

Summary
The pathogenesis of cerebral ischaemia reperfusion injury (IRI) has not been fully described. Accordingly, there is little effective drug available for the treatment of cerebral IRI. The aim of our study was to explore the exact role played by Mfn1‐mediated mitochondrial protection in cerebral IRI and evaluate the beneficial action of resveratrol on reperfused brain. Our study demonstrated that hypoxia‐reoxygenation (HR) injury caused N2a cell apoptosis and this process was highly affected by mitochondrial dysfunction. Decreased mitochondrial membrane potential, increased mitochondrial oxidative stress, and an activated mitochondrial apoptosis pathway were noted in HR‐treated N2a cells. Interestingly, resveratrol treatment could attenuate N2a cell apoptosis via sustaining mitochondrial homeostasis. Further, we found that resveratrol modulated mitochondrial performance via activating the Mfn1‐related mitochondrial protective system. Knockdown of Mfn1 could abolish the beneficial effects of resveratrol on HR‐treated N2a cells. Besides, we also report that resveratrol regulated Mfn1 expression via the AMPK pathway; inhibition of AMPK pathway also neutralized the anti‐apoptotic effect of resveratrol on N2a cells in the setting of cerebral IRI. Taken together our results show that mitochondrial damage is closely associated with the progression of cerebral IRI. In addition we also demonstrate the protective action played by resveratrol on reperfused brain and show that this effect is achieved via activating the AMPK‐Mfn1 pathway.
Keywords: AMPK pathway, cerebral IR injury, Mfn1, mitochondrial stress, resveratrol
1. INTRODUCTION
Acute ischaemic stroke causes extensive death of neurons which increases the resultant disability and mortality significantly.1, 2 Accordingly, timely reperfusion therapy is important as an effective approach to reverse the damage to brain function and thus improve the prognosis in patients with ischaemia stroke.3, 4 However, reperfusion itself brings about additional damage to ischaemic brain tissue, and this is termed as ‘ischaemia reperfusion injury’ which accounts for increased perioperative mortality.5, 6 Although many experiments have been conducted over several decades in this field the molecular features, the underlying cerebral ischaemia reperfusion injury (IRI) have not been described comprehensively.7, 8
At the molecular levels, neuronal death is primarily modulated by three signalling pathways. These are mitochondrial apoptosis, endoplasmic reticulum‐related apoptosis and Fas‐mediated death.9, 10 Notably, brain tissues contain abundant mitochondria which consistently supply energy to sustain brain function such as neurotransmitters release and neuronal survival.11, 12 Mitochondrial apoptosis has been reported in the progression of cerebral IRI through a poorly understood mechanism.13, 14 At reperfusion stage, mitochondria produce the majority of ROS that are generated, and the latter alters the mitochondrial membrane potential.15, 16 Decreased mitochondrial potential impairs mitochondrial energy metabolism and further promotes mitochondrial apoptosis activation via induction of pro‐apoptotic factors leakage from the mitochondria into the cytoplasm.17, 18 However, a protective system for mitochondrial apoptosis has not been identified.19, 20 Recently, Mfn1‐mediated mitochondrial fusion has been recognised as a possible primary mechanism to reverse to mitochondrial homeostasis. Mfn1 promotes the fusion between fragmented mitochondria and sustains mitochondrial membrane potential.21, 22 In addition, Mfn1‐mediated mitochondrial protection also inhibits the mitochondrial apoptosis in several disease models such as atherosclerotic lesions, heart failure, ageing and kidney injury.23, 24 Interestingly, this concept has not been verified in the model of cerebral IRI.
From a pharmacological perspective, several drugs could be used to activate Mfn1 and thus reverse mitochondrial function in the setting of cerebral IRI. These include melatonin and liraglutide.25, 26 Resveratrol (3,5,4′‐trihydroxy‐trans‐stilbene) belongs to a class of compounds called plant polyphenols and is also a phytoalexin produced by plants during environmental stress and infections to help overcome their adverse effects.27, 28 This inherent ability to provide protection against stress conditions is the main feature that makes resveratrol an attractive candidate for prevention and treatment of human diseases. Ample evidence supports its therapeutic effects on reperfused brains. Resveratrol reduces neuronal oxidative stress, attenuates calcium overload, inhibits endoplasmic reticulum (ER) stress and blocks mitochondrial apoptosis.29, 30 In addition, the inhibitory action of resveratrol on mitochondrial fission has been reported in several careful studies. Interestingly, no studies have investigated the role of resveratrol in Mfn1‐mediated mitochondrial protection.
Mechanistically, several pathways have been reported to be the upstream regulator of mitochondrial homeostasis. Notably, AMPK has been reported to be associated with Mfn1 activation. For examples, in diabetic cardiomyopathy, activation of AMPK promotes Mfn1 upregulation and the latter reduces mitochondrial oxidative stress and inhibits caspase‐9 apoptosis. In cardiac ischaemia reperfusion, AMPK activation promotes Mfn1 upregulation and sustains mitochondrial function. Notably, no evidence has been shown thus far to establish the influence of AMPK on Mfn1 in the setting of cerebral IRI. Accordingly, the goal of our study is to explore the protective effect of resveratrol in cerebral reperfusion injury and mitochondrial function and verify whether resveratrol modulates mitochondrial protection and neuronal viability via the AMPK‐Mfn1 pathways.31, 32
2. MATERIALS AND METHODS
2.1. Cellular culture and treatment
The in vitro N2a cell line (American Type Culture Collection, ATCC® CL‐101™) was used for the hypoxia‐reoxygenation (HR) model to mimic IR injury in vivo. In the present study, 30 minutes of hypoxia and 4 hours of reoxygenation were used to induce cell damage. To suppress the AMPK pathway, compound C (CC; Selleck Chemicals) was added into the cell culture medium for two hours.33, 34
2.2. Ethical approval
Experimental protocol was approved by PLA Army General Hospital. All animal researches were carried out according to the guidelines of Animal Care and Use Committee of PLA Army General Hospital. The ethical approval number is PLA301‐ 2017–88JGH211.
