT cells as previously described (Johnson et al., 2018). Briefly, the day before transfection, cells were seeded onto poly-L-lysine (MP Biomedicals) -coated 15 cm plates to be 70%–80% at the time of transfection. Cells were transfected with a total of 22.5 μg DNA using PEImax (Polysciences, Inc.) at a ratio of 1:2 (DNA:PEI). For lentiviral vectors, plasmid amounts were 3.4 μg CMV-VSV-G (Addgene plasmid #8454), 9 μg psPax2 (Addgene plasmid #12260), and 10.1 μg transgene (LKO.1 controls or shRNA construct). For HIV reporter viruses plasmid amounts were 3.4 μg CMV-VSV-G and 19.1 μg HIV cassette. Virus-like particles containing Vpx were produced using 3.4 μg CMV-VSV-G and 19.1 μg pSIV3+ (Mangeot et al., 2000). The morning after transfection, cells were washed once and replenished with 30 mL of fresh media. Supernatants were harvested 32-36 h after feeding 293FTs and then passed through 0.45 μm syringe filters (Corning) to remove debris. Supernatants of reporter virus and lentiviral vectors were either used fresh for transduction or concentrated by ultracentrifugation by spinning in conical tubes (Beckman) at 25 K rpm for 2 h at 4°C in an SW32 swing-bucket rotor (Beckman). Lentiviral pellets were thoroughly resuspended (~30-fold concentrated) in MDDC media without cytokines. Before transducing cells, insoluble material was clarified from lentivirus stocks by centrifuging at 300 rcf. for 3 min at 4C. Viral stocks were frozen at −80°C and used as indicated below.
Perturbation of DCs
To genetically modify MDDCs, we isolated CD14+ monocytes from buffy coats as described above and transduced them with lentiviral vectors in the presence of virus-like particles packaging Vpx similar to previously described protocols (Johnson et al., 2018; Manel et al., 2010). Briefly, monocytes were resuspended in complete MDDC media containing GM-CSF (10 ng/ml), IL-4 (50 ng/ml), polybrene (Sigma, 1 μg/ml), and supernatants containing Vpx particles (1 mL supernatant for 1 × 10^7 cells) and then were plated in either 10 cm dishes (6 × 10^6 cells in 9 mL media) or 96 well plates (160,000 cells in 150 μL media). Roughly 30 min after plating, clarified concentrated lentiviral stocks were added to transduce cells. We routinely observed that 95%–99% of cells could be transduced using 150 μL concentrated vector in 10 cm plates or 5 μL concentrated vector per well in 96 well plates.
Infections and stimulations
MDDCs were infected or stimulated with innate agonists between day 4 and day 6 after differentiation. For infections with HIV-1 and related lentiviruses, except where noted otherwise, Vpx+ virus-like particles were used to overcome virus restriction (1 mL supernatant for 1 × 10^7 cells) (Johnson et al., 2018; Manel et al., 2010). MDDCs were counted on day 4 and resuspended at 800,000 cell per ml in fresh medium with GM-CSF, IL-4, and polybrene (1 μg/ml) and then reseeded into appropriate culture vessels. For most assays, MDDCs were infected at a density of 800,000 cells per ml by diluting virus in MDDC media (without cytokines or polybrene) to a final volume normalized to controls. Innate and inflammatory stimuli were used at the indicated concentrations: poly(I:C) (InvivoGen, 10 μg/ml); LPS (List Biological Laboratories, INC, 1 ng/ml).
Flow cytometry
Infected or stimulated MDDCs were washed with phosphate buffered saline (PBS, Corning) and then exposed to LIVE/DEAD violet (ThermoFisher) in PBS for 15 min at 4°C in the dark. Cells were either simultaneously stained for surface markers or first washed with PBS and then stained in FACS buffer containing 1% Bovine Serum Albumin (BSA, Roche) and 1mM EDTA in PBS for 15-30 min in the dark at 4°C. Cells were then washed with PBS and fixed with 0.5% paraformaldehyde (Electron Microscopy Sciences) diluted in PBS. Cells were either sorted on a FACSAria II or data was acquired on an LSR II flow cytometer (BD Biosciences). Data were then analyzed using FlowJo software (FlowJo LLC).
RNA-seq
MDDCs from multiple donors were infected or stimulated as indicated above, stained with anti-human CD86 and a live-dead viability dye and then sorted into serum on a FACSAria II (BD Biosciences). Cells were lysed in TRIzol reagent (Thermo Fisher), RNA was isolated according the manufacturer’s instructions, and then samples were submitted to HudsonAlpha Institute for Biotechnology (https://hudsonalpha.org/) for library preparation and RNA-sequencing.
50bp-length single-end sequences were aligned to the human genome (hg38) using STAR version 2.4.2a. Samtools 0.1.19 was used to filter alignments to a MAPQ score threshold of 30. Counts per gene were called using featureCounts version 1.4.6 and gencode v24 genome annotation. Samtools 0.1.19 was used to filter alignments to a MAPQ score threshold of 30. We confirmed the presence of HIV-1 or HIV-2 sequences in infected samples yet these sequences were not taken into account during differential expression analysis. Differential expression analysis was performed separately for each RNA-seq experiment using DESeq2 version 1.10.1 and R version 3.2.3, using time and treatment together as the design parameter. Genes were considered differentially expressed in a given comparison if the FDR-adjusted p value was below 0.1.
ATAC-seq
MDDCs from three unique donors were infected in the presence of Vpx to match infection conditions as performed for RNA-seq at 2, 8, 24, and 48 h after infection. Cells were stained for LIVE/DEAD violet and CD86 to assess activation status and sorted on a FACSAria II (BD Biosciences) according to the gating strategy established for RNA-seq. 50,000 sorted MDDCs from each condition were immediately prepped for ATAC-seq (Buenrostro et al., 2013). Cells were pelleted by spinning at 500 g for 5 min at 4°C, resuspended in cold PBS, and then centrifuged again at 500 g for 5 min at 4°C. Cell pellets were lysed in cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1% IGEPAL CA-630) and immediately spun at 500 g for 10 min at 4°C. The nuclear pellet was used directly for the transposition reaction by resuspending in a reaction mix (20 μL 2 × TD buffer, 2 μL Tn5 transposase (Nextera DNA Library Prep Kit, Illumina) and 18 μL nuclease-free water). Transposition was performed at 37°C for 30 min and then samples were immediately processed using a MinElute kit (QIAGEN) to isolate DNA.
