

Abstract
Fibroblast growth factor-2 (FGF2) protects the heart from ischemia–reperfusion (I-R) injury via a vast network of protein kinases. In the heart, downstream effectors of these FGF2-triggered signals have not yet been identified. It is hypothesized that nitric oxide (NO) signaling and ATP-sensitive potassium (KATP) channel activity are key effectors of protein kinases activated by FGF2-mediated cardioprotection. Hearts with a cardiac-specific overexpression of FGF2 (FGF2 Tg) were subjected to I-R injury in the absence or the presence of selective inhibitors of NO synthase (NOS) isoforms or sarcolemmal (sarcKATP) and mitochondrial (mitoKATP) KATP channels. Multiple NOS isoforms are necessary for FGF2-mediated cardioprotection, and nitrite levels are significantly reduced in FGF2 Tg hearts upon inhibition of protein kinase C or mitogen-activated protein kinases. Likewise, sarcKATP and mitoKATP channels are important for cardioprotection elicited by endogenous FGF2. These findings suggest that FGF2-induced cardioprotection occurs via protein kinase-NOS pathways as well as KATP channel activity.
Keywords: Growth factor, nitric oxide synthase, extracellular signal regulated kinase (ERK), p38, PKC, transgenic mouse, ischemia–reperfusion
Introduction
Since it was established that brief periods of ischemia are capable of protecting the heart against subsequent injury (Murry et al. 1986), a potential therapeutic intervention for patients with coronary (ischemic) heart disease that can mimic this phenomenon has been the subject of many investigations (Kharbanda 2010). Among these potential therapies is fibroblast growth factor-2 (FGF2).
FGF2 has been implicated in multiple protective pathways related to ischemia–reperfusion (I-R) injury including cardioprotection, the inflammatory response, angiogenesis, and vascular remodeling (Detillieux et al. 2003; Liao 2008). A number of pharmacological and in vitro studies have suggested that FGF2 can act directly on cardiomyocytes (Padua et al. 1995; Jiang et al. 2002; Jiang et al. 2004) to maintain the integrity and function of the myocardium during I-R injury. Our laboratory has demonstrated in an ex vivo mouse model of I-R injury that cardiac overexpression of human FGF2 decreases ischemic damage, as measured by the recovery of post-ischemic ventricular function and myocardial infarct size (House et al. 2003). These cardioprotective effects of FGF2 have been described by findings in other laboratories as well (Hampton et al. 2000; Sheikh et al. 2001; Jiang et al. 2002). Additionally, previous findings from our laboratory (House et al. 2003, 2005, 2007) and others (Jiang et al. 2002) have shown involvement of both the mitogen-activated protein kinase (MAPK) and protein kinase C (PKC) pathways in FGF2-induced cardioprotection. These kinase pathways activated by FGF2 are also involved in other modes of cardioprotection (Barancik et al. 2000; Strohm et al. 2000; Lips et al. 2004; Budas et al. 2007). However, additional pathways, which may be independent of or interact with kinase signaling, may also play a role in the mediation of the protective effect of FGF2.
Certain biological actions, in particular vascular and angiogenic functions, of FGFs have been shown to occur through nitric oxide (NO) signaling (Huang et al. 1997; Tiefenbacher and Chilian 1997; Cuevas et al. 1999; Hampton et al. 2000; Parenti et al. 2001) or ATP-sensitive potassium (KATP) channel activity (Cuevas et al. 1991; Boussairi and Sassard 1994; Tiefenbacher and Chilian 1997; Kajita et al. 2001). Hampton and colleagues determined that exogenously FGF2 applied prior to ischemia was able to protect the heart from post-ischemic dysfunction in hearts in an inducible nitric oxide synthase (iNOS)-dependent manner (Hampton et al. 2000). However, it remains unclear whether the reduction in infarct size and improvement in cardiac function mediated by endogenously expressed FGF2 in ischemic cardiomyocytes relies on the activity of any species of NOS.
Vascular studies have implicated KATP channel activation in the signal transduction and biological activity of FGF1 and FGF2 in angiogenesis and vasodilation (Cuevas et al. 1991; Boussairi and Sassard 1994; Tiefenbacher and Chilian 1997; Kajita et al. 2001). However, there is no evidence, to date, as to the involvement of KATP channels (whether mitochondrial or sarcolemmal) in the cardioprotective effect mediated by FGF2.
In this study, mouse hearts with a cardiac-specific overexpression of human FGF2 were subjected to ischemia and reperfusion in the presence and the absence of NOS inhibitors selective against iNOS and neuronal (nNOS) as well as inhibitors selective for sarcolemmal (sarcKATP) or mitochondrial (mitoKATP) KATP channels, and cardiac function, infarct size, and NO release were measured to test the hypothesis that cardioprotection induced by overexpression of FGF2 is linked to NOS-associated pathways and sarcKATP and mitoKATP channels during I-R injury.
Methods
Mice were housed in a pathogen-free facility and handled in accordance with standard use protocols, animal welfare regulations, and the NIH Guide for the Care and Use of Laboratory Animals. All protocols were approved by the University of Cincinnati Institutional Animal Care and Use Committee. Non-transgenic (NTg) mice and mice with a cardiac-specific overexpression of all FGF2 isoforms (FGF2 Tg MHC20 and FGF2 Tg MHC 25), on a FVB/N background, were randomly assigned to the studies. A total of 17 mice were excluded from the I-R injury or pharmacological inhibition studies. For each group, 11 NTg mice and 15 FGF2 Tg mice completed the I-R studies or the I-R +pharmacological NOS isoform inhibition studies, six to eleven mice of each genotype completed the I-R +pharmacological protein kinase inhibition studies or the I-R +pharmacological KATP channel inhibition studies. Exclusion from any I-R study was based on the signs of aortic or pulmonary vein leak in the working heart preparation. Aortic leak was represented as an aortic pressure <60 mmHg on Langendorff, retrograde perfusion mode. Pulmonary vein leak was demonstrated as an aortic flow <2.0 ml/min, low (<4 mmHg) atrial pressure, and a perfusate gas pO2 >380 mmHg or a visible leak (i.e. hole in ventricle or atrium) in the heart.
Model of low-flow global ischemia in isolated work-performing heart preparation
After a 30 min equilibration period at a basal cardiac workload of 250 (ml/min)*mmHg, the venous return was quickly (<30 s) reduced by 1 ml increments to a flow of 1 ml/min for 60 min to elicit a low-flow, global ischemic insult, leading to irreversible injury (i.e. cardiac dysfunction and myocardial infarction). Coronary flow was reduced by approximately 90% of its original rate (from 2.3 to 0.2 ml/min) during this global ischemia, mimicking a severe coronary artery stenosis. To maintain the work demand on the heart during ischemia, the heart was paced 10–15 beats per minute above its intrinsic heart rate. After 60 min of low-flow ischemia, venous return was quickly (<30 s) increased by 1 ml increments to a flow of 5 ml/min, and reperfusion occurred for 120 min. Functional data, perfusate gases, and coronary effluent were obtained at designated time points of baseline, low-flow ischemia, and reperfusion. Indices of diastolic and systolic function (e.g. end-diastolic pressure, half relaxation time, and time to peak pressure), percent recovery of function, and left ventricular developed pressure as well as coronary flow changes and myocardial oxygen consumption were assessed at baseline, ischemia, and reperfusion.
Pharmacological assessment of NO signaling
NG-nitro-l-arginine methyl ester hydrochloride (L-NAME), a non-selective NOS inhibitor (Huang et al. 1997), N6-(1-iminoethyl)-l-lysine hydrochloride (L-NIL), a NOS inhibitor selective for the inducible NOS isoform with minimal effects on other NOS isoforms (iNOS) (Moore et al. 1994), 2-(1-(3-Dimethylaminopropyl)-1H-indol-3-yl)-3-(1H-indol-3-yl)maleimide-HCl (bisindolylmaleimide also known as GF109203x or GFX) (GFX), a nonspecific inhibitor of multiple PKC isoforms at their catalytic ATP-binding sites (Bit et al. 1993), or SB203580 hydrochloride (4-(4-fluorophenyl)-2-(4-methyl-sulfinylphenyl)-5-(4-pyridyl)-1H-imidazole), a p38 inhibitor (Cuenda et al. 1995) were dissolved in Milli-Q water. 1-(2-Trifluoromethyl-phenyl) imidazole (TRIM), a NOS inhibitor selective for the nNOS isoform with some selectivity for iNOS and no activity for endothelial NOS (eNOS) (Handy and Moore 1997) was dissolved in ethanol. U0126 (1,4-diamino-2,3-dicyano-1, 4-bis[2-aminophenylthio]butadiene), an inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK)1/2, upstream kinases of ERK activation (Favata et al. 1998) was dissolved in dimethyl sulfoxide (DMSO). All drugs were diluted in Kreb’s solution to obtain final concentrations of L-NAME, 400 μM; L-NIL, 400 μM; TRIM, 100 μM; GFX, 1 μM; U0126, 2.5 μM; SB203580, 2 μM.
Concentrations for NOS or PKC or MAPK inhibitors were chosen because these drug concentrations blocked NOS or protein kinase activation, respectively, as previously published (Martiny-Baron et al. 1993; Cuenda et al. 1995; Handy and Moore 1997; Favata et al. 1998; Hampton et al. 2000; Zhao et al. 2000) without any adverse effects (i.e. cardiac arrhythmias or increased left atrial pressure) which were observed at higher drug levels (unpublished observations with 10 μM GFX, 5 μM U0126, 5 μM SB203580, and 400 μM TRIM). These pharmacological agents were administered for the last 15 min of baseline and the first 15 min of ischemia and again for the last 15 min of ischemia and the first 15 min of reperfusion (Figure 1). Vehicle controls were also performed. Previous work by Kardami and colleagues (Jiang et al. 2004) demonstrated that FGF2 protects the heart against myocardial cell injury/death when it is given prior to or following ischemia; therefore, the timing of inhibitor treatment was for the purpose of blocking FGF2 signaling at key time points of the I-R protocol.
Figure 1.
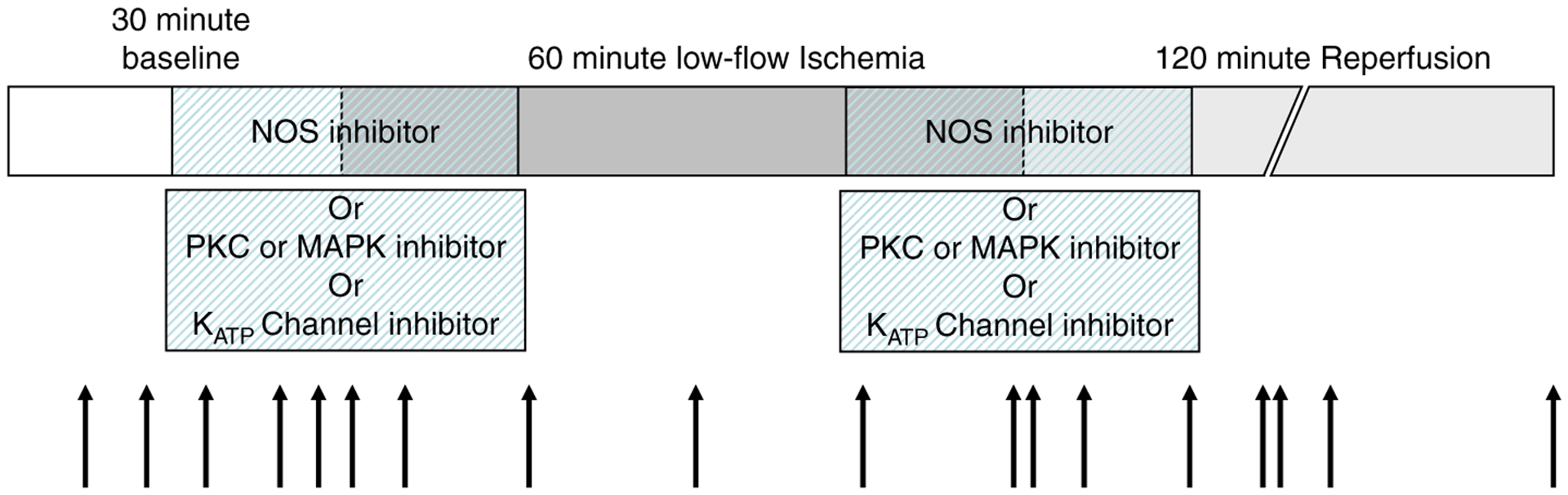
Schematic of low-flow I-R protocol. Functional measurements and perfusate gases were taken at baseline, low-flow ischemia, and reperfusion (black arrows). Inhibitors were administered 15 min prior to ischemia and/or during the first 15 min of ischemia as well as during the last 15 min of ischemia and first 15 min of reperfusion (hashed bars).