2.3. TUNEL staining and 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT) assay
Cell death was measured via a TUNEL assay using an in situ cell death detection kit (Roche). The TUNEL kit was used to stain nuclei containing fragmented DNA. After the HR injury, the cells were fixed with 3.7% paraformaldehyde for 30 minutes at room temperature.35, 36 Subsequently, the samples were incubated with equilibration buffer, nucleotide mix and rTdT enzyme at 37°C for 60 minutes. Then, a saline‐sodium citrate buffer was used to stop the reaction. After being loaded with DAPI, the samples were visualized via fluorescence microscopy (Olympus BX‐61). In addition, an MTT assay was performed to analyse the cell viability according to methods described in a previous study. Absorbance was determined at 570 nm. The relative cell viability was recorded as a ratio to that in the control group.37, 38
2.4. Caspase activity detection and ELISA
Caspase‐3 and caspase‐9 activities were determined using commercial kits (Beyotime Institute of Biotechnology). The levels of antioxidant factors, including glutathione peroxidase (GPX), superoxide dismutase (SOD) and glutathione (GSH), were measured with ELISA kits purchased from Beyotime Institute of Biotechnology.39, 40
2.5. Transfection
Transfection with siRNA was used to inhibit Mfn1 expression in resveratrol‐treated N2a cells. Briefly, siRNA against Mfn1 (Yangzhou Ruibo Biotech Co., Ltd.) was used to infect cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.41, 42 The negative control group was transfected with negative control siRNA. Transfection was performed for approximately 48 hours. Then, Western blotting was used to observe the knockdown efficiency43 after harvesting the transfected cells.44, 45
2.6. Immunofluorescence analysis of mROS
Immunofluorescence analysis was used to analyse mitochondrial ROS (mROS) production. After the HR injury, the cells were washed three times with PBS and then resuspended in PBS using 0.25% trypsin. Subsequently, the cells were incubated with the MitoSOX red mitochondrial superoxide indicator (Molecular Probes, USA) for 15 minutes at 37°C in the dark. After three washes with PBS, mROS production was analysed via immunofluorescence analysis.46, 47
2.7. Western blotting
Each group of samples was collected, the cell lysates were prepared, and the total protein was extracted and quantified using a BCA protein concentration kit according to the manufacturer's instructions. The proteins were separated by sodium dodecyl sulphate‐polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membranes were blocked with 5% BSA in Tris‐buffered saline with Tween‐20 (TBST) for 2 hours at room temperature and incubated with primary antibodies overnight at 4°C. The membranes were washed three times with PBS; IRDye 800CW goat anti‐rabbit IgG (H + L) (926‐32211; Licor) was added; and the membranes were incubated at 37°C for 1 hour. The bands were detected using a chemiluminescent imaging system. The primary antibodies used in the present study were as follows: pro‐caspase 3 (1:1000; Abcam, #ab13847), cleaved caspase 3 (1:1000; Abcam, #ab49822), Bcl2 (1:1000; Cell Signaling Technology, #3498), Bax (1:1000; Cell Signaling Technology, #2772), caspase 9 (1:1000; Cell Signaling Technology, #9504), c‐IAP (1:1000; Cell Signaling Technology, #4952), cyt‐c (1:1000; Abcam; #ab90529), Drp1 (1:1000; Abcam, #ab56788), Opa1 (1:1000; Abcam, #ab42364), Mfn2 (1:1000; Abcam, #ab57602), Mff (1:1000; Cell Signaling Technology, #86668), p‐ERK (1:1000; Abcam, #ab176660), and t‐ERK (1:1000; Abcam #ab54230). Band intensities were normalized to the respective internal standard signal intensity GAPDH (1:1000; Cell Signaling Technology, #5174) and/or β‐actin (1:1000; Cell Signaling Technology, #4970) using quantity one Software (version 4.6.2; Bio‐Rad Laboratories, Inc).48, 49
2.8. Immunofluorescent staining and mitochondrial potential detection
After the HR injury, first, the cells were fixed with 4% paraformaldehyde for 30 minutes at room temperature. After a 10‐minute incubation with 3% hydrogen peroxide to block endogenous peroxidase activity, the samples were incubated with primary antibodies overnight at 4°C. Then, the slides were washed with PBS and incubated with a secondary antibody (1:500; Invitrogen) at room temperature for 45 minutes.50, 51 The nuclei were stained using DAPI. The images were acquired via fluorescence microscopy (Olympus BX‐61). The following primary antibodies were used in the present study: OPA1 (1:1000; Abcam, #ab42364), p‐ERK (1:1000; Abcam, #ab176660), and cyt‐c (1:1000; Abcam; #ab90529). The mitochondrial potential was measured using a JC‐1 kit (Beyotime Institute of Biotechnology). After the HR injury, the cells were washed three times with PBS and then incubated with fresh medium supplemented with 10 mg/mL JC‐1. After 30 minutes, the samples were washed three times with PBS to remove the free probe, and then, fresh medium was added. The samples were observed via fluorescence microscopy (Olympus BX‐61). The red/green fluorescence of JC‐1 was analysed using image‐pro plus version 4.5 (Media Cybernetics, Inc).47, 52
2.9. Statistical analysis
The statistical analyses were performed using spss 16.0 (SPSS, Inc). All results in the present study were obtained by one‐way analysis of variance, followed by Tukey's test. P < .05 was considered statistically significant.
3. RESULTS
3.1. Resveratrol sustains N2a cell survival under IRI
In the present study, N2a cells were used and an HR model was established to mimic the cerebral IRI in vitro. Cell viability was determined using an MTT assay. Compared to the control group, HR treatment reduced cell viability in N2a cells. Subsequently, to support the protective effect of resveratrol on N2a cells high and lower doses of resveratrol were added into the medium in the presence of HR injury. Then, cell viability was determined via the MTT assay. Compared to the HR injury group, low dose of resveratrol cannot improve the cell viability whereas high dose of resveratrol significantly increased cell viability in presence of HR injury (Figure 1A), indicative of the protective effect of resveratrol on the injured and reperfused N2a cells. This result was further supported via lactate dehydrogenase (LDH) release into the cell culture medium. Compared to the control group, HR injury elevated the cell death ratio and this effect was observed at high rather than low dosage resveratrol (Figure 1B).
Figure 1.
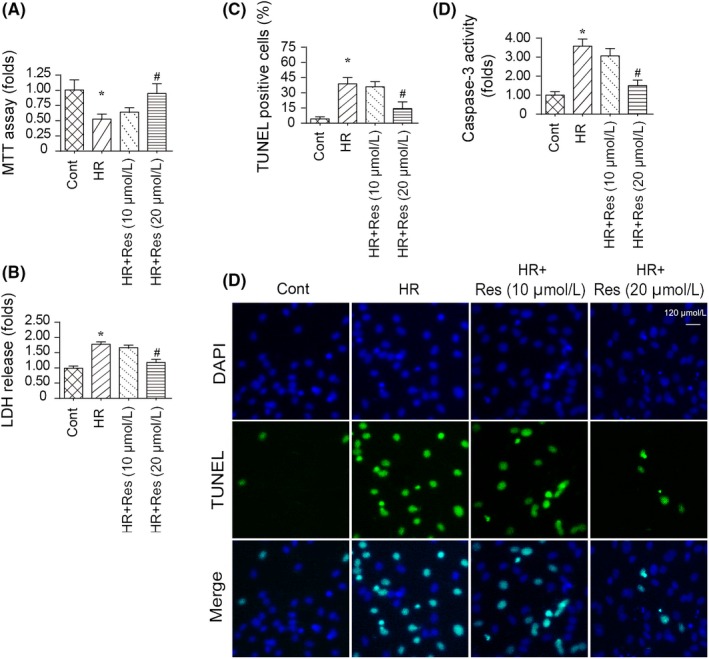
Resveratrol sustains N2a cell viability in response to hypoxia‐reoxygenation (HR) injury. A, MTT assay was used to confirm the alterations of N2a cell viability. Resveratrol was added into the medium of N2a cell at low and high dose. N2a cell was treated with HR injury. B, Lactate dehydrogenase (LDH) release assay was used to detect cell death in answer to HR injury. Low dose and high dose of resveratrol were used to incubate with N2a cell in the presence of HR injury. C‐D, TUNEL assay for apoptotic cell. The apoptotic N2a cells were stained by TUNEL‐positive cells, and then, the number of TUNEL‐positive cell was recorded. E, Caspase‐3 activity was measured using ELISA. Resveratrol was added into the medium of N2a cell at low and high dose. N2a cell was treated with HR injury. *P < .05 vs control group; # P < .05 vs HR group
To further support the protective action of resveratrol on HR injured N2a cells, a TUNEL assay was performed to quantify cell death.53, 54 As shown in Figure 1C and D, compared to the control group, HR injury increased the ratio of TUNEL‐positive cells, and this effect was reversed by high dose resveratrol. In addition, cell death was also monitored via measuring caspase‐3 activity. Compared to the control group, the activity of caspase‐3 was increased in response to HR injury (Figure 1E), indicative of caspase‐3 activation at the stage of reperfusion injury. Interestingly, resveratrol at high dose had the ability to prevent caspase‐3 activation. Altogether, these data verify the beneficial impact of resveratrol on N2a cells in the setting of HR injury.
