

ABSTRACT
The McMurdo Dry Valleys (MDV) in Antarctica harbor a diverse assemblage of mat-forming diazotrophic cyanobacteria that play a key role in nitrogen cycling. Prior research showed that heterotrophic diazotrophs also make a substantial contribution to nitrogen fixation in MDV. The goals of this study were to survey autotrophic and heterotrophic diazotrophs across the MDV to investigate factors that regulate the distribution and relative ecological roles of each group. Results indicated that diazotrophs were present only in samples with mats, suggesting a metabolic coupling between autotrophic and heterotrophic diazotrophs. Analysis of 16S rRNA and nifH gene sequences also showed that diazotrophs were significantly correlated to the broader bacterial community, while co-occurrence network analysis revealed potential interspecific interactions. Consistent with previous studies, heterotrophic diazotrophs in MDV were diverse, but largely limited to lakes and their outlet streams, or other environments protected from desiccation. Despite the limited distribution, heterotrophic diazotrophs may make a substantial contribution to the nitrogen budget of MDV due to larger surface area and longer residence times of lakes. This work contributes to our understanding of key drivers of bacterial community structure in polar deserts and informs future efforts to investigate the contribution of nitrogen fixation to MDV ecosystems.
Keywords: McMurdo Dry Valleys, Antarctica, co-occurrence network, cyanobacteria, heterotrophic diazotrophy
A survey of N-fixing autotrophic and heterotrophic bacteria in the McMurdo Dry Valleys suggests a substantial role for heterotrophic diazotrophs in N cycling and reveals distinct distributional patterns and potential associations with the broader microbial community.
INTRODUCTION
The biological reduction of dinitrogen (N2) to ammonium by nitrogen-fixing diazotrophs is the primary pathway by which bioavailable nitrogen enters the Earth's biosphere (Vitousek et al. 2013). Diazotrophic species include a diverse group of autotrophic and heterotrophic bacteria found free-living in terrestrial and aquatic environments, as well as in symbiotic association with plants, insects, lichen and phytoplankton (reviewed by Zehr et al. 2003). The contribution of nitrogen supplied by diazotrophs is of particular importance in N-limited environments such as deserts (reviewed by Elbert et al. 2012; Van Goethem and Cowan 2019). In the desert southwest of the United States, for example, N-fixing cyanobacteria contribute substantial amounts of nitrogen (up to 51 mg N2 m−2 h−1) to the benthic zone of desert streams (Grimm and Petrone 1997), while the N-fixing activity of heterotrophic diazotrophs is associated with perennial shrubs in Tehuacán Desert, Mexico, and is thought to support the fertility of soil biota under the shrubs (Rodriguez-Zaraqoza et al. 2008).
One of the most extreme environments on Earth is the McMurdo Dry Valleys (MDV) desert system of Antarctica. The MDV comprise the largest ice-free region on the continent (reviewed by Cary et al. 2010), receiving only 50 to 100 mm of precipitation annually (Keys 1980; Doran et al. 2002). Soils in the MDV are characterized by <2% water content, along with low organic matter, high salt content and high pH (Claridge and Campbell 1977; Bockheim 1997; Barrett et al. 2007). In spite of the extreme conditions, the application of culture-independent methods has revealed a higher than expected level of microbial diversity and biomass in the MDV (Cowan et al. 2002, 2010; Yergeau et al. 2007; Cary et al. 2010; Van Horn et al. 2016). Similar to desert ecosystems in temperate regions, ephemeral meltwater streams generated by glacial runoff constitute microbial hotspots of productivity and biomass in the MDV (Barrett et al. 2006, Stanish et al. 2013; Niederberger et al. 2015a,b), but differ in the high level of illumination and ultraviolet irradiation received by MDV streams during the austral summer.
Microbial mats, composed largely of filamentous photosynthetic cyanobacteria and often dominated by diazotrophic Nostoc spp., play a key role in biogeochemical cycling within the MDV (Vincent 2000; Gooseff et al. 2004; McKnight et al. 2004; Niederberger et al. 2015b). Kohler et al. (2018), for example, demonstrated the contribution of fixed nitrogen derived from diazotrophic cyanobacteria to downstream microbial communities in MDV streams. In addition to providing pond and stream ecosystems with biologically accessible carbon (C) and N during the austral summer, it has been hypothesized that aeolian transport of cyanobacteria mat detritus is a major source of nutrients to the oligotrophic arid soils of the MDV (Hopkins et al. 2006a,b; Barrett et al. 2007).
The highly conserved nifH gene, which encodes the Fe-protein reductase component of the nitrogenase enzyme complex, is a commonly used marker in molecular investigations of diazotroph diversity (see reviews by Zehr et al. 2003; Sohm, Webb and Capone 2011; Hoffman et al. 2014; Santos and Dean 2017). This gene shares a rough concordance with rRNA gene sequence phylogeny across several prokaryotic phyla, allowing for resolution of taxonomic identity from nifH sequences (Zehr et al. 2003). NifH gene sequences have been used as a proxy for diazotroph diversity in a wide range of aquatic (e.g. Short and Zehr 2005; Steppe and Paerl 2005; Zhang et al. 2017) and terrestrial environments (e.g. Rosch and Bothe 2005; Tu et al. 2016). While research on diazotroph diversity and activity in Antarctica has largely focused on cyanobacteria species (e.g. Pandey et al. 2004; Cowan et al. 2011; Van Goethem and Cowan 2019), the relative importance of heterotrophic diazotrophs has also been noted. NifH sequence analysis of hypolithic communities from the MDV, for example, revealed that all putative diazotrophs were Proteobacteria (Lacap-Bugler et al. 2017), in spite of the dominance of non-diazotrophic cyanobacteria species in the assemblage. In another recent study, nifH sequence libraries from sediments collected in Miers Valley in the MDV by Niederberger et al. (2012) demonstrated the presence of highly diverse assemblages of heterotrophic diazotrophs along with cyanobacteria. Results of Niederberger et al. (2012) showed that heterotrophic N-fixing species contributed up to 50% of total nitrogen fixed, indicating that heterotrophic diazotrophs may constitute a significant source of fixed N to the MDV ecosystem.
The relative ecological roles of autotrophs and heterotrophs within the MDV may have important implications for understanding C and N cycling in this system; although both contribute N to the system, cyanobacteria are a source of fixed carbon while heterotrophs are a sink. The contribution of each group of microbes is likely dynamic, as light and temperature regimes change seasonally or with anticipated warming due to climate variability (Jung et al. 2011; Buelow et al. 2016; Makhalanyane, Van Goethem and Cowan 2016; Gooseff et al. 2017; Niederberger et al. 2019). Ecosystem-level disturbance may also lead to the loss of key species and instability of diazotroph co-occurrence patterns (Wang et al. 2017), affecting nutrient cycling and ecological networks of microorganisms in the MDV that benefit from their association with diazotrophs.
The overall goals of the current study were to investigate the diversity, composition and relative distributions of heterotrophic and autotrophic N-fixing bacterial assemblages in the MDV, as well as potential relationships between diazotrophic bacteria (based on nifH sequence data) and the broader microbial community (based on 16S rRNA gene sequence data). These relationships were investigated using multivariate statistical methods, both at the whole community level and for subsets of the community. Positive correlations in relative abundance were then inferred from co-occurrence network analysis to identify potential interspecific relationships between diazotrophs and the other members of the microbial community.
MATERIALS AND METHODS
Sixty-nine surface sediment samples were collected from 23 sites in the MDV over two field seasons (Fig. 1; Table 1). With some exceptions noted below, each site was sampled along a transect, with samples collected from submerged sediments at edges of ponds/lakes or streams (designated by site_number.1), in the wetted region adjacent to water bodies (designated by site_number.2) and in an adjacent dry area (designated by site_number.3). At each site, surface samples were collected from two to three randomly placed 10 cm × 10 cm quadrants to a depth of ∼1 cm. Sediments from each quadrant were combined in a sterile polyethylene bag (Whirl-Pak®; Nasco, Fort Atkinson, WI USA). Composite samples were mixed and stored at ambient temperature until processing within 2–6 h of collection.
Figure 1.

Map of sites sampled in 2014 (Sites 1–12) and 2015 (Sites 13–23). Main panel: MDV with boxes indicating valleys that were sampled, Antarctica (inset box); (A) Victoria Valley sample sites; (B) Taylor Valley sample sites; (C) Miers Valley, Garwood Valley and Nostoc Flats sample sites with boxed area from panel (D); and (D) Upper Garwood Valley sample sites. (Satellite imagery provided by Polar Geospatial Center)
Table 1.
Site location, collection date and mat characteristics.