Inhibition of cardiac KATP channels
HMR1098 (10 μM, a kind gift from Sonofi-Aventis, Frankfurt, Germany) was used to inhibit sarcolemmal (sarc) KATP channels. 5-Hydroxydecanoic acid (5-HD, 100 μM) was used to inhibit mitochondrial (mito) KATP channels. Concentrations for KATP channel inhibitors were chosen because these drug concentrations blocked sarcKATP or mitoKATP channels as previously published (Grover and Garlid 2000; Sato et al. 2000) without any adverse effects, which were observed at higher drug levels (unpublished observations with 100 μM HMR1098 and 300 μM 5-HD). To distinguish possible roles for cardiac KATP channels as early triggers or late effectors of cardioprotection initiated by FGF2, these pharmacologic agents were administered at one of the two time points during the I-R protocol: for the last 15 min of baseline and the first 15 min of ischemia, or for the last 15 min of ischemia and the first 15 min of reperfusion (Figure 1). Vehicle controls were also performed.
Measurement of myocardial infarct size
Infarct size was determined by the histochemical stain, 2,3,5-triphenyltetrazolium chloride (TTC), which delineates viable vs. necrotic tissue as described previously by our laboratory (House et al. 2003). After the I-R study, NTg and FGF2 Tg hearts were perfused with warmed 1% TTC stain (pH 7.4) via the aortic cannula. The hearts were frozen, sliced transversely, digitally photographed, and weighed. The area at risk (whole heart) and the infarct zone were determined by computer morphometry (NIH imaging software, 1.61 version). Infarct size was depicted as a percent of the entire heart.
Measurement of NO release into coronary effluent
To assess the amount of NO released in response to cardiac-specific overexpression of FGF2 in the context of I-R injury, coronary effluent was collected from NTg and FGF2 Tg hearts subjected to I-R injury in the absence or the presence of NOS inhibitors. Additionally, to determine the interaction between protein kinases (e.g. PKC and MAPK) and NOS pathways in FGF2-induced cardioprotection, coronary effluent samples were collected at designated time points from NTg and FGF2 Tg hearts subjected to I-R injury in the absence or the presence of protein kinase inhibitors. Time points collected included: every 2 min for the last 10 min of baseline, for the first 30 min and last 15 min of ischemia, during the increase in flow (from 2 to 4 ml/min), and every 2 min for the for the first 14 min and last 10 min of reperfusion. Quantitative determination of NO released at various time points of baseline, ischemia, and reperfusion was determined through the use of a Fluorometric Nitric Oxide Assay Kit (Calbiochem, Darmstadt, Germany). The basis for this reaction is the addition of 2,3-diaminonaphthalene (DAN) and then sodium hydroxide which converts nitrite to a fluorescent compound, 1(H)-naphthotriazole. Measurement of the fluorescence of 1(H)-naphthotriazole provides an accurate assessment of nitrite concentration in a sample.
Two hundred microliters of each coronary effluent sample were aliquoted into wells on a 96-well plate. Ten microliters of DAN were added to each well and incubated for 10 min and 20 μl of NaOH was then added to each well, and the plate was read in a GENios fluorometer (Tecan, Durham, North Carolina, USA) using an excitation wavelength of 365 nm and an emission wavelength of 450 nm. A standard curve was calculated using standards of 0.078 to 5 μM of nitrite (Calbiochem). The concentration of the nitrite in coronary effluent samples was calculated using Prism 3.0 software and was normalized to coronary flow and heart weight.
Western blot analysis of NOS isoform expression
Non-ischemic NTg and FGF2 Tg hearts were subjected to Western immunoblot analysis to determine the expression of the level of eNOS, nNOS, and iNOS isoforms in response to cardiac-specific overexpression of FGF2. Non-ischemic NTg and FGF2 Tg hearts were snap frozen in liquid nitrogen and homogenized as previously described by our laboratory (House et al. 2003, 2005, 2007). One hundred and fifty micrograms of each heart homogenate were subjected to Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS–PAGE), and Western blot analysis was performed using antibodies specific for eNOS (1:500 dilution, Santa Cruz Biotechnologies, Santa Cruz, California, USA), nNOS (1:500 dilution, BD Transduction Labs, San Jose, California, USA) or iNOS isoforms (1:500 dilution, BD Transduction Labs, San Jose, California, USA). Equal protein loading was assessed via Ponceau S (Sigma Aldrich, St. Louis, Missouri, USA) staining of total protein content. Densitometry of protein bands was performed using a Fluorchem 8800 gel imager (Alpha Innotech, San Leandro, California, USA).
Statistical analysis
All values are expressed as mean ± Standard Error of the Mean (SEM). Percent functional recovery and myocardial infarct size were compared using a oneway ANOVA followed by post-hoc analysis using the Student’s t-test. Differences between nitrite release at various time points were compared using two-way ANOVA for time and treatment with repeated measures using the post-hoc test of Fisher’s least significant difference. Western blot data was compared using a Student’s t-test. Statistical differences were considered significant when p < 0.05.
Results
Effect of cardiac-specific overexpression of FGF2 on NOS isoform expression
The protein expression level of eNOS, iNOS, and nNOS was determined in non-ischemic NTg and FGF2 Tg hearts to determine if overexpression of FGF2 primes the heart for ischemia-reperfusion insult through upregulation of NOSs, as these have been implicated in a variety of models of cardioprotection (Jones and Bolli 2006; Otani 2009). FGF2 Tg hearts showed significantly increased expression of iNOS compared to NTg hearts (p < 0.05, Figure 2). However, there was no significant difference in the protein expression level of eNOS or nNOS in non-ischemic FGF2 Tg or NTg hearts.
Figure 2.
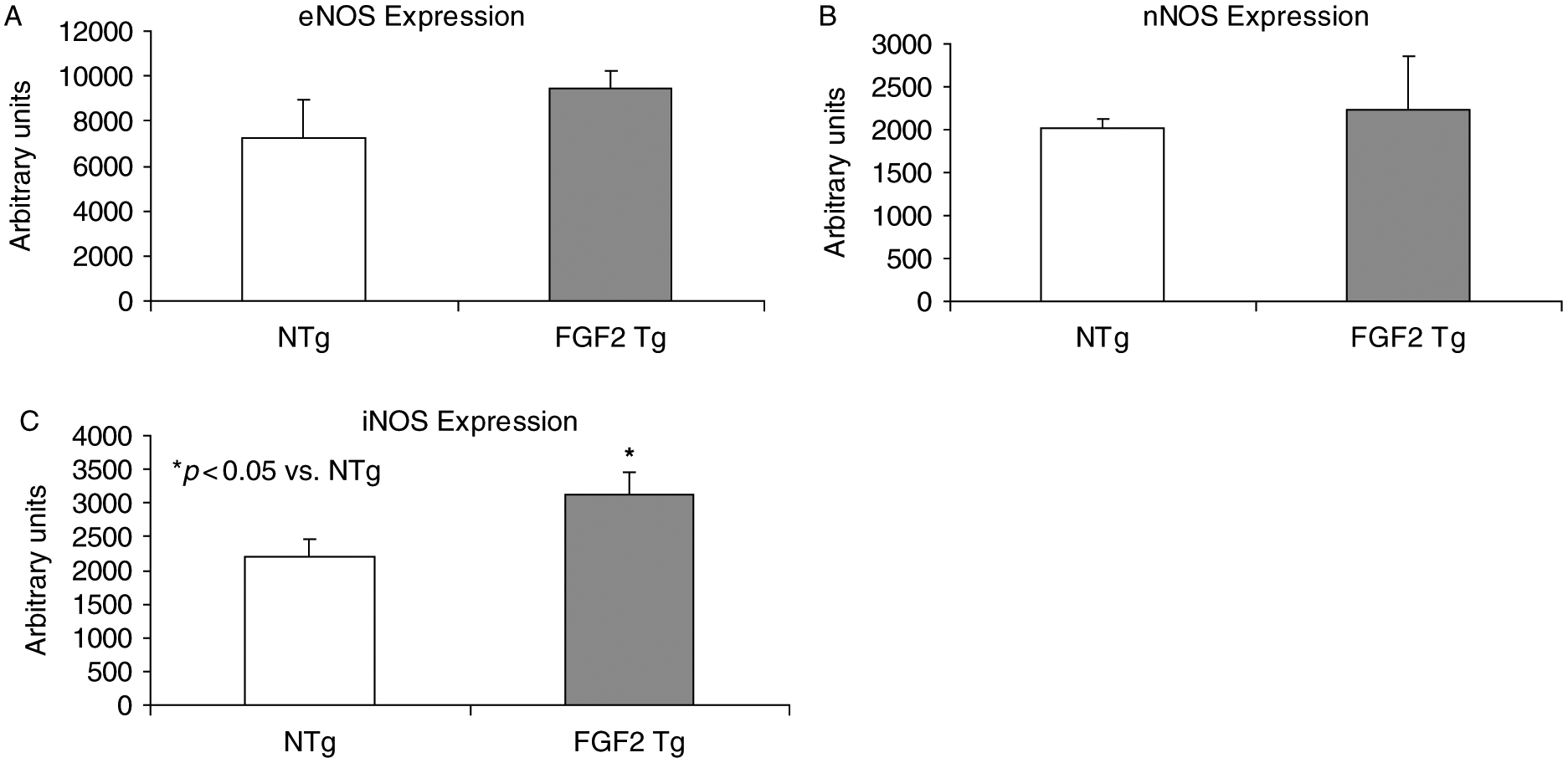
Level of NOS isoforms [eNOS (Panel A), nNOS (Panel B), and iNOS (Panel C)] in non-ischemic NTg and FGF2 Tg hearts. Overexpression of FGF2 resulted in an increase in iNOS expression compared to NTg hearts, suggesting that overexpressing FGF2 in the heart modulates signaling pathways such as NOS and NO which are involved in cardioprotection. n = 4–6 hearts per group. *p < 0.05 vs. NTg.
Inducible NOS and activation mediates FGF2-induced cardioprotection from myocardial infarction but not post-ischemic contractile dysfunction
The importance of NOS activity in FGF2-induced cardioprotection was examined in the presence of pharmacological agents that inhibit NOS activity, including L-NAME (400 μM), L-NIL (400 μM), and TRIM (100 μM). FGF2 Tg and NTg hearts were subjected to global, low-flow ischemia for 60 min, followed by reperfusion for 120 min and treated with NOS inhibitor or vehicle (Figure 1). In the vehicle-treated hearts, significantly higher recovery of cardiac function and lower myocardial infarct volume were observed in FGF2 Tg hearts when compared to NTg hearts (p < 0.05, Figure 3A,B) as previously demonstrated by our laboratory (House et al. 2003). Upon NOS inhibition with L-NAME, L-NIL, or TRIM, recovery of post-ischemic cardiac function in FGF2 Tg hearts was not different from vehicle-treated cohort hearts (Figure 3A). However, NOS inhibition with either L-NAME, L-NIL, or TRIM significantly abrogated the reduction of infarct size in FGF2 Tg hearts vs. vehicle treatment (p < 0.05, Figure 3B).
Figure 3.