3.2. Resveratrol inhibits mitochondrial apoptosis in N2a cell
There are three pathways that are potentially implicated in N2a cell apoptosis at the stage of reperfusion. These are the mitochondria‐related caspase‐9 apoptosis axis, the ER‐mediated caspase‐12 death pathway and the Fas‐dependent necrosis cascade. In the present study, mitochondrial apoptosis was determined in response to resveratrol treatment.55, 56 Firstly, capsase‐9 activity was determined via ELISA. As shown in Figure 2A, compared to the control group, HR injury elevated the caspase‐9 activity, implicating caspase‐9 at the stage of reperfusion injury. Interestingly, resveratrol at high dose had the ability to reduce capsase‐9 activity (Figure 2A). To further explain the mitochondria‐protective effect of resveratrol in HR‐attacked N2a cells, Western blotting was performed and the expression of mitochondrial apoptotic proteins was analysed. Compared to the control group, expression of both Bax and caspase‐9 was activated by HR injury whereas the levels of Bcl‐2 and survivin were significantly downregulated injury (Figure 2B‐F). This indicated that HR injury caused an imbalance between mitochondrial apoptosis‐related factors. Interestingly, resveratrol treatment reversed the levels of anti‐apoptotic proteins and reduced the pro‐apoptotic factors in the presence of HR injury (Figure 2B‐F), suggesting that mitochondrial apoptosis seems to be powerfully inhibited by resveratrol in N2a cells.
Figure 2.
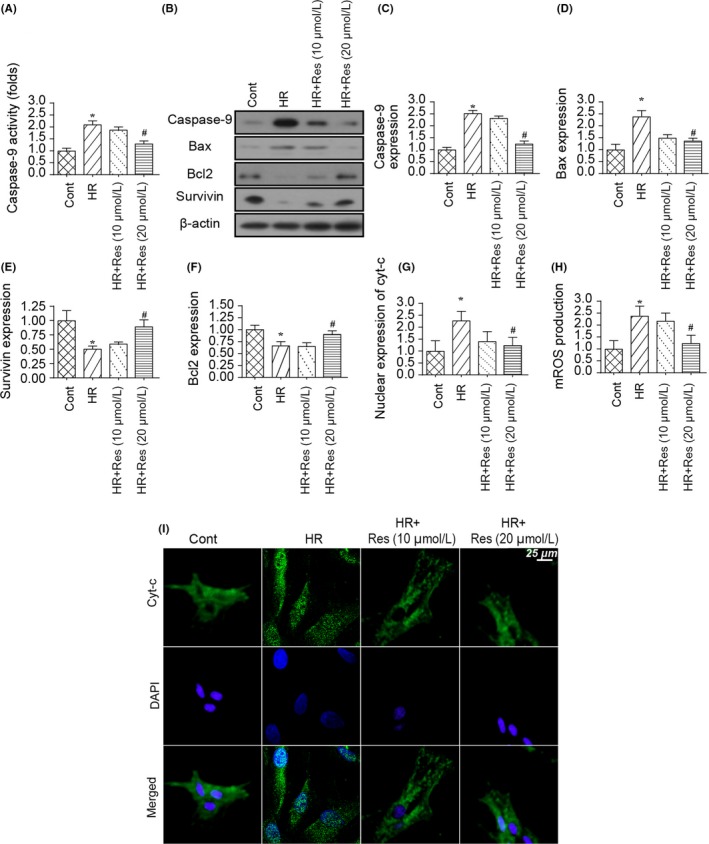
Mitochondrial apoptosis is inhibited by resveratrol in N2a cell. A, Caspase‐9 activity was determined using ELISA. Resveratrol was added into the medium of N2a cell at low and high dose. N2a cell was treated with hypoxia‐reoxygenation (HR) injury. B‐F, Western blotting was performed to observe the changes in mitochondrial apoptosis‐related proteins. Pro‐apoptotic proteins such as Bax and caspase‐9 were upregulated in response to HR injury and were reduced to near‐normal levels with resveratrol treatment. In contrast, anti‐apoptotic proteins such as Bcl‐2 and survivin were inhibited by HR injury and were reversed to near‐normal levels with resveratrol treatment. G‐H, Immunofluorescence assay for cyt‐c translocation from cytoplasm into nucleus. The expression of nuclear cyt‐c was determined. I, Mitochondrial permeability transition pore (mPTP) opening rate was determined using ELISA, and the mPTP opening was negatively modulated by resveratrol in the setting of cerebral IR injury. *P < .05 vs control group; # P < .05 vs HR group
Based on previous studies, the upstream trigger of mitochondrial apoptosis is the release of cyt‐c from mitochondria into the nucleus. To verify the mitochondrial cyt‐c translocation, an immunofluorescence assay was used. As shown in Figure 2G‐H, compared to the control group, HR injury promoted the migration of cyt‐c from cytoplasm into the nucleus, and this effect could be impaired by resveratrol treatment at high dose. Further, cyt‐c translocation was highly dependent on mitochondrial permeability transition pore (mPTP) opening.57, 58 Interestingly, mPTP opening rate could be augmented by HR injury and was inhibited by resveratrol in the setting of HR injury (Figure 2I). Altogether, these data inform us that mitochondrial apoptosis is activated by HR injury in N2a cells whereas resveratrol treatment sends a pro‐survival signal for N2a cells via inhibition of the mitochondrial apoptosis pathway.
3.3. Resveratrol modulates mitochondrial function and energy metabolism in the setting of IRI
Mitochondrial dysfunction and disorder of bioenergetics have been acknowledged as the early events for mitochondrial apoptosis. Accordingly, mitochondrial function and energy metabolism were determined in N2a cell in the presence of HR injury. As shown in Figure 3A,B, compared to the control group, mitochondrial membrane potential was reduced to near‐normal levels. Interestingly, resveratrol treatment reversed mitochondrial membrane potential, as evidenced by increased red‐to‐green fluorescence ratio.59, 60 Besides, mitochondrial function was also determined via measuring adenosine triphosphate (ATP) content in N2a cells. As shown in Figure 3C, compared to the control group, HR injury reduced ATP production in N2a cells, and this alteration could be reversed by high dose resveratrol. Thus demonstrating the beneficial effects of resveratrol on mitochondria at the stage of reperfusion injury.
Figure 3.
