

Abstract
Recently, we identified a novel fatty acid amide hydrolase (FAAH) inhibitor, PKM‐833 [(R)‐N‐(pyridazin‐3‐yl)‐4‐(7‐(trifluoromethyl)chroman‐4‐yl)piperazine‐1‐carboxamide]. The aim of the present study is to characterize the pharmacological profile of PKM‐833 in vitro and in vivo. PKM‐833 showed potent inhibitory activities against human and rat FAAH with IC50 values of 8.8 and 10 nmol/L, respectively, 200‐fold more selectivity against other 137 molecular targets, and irreversible mode of action. In pharmacokinetic and pharmacodynamic studies, PKM‐833 showed excellent brain penetration and good oral bioavailability, and elevated anandamide (AEA) concentrations in the rat brain. These data indicate that PKM‐833 is a potent, selective, orally active, and brain‐penetrable FAAH inhibitor. In behavioral studies using rat models, PKM‐833 significantly attenuated formalin‐induced pain responses (3 mg/kg) and improved mechanical allodynia in complete freund's adjuvant (CFA)‐induced inflammatory pain (0.3‐3 mg/kg). On the other hand, PKM‐833 did not show the analgesic effects against mechanical allodynia in chronic constriction injury (CCI)‐induced neuropathic pain up to 30 mg/kg. Regarding side effects, PKM‐833 had no significant effects on catalepsy and motor coordination up to 30 mg/kg. These results indicate that PKM‐833 is a useful pharmacological agent that can be used to investigate the role of FAAH and may have therapeutic potential for the treatment of inflammatory pain without undesirable side effects.
Keywords: analgesia, anandamide, endocannabinoid, FAAH, PKM‐833
Abbreviations
- AA‐5‐HT
N‐arachidonoyl‐serotonin
- AAMCA
arachidonyl‐7‐amino, 4‐methyl coumarin amide
- AEA
anandamide
- AMC
7‐amino‐4‐methyl coumarin
- ASP8477
3‐pyridyl 4‐(phenylcarbamoyl)piperidine‐1‐carboxylate
- CB
cannabinoid
- CCI
chronic constriction injury
- CFA
complete freund's adjuvant
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- Naproxen
(S)‐(+)‐6‐methoxy‐α‐methyl‐2‐naphthaleneacetic acid
- PF‐04457845
N‐pyridazin‐3‐yl‐4‐(3‐{[5‐(trifluoromethyl)pyridin‐2‐yl]oxy}benzylidene)piperidine‐1‐carboxamide
- PF‐3845
N‐3‐pyridinyl‐4‐[[3‐[[5‐(trifluoromethyl)‐2‐pyridinyl]oxy]phenyl]methyl]‐1‐piperidinecarboxamide
- PKM‐833
[(R)‐N‐(pyridazin‐3‐yl)‐4‐(7‐(trifluoromethyl)chroman‐4‐yl)piperazine‐1‐carboxamide]
- Pregabalin
3(S)‐(aminomethyl)‐5‐methylhexanoic acid
- PWT
paw withdrawal threshold
- URB597
(3'‐(aminocarbonyl)[1,1'‐biphenyl]‐3‐yl)‐cyclohexylcarbamate
- WIN 55,212‐2 mesylate
(R)‐(+)‐(2,3; dihydro‐5‐methyl‐3‐(4‐morpholinylmethyl)pyrrolo[1.2.3‐de]‐1,4‐benzoxazin‐6‐yl)‐1‐naphthalenylmethanone mesylate
1. INTRODUCTION
Anandamide (AEA) and 2‐arachidonylglycerol are dominant and representative endocannabinoids that bind to two G protein‐coupled cannabinoid (CB) receptors, CB1 and CB2. The CB1 receptor is predominantly expressed in the central nervous system, while CB2 receptor is mainly expressed in immune cells.1 Classical CB agonists are well known to be effective for several disorders such as multiple sclerosis, chemotherapy‐induced allodynia, migraine, and neuropathic pain.2, 3, 4, 5 However, they also elicit undesirable side effects associated with the direct activation of the CB1 receptor. Fatty acid amide hydrolase (FAAH) is an enzyme that predominantly hydrolyzes AEA expressed in both central nervous system and peripheral tissues.6, 7 Therefore, pharmacological inhibition of FAAH can represent a unique approach that does not involve the direct activation of CB1 receptor. Preclinical studies investigated the role of FAAH using FAAH inhibitors and FAAH knockout mice.8, 9, 10, 11 Several FAAH inhibitors with brain penetration have been subjected to pharmacological analysis12, 13, 14, 15, 16, 17, 18, 19, 20 and some of them showed analgesic effects on inflammatory and neuropathic pain without undesirable side effects in rat models.13, 17 They have reached clinical trials to obtain the proof of concept.21, 22
Recently, we identified a novel FAAH inhibitor, PKM‐833 [(R)‐N‐(pyridazin‐3‐yl)‐4‐(7‐(trifluoromethyl)chroman‐4‐yl) piperazine‐1‐carboxamide] in the course of the extensive chemical optimization of existing urea‐structured FAAH inhibitors to find a highly brain‐penetrable compound. The aim of the present study is to characterize in vitro and in vivo pharmacological profiles of PKM‐833. PKM‐833 is a potent, selective, and irreversible FAAH inhibitor with good oral bioavailability and brain penetration. The in vivo effects of PKM‐833 in rats were tested via the formalin test, CFA‐induced inflammatory pain, and CCI‐induced neuropathic pain models. In addition, we examined whether PKM‐833 induced catalepsy and/or motor coordination impairment in comparison with those of reference compounds in rats. To examine the target engagement of PKM‐833 in rat brain, AEA concentrations were measured in the brain.
2. MATERIAL AND METHODS
2.1. Animals
Male Sprague‐Dawley rats (5‒6 weeks old, Charles River Laboratories Japan or 7 weeks old, Japan SLC, Inc) and male Wistar rats (5 weeks old, Japan SLC, Inc) were purchased. All rats were group housed or individually housed in an air‐conditioned room (room temperature, 23 ± 3°C; humidity, 55% ± 15%) with a 12‐h light‐dark cycle (lights on: 07:30‒19:30). Each rat had free access to standard chow (CE‐2, CLEA Japan, Inc) and tap water. At least 7 days were allowed to acclimatize to the facility prior to starting the experiments.