| Site ID | Date | Latitude | Longitude | Valley | Location | Mat characteristics |
|---|---|---|---|---|---|---|
| 1 | 1/11/2014 | −77.6598 | 163.1160 | Taylor | Spaulding Pond | Cyanobacteria |
| 2 | 1/13/2014 | −77.6518 | 163.1019 | Taylor | Upper Delta Stream | Cyanobacteria |
| 3 | 1/14/2014 | −77.6613 | 163.0900 | Taylor | Base of Howard Glacier | Cyanobacteria |
| 4 | 1/15/2014 | −77.6440 | 162.7568 | Taylor | Edge of Lake Chad | No mat |
| 5 | 1/17/2014 | −77.3376 | 161.6291 | Victoria | Downstream of Victoria Upper Lake | No mat |
| 6 | 1/17/2014 | −77.3280 | 161.6118 | Victoria | Victoria Upper Lake | Cyanobacteria |
| 7 | 1/18/2014 | −77.6579 | 163.0963 | Taylor | Howard Glacier flood plain | Cyanobacteria |
| 8 | 1/20/2014 | −77.6442 | 163.1744 | Taylor | Lower Delta Stream | Cyanobacteria |
| 9 | 1/21/2014 | −77.6248 | 163.0655 | Taylor | Green Creek | Cyanobacteria |
| 10 | 1/22/2014 | −77.5844 | 163.5916 | Taylor | Explorer's Cove | Cyanobacteria/moss |
| 11 | 1/23/2014 | −77.6423 | 163.2060 | Taylor | Crescent Stream | Cyanobacteria |
| 12 | 1/24/2014 | −77.5968 | 163.2738 | Taylor | Base of Commonwealth Glacier | Cyanobacteria |
| 13 | 1/12/2015 | −78.0254 | 163.8333 | Garwood | Base of Joyce Glacier | No mat |
| 14 | 1/17/2015 | −78.0233 | 163.9175 | Garwood | Southern Hillside | Cyanobacteria |
| 15 | 1/17/2015 | −78.0231 | 163.9156 | Garwood | Southern Hillside | Cyanobacteria |
| 16 | 1/18/2015 | −78.0230 | 163.9019 | Garwood | South side of Garwood Lake | Cyanobacteria |
| 17 | 1/19/2015 | −78.0270 | 164.1370 | Garwood | Garwood River at Mid Valley | No mat |
| 18 | 1/20/2015 | −77.6610 | 163.0918 | Taylor | Base of Howard Glacier | Cyanobacteria |
| 19 | 1/20/2015 | −77.6572 | 163.1316 | Taylor | Delta Stream Tributary | Cyanobacteria |
| 20 | 1/21/2015 | −78.0458 | 163.6748 | Marshall | Nostoc Flats | Cyanobacteria |
| 21 | 1/21/2015 | −78.0936 | 163.8319 | Miers | North side of Lake Miers | Cyanobacteria |
| 22 | 1/22/2015 | −78.0242 | 163.8882 | Garwood | Southern Hillside control site | No mat |
| 23 | 1/26/2015 | −78.0289 | 164.2506 | Garwood | Pond at Lower Garwood | Cyanobacteria |
In January 2014, sediment samples were collected from 12 sites (Sites 1–4 and 7–12) in Taylor Valley and two sites (Sites 5 and 6) in Victoria Valley (Fig. 1; Table 1). In January 2015, samples were collected from Sites 13–17 and 22–23 in Garwood Valley, Sites 18 and 19 in Taylor Valley, Site 20 in Marshall Valley and Site 21 in Miers Valley, for a total of 11 sites (Fig. 1; Table 1). At sites with extensive wetted regions (Sites 1, 7, 16 and 20), two samples were collected from wetted soils, yielding a transect with four samples instead of three. A single ‘control’ sample was collected at Site 22 from a dry region on a steep hillside in Garwood Valley. Site 17 was from a rocky ravine in Garwood Valley without adjacent dry soil, providing only submerged and wetted samples. In addition to surface samples, a subsurface sample was also collected for samples 3.1 and 7.1 (designated 3.1.sub and 7.1.sub) from Taylor Valley. Site 18 collected from Taylor Valley in 2015 was at the same location as Site 3 collected in 2014. A single stream sample 19.1 was collected just downstream of Site 18.
Water samples for pH were collected from the overlying water in the submerged regions and from pore water in submerged and wetted regions, and filter-sterilized through 0.2 µm polycarbonate (PC) filters. Samples were stored cold in polypropylene centrifuge tubes, and the pH was later measured with a Hanna Instruments model 2213 pH/ORP meter (Hanna Instruments, Smithfield, RI).
Three replicate 1 cm3 sediment cores were collected for chlorophyll-a determination. Cored samples were transferred into Whirl-Pak bags in the field and later transferred to 50 mL conical centrifuge tubes and stored frozen in the dark until analysis. Chlorophyll-a was extracted over 24 h at −20°C using 90% acetone, and then measured on a Turner 10AU fluorometer and corrected for phaeophytin (Holm-Hansen et al. 1965; Vincent et al. 1993).
Water samples were collected from surface water where available and from pore water at submerged and wetted sites for dissolved nutrient as well as dissolved organic and inorganic carbon analyses. Pore water was obtained by driving a 50 mL conical centrifuge tube, modified with several drill holes, into the sediments and allowing pore water to fill the tube. Water was extracted from the tube by inserting acid-washed silicone tubing into the centrifuge tube, fitted with a 50 cc barrel syringe that allowed collection of the water. Pore water was first collected in 250 mL high-density polyethylene bottles and transported back to camp for further processing. Pore water was filtered through 25 mm Whatman GF/F filters and subsampled for nutrient concentrations: nitrate + nitrite (NO3+ NO2), orthophosphate (PO4), silicate [Si(OH)4] and ammonium (NH4), and dissolved organic carbon (DOC). All subsamples were stored frozen.
Nutrient concentrations were measured on a Bran and Luebbe AutoAnalyzer II with MT-19 manifold chemistry module for NO3 and NO2 (Whitledge et al. 1981; Bran and Luebbe 1997; Bran and Luebbe Method G-172-96), PO4 (Bran and Luebbe Method G-175-96) and Si(OH)4 (Bran and Luebbe Method G-177-96) (Bran and Luebbe Inc. 1997). NH4 was analyzed according to Solorzano (1969). Pore water subsamples for DOC were acidified prior to analysis using a Shimadzu TOC-V high-temperature combustion instrument (Sharp et al. 2002)). Other soil characteristics including % moisture, conductivity and elemental analysis were determined as previously described by Lee et al. (2019).
DNA extraction
Composite samples were subsampled for DNA extraction using the Mo Bio Powersoil DNA Isolation Kit (Mo Bio Laboratories, Carlsbad, CA). Replicate samples were extracted from sample 14.1 (designated 14.1B and 14.1C) collected in Garwood Valley to evaluate representativeness of subsamples. Extracted DNA was stored on dry ice in the field until return to Crary Lab at McMurdo Station, after which samples were stored at −80°C.
NifH amplification
Samples were amplified by polymerase chain reaction (PCR) using primers targeting conserved regions in the nifH gene to identify communities harboring diazotrophs. PCR reactions consisted of 2 µL DNA (5–10 ng), 1× polymerase buffer, 0.2 mM dNTPs, 2.5 mM Mg2+, 0.2 µM forward primer IGK3 [5′-GCIWTHTAYGGIAARGGIGGIATHGGIA-3′; Ando et al. (2005)], 0.2 µM reverse primer nifH2 [5′-ADNGCCATCATYTCNCC-3′; Zani et al. (2000)] and 0.625 units Jump Start Taq Polymerase (Sigma-Aldrich Corp., St Louis, MO) in a 50 µL reaction. PCR reactions were incubated at 94°C for 2 min, and amplified for 10 cycles of 94°C for 45 s, 60°C for 45 s, decreasing 0.5°C per cycle, and 72°C for 1.5 min, followed by 25 cycles of 94°C for 45 s, 51°C for 45 s and 72°C for 1.5 min.
Samples from the submerged and wetted regions at Sites 1–3 and 7–12, collected in Taylor Valley in 2014, were then amplified in nested PCR reactions for nifH amplicon sequencing by Ion Torrent (Thermo Fisher Scientific). A nested PCR reaction was necessary to minimize amplification of chlorophyllide reductase gene sequences, Group V nitrogenase homologs (Raymond et al. 2004). The initial PCR reaction consisted of 2 µL template DNA (5–10 ng), 1× polymerase buffer, 0.2 mM dNTPs, 2.5 mM Mg2+, 0.2 µM primer IGK3, 0.2 µM primer nifH2 and 0.625 units Jump Start Taq Polymerase (Sigma-Aldrich Corp., St Louis, MO) in a 50 µL reaction. PCR reactions were incubated at 94°C for 2 min, and amplified for 10 cycles of 94°C for 45 s, 60°C for 45 s, decreasing 0.5°C per cycle, and 72°C for 1.5 min, followed by five cycles of 94°C for 45 s, 51°C for 45 s and 72°C for 1.5 min. The PCR reaction was then diluted with three volumes of sterile water and subjected to a second PCR reaction. Triplicate reactions for each sample included 4 µL of diluted PCR reaction, 1× polymerase buffer, 0.2 mM dNTPs, 2.5 mM Mg2+, 0.2 µM forward primer IGK3-92, -93, -94 or -95, modified at the 5′ end with a barcode nucleotide sequence required for sequencing by Ion Torrent, 0.2 µM reverse primer DVV-Ion_Torrent [5′-ATIGCRAAICCICCRCAIACIACRTC-3′; Ando et al. (2005)], modified at the 5′ end with a barcode nucleotide sequence required for sequencing by Ion Torrent and 0.625 units Jump Start Taq Polymerase (Sigma-Aldrich Corp.). PCR reactions were incubated at 94°C for 2 min, and amplified for 30 cycles of 94°C for 30 s, 58°C for 30 s and 72°C for 1 min. Products of triplicate reactions were pooled and purified using SPRISelect Beads (Beckman Coulter, Indianapolis, IN) to select for amplicons > 200 bp. Purified PCR products were then precipitated with ethanol and sequenced using an Ion Torrent PGM DNA sequencer (ThermoFisher) at the University of Waikato DNA Sequencing Facility (Hamilton, New Zealand). The resulting sequencing reads were processed using mothur v.1.35.1 (Schloss et al. 2009) to discard all reads shorter than 300 bp (the anticipated amplicon size was around 395 bp), all reads longer than the 97.5th percentile and all reads with homopolymers more than the 97.5th percentile. All reads with mismatches to the forward PCR primer or with expected error rates higher than 1% (-fastq_maxee_rate 0.01) were discarded using USEARCH (Edgar 2010)), and all reads were truncated to 270 bp. The resulting reads were de-replicated, singleton reads found only once across all samples were discarded and the remaining reads were clustered at 95% identity.
Phylogenetic analysis
The translated amino acid sequences of 46 nifH operational taxonomic units (OTUs) representing sequences that were present at greater than 1% of the nifH library were subjected to phylogenetic analysis. Amino acid sequences were aligned in Geneious v.10.2.2 (http://www.geneious.com/) along with nifH sequences for relevant taxa and closely related environmental isolates in GenBank. The alignment was imported into MEGA v.7 (Sudhir, Stecher and Tamura 2016) for phylogenetic analysis using the neighbor-joining method (Saitou and Nei 1987). The evolutionary distances were computed using the p-distance method and the rate variation among sites was modeled with a gamma distribution.
16S rRNA gene sequence analysis
DNA was submitted to MR DNA Laboratory (Shallowater, TX) for 16S rRNA gene sequencing. DNA was quality-checked and the V1–V3 region of the 16S rRNA gene was amplified using Eubacterial primers ill27Fmod (AGRGTTTGATCMTGGCTCAG; Kuske 2006) and ill519Rmod (5′-GTNTTACNGCGGCKGCTG-3′), modified with barcode sequences for paired-end sequence analysis (2 × 300) on the Illumina MiSeq platform (Illumina, Inc., San Diego, CA). Paired-end reads exceeding the Illumina Q30 quality standard were joined and chimeras removed before further analysis using the QIIME v.1.9.1 (Caporaso et al. 2010) pipeline. Sequences were clustered into OTUs at 97% similarity and taxonomy assigned using the pick_open_reference_otus.py script. Chloroplast sequences and 16S rRNA OTUs containing <10 sequences across all 69 samples were removed. Three groups of OTU libraries were then generated for analysis: (i) No_chlor containing all remaining 16S rRNA OTUs, (ii) No_cyano OTUs in which cyanobacteria sequences were removed and (iii) Cyano_only OTUs that contained only cyanobacteria sequences. The No_chlor OTU libraries were rarefied to a sampling depth of 33 531 and diversity analysis was conducted on rarefied libraries using the core_diversity_analyses.py script in QIIME v.1.9.1 (Caporaso et al. 2010).