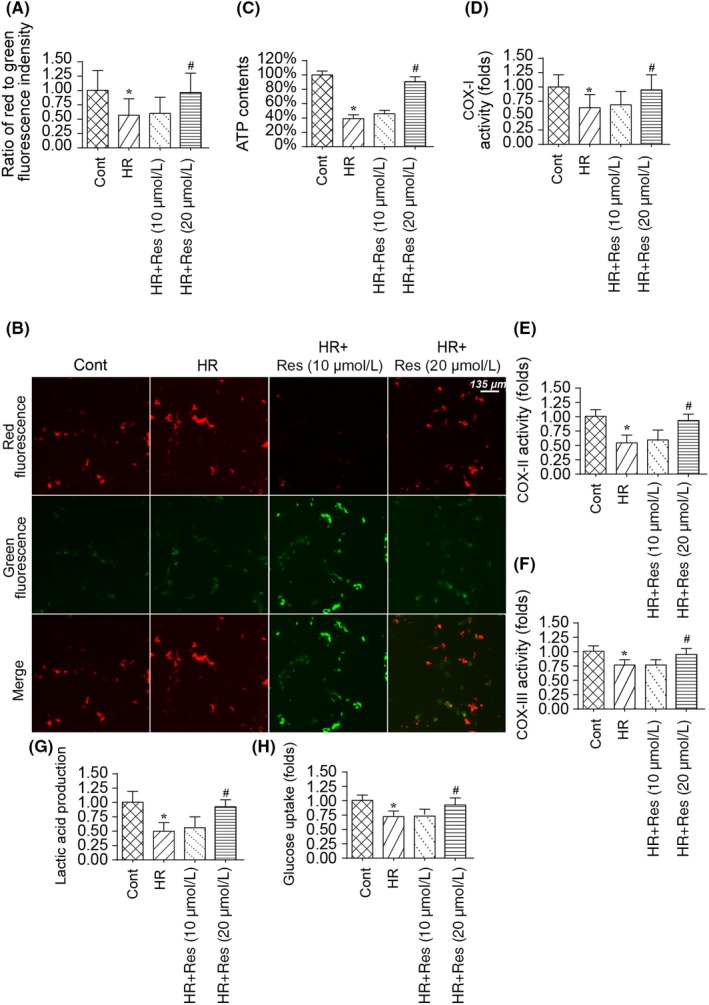
Resveratrol improves mitochondrial energy metabolism in hypoxia‐reoxygenation (HR)‐treated N2a cells. A‐B, JC‐1 probe was used to label mitochondrial membrane potential. Red fluorescence intensity indicates the normal mitochondrial membrane potential and green fluorescence means the damage mitochondrial membrane potential. C, Adenosine triphosphate (ATP) production was determined via ELISA. Resveratrol was added into the medium of N2a cell at low and high dose. N2a cell was treated with HR injury. D‐F, Mitochondrial respiratory complex activity was detected via ELISA. HR injury elevated the activity of mitochondrial respiratory complex activity and this effect could be inhibited by resveratrol treatment. G‐H, The uptake of glucose and lactic acid production was measured using ELISA. Resveratrol was added into the medium of N2a cell at low and high dose. N2a cell was treated with HR injury. *P < .05 vs control group; # P < .05 vs HR group
Subsequently, mitochondrial metabolism was determined via analysing the activity of the mitochondrial respiratory complex. As shown in Figure 3D‐F, compared to the control group, HR injury reduced the activity of the mitochondrial respiratory complex, and their phenotypic alterations could be reversed by resveratrol at high dose. This information indicates that resveratrol sustains mitochondrial respiratory function in the setting of reperfusion injury.61, 62 In addition to mitochondrial respiratory function, we also measured glucose uptake and lactic acid production. As shown in Figure 3C‐H, compared to the control group, HR injury reduced glucose uptake and inhibited lactic acid production, suggestive of inhibition of glucose metabolism. Interestingly, resveratrol treatment reversed the glucose uptake and promoted lactic acid production in N2a cells at the stage of reperfusion injury. Altogether, these results support the functional importance of resveratrol on mitochondrial protection in reperfused‐attacked N2a cells.
3.4. Resveratrol inhibits mitochondrial oxidative stress induced by IRI
In addition to the mitochondrial energy metabolism disorder, we also observed alterations in mitochondrial oxidative stress with resveratrol treatment. Firstly, mitochondrial ROS production was determined via immunofluorescence.63 As shown in Figure 4A,B, compared to the control group, the ROS levels were significantly increased in HR‐treated N2a cells, and this effect could be reversed by high dose of resveratrol in the presence of HR injury, suggestive of the antioxidative action of resveratrol on reperfused‐treated N2a cells. Furthermore, to provide more evidence for the alterations of cell redox balance, an ELISA assay was used to measure the activity of cell antioxidant. Compared to the control group, HR injury reduced the levels of SOD, GSH and MDA (Figure 4C‐E). Interestingly, resveratrol treatment reversed the content of SOD, GSH and MDA in the presence of HR injury (Figure 4C‐E). Altogether, this information indicated that resveratrol modulated mitochondrial oxidative stress in the setting of cerebral IR injury.
Figure 4.
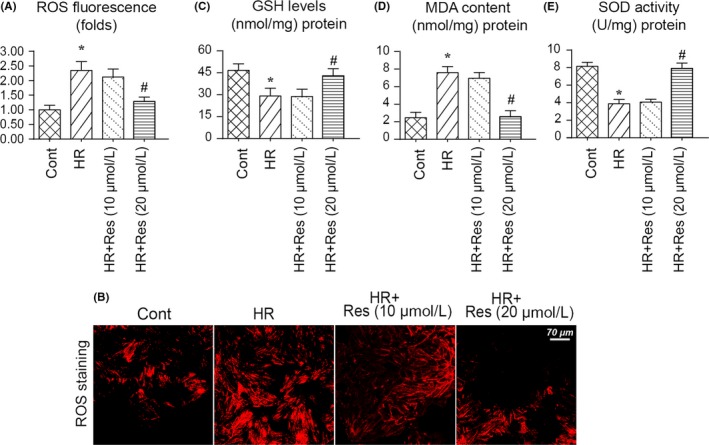
Resveratrol treatment attenuates hypoxia‐reoxygenation (HR)‐mediated mitochondrial oxidative stress in N2a cell. A‐B, Mitochondrial oxidative stress was evaluated via immunofluorescence. C‐E, ELISA was used to measure the change in cellular antioxidants such as superoxide dismutase (SOD), glutathione (GSH) and glutathione peroxidase (GPX). *P < .05 vs control group; # P < .05 vs HR group
3.5. Resveratrol activates Mfn1‐related mitochondrial protective system
Subsequently, we explored the protective mechanism by which resveratrol affected the mitochondrial homeostasis and N2a cell viability.64 Mfn1‐related mitochondrial protective system has been considered as a potential regulatory mechanism sustaining mitochondrial homeostasis via multiple mechanisms. Therefore, we asked whether a Mfn1‐related mitochondrial protection system is activated by resveratrol and promoted mitochondrial homeostasis. First, Western blotting was used to detect the expression of Mfn1 in response to resveratrol treatment. Compared to the control group, HR injury reduced the expression of Mfn1 (Figure 5A,B), indicative of Mfn1 inhibition by HR injury. Interestingly, resveratrol treatment reversed the expression of Mfn1 in the presence of HR injury (Figure 5A,B). To confirm the necessary role of Mfn1 in mitochondrial protection and N2a cell viability, siRNA against Mfn1 was transfected into resveratrol‐treated N2a cells. The knockdown efficiency was verified via Western blotting. As shown in Figure 5A,B, transfection of Mfn1 siRNA prevented resveratrol‐mediated Mfn1 upregulation in the presence of HR injury. Subsequently, N2a cell viability was determined in response to Mfn1 knockdown. As shown in Figure 5C, compared to the control group, HR injury reduced N2a cell viability and this alteration could be reversed by resveratrol. Interestingly, loss of Mfn1 abolished the protective effect of resveratrol on N2a cell viability. Similar results were also obtained via the LDH release assay. HR‐mediated LDH release could be inhibited by resveratrol: and this effect was negated by Mfn1 siRNA (Figure 5D).