All experimental procedures were approved by the Institutional Animal Care and Use Committee complied with the Japanese law “Act on Welfare and Management of Animals” and the guidelines from the Ministry of Health, Labor and Welfare of Japan.
2.2. Drugs
The structure of PKM‐833 is shown in Figure 1. PKM‐833 was synthesized in‐house and used as a free base in all experiments. PKM‐833 and URB597 (Cayman Chemical), used as a reference compound in irreversible inhibition test were dissolved in dimethyl sulfoxide (Wako Pure Chemical Industries) and stored at –20°C to perform in vitro assays. PKM‐833 was suspended in 0.5% hydroxypropyl methylcellulose solution (Sigma‐Aldrich Co. LLC), except that 20% gerucire 44/14 (Gattefosse) was used in catalepsy test, and orally administered at a volume of 10 mL/kg. When PKM‐833 was intravenously administered at a volume of 1 mL/kg in the pharmacokinetic study, it was dissolved in 40% (v/v) N, N‐dimethylacetamide (Wako Pure Chemical Industries), 40% (v/v) polyethylene glycol 400 (Wako Pure Chemical Industries), and 20% distilled water. Naproxen sodium (Sigma‐Aldrich), used as a reference compound in CFA‐induced inflammatory pain model was dissolved in 0.5% methylcellulose solution (Wako Pure Chemical Industries) and orally administered at a volume of 10 mL/kg. Pregabalin (US Pharmacopeia Convention), used as a reference compound in formalin and rotarod tests, and CCI‐induced neuropathic pain model was dissolved in saline and orally administered at a volume of 10 mL/kg. WIN 55,212‐2 mesylate (Wako Pure Chemical Industries) as a reference compound in catalepsy test was dissolved in 10% dimethyl sulfoxide (Wako Pure Chemical Industries), 10% Tween 80 (Wako Pure Chemical Industries), and 80% distilled water, and subcutaneously administered at a volume of 2 mL/kg.
Figure 1.
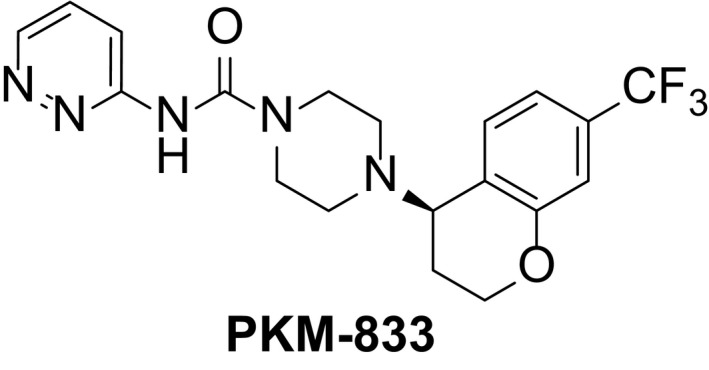
Chemical structure of PKM‐833
2.3. In vitro assays
Human or rat microsomes were prepared by Oriental Yeast Co., Ltd. using human leukemic monocyte lymphoma, U937 (ATCC No. CRL‐1593.2) and brain tissues of male Sprague‐Dawley rats (Charles River Laboratories Japan), respectively.23 The inhibitory activity and potency of PKM‐833 on each microsome was determined by measuring the conversion of the FAAH substrate converted from arachidonyl‐7‐amino, 4‐methyl coumarin amide (AAMCA) to 7‐amino‐4‐methyl coumarin (AMC) using a fluorescence imaging plate reader (EnVision2000), as described by Kage et al (2007)24 with a minor modification. For IC50 determination, PKM‐833 was incubated with each microsome for 5 minutes at room temperature. After addition of AAMCA, the first fluorescence intensity at 355 nm/460 nm was measured, followed by incubation for 120 minutes (human) or 30 minutes (rat). Then, the second fluorescence at 355 nm/460 nm was measured. Based on the difference between the first and the second fluorescence, maximal inhibition (100%) and 0% activity in the assay were defined as no enzyme control and no compound control, respectively. Each assay was performed in triplicate at eight PKM‐833 concentrations (0.1‐300 nmol/L). The inhibition (%) and IC50 values were calculated using Assay Explorer (Accelrys, Inc). For Kinact/Ki determination, PKM‐833 (1.25‐80 nmol/L) or URB597 (0.47‐30 nmol/L) in triplicate at seven concentrations was incubated with each microsome and AAMCA. The reaction progress curve was collected for 120 minutes (human) or 60 minutes (rat) with readings taken in 90 seconds intervals, respectively. The K inact/K i value was calculated using GraphPad Prism Software, version 5 (GraphPad Software Inc), as described by Mileni et al (2008).25
2.4. Off‐target activity
The effect of PKM‐833 at a concentration of 30 μmol/L on the activity of human monoacylglycerol lipase (MAGL) was tested using the MAGL inhibitor screening assay kit (Cayman Chemical). The selectivity of PKM‐833 at a concentration of 20 μmol/L was further assessed against 137 molecular targets, including neurotransmitter receptors, enzymes, ion channels, and transporters, by using the Cerep BioPrint In Vitro Pharmacology Profiling (Cerep SA).
2.5. Pharmacokinetic study
The pharmacokinetic profile of PKM‐833 was examined using male Sprague‐Dawley rats (Japan SLC). PKM‐833 was intravenously administered at a dose of 1 mg/kg or orally administered at doses of 0.3, 1, and 3 mg/kg to the rats. Blood samples were collected at appropriate time intervals and centrifuged at 2,150g for 15 minutes at 4°C. Components of each plasma sample were extracted by using ethyl acetate and the 96‐well Isolute SLE + plate (Biotage AB). The extracts were then evaporated to dryness, reconstituted in 45% (v/v) acetonitrile and 55% (v/v) 10 mmol/L ammonium acetate, and analyzed by LC‐MS/MS. To assess brain penetration, PKM‐833 was orally administered at a dose of 1 mg/kg to the rats. After 1 and 4 hours of PKM‐833 administration, blood samples and the cerebral cortex were collected. The collected tissue was homogenized in five times its volume of water using a Micro Smash TM‐100 (Tomy Seiko Co., Ltd.) and the internal standard was added. The homogenates were centrifuged at 17,800g for 15 minutes at 4°C and supernatants were analyzed by LC‐MS/MS. LC‐MS/MS was performed as described in the supplemental methods (Methods S1). Pharmacokinetic parameters were calculated via noncompartmental analysis using WinNolin version 6.1 (Pharsight).