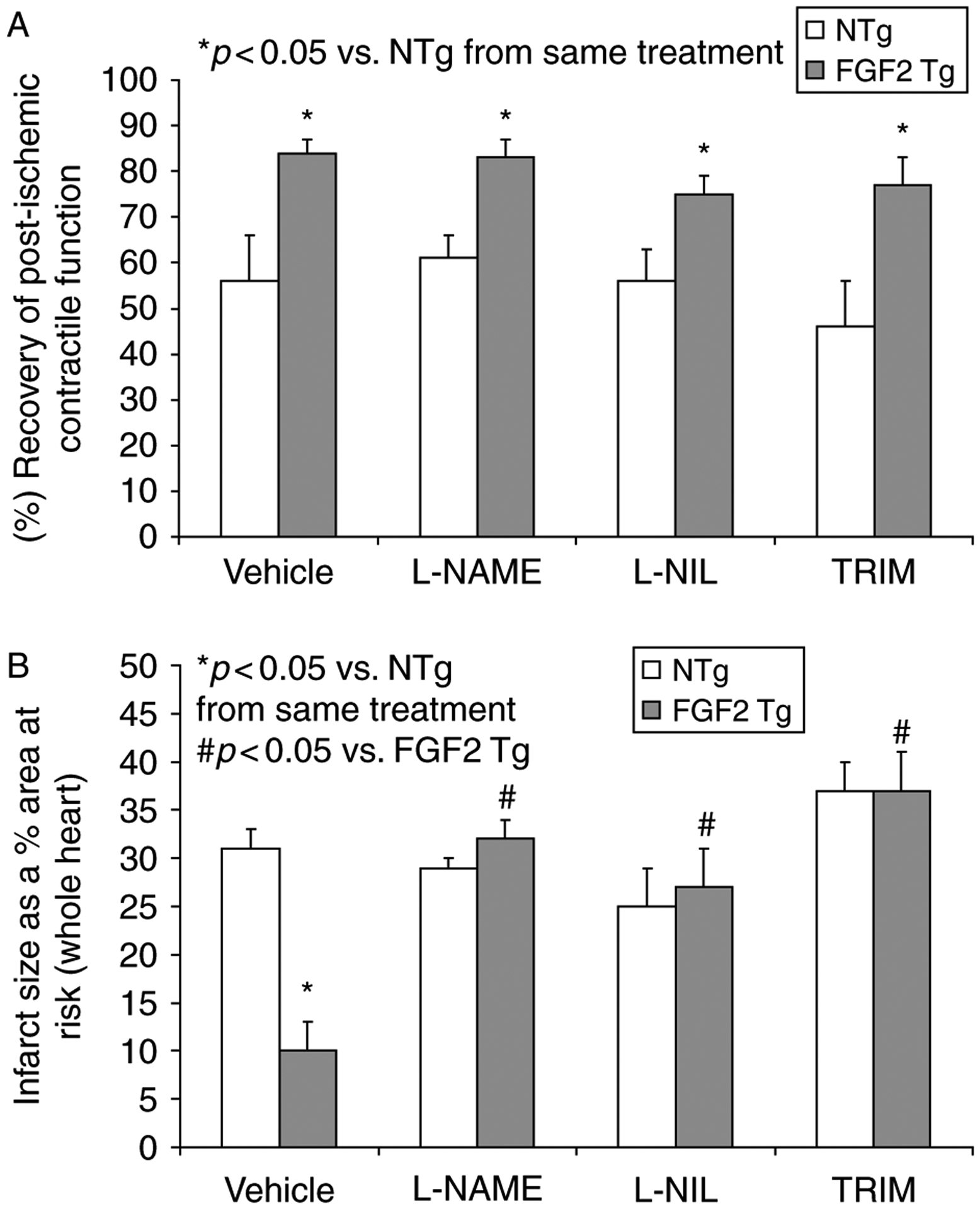
(A) Percent recovery of post-ischemic contractile function and (B) myocardial infarct size (as a % of the whole heart) in vehicle- and NOS inhibitor-treated NTg and FGF2 Tg hearts. (A) FGF2 Tg hearts had a significantly improved recovery of post-ischemic contractile function compared to NTg hearts. L-NAME, L-NIL, or TRIM treatment to FGF2 Tg hearts did not affect the cardioprotective effect of FGF2 overexpression on contractile dysfunction. Percent recovery of post-ischemic contractile function, + dP/dt following 60 min of ischemia and 120 min of reperfusion compared to baseline + dP/dt. (B) Overexpression of FGF2 significantly reduced infarct size (>50% reduction) compared to NTg hearts. L-NAME, L-NIL, or TRIM treatment to FGF2 Tg hearts abolished the cardioprotective effect of FGF2. n = 11–15 hearts per group. *p < 0.05 vs. NTg from same treatment. #p < 0.05 vs. vehicle treatment.
NO release into coronary effluent during I-R injury
Since iNOS expression is significantly increased in FGF2 Tg hearts (Figure 2), production of NO may be increased in this group. To assess the level of NO released by FGF2 Tg and NTg hearts during I-R injury, coronary effluent samples were collected during designated time points of equilibration, ischemia, and reperfusion. Levels of nitrite , a product of NOS activity in vivo and a reflection of NO release by the heart were measured in these samples. Nitrite concentration was significantly higher in FGF2 Tg hearts compared to NTg hearts both prior to and following I-R injury (p < 0.05, Figure 4), indicating a higher NO release coupled with FGF2 overexpression. A significant (p < 0.05) reduction in nitrite release was observed in FGF2 Tg hearts subjected to NOS inhibition compared to vehicle-treated hearts, and this decrease in nitrite release was similar for L-NAME (Figure 4A), L-NIL (Figure 4B) or TRIM treatment (Figure 4C), demonstrating that both inducible isoform (iNOS) and/or the neuronal isoform (nNOS) were necessary for the increase in NO production seen in FGF2 overexpressing hearts.
Figure 4.
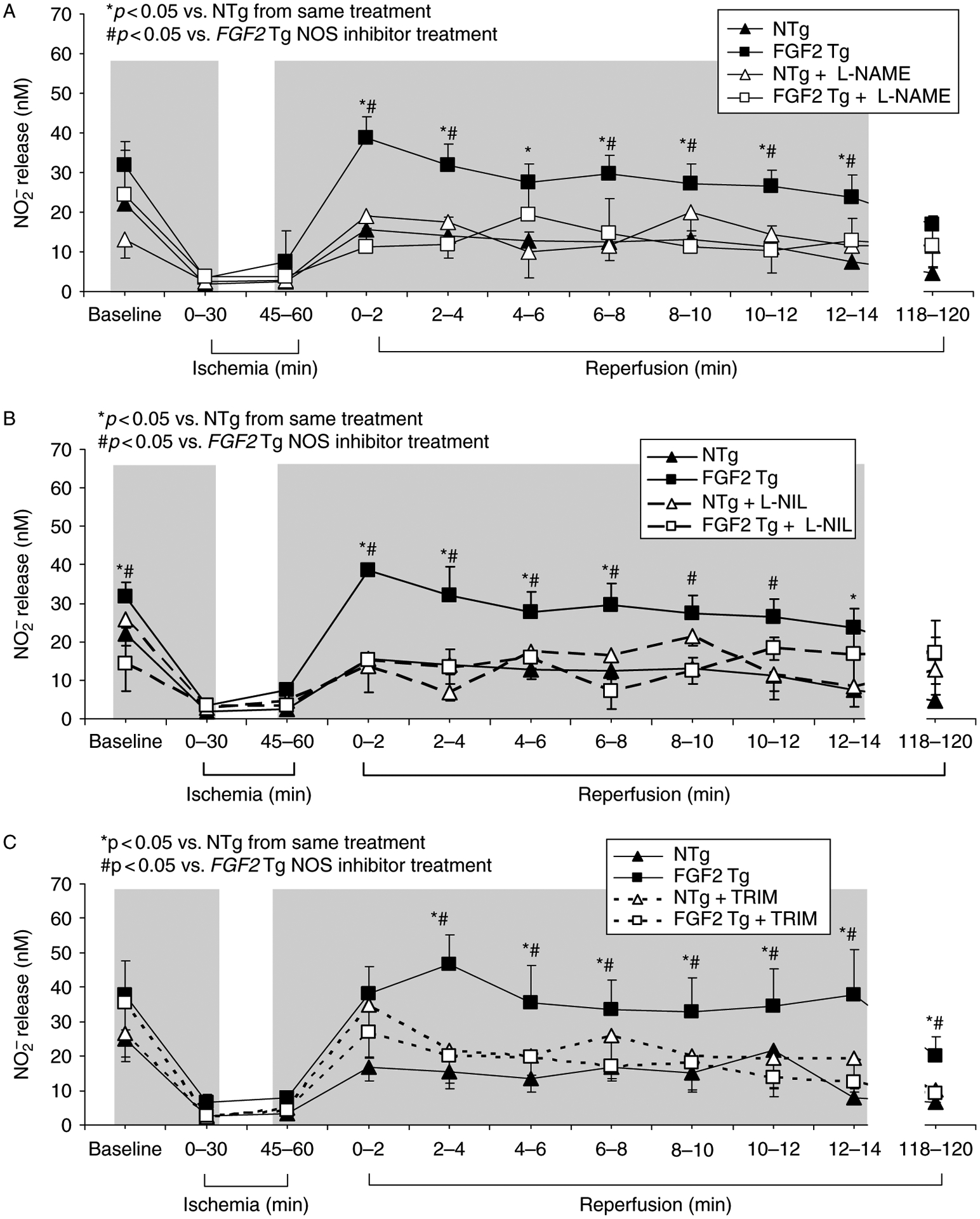
Effect of NO release, as measured by nitrite levels in vehicle- and NOS inhibitor-treated NTg and FGF2 Tg hearts. Nitrite release was significantly elevated in FGF2 Tg hearts compared to NTg hearts. L-NAME treatment resulted in a decreased level of nitrite release FGF2 Tg hearts, indicating that NOS activity was inhibited (A), L-NIL treatment (B) or TRIM administration (C) resulted in a decreased level of nitrite release in FGF2 Tg hearts, indicating that NOS activity was inhibited and that iNOS and/or nNOS activity was important for NO production leading to protection against I-R injury. NTg, + black triangle; NTg + L-NAME, open triangle; NTg + L-NIL, open triangle/dashed line; NTg + TRIM, open triangle/gray dashed line; FGF2 Tg, black square; FGF2 Tg + L-NAME, open square; FGF2 Tg + L-NIL, open square/dashed line; and FGF2 Tg + TRIM, open square/gray dashed line. Gray region, time points of inhibitor treatment (15 min prior to through first 15 min of ischemia and last 15 min of ischemia through first 15 min of reperfusion). n = 11–15 hearts per group. *p < 0.05 vs. NTg from same treatment. #p < 0.05 vs. FGF2 Tg NOS inhibitor treatment.
Protein kinases involved in NOS activation and FGF2-induced cardioprotection
Our laboratory has previously demonstrated that PKC and ERK activation is necessary for FGF2-induced cardioprotection against both cardiac dysfunction and myocardial infarction following I-R injury (House et al. 2005, 2007), and in this study, the possibility of crosstalk between these kinases and NOS activity to evoke the cardioprotective response of FGF2 is explored. There are two major forms of post-translational regulation which alter NOS isoform activity: subcellular targeting and phosphorylation (Massion and Balligand 2003). Modulation of NOS isoform activity can be mediated by a number of kinases, including PKCs and MAPKs (Forstermann et al. 1995; Forstermann 2010). Since FGF2 activates both these pathways to protect the heart from I-R injury (Jiang et al. 2002; House et al. 2005, 2007), it follows that NO may evoke the cardioprotective response of FGF2 by activation of MAPKs or PKC. Coronary effluent samples from hearts subjected to PKC or MAPK inhibition were evaluated for levels of nitrite, a product of NOS activity, released during I-R injury to determine whether PKC or MAPK signaling stimulates NOS activity, leading to FGF2-induced cardioprotection. A significant reduction in nitrite release was observed in FGF2 Tg hearts receiving bisindolylmaleimide (GFX, 1 μM), a non-selective PKC inhibitor (p < 0.05, Figure 5A), suggesting that PKC is upstream of NOS in FGF2-induced cardioprotection. Nitrite levels were also assessed in coronary effluent samples from hearts subjected to MAPK (MEK/ERK or p38) inhibition to determine whether MAPK signaling stimulates NOS activity, leading to FGF2-induced cardioprotection. A significant reduction in nitrite release was observed in FGF2 Tg hearts receiving U0126 (2.5 μM), the MEK/ERK inhibitor or SB203580 (2 μM), the p38 inhibitor, (p < 0.05, Figure 5B,C), suggesting that both ERK and p38 are upstream of iNOS and/or nNOS in FGF2-induced cardioprotection against myocardial infarction.