Multivariate statistical analysis
Multivariate statistical analysis was conducted in PRIMER v.7 (Clarke and Gorley 2015). OTU libraries were standardized and square root transformed before computing Bray–Curtis dissimilarity matrices. Cluster analysis was used to generate dendrograms showing similarities of nifH OTU abundances representing greater than 1% of the nifH library and 16S rRNA gene OTUs. Shade plots were then constructed for visualization of OTU abundances of the nifH library and class-level distribution of taxa based on 16S rRNA gene. The Spearman rank correlation method in the RELATE function in PRIMER was used to test the null hypothesis that there was no correlation between multivariate patterns generated by the No_cyano and Cyano_only groups for the 16S rRNA gene or nifH OTU abundance data. The analysis of similarity (ANOSIM) function in PRIMER was applied to 16S rRNA gene OTU abundance data to test the null hypotheses that (i) there were no differences in assemblages for samples collected in submerged versus wetted versus dry regions, (ii) there were no differences in assemblages for samples collected from different valleys, (iii) there were no differences in assemblages for samples collected in areas with cryptogamic mats compared to areas with no visible mat present and (iv) there were no differences in assemblages for samples that were positive for nifH compared to those that were negative for nifH. Permutation tests randomly resampled data 999 times.
Environmental data were fourth root transformed and normalized. Principal component analysis (PCA) was conducted using PRIMER v.7 (Clarke and Gorley 2015) to identify environmental variables in submerged and wetted regions that contributed to the variability between sites. The Biota-Environment STepwise (BEST) matching function in PRIMER was used to identify environmental variables that were correlated to changes in microbial community structure based on both 16S rRNA gene and nifH OTU abundances for submerged and wetted regions. Null hypotheses to test correlations between a set of environmental variables and the assemblage were then examined with BEST using permutation tests that randomly resampled data 99 times.
Network inference
Correlations between taxon relative abundances in the 16S rRNA gene library and nifH sequences were investigated using CoNet (Faust and Raes 2016). The 16S rRNA gene library included samples from sites that were sequenced for nifH and for which taxonomy was assigned using the pick_closed_reference_otus.py program in QIIME at 97% similarity based on Greengenes 13_5 16S rRNA gene database (McDonald et al. 2012). 16S rRNA OTUs with fewer than 100 counts across all samples were excluded from analysis, leaving 598 taxa. The nifH library included the 46 most highly abundant sequences as described above. Sequences that were present at only one site were excluded from analysis. A distribution of pairwise scores was calculated for each of the following similarity measures: Bray–Curtis dissimilarity, Kullbacker–Leibler dissimilarity and Pearson and Spearman correlations. Initial thresholds were selected so that each of these measures contributed 1000 positive edges and 1000 negative edges to the network. The initial network was then used to generate 1000 renormalized permutations and 1000 bootstrap scores and a P-value calculated for each similarity measure. P-values were merged taking into account any correlations between similarity measures (Brown 1975). Multiple-test corrections were conducted using Benjamini–Hochberg's method (Benjamini and Hochberg 1995) such that any edges with merged P-values below 0.05 were retained. The inferred network including only positive correlations was visualized in Cytoscape v. 3.6.1 (Shannon et al. 2003).
RESULTS
Environmental conditions
Environmental context is provided for submerged, wetted and dry soil habitats through measurements of soil moisture, conductivity, chlorophyll-a and inorganic and organic nutrients summarized in Table S1 (Supporting Information). Not surprisingly, soil moisture was highest in soils from submerged sites (13–75%), followed by wetted soils (13–57%) and finally ‘dry’ soil habitats (2–18%). Chlorophyll-a concentrations were similar for submerged and wetted habitats (range 0.0023–1.32 ug/g), and roughly an order of magnitude higher than found in dry soils. Pore water pH and nutrients were collected at submerged and wetted habitats. pH was more variable at submerged habitats (5.90–7.43) compared to wetted soil habitats (6.16–7.41), with average pore water pH slightly acidic in both habitats. At submerged habitat sites, pore water NOx, PO4, SiO2 and DOC were roughly 2-fold higher than the overlying water. Comparisons between pore water nutrients at submerged versus wetted sites suggest that wetted sites were elevated (∼2-fold) for NOx and SiO2 but more similar to submerged sites for PO4. Average NH4 was >10-fold higher in wetted pore water compared to submerged soil habitats, although much of this difference was due to a single site (Site 16, data not shown). PCA analysis of environmental attributes for submerged and wetted samples showed that 47.3% of the variability between samples could be represented on PC1 and PC2 (Figure S1, Supporting Information). Pore water pH, conductivity and NOx concentrations contributed the most to variability between samples.
NifH amplification and sequence analysis
PCR amplification of nifH yielded positive results for samples collected in submerged and wetted regions of transects, except for Site 4 in Taylor Valley, Sites 5 and 6 in Victoria Valley and Sites 13, 17 and 20 in Garwood Valley. A total of 18 samples representing paired submerged and associated wetted regions of Sites 1–3 and 7–12 collected from Taylor Valley were submitted for amplicon sequencing. The number of sequences obtained varied from 258 for sample 9.2 to over 41 000 sequences for sample 12.2 (Table 2). Binning at 95% similarity resulted in 102 nifH OTUs ranging from 13 to 67 OTUs per sample. Diversity must be viewed with caution, however, due to the large difference in library sizes (Gihring, Green and Schadt 2012). The number of nifH OTUs that would be obtained with additional sequencing effort was estimated using the Chao–Shen index (Chao and Shen 2003; Table 2).
Table 2.
The total number of nifH sequences, OTUs (binned at 95% similarity) and sequence distribution between cyanobacteria and heterotroph lineages (calculated as a % of total sequences) for samples collected in 2014 from Taylor Valley. Chao–Shen index estimated the number of additional OTUs generated by obtaining an additional 250 sequences.
| Sample ID | Total sequences | OTUs | Cyanobacteria (%) | Heterotrophs (%) | Chao–Shen index |
|---|---|---|---|---|---|
| 1.1 | 768 | 23 | 0.26 | 99.7 | 2.229 |
| 1.2 | 618 | 19 | 21.5 | 78.5 | 1.254 |
| 2.1 | 19826 | 67 | 49.2 | 50.8 | 0.050 |
| 2.2 | 2731 | 26 | 89.0 | 11.0 | 0.819 |
| 3.1 | 7408 | 33 | 90.9 | 9.1 | 0.130 |
| 3.2 | 6308 | 25 | 93.5 | 6.5 | 0.272 |
| 7.1 | 555 | 13 | 73.3 | 26.7 | 2.052 |
| 7.2 | 19463 | 24 | 51.2 | 48.8 | 0.025 |
| 8.1 | 9013 | 25 | 99.1 | 0.89 | 0.082 |
| 8.2 | 13939 | 17 | 99.8 | 0.20 | 0.053 |
| 9.1 | 2091 | 20 | 99.2 | 0.76 | 0.675 |
| 9.2 | 258 | 13 | 90.4 | 9.6 | Failed |
| 10.1 | 4253 | 38 | 70.4 | 29.6 | 0.113 |
| 10.2 | 4300 | 26 | 57.6 | 42.4 | 0.168 |
| 11.1 | 1020 | 13 | 99.6 | 0.39 | 0.635 |
| 11.2 | 3104 | 17 | 98.5 | 1.48 | 0.150 |
| 12.1 | 561 | 14 | 98.6 | 1.43 | 2.045 |
| 12.2 | 41121 | 26 | 99.96 | 0.041 | 0.012 |
A single cyanobacteria nifH OTU (RM_252) occurred in every sample and was identical to nifH OTU43 (GenBank Accession number HM140768; Table 3) from Niederberger et al. (2012). Differences in choice of degenerate PCR primers used in Niederberger et al. (2012) and this study may be expected to result in differential amplification of nifH (Angel et al. 2018). In addition, nifH sequences reported in Niederberger et al. (2012) were isolated from Miers Valley, Antarctica, while those identified in this study were all from Taylor Valley. In spite of the difference in primers and distance between Miers Valley and Taylor Valley (Fig. 1), the nifH libraries sequenced in this study included eleven OTUs that shared 99% or greater similarity with nifH OTUs detected in Niederberger et al. (2012; Table 3), with four OTUs identical between the two libraries.
Table 3.
NifH sequences from this study having ≥99% identity with nifH sequences reported in Niederberger et al. (2012). Shared sequences included Alphaproteobacteria (Alpha), Deltaproteobacteria Group I (Delta GrI), Deltaproteobacteria Group II (Delta GrII), Gammaproteobacteria (Gamma), unclassified lineages and cyanobacteria (Cyano).
| OTU | Phylogenetic identity | Accession | E-value | % Identity | Sequence IDa |
|---|---|---|---|---|---|
| R22_5549 | Unclassified | HM140726 | 1.64E-119 | 99.6 | OTU1 |
| R22_66430 | Unclassified | HM140731 | 6.07E-99 | 100.0 | OTU10 |
| Rn01_112 | Unclassified | HM140757 | 3.58E-101 | 100.0 | OTU32 |
| R22_5253 | Delta GrI | HM140738 | 2.77E-102 | 99.0 | OTU13 |
| R21_1235 | Delta GrII | HM140744 | 2.16E-98 | 99.5 | OTU19 |
| R21_902 | Delta GrII | HM140747 | 2.77E-102 | 99.0 | OTU22 |
| R21_203 | Delta GrII | HM140752 | 2.77E-102 | 99.0 | OTU27 |
| R21_2604 | Gamma | HM140743 | 2.77E-102 | 99.0 | OTU18 |
| R22_9 | Gamma | HM140763 | 1.00E-96 | 99.0 | OTU38 |
| R23_18720 | Alpha | HM140765 | 1.69E-99 | 100.0 | OTU40 |
| RM_252 | Cyano | HM140768 | 7.28E-143 | 100.0 | OTU43 |
From Niederberger et al. (2012).
Of the 102 nifH OTUs, 31 were most closely related to cyanobacteria nifH sequences and represented 0.26% (sample 1.1) to 99.96% (sample 12.2) of the nifH libraries (Table 2). Phylogenetic analysis of the 46 most highly abundant amino acid sequences revealed the presence of a diverse community of heterotrophic (Fig. 2A) and autotrophic (Fig. 2B) diazotrophs. Two OTUs had <95% similarity to nifH amino acid sequences from known diazotrophs and are labeled as ‘unclassified’ (Fig. 2A). The remaining nifH sequences clustered with heterotrophic bacteria within the Bacteroidetes and Proteobacteria (Fig. 2A).
Figure 2.

Phylogenetic analysis of deduced heterotrophic (A) and cyanobacterial (B) nifH amino acid sequences. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (100 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and are in the units of the number of amino acid differences per site. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). All positions with <75% site coverage were eliminated. That is, fewer than 25% alignment gaps, missing data and ambiguous bases were allowed at any position. There were a total of 89 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 (Kumar, Stecher and Tamura 2016).
ANOSIM analysis of nifH sequence distribution revealed no significant differences in submerged versus wetted assemblages. Cluster analysis of samples based on nifH OTUs (Fig. 3) revealed a topology loosely correlated to sample location. The submerged sample from Spaulding Pond (sample 1.1), for example, was distinct from stream-collected samples. The nifH assemblages at Sites 9 and 12 from Green Creek and the base of Commonwealth Glacier on the north side of Taylor Valley were more similar to each other than they were to samples collected from the south side of the valley near Howard and Crescent Glaciers (Sites 2, 3, 7 and 11). The nifH assemblage in sample 2.1, located just downstream of Spalding Pond, was most closely related to sample 1.2, at the edge of the pond. Conversely, the nifH assemblage in samples collected at the wetted region of Site 2 (sample 2.2) was most similar to samples collected at Explorer's Cove (Site 10), possibly related to the higher diversity of Cyanobacteria nifH OTUs from these samples.
Figure 3.

(A) Cluster analysis samples based on nifH OTUs. (B) Shade plot of standardized, square root-transformed OTU abundance. OTUs in the shade plot are arranged according to prokaryotic diversity.
The nifH sequence assemblages were not significantly correlated to environmental variables (P = 0.31). When evaluated separately, however, there was a significant correlation between cyanobacteria nifH assemblages and environmental variables (Rho = 0.398; P = 0.03), with pore water pH, pore water magnesium, pore water potassium and pore water DOC concentrations contributing most to the relationship. There was no significant correlation between the heterotrophic nifH assemblage and environmental variables.
16S rRNA gene sequence analysis
Altogether, 69 libraries of 16S rRNA gene amplicon sequences from 23 sites were generated. After removal of sequences identified as chloroplast 16S rRNA gene, the number of remaining sequences ranged from 33531 to 167085 (median = 97430; Table 4) per sample. The number of observed 16S rRNA gene OTUs generated with a 97% similarity cutoff ranged from 1192 to 8140 per sample. The number of 16S rRNA OTUs in submerged and wetted regions was significantly greater than the number of OTUs in dry regions (Table 4; P < 0.05). After rarefaction to 33531, Chao1 diversity was greatest in wetted regions and was significantly higher in both submerged and wetted regions compared to dry regions ( P < 0.05; Table 4).
Table 4.
16S rRNA library statistics for submerged, wetted and dry regions. Sequences identified as chloroplast 16S rRNA were removed. Chao1 diversity was generated after rarefaction to 33531 sequences.
| Submerged | Wetted | Dry | |
|---|---|---|---|
| Number of sequences: | |||
| Range (mean) | 49943–167085 (97761) | 61764–146418 (93153) | 33531–142675 (97314) |
| Observed OTUs: | |||
| Range (mean) | 1192–7514 (5376) | 1862–8140 (5445) | 1292–6650 (3838) |
| Chao 1 diversity: | |||
| Mean (std. deviation) | 5.521 (1.284) | 5.999 (1.574) | 4.096 (1.475) |
Few 16S rRNA gene OTUs were shared between all libraries (Figure S2 and Table S2, Supporting Information). Samples collected from submerged regions shared 134 OTUs, with 34 OTUs unique to those sites, including species within Cyanobacteria, Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Nitrospirae, Proteobacteria and Verrucomicrobia phyla. Wetted samples shared 164 OTUs, of which 58 were unique to wetted libraries. Unique 16S rRNA gene OTUs in this region included species from the same phyla as submerged regions, as well as species within the Planctomycetes phylum. Samples from dry regions of transects shared the fewest OTUs (77), with 22 OTUs unique to this region, including species within Acidobacteria, Actinobacteria, Chloroflexi, Gemmatimonadetes and Deinococcus–Thermus phyla.
Multivariate statistical analysis
ANOSIM analysis of 16S rRNA gene library revealed a significant difference in bacterial assemblages based on location within a transect (submerged, wetted and dry; R = 0.341; P < 0.05), and between sites with visible mats versus samples with no visible mat (R = 0.470 and R = 0.362, respectively; P < 0.05). Bacterial communities collected from Taylor Valley versus Garwood Valley were also significantly different (R = 0.289; P < 0.05).
Cluster analysis of 16S rRNA gene libraries revealed further relationships in community structure loosely based on sample type (submerged, wetted or dry), location and field season (Fig. 4A, see Clusters I–XII). Notably, bacterial assemblages from samples that were also positive for nifH clustered together, while samples that were negative for nifH formed distinct clusters independent of position within the transect. Samples collected from submerged and wetted regions of transects that were negative for nifH (Sites 4, 5, 6, 13 and 17; Clusters III, X, XI and XII), for example, were more highly correlated to each other and/or to samples collected from dry regions than to samples that were positive for nifH. Samples collected from ponds or lakes (Sites 1, 16, 21 and 23) formed two distinct clusters (Clusters I and II). Samples collected in Victoria Valley (Sites 5 and 6) formed Cluster III, while Clusters VI and VII collected on the south side of Taylor Valley (Sites 2, 3, 7 and 11) were distinct from bacterial assemblages collected on the north side of the Valley (Sites 9 and 12; Cluster VIII).
Figure 4.

(A) Cluster analysis of samples based on 16S rRNA OTUs. Samples are identified based on collection point along a transect including submerged (closed diamonds), wetted (open triangles) and dry (open circles) samples. Twelve clusters were identified loosely based on absence of visible mat or location (see text for details). Clusters outlined in black and shaded in gray indicate sites that were also positive for nifH. (B) Shade plot of standardized and square root-transformed 16S rRNA OTU relative abundance within a taxonomic class and arranged according to phylum.
A shade plot depicting the relative abundance of selected bacterial classes revealed the basis for patterns observed in the cluster diagram (Fig. 4B). Bacterial communities from dry samples generally had a higher relative abundance of Actinobacteria, Acidobacteria and Gemmatamonidetes, while samples collected from submerged or wetted regions generally had higher relative abundances of Cyanobacteria and/or Proteobacteria, as well as specific classes of bacteria within the Chloroflexi, Planctomycetes and Bacteroidetes. Samples collected from dry regions during the 2015 field season had higher relative abundances of Cyanobacteria, Planctomycetes and Verrucomicrobia compared to samples collected in 2014. In addition, Cluster I with samples collected from submerged and wetted regions of lakes or ponds had a high relative abundance of Bacteroidia and Anaerolineae compared to samples from streams or dry regions.
ANOSIM analysis of the heterotrophic community alone (No_cyanos) or the cyanobacterial community alone (Cyanos_only) revealed significant differences in bacterial assemblages based on location within the transect (R = 0.352 and 0.166, respectively; P < 0.05). When evaluated by region within a transect, the heterotrophic bacterial population was highly and significantly correlated to the cyanobacterial population (Rho = 0.736; P < 0.05). Furthermore, ANOSIM analysis of the 16S rRNA gene sequence libraries revealed significant differences between assemblages collected from submerged and wetted regions that were positive for nifH versus those that were negative (R = 0.434; P = 0.001).
BEST analysis of submerged and wetted samples indicated no significant relationship between bacterial assemblages and environmental variables. Analysis of specific subsets of bacterial assemblages (i.e. based on year, location within a transect, Only_cyano and No_cyanos groups) also demonstrated no significant correlation with environmental variables measured here.
Comparisons between nifH and 16S rRNA gene community structure
The community structure of nifH OTUs was highly and significantly correlated to the bacterial assemblages based on 16S rRNA gene sequences (Rho = 0.614; P = 0.01). This relationship was primarily driven by nifH OTUs within the cyanobacteria clade. When divided into cyanobacteria and heterotrophic nifH libraries, the structure of cyanobacteria nifH library was significantly correlated to community structure of the entire 16S rRNA gene library (Rho = 0.403; P = 0.001), as well as to the Only_cyanos (Rho = 0.474; P = 0.001) and the No_cyanos (Rho = 0.391; P = 0.001) 16S rRNA gene libraries. There was no significant correlation between heterotrophic nifH and 16S rRNA gene assemblages.
Network analysis
Network analysis identified 41 nifH OTUs with significant positive correlations to 351 16S rRNA gene OTUs in 18 samples (Fig. 5; Table S3, Supporting Information). Of these, 22 heterotrophic nifH OTUs and 19 cyanobacteria nifH OTUs were positively correlated to 136 and 220 16S rRNA gene OTUs, respectively (Table S3, Supporting Information). Cyanobacterial nifH OTUs represented in the network were distributed in up to 18 samples (indicated as counts; Table S3, Supporting Information), and were significantly correlated with as few as 3 and up to 44 16S rRNA gene OTU abundances (indicated as degrees; Table S3, Supporting Information). Heterotrophic nifH OTUs were significantly correlated with 2 to 39 16S rRNA gene OTUs. It might be expected that sample counts were inversely related to the degree. That is, samples with restricted distribution may be randomly correlated to a larger number of 16S rRNA gene sequences, but there was no correlation between the count and degree for nifH OTUs (R2 = 0.0003).
Figure 5.
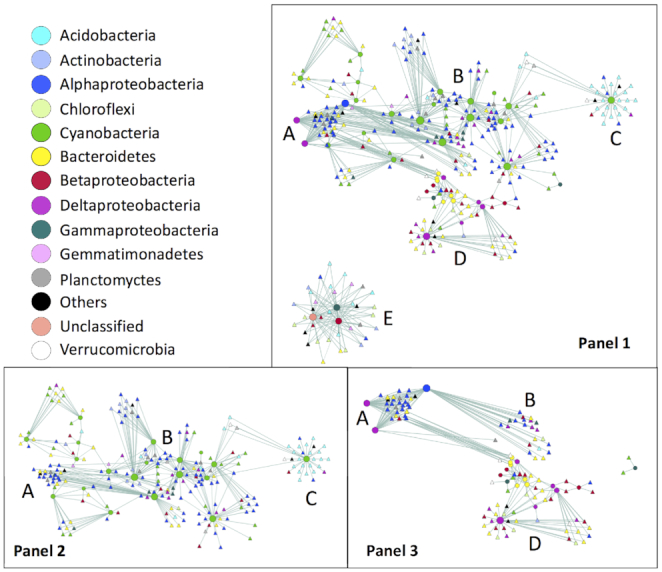
Network analysis illustrating significant correlations between nifH and 16S rRNA OTU abundance for 18 sites included in the nifH library. Node color indicates phylum except for Proteobacteria, which are identified to the class level. nifH (circles) and 16S rRNA OTUs (triangles) are connected by edges, where the edge thickness is proportional to the significance of the correlation (merged P-value) and the sizes of the nifH nodes are proportional to the number of edges connecting them. Panel 1: all nodes; Panel 2: Cyanobacteria nifH and associated 16S rRNA nodes only; Panel 3: heterotrophic nifH and associated 16S rRNA nodes only. A, B, C, D and E are for orientation and refer to groups of nodes described in greater detail in the text.
Five groups or clusters within the network were labeled A–E on Fig. 5, based on shared 16S rRNA gene nodes (triangles) and/or the relative positions of heterotrophic versus cyanobacteria nifH nodes (circles) in the network (Fig. 5, Panels 2 and 3). Cluster A included both heterotrophic and cyanobacteria nifH OTU nodes, but there were few 16S rRNA gene OTUs shared between the autotrophic and heterotrophic diazotrophs in this cluster. Cyanobacterial nifH OTUs in Cluster A were broadly distributed with generally higher relative abundances at Lower Delta Stream (Site 8) and at Green Creek and the base of Commonwealth Glacier (Sites 9 and 12) on the north side of Taylor Valley. The 16S rRNA gene OTUs that correlated with cyanobacterial nifH OTUs in Cluster A were primarily within the Bacteroidetes, Cyanobacteria (Nostocaceae) and Proteobacteria (Table S3, Supporting Information).
Two heterotrophic nifH nodes in Cluster A shown on Fig. 5 (Panel 3) represented OTUs (R22_5253 and R22_50498) from sulfate-reducing Deltaproteobacteria Group I within the Desulfovibrio and Desulfobulbus genera (Fig. 5, Panel 3; Table S3, Supporting Information), found at Spaulding Pond (Site 1), upper Delta Stream (Site 2) and Explorer's Cove (Site 10) (Fig. 3). Cluster A also included the only Alphaproteobacteria nifH node in the network (OTU Rn04_89), found primarily at Explorer's Cove (Site 10) and the sites near Howard Glacier (Sites 3 and 7). The 16S rRNA gene OTUs that correlated with heterotrophic nifH OTUs in Cluster A were dominated by Bacteroidetes and Alphaproteobacteria (Table S3, Supporting Information). Several Chlorobi and Planctomycetes 16S rRNA gene OTUs were also significantly correlated with heterotrophic nifH OTUs in Cluster A. Notably absent were cyanobacterial 16S rRNA gene OTUs correlated to heterotrophic nifH OTUs in this cluster, although the heterotrophic nifH nodes shared several 16S rRNA gene nodes with cyanobacterial nifH nodes in Cluster B (Fig. 5, Panels 2 and 3).
Cluster B shown in Fig. 5 (Panel 2) included only cyanobacterial nifH OTUs (Table S3, Supporting Information). These OTUs were broadly distributed with generally lower relative abundance at Lower Delta Stream (Site 8), and on the north side of Taylor Valley at Green Creek (Site 9) and the base of Commonwealth Glacier (Site 12). 16S rRNA gene OTUs that correlated with cyanobacterial nifH OTUs in Cluster B were dominated by Proteobacteria. Cyanobacterial nifH OTUs with higher abundance at the base of Howard Glacier (Sites 3 and 7) and Crescent Stream (Site 11) were significantly correlated with several Acidobacterial 16S rRNA gene OTUs, while those that were found in higher abundance in Upper Delta Stream (Site 2) and Explorer's Cove (Site 10) were correlated with Actinobacterial 16S rRNA gene OTUs.
Cluster C included a single cyanobacteria nifH node found primarily at Green Creek (Site 9) and Crescent Stream (Site 11), and was positively correlated with a number of Acidobacteria 16S rRNA gene OTUs within the photosynthetic Chloracidobacteria genus. Notably, no Actinobacteria 16S rRNA gene OTUs were significantly correlated with this nifH node.
Cluster D shown in Fig. 5 (Panel 3) included a large number of heterotrophic nifH nodes within the Betaproteobacteria, Gammaproteobacteria and Group 2 Deltaproteobacteria, as well as a number of Bacteroidetes nifH and an unclassified nifH node, representing sequences that were broadly distributed across sites. There were no cyanobacterial nifH nodes in this cluster. NifH OTUs in this cluster were positively correlated with 16S rRNA gene OTUs within the Bacteroidetes and Proteobacteria, primarily Betaproteobacteria. There were no Acidobacteria 16S rRNA gene nodes and few Cyanobacteria 16S rRNA gene nodes in Cluster D. Those cyanobacterial 16S rRNA gene OTUs that were positively correlated with heterotrophic nifH nodes in this Cluster were phylogenetically associated with Leptolyngbya, Pseudoanabaena and Phormidium genera.
Cluster E included heterotrophic nifH OTUs within the Gammaproteobacteria (R21_2604) and Betaproteobacteria (R21_4204), as well as an unclassified nifH (R21_3689) from Spaulding Pond (Site 1) and from the base of Howard Glacier (Site 3). None of the 16S rRNA gene nodes were shared with other clusters. Acidobacteria, Actinobacteria, Chloroflexi and Gemmatimonadetes 16S rRNA OTUs dominated this cluster, while cyanobacterial 16S rRNA gene OTUs were not present.
DISCUSSION
Nitrogen input to MDV sediments is typically low, driven by atmospheric deposition of nitrate or through biological nitrogen fixation of nitrogen gas to ammonia (reviewed by Van Goethem and Cowan 2019). Research efforts have primarily focused on N-fixation by autotrophic cyanobacteria, primarily Nostoc spp., and the significance of cyanobacteria-derived N to the nutrient budget and ecology of microbial communities in MDV ecosystems (Kohler et al. 2018; reviewed by Van Goethem and Cowan 2019). A study by Niederberger et al. (2012), however, demonstrated the sizeable contribution of heterotrophic bacteria to N-fixation in sediment samples collected from ponds and outlet streams in Miers Valley, Antarctica, suggesting that nitrogen input through biological N-fixation to this desert environment may be substantially greater than expected based on estimates from cyanobacteria biomass alone. The current study extends this work to examine the distribution of heterotrophic diazotrophs across several ecosystems (submerged or wetted regions of streams, ponds or lakes, and cyanobacteria- or bryophyte-dominated mats versus no visible mat) of the MDV and to investigate relationships between diazotrophs and the broader microbial community.
Detection of the nifH gene by PCR amplification was restricted to samples with visible cyanobacteria or cryptogamic mats. Previous studies suggested that cyanobacteria are primary colonizers of sediments and soils (reviewed by Rossi and De Philippis 2015), including polar desert sediments that have been newly exposed during deglaciation (Liu et al. 2016; Pessi et al. 2019) or in response to an induced wetting event (Niederberger et al. 2019). As ‘ecosystem engineers’ (Chrismas, Anesio and Sanchez-Baracaldo 2018), cyanobacteria may facilitate recruitment and colonization of heterotrophic bacteria by improving soil structure and providing a labile pool of carbon and nitrogen to oligotrophic environments (Schmidt et al. 2008; Duc et al. 2009). Here, samples collected from ephemeral streams produced during summer months from glacial runoff likely represent early stages of community development. These sites were dominated by cyanobacteria nifH sequences, contributing up to 99.96% of sequences obtained within a site (Table 2).
Cyanobacteria nifH OTUs were exclusively assigned to the Nostocales (Fig. 2B). The absence of nifH sequences from Oscillatoriales, including Lyngbya, Phormidium and Plectonema spp., was surprising given the presence and dominance of this species complex at several sites. Greater than 24% of the 16S rRNA gene OTUs from the surface sample collected at the base of Howard Glacier (sample 3.1), for example, were identified as Phormidium. The absence of nitrogenase from Phormidium was noted in a previous study of nifH sequence diversity and nitrogenase activity in meltwater ponds of the McMurdo Ice Shelf, where Oscillatoriales dominated the cyanobacteria assemblage (Jungblut and Neilan 2009). These results suggest that Phormidium present in samples collected in the MDV is a non-N-fixing species or strain (e.g. Prabaharan, Sumathi and Subramanian 1994; Chamizo et al. 2018), or has accumulated mutations in the nifH gene that impact amplification with the primers used in this and other studies.
Cyanobacteria nifH OTU RM_252 was the only OTU detected at all sites in Taylor Valley that were positive for nifH (Fig. 3). This OTU was closely related to Nostoc sp. cyanobiont of the lichen, Peltigera membranacea and shared 100% identity with OTU43 in Niederberger et al. (2012) as well as with a nifH sequence isolated from the Tibetan Plateau (GenBank Accession No. KR819439.1). Other putative cyanobionts detected in this investigation included nifH OTUs RM_5554 and R22_140654 based on phylogenetic analysis (Fig. 2B). Zuniga et al. (2017) demonstrated that the most abundant Peltigera cyanobionts collected from Antarctic soils were also the most abundant phylotypes in substrate from the same collection site, suggesting that the substrate included a high abundance of lichen propagules or fragments, and/or that free-living Nostoc sp. was selected for lichenization. Both RM_252 and RM_5554 nifH OTUs were broadly distributed in samples collected in the study described here, and dominated the nifH library at some sites (Fig. 3). The broad distribution and high relative abundance of these nifH OTUs suggest that putative cyanobionts may be key constituents of diazotroph assemblages in the MDV while also contributing to the colonization success and diversity of lichens in MDV (Perez-Ortega et al. 2012).
Other nitrogen-fixing cyanobacteria were more limited in their distribution and may have been constrained by environmental filtering. Wang et al. (2017) showed that soil pH was the strongest driver of diazotrophic community structure across desert and non-desert environments in Mongolia, China, while others (e.g. Geyer et al. 2014; Feeser et al. 2018) also observed correlations in pH or conductivity with the relative abundance and diversity of microbial taxa in the MDV, Antarctica. In results presented here, the diazotroph community structure was significantly correlated to pore water pH. OTU RM_4765, for example, was abundant in nifH libraries of samples collected on the north side of Taylor Valley at the base of Commonwealth Glacier (Site 12) and from Green Creek (Site 9), where it made up >50% of the nifH library in the sample 9.1. These sites were characterized by the highest pH and lowest conductivity levels of samples with positive amplification of nifH. Phylogenetic analysis of OTU RM_4765 shows a close relationship to nifH from Anabaena cylindrica, a freshwater cyanobacteria species that is primarily found at neutral to basic pH and has a low tolerance to salinity (Bhadauriya et al. 2007). Likewise, cyanobacteria nifH OTU R23_18734 was detected only at the base of Howard Glacier, where the lowest pore water pH (5.9) and highest PO4 concentrations were measured. While conductivity was not significantly correlated to the diazotroph community structure, cyanobacteria nifH OTU R23_38659, most closely related to the euryhaline genus, Rivularia, was found primarily in Lower Delta Stream (Site 8) and Explorer's Cove (Site 10) with high conductivity levels ranging from 400 to >1600 µS/cm.
Although cyanobacteria nifH dominated at most sites, a larger number of heterotrophic nifH sequences were retrieved during this investigation. Of the 102 nifH OTUs, 71 were phylogenetically related to heterotrophic species. Heterotrophic nifH gene sequences were detected in samples collected from sites with visible mat, suggesting that heterotrophic members of the diazotroph community were tightly coupled with photosynthetic mat-forming species (Smith et al. 2017). Cyanobacteria provide structure and produce extracellular polymeric substances (EPS) that help to stabilize sediments and reduce desiccation, providing a favorable environment for colonization of heterotrophic species (Mazor et al. 1996; Mager and Thomas 2011). Nitrogen fixation is also energetically costly, and heterotrophic species may rely on cyanobacteria as an external C source (Mager and Thomas 2011; Rossi and De Philippis 2015; Smith et al. 2017). Rahav, Giannetto and Bar-Zeev (2016), for example, demonstrated a 10-fold increase in activity and a doubling of heterotrophic diazotrophs after addition of EPS to water sample incubations. In addition to supplying carbon to the heterotrophic community, the anaerobic environment associated with cyanobacteria-produced EPS may provide protection for the nitrogenase complex of heterotrophic species (Rahav, Giannetto and Bar-Zeev 2016).
The abundances of heterotrophic nifH OTUs relative to cyanobacteria were greatest at a permanent water body, Spaulding Pond (Site 1), as well as in the outflow stream, Upper Delta Stream (Site 2), where they contributed 99.7% (Site 1) and 50.8% (Site 2) of nifH sequences at those sites (Table 2). The diazotroph assemblage at Spaulding Pond may be representative of a more established community, where development of a stable anaerobic environment served to structure the heterotrophic community. Several heterotrophic nifH OTUs at this site included sequences closely related to nifH homologs from obligate anaerobes within the Deltaproteobacteria Geobacter sp. (e.g. Duc et al. 2009), several of which share 99% identity with nifH OTUs reported in Niederberger et al. (2012) (Table 3; Fig. 3). Results also provide evidence that the heterotrophic diazotroph community at Upper Delta Stream was seeded by outflow from Spaulding Pond, as many of the shared sequences in wetted and submerged samples of Sites 1 and 2, as well as Site 3 (inlet stream to Spaulding Pond from base of Howard Glacier), but rarely occurred at other sites (Fig. 3).
Submerged sediments at Explorer's Cove (Site 10) also included a diverse assemblage of heterotrophic nifH sequences. This site was in close proximity to McMurdo Sound and was characterized by a thick layer of moss, representing a late successional biocrust stage (Dojani et al. 2011; Maier et al. 2018). The increased complexity of this mat environment may have supported the formation of anoxic microniches that allowed for recruitment over time of anaerobic diazotrophs, similar to Site 1 at Spaulding Pond. Indeed, diazotrophic anaerobes classified as Bacteroidetes together with Deltaproteobacteria Groups I and II made up the largest proportion of heterotrophic diazotrophs at Site 10, while aerobic species within the Beta and Gammaproteobacteria clades were not detected among heterotrophic diazotrophs at this site (Fig. 3). NifH OTU R22_65896, phylogenetically related to nitrogenase in the Purple Non-Sulfur Bacteria group, was detected only at this site. Purple Non-Sulfur Bacteria are facultative or obligate phototrophs that typically occupy anaerobic environments. This OTU was most similar to sequences from Rhodopseudomonas palustris, a common member of marine coastal sediment microbial communities and widely known for its metabolic versatility (reviewed by Larimer et al. 2004). As a member of the Alphaproteobacteria, its phylogenetic position in Fig. 2A supports the hypothesis that the nifH gene in this species was acquired through lateral gene transfer (Cantera, Kawasaki and Seki 2004).
The diversity of heterotrophic diazotrophs in stable water features of the MDV supports results of Niederberger et al. (2012), which noted the large contribution of this physiological group to N-fixation in samples collected from lakes and outlet streams of Miers Valley. Although many of the nifH sequences reported in Niederberger et al. (2012) are shared in this study (Table 3), there was a notable absence of nifH sequences from Bacteroides in Niederberger et al. (2012). Prior investigations (Lee et al. 2012; Kwon et al. 2017) identified a high level of intervalley heterogeneity in bacterial taxa from MDV lakes, and specifically Bacteroides (Lee et al. 2012), suggesting potential endemism within the bacterial community. This is further supported here by the lack of Bacteroides 16S rRNA gene from samples collected at Lake Miers (sample 21.1; Fig. 4B) as well as Victoria Valley (Sites 5 and 6). In contrast, diazotrophs within the Bacteroides genus and other member of the Bacteriodales were broadly distributed in Taylor Valley (Figs 3 and 4) and dominated nifH OTUs from submerged samples at Spaulding Pond (Site 1; Fig. 3). This may have important consequences with respect to nutrient cycling in MDV ecosystems, as Bacteroides are capable of metabolizing cellulosic biomass (Leschine 1995), providing a conduit of accessible carbon and fixed nitrogen to other members of the community.
Analysis of the microbial community structure based on 16S rRNA gene profiles showed that the heterotrophic community was highly correlated to cyanobacterial community structure, providing further evidence that bacterial community structure and diversity in the MDV are controlled in part by the presence and relative abundance of cyanobacteria (Geyer et al. 2017; Lee et al. 2019). Similar to nifH OTU structure, 16S rRNA gene OTUs representing the broader microbial community appeared to be loosely structured in the MDV according to location (Fig. 4), with communities isolated from ponds and lakes forming distinct clusters, and samples collected on the north side of Taylor Valley different from those on the south side. Although the community structure based on 16S rRNA gene was not correlated to environmental variables, there was a significant correlation between 16S rRNA gene and nifH community structure. Likewise, samples that were negative for nifH by PCR were more closely related to each other regardless of location within a transect (dry versus submerged or wetted), suggesting that the presence of diazotrophs may have played a larger role in structuring the bacterial community than the presence of water.
Network analysis revealed additional interspecific relationships between diazotrophs and members of the larger microbial community. Networks involving cyanobacteria members of the diazotroph community in Clusters A, B and C (Fig. 5) showed significant correlations to other members of the community, ranging from a minimum of 2 (for R22_116588) to a maximum of 44 (for R22_53758) first neighbors (median = 9 first neighbors; Table S3, Supporting Information). The ubiquitous putative lichen cyanobiont represented by nifH OTU_252 (Fig. 5; Table S3, Supporting Information) was correlated to only three 16S rRNA gene OTUs in the network, in spite of its broad distribution in the MDV. Network analysis presented in Tu et al. (2016) also resulted in few correlations between the symbiotic diazotroph, Bradyrhizobium, and other members of the microbial community in a grassland ecosystem. This may be due to the physical isolation of symbiotic species, which would not be subject to the same biotic and abiotic environmental conditions that free-living species experience (Tu et al. 2016). While the distribution of heterotrophic diazotrophs was limited to sites with visible mat, results of co-occurrence network analysis presented here also indicated that heterotrophic diazotrophs participate in potential syntrophic partnerships with other members of the heterotrophic community, independent of cyanobacteria diazotroph abundance. The 17 heterotrophic nifH OTUs comprising Cluster D (Fig. 5; Table S3, Supporting Information), for example, were significantly correlated with up to 35 other species dominated by Alphaproteobacteria, Betaproteobacteria and Bacteroidia. Interspecific relationships shared among multiple heterotrophic diazotroph and non-diazotrophic species may expand the functional capacity of this group beyond the contribution of reduced nitrogen, and contribute to functional redundancy of the bacterial community (de Scally et al. 2016). In addition, significant interspecific interactions may also serve to stabilize relationships in late successional communities through eco-evolutionary feedbacks that occur over varying time scales (Patel, Cortez and Schreiber 2018) or in response to environmental change.
The ecological roles of heterotrophic diazotrophs relative to autotrophic diazotrophs in the MDV likely represent a dynamic relationship. As noted above, results indicate that cyanobacteria are primary colonizers of ephemeral streams, while the relative abundance of heterotrophic diazotroph OTUs and their diversity in MDV ecosystems increased at sites with established biomass (such as Site 10) or a more stable environment (such as Site 1). Although seasonal variations in diazotroph communities were not examined here, variations in light and temperature that promote growth and dominance of autotrophic cyanobacteria during the Austral summer may also limit autotrophs during late summer and into fall, when heterotrophic diazotrophs would play a larger role in the ecology of the system. Anticipated climate change conditions resulting in elevated temperatures have been shown to increase overall nifH gene abundance in Antarctic soils (Jung et al. 2011). However, the increased glacial runoff and release of stored carbon (Chen et al. 2015; Smith et al. 2017) over an extended warm season with diminishing light levels may tip the balance between heterotrophic and autotrophic species to favor growth of heterotrophic diazotrophs.
Overall, results of this study provide a better understanding of ecological roles of autotrophic and heterotrophic diazotrophs and factors regulating their distribution in the MDV. As ecosystem engineers, cyanobacteria exert some control over the structure of the microbial community, including heterotrophic diazotrophs, while interspecific relationships with N-fixing heterotrophic species that develop over time likely extend the functional capacity and stability of the community at some sites. Consistent with Niederberger et al. (2012), results of this study indicate that heterotrophic diazotrophs in the MDV are diverse, but largely limited to lakes or ponds and their outlet streams, as well as other stable environments that offer protection from desiccation such as the mossy environment at Explorer's Cove (Site 10). Despite the limited distribution, however, contributions by heterotrophic diazotrophs to the N-budget of MDV ecosystems could be substantial due to the larger surface area and long residence times of lakes compared to ephemeral streams (e.g. Marcarelli and Wurtsbaugh 2009). Results of this investigation will help guide further work evaluating the relative ecological roles of N-fixing heterotrophic versus autotrophic species and their contributions to microbial commotuunity dynamics in the MDV ecosystem as temperature and light regimes change over the course of a season or with anticipated changes in climate.
Supplementary Material
ACKNOWLEDGEMENTS
Nutrient analysis was completed by Edmund Antell and Sarah Blaser at the Wilkerson/Dugdale research laboratory at San Francisco State University. Map (Fig. 1) was created with assistance from Michael Wethington, Polar Geospatial Center. We would like to thank the staff of the United States Antarctic Program and Antarctica New Zealand for exceptional logistical support during each field season. We are also indebted to Antarctica New Zealand and members of the NZTABS team for their assistance during the entire season that made this work possible.
FUNDING
This research was supported by the National Science Foundation (NSF) grants ANT 0739648 and 1246292 (to SCC), ANT 0739633 (to DGC) and ANT 0739640 (to EJC). Geospatial support for this work provided by the Polar Geospatial Center under NSF PLR awards 1043681 and 1559691. The work was also supported by logistics grants from Antarctica New Zealand, from the New Zealand Foundation for Research, Science and Technology (UOWX0710) and the New Zealand Ministry of Business, Innovation and Employment (UOWX1401) to SCC in support of the New Zealand Terrestrial Antarctic Biocomplexity Survey (NZTABS) program.
Conflicts of interest. None declared.
REFERENCES
- Ando S, Goto M, Meunchang S et al.. Detection of nifH sequences in sugarcane (Saccharum officinarum L.) and pineapple (Ananas comosus [L.] Merr.). J Soil Sci Plant Nutr. 2005;51:303–8. [Google Scholar]
- Angel R, Nepel M, Panholzl C et al.. Evaluation of primers targeting the diazotroph functional gene and development of NifMAP—a bioinformatics pipeline for analyzing nifH amplicon data. Front Microbiol. 2018;9:703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JE, Virginia RA, Lyons WB et al.. Biogeochemical stoichiometry of Antarctic Dry Valley ecosystems. J Geophys Res. 2007;112:1–12. [Google Scholar]
- Barrett JE, Virginia RA, Wall DH et al.. Co-variation in soil biodiversity and biogeochemistry in northern and southern Victoria Land, Antarctica. Antarct Sci. 2006;18:535–48. [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Bhadauriya P, Gupta R, Singh S et al.. Physiological and biochemical alterations in a diazotrophic cyanobacterium Anabaena cylindrica under NaCl stress. Curr Microbiol. 2007;55:334–8. [DOI] [PubMed] [Google Scholar]
- Bockheim JG. Properties and classification of cold desert soils from Antarctica. Science. 1997;231:224–31. [Google Scholar]
- Bran and Luebbe, Inc. Bran Luebbe AutoAnalyzer Applications: AutoAnalyzer Method No. G-172-96 Nitrate and Nitrite in Water and Seawater. Buffalo Grove: Bran Luebbe, Inc, 1997. [Google Scholar]
- Brown MB. A method for combining non-independent, one-sided tests of significance. Biometrics. 1975;31:987–92. [Google Scholar]
- Buelow HN, Winter AS, Van Horn DJ et al.. Microbial community responses to increased water and organic matter in the arid soils of the McMurdo Dry Valleys, Antarctica. Front Microbiol. 2016;7:1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantera JJL, Kawasaki H, Seki T. The nitrogen-fixing gene (nifH) of Rhodopseudomonas palustris: a case of lateral gene transfer? Microbiol. 2004;150:2237–46. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary SC, McDonald IR, Barrett JE et al.. On the rocks: the microbiology of Antarctic Dry Valley soils. Nat Rev Microbiol. 2010;8:129–38. [DOI] [PubMed] [Google Scholar]
- Chamizo S, Mugnai G, Rossi F et al.. Cyanobacteria inoculation improves soil stability and fertility on different textured soils: gaining insights for applicability in soil restoration. Front Environ Sci. 2018;6:49. [Google Scholar]
- Chao A, Shen T-J. Nonparametric estimation of Shannon's index of diversity when there are unseen species in sample. Environ Ecol Stat. 2003;10:429–43. [Google Scholar]
- Chen J, Luo Y, Xia J et al.. Stronger warming effects on microbial abundances in colder regions. Sci Rep. 2015;5:18032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrismas NAM, Anesio AM, Sanchez-Baracaldo P. The future of genomics in polar and alpine cyanobacteria. FEMS Microbiol Ecol. 2018;94:fiy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claridge GGC, Campbell IB. The salts in Antarctic soils, their distribution and relationship to soil processes. Soil Sci. 1977;123:377–84. [Google Scholar]
- Clarke KR, Gorley RN. PRIMER v7:User Manual/Tutoria. Plymouth: PRIMER-E, 2015. [Google Scholar]
- Cowan DA, Khan N, Pointing S et al.. Diverse hypolithic refuge communities in Antarctic Dry Valleys. Antarct Sci. 2010;22:714–20. [Google Scholar]
- Cowan DA, Russell NJ, Mamais A et al.. Antarctic Dry Valley mineral soils contain unexpectedly high levels of microbial biomass. Extremophiles. 2002;6:431–6. [DOI] [PubMed] [Google Scholar]
- Cowan DA, Sohm JA, Makhalanyane T et al.. Hypolithic communities: important nitrogen sources in Antarctic desert soils. Environ Microbiol Rep. 2011;3:581–6. [DOI] [PubMed] [Google Scholar]
- de Scally S, Makhalanyane T, Frossard A et al.. Antarctic microbial communities are functionally redundant, adapted and resistant to short-term temperature perturbations. Soil Biol Biochem. 2016;103:160–70. [Google Scholar]
- Dojani S, Budel B, Deutschewitz K et al.. Rapid succession of biological soil crusts after experimental disturbance in the Succulent Karoo, South Africa. Appl Soil Ecol. 2011;48: 263–9. [Google Scholar]
- Doran PT, McKay CP, Clow GD et al.. Valley floor climate observations from the McMurdo Dry Valleys, Antarctica, 1986–2000. J Geophys Res-Atmos. 2002;107:ACL 13–1-13-12. [Google Scholar]
- Duc L, Noll M, Meier B et al.. High diversity of diazotrophs in the forefield of a receding alpine glacier. Microb Ecol. 2009;57:179–90. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. [DOI] [PubMed] [Google Scholar]
- Elbert W, Weber B, Burrows S et al.. Contribution of cryptogamic covers to the global cycles of carbon and nitrogen. Nat Geosci. 2012;5:459–62. [Google Scholar]
- Faust K, Raes J. CoNet app: inference of biological association networks using Cytoscape. F1000Res. 2016;5:1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeser KL, Van Horn DJ, Buelow HN et al.. Local and regional scale heterogeneity drive bacterial community diversity and composition in a polar desert. Front Microbiol. 2018;9:1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer KM, Altrichter AE, Takacs-Vesbach CD et al.. Bacterial community composition of divergent soil habitats in a polar desert. FEMS Microbiol Ecol. 2014;89:490–4. [DOI] [PubMed] [Google Scholar]
- Geyer KM, Takacs-Vesbach CD, Gooseff MN et al.. Primary productivity as a control over soil microbial diversity along environmental gradients in a polar desert ecosystem. PeerJ. 2017;5:e3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gihring TM, Green SJ, Schadt CW. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ Microbiol. 2012;14:285–90. [DOI] [PubMed] [Google Scholar]
- Gooseff MN, Barrett JE, Adams BJ et al.. Decadal ecosystem response to an anomalous melt season in a polar desert in Antarctica. Nat Ecol Evol. 2017;1:1334–8. [DOI] [PubMed] [Google Scholar]
- Gooseff MN, McKnight DM, Runkel RL et al.. Denitrification and hydrologic transient storage in a glacial meltwater stream, McMurdo Dry Valleys, Antarctica. Limnol Oceanogr. 2004;49:1884–95. [Google Scholar]
- Grimm NB, Petrone KC. Nitrogen fixation in a desert stream ecosystem. Biogeochemistry. 1997;37:33–61. [Google Scholar]
- Hoffman BM, Lukoyanov D, Yang Z-Y et al.. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem Rev. 2014;114:4041–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm-Hansen O, Lorenzen CJ, Holmes RW et al.. Fluorometric determination of chlorophyll. J Cons Int Explor Mer. 1965;30:3–5. [Google Scholar]
- Hopkins DW, Sparrow AD, Elberling B et al.. Carbon, nitrogen and temperature controls on microbial activity in soils from an Antarctic Dry Valley. Soil Biol Biochem. 2006a;38:3130–40. [Google Scholar]
- Hopkins DW, Sparrow AD, Novis PM et al.. Controls on the distribution of productivity and organic resources in Antarctic Dry Valley soils. Proc R Soc B. 2006b;273:2687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungblut AD, Neilan BA. NifH gene diversity and expression in a microbial mat community on the McMurdo Ice Shelf, Antarctica. Antarct Sci. 2009;22:117–22. [Google Scholar]
- Jung J, Yeom J, Kim J et al.. Change in gene abundance in the nitrogen biogeochemical cycle with temperature and nitrogen addition in Antarctic soils. Res Microbiol. 2011;162:1018–26. [DOI] [PubMed] [Google Scholar]
- Keys J. Air Temperature, Wind, Precipitation and Atmospheric Humidity in the McMurdo Region, Antarctica. Geology Dept., Victoria Univ, Wellington, NZ: Antarctica Pub, 1980. [Google Scholar]
- Kohler TJ, Stanish LF, Liptzin D et al.. Catch and release: hyporheic retention and mineralization of N-fixing Nostoc sustains downstream microbial mat biomass in two polar desert streams. Limnol Oceanog Lett. 2018;3:357–64. [Google Scholar]
- Kumar S, Stecher Gand Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol. 2016;33:1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuske CR. Environmental survey for four pathogenic bacteria and closely related species using phylogenetic and functional genes. J Forensic Sci. 2006;51:548–58. [DOI] [PubMed] [Google Scholar]
- Kwon M, Kim M, Takacs-Vesbach C et al.. Niche specialization of bacteria in permanently ice-covered lakes of the McMurdo Dry Valleys, Antarctica. Environ Microbiol. 2017;19:2258–71. [DOI] [PubMed] [Google Scholar]
- Lacap-Bugler DC, Lee KK, Archer S et al.. Global diversity of desert hypolithic cyanobacteria. Front Microbiol. 2017;8:867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer FW, Chain P, Hauser L et al.. Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat Biotechnol. 2004;22:55–61. [DOI] [PubMed] [Google Scholar]
- Lee CK, Barbier BA, Bottos EM et al.. The inter-valley soil comparative survey: the ecology of Dry Valley edaphic microbial communities. ISME J. 2012;6:1046–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Laughlin DC, Bottos EM et al.. Biotic interactions are an unexpected yet critical control on the complexity of an abiotically driven polar ecosystem. Commun Biol. 2019;2:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leschine SB. Cellulose degradation in anaerobic environments. Ann Rev Microbiol. 1995;49:399–426. [DOI] [PubMed] [Google Scholar]
- Liu JB, Kong WD, Zhang GS et al.. Diversity and succession of autotrophic microbial community in high-elevation soils along deglaciation chronosequence. FEMS Microbiol Ecol. 2016;92:fiw160. [DOI] [PubMed] [Google Scholar]
- Mager DM, Thomas AD. Extracellular polysaccharides from cyanobacterial soil crusts: a review of their role in dryland soil processes. J Arid Environ. 2011;75:91–7. [Google Scholar]
- Maier S, Tamm A, Wu DM et al.. Photoautotrophic organisms control microbial abundance, diversity, and physiology in different types of biological soil crusts. ISME J. 2018;12:1032–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhalanyane TP, Van Goethem MW, Cowan DA. Microbial diversity and functional capacity in polar soils. Curr Opin Biotech. 2016;38:159–66. [DOI] [PubMed] [Google Scholar]
- Marcarelli AM, Wurtsbaugh WA. Nitrogen fixation varies spatially and seasonally in linked stream–lake ecosystems. Biogeochem. 2009;94:95–110. [Google Scholar]
- Mazor G, Kidron GJ, Vonshak A et al.. The role of cyanobacterial exopolysaccharides in structuring desert microbial crusts. FEMS Microbiol Ecol. 1996;21:121–30. [Google Scholar]
- McDonald D, Price MN, Goodrich J et al.. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight DM, Runkel RL, Tate CM et al.. Inorganic N and P dynamics of Antarctic glacial meltwater streams as controlled by hyporheic exchange and benthic autotrophic communities. J N Am Benthol Soc. 2004;23:171–88. [Google Scholar]
- Niederberger TD, Bottos EM, Sohm JA et al.. Rapid microbial dynamics in response to an induced wetting event in Antarctic Dry Valley soils. Front Microbiol. 2019;10:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederberger TD, Sohm JA, Gunderson TE et al.. Microbial community composition of transiently wetted Antarctic Dry Valley soils. Front Microbiol. 2015b;6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederberger TD, Sohm JA, Gunderson T et al.. Carbon-fixation rates and associated microbial communities residing in arid and ephemerally wet Antarctic Dry Valley soils. Front Microbiol. 2015a;6:1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederberger TD, Sohm JA, Tirindelli J et al.. Diverse and highly active diazotrophic assemblages inhabit ephermally wetted soils of the Antarctic Dry Valleys. FEMS Microbiol Ecol. 2012;82:376–90. [DOI] [PubMed] [Google Scholar]
- Pandey KD, Shukla SP, Shukla PN et al.. Cyanobacteria in Antarctica: ecology, physiology and cold adaptation. Cell Mol Biol. 2004;50: 575–84. [PubMed] [Google Scholar]
- Patel S, Cortez MH, Schreiber SJ. Partitioning the effects of eco-evolutionary feedbacks on community stability. Am Nat. 2018;191:381–94. [Google Scholar]
- Perez-Ortega S, Ortiz-Alvarez R, Allan Green TG et al.. Lichen myco- and photobiont diversity and their relationships at the edge of life (McMurdo Dry Valleys, Antarctica). FEMS Microbiol Ecol. 2012;82:429–48. [DOI] [PubMed] [Google Scholar]
- Pessi IS, Pushkareva E, Lara Y et al.. Marked succession of cyanobacterial communities following glacier retreat in the high Arctic. Microb Ecol. 2019;77:136–47. [DOI] [PubMed] [Google Scholar]
- Prabaharan D, Sumathi M, Subramanian G. Ability to use ampicillin as a nitrogen-source by the marine cyanobacterium Phormidium valderianum BDU-30501. Curr Microbiol. 1994;28:315–20. [Google Scholar]
- Rahav E, Giannetto MJ, Bar-Zeev E. Contribution of mono and polysaccharides to heterotrophic N2 fixation at the eastern Mediterranean coastline. Sci Rep. 2016;6:27858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond J, Siefert JL, Staples CR et al.. The natural history of nitrogen fixation. Mol Biol Evol. 2004;21:541–54. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Zaraqoza S, Gonzalez-Ruiz T, Gonzalez-Lozano E et al.. Vertical distribution of microbial communities under the canopy of two legume bushes in the Tehuacán Desert, Mexico. Eur J Soil Biol. 2008;44:373–80. [Google Scholar]
- Rosch C, Bothe H. Improved assessment of denitrifying, N2-fixing, and total-community bacteria by terminal restriction fragment length polymorphism analysis using multiple restriction enzymes. Appl Environ Microb. 2005;71:2026–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F, De Philippis R. Role of cyanobacterial exopolysaccharides in phototrophic biofilms and in complex microbial mats. Life-Basel. 2015;5:1218–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. [DOI] [PubMed] [Google Scholar]
- Santos PC, Dean DR. FeS cluster assembly: NIF system in nitrogen-fixing bacteria. In: Scott RA.(ed). Encyclopedia of Inorganic and Bioinorganic Chemistry. Hoboken: John Wiley and Sons, Ltd, 2017, 1–13. [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T et al.. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb. 2009;75:7537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SK, Reed SC, Nemergut DR et al.. The earliest stages of ecosystem succession in high-elevation (5000 metres above sea level), recently deglaciated soils. Proc Biol Sci. 2008;275:2793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O et al.. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp JH, Carlson CA, Peltzer ET et al.. Final dissolved organic carbon broad community intercalibration and preliminary use of DOC reference materials. Mar chem. 2002;77:239–53. [Google Scholar]
- Short SM, Zehr JP. Quantitative analysis of nifH genes and transcripts from aquatic environments. Methods Enzymol. 2005;397:380–94. [DOI] [PubMed] [Google Scholar]
- Smith HJ, Foster RA, McKnight DM et al.. Microbial formation of labile organic carbon in Antarctic glacial environments. Nat Geosci. 2017;10:356–9. [Google Scholar]
- Sohm JA, Webb EA, Capone DG. Emerging patterns of marine nitrogen fixation. Nat Rev Microbiol. 2011;9:499–508. [DOI] [PubMed] [Google Scholar]
- Solorzano L. Determination of ammonia in natural waters by the phenolhypochlorite method. Limnol Oceanogr. 1969;14:99–801. [Google Scholar]
- Stanish LF, O'Neill SP, Gonzalez A et al.. Bacteria and diatom co-occurrence patterns in microbial mats from polar desert streams. Environ Microbiol. 2013;15:1115–31. [DOI] [PubMed] [Google Scholar]
- Steppe TF, Paerl HW. Nitrogenase activity and nifH expression in a marine intertidal microbial mat. Microb Ecol. 2005;49:315–24. [DOI] [PubMed] [Google Scholar]
- Sudhir K, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Q, Zhou X, He Z et al.. The diversity and co-occurrence patterns of N2-fixing communities in a CO2-enriched grassland ecosystem. Microb Ecol. 2016;71:604–15. [DOI] [PubMed] [Google Scholar]
- Van Goethem MW, Cowan DA. Role of cyanobacteria in the ecology of polar environments. In: Castro-Sowinski S.(ed). The Ecological Role of Micro-organisms in the Antarctic Environment. New York: Springer International Publishing, 2019, 3–23. [Google Scholar]
- Van Horn DJ, Wolf CR, Colman DR et al.. Patterns of bacterial biodiversity in the glacial meltwater streams of the McMurdo Dry Valleys, Antarctica. FEMS Microbiol Ecol. 2016;92:fiw148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent WF, Downes MT, Castenholz RW et al.. Community structure and pigment organisation of cyanobacteria-dominated microbial mats in Antartictca. Eur J Phycol. 1993;28:213–21. [Google Scholar]
- Vincent WF. Cyanobacterial dominance in the polar regions. In: Whitton BA, Potts M.(eds). The Ecology of Cyanobacteria. Dordrecht: Springer, 2000, 321–40. [Google Scholar]
- Vitousek PM, Menge DNL, Reed SC et al.. Biological nitrogen fixation: rates, patterns and ecological controls in terrestrial ecosystems. Philos T R Soc B. 2013;368:20130119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li H, Li J et al.. The diversity and co-occurrence patterns of diazotrophs in the steppes of Inner Mongolia. Catena. 2017;157:130–8. [Google Scholar]
- Whitledge TE, Malloy SC, Patton CJ et al.. Automated nutrient analyses in seawater. Brookhaven Natinoal Laboratory Technical Report BNL 51398. 1981, doi: 10.2172/5433901. [DOI] [Google Scholar]
- Yergeau E, Newsham KK, Pearce DA et al.. Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ Microbiol. 2007;9:2670–82. [DOI] [PubMed] [Google Scholar]
- Zani S, Mellon MT, Collier JL et al.. Expression of nifH genes in natural microbial assemblages in Lake George, New York, detected by reverse transcriptase PCR. Appl Environ Microb. 2000;66:3119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehr JP, Jenkins BD, Short SM et al.. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ Microbiol. 2003;5:539–54. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yang Q, Ling J et al.. Diversity and structure of diazotrophic communities in mangrove rhizosphere, revealed by high-throughput sequencing. Front Microbiol. 2017;8: 2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga C, Leiva D, Caru M et al.. Substrates of Peltigera lichens as a potential source of cyanobionts. Microb Ecol. 2017;74:561–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.