To generate ATAC-seq libraries for sequencing, DNA fragments from MinElute samples were amplified by PCR using NEBNext PCR master mix (New England Biolabs) and custom Nextera primers (Buenrostro et al., 2013) (Table S7) using the following conditions: 72°C for 5 min; 98°C for 30 s; and then cycling at 98°C for 10 s, 63°C for 30 s and 72°C for 1 min. To stop amplification before saturation in order to reduce bias, we monitored the PCR reaction using SYBR Green (Thermo Fisher) during qPCR. Samples were cycled a total of 12-15 times. Libraries were sequenced on a NextSeq 500 (Illumina) using a 150 cycle high output kit. Unique sequence read pairs were aligned to the human genome (hg38) using bowtie2 (2.2.3), filtered based on mapping score (MAPQ > 30, Samtools (0.1.19)), and duplicates removed (Picard version 1.120). Only pairs that aligned uniquely and concordantly to non-mitochondrial human chromosomes were retained.
For establishing the network Prior, a merged ATAC-seq Sam file was generated by combining all ATAC-seq conditions. Peak calling was performed with Peakdeck version 1.1 (parameters -bin 160, -STEP 25, -back 5000, -npBack 10000) using the start and end locations of the pairs to define fragment lengths, with a p value of 1e-4 as the cutoff, outputting 87,681 unique, non-overlapping, high-confidence peaks. Putative binding events were discovered by finding motif occurrences using HOCOMOCO version 10 (Kulakovskiy et al., 2013) as the motif database, with position weight matrices for 601 of 640 human TFs discovered using FIMO (Memesuite version 4.10.1) with a p value cutoff of 1e-4. When scored against our gold standards (see below) HOCOMOCO performed better than the curated CisBP 2.0 motif database (Weirauch et al., 2014) composed of JASPAR (Mathelier et al., 2016), TRANSFAC (Matys et al., 2006), and other motif collections, possibly due to low redundancy in motif position weight matrices. Bedtools (version 2.25) was used to generate a Markov model of order 1 from all peak intervals to use as a background peak file (parameter “–bgfile”). For each TF, the target set was further refined by filtering out binding sites below the top quartile, ranked by the FIMO p value. TF binding sites were linked to a target gene if the peak fell within a 1kb window of the gene body, creating a Prior matrix with 601 columns and 24909 rows (Table S4). Entries at row “I” column “J” were set to 1 to denote a putative regulatory event between TF “i” and target gene “j.” All other entries were set to 0, which penalizes, but does not completely prevent a regulatory edge connecting “i” and “j.”
Expression Normalization
Expression data was normalized using DESeq2 to remove the dependence of the variance on the mean. We explored linear models for batch correction such as ComBat and limma, but these models were ruled out as they superimpose each experiment’s center of mass - essentially placing different experiments on top of one another in PCA space—experiments which should have unique, nonoverlapping centers of mass (due to having different ratios of stimulus conditions as compared to mock conditions). Thus, we used DESeq2 rlog- transformed expression data to normalized gene expression values relative to mock conditions for each time point and for each donor by doing a vector subtraction. In our time series RNA-seq experiment, 3 samples were identified as outliers in gene expression biclusters from DESeq2 analysis (noted in Metadata, Table S1), consistently clustering away from the rest of the 93 samples across nearly all condition contrasts in PCA plots. Since network inference is sensitive to outliers due to a z-scoring step, and because follow-up RNA-seq experiments under the same conditions supported the idea that these samples were outliers, these 3 samples were removed from the network inference procedure.
Estimation of TF activities
The network inference methodology used here builds directly upon our previous platform, the Inferelator, in which the log gene expression is modeled as the linear combination of TF predictor variables (Bonneau et al., 2006). Earlier versions of the inferelator model used the TF expression as the predictor, but current methods use a latent variable, the estimated TF activity, as the predictor (Greenfield et al., 2013). The activity of a TF is inferred from the changes of the TF’s putative target genes and has been shown to be able to uncover more known relationships between TFs and target genes than using TF expression as the predictor (Arrieta-Ortiz et al., 2015).
In the Inferelator, we define the equation for inferring a network as X = BA. “X” is the gene expression matrix, “A” is the transcription factor activity matrix, and “B” is the betas matrix, which can also be thought as the connectivity network. If beta is found to be nonzero, then we have inferred an association between the gene and the TF. This linear equation appears straightforward, but it cannot be immediately solved because there are two unknowns, both “B,” the network beta value, and “A,” the TF activity. We therefore used a two-step process. First, we solved for “A,” using the Prior network, with the equation X = PA. In this equation, the Prior network “P” is known. However, because the confidence in any one of its chromatin accessibility derived entries is low, we randomly left out half of the entries of P, and solved the linear equation in 400 iterations, deriving 400 estimates for the activities, “A.” We solved the inverse problem X = PA by multiplying “X” by the pseudoinverse of the Prior matrix, “P.” The second step of the process is to solve the linear regression problem X = BA, with “B” as the unknown, which was done as described below using stability selection with elastic net regularization.
Network inference with subsampled Priors
Since the Prior matrix was built with thresholding at fixed p values, both at the level of peak finding and the level of motif binding, any single entry in the Prior matrix cannot be considered accurate. In order to improve computational reproducibility, we use a subsampling strategy, which was recently pioneered in a different context to increase prediction accuracy when assessing whether pathway perturbations hold true across different animal species (Hafemeister et al., 2015). This subsampling strategy decreases the density of the Prior matrix, which is a desideratum when the number of targets per TF in the Prior is more than 2000 on average. Therefore, for each of the 400 computational estimations of the transcription factor activity, we sampled 50% of the nonzero entries in the Prior without replacement. This enabled TFs with similar motif preferences and subtle differences in target sets to be teased apart over the course of many Inferelator runs. For example, in a single Inferelator run, a number of different IRF family members could be assigned at random to have undue emphasis on IFN and ISG expression, since many IRFs overlap in motif preference. In some extreme single run cases, due to inherent randomness in the inference procedure, no IRFs were assigned to IFNB1 or IFNL1. In contrast, after multiple runs where estimated TF activities were subsampled, the ATAC-seq-based network correctly predicted a strong influence of IRF3 on IFNB1, IFNL1, and core ISGs target sets.