Figure 5.
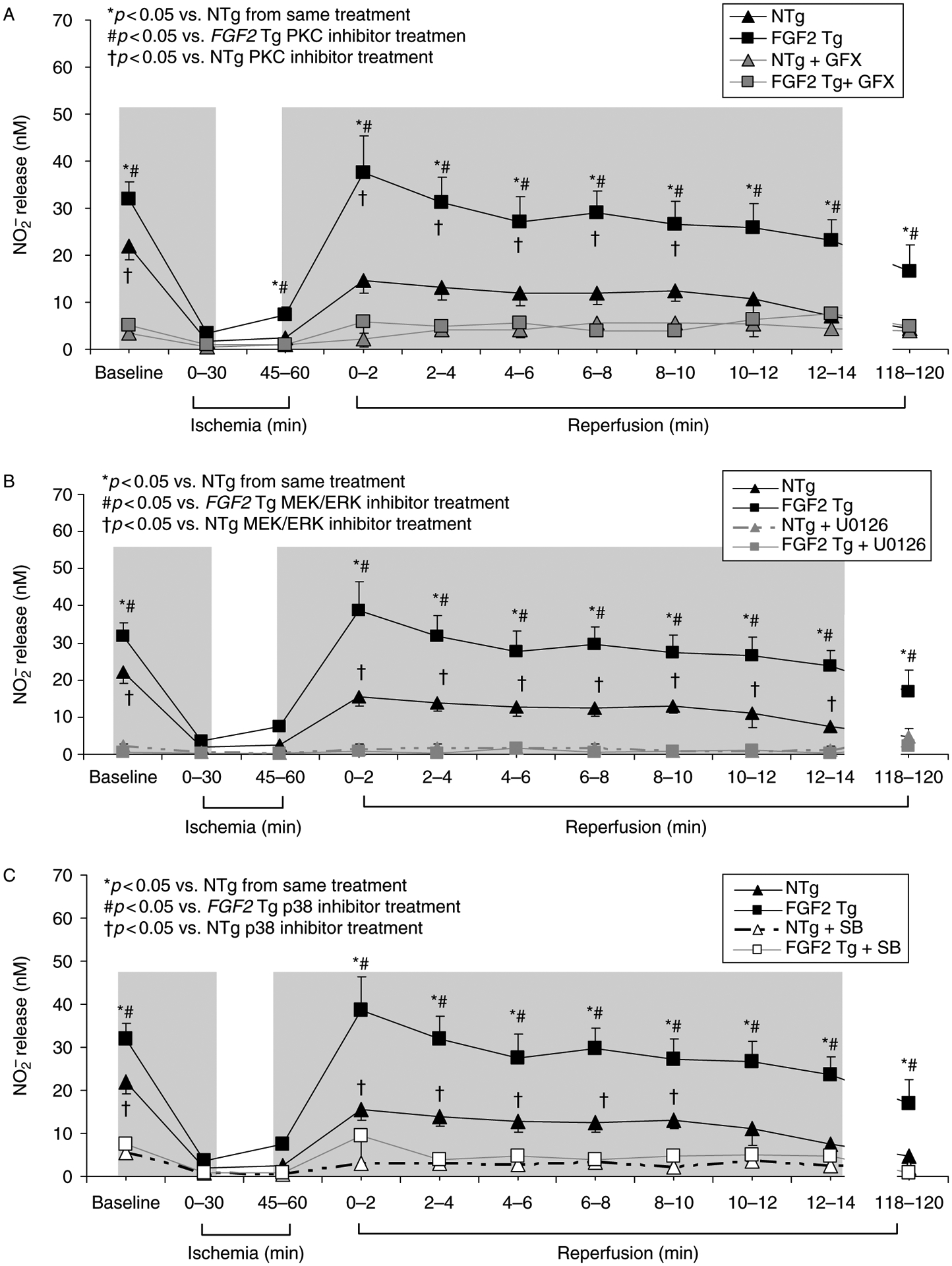
Effect of NO release, as measured by nitrite levels in vehicle- and protein kinase (PKC, ERK or p38) inhibitor-treated NTg and FGF2 Tg hearts. Nitrite release was significantly elevated in FGF2 Tg hearts compared to NTg hearts. PKC inhibition (GFX treatment, 1 μM) resulted in a decreased level of nitrite release in both FGF2 Tg and NTg hearts, indicating that NOS activity was regulated by PKC signaling (A). MEK/ERK inhibition (U0126, 2.5 μM) (B) or p38 inhibition (SB203580, 2 μM) (C) resulted in a decreased level of nitrite release in both FGF2 Tg and NTg hearts, indicating that NOS activity was inhibited and that protein kinases regulate NOS activity and NO production leading to protection against I-R injury. NTg, black triangle; NTg + GFX, gray triangle; NTg + U0126, gray triangle/dashed gray line; NTg + SB, open triangle/dashed line; FGF2 Tg, black square; FGF2 Tg + GFX, gray square; FGF2 Tg + U0126, gray square/dashed gray line; and FGF2 Tg + SB, open square/gray line. Gray region, time points of inhibitor treatment (15 min prior to through first 15 minutes of ischemia and last 15 minutes of ischemia through first 15 min of reperfusion). n = 6–11 hearts per group. *p < 0.05 vs. NTg from same treatment. #p < 0.05 vs. FGF2 Tg protein kinase inhibitor treatment. †p < 0.05 vs. NTg protein kinase inhibitor treatment.
Sarcolemmal and mitochondrial KATP channel involvement in FGF2-induced cardioprotection against myocardial dysfunction and infarction
Vascular studies have demonstrated that the vasoactive action of FGF2 is mediated in part by KATP channels (Cuevas et al. 1991; Boussairi and Sassard 1994; Tiefenbacher and Chilian 1997; Kajita et al. 2001). Both cardiac sarcKATP and mitoKATP channels have been implicated in I-R injury and to be important effectors of cardioprotection, including ischemic preconditioning (Schultz et al. 1997; Gross and Fryer 1999; Grover and Garlid 2000; O’Rourke 2000). Yet, the role of KATP channel in FGF2-mediated cardioprotection is not known. The loss of sarcKATP channel function caused by 10 μM HMR1098 treatment during the last 15 min of baseline and the first 15 min of ischemia eliminated the protective effect of FGF2 overexpression in the heart on post-ischemic recovery of function (p < 0.05, Figure 6A) and prevention of myocardial infarction (p < 0.05, Figure 6B). HMR1098 given during the last 15 min of ischemia and the first 15 min of reperfusion affected neither final post-ischemic functional recovery (Figure 6C) nor infarct size (Figure 6D). This suggests that the sarcKATP channel is a trigger for FGF2-induced cardioprotection.
Figure 6.
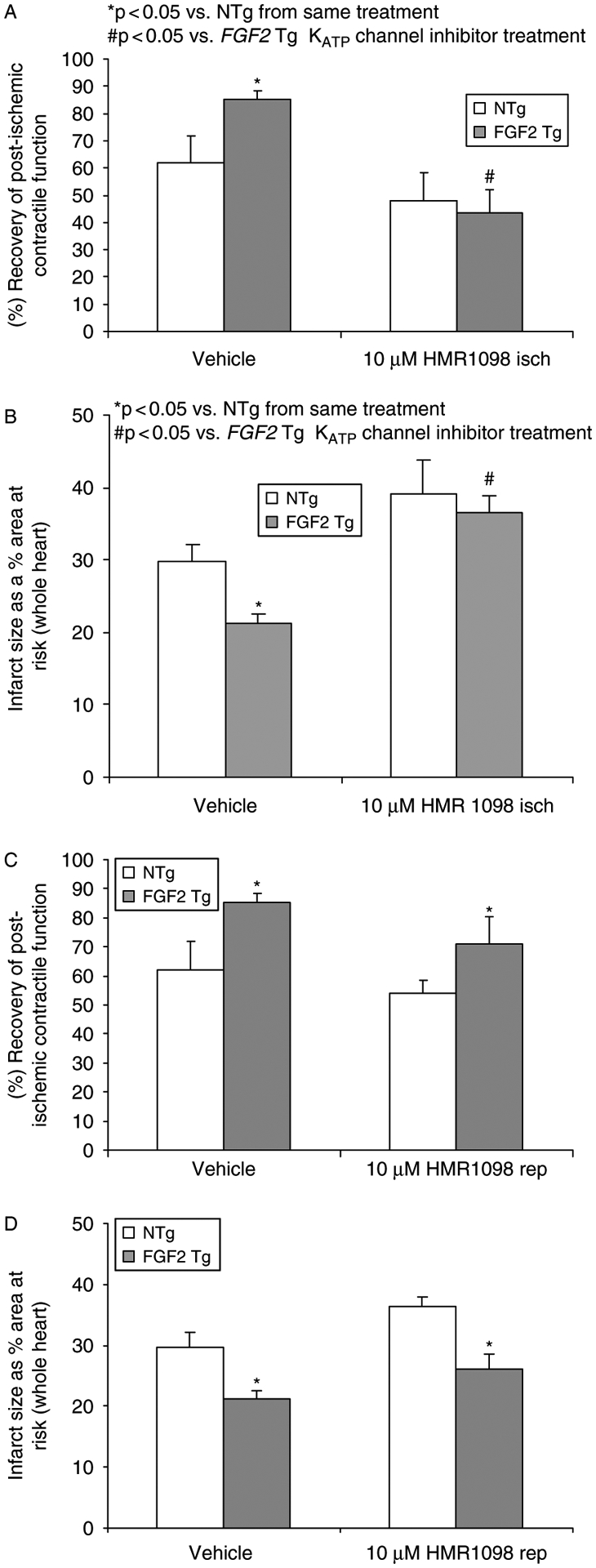
Effects of sarcolemmal KATP channels in endogenous FGF2-induced cardioprotection on post-ischemic recovery of contractile function (A and C) and myocardial infarct size (B and D) during ischemia and reperfusion. (Panels A and B) Mouse hearts overexpressing FGF2 (FGF2 Tg) and their wildtype cohort (NTg) were treated with vehicle or the sarcolemmal KATP channel inhibitor HMR1098 (10 μM) during the last 15 min of baseline equilibration and the first 15 min of ischemia and assessed for the ability to recover contractile function or reduce infarct size following 60 min of low-flow ischemia and 120 min of reperfusion. Inhibition of the sarcKATP channel at ischemia in FGF2 Tg hearts lose cardioprotection against cardiac dysfunction and infarction. (Panels C and D) FGF2 Tg and NTg hearts were treated with vehicle or the sarcolemmal KATP channel inhibitor HMR1098 (10 μM) during the last 15 min of ischemia and the first 15 min of reperfusion. Inhibition of the sarcKATP channel at reperfusion had no effect on FGF2-induced cardioprotection suggesting that this channel is most likely not an end-effector in this protective phenotype. n = 6–11 hearts per group. *p < 0.05 vs. NTg from same treatment. #p < 0.05 vs. FGF2 Tg KATP channel inhibitor treatment.
Administration of 100 μM 5-HD had a deleterious effect on post-ischemic recovery of cardiac function and increased myocardial infarction in both NTg and FGF2 Tg hearts when given during the last 15 min of baseline and first 15 min of ischemia (p < 0.05, Figures 7A,B). This result suggests that mitoKATP channel opening during the onset of ischemia is essential for the ability of the heart to survive an irreversible ischemic insult. Additionally, 5-HD administration during the last 15 min of ischemia and the first 15 min of reperfusion resulted in diminished functional recovery in FGF2 Tg hearts (p < 0.05, Figure 7C) and significantly increased infarct size (Figure 7D). These results also suggest that mitoKATP is an end-effector of FGF2-induced cardioprotection.
Figure 7.