Figure 5.
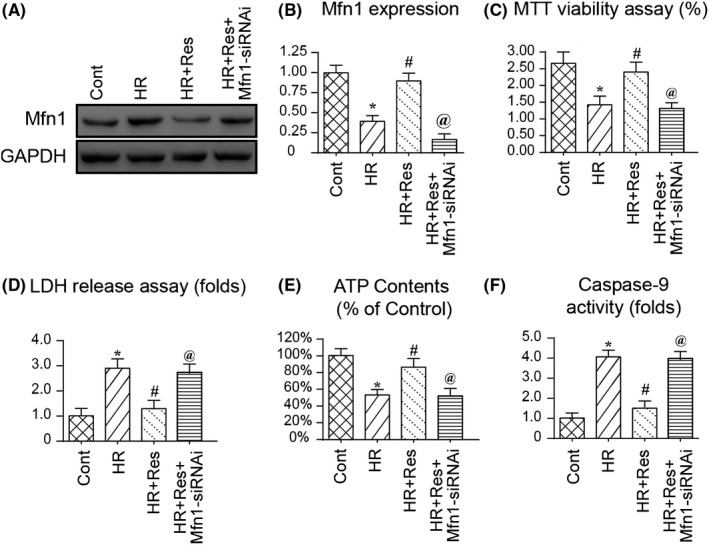
Mfn1‐mediated mitochondrial protection system is activated by resveratrol in the presence of hypoxia‐reoxygenation (HR) injury. A‐B, Western blotting was used to observe the alterations of Mfn1 expression in response to HR injury and/or resveratrol treatment. Besides, siRNA against Mfn1 was transfected into N2a cell and the knockdown efficiency of N2a cell was determined via Western blotting. C, Cell viability was measured via MTT assay. D, Lactate dehydrogenase (LDH) release assay was used to verify the cell death in response to Mfn1 knockdown. E, Adenosine triphosphate (ATP) production was detected via ELISA. Resveratrol was added into the medium of N2a cell at low and high dose. N2a cell was treated with HR injury. F, Caspase‐9 activity was measured to reflect the mitochondrial damage in response to Mfn1 deletion. *P < .05 vs control group; # P < .05 vs HR group; @ P < .05 vs HR + Res group
With respect to mitochondrial function, ATP production was determined in response to Mfn1 knockdown. As shown in Figure 5E, compared to the control group, HR‐repressed ATP production could be reversed by resveratrol. Interestingly, knockdown of Mfn1 impaired resveratrol‐mediated ATP upregulation in the presence of HR injury. In addition, caspase‐9 activity was activated by HR injury and was reversed to near‐normal levels with resveratrol treatment (Figure 5F). Interestingly, the inhibitory effect of resveratrol on caspase‐9 was dependent on Mfn1 because loss of Mfn1 abolished the regulatory effects of resveratrol on caspase‐9. Altogether, the above information indicates that Mfn1 is required for resveratrol‐mediated mitochondrial protection and N2a cell survival.
3.6. Resveratrol affects Mfn1 via AMPK pathway
To the end, we asked the molecular mechanism by which resveratrol regulated Mfn1. Based on a previous study, the AMPK pathway is involved in Mfn1 regulation in several disease models such as cardiac microvascular IR injury and cardiac cardiomyopathy. Therefore, in the present study, we asked whether AMPK pathway was involved in Mfn1 regulation in cerebral IR injury.62 Compared to the control group, HR injury reduced the activity of AMPK, as evidenced by decreased p‐AMPK expression (Figure 6A‐C). Besides, Mfn1 expression was also downregulated in response to HR injury (Figure 6A‐C). Interestingly, resveratrol treatment reversed p‐AMPK expression, an effect that was accompanied with an increase in Mfn1 in the presence of HR injury (Figure 6A‐C). To confirm whether AMPK was involved in Mfn1 regulation, the pathway blocker was added into resveratrol‐treated N2a cells. Notably, treatment with compound c could prevent resveratrol‐mediated AMPK activation (Figure 6A‐C), an effect that was accompanied with a drop in Mfn1. This information indicates that resveratrol activates Mfn1 in a manner dependent on AMPK pathway.
Figure 6.
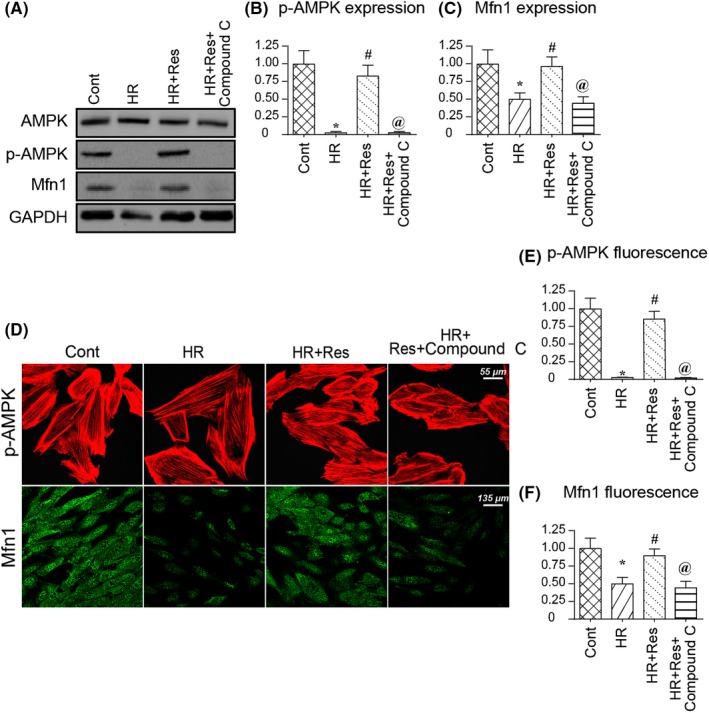
Resveratrol modulates Mfn1 expression via AMPK pathway. A‐C, Western blotting was used to confirm the alterations of Mfn1 and p‐AMPK. Compound C (CC) was used to prevent AMPK activation induced by resveratrol in the presence of hypoxia‐reoxygenation (HR) injury. D‐F, Immunofluorescence assay was used to detect the change in Mfn1 and p‐AMPK in response to resveratrol and CC. The relative expression of fluorescence intensity was recorded to reflect the alterations of p‐AMPK and Mfn1. *P < .05 vs control group; # P < .05 vs HR group; @ P < .05 vs HR + Res group
To provide more evidence for the alterations of Mfn1 and p‐AMPK in response to resveratrol treatment,61 immunofluorescence assay was used to determine the alteration of Mfn1 and p‐AMPK expression. As shown in Figure 6D‐F, compared to the control group, HR injury reduced the fluorescence intensity of Mfn1 and p‐AMPK. Interestingly, resveratrol treatment reversed the levels of p‐AMPK and Mfn1, and these alterations could be repressed by compound c (Figure 6D‐F). Altogether, our information confirmed that resveratrol modulated Mfn1 expression via activating the AMPK pathway.