2.6. AEA concentrations
Male Sprague‐Dawley rats (Charles River Laboratories Japan) were subjected to measure AEA concentrations. PKM‐833 was orally administered 1 hour before decapitation at doses of 0.3, 1, and 3 mg/kg. In the time‐course study, PKM‐833 was orally administered to rats 1, 2, 4, 8, 24, or 48 hours before decapitation at a dose of 1 mg/kg. The rats were decapitated using a guillotine and brain tissues were rapidly dissected on an ice‐cold dish. The cerebral cortex, as a representative region in the brain, was immediately weighted and frozen in liquid nitrogen until measuring AEA concentrations. AEA concentrations were measured according to the protocol described in Richardson et al (2007) with a minor modification.26 AEA‐d8 (Cayman Chemical), as an internal standard, was added into each cerebral cortex tissue and homogenized with ice cold ethyl acetate/hexane (9:1, v/v) to extract AEA. The homogenates were washed three times with approximately 20% of its volume of water and centrifuged at 7,000g for 15 minutes at 4°C. The supernatant of each sample was collected in a pear‐shaped glass flask (Thermo Fisher Scientific Inc), evaporated to dryness under nitrogen gas at room temperature for approximately 3 hours, solubilized in 1 mL hexane/chloroform (3:1, v/v), and loaded onto the Sep Pak Silica cartridges (Waters) using extraction manifold (Waters). Then, the cartridges were washed three times with hexane/chloroform (3:1, v/v) and the eluted extracts were collected using 2% methanol/0.2% triethylamine/chloroform buffer at a volume of 4 mL. The eluates were evaporated to dryness under nitrogen gas at room temperature for 1 hour, solubilized in 1 mL acetonitrile, and measured AEA concentrations by LC‐MS/MS analysis (Methods S2).
2.7. Formalin test
The formalin‐induced pain responses were assessed in male Wistar rats (Japan SLC) using transparent plastic boxes.27, 28 Formalin 0.5% (v/v) (Wako Pure Chemical Industries) was injected into the plantar surface of the left hind paw at a volume of 50 μL, and then rats were placed into the box. The duration of pain responses (flinching, licking, and biting) of the injected paw was manually recorded during a 45 minutes period. PKM‐833 at doses of 0.3, 1, and 3 mg/kg or pregabalin at a dose of 30 mg/kg was orally administered 1 hour before the injection of formalin. For the analysis of anti‐nociceptive effects of the test compounds, the periods from 0 to 10 minutes, and from 10 to 45 minutes after the injection of formalin were defined as early phase and late phase, respectively.
2.8. CFA‐induced inflammatory pain
The CFA‐induced inflammatory pain was assessed in male Sprague‐Dawley rats (Charles River Laboratories Japan) using von Frey filaments (Stoelting Co). Fifty percent of CFA was injected into the plantar surface of the right hind paw at a volume of 0.1 mL, and 5 days later, the paw withdrawal threshold (PWT) was determined by the up‐down method described in Chaplan et al (1994).29 PKM‐833 at doses of 0.3, 1, and 3 mg/kg or naproxen at a dose of 15 mg/kg was orally administered 1 or 4 hours before the measurement of PWT, respectively.
2.9. CCI‐induced neuropathic pain
The CCI‐induced neuropathic pain was assessed in male Sprague‐Dawley rats (Charles River Laboratories Japan) using von Frey filaments (Stoelting Co). CCI surgery was performed according to the modified method of Bennett et al (1988).30 Briefly, the left sciatic nerves were exposed under anesthesia and loosely ligated with four ligatures (4‐0 blade silk: Matsuda Ika Kogyo Co., Ltd.) around the sciatic nerve at intervals of 1 mm. As a sham operation, the right sciatic nerve was isolated, but it was not ligated. The PWT was determined by the up‐down method described in Chaplan et al (1994).29 On day 14 after surgery, PKM‐833 at doses of 3, 10, and 30 mg/kg or pregabalin at a dose of 30 mg/kg was orally administered 4 hours before the measurement of PWT.
2.10. Catalepsy
Male Sprague‐Dawley rats (Japan SLC) were subjected to catalepsy. PKM‐833 at doses of 10 and 30 mg/kg or WIN 55,212‐2 at a dose of 3 mg/kg was orally or subcutaneously administered 1 or 4 hours before the test, respectively. Catalepsy was assessed by placing both rat forepaws on a horizontal bar raised approximately 10 cm above the floor.31 The latencies required for the rat to remove its forepaws, move a hindlimb, and climb down from the bars into a normal posture were recorded with a cut‐off time of 90 seconds.
2.11. Motor coordination
The motor coordination was evaluated in male Sprague‐Dawley rats (Charles River Laboratories Japan) using a rotarod apparatus (Ugo Basile, SRL). All rats were trained once a day on the apparatus for 2 consecutive days. In the first training session, the rats were trained on the rod and the rotation speed was gradually increased from 4 to 20 rpm during a 180 seconds period, followed by an additional training on the rod at the constant speed of 20 rpm for 200 seconds. On the next day after the first training session, the second training session was conducted. The rats were trained on the rod at 20 rpm up to five times until the rats can walk for at least 200 seconds without falling off. Only the rats met these criteria were used for the subsequent experiment. On the day of testing, the selected rats were trained on the rod at the same condition in the two consecutive sessions again and the rats that met the criteria at least once were finally selected to use in the test session with drug treatment. In the test session, the duration that rats walked on the rod was measured 1 or 2 hours after the oral administration of PKM‐833 at doses of 10 and 30 mg/kg or pregabalin at doses of 10 and 30 mg/kg, respectively, with a cut‐off time of 200 seconds.
2.12. Statistical analysis
The effect of PKM‐833 was analyzed by one‐way analysis of variance and post‐hoc comparisons were performed by Dunnett's (AEA concentrations and formalin test) and Steel's tests (CFA‐induced inflammatory pain, CCI‐induced neuropathic pain, catalepsy, and rotarod test). Wilcoxon test was used to analyze the effect of naproxen in CFA‐induced inflammatory pain, pregabalin in CCI‐induced neuropathic pain, and WIN 55,212‐2 in catalepsy. Student's t‐test or Steel's test was used to analyze the effect of pregabalin in formalin test or rotarod test, respectively. A probability level of <.05 was considered statistically significant.