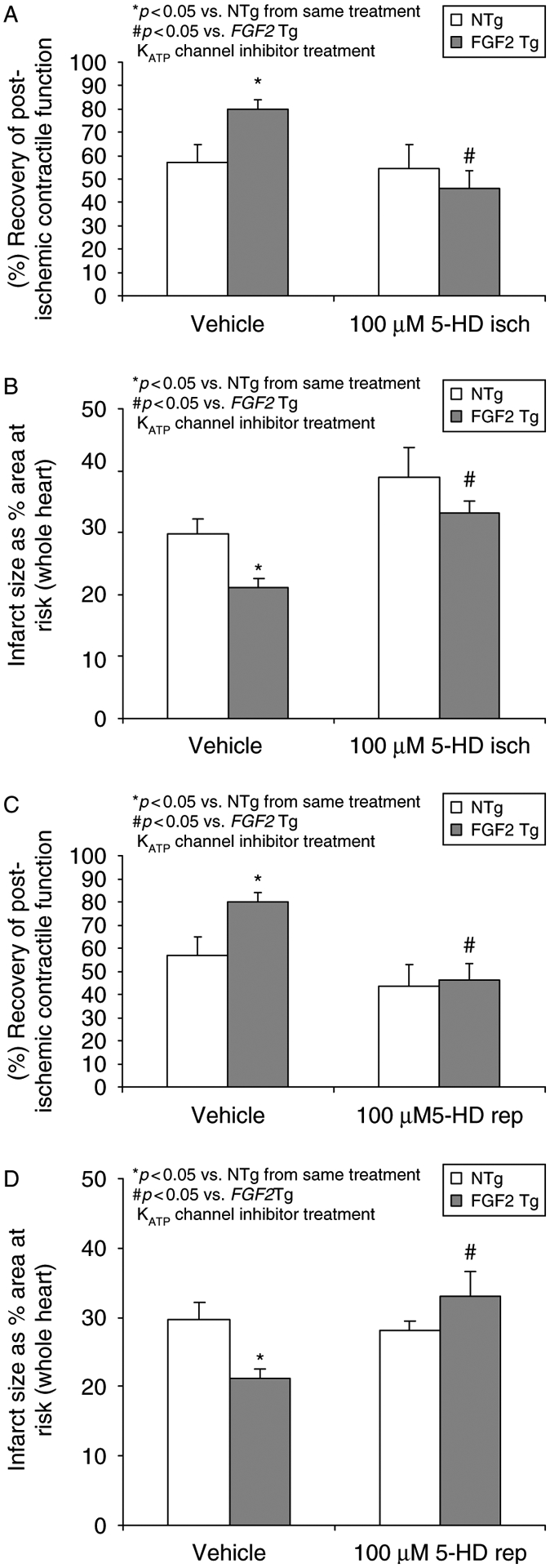
Effects of mitochondrial KATP channels in endogenous FGF2-induced cardioprotection on post-ischemic recovery of contractile function (A and C) and myocardial infarct size (B and D) during ischemia and reperfusion. (Panels A and B) Mouse hearts overexpressing FGF2 (FGF2 Tg) and their wildtype cohort (NTg) were treated with vehicle or the mitochondrial KATP channel inhibitor 5-HD (100 μM) during the last 15 min of baseline equilibration and the first 15 min of ischemia and assessed for the ability to recover contractile function or reduce infarct size following 60 min of low-flow ischemia and 120 min of reperfusion. Inhibition of the mitoKATP channel at ischemia in FGF2 Tg hearts lose cardioprotection against cardiac dysfunction and infarction. (Panels C and D) FGF2 Tg and NTg hearts were treated with vehicle or the mitochondrial KATP channel inhibitor 5-HD (100 μM) during the last 15 min of ischemia and the first 15 min of reperfusion. Similarly, inhibition of the mitoKATP channel at reperfusion blocked FGF2-induced cardioprotection suggesting that this channel is most likely a trigger and an end-effector in this protective phenotype. n = 6–11 hearts per group. *p < 0.05 vs. NTg from same treatment. #p < 0.05 vs. FGF2 Tg KATP channel inhibitor treatment.
Discussion and conclusions
These data suggest that FGF2 overexpression modulates both NOS expression and NOS activity in the heart, and more importantly, that FGF2-induced cardioprotection against myocardial infarction is mediated via a NOS-dependent pathway. These findings indicate that NO signaling is a potential therapeutic target of endogenously expressed FGF2-induced cardioprotection against myocardial infarction. In addition, the application of pharmacologic blockers of KATP channel function reveals that operation of these channels early in ischemia are crucial to the ability of the FGF2 Tg heart to enhance recovery of function and minimize infarct size as well as in reperfusion to reduce myocardial infarction. This is the first demonstration that KATP channels are involved in the cardioprotective effect of FGF2.
ATP-sensitive potassium (KATP) channels are present in the mammalian heart, and experimental and clinical studies have identified the sarcolemmal (sarcKATP) and/or mitochondrial (mitoKATP) channel as an end-effector in the cardioprotective pathway of ischemic and pharmacological preconditioning (Gross and Fryer 1999; Grover and Garlid 2000; O’Rourke 2000). Yet, based on pharmacological studies using selective channel openers (such as diazoxide) and genetic knockout studies (involving ablation of the Kir6.2 subunit), there is disagreement in the field on the relative importance of sarcKATP vs. mitoKATP in cardioprotection (Grover and Garlid 2000; O’Rourke 2000). Some studies suggest that, although sarcKATP is more important during preconditioning, mitoKATP is more important for protection during long I-R injury (Wang et al. 2001; Suzuki et al. 2002; Marinovic et al. 2006). It appears, by way of consensus, that while neither sarcKATP nor mitoKATP is sufficient for the full cardioprotective phenotype, at least one of the two channels is necessary. Our present work has demonstrated that the sarcKATP channel is an important trigger in the cardioprotective effect of FGF2 in the heart as inhibition of this channel in FGF2 Tg resulted in poorer post-ischemic contractile dysfunction and larger infarct compared to vehicle-treated FGF2 Tg (Figure 6A,B). The role of sarcKATP channels in modulating cardiac function in diseased hearts (i.e. I-R injury or heart failure) is controversial such that similar observations were made in isolated perfused rat hearts subjected to low-flow ischemia by Gögelein and colleagues (2001); whereas no significant cardiodepressant effects were observed in anesthetized rats subjected to ischemia-reperfusion. Furthermore, evidence from mice deficient in (Suzuki et al. 2002) or with a mutated form (Rajashree et al. 2002) of the inward-rectifying K+ channel subunit (Kir6.2), a component of the cardiac sarcKATP channel, revealed that this particular KATP channel was important for normal myocardial function, and perturbations of this channel led to a poor recovery of cardiac function following ischemic injury. On the other hand, Pasdois et al. (2007) reported that 10 μM HMR1098 improved the recovery of cardiac function in Langendorff perfused rat hearts. Also, HMR1098 had no effect on cardiac function in in vivo failing canine hearts (Saavedra et al. 2002). The mitochondrial KATP channel is essential for the ability of the heart to survive an irreversible ischemic insult as well as plays an accessory role as an end-effector of FGF2-induced cardioprotection against cardiac dysfunction and infarction (Figure 7). The importance of mitoKATP channels in protecting the heart during I-R injury is well documented (reviewed in O’Rourke 2004), but this is the first data demonstrating its role in FGF2-mediated protection.
All three NOS isoforms, eNOS, iNOS, and nNOS, have been detected in the myocardium (Forstermann et al. 1994). There is also a growing body of evidence that NOS isoforms play prominent roles in cardiomyocyte regulation in physiological and disease states. It has been well established that nNOS participates in the regulation of cardiac function (Sears et al. 2003; Zhang et al. 2008) and, after I-R, can impact cardiac function, infarct development, and remodeling (Dawson et al. 2005; Saraiva et al. 2005; Otani 2009; Burkard et al. 2010). Additionally, it is well documented that iNOS modulates the disease state of cardiac I-R injury and is necessary or instrumental in cardioprotection (Jones and Bolli 2006). Evidence has indicated that exogenously added FGF2 reduced myocardial stunning in the mouse heart via an iNOS-dependent mechanism (Hampton et al. 2000). Here, we examine if iNOS and/or nNOS are responsible for the protective effects of FGF2, since several biological actions of FGFs have been shown to occur through NO signaling (Huang et al. 1997; Cuevas et al. 1999).
This study supports a role for iNOS in mediating FGF2-induced cardioprotection against myocardial infarction (Figure 3B). Other laboratories have demonstrated upregulation of iNOS and eNOS in the cardiovascular system in response to FGF2 (Kostyk et al. 1995; Tiefenbacher and Chilian 1997; Hampton et al. 2000), and our investigations have confirmed that cardiac-specific overexpression of FGF2 resulted in an upregulation of iNOS (Figure 2), suggesting a priming effect of FGF2 on myocardial tissue through induction of iNOS expression prior to the onset of ischemia. While iNOS was the only isoform with upregulated expression in mice overexpressing FGF2 in the heart, FGF2 has previously been shown to increase the expression of eNOS in endothelial cells (Kostyk et al. 1995); however, there is no significant induction of eNOS in the heart by cardiac-specific overexpression of FGF2 (Figure 2).
Although cardiac-specific FGF2 overexpression does not change the levels of nNOS in the non-ischemic heart, the data presented here suggest that nNOS activity may be necessary for FGF2-mediated cardioprotection (Figure 4B). nNOS localizes to the sarcoplasmic reticulum in cardiomyocytes and alters SR Ca2+ uptake (Xu et al. 1999) as well as has an impact on the survival of mice after I-R injury (Saraiva et al. 2005), which is associated with a decreased infarct size and production of reactive oxygen species in post-ischemic hearts (Burkard et al. 2010). Overall, these findings suggest that nNOS may adjust calcium cycling in the heart to protect the heart from injury associated with Ca2+ overload. Our data give credit to this hypothesis, indicating that FGF2-mediated protection from cardiomyocyte injury/death may require nNOS activity.
This study shows a role for NO in FGF2-mediated protection from infarct development (Figure 3B), but not post-ischemic contractile dysfunction, as NOS-inhibitor-treated FGF2 Tg hearts have the same functional recovery following I-R injury as FGF2 Tg hearts without NOS inhibition (Figure 3). This uncoupling of irreversible tissue death (infarct development) and reversible acute functional injury (stunning) has been seen previously in other models of cardioprotection (Mitchell et al. 1993; Cohen et al. 1999) and is further evident that distinct biological pathways are responsible for each (Bolli 1990). There is a complex relationship between stunning and infarction, and while both can occur as a result of I-R, the pathways leading to each are biochemically distinct (reviewed in Buja and Entman 1998; Bolli and Marban 1999; Pomblum et al. 2010). It should be noted that, while irreversible cardiac dysfunction can and may develop in response to tissue death in the long term (Bolli 1990), the function of the heart immediately after reperfusion, a measure of stunning, was not affected. The data presented here suggest that NOS isoforms activated by overexpression of FGF2 favors pathways that alter cell survival, instead of those that affect post-ischemic function. This has been documented in other models of NO-mediated cardioprotection, where inhibition of NO caused no difference in functional parameters of the isolated heart, but resulted in a change in infarct size (Cohen et al. 2010), although it should be noted that these findings are not unequivocal (Schulz et al. 2004).