Ensemble Model and AUPR curves
The 400 individual models that were generated by the inferelator were combined by ranking each interaction by summed beta values to generate an ensemble network (EN-ATAC x400). While this ensemble model predicted over 2 million interactions, we have provided a table to accompany this manuscript that lists the top 500,000 edges, selected based on absolute value of the beta sign (Table S5). Networks of this size cannot be reasonably visualized or efficiently analyzed by current computational resources, so for this work we chose to display the top 75,000 edges in the ensemble network. At this choice of a cutoff in ranked interactions, there are 542 TFs assigned as regulators for 21862 targets. This ranking was used to compute an area under the precision-recall curve (AUPR) by validating the predicted interactions against “gold standard” TF-to-gene-target connections that have been reported in the literature.
Proportional Venn Diagrams
Venn diagrams were generated using BioVenn (Hulsen et al., 2008). IRFs 1, 3, 5, 7, 8, and 9 and the NF-κB family members RELA, RELB, REL, NFKB1, and NFKB2 are known to be important for primary and secondary IFN responses and influence ISG expression, so we determined whether the targets of these IRFs and the NF-κB family members (Figure S3E), or specific targets of IRF3 that were predicted by the EN-ATAC x400 network (75k edges) or the BBSR network (~125k edges) (Figure S3F) overlapped with a set of “core” mammalian ISGs (Shaw et al., 2017).
Hypergeometric tests for TF enrichment
With the network model established, we can then ask whether any given Transcription Factor is associated with differential gene expression through its network targets. For every contrast, we used the hypergeometric test for enrichment to query whether the set of differentially expressed genes for a given contrast was enriched for the targets of any of the transcription factors. We used a Bonferroni correction, dividing by the number of transcription factors, when estimating these p values. For finding a global ranking of enrichment across multiple differential gene expression contrasts, the weighted z-method for combining probabilities was used with equal weights following a previously defined method (Whitlock, 2005).
Modularity and similarity K-means clustering
In order to interpret the results of the network inference, which yielded a network with 75,000 edges, we asked whether there existed large-scale clusters in the networks. To this end we used the python-louvain clustering module, which implements the modularity clustering algorithm, maximizing an objective function that sums edges within communities while subtracting edges between communities (Blondel et al., 2008). Due to the stochasticity of this algorithm, where the modularity score and even the number of clusters varies with each run, we repeated the run until convergence, which occurred on the order of a thousand runs. This computational approach allowed us not only to create clusters for large-scale communities, but also build a dendogram of pairwise similarities between every gene in the network, where the pairwise entry corresponds to the number of times in the thousand runs that the two genes were clustered together. This dendogram allowed us to identify “unclustered” nodes in the network: genes that do not consistently cluster with any of the main groups. The dendogram was split into 500, which resulted in 10 major clusters, defined as a cluster with more than 1% of the total gene set. These clusters were then labeled with identifiers from Enrichr pathway analysis. For complete pathway analysis results for the top 10 clusters, please see:
Cluster 1 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=63e7351cf48039d06d31d38ef57d6820
Cluster 2 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=f1f6b5d0849162056eceda2ad94ccf2c
Cluster 3 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=cf8ef3f0a7b3dcc1fe955143dfd64c57
Cluster 4 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=6e7b1a6ad9531df258d054af8a0fa52b
Cluster 5 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=d145ebd559a7567ab49efc93d82f178c
Cluster 6 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=0667b6ade824cdb73caba77d360ce8cb
Cluster 7 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=71b1b78f3d963438cb893c008c5bdd46
Cluster 8 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=59396b1d18359cc4aa6fecffc4044fd6
Cluster 9 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=e41b8f52efcf611c64467d84dfdf207c
Cluster 10 https://amp.pharm.mssm.edu/Enrichr/enrich?dataset=6181b245abf99ba27c0b8c822688b424
Network Visualization
Network visualization software Gephi Version 0.9.1 (Bastian et al., 2009) was used to visualize the network using the Force Atlas2 layout algorithm (Jacomy et al., 2014). For visualizing smaller network components with louvain clustering, we used a jp_gene_viz visualization tool developed at the Simons Foundation. This tool is publicly accessible online using Binder as a resource to complement this publication (https://mybinder.org/v2/gh/flatironinstitute/dc_network/master).
Instructions for Visualizing the Gephi-formatted MDDC HIV-Response Network
Download the Gephi open graph visualization platform from the following web address: https://gephi.org/. Download the EN-ATAC x400 final network from the following link: https://www.dropbox.com/s/4pzwdnn340ary49/EN_ATAC_400_network_final.gephi?dl=0. Once the Gephi software has been installed, start the program and open the downloaded EN_ATAC network file. The graph window will show the top 75,000 edges in the ensemble network, with clusters color-coded as indicated in the manuscript. Zoom in or out by scrolling up or down while the cursor is positioned in the graph window. The graph can be re-centered by clicking and holding the control button and then moving the cursor. To view a specific gene and its connections predicted in the top 75k edges, in the “Filters” window, click on the folder, “Topology,” and then double-click on “Ego Network.” In the bottom box that appears, labeled “Node ID,” enter your gene of interest, then click “OK” and click “Filter.” Differentially expressed genes for a given contrast can be highlighted by clicking the folder, “Partition,” in the same “Filters” window. Double-click on the contrast of interest and then in the bottom Partition Settings box, select the box for 1.0 to show those genes that are differentially expressed under the given condition. A number of different layouts and exploratory tools are also available. For more information, see https://gephi.org/users/.
Instructions for Launching the Jupyter Subnetworks Widget
Navigate to https://mybinder.org/v2/gh/flatironinstitute/dc_network/master and click “launch binder.” Once the binder has loaded, click on “notebooks.” First, unlock functionality of the notebooks by selecting “Decrypt data.ipynb.” Click the “Run” button on the browser tab that opens until the cell, “encrypt_files.decrypt_widget()” is reached. Enter “nick” into the key box and then click “decrypt.” When the decrypt script has finished running, navigate back to the Home tab to select the binder file “Visualize network.ipynb” or a network browswer with appended heatmaps, “Network and Heatmap.ipynb.” These binder files will open additional browser tabs. Click the “Run” button to run each step of the widget down to the cell that contains “L.gene_click().” The subnetwork browser will load after several seconds, which can then be explored by entering a gene or multiple genes into the “match” box and selecting a variety of layouts and regulatory connections.