3. RESULTS
3.1. In vitro and pharmacokinetic profile of PKM‐833
The inhibitory activity and mode of action of PKM‐833 were examined using human and rat microsomes containing FAAH. PKM‐833 inhibited human and rat FAAH with IC50 values of 8.8 ± 3.0 and 10 ± 3 nmol/L, respectively. PKM‐833 showed human and rat K inact/K i values with 34 300 ± 10 800 and 128 000 ± 39 800 mol−1 L−1 s−1, respectively, while URB597 exhibited them with 160 000 ± 30 400 and 1 170 000 ± 304 000 mol−1 L−1 s−1, respectively. These results indicate that PKM‐833 shows lower irreversible potency than URB597.
The selectivity of PKM‐833 was examined against human MAGL. PKM‐833 did not inhibit human MAGL even at a concentration of 30 μmol/L. The selectivity of PKM‐833 was also examined against other 137 molecular targets (Table S1). PKM‐833 exhibited weak inhibitory effects (greater than 50%) in five receptor binding assays [PFA, 5‐HT2B, sigma (nonselective), Na+ channel (site 2) and Cl‐ channel (GABA gated)] at a concentration of 20 μmol/L. In the subsequent follow‐up assays, each IC50 value was calculated as 2.3, 6.4, 14, 11, and 23 μmol/L, respectively, suggesting that PKM‐833 has more than 200‐fold selectivity against other molecular targets.
PKM‐833 showed a plasma half‐life of 5.8 hours at an intravenous dose of 1 mg/kg and good oral bioavailability of 41 to 91% at dose ranges from 0.3 to 3 mg/kg in rats (Table S2). PKM‐833 at a dose of 1 mg/kg also exhibited excellent brain penetration 1 and 4 hours after the oral administration in rats (Table S2).
3.2. AEA concentrations
We examined both dose‐ and time‐dependent effects of PKM‐833 on AEA concentrations in rat brain. AEA concentrations elevated in a dose‐dependent manner and showed significant effects at doses of 1 and 3 mg/kg, 1 hour after the administration of PKM‐833 (Figure 2A). In a study of the time‐course, AEA concentrations significantly elevated for 8 hours but not at 24 and 48 hours after the administration of PKM‐833 (Figure 2B).
Figure 2.
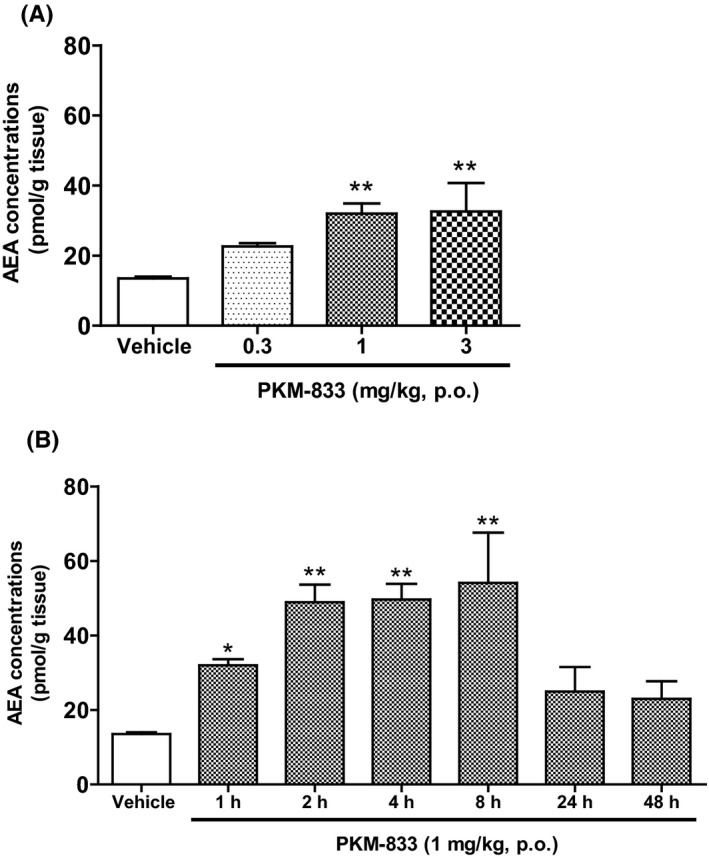
Dose‐ (A) and time‐ (B) dependent effects of PKM‐833 on AEA concentrations in rat brain. (A) PKM‐833 (0.3‐3 mg/kg, p.o.) was administered 1 hour before decapitation. (B) PKM‐833 (1 mg/kg, p.o.) was administered from 1 to 48 hours before decapitation. Data are expressed as the mean ± SEM of three animals. Asterisks represent a significant difference between PKM‐833 treatment and vehicle groups, *P < .05, **P < .01, (Dunnett's test)
3.3. Formalin test
We examined the effects of PKM‐833 and pregabalin via the formalin test in rats. PKM‐833 at a dose of 3 mg/kg significantly reduced formalin‐induced pain responses in the late phase as well as pregabalin at a dose of 30 mg/kg (Figure 3B). Neither PKM‐833 nor pregabalin changed formalin‐induced pain responses in the early phase (Figure 3A).
Figure 3.
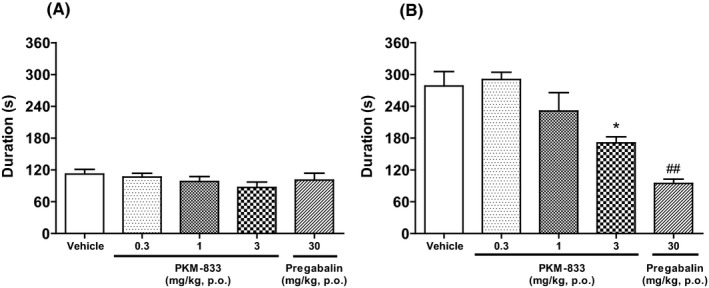
Effects of PKM‐833 and pregabalin on the early phase (A) and late phase (B) of formalin‐induced pain responses in rats. PKM‐833 (0.3‐3 mg/kg, p.o.) or pregabalin (30 mg/kg, p.o.) was administered 1 hour before the intraplantar injection of 0.5% formalin. Data are expressed as the mean ± SEM of six animals. Asterisks represent a significant difference between treatment and vehicle groups, *P < .05 (Dunnett's test). Sharps represent a significant difference between treatment and vehicle groups, ## P < .01 (Student's t‐test)
3.4. CFA‐induced inflammatory pain
We examined the effects of PKM‐833 and naproxen on CFA‐induced inflammatory pain in rats. PKM‐833 improved mechanical allodynia in a dose‐dependent manner and significantly suppressed it 1 and 4 hours after the administration at doses ranging from 0.3 to 3 mg/kg (Figure 4A and B). Naproxen at a dose of 15 mg/kg also significantly suppressed mechanical allodynia 1 and 4 hours after the administration. (Figure 4A and B).