Multiple potential mechanisms may mediate the protection from I-R injury provided by enhanced NO production in FGF2 Tg hearts. Our laboratory has demonstrated that PKC and ERK activation acutely mediate FGF2-induced cardioprotection against both cardiac dysfunction and myocardial infarction following I-R injury (House et al. 2005, 2007), which may in turn modulate substrates that have relevance for myocardial function or protection from I-R injury, including NOS isoform (Forstermann et al. 1995; Forstermann 2010) and KATP channel (Light et al. 2000; Jaburek et al. 2006) expression and activity. While the effects of phosphorylation on nNOS are residue-specific, it should be noted that phosphorylation of nNOS at S847 (Rameau et al. 2004), as well as S1412 (Adak et al. 2001; Rameau et al. 2007) increases the activity of the enzyme. The findings presented here implicate PKC, ERK1/2 and p38 as regulators of NO release in the ischemic heart during I-R injury (Figure 5). Interestingly, FGF2 activation of PKC-α has been shown to result in an increase in the phosphorylation of eNOS at S1179, and a subsequent increase in NO production in endothelial tissue (Partovian et al. 2005); this is of particular importance, as this site is analogous to the positive nNOS regulator S1412 (Adak et al. 2001). There is also evidence demonstrating that the activation of PKC-ϵ associated with the cardioprotective effect of ischemic preconditioning is prevented by the inhibition of NO pathway (Ping et al. 1999). Furthermore, PKC-mediated post-translational modification is linked to the regulation of sarcKATP and mitoKATP channels (Kwak et al. 1996; Light et al. 2000). There is evidence that PKC-δ and -ε are necessary for sarcKATP or mitoKATP channel opening in cardioprotection and other cellular functions (Ohnuma et al. 2002; Harada et al. 2004). MitoKATP opening and inhibition of the associated pro-apoptotic cardiac mitochondrial permeability transition pore via the PKC-ε pathway prevents cardiomyocyte apoptosis in a model of cardioprotection (Baines et al. 2003). Previous studies in our laboratory demonstrated that PKC isoforms α, δ, and ε are localized to the sarcolemma or mitochondria in FGF2 Tg hearts at either baseline or various time points of I-R injury (House et al. 2007). Since these pathways operate downstream of FGF2 (Jiang et al. 2002; House et al. 2005; Liao et al. 2007) and upstream of KATP channel opening (Ohnuma et al. 2002), it is likely that at least one kinase cascade, attributed to cardioprotective FGF2 signaling, terminates at the mitoKATP and/or sarcKATP channel to influence its opening and induce physiological changes that protect the heart from I-R injury.
Subfamilies of the MAPK pathway have also been implicated in modulating NOS signaling in the heart (Singh et al. 1996), and recently it has been shown that MAPK inhibition abrogates iNOS activity in sildenifil-induced cardioprotection (Das et al. 2009). In addition, the ERK pathway has been implicated in FGF2-induced cytoprotection against iNOS-mediated apoptosis (Iwai-Kanai et al. 2002). These findings support the conclusions presented here that MAPK and PKC both regulate NOS function in the heart during I-R, in hearts with increased expression of FGF2. With the finding that NOS and NO signaling are downstream of kinase activation (Figure 5), resulting in decreased myocardial infarct size following I-R injury, but not post-ischemic cardiac dysfunction, these data suggest that the kinase-NOS pathway is involved in the reduction of infarct development. Therefore, the preserved cardiac function in the presence of NOS inhibition (Figure 3A), but not PKC or MAPK inhibition (see references House et al. 2005, 2007), is most likely due to kinase activation and downstream changes in activity of proteins involved in calcium handling and/or myofibril function (Bolli and Marban 1999) independent of NO signaling. Ongoing studies are being performed to identify these mechanisms involved in the improvement of post-ischemic cardiac function induced by FGF2 overexpression.
This study, for the first time, demonstrates involvement of NO signaling and KATP channel activity in the cardioprotective effect elicited by endogenous FGF2. This is similar to signaling cascades activated by other tyrosine kinase receptor ligands such as epithelial growth factor (Namiki and Akatsuka 1990; Philipp et al. 2006) and insulin-like growth factor (Haylor et al. 1991; Tsukahara et al. 1994) in vascular function. In addition, our data confirm the direct or indirect interplay between NO and KATP channels in cardioprotection and I-R injury. For example, there is well-documented evidence that many pharmacological cardioprotective agents elicit their actions via NO and KATP channel function (Uchiyama et al. 2003; Xu et al. 2004; Cohen et al. 2006; Bai et al. 2011). Furthermore, evidence by Tsuura and colleagues (1994) and that of Kawano and colleagues (2009) demonstrate that NO, by modulation of the glycolytic pathway in pancreas or direct S-nitrosylation of channel in sensory neurons, respectively, can affect KATP channel activity and function.
Limitations.
An important limitation of these studies is that the role of eNOS in FGF2-mediated protection remains to be elucidated. The effects of eNOS on the infarcted heart have been the subject of some controversy, and there have been conflicting reports regarding eNOS’s role in cardioprotection (Sharp et al. 2002; Guo et al. 2008). This study does not rule out the possibility that eNOS may provide some of the cardioprotection induced by FGF2 overexpression in the heart. Regrettably, the tools for studying eNOS are currently limited, as there are no selective eNOS inhibitors, and two distinct strains of eNOS knockout mice have produced incongruous data in the hands of different groups (Bell and Yellon 2001; Sharp et al. 2002). While it has been demonstrated here that iNOS and/or nNOS are necessary for FGF2-mediated cardioprotection, the question of whether eNOS is also involved will be the subject of future studies as better tools are developed, and will be necessary to completely understand NO signaling in FGF2-induced cardioprotection. An additional limitation of this study is the relative selectivity of the nNOS inhibitor TRIM. It has been shown that TRIM may also inhibit iNOS at concentrations sufficient to inhibit nNOS (Handy et al. 1996), although TRIM has been used previously to distinguish nNOS from iNOS in ischemia studies in pigs (Adams et al. 2007). However, our studies examining the effect of protein kinase inhibition on nitrite levels suggest that a phosphorylatable NOS, in addition to iNOS, has a role in FGF2-induced cardioprotection. Further delineation of nNOS and eNOS in cardioprotection triggered by FGF2-protein kinase signaling is warranted. A limitation in the field of KATP channel function in I-R injury is the selectivity of the pharmacological inhibitors for sarcKATP and mitoKATP channels; yet, the main tool for discriminating these two K+ channels is pharmacological manipulation. While HMR1098 is indeed highly selective for sarcKATP, there is controversy regarding selectivity of 5-HD for mitoKATP vs. possible direct effects on succinate dehydrogenase activity (Hanley and Daut 2005).
In summary, the data suggest that activation of NOSs and the production of NO during I-R injury mediate FGF2-induced cardioprotection from post-ischemic myocardial cell death, but not post-ischemic cardiac dysfunction (Figure 3). This study shows that cardiac-specific overexpression of FGF2 results in a NOS-dependent increase in NO production, which is acutely regulated by PKC, ERK, and p38 (Figure 5). Furthermore, for the first time, these findings indicate a role of sarcolemmal and mitochondrial KATP channels in FGF2-induced cardioprotection against cardiac dysfunction and myocardial infarction (Figures 6 and 7). NO and KATP channels are part of a complex network of intracellular signals, which currently include PKC isoforms and MAPKs, activated by FGF2 to protect the heart against cardiac dysfunction and myocardial infarction during I-R injury (Figure 8).
Figure 8.
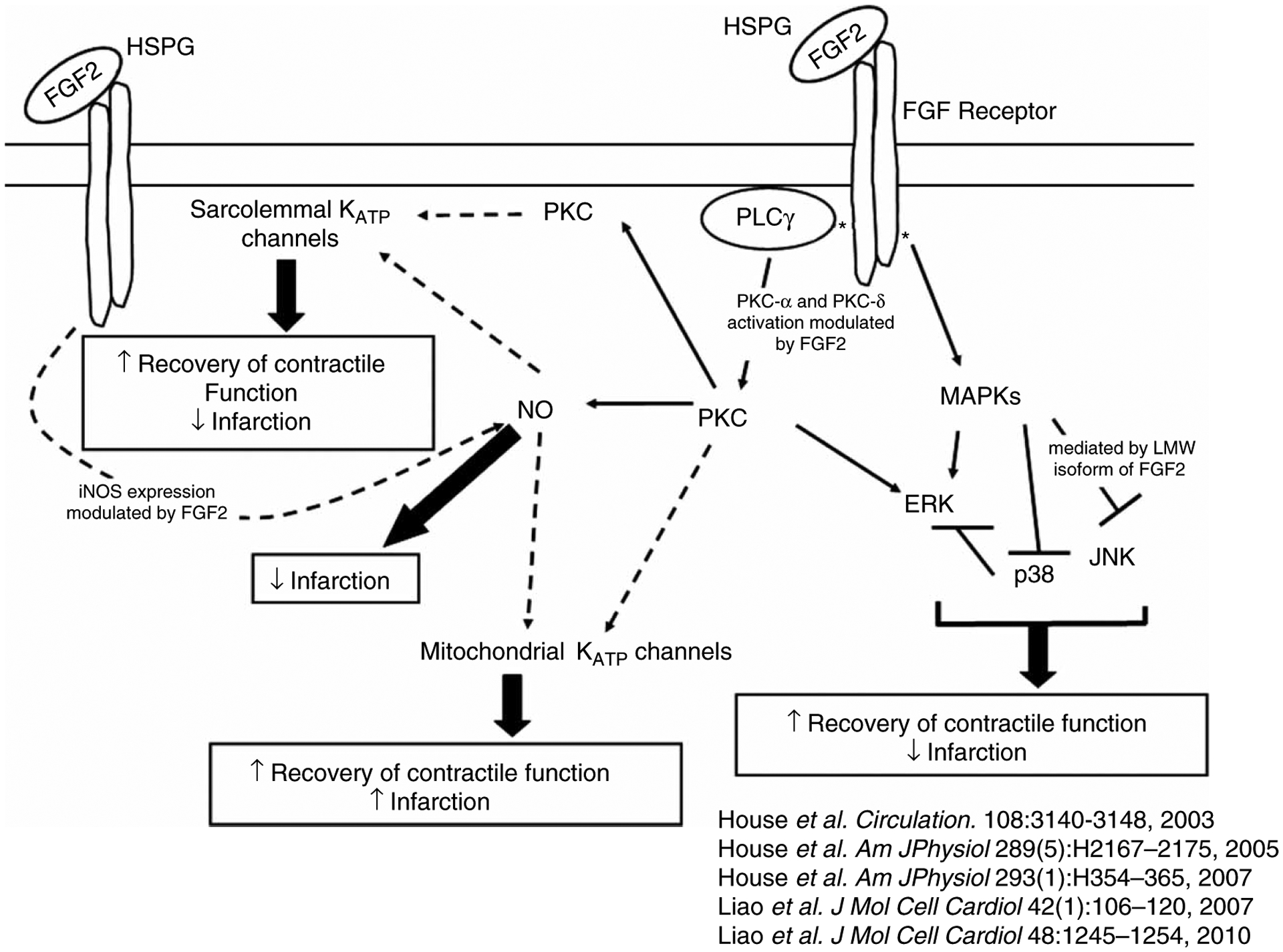
Schematic depicting the intracellular signaling network of endogenous FGF2-induced cardioprotection against cardiac dysfunction and myocardial infarction. Our laboratory previously demonstrated that endogenously expressed FGF2 in heart resulted in cardioprotection (House et al. 2003; Liao et al. 2007, 2010) via PKC and ERK activation (House et al. 2005) and p38 (House et al. 2005) as well as JNK inhibition (Liao et al. 2007). This protein kinase activation or inhibition was modulated by cross-talk between PKC and MAPKs (House et al. 2005; House et al. 2007). Our recent data demonstrate an involvement of NO and KATP channels, directly or indirectly, to enhance post-ischemic recovery of cardiac function and/or reduce infarct size elicited by endogenous FGF2. Solid line indicates documented involvement of signaling cascade. Dashed line indicates potential pathway of protection. Upward arrow indicates improvement in recovery of post-ischemic contractile function and downward arrow indicates a reduction in infarct size.