4. DISCUSSION
In the present study we report that cerebral IR injury was associated with mitochondrial stress, as evidenced by decreased mitochondrial membrane potential, increased mitochondrial oxidative stress, reduced mitochondrial energy metabolism, and activated mitochondrial apoptosis. Interestingly, resveratrol treatment could attenuate reperfusion‐mediated mitochondrial stress and this beneficial effect seems to be associated with Mfn1‐mediated mitochondrial protection.65 Loss of Mfn1 abolished mitochondrial protection exerted by resveratrol. In addition, we also found that resveratrol modulated Mfn1 in a manner dependent on the AMPK pathway; and blockade of AMPK abolished the beneficial effect of resveratrol on Mfn1 upregulation. Therefore, the key finding of our study is that mitochondria damage is the potential target of cerebral IR injury and resveratrol has the ability to sustain neuronal viability via improving mitochondrial performance by modulating the AMPK‐Mfn1 pathway.66 As far as we know, this is the first study to investigate the detailed role played by mitochondrial stress in the progression of cerebral IRI. Meanwhile, we also explained the regulatory mechanism exerted by resveratrol on mitochondrial injury and neuronal viability. This finding pave a new way to explore an effective approach for the treatment of the cerebral IRI in the future.
The influence of mitochondrial stress on neuronal injury has been extensively explored. For example, in spinal cord injury, mitochondrial dysfunction is associated with neuronal apoptosis in a manner dependent on Akt‐GSK3β pathway.67, 68 In hippocampal neuron development, mitochondrial dysfunction promotes chronic energy depletion due to iron deficiency. Besides, attenuation of mitochondrial oxidative stress via antioxidant mitoQ improves RPS‐mediated sensorineural hearing loss. In ischaemia stroke, activation of mitochondrial autophagy sustains cerebral function via modulating neuronal viability. More importantly, blood‐brain barrier dysfunction, chronic neurodegeneration, Alzheimer's disease, Huntington's disease, and mitochondrial stress are all interlinked through various pathways with neuronal viability. In the present study, our data established a centre role played by mitochondria in brain ischaemia reperfusion stress. This information provides a piece of evidence to support the functional importance of mitochondria in cerebral homeostasis.
In the present study, we found that resveratrol effectively attenuated reperfusion‐mediated neuronal, injury. This finding was similar to the previous studies. For examples, in neurodegeneration‐mediated microglia polarization, resveratrol is shown to promote microglia survival in a manner dependent on autophagy. In Alzheimer's disease, resveratrol exerts neuroprotective activation via upregulating Sirt1 expression. In the models of cerebral ischaemia, stress‐induced depressive‐like behaviour, neuropathic pain, diabetic retinopathy, early brain injury after subarachnoid haemorrhage and memory dysfunction, the therapeutic effect of resveratrol has been widely reported.69, 70 In the present study, we found that resveratrol regulated cerebral IR injury via modulating mitochondrial function. Resveratrol reduced mitochondrial oxidative stress, improved mitochondrial energy metabolism, repressed mitochondrial apoptosis, finally sending a pro‐survival signals for reperfused neurons. Overall our results, combined with the previous studies, comprehensively highlight the neuroprotective actions of resveratrol. Notably, more additional experiments should be performed to support our findings in clinical trials.
At the molecular levels, resveratrol regulates mitochondrial function via the AMPK‐Mfn1 pathway. AMPK is a pro‐survival signalling in various disease models.43, 71 For example, AMPK affects Drp1 stabilization and consequently sustains mitochondrial function in diabetic cardiomyopathy. Besides, in oxidative stress‐mediated liver injury, the AMPK pathway could modulate mitochondrial redox balance via CaMKK2 pathway. Similarly, in myocardial ischaemia reperfusion injury, AMPK activates OPA‐required mitochondrial fusion and the latter inhibits mitochondrial apoptosis. Furthermore, in liver cancer, the AMPK pathway modulates mitochondrial homeostasis via upregulating PGC1 expression.72, 73 In addition to Mfn1, several research studies have established the role of Mfn1 in mitochondrial damage. For example, Mfn1 activation is associated with mitochondrial autophagy activation. Besides, Mfn1 also modulates mitochondrial fusion and the latter represses mitochondrial fission. Moreover, Mfn1 activation promotes mitochondrial synthesis and energy metabolism. In the present study, we report that the AMPK‐Mfn1 pathway is closely regulated by resveratrol.74, 75 This finding provides an easy and effective approach to activate protective the AMPK‐Mfn1 pathway in the setting of cerebral IR injury.76
5. CONCLUSION
Overall, our data demonstrate the pathogenesis of cerebral reperfusion injury is linked to mitochondrial stress77, 78 including mitochondrial redox imbalance and mitochondrial apoptosis. However, resveratrol treatment could activate the AMPK‐Mfn1 axis and therefore sustains mitochondrial function, favouring neuron survival in the context of cerebral IR injury.79
CONFLICT OF INTEREST
The authors have declared that they have no conflicts of interest.
AUTHORS' CONTRIBUTION
JBG and YJL conceived the research; WDL and HJW performed the experiments; all authors participated in discussing and revising the manuscript.
ACKNOWLEDGEMENTS
Not applicable.
Gao J, Wang H, Li Y, Li W. Resveratrol attenuates cerebral ischaemia reperfusion injury via modulating mitochondrial dynamics homeostasis and activating AMPK‐Mfn1 pathway. Int J Exp Path. 2020;100:337–349. 10.1111/iep.12336
Gao and Wang contributed equally to this work.
Funding information
This work was supported by the National Natural Science Foundation of China (Grant/Award Numbers: 81771347) and Key Military Research Projects On Equipment.
REFERENCES
- 1. Aanhane E, Schulkens IA, Heusschen R, et al. Different angioregulatory activity of monovalent galectin‐9 isoforms. Angiogenesis. 2018;21:545‐555. [DOI] [PubMed] [Google Scholar]
- 2. Araki M, Hisamitsu T, Kinugasa‐Katayama Y, et al. Serum/glucocorticoid‐regulated kinase 1 as a novel transcriptional target of bone morphogenetic protein‐ALK1 receptor signaling in vascular endothelial cells. Angiogenesis. 2018;21:415‐423. [DOI] [PubMed] [Google Scholar]
- 3. Abukar Y, Ramchandra R, Hood SG, et al. Increased cardiac sympathetic nerve activity in ovine heart failure is reduced by lesion of the area postrema, but not lamina terminalis. Basic Res Cardiol. 2018;113:35. [DOI] [PubMed] [Google Scholar]
- 4. Botker HE, Hausenloy D, Andreadou I, et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol. 2018;113:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Afonso MB, Rodrigues PM, Simao AL, et al. miRNA‐21 ablation protects against liver injury and necroptosis in cholestasis. Cell Death Differ. 2018;25:857‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alvarez‐Fernandez M, Sanz‐Flores M, Sanz‐Castillo B, et al. Therapeutic relevance of the PP2A‐B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ. 2018;25:828‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abeysuriya RG, Lockley SW, Robinson PA, et al. A unified model of melatonin, 6‐sulfatoxymelatonin, and sleep dynamics. J Pineal Res. 2018;64:e12474. [DOI] [PubMed] [Google Scholar]
- 8. Zhou H, Ma Q, Zhu P, et al. Protective role of melatonin in cardiac ischemia-reperfusion injury: from pathogenesis to targeted therapy. J Pineal Res. 2018;64(3):e12471. [DOI] [PubMed] [Google Scholar]
- 9. Angelova PR, Barilani M, Lovejoy C, et al. Mitochondrial dysfunction in Parkinsonian mesenchymal stem cells impairs differentiation. Redox Biol. 2018;14:474‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Armartmuntree N, Murata M, Techasen A, et al. Prolonged oxidative stress down‐regulates Early B cell factor 1 with inhibition of its tumor suppressive function against cholangiocarcinoma genesis. Redox Biol. 2018;14:637‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crist AM, Lee AR, Patel NR, et al. Vascular deficiency of Smad4 causes arteriovenous malformations: a mouse model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis. 2018;21:363‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Daniel E, Azizoglu DB, Ryan AR, et al. Spatiotemporal heterogeneity and patterning of developing renal blood vessels. Angiogenesis. 2018;21:617‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coverstone ED, Bach RG, Chen L, et al. A novel genetic marker of decreased inflammation and improved survival after acute myocardial infarction. Basic Res Cardiol. 2018;113:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davidson SM, Arjun S, Basalay MV, et al. The 10th Biennial Hatter Cardiovascular Institute workshop: cellular protection‐evaluating new directions in the setting of myocardial infarction, ischaemic stroke, and cardio‐oncology. Basic Res Cardiol. 2018;113:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bagati A, Bianchi‐Smiraglia A, Moparthy S, et al. FOXQ1 controls the induced differentiation of melanocytic cells. Cell Death Differ. 2018;25:1040‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bellomo C, Caja L, Fabregat I, et al. Snail mediates crosstalk between TGFbeta and LXRalpha in hepatocellular carcinoma. Cell Death Differ. 2018;25:885‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brazao V, Colato RP, Santello FH, et al. Effects of melatonin on thymic and oxidative stress dysfunctions during Trypanosoma cruzi infection. J Pineal Res. 2018;65:e12510. [DOI] [PubMed] [Google Scholar]
- 18. Ding M, Ning J, Feng N, et al. Dynamin‐related protein 1‐mediated mitochondrial fission contributes to post‐traumatic cardiac dysfunction in rats and the protective effect of melatonin. J Pineal Res. 2018;64:e12447. [DOI] [PubMed] [Google Scholar]
- 19. Ba X, Boldogh I. 8‐Oxoguanine DNA glycosylase 1: beyond repair of the oxidatively modified base lesions. Redox Biol. 2018;14:669‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Biernacki M, Ambrozewicz E, Gegotek A, et al. Redox system and phospholipid metabolism in the kidney of hypertensive rats after FAAH inhibitor URB597 administration. Redox Biol. 2018;15:41‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dingle AM, Yap KK, Gerrand YW, et al. Characterization of isolated liver sinusoidal endothelial cells for liver bioengineering. Angiogenesis. 2018;21:581‐597. [DOI] [PubMed] [Google Scholar]
- 22. Farber G, Hurtado R, Loh S, et al. Glomerular endothelial cell maturation depends on ADAM10, a key regulator of Notch signaling. Angiogenesis. 2018;21:335‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DeLeon‐Pennell KY, Mouton AJ, Ero OK, et al. LXR/RXR signaling and neutrophil phenotype following myocardial infarction classify sex differences in remodeling. Basic Res Cardiol. 2018;113:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deussen A. Mechanisms underlying coronary autoregulation continue to await clarification. Basic Res Cardiol. 2018;113:34. [DOI] [PubMed] [Google Scholar]
- 25. Cabon L, Bertaux A, Brunelle‐Navas MN, et al. AIF loss deregulates hematopoiesis and reveals different adaptive metabolic responses in bone marrow cells and thymocytes. Cell Death Differ. 2018;25:983‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Capece D, D'Andrea D, Verzella D, et al. Turning an old GADD get into a troublemaker. Cell Death Differ. 2018;25:640‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fan T, Pi H, Li M, et al. Inhibiting MT2‐TFE3‐dependent autophagy enhances melatonin‐induced apoptosis in tongue squamous cell carcinoma. J Pineal Res. 2018;64:e12457. [DOI] [PubMed] [Google Scholar]
- 28. Fernandez Vazquez G, Reiter RJ, Agil A. Melatonin increases brown adipose tissue mass and function in Zucker diabetic fatty rats: implications for obesity control. J Pineal Res. 2018;64:e12472. [DOI] [PubMed] [Google Scholar]
- 29. Cheignon C, Tomas M, Bonnefont‐Rousselot D, et al. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018;14:450‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen T, Dai SH, Li X, et al. Sirt1‐Sirt3 axis regulates human blood‐brain barrier permeability in response to ischemia. Redox Biol. 2018;14:229‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Care A, Bellenghi M, Matarrese P, et al. Sex disparity in cancer: roles of microRNAs and related functional players. Cell Death Differ. 2018;25:477‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen H, Kankel MW, Su SC, et al. Exploring the genetics and non‐cell autonomous mechanisms underlying ALS/FTLD. Cell Death Differ. 2018;25:646‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Edwards KS, Ashraf S, Lomax TM, et al. Uncoupling protein 3 deficiency impairs myocardial fatty acid oxidation and contractile recovery following ischemia/reperfusion. Basic Res Cardiol. 2018;113:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jung M, Dodsworth M, Thum T. Inflammatory cells and their non‐coding RNAs as targets for treating myocardial infarction. Basic Res Cardiol. 2018;114:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Y, Liu K, Shi Y, et al. The tango of ROS and p53 in tissue stem cells. Cell Death Differ. 2018;25:637‐639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cieri D, Vicario M, Giacomello M, et al. SPLICS: a split green fluorescent protein‐based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018;25:1131‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hardeland R. Melatonin and inflammation‐story of a double‐edged blade. J Pineal Res. 2018;65:e12525. [DOI] [PubMed] [Google Scholar]
- 38. Hobson SR, Gurusinghe S, Lim R, et al. Melatonin improves endothelial function in vitro and prolongs pregnancy in women with early‐onset preeclampsia. J Pineal Res. 2018;65:e12508. [DOI] [PubMed] [Google Scholar]