Figure 4.
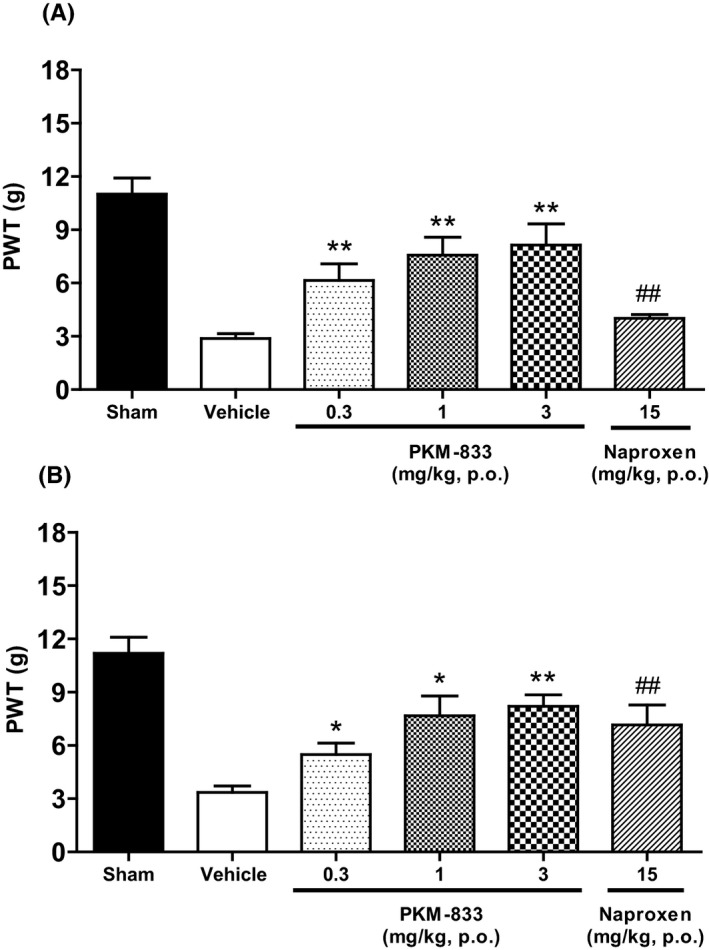
Effects of PKM‐833 and naproxen on CFA‐induced inflammatory pain in rats. PKM‐833 (0.3‐3 mg/kg, p.o.) or naproxen (15 mg/kg, p.o.) was administered 1 (A) or 4 hours (B) before measuring the PWT, respectively. Data are expressed as the mean ± SEM of nine animals. Asterisks represent a significant difference between treatment and vehicle groups, *P < .05, **P < .01 (Steel's test). Sharps represent a significant difference between treatment and vehicle groups, ## P < .01 (Wilcoxon test)
3.5. CCI‐induced neuropathic pain
We examined the effects of PKM‐833 and pregabalin on CCI‐induced neuropathic pain model in rats. PKM‐833 at a dose of 30 mg/kg did not attenuate mechanical allodynia, while pregabalin at a dose of 30 mg/kg significantly antagonized it (Figure 5).
Figure 5.
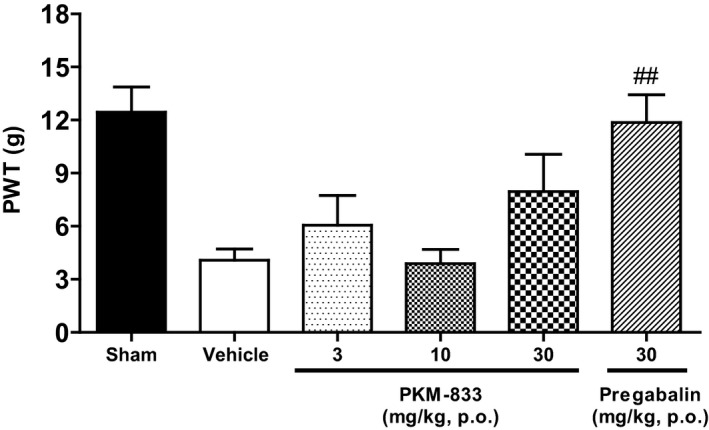
Effects of PKM‐833 and pregabalin on CCI‐induced neuropathic pain in rats. PKM‐833 (3‐30 mg/kg, p.o.) or pregabalin (30 mg/kg, p.o.) was administered 4 hours before measuring the PWT. Data are expressed as the mean ± SEM of eight animals. Sharps represent a significant difference between treatment and vehicle groups, ## P < .01 (Wilcoxon test)
3.6. Catalepsy
The cataleptic effect of PKM‐833 was tested at doses of 10 and 30 mg/kg in rats. PKM‐833 did not induce catalepsy at any tested doses, while WIN 55,212‐2, as a reference compound caused catalepsy at a dose of 3 mg/kg (Table 1).
Table 1.
Effects of PKM‐833 and WIN 55,212‐2 on catalepsy in rats
| Dose | Duration (s) | ||
|---|---|---|---|
| 1 h | 4 h | ||
| Vehicle | 0.7 ± 0.1 | 1.9 ± 0.7 | |
| PKM‐833 | 10 mg/kg, p.o. | 0.5 ± 0.1 | 3.7 ± 1.9 |
| PKM‐833 | 30 mg/kg, p.o. | 0.8 ± 0.2 | 1.0 ± 0.3 |
| WIN 55,212‐2 | 3 mg/kg, s.c. | 13 ± 2## | 77 ± 7## |
Data are expressed as the mean ± SEM of eight animals. Sharps represent a significant difference between treatment and vehicle groups, ## P<.01 (Wilcoxon test).