Acknowledgments
This work was supported by grants from the American Heart Association (SDG 23004N), the Pharmaceutical Research and Manufacturers of America (Research Starter Grant), NIH/NHLBI R01 (HL075633) to J. Schultz, an Undergraduate Student Summer Fellowship from the American Heart Association, Ohio Valley Association to D. Porter, and a NIH Training Grant (T35 DK060444) Medical Student Summer Fellowship to G. Carpenter. The authors gratefully acknowledge Dr Heinz Gögelein and Sanofi-Aventis for the gift of HMR1098. The authors would like to acknowledge M. Bender and A. Whitaker for their excellent animal husbandry, G. Newman for assistance with the isolated heart apparatus and I-R studies, David Palacios for assistance with the I-R + KATP channel inhibition studies, Laura Moon for assistance with measurement of nitrite levels, and N. Vatamaniuc for assistance with cardiac function data analysis.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Adak S, Santolini J, Tikunova S, Wang Q, Johnson JD, Stuehr DJ. 2001. Neuronal nitric-oxide synthase mutant (Ser-1412 → Asp) demonstrates surprising connections between heme reduction, NO complex formation, and catalysis. J Biol Chem 276: 1244–1252. [DOI] [PubMed] [Google Scholar]
- Adams JA, Wu D, Bassuk J, Arias J, Lozano H, Kurlansky P, Lamas GA. 2007. Nitric oxide synthase isoform inhibition before whole body ischemia reperfusion in pigs: Vital or protective? Resuscitation 74:516–525. [DOI] [PubMed] [Google Scholar]
- Bai YM, Murakami H, Iwasa M, Sumi S, Yamada Y, Ushikoshi H, Aoyama T, Nishigaki K, Takemura G, Uno B, Minatoguchi S. 2011. Cilostazol protects the heart against ischaemia reperfusion injury in a rabbit model of myocardial infarction: Focus on adenosine, nitric oxide and mitochondrial ATP-sensitive potassium channels. Clin Exp Pharmacol Physiol 38:658–665. [DOI] [PubMed] [Google Scholar]
- Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. 2003. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res 92:873–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barancik M, Htun P, Strohm C, Kilian S, Schaper W. 2000. Inhibition of the cardiac p38-MAPK pathway by SB203580 delays ischemic cell death. J Cardiovasc Pharmacol 35:474–483. [DOI] [PubMed] [Google Scholar]
- Bell RM, Yellon DM. 2001. The contribution of endothelial nitric oxide synthase to early ischaemic preconditioning: The lowering of the preconditioning threshold. An investigation in eNOS knockout mice. Cardiovasc Res 52:274–280. [DOI] [PubMed] [Google Scholar]
- Bit RA, Davis PD, Elliott LH, Harris W, Hill CH, Keech E, Kumar H, Lawton G, Maw A, Nixon JS, Vesey DR, Wadsworth J, Wilkinson SE. 1993. Inhibitors of protein kinase C.3. Potent and highly selective bisindolylmaleimides by conformational restriction. J Med Chem 36:21–29. [DOI] [PubMed] [Google Scholar]
- Bolli R 1990. Mechanism of myocardial “stunning”. Circulation 82:723–738. [DOI] [PubMed] [Google Scholar]
- Bolli R, Marban E. 1999. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 79:609–634. [DOI] [PubMed] [Google Scholar]
- Boussairi EH, Sassard J. 1994. Cardiovascular effects of basic fibroblast growth factor in rats. J Cardiovasc Pharmacol 23: 99–102. [DOI] [PubMed] [Google Scholar]
- Budas GR, Churchill EN, Mochly-Rosen D. 2007. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia–reperfusion injury. Pharmacol Res 55:523–536. [DOI] [PubMed] [Google Scholar]
- Buja LM, Entman ML. 1998. Modes of myocardial cell injury and cell death in ischemic heart disease. Circulation 98:1355–1357. [DOI] [PubMed] [Google Scholar]
- Burkard N, Williams T, Czolbe M, Blomer N, Panther F, Link M, Fraccarollo D, Widder JD, Hu K, Han H, Hofmann U, Frantz S, Nordbeck P, Bulla J, Schuh K, Ritter O. 2010. Conditional overexpression of neuronal nitric oxide synthase is cardioprotective in ischemia/reperfusion. Circulation 122:1588–1603. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. 2006. Nitric oxide is a preconditioning mimetic and cardioprotectant and is the basis of many available infarct-sparing strategies. Cardiovasc Res 70: 231–239. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. 1999. Smaller infarct after preconditioning does not predict extent of early functional improvement of reperfused heart. Am J Physiol 277: H1754–H1761. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Liu Y, Solenkova NV, Downey JM. 2010. Cardioprotective PKG-independent NO signaling at reperfusion. Am J Physiol Heart Circ Physiol 299:H2028–H2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. 1995. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett 364:229–233. [DOI] [PubMed] [Google Scholar]
- Cuevas P, Carceller F, Martinez-Coso V, Cuevas B, Fernandez-Ayerdi A, Reimers D, Asin-Cardiel E, Gimenez-Gallego G. 1999. Cardioprotection from ischemia by fibroblast growth factor: Role of inducible nitric oxide synthase. Eur J Med Res 4: 517–524. [PubMed] [Google Scholar]
- Cuevas P, Carceller F, Ortega S, Zazo M, Nieto I, Gimenez-Gallego G. 1991. Hypotensive activity of fibroblast growth factor. Science 254:1208–1210. [DOI] [PubMed] [Google Scholar]
- Das A, Salloum FN, Xi L, Rao YJ, Kukreja RC. 2009. ERK phosphorylation mediates sildenafil-induced myocardial protection against ischemia–reperfusion injury in mice. Am J Physiol Heart Circ Physiol 296:H1236–H1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson D, Lygate CA, Zhang MH, Hulbert K, Neubauer S, Casadei B. 2005. nNOS gene deletion exacerbates pathological left ventricular remodeling and functional deterioration after myocardial infarction. Circulation 112:3729–3737. [DOI] [PubMed] [Google Scholar]
- Detillieux KA, Sheikh F, Kardami E, Cattini PA. 2003. Biological activities of fibroblast growth factor-2 in the adult myocardium. Cardiovasc Res 57:8–19. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. 1998. Identification of a novel inhibitor of mitogen-activated protein kinase. J Biol Chem 273:18623–18632. [DOI] [PubMed] [Google Scholar]
- Forstermann U 2010. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch 459:923–939. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, Kleinert H. 1994. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 23:1121–1131. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Gath I, Schwarz P, Closs EI, Kleinert H. 1995. Isoforms of nitric oxide synthase. Properties, cellular distribution and expressional control. Biochem Pharmacol 50: 1321–1332. [DOI] [PubMed] [Google Scholar]
- Gogelein H, Ruetten H, Albus U, Englert HC, Busch AE. 2001. Effects of the cardioselective KATP channel blocker HMR 1098 on cardiac function in isolated perfused working rat hearts and in anesthetized rats during ischemia and reperfusion. Naunyn Schmiedebergs Arch Pharmacol 364:33–41. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Fryer RM. 1999. Sarcolemmal versus mitochondrial ATP-sensitive K + channels and myocardial preconditioning. Circ Res 84:973–979. [DOI] [PubMed] [Google Scholar]
- Grover GJ, Garlid KD. 2000. ATP-sensitive potassium channels: A review of their cardioprotective pharmacology. J Mol Cell Cardiol 32:677–695. [DOI] [PubMed] [Google Scholar]
- Guo Y, Li Q, Wu WJ, Tan W, Zhu X, Mu J, Bolli R. 2008. Endothelial nitric oxide synthase is not necessary for the early phase of ischemic preconditioning in the mouse. J Mol Cell Cardiol 44:496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton TG, Amende I, Fong J, Laubach VE, Li J, Metais C, Simons M. 2000. Basic FGF reduces stunning via a NOS2-dependent pathway in coronary-perfused mouse hearts. Am J Physiol Heart Circ Physiol 279:H260–H268. [DOI] [PubMed] [Google Scholar]
- Handy RL, Harb HL, Wallace P, Gaffen Z, Whitehead KJ, Moore PK. 1996. Inhibition of nitric oxide synthase by 1-(2-trifluoromethylphenyl) imidazole (TRIM) in vitro: Antinociceptive and cardiovascular effects. Br J Pharmacol 119:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handy RL, Moore PK. 1997. Mechanism of the inhibition of neuronal nitric oxide synthase by 1-(2-trifluoromethylphenyl) imidazole (TRIM). Life Sci 60:PL389–PL394. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Daut J. 2005. K(ATP) channels and preconditioning: A re-examination of the role of mitochondrial K(ATP) channels and an overview of alternative mechanisms. J Mol Cell Cardiol 39:17–50. [DOI] [PubMed] [Google Scholar]
- Harada N, Miura T, Dairaku Y, Kametani R, Shibuya M, Wang R, Kawamura S, Matsuzaki M. 2004. NO donor-activated PKC-delta plays a pivotal role in ischemic myocardial protection through accelerated opening of mitochondrial K-ATP channels. J Cardiovasc Pharmacol 44:35–41. [DOI] [PubMed] [Google Scholar]
- Haylor J, Singh I, el Nahas AM. 1991. Nitric oxide synthesis inhibitor prevents vasodilation by insulin-like growth factor I. Kidney Int 39:333–335. [DOI] [PubMed] [Google Scholar]
- House SL, Bolte C, Zhou M, Doetschman T, Klevitsky R, Newman G, Schultz Jel J. 2003. Cardiac-specific overexpression of fibroblast growth factor-2 protects against myocardial dysfunction and infarction in a murine model of low-flow ischemia. Circulation 108:3140–3148. [DOI] [PubMed] [Google Scholar]
- House SL, Branch K, Newman G, Doetschman T, Schultz Jel J. 2005. Cardioprotection induced by cardiac-specific overexpression of fibroblast growth factor-2 is mediated by the MAPK cascade. Am J Physiol Heart Circ Physiol 289: H2167–H2175. [DOI] [PubMed] [Google Scholar]
- House SL, Melhorn SJ, Newman G, Doetschman T, Schultz JE. 2007. The protein kinase C pathway mediates cardioprotection induced by cardiac-specific overexpression of fibroblast growth factor-2. Am J Physiol Heart Circ Physiol 293:H354–H365. [DOI] [PubMed] [Google Scholar]
- Huang Z, Chen K, Huang PL, Finklestein SP, Moskowitz MA. 1997. bFGF ameliorates focal ischemic injury by blood flow-independent mechanisms in eNOS mutant mice. Am J Physiol 272:H1401–H1405. [DOI] [PubMed] [Google Scholar]
- Iwai-Kanai E, Hasegawa K, Fujita M, Araki M, Yanazume T, Adachi S, Sasayama S. 2002. Basic fibroblast growth factor protects cardiac myocytes from iNOS-mediated apoptosis. J Cell Physiol 190:54–62. [DOI] [PubMed] [Google Scholar]
- Jaburek M, Costa AD, Burton JR, Costa CL, Garlid KD. 2006. Mitochondrial PKC epsilon and mitochondrial ATP-sensitive K + channel copurify and coreconstitute to form a functioning signaling module in proteoliposomes. Circ Res 99:878–883. [DOI] [PubMed] [Google Scholar]
- Jiang ZS, Padua RR, Ju H, Doble BW, Jin Y, Hao J, Cattini PA,Dixon IM, Kardami E. 2002. Acute protection of ischemic heart by FGF-2: Involvement of FGF-2 receptors and protein kinase C. Am J Physiol Heart Circ Physiol 282:H1071–H1080. [DOI] [PubMed] [Google Scholar]
- Jiang ZS, Srisakuldee W, Soulet F, Bouche G, Kardami E. 2004. Non-angiogenic FGF-2 protects the ischemic heart from injury, in the presence or absence of reperfusion. Cardiovasc Res 62: 154–166. [DOI] [PubMed] [Google Scholar]
- Jones SP, Bolli R. 2006. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol 40:16–23. [DOI] [PubMed] [Google Scholar]
- Kajita Y, Takayasu M, Yoshida J, Dietrich HH, Dacey RG Jr., 2001. Vasodilatory effect of basic fibroblast growth factor in isolated rat cerebral arterioles: Mechanisms involving nitric oxide and membrane hyperpolarization. Neurol Med Chir (Tokyo) 41: 177–185, discussion 185–176. [DOI] [PubMed] [Google Scholar]
- Kawano T, Zoga V, Kimura M, Liang MY, Wu HE, Gemes G, McCallum JB, Kwok WM, Hogan QH, Sarantopoulos CD. 2009. Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: Action by direct S-nitrosylation. Mol Pain 5:12–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda RK. 2010. Cardiac conditioning: A review of evolving strategies to reduce ischaemia-reperfusion injury. Heart 96: 1179–1186. [DOI] [PubMed] [Google Scholar]
- Kostyk SK, Kourembanas S, Wheeler EL, Medeiros D, McQuillan LP, D’Amore PA, Braunhut SJ. 1995. Basic fibroblast growth factor increases nitric oxide synthase production in bovine endothelial cells. Am J Physiol 269:H1583–H1589. [DOI] [PubMed] [Google Scholar]
- Kwak YG, Park SK, Cho KP, Chae SW. 1996. Reciprocal modulation of ATP-sensitive K + channel activity in rat ventricular myocytes by phosphorylation of tyrosine and serine/threonine residues. Life Sci 58:897–904. [DOI] [PubMed] [Google Scholar]
- Liao S 2008. The role of fibroblast growth factor-2 isoforms in ischemia–reperfusion injury and cardioprotection. Cincinnati: University of Cincinnati. [Google Scholar]
- Liao S, Porter D, Scott A, Newman G, Doetschman T, Schultz Jel J. 2007. The cardioprotective effect of the low molecular weight isoform of fibroblast growth factor-2: The role of JNK signaling. J Mol Cell Cardiol 42:106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S, Bodmer JR, Azhar M, Newman G, Coffin JD, Doetschman T, Schultz Jel J. 2010. The influence of FGF2 high molecular weight (HMW) isoforms in the development of cardiac ischemia-reperfusion injury. J Mol Cell Cardiol 48:1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ. 2000. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc Natl Acad Sci USA 97: 9058–9063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips DJ, Bueno OF, Wilkins BJ, Purcell NH, Kaiser RA, Lorenz JN, Voisin L, Saba-El-Leil MK, Meloche S, Pouyssegur J, Pages G, De Windt LJ, Doevendans PA, Molkentin JD. 2004. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation 109:1938–1941. [DOI] [PubMed] [Google Scholar]
- Marinovic J, Bosnjak ZJ, Stadnicka A. 2006. Distinct roles for sarcolemmal and mitochondrial adenosine triphosphate-sensitive potassium channels in isoflurane-induced protection against oxidative stress. Anesthesiology 105:98–104. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. 1993. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem 268:9194–9197. [PubMed] [Google Scholar]
- Massion PB, Balligand JL. 2003. Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): Lessons from genetically modified mice. J Physiol 546: 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell MB, Winter CB, Banerjee A, Harken AH. 1993. The relationship between ischemia–reperfusion injury, myocardial stunning and cardiac preconditioning. Surg Gynecol Obstet 177: 97–114. [PubMed] [Google Scholar]
- Moore WM, Webber RK, Jerome GM, Tjoeng FS, Misko TP, Currie MG. 1994. L-N6-(1-iminoethyl)lysine: A selective inhibitor of inducible nitric oxide synthase. J Med Chem 37: 3886–3888. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. 1986. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 74:1124–1136. [DOI] [PubMed] [Google Scholar]
- Namiki A, Akatsuka N. 1990. Vascular smooth muscle relaxation induced by epidermal growth factor is endothelium-dependent. Eur J Pharmacol 180:247–254. [DOI] [PubMed] [Google Scholar]
- O’Rourke B 2004. Evidence for mitochondrial K + channels and their role in cardioprotection. Circ Res 94:420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke B 2000. Myocardial K(ATP) channels in preconditioning. Circ Res 87:845–855. [DOI] [PubMed] [Google Scholar]
- Ohnuma Y, Miura T, Miki T, Tanno M, Kuno A, Tsuchida A, Shimamoto K. 2002. Opening of mitochondrial K(ATP) channel occurs downstream of PKC-epsilon activation in the mechanism of preconditioning. Am J Physiol Heart Circ Physiol 283:H440–H447. [DOI] [PubMed] [Google Scholar]
- Otani H 2009. The role of nitric oxide in myocardial repair and remodeling. Antioxid Redox Signal 11:1913–1928. [DOI] [PubMed] [Google Scholar]
- Padua RR, Sethi R, Dhalla NS, Kardami E. 1995. Basic fibroblast growth factor is cardioprotective in ischemia–reperfusion injury. Mol Cell Biochem 143:129–135. [DOI] [PubMed] [Google Scholar]
- Parenti A, Morbidelli L, Ledda F, Granger HJ, Ziche M. 2001. The bradykinin/B1 receptor promotes angiogenesis by up-regulation of endogenous FGF-2 in endothelium via the nitric oxide synthase pathway. Faseb J 15:1487–1489. [PubMed] [Google Scholar]
- Partovian C, Zhuang Z, Moodie K, Lin M, Ouchi N, Sessa WC, Walsh K, Simons M. 2005. PKCalpha activates eNOS and increases arterial blood flow in vivo. Circ Res 97:482–487. [DOI] [PubMed] [Google Scholar]
- Pasdois P, Beauvoit B, Costa AD, Vinassa B, Tariosse L, Bonoron-Adele S, Garlid KD, Dos Santos P. 2007. Sarcoplasmic ATP-sensitive potassium channel blocker HMR1098 protects the ischemic heart: Implication of calcium, complex I, reactive oxygen species and mitochondrial ATP-sensitive potassium channel. J Mol Cell Cardiol 42:631–642. [DOI] [PubMed] [Google Scholar]
- Philipp S, Critz SD, Cui L, Solodushko V, Cohen MV, Downey JM. 2006. Localizing extracellular signal-regulated kinase (ERK) in pharmacological preconditioning’s trigger pathway. Basic Res Cardiol 101:159–167. [DOI] [PubMed] [Google Scholar]
- Ping P, Takano H, Zhang J, Tang XL, Qiu Y, Li RC, Banerjee S, Dawn B, Balafonova Z, Bolli R. 1999. Isoform-selective activation of protein kinase C by nitric oxide in the heart of conscious rabbits: A signaling mechanism for both nitric oxide-induced and ischemia-induced preconditioning. Circ Res 84: 587–604. [DOI] [PubMed] [Google Scholar]
- Pomblum VJ, Korbmacher B, Cleveland S, Sunderdiek U, Klocke RC, Schipke JD. 2010. Cardiac stunning in the clinic: The full picture. Interact Cardiovasc Thorac Surg 10:86–91. [DOI] [PubMed] [Google Scholar]
- Rajashree R, Koster JC, Markova KP, Nichols CG, Hofmann PA. 2002. Contractility and ischemic response of hearts from transgenic mice with altered sarcolemmal K(ATP) channels. Am J Physiol Heart Circ Physiol 283:H584–H590. [DOI] [PubMed] [Google Scholar]
- Rameau GA, Chiu LY, Ziff EB. 2004. Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-D-aspartate receptor. J Biol Chem 279: 14307–14314. [DOI] [PubMed] [Google Scholar]
- Rameau GA, Tukey DS, Garcin-Hosfield ED, Titcombe RF, Misra C, Khatri L, Getzoff ED, Ziff EB. 2007. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J Neurosci 27:3445–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra WF, Paolocci N, Kass DA. 2002. Effects of cardioselective KATP channel antagonism on basal, stimulated, and ischaemic myocardial function in in vivo failing canine heart. Br J Pharmacol 135:657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva RM, Minhas KM, Raju SV, Barouch LA, Pitz E, Schuleri KH, Vandegaer K, Li D, Hare JM. 2005. Deficiency of neuronal nitric oxide synthase increases mortality and cardiac remodeling after myocardial infarction: Role of nitroso-redox equilibrium. Circulation 112:3415–3422. [DOI] [PubMed] [Google Scholar]
- Sato T, Sasaki N, Seharaseyon J, O’Rourke B, Marban E. 2000. Selective pharmacological agents implicate mitochondrial but not sarcolemmal K(ATP) channels in ischemic cardioprotection. Circulation 101:2418–2423. [DOI] [PubMed] [Google Scholar]
- Schultz JE, Qian YZ, Gross GJ, Kukreja RC. 1997. The ischemia-selective KATP channel antagonist, 5-hydroxydecanoate, blocks ischemic preconditioning in the rat heart. J Mol Cell Cardiol 29: 1055–1060. [DOI] [PubMed] [Google Scholar]
- Schulz R, Kelm M, Heusch G. 2004. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res 61:402–413. [DOI] [PubMed] [Google Scholar]
- Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, Wallis HL, Neubauer S, Terrar DA, Casadei B. 2003. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res 92:e52–e59. [DOI] [PubMed] [Google Scholar]
- Sharp BR, Jones SP, Rimmer DM, Lefer DJ. 2002. Differential response to myocardial reperfusion injury in eNOS-deficient mice. Am J Physiol Heart Circ Physiol 282:H2422–H2426. [DOI] [PubMed] [Google Scholar]
- Sheikh F, Sontag DP, Fandrich RR, Kardami E, Cattini PA. 2001. Overexpression of FGF-2 increases cardiac myocyte viability after injury in isolated mouse hearts. Am J Physiol Heart Circ Physiol 280:H1039–H1050. [DOI] [PubMed] [Google Scholar]
- Singh K, Balligand JL, Fischer TA, Smith TW, Kelly RA. 1996. Regulation of cytokine-inducible nitric oxide synthase in cardiac myocytes and microvascular endothelial cells. Role of extracellular signal-regulated kinases 1 and 2 (ERK1/ERK2) and STAT1 alpha. J Biol Chem 271:1111–1117. [DOI] [PubMed] [Google Scholar]
- Strohm C, Barancik T, Bruhl ML, Kilian SA, Schaper W. 2000. Inhibition of the ER-kinase cascade by PD98059 and UO126 counteracts ischemic preconditioning in pig myocardium. J Cardiovasc Pharmacol 36:218–229. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Sasaki N, Miki T, Sakamoto N, Ohmoto-Sekine Y, Tamagawa M, Seino S, Marban E, Nakaya H. 2002. Role of sarcolemmal K(ATP) channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest 109: 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiefenbacher CP, Chilian WM. 1997. Basic fibroblast growth factor and heparin influence coronary arteriolar tone by causing endothelium-dependent dilation. Cardiovasc Res 34:411–417. [DOI] [PubMed] [Google Scholar]
- Tsukahara H, Gordienko DV, Tonshoff B, Gelato MC, Goligorsky MS. 1994. Direct demonstration of insulin-like growth factor-I-induced nitric oxide production by endothelial cells. Kidney Int 45:598–604. [DOI] [PubMed] [Google Scholar]
- Tsuura Y, Ishida H, Hayashi S, Sakamoto K, Horie M, Seino Y. 1994. Nitric oxide opens ATP-sensitive K+ channels through suppression of phosphofructokinase activity and inhibits glucose-induced insulin release in pancreatic beta cells. J Gen Physiol 104:1079–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama Y, Otani H, Okada T, Uchiyama T, Ninomiya H, Kido M, Imamura H, Nakao S, Shingu K. 2003. Integrated pharmacological preconditioning in combination with adenosine, a mitochondrial KATP channel opener and a nitric oxide donor. J Thorac Cardiovasc Surg 126:148–159. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kudo M, Xu M, Ayub A, Ashraf M. 2001. Mitochondrial KATP channel as an end effector of cardioprotection during late preconditioning: Triggering role of nitric oxide. J Mol Cell Cardiol 33:2037–2046. [DOI] [PubMed] [Google Scholar]
- Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. 1999. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci USA 96:657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Ji X, Boysen PG. 2004. Exogenous nitric oxide generates ROS and induces cardioprotection: Involvement of PKG, mitochondrial KATP channels, and ERK. Am J Physiol Heart Circ Physiol 286:H1433–H1440. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Zhang MH, Sears CE, Emanuel K, Redwood C, El-Armouche A, Kranias EG, Casadei B. 2008. Reduced phospholamban phosphorylation is associated with impaired relaxation in left ventricular myocytes from neuronal NO synthase-deficient mice. Circ Res 102:242–249. [DOI] [PubMed] [Google Scholar]
- Zhao T, Xi L, Chelliah J, Levasseur JE, Kukreja RC. 2000. Inducible nitric oxide synthase mediates delayed myocardial protection induced by activation of adenosine A(1) receptors: Evidence from gene-knockout mice. Circulation 102:902–907. [DOI] [PubMed] [Google Scholar]