Gene Set Enrichment Analysis (GSEA)
GSEA was performed by comparing each condition in the time series RNA-seq data to the corresponding mock time point. 36883 gene features for each condition were ranked by the signal to noise metric of GSEA and the analysis was performed using the standard weighted enrichment statistic against 3815 human gene sets contained in the Molecular Signatures Database that included all (H) Hallmark gene sets, (C2) curated gene sets, and (C3) motif gene sets. In a first pass analysis, the normalized enrichment score (NES) was calculated using 500 gene set permutations and once relevant gene sets were identified a more stringent statistical analysis was performed using 1000 phenotype permutations. Full GSEA results can be accessed through the following link: https://www.dropbox.com/sh/2u4psikt2tyz4r0/AAB4Re3YKtOA5lqPEEAwfR7Aa?dl=0
Immunoblotting
1 million cells were lysed in 100 μL of RIPA buffer (50mM Tris HCl, 150mM NaCl, 0.1% SDS, 0.5% DOC, 1% NP-40, Protease inhibitor (Roche; 1187358001)). Lysis was performed on ice for 30 minutes. Lysates were cleared by centrifugation at 8000 g for 8 minutes at 4°C, and 20 μl of Laemmli 6X (12% SDS, 30% Glycerol, 0.375M Tris-HCl pH6.8, 30% 2-mercaptoehtanol, 1% bromophenol blue) was added and samples were boiled at 95°C for 15min. Lysates were resolved on Criterion or 4%–20% Biorad precast SDS-PAGE gels and transferred on PVDF membrane. Membranes were saturated and proteins were blotted with antibodies (listed in Key Resources Table) in 5% non-fat dry milk, PBS, 0.1% Tween buffer. ECL signal was recorded on the ChemiDoc-XRS or ChemiDoc Touch Biorad Imager. Data was analyzed and quantified with the Image Lab software (Biorad).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Human CD86 (B7-2) PE-Cyanine5 | eBioscience | Cat# 15-0869-42; RRID:AB_11042003 |
| Anti-Human CD86 (B7-2) PE | Thermo Fisher | Cat#12-0869-42; RRID: AB_10732345 |
| Anti-Human CD11c-APC | Thermo Fisher | Cat# 17-0116-42; RRID:AB_1659668 |
| Anti-Human HLA-DR PE-Cyanine5 | BioLegend | Cat# 307607; RRID:AB_314685 |
| Anti-Human DC-SIGN PE | R&D Systems | Cat# FAB161P; RRID:AB_357064 |
| Anti-Human Actin | Millipore | Cat# MAB1501; RRID: AB_2223041 |
| Anti-Human Vinculin (Clone hVIN-1) | Sigma | Cat# V9264; RRID: AB_10603627 |
| Anti-Human PRDM1 Blimp-1 (Clone C14A4) | Cell Signaling Technology | Cat# 9115; RRID: AB_2169699 |
| Anti-Human NONO | Sigma | Cat# N8664; RRID: AB_10601034 |
| True Blot anti-mouse Ig HRP | Thermo Fisher | Cat# 18-8817-33; RRID: AB_10126892 |
| True Blot anti-rabbit Ig HRP | Thermo Fisher | Cat# 18-8816-31; RRID: AB_469528 |
| Bacterial and Virus Strains | ||
| Stbl3 competent E. coli | Thermo Fisher | Cat# C737303 |
| Biological Samples | ||
| Human leukocytes from normal donors | Bloodworks Northwest | Product code: 2490-03 http://www.bloodworksnw.org/ |
| Human leukocytes from normal donors | New York Blood Center | N/A |
| Human leukocytes from normal donors | ARUP Blood Services | N/A |
| Human leukocytes from normal donors | venipunctures approved by the Institut National de la Santé et de la Recherche Médicale ethics committee, Paris, France | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ficoll-Paque Plus | GE Healthcare | Cat# 17-1440-02 |
| Fetal Bovine Serum | Peak Serum Inc | (Lot#125N16) |
| Fetal Bovine Serum | GIBCO | (Lot #1982147) |
| Bovine Serum Albumin | Roche | Cat# 03116956001 |
| Recombinant Human IL-4 | Preprotech | Cat# 200-04 |
| Recombinant Human IL-4 | Miltenyi Biotech | Cat# 130-093-922 |
| Recombinant Human GM-CSF | Preprotech | Cat# 300-03 |
| Recombinant Human GM-CSF | Miltenyi Biotech | Cat# 130-093-867 |
| HEPES | Sigma | Cat# H3537 |
| Phosphate Buffer Saline (PBS) | Corning | Cat# 45000-446 |
| EDTA 0.5M | Sigma | Cat# E7889 |
| Penicillin-Streptomycin | Thermo Fisher | Cat# 15140-122 |
| 2-Mercaptoethanol | Thermo Fisher | Cat# 21985023 |
| L-glutamine | Thermo Fisher | Cat# 25030081 |
| MEM non-essential amino acids | Thermo Fisher | Cat# 11140050 |
| Sodium Pyruvate | Thermo Fisher | Cat# 11360070 |
| RPMI 1640 medium | Thermo Fisher | Cat# 11875-119 |
| DMEM | Thermo Fisher | Cat# 11965-118 |
| HAT supplement | Thermo Fisher | Cat# 21060-017 |
| poly-L-lysine hydrobromide | MP Biomedicals | Cat# 0219454405 |
| Polyinosinic-polycytidylic acid (poly(I:C)) high molecular weight | InvivoGen | Cat# tlrl-pic |
| 2′3′-cGAMP (cyclic [G(2′,5′)pA(3′,5′)p] | InvivoGen | Cat# tlrl-cga23 s |
| Recombinant Human Interferon-alpha 2 alpha | Thermo Fisher | Cat# 111001 |
| Recombinant Human Interferon-alpha 2 alpha | ImmunoTools | Cat# 11343506, (No CAS) |
| LPS – ultra pure from Salmonella minnesota R595 | List Biological Laboratories, INC. | Cat# 434 |
| R848 | Invivogen | Cat# tlrl-r848, CAS: 144875-48-9 |
| Poly(dG:dC) | Invivogen | Cat# tlrl-pgcn, CAS: 90385-88-9 |
| Cyclosporine A | Euromedex | Cat# S2286; CAS: 59865-13-3 |
| Am80 | Tocris | Cat# 3507 |
| BMS 195614 | Tocris | Cat# 3660 |
| all-trans retinoic acid (ATRA) | Tocris | Cat# 0695 |
| TRIzol reagent | Thermo Fisher | Cat# 15596026 |
| GlycoBlue Coprecipitate | Thermo Fisher | Cat# AM9516 |
| Polyethylenimine “Max,” (Mw 40,000) - High Potency Linear PEI | Polysciences, Inc. | Cat# 24765-1 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15713-S |
| Polybrene | Sigma | Cat# TR-1003-G |
| Puromycin | Invivogen | Cat# ant-pr-1 |
| TaqMan Fast Universal PCR Master Mix | Thermo Fisher | Cat# 4352402 |
| NEBNext® High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat# M0541S |
| SYBR Green I Nucleic Acid Stain | Thermo Fisher | Cat# S-7563 |
| cOmplete, EDTA-free, Protease inhibitor cocktails tablets | Roche | Cat# 11873580001 |
| Critical Commercial Assays | ||
| LIVE/DEAD® Fixable Violet Dead Cell Stain Kit | Molecular Probes | Cat# L34955 |
| Complete kit (Universal) KAPA Library Quantification Kits for Next-Generation Sequencing | Kappa Biosystems | Cat# KK4824 |
| Nextera DNA Library Prep Kit | Illumina Inc. | Cat# FC-121-1030 |
| Plasmid DNA Purification Nucleobond Xtra Maxi Kit | Takara | Cat# 740414.100 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69504 |
| Minelute Kit | QIAGEN | Cat# 28004 |
| Luciferase assay system | Promega | Cat# E4530 |
| LEGENDplex Human Type 1/2/3 Interferon Panel (5-plex) | Ozyme | Cat# BLE740396B |
| Deposited Data | ||
| RNA-seq on stimulated DCs | This paper | GEO: GSE125817 |
| ATAC-seq on stimulated DCs | This paper | GEO: GSE125918 |
| Experimental Models: Cell Lines | ||
| HEK293FT cells | Thermo Fisher | Cat# R70007, RRID:CVCL_6911 |
| HL116 reporter cells | (Uzé et al., 1994) | Gift from Sandra Pellegrini |
| Oligonucleotides | ||
| ATAC-seq sequencing primers | (Buenrostro et al., 2013) | (see Table S7) |
| TaqMan GAPDH assay | ThermoFisher | Cat# Hs02758991_g1 |
| TaqMan ISG15 assay | ThermoFisher | Cat# Hs01921425_s1 |
| TaqMan CXCL10 assay | ThermoFisher | Cat# Hs01124251_g1 |
| TaqMan cGAS/MB21D1 assay | ThermoFisher | Cat# Hs00403553_m1 |
| TaqMan eGFP assay | ThermoFisher | Cat# Mr04329676_mr |
| TaqMan IRF3 assay | ThermoFisher | Cat# Hs01547283_m1 |
| TaqMan HIVEP1 assay | ThermoFisher | Cat# Hs00172428_m1 |
| TaqMan KLF13 assay | ThermoFisher | Cat# Hs00429818_m1 |
| TaqMan RARA assay | ThermoFisher | Cat# Hs00940446_m1 |
| TaqMan STAT2 assay | ThermoFisher | Cat# Hs01013123_m1 |
| TaqMan IFNB1 assay | ThermoFisher | Cat# Hs01077958_s1 |
| TaqMan IFNL1 assay | ThermoFisher | Cat# Hs00601677_g1 |
| Recombinant DNA | ||
| HIV-1-GFP | (Manel et al., 2010) | HIV-1 GFP-reporter virus |
| HIV-2-GFP | (Manel et al., 2010) | HIV-2 GFP-reporter virus |
| LKO-GFP | (Manel et al., 2010) | Non-replicating lentivector |
| P86-HA | (Lahaye et al., 2013) | Mutagenized virus HIVac-2 |
| NL(AD8) | (Manel et al., 2010) | wild type HIV-1 molecular clone |
| JK7312AS | Gift from Beatrice Hahn (Lahaye et al., 2018) | wild type HIV-2 molecular clone |
| pSIV3+ (for production of virus-like particles containing Vpx) | (Mangeot et al., 2000) | SIVmac251 GenBank acc. no. M19499 |
| pLKO.1 – TRC cloning vector | Gift from David Root (Moffat et al., 2006) | Addgene plasmid #10878 |
| psPAX2 | Gift from Didier Trono | Addgene plasmid #12260 |
| pCMV-VSV-G | Gift from Bob Weinberg (Stewart et al., 2003) | Addgene plasmid #8454 |
| control shRNA | Sigma | SHC002 |
| LacZ shRNA | Sigma | TRCN0000072229 |
| NONO shRNA | Sigma | TRCN0000074562 |
| cGas shRNA | Sigma | TRCN0000149984 |
| IRF3 sh | (Johnson et al., 2018) | N/A |
| HIVEP1 sh1 | Sigma | TRCN0000235474 |
| HIVEP1 sh3 | Sigma | TRCN0000235476 |
| KLF13 sh1 | Sigma | TRCN0000016923 |
| KLF13 sh2 | Sigma | TRCN0000016925 |
| STAT2 sh1 | Sigma | TRCN0000364399 |
| STAT2 sh2 | Sigma | TRCN0000364400 |
| RARA sh1 | Sigma | TRCN0000275554 |
| RARA sh2 | Sigma | TRCN0000020370 |
| PRDM1 sh2 | Sigma | TRCN0000013609 |
| PRDM1 sh3 | Sigma | TRCN0000013610 |
| PRDM1 sh5 | Sigma | TRCN0000013612 |
| Software and Algorithms | ||
| R | N/A | https://www.r-project.org/ |
| Integrative Genomics Viewer (IGV) | N/A | http://software.broadinstitute.org/software/igv/ |
| FlowJo Version 8.7 | FlowJo LLC | https://www.flowjo.com/ |
| FlowJo Version 9.9.4 | FlowJo LLC | https://www.flowjo.com/ |
| LEGENDplex Software – Version 8.0 | LEGENDplex | http://www.vigenetech.com/LEGENDplex7.htm |
| Image Lab software – Version 5.2.1 | BioRad | http://www.bio-rad.com/fr-fr/product/image-lab-software?ID=KRE6P5E8Z |
| GraphPad Prism 6.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Enrichr | (Chen et al., 2013; Kuleshov etal., 2016) | http://amp.pharm.mssm.edu/Enrichr/ |
| Inferelator | (Bonneau et al., 2006) | N/A |
| Gephi | (Bastian et al., 2009) | https://gephi.org/ |
| BioVenn | (Hulsen et al., 2008) | http://www.biovenn.nl/ |
| Other | ||
| LS Columns | Miltenyi Biotec, Inc | 130-042-401 |
| SW 28 Ti Rotor, Swinging Bucket | Beckman | Cat# 342204 |
| Thinwall Polyallomer, Conical Tubes; Size:25 × 89 | Beckman | Cat# 358126 |
| 0.45 μm syringe filters | Corning | Cat# 28200-026 |
| QuadroMACS Separator | Miltenyi Biotec, Inc | 130-090-976 |
| CD14 MicroBeads, human | Miltenyi Biotec, Inc | 130-050-201 |
| LSR II Flow Cytometer | BD Biosciences | N/A |
| FACSAria II Cell Sorter | BD Biosciences | N/A |
| CFX96 thermal cycler | Biorad | Model T100 |
| Lightcycler 480 System | Roche | Product# 05015278001 |
Quantitative PCR
50,000 to 200,000 MDDCs were lysed in TRIzol reagent (Thermo Fisher) and then RNA was isolated following the manufacturer’s instructions with minor modifications. In brief, we performed two sequential chloroform extractions and added Glycoblue (Thermo Fisher) as a carrier prior to precipitation with isopropanol. RNA pellets were washed in 75% ethanol and resuspended in 20 μL of DNase- and RNase-free water. 500 μg of RNA was converted into cDNA using Superscript III (ThermoFisher). Quantitative PCR reactions were carried out using TaqMan primer probes (ABI) and TaqMan Fast Universal PCR Master Mix (ThermoFisher) in either a Lightcycler (Roche) or a CFX96 thermocycler (BioRad) in a volume of 10 μL according to the following cycling conditions: 50°C for 2 min, 95°C for 2 min, then 55 cycles each of 95°C for 3 s, to 60°C for 30 s, followed by 95°C for 5 s. A melting curve analysis was then performed going from 65°C to 95°C in 0.5°C intervals every 5 s. Data were plotted as expression relative to GAPDH x 1000.
IFNL1 Protein Quantification
IFNL1 protein concentrations were measured on supernatants from infected or treated MDDCs using a LEGENDplex Human Anti-Virus Response assay (BioLegend) according to the manufacturer’s protocol. Data were acquired on a BD FACSVerse (BD) and analyzed with LEGENDplex Software (BioLegend).
Bioassays for type I IFN
To quantify IFN activity from infected or stimulated cells we assayed supernatants with HL116 reported cells that contain firefly luciferase gene under control of the IFN-inducible 6-16 promoter (Uzé et al., 1994). Supernatants from treated or untreated MDDCs were transferred to 20,000 HL116 cells in 96 well plates. After 7 h, HL116 cells were lysed in passive lysis buffer and subsequently scored for luciferase activity using a luciferin-based method (Promega). Relative light units were converted to units per ml of IFN using a standard curve that was generated from serial dilutions of recombinant human IFNa2a, with HL116 cells responding in a linear range between 2 and 200 U/ml of IFN.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses incorporated into the computational implementation of network inference were performed as stated above in the STAR Methods section. Otherwise, statistical tests were performed as indicated in the figure legends or using Prism 6.0 (GraphPad) to calculate either a two-way ANOVA with Sidak’s multiple comparisons test or a two-tailed t test using paired samples. The number of unique donors for MDDC experiments is also listed in the figure legends, representing the number of biological replicates performed for a given experiment. Data reflects pooled data from multiple experiments where indicated.
DATA AND CODE AVAILABILITY
The RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) database under ID code GEO: GSE125817. The ATAC-seq data have been deposited under ID code GEO: GSE125918. Both can be accessed from the series code GEO: GSE125919.
Supplementary Material
Highlights.
Network inference reveals a transcriptional map of the innate immune response to HIV
Integrating cell-specific chromatin accessibility data improves network inference
Immune responses coincide with network enrichment of IRF, STAT, and NF-κB factors
RARA and PRDM1 are validated as additional regulators of the innate response to HIV
ACKNOWLEDGMENTS
This project was supported by the Howard Hughes Medical Institute (to D.R.L.) and the Helen and Martin Kimmel Center for Biology and Medicine (to D.R.L.), grants from the National Institutes of Health (F32AI093231 to J.S.J., R21AI084633 to D.R.L., and R01AI025032, R01AI032972, and U19AI100627 to A.A.), Sidaction (VIH2016126002 to N.M. and 17-1-AAE-11097-2 to N.M.), ANRS (ECTZ36691, ECTZ25472, and ECTZ71745 to N.M.), state funding from the Agence Nationale de la Recherche under the Investissements d’avenir program (ANR-10-IAHU-01 to M.M.M.), and the ATIP-Avenir INSERM program (to M.M.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.054.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahn YT, Huang B, McPherson L, Clayberger C, and Krensky AM (2007). Dynamic interplay of transcriptional machinery and chromatin regulates “late” expression of the chemokine RANTES in T lymphocytes. Mol. Cell. Biol 27, 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcántara-Hernández M, Leylek R, Wagar LE, Engleman EG, Keler T, Marinkovich MP, Davis MM, Nolan GP, and Idoyaga J (2017). High-Dimensional Phenotypic Mapping of Human Dendritic Cells Reveals Interindividual Variation and Tissue Specialization. Immunity 47, 1037–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, et al. (2009). Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 326, 257–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrieta-Ortiz ML, Hafemeister C, Bate AR, Chu T, Greenfield A, Shuster B, Barry SN, Gallitto M, Liu B, Kacmarczyk T, et al. (2015). An experimentally supported model of the Bacillus subtilis global transcriptional regulatory network. Mol. Syst. Biol 11, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, and Palucka K (2000). Immunobiology of dendritic cells. Annu. Rev. Immunol 18, 767–811. [DOI] [PubMed] [Google Scholar]
- Bastian M, Heymann S, and Jacomy M (2009). Gephi: an open source software for exploring and manipulating networks. In Proceedings of the Third International Conference on Weblogs and Social Media, Adar E, Hurst M, Finin T, Glance N, Nicolov N, and Tseng B, eds. (AAAI Press; ), pp. 361–362. [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. J. Stat. Mech 2008, P10008. [Google Scholar]
- Bönelt P, Wöhner M, Minnich M, Tagoh H, Fischer M, Jaritz M, Kavirayani A, Garimella M, Karlsson MC, and Busslinger M (2019). Precocious expression of Blimp1 in B cells causes autoimmune disease with increased self-reactive plasma cells. EMBO J. 38, e100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau R, Reiss DJ, Shannon P, Facciotti M, Hood L, Baliga NS, and Thorsson V (2006). The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 7, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X (2016). Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol 16, 35–50. [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier N, Mertins P, Artyomov MN, Shalek AK, Iannacone M, Ciaccio MF, Gat-Viks I, Tonti E, DeGrace MM, Clauser KR, et al. (2011). Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell 147, 853–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, et al. (2012). A validated regulatory network for Th17 cell specification. Cell 151, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. ; Cancer Genome Atlas Analysis Network (2018). The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L, Matson V, Flood B, Spranger S, and Gajewski TF (2017). Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 27, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham TJ, and Duester G (2015). Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol 16, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, et al. (2018). A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnewski P, Das S, Parigi SM, and Villablanca EJ (2017). Retinoic Acid and Its Role in Modulating Intestinal Innate Immunity. Nutrients 9, E68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks SG, Tracy R, and Douek DC (2013). Systemic effects of inflammation on health during chronic HIV infection. Immunity 39, 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delescluse C, Cavey MT, Martin B, Bernard BA, Reichert U, Maignan J, Darmon M, and Shroot B (1991). Selective high affinity retinoic acid receptor alpha or beta-gamma ligands. Mol. Pharmacol 40, 556–562. [PubMed] [Google Scholar]
- DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W, Marietta EV, Kasarda DD, Waldmann TA, Murray JA, et al. (2011). Coadjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature 471, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbarth SC (2019). Dendritic cell subsets in T cell programming: location dictates function. Nat. Rev. Immunol 19, 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez S, Tanaskovic S, Helbig K, Rajasuriar R, Kramski M, Murray JM, Beard M, Purcell D, Lewin SR, Price P, and French MA (2011). CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J. Infect. Dis 204, 1927–1935. [DOI] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, and Chen ZJ (2013). Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Revy P, Brousse N, Lepelletier Y, Folli C, Durandy A, Chambon P, and Dy M (2003). Retinoids regulate survival and antigen presentation by immature dendritic cells. J. Exp. Med 198, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Roach JC, Kennedy K, Hai T, Bolouri H, and Aderem A (2006). Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173–178. [DOI] [PubMed] [Google Scholar]
- Goubau D, Deddouche S, and Reis e Sousa C (2013). Cytosolic sensing of viruses. Immunity 38, 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudot C, Coillard A, Villani AC, Gueguen P, Cros A, Sarkizova S, Tang-Huau TL, Bohec M, Baulande S, Hacohen N, et al. (2017). Aryl Hydrocarbon Receptor Controls Monocyte Differentiation into Dendritic Cells versus Macrophages. Immunity 47, 582–596. [DOI] [PubMed] [Google Scholar]
- Goujon C, Jarrosson-Wuillème L, Bernaud J, Rigal D, Darlix JL, and Cimarelli A (2006). With a little help from a friend: increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(MAC). Gene Ther. 13, 991–994. [DOI] [PubMed] [Google Scholar]
- Granelli-Piperno A, Golebiowska A, Trumpfheller C, Siegal FP, and Steinman RM (2004). HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc. Natl. Acad. Sci. USA 101, 7669–7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield A, Madar A, Ostrer H, and Bonneau R (2010). DREAM4: Combining genetic and dynamic information to identify biological networks and dynamical models. PLoS ONE 5, e13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield A, Hafemeister C, and Bonneau R (2013). Robust data-driven incorporation of prior knowledge into the inference of dynamic regulatory networks. Bioinformatics 29, 1060–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, Romero R, Bilal E, Meyer P, Norel R, Rhrissorrakrai K, Bonneau R, and Tarca AL (2015). Inter-species pathway perturbation prediction via data-driven detection of functional homology. Bioinformatics 31, 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E, et al. (2018). TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46 (D1), D380–D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, and Latz E (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol 14, 463–477. [DOI] [PubMed] [Google Scholar]
- Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, and Skowronski J (2011). Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsen T, de Vlieg J, and Alkema W (2008). BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima N, Thompson JM, and Iwasaki A (2008). Dendritic cells and macrophages in the genitourinary tract. Mucosal Immunol. 1, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB, and Donlin LT (2014). Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A (2012). A virological view of innate immune recognition. Annu. Rev. Microbiol 66, 177–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, and Medzhitov R (2015). Control of adaptive immunity by the innate immune system. Nat. Immunol 16, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacomy M, Venturini T, Heymann S, and Bastian M (2014). ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS ONE 9, e98679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski A, Tiuryn J, and Prabhakar S (2016). Romulus: robust multistate identification of transcription factor binding sites from DNase-seq data. Bioinformatics 32, 2419–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JS, Lucas SY, Amon LM, Skelton S, Nazitto R, Carbonetti S, Sather DN, Littman DR, and Aderem A (2018). Reshaping of the Dendritic Cell Chromatin Landscape and Interferon Pathways during HIV Infection. Cell Host Microbe 23, 366–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jønsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, Egedal JH, Kjolby M, Mohammadi P, Prabakaran T, et al. (2017). IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun 8, 14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Zang TM, Rihn SJ, Zhang F, Kueck T, Alim M, Schoggins J, Rice CM, Wilson SJ, and Bieniasz PD (2016). Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell Host Microbe 20, 392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller AD, and Maniatis T (1991). Identification and characterization of a novel repressor of beta-interferon gene expression. Genes Dev. 5, 868–879. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Gregersen PK, and Diamond B (2013). Regulation of dendritic cell activation by microRNA let-7c and BLIMP1. J. Clin. Invest 123, 823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko YA, Chan YH, Liu CH, Liang JJ, Chuang TH, Hsueh YP, Lin YL, and Lin KI (2018). Blimp-1-Mediated Pathway Promotes Type I IFN Production in Plasmacytoid Dendritic Cells by Targeting to Interleukin-1 Receptor-Associated Kinase M. Front. Immunol 9, 1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy IV, Medvedeva YA, Schaefer U, Kasianov AS, Vorontsov IE, Bajic VB, and Makeev VJ (2013). HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 41, D195–D202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Ségéral E, Yatim A, Emiliani S, Schwartz O, and Benkirane M (2011). SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, El Marjou A, Lacabaratz C, Lelièvre JD, and Manel N (2013). The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 39, 1132–1142. [DOI] [PubMed] [Google Scholar]
- Lahaye X, Gentili M, Silvin A, Conrad C, Picard L, Jouve M, Zueva E, Maurin M, Nadalin F, Knott GJ, et al. (2018). NONO Detects the Nuclear HIV Capsid to Promote cGAS-Mediated Innate Immune Activation. Cell 175, 488–501. [DOI] [PubMed] [Google Scholar]
- Lee MN, Roy M, Ong SE, Mertins P, Villani AC, Li W, Dotiwala F, Sen J, Doench JG, Orzalli MH, et al. (2013). Identification of regulators of the innate immune response to cytosolic DNA and retroviral infection by an integrative approach. Nat. Immunol 14, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Marié IJ, and Durbin JE (2011). Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol 1, 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madar A, Greenfield A, Vanden-Eijnden E, and Bonneau R (2010). DREAM3: network inference using dynamic context likelihood of relatedness and the inferelator. PLoS ONE 5, e9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado RA, and von Andrian UH (2010). How tolerogenic dendritic cells induce regulatory T cells. Adv. Immunol 108, 111–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, and Littman DR (2010). A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 467, 214–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeot PE, Nègre D, Dubois B, Winter AJ, Leissner P, Mehtali M, Kaiserlian D, Cosset FL, and Darlix JL (2000). Development of minimal lentivirus vectors derived from simian immunodeficiency virus (SIVmac251) and their use for gene transfer into human dendritic cells. J. Virol 74, 8307–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, et al. (2016). JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 44 (D1), D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. (2006). TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al. (2017). Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraldi ER, Pokrovskii M, Watters A, Castro DM, De Veaux N, Hall JA, Lee JY, Ciofani M, Madar A, Carriero N, et al. (2019). Leveraging chromatin accessibility for transcriptional regulatory network inference in T Helper 17 Cells. Genome Res 29, 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Fairfax KA, and Kallies A (2007). BLIMP1 guides the fate of effector B and T cells. Nat. Rev. Immunol 7, 923–927. [DOI] [PubMed] [Google Scholar]
- Ramsey SA, Klemm SL, Zak DE, Kennedy KA, Thorsson V, Li B, Gilchrist M, Gold ES, Johnson CD, Litvak V, et al. (2008). Uncovering a macrophage transcriptional program by integrating evidence from motif scanning and expression dynamics. PLoS Comput. Biol 4, e1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman A, Hemmert KC, Ochi A, Jamal M, Henning JR, Barilla R, Quesada JP, Zambirinis CP, Tang K, Ego-Osuala M, et al. (2013). Role of fatty-acid synthesis in dendritic cell generation and function. J. Immunol 190, 4640–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander J, Schmidt SV, Cirovic B, McGovern N, Papantonopoulou O, Hardt AL, Aschenbrenner AC, Kreer C, Quast T, Xu AM, et al. (2017). Cellular Differentiation of Human Monocytes Is Regulated by Time-Dependent Interleukin-4 Signaling and the Transcriptional Regulator NCOR2. Immunity 47, 1051–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, Levin D, Wijeyesinghe S, Makamdop KN, del Prete GQ, et al. (2014). Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 511, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AE, Hughes J, Gu Q, Behdenna A, Singer JB, Dennis T, Orton RJ, Varela M, Gifford RJ, Wilson SJ, and Palmarini M (2017). Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol. 15, e2004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siahpirani AF, and Roy S (2017). A prior-based integrative framework for functional transcriptional regulatory network inference. Nucleic Acids Res. 45, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvin A, Yu CI, Lahaye X, Imperatore F, Brault JB, Cardinaud S, Becker C, Kwan WH, Conrad C, Maurin M, et al. (2017). Constitutive resistance to viral infection in human CD141+ dendritic cells. Sci. Immunol 2, eaai8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smed-Sörensen A, Loré K, Vasudevan J, Louder MK, Andersson J, Mascola JR, Spetz AL, and Koup RA (2005). Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J. Virol 79, 8861–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari I, and Nagy L (2008). Nuclear receptor signalling in dendritic cells connects lipids, the genome and immune function. EMBO J. 27, 2353–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry C, and Amigorena S (2001). The cell biology of antigen presentation in dendritic cells. Curr. Opin. Immunol 13, 45–51. [DOI] [PubMed] [Google Scholar]
- Uzé G, Di Marco S, Mouchel-Vielh E, Monneron D, Bandu MT, Horisberger MA, Dorques A, Lutfalla G, and Mogensen KE (1994). Domains of interaction between alpha interferon and its receptor components. J. Mol. Biol 243, 245–257. [DOI] [PubMed] [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, et al. (2017). Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada J, and Makino H (2016). Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol 12, 13–26. [DOI] [PubMed] [Google Scholar]
- Walker B, and McMichael A (2012). The T-cell response to HIV. Cold Spring Harb. Perspect. Med 2, a007054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GP, Ciuffi A, Leipzig J, Berry CC, and Bushman FD (2007). HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 17, 1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock MC (2005). Combining probability from independent tests: the weighted Z-method is superior to Fisher’s approach. J. Evol. Biol 18, 1368–1373. [DOI] [PubMed] [Google Scholar]
- Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, Patterson A, Smith AM, Chang CH, Liu Z, et al. (2016). Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity 44, 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Schneider M, Seifried J, Soonthornvacharin S, Akleh RE, Olivieri KC, De Jesus PD, Ruan C, de Castro E, Ruiz PA, et al. (2015). PQBP1 Is a Proximal Sensor of the cGAS-Dependent Innate Response to HIV-1. Cell 161, 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, Gray EE, Zhen A, Wu NC, Yamada DH, et al. (2015). Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 163, 1716–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) database under ID code GEO: GSE125817. The ATAC-seq data have been deposited under ID code GEO: GSE125918. Both can be accessed from the series code GEO: GSE125919.