3.7. Motor coordination
We examined the effects of PKM‐833 and pregabalin on motor coordination in the rotarod test. PKM‐833 did not affect rotarod performance at doses of 10 and 30 mg/kg, while pregabalin significantly impaired motor coordination at a dose of 30 mg/kg (Table 2).
Table 2.
Effects of PKM‐833 and pregabalin on motor coordination in rats
| Dose | Duration (s) | ||
|---|---|---|---|
| 1 h | 2 h | ||
| Vehicle | 200 ± 0 | ||
| PKM‐833 | 10 mg/kg, p.o. | 200 ± 0 | |
| PKM‐833 | 30 mg/kg, p.o. | 195 ± 6 | |
| Vehicle | 200 ± 0 | ||
| Pregabalin | 10 mg/kg, p.o. | 199 ± 1 | |
| Pregabalin | 30 mg/kg, p.o. | 167 ± 14# | |
Data are expressed as the mean ± SEM of ten animals. Sharps represent a significant difference between treatment and vehicle groups, # P<.05 (Steel's test).
4. DISCUSSION
We identified a novel potent, selective, orally active, and brain‐penetrable FAAH inhibitor, PKM‐833. PKM‐833 showed potent inhibitory activities on human and rat FAAH with IC50 values of 8.8 and 10 nmol/L, respectively, and had no significant off‐target activity against MAGL or other pharmacological targets, including neurotransmitter receptors, enzymes, ion channels, and transporters. PKM‐833 also showed lower irreversible potency than URB597 as a representative FAAH inhibitor. In addition, PKM‐833 showed favorable pharmacokinetic profiles with good oral bioavailability and brain penetration, and elevated brain AEA concentrations in rats. These results indicate that PKM‐833 is a good pharmacological agent for investigating the in vitro and in vivo roles of FAAH.
In the present study, we compared the effects of PKM‐833 with those of currently available analgesic agents, pregabalin and naproxen, via the formalin test and CFA‐induced inflammatory pain model in rats, respectively. Formalin‐induced pain responses after intraplantar injection are well known to consist of the biphaic phases, that is, early and late phases. Because currently available analgesic agents, such as pregabalin and morphine attenuate formalin‐induced pain responses, the formalin test has been commonly used to examine the analgesic potential of test compounds in rats.32, 33 PKM‐833 significantly reduced formalin‐induced pain responses in the late phase but not in the early phase in rats. These results were consistent with those previously reported by Maione et al (2007), which showed that the structurally unrelated FAAH inhibitor, AA‐5‐HT significantly decreased formalin‐induced pain responses only in the late phase in rats.34 However, there were previous reports that FAAH knockout mice were effective in both early and late phases.8, 10 The interpretation of phenotype analysis using genetically manipulated FAAH deficient mice might be limited by gene compensation, developmental effect, and strain variance. Therefore, the pharmacological treatment of FAAH by PKM‐833 is useful to explore the role of FAAH. Hence, the present results suggest that PKM‐833 can have analgesic effects in rats.
The intraplantar injection of CFA induces strong inflammatory pain in rats and one of clinically effective nonsteroidal anti‐inflammatory drugs, naproxen is well known to show analgesic effects on CFA‐induced inflammatory pain in rats.12 Therefore, CFA‐induced inflammatory pain is used as an animal model to examine the analgesic effect accompanied with anti‐inflammation. In the present study, PKM‐833 significantly improved mechanical allodynia on CFA‐induced inflammatory pain at doses ranging from 0.3 to 3 mg/kg and the effect was comparable with that of naproxen in rats. These results were also consistent with the previous findings that other FAAH inhibitors, PF‐3845 and PF‐04457845 improved mechanical allodynia on CFA‐induced inflammatory pain similar to that of naproxen in rats.12, 13 Therefore, these findings indicate that FAAH inhibitors may have the potential to improve inflammatory pain similarly to naproxen.
In the present study, we evidenced that PKM‐833 significantly attenuated formalin‐induced pain responses and improved mechanical allodynia in CFA‐induced inflammatory pain in rats. However, it was unclear whether these effects were mediated by the inhibition of FAAH in the rat brain. Therefore, we examined whether PKM‐833 elevated AEA concentrations to investigate the biochemical effect of FAAH inhibition in the brain. PKM‐833 increased AEA concentrations in the brain in a dose‐dependent manner, indicating that PKM‐833 exhibited intracellular and brain penetration. The efficacy of PKM‐833 on formalin‐induced pain responses and CFA‐induced inflammatory pain was clearly observed in the dose ranges to elevate AEA concentrations in the brain. Therefore, these results indicate that the analgesic effects of PKM‐833 may be exerted through the inhibition of FAAH in the rat brain. Further studies with several structurally unrelated FAAH inhibitors will be necessary to clarify the increasing rate of AEA concentrations required to exhibit analgesic responses.
We also examined the effect of PKM‐833 against neuropathic pain in rats. CCI of the sciatic nerve is well known to result in mechanical allodynia of the hindlimb in rats and pregabalin is markedly effective against neuropathic pain in both animals and humans.35, 36 In the present study, pregabalin significantly improved CCI‐induced mechanical allodynia. On the other hand, PKM‐833 did not have clear analgesic effects up to 30 mg/kg, which was 10‐fold higher than the effective dose (3 mg/kg) in both formalin‐induced pain responses and CFA‐induced inflammatory pain. These results indicate that PKM‐833 exhibits a more predominant effect on inflammatory pain than on neuropathic pain.
The effects of FAAH inhibitors on CCI‐induced neuropathic pain in rats have been reported. Although the acute effects of systemic administration of FAAH inhibitors on CCI‐induced mechanical allodynia in rats have not been determined yet, systemically repeated administration for 7 and 14 days of a brain‐penetrable FAAH inhibitor, URB597 to rats showed alleviation.34, 37 Taken together our results with the previous reports, we suggest that the repeated administration of FAAH inhibitors to rats can exert more prominent effects against neuropathic pain than a single administration of them.
The effects of FAAH inhibitors have been also examined on partial sciatic nerve or entire spinal nerve ligation‐induced neuropathic pain in rats. URB597 had no improvement on partial sciatic nerve ligation‐induced mechanical allodynia,38 similar to our finding that PKM‐833 did not significantly attenuate mechanical allodynia in CCI‐induced neuropathic pain. However, brain‐penetrable FAAH inhibitors including URB597 improved spinal nerve ligation‐induced mechanical allodynia at a single systemic administration in rats.15, 16, 17, 20 Because the effects of FAAH inhibitors on neuropathic pain remain inconclusive, further studies to assess whether a single and/or repeated administration of PKM‐833 can have analgesic potential on other animal models reflecting neuropathic pain accompanied with nerve ligation will be therefore worthwhile.