- 39. Cortese‐Krott MM, Mergia E, Kramer CM, et al. Identification of a soluble guanylate cyclase in RBCs: preserved activity in patients with coronary artery disease. Redox Biol. 2018;14:328‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cremonini E, Wang Z, Bettaieb A, et al. (‐)‐Epicatechin protects the intestinal barrier from high fat diet‐induced permeabilization: implications for steatosis and insulin resistance. Redox Biol. 2018;14:588‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Herz K, Becker A, Shi C, et al. Visualization of endothelial cell cycle dynamics in mouse using the Flt‐1/eGFP‐anillin system. Angiogenesis. 2018;21:349‐361. [DOI] [PubMed] [Google Scholar]
- 42. Klingler M, Decristoforo C, Rangger C, et al. Site‐specific stabilization of minigastrin analogs against enzymatic degradation for enhanced cholecystokinin‐2 receptor targeting. Theranostics. 2018;8(11):2896‐2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mensink M, Anstee NS, Robati M, et al. Anti‐apoptotic A1 is not essential for lymphoma development in Emicro‐Myc mice but helps sustain transplanted Emicro‐Myc tumour cells. Cell Death Differ. 2018;25:795‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou H, Li N, Yuan Y, et al. Activating transcription factor 3 in cardiovascular diseases: a potential therapeutic target. Basic Res Cardiol. 2018;113:37. [DOI] [PubMed] [Google Scholar]
- 45. Ter Horst EN, Krijnen PAJ, Hakimzadeh N, et al. Elevated monocyte‐specific type I interferon signalling correlates positively with cardiac healing in myocardial infarct patients but interferon alpha application deteriorates myocardial healing in rats. Basic Res Cardiol. 2018;114:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang M, Zhang Y, Xu E, et al. Rbm24, a target of p53, is necessary for proper expression of p53 and heart development. Cell Death Differ. 2018;25:1118‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Y, Chen Y, Guan L, et al. Carnitine palmitoyltransferase 1C regulates cancer cell senescence through mitochondria‐associated metabolic reprogramming. Cell Death Differ. 2018;25:733‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou H, Du W, Li Y, et al. Effects of melatonin on fatty liver disease: the role of NR4A1/DNA‐PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res. 2018;64:e12450. [DOI] [PubMed] [Google Scholar]
- 49. Zhao Z, Lu C, Li T, et al. The protective effect of melatonin on brain ischemia and reperfusion in rats and humans: in vivo assessment and a randomized controlled trial. J Pineal Res. 2018;65:e12521. [DOI] [PubMed] [Google Scholar]
- 50. Zhou YQ, Liu DQ, Chen SP, et al. Reactive oxygen species scavengers ameliorate mechanical allodynia in a rat model of cancer‐induced bone pain. Redox Biol. 2018;14:391‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou L, Zhang H, Davies KJA, et al. Aging‐related decline in the induction of Nrf2‐regulated antioxidant genes in human bronchial epithelial cells. Redox Biol. 2018;14:35‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu R, Garcia‐Barros M, Wen S, et al. Tumor suppressor p53 links ceramide metabolism to DNA damage response through alkaline ceramidase 2. Cell Death Differ. 2018;25:841‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X, Ha T, Liu L, et al. TLR3 mediates repair and regeneration of damaged neonatal heart through glycolysis dependent YAP1 regulated miR‐152 expression. Cell Death Differ. 2018;25:966‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang L, Tu Z, Liu C, et al. Dual roles of TRF1 in tethering telomeres to the nuclear envelope and protecting them from fusion during meiosis. Cell Death Differ. 2018;25:1174‐1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meyer IS, Leuschner F. The role of Wnt signaling in the healing myocardium: a focus on cell specificity. Basic Res Cardiol. 2018;113:44. [DOI] [PubMed] [Google Scholar]
- 56. Merz J, Albrecht P, von Garlen S, et al. Purinergic receptor Y2 (P2Y2)‐ dependent VCAM‐1 expression promotes immune cell infiltration in metabolic syndrome. Basic Res Cardiol. 2018;113:45. [DOI] [PubMed] [Google Scholar]
- 57. Skobowiat C, Brozyna AA, Janjetovic Z, et al. Melatonin and its derivatives counteract the ultraviolet B radiation‐induced damage in human and porcine skin ex vivo. J Pineal Res. 2018;65:e12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sinha B, Wu Q, Li W, et al. Protection of melatonin in experimental models of newborn hypoxic‐ischemic brain injury through MT1 receptor. J Pineal Res. 2018;64:e12443. [DOI] [PubMed] [Google Scholar]
- 59. Yang T, Cao C, Yang J, et al. miR‐200a‐5p regulates myocardial necroptosis induced by Se deficiency via targeting RNF11. Redox Biol. 2018;15:159‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tan SWS, Yip GW, Suda T, et al. Small Maf functions in the maintenance of germline stem cells in the Drosophila testis. Redox Biol. 2018;15:125‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mehra P, Guo Y, Nong Y, et al. Cardiac mesenchymal cells from diabetic mice are ineffective for cell therapy‐mediated myocardial repair. Basic Res Cardiol. 2018;113:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li J, Cai SX, He Q, et al. Intravenous miR‐144 reduces left ventricular remodeling after myocardial infarction. Basic Res Cardiol. 2018;113:36. [DOI] [PubMed] [Google Scholar]
- 63. Stutz MD, Ojaimi S, Allison C, et al. Necroptotic signaling is primed in Mycobacterium tuberculosis‐infected macrophages, but its pathophysiological consequence in disease is restricted. Cell Death Differ. 2018;25:951‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sorrentino G, Mantovani F, Del Sal G. The stiff RhoAd from mevalonate to mutant p53. Cell Death Differ. 2018;25:643‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Serrano BP, Hardy JA. Phosphorylation by protein kinase A disassembles the caspase‐9 core. Cell Death Differ. 2018;25:1025‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Qiu X, Zhang Y, Han J. RIP3 is an upregulator of aerobic metabolism and the enhanced respiration by necrosomal RIP3 feeds back on necrosome to promote necroptosis. Cell Death Differ. 2018;25:821‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhou H, Li D, Zhu P, et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J Pineal Res. 2017;63(4):e12438. [DOI] [PubMed] [Google Scholar]
- 68. Li R, Xin T, Li D, et al. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018;18:229‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhu P, Hu S, Jin Q, et al. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhou H, Wang S, Zhu P, et al. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lim J, Murthy A. Controlling inflammation by selective autophagy. Cell Death Differ. 2018;25:825‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Linder M, Hecking M, Glitzner E, et al. EGFR controls bone development by negatively regulating mTOR‐signaling during osteoblast differentiation. Cell Death Differ. 2018;25:1094‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ko A, Han SY, Choi CH, et al. Oncogene‐induced senescence mediated by c‐Myc requires USP10 dependent deubiquitination and stabilization of p14ARF. Cell Death Differ. 2018;25:1050‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jain R, Mintern JD, Tan I, et al. How do thymic epithelial cells die? Cell Death Differ. 2018;25:1002‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hockings C, Alsop AE, Fennell SC, et al. Mcl‐1 and Bcl‐xL sequestration of Bak confers differential resistance to BH3‐only proteins. Cell Death Differ. 2018;25:719‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gong H, Zhang Y, Jiang K, et al. p73 coordinates with Delta133p53 to promote DNA double‐strand break repair. Cell Death Differ. 2018;25:1063‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hao Z, Dandan L, Pingjun Z, et al. Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia-reperfusion injury. J Pineal Res. 2018;65(3):e12503. [DOI] [PubMed] [Google Scholar]
- 78. Qinhua J, Ruibing L, Nan H, et al. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018;14:576‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hao Z, Wenjuan D, Ye L, et al. Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res. 2018;64(1):e12450. [DOI] [PubMed] [Google Scholar]