To examine the potential liability of PKM‐833 that could be produced along with its analgesic effects, we examined the effects of PKM‐833 on catalepsy and motor coordination in rats. Classical CB agonists such as tetrahydrocannabinol and WIN 55,212‐2 are known to cause catalepsy in rats and mainly by the direct agonism of the CB1 receptor in the brain.39, 40 Unlike WIN 55,212‐2, PKM‐833 did not induce catalepsy up to 30 mg/kg, which was 30‐fold higher than the efficacious dose (1 mg/kg) equal to that of naproxen in CFA‐induced inflammatory pain. These results were nearly consistent with the previous finding by Ahn et al (2011) that PF‐04457845 had no effects on catalepsy up to 100‐fold higher dose than the minimum effective dose of 0.1 mg/kg in CFA‐induced inflammatory pain.13 Taken together, we conclude that the analgesic effects of PKM‐833 in rats are mediated by specific inhibition of FAAH.
Motor impairments such as somnolence and wobbliness are one of serious adverse effects of pregabalin in the clinic.41 Therefore, we compared the effect of PKM‐833 on motor coordination with that of pregabalin by using a rotarod test in rats. PKM‐833 did not affect rotarod performance up to 30 mg/kg, which was 30‐fold higher than the efficacious dose (1 mg/kg) in CFA‐induced inflammatory pain. On the other hand, pregabalin impaired rotarod performance at the same dose (30 mg/kg) as that required for treating formalin‐induced pain responses and CCI‐induced neuropathic pain. These results were consistent with the previous report by Watabiki et al (2017) that a brain‐penetrable FAAH inhibitor, ASP8477 did not affect motor coordination up to 30 mg/kg with a wide safety margin.17 Overall, these findings indicate that PKM‐833 has less liability on muscle tones in rats.
In the present study, we confirmed that PKM‐833 was a potent, selective, orally active, and brain‐penetrable FAAH inhibitor with irreversible mode of action. PKM‐833 reduced formalin‐induced pain responses and CFA‐induced inflammatory pain without cataleptic effects and motor impairments in rats. On the other hand, PKM‐833 did not exhibit the analgesic effects against CCI‐induced neuropathic pain in rats. Moreover, we demonstrated that PKM‐833 elevated AEA concentrations in the brain. Our findings suggest that PKM‐833 is a good pharmacological agent to assess the roles of FAAH and may have therapeutic potential for the treatment of inflammatory pain without undesirable side effects.
DISCLOSURE
The authors declare that there is no conflict of interest.
AUTHOR CONTRIBUTION
All authors are employees of Mochida Pharmaceutical Co., Ltd. Endo, Takeuchi, and Maehara participated in research design. Endo and Takeuchi conducted experiments and performed data analysis. Endo and Maehara wrote or contributed to the writing of the manuscript.
Supporting information
ACKNOWLEDGEMENTS
We thank Ayako Okamoto and Keiko Taguchi for assistance with in vitro experiments. We thank Junko Kamiya, Yuka Fukudome, and Koutarou Hoshida for assistance with in vivo experiments.
Endo T, Takeuchi T, Maehara S. Pharmacological characterization of a novel, potent, selective, and orally active fatty acid amide hydrolase inhibitor, PKM‐833 [(R)‐N‐(pyridazin‐3‐yl)‐4‐(7‐(trifluoromethyl)chroman‐4‐yl) piperazine‐1‐carboxamide] in rats: Potential for the treatment of inflammatory pain. Pharmacol Res Perspect. 2020;8:e00569 10.1002/prp2.569
REFERENCES
- 1. Kendall DA, Yudowski GA. Cannabinoid receptors in the central nervous system: their signaling and roles in disease. Front Cell Neurosci. 2017;4:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gado F, Digiacomo M, Macchia M, Bertini S, Manera C. Traditional uses of cannabinoids and new perspectives in the treatment of multiple sclerosis. Medicines. 2018;5:E91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Masocha W. Targeting the endocannabinoid system for prevention or treatment of chemotherapy‐induced neuropathic pain: studies in animal models. Pain Res Manag. 2018;5234943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagy‐Grócz G, Zádor F, Dvorácskó S, et al. Interactions between the kynurenine and the endocannabinoid system with special emphasis on migraine. Int J Mol Sci. 2017;18:E1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davis MP. Cannabinoids in pain management: CB1, CB2 and non‐classic receptor ligands. Expert Opin Investig Drugs. 2014;23:1123‐1140. [DOI] [PubMed] [Google Scholar]
- 6. Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty‐acid amides. Nature. 1996;384:83‐87. [DOI] [PubMed] [Google Scholar]
- 7. Egertová M, Cravatt BF, Elphick MR. Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience. 2003;119:481‐496. [DOI] [PubMed] [Google Scholar]
- 8. Cravatt BF, Demarest K, Patricelli MP, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371‐9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc Natl Acad Sci USA. 2004;101:10821‐10826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor‐mediated phenotypic hypoalgesia. Pain. 2004;109:319‐327. [DOI] [PubMed] [Google Scholar]
- 11. Chang L, Luo L, Palmer JA, et al. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahn K, Johnson DS, Mileni M, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16:411‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahn K, Smith SE, Liimatta MB, et al. Mechanistic and pharmacological characterization of PF‐04457845: a highly potent and selective fatty acid amide hydrolase inhibitor that reduces inflammatory and noninflammatory pain. J Pharmacol Exp Ther. 2011;338:114‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caprioli A, Coccurello R, Rapino C, et al. The novel reversible fatty acid amide hydrolase inhibitor ST4070 increases endocannabinoid brain levels and counteracts neuropathic pain in different animal models. J Pharmacol Exp Ther. 2012;342:188‐195. [DOI] [PubMed] [Google Scholar]
- 15. Chobanian HR, Guo Y, Liu P, et al. Discovery of MK‐4409, a novel oxazole FAAH inhibitor for the treatment of inflammatory and neuropathic pain. ACS Med Chem Lett. 2014;5:717‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keith JM, Jones WM, Tichenor M, et al. Preclinical characterization of the FAAH inhibitor JNJ‐42165279. ACS Med Chem Lett. 2015;6:1204‐1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watabiki T, Tsuji N, Kiso T, Ozawa T, Narazaki F, Kakimoto S. In vitro and in vivo pharmacological characterization of ASP8477: a novel highly selective fatty acid amide hydrolase inhibitor. Eur J Pharmacol. 2017;815:42‐48. [DOI] [PubMed] [Google Scholar]
- 18. Griebel G, Stemmelin J, Lopez‐Grancha M, et al. The selective reversible FAAH inhibitor, SSR411298, restores the development of maladaptive behaviors to acute and chronic stress in rodents. Sci Rep. 2018;8:2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kiss LE, Beliaev A, Ferreira HS, et al. Discovery of a potent, long‐acting, and CNS‐active inhibitor (BIA 10–2474) of fatty acid amide hydrolase. ChemMedChem. 2018;13:2177‐2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karbarz MJ, Luo L, Chang L, et al. Biochemical and biological properties of 4‐(3‐phenyl‐[1,2,4] thiadiazol‐5‐yl)‐piperazine‐1‐carboxylic acid phenylamide, a mechanism‐based inhibitor of fatty acid amide hydrolase. Anesth Analg. 2009;108:316‐329. [DOI] [PubMed] [Google Scholar]
- 21. Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo‐controlled clinical trial with the irreversible fatty acid amide hydrolase‐1 inhibitor PF‐04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837‐1846. [DOI] [PubMed] [Google Scholar]
- 22. Bradford D, Stirling A, Ernault E, et al. The MOBILE study‐A phase IIa enriched enrollment randomized withdrawal trial to assess the analgesic efficacy and safety of ASP8477, a fatty acid amide hydrolase inhibitor, in patients with peripheral neuropathic pain. Pain Med. 2017;18:2388‐2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramarao MK, Murphy EA, Shen MWH, et al. A fluorescence‐based assay for fatty acid amide hydrolase compatible with high‐throughput screening. Anal Biochem. 2005;343:143‐151. [DOI] [PubMed] [Google Scholar]
- 24. Kage KL, Richardson PL, Traphagen L, et al. A high throughput fluorescent assay for measuring the activity of fatty acid amide hydrolase. J Neurosci Methods. 2007;161:47‐54. [DOI] [PubMed] [Google Scholar]
- 25. Mileni M, Johnson DS, Wang Z, et al. Structure‐guided inhibitor design for human FAAH by interspecies active site conversion. Proc Natl Acad Sci USA. 2008;105:12820‐12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Richardson D, Ortori CA, Chapman V, Kendall DA, Barrett DA. Quantitative profiling of endocannabinoids and related compounds in rat brain using liquid chromatography‐tandem electrospray ionization mass spectrometry. Anal Biochem. 2007;360:216‐226. [DOI] [PubMed] [Google Scholar]
- 27. Dubuisson D, Dennis SG. The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain. 1977;4:161‐174. [DOI] [PubMed] [Google Scholar]
- 28. Akada Y, Mori R, Matsuura K, et al. Pharmacological profiles of the novel analgesic M58996 in rat models of persistent and neuropathic pain. J Pharmacol Sci. 2006;102:205‐212. [DOI] [PubMed] [Google Scholar]
- 29. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55‐63. [DOI] [PubMed] [Google Scholar]
- 30. Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87‐107. [DOI] [PubMed] [Google Scholar]
- 31. Arakawa K, Maehara S, Yuge N, et al. Pharmacological characterization of a novel potent, selective, and orally active phosphodiesterase 10A inhibitor, PDM‐042 [(E)‐4‐(2‐(2‐(5,8‐dimethyl‐[1,2,4]triazolo[1,5‐a]pyrazin‐2‐yl)vinyl)‐6‐(pyrrolidin‐1‐yl)pyrimidin‐4‐yl)morpholine] in rats: potential for the treatment of schizophrenia. Pharmacol Res Perspect. 2016;4:e00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Field MJ, Oles RJ, Lewis AS, McCleary S, Hughes J, Singh L. Gabapentin (neurontin) and S‐(+)‐3‐isobutylgaba represent a novel class of selective antihyperalgesic agents. Br J Pharmacol. 1997;121:1513‐1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Munro G, Christensen JK, Erichsen HK, et al. NS383 selectively inhibits acid‐sensing ion channels containing 1a and 3 subunits to reverse inflammatory and neuropathic hyperalgesia in rats. CNS Neurosci Ther. 2016;22:135‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maione S, De Petrocellis L, de Novellis V, et al. Analgesic actions of N‐arachidonoyl‐serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol. 2007;150:766‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Field MJ, Bramwell S, Hughes J, Singh L. Detection of static and dynamic components of mechanical allodynia in rat models of neuropathic pain: are they signalled by distinct primary sensory neurones? Pain. 1999;83:303‐311. [DOI] [PubMed] [Google Scholar]
- 36. van Seventer R, Feister HA, Young JP Jr, Stoker M, Versavel M, Rigaudy L. Efficacy and tolerability of twice‐daily pregabalin for treating pain and related sleep interference in postherpetic neuralgia: a 13‐week, randomized trial. Curr Med Res Opin. 2006;22:375‐384. [DOI] [PubMed] [Google Scholar]
- 37. Jiang H‐X, Ke B‐W, Liu J, et al. Inhibition of fatty acid amide hydrolase improves depressive‐like behaviors independent of its peripheral antinociceptive effects in a rat model of neuropathic pain. Anesth Analg. 2019;129:587‐597. [DOI] [PubMed] [Google Scholar]
- 38. Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Craft RM, Wakley AA, Tsutsui KT, Laggart JD. Sex differences in cannabinoid 1 vs. cannabinoid 2 receptor‐selective antagonism of antinociception produced by delta9‐tetrahydrocannabinol and CP55,940 in the rat. J Pharmacol Exp Ther. 2012;340:787‐800. [DOI] [PubMed] [Google Scholar]
- 40. Fox A, Kesingland A, Gentry C, et al. The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92:91‐100. [DOI] [PubMed] [Google Scholar]
- 41. Parsons B, Fujii K, Nozawa K, Yoshiyama T, Ortiz M, Whalen E. The efficacy of pregabalin for the treatment of neuropathic pain in Japanese subjects with moderate or severe baseline pain. J Pain Res. 2019;12:1061‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials