

Abstract
The N-methyl-D-aspartate receptor (NMDAR), a ligand-gated ion channel activated by L-glutamate and glycine, plays a major role in the synaptic plasticity underlying learning and memory. NMDARs are involved in neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease and NMDAR hypofunction is implicated in schizophrenia. Herein we describe structure-activity relationship (SAR) studies on 2-naphthoic acid derivatives to investigate structural requirements for positive and negative allosteric modulation of NMDARs. These studies identified compounds such as UBP684 (14b), which act as pan potentiators by enhancing NMDAR currents in diheteromeric NMDAR tetramers containing GluN1 and GluN2A-D subunits. 14b and derivatives thereof are useful tools to study synaptic function and have potential as leads for the development of drugs to treat schizophrenia and disorders that lead to a loss of cognitive function. In addition, SAR studies have identified a series of styryl substituted compounds with partial NAM activity and a preference for inhibition of GluN2D versus the other GluN2 subunits. In particular, the 3-and 2-nitrostyryl derivatives UBP783 (79i) and UBP792 (79h) had IC50s of 1.4 μM and 2.9 μM, respectively, for inhibition of GluN2D but showed only 70–80% maximal inhibition. GluN2D has been shown to play a role in excessive pain transmission due to nerve injury and potentially in neurodegenerative disorders. Partial GluN2D inhibitors may be leads for the development of drugs to treat these disorders without the adverse effects observed with full NMDAR antagonists.
Keywords: N-Methyl-D-aspartate receptor, NMDA, GluN2, positive allosteric modulator, negative allosteric modulator, 2-naphthoic acid
Introduction
N-Methyl-D-aspartic acid receptors (NMDARs) are members of the ionotropic glutamate receptor family, which are L-glutamate-gated ion channels that mediate fast excitatory synaptic transmission in the central nervous system (CNS). NMDARs are tetramers assembled from two GluN1 subunits, each containing a binding site for the co-agonist glycine and two GluN2A-D subunits, containing a binding site for L-glutamate in the ligand binding domain (LBD) region on each subunit. In some areas of the CNS, GluN3A and GluN3B subunits are incorporated into the tetramer [1–3]. An important function of NMDARs is to initiate the synaptic plasticity that occurs during CNS development and in learning and memory [4].
NMDARs have been the subject of intense investigations into the development of drugs that can modulate their activity. This is due to the involvement of these receptors in neurological disorders such as epilepsy, chronic pain, depression and schizophrenia and neurodegenerative disorders such as ischemia, Alzheimer’s and Parkinson’s diseases [5]. Very few compounds have made it into the clinic, with low affinity channel blockers such as memantine, which is used to treat moderate to severe Alzheimer’s disease, being the exception. Antagonists interacting with the glutamate binding site on GluN2 subunits, the glycine binding site on GluN1 subunits, the ion channel with high affinity and the N-terminal domain of the GluN2B subunit have so far failed in clinical trials for stroke, head injury and epilepsy [1,5,6]. This is largely due to lack of efficacy and/or intolerable side effects such as amnesia, ataxia and psychotomimetic effects. More recently, attention has focused on the development of allosteric modulators, which may be able to treat neurological disorders without interfering with the normal physiological functions of NMDARs, thereby reducing side effects [6]. Furthermore, most efforts in NMDAR drug development have focused on inhibitors of activity. However, for treating schizophrenia, which is associated with NMDAR hypofunction, NMDAR positive allosteric modulators (PAMs) may have therapeutic benefits over currently available antipsychotics [6]. NMDAR PAMs may also have a role in treating the cognitive deficits observed in patients with other disorders such as Alzheimer’s and Parkinson’s disease [6].
We have reported a series of phenanthrene derivatives that can act as negative allosteric modulators (NAMs) and/or PAMs by binding to a site(s) distinct from those known for previous NMDAR ligands [7]. For example, UBP512 (1) (Figure 1) potentiated NMDARs containing GluN2A, had no effect on GluN2B and inhibited GluN2C and GluN2D in an electrophysiological assay. Others have reported compounds that potentiate GluN2C and GluN2D (2, CIQ) [8] or specifically potentiate GluN2C (3) [9] (Figure 1). Recently, a GluN2A selective PAM (6) has been reported, which was developed from the lead compounds GNE-6901 (4) and GNE-8324 (5) (Figure 1) [10]. X-ray crystallography has revealed that these compounds bind at the dimer interface of the ligand binding domains (LBDs) of GluN1 and GluN2A [10]. Endogenous neurosteroids such as pregnenolone sulfate and synthetic derivatives thereof also potentiate NMDAR responses [11,12].
Figure 1.

Structures of known NMDAR positive and negative allosteric modulators.
Removal of the terminal aromatic ring in 1 and modification of the resultant 2-naphthoic acid core afforded a series of derivatives, such as UBP617 (7) (Figure 1), which non-selectively inhibited receptors containing different GluN2 subunits [13]. Interestingly, 7 and several structurally related analogues showed submaximal inhibition of NMDARs making them potential leads for neurological conditions associated with overactivation of the receptor, such as neuropathic pain, where a complete block of activity could have adverse effects due to inhibition of essential brain functions. Others have reported compounds that selectively antagonise GluN2C and GluN2D (QNZ46, 8, Figure 1) [14] and a series of compounds that act as partial negative allosteric modulators (NAMs) [15]. NAMs that specifically inhibit GluN2A (TCN-201, MPX-004, 9, Figure 1) have been reported [16,17]. Inhibition of GluN1/GluN2A by 9 has been shown to be dependent on the glycine concentration and X-ray crystallography has revealed that it binds to the dimer interface of the ligand binding domains of GluN1 and GluN2A [17].
Herein we describe structure-activity relationship (SAR) studies on 2-naphthoic acid derivatives as analogues of our previously reported phenanthrene [7] and naphthalene series [7a,13] with the aim of understanding the structural requirements for NMDAR NAMs and PAMs.
Results
Chemistry
The synthesis of the naphthalene derivatives described in this report is outlined in Schemes 1–10. Heck coupling between methyl 6-bromo-2-naphthoate (10) and the appropriate alkene afforded 11a-b (Scheme 1). To aid SAR studies, a small amount of the trans-alkene (11a-b) was hydrolysed to its corresponding acid (12a-b) using base. However, the majority was taken forward and hydrogenated before being hydrolysed with base to yield saturated acids 14a-b. The 6-cyclopropyl derivative (17) was synthesized from 10 in three steps (Scheme 1). Firstly, Stille coupling between 10 and tri-n-butyl(vinyl)tin afforded 15. The cyclopropyl ring was then formed via a Simmons-Smith reaction and the resultant ester (16) hydrolysed to the desired acid (17) using base.
Scheme 1a.

aReagents and conditions: (a) Alkene, P(o-tolyl)3, TEA, Pd(OAc)2, DMF, 100 °C, 18 h; (b) H2, 10% Pd/C, rt, 18 h; (c) (i) LiOH or NaOH (aq), THF, 65 °C or dioxane, 80 °C, (ii) 1 M HCI (aq); (d) (n-Bu)3SnCH=CH2, Pd(PPh3)4, toluene, reflux, 4 h; (e) CH2I2, Et2Zn, DCM, 0 °C, 18 h; (f) (i) NaOH (aq), THF/H20, rt, 18 h, (ii) 1 M HCI (aq).
In order to gather SAR information, several analogues of 14a were synthesized (Scheme 2). Reduction of the 3-carboxy moiety using LiAlH4 proceeded smoothly, giving the 3-hydroxymethyl derivative 18 in good yield. Reaction of 18 with PBr3 afforded 3-bromomethyl derivative 19 which was in turn converted to nitrile 20 by reaction with NaCN under phase transfer conditions. Acidic hydrolysis of the nitrile yielded the corresponding 3-acetic acid derivative 21. The 5-bromo analogue 22 was readily synthesized by reacting 14a with a solution of bromine in glacial acetic acid (Scheme 2). Alkylation of 6-hydroxy-2-naphthoic acid (23) with 1-bromobutane afforded the 6-butoxy derivative 24 in good yield (Scheme 2).
Scheme 2a.

aReagents and conditions: (a) LiAIH4, THF, 0 °C then 65 °C, 4 h; (b) PBr3, 0 °C then rt, 1 h; (c) NaCN, TBAB, H20/DCM (1:1), rt, 48 h; (d) H2S04, Ac0H/H20 (1:1), 118 °C, 18 h; (e) Br2, AcOH, 50 °C; (f) (i) 1-Bromobutane, NaOH, Et0H/H20 (3:1), 78 °C, 18 h, (ii) 10% NaOH, 78 °C, 2 h, (iii) 2 M HCI (aq).
Methyl 6-bromo-2-naphthoate (10) was utilised as a starting material in the synthesis of various analogues of 14b (Schemes 3 and 4). Heck coupling between 10 and the appropriate alkene led to the synthesis of trans alkene intermediates 25a-b and 30 (Scheme 3). To aid SAR studies, a sample of 30 was hydrolysed using base to yield unsaturated acid 31. Hydrogenation of 25a-b and 30 gave the corresponding alkyl esters which were subjected to base hydrolysis to give acids 27a-b and 33 (Scheme 3). Sonogashira coupling between 10 and 4-methylpent-1-yne afforded ester 28 which was readily hydrolysed to acid 29 (Scheme 3). Using transesterification under acidic conditions, 10 was converted to isobutyl ester 34 in reasonable yield. Palladium catalysed carboxylation using carbon monoxide subsequently afforded acid 35 (Scheme 4). A Claisen condensation between 10 and ethyl 4-methylvalerate followed by intramolecular decarboxylation yielded ketone 36. Palladium catalysed carboxylation using carbon monoxide gave ester 37 which was hydrolysed using base to afford acid 38 (Scheme 4). Sonogashira coupling between 10 and 4-pentyn-2-ol led to the synthesis of alcohol 39. Hydrogenation of the alkyne bond followed by oxidation of the alcohol using Dess-Martin periodinane (DMP) afforded ketone 40 in good yield (Scheme 4). The introduction of a carboxymethyl group was achieved by reacting ketone 40 with methyl diethylphosphonoacetate under Wittig reaction conditions. Hydrogenation of the resultant alkene gave ester (41) which was readily hydrolysed to di-acid 42 using base (Scheme 4).
Scheme 3a.

aReagents and conditions: (a) Alkene, P(o-tolyl)3, TEA, Pd(OAc)2, DMF, 100 °C, 18 h; (b) H2,10% Pd/C, rt, 18 h; (c) (i) NaOH, THF/H20, 65 °C, (ii) 1 M HCI (aq); (d) 4-methylpent-1-yne, NHEt2, Cui, Pd(Ph3)2CI2, THF, 50 °C, 18 h; (e) (i) LiOH, dioxane/HzO, rt, 18 h, (ii) 1 M HCI (aq); (f) (i) NaOH, dioxane/HzO, rt, 18 h, (ii) 1 M HCI (aq).
Scheme 4a.

aReagents and conditions: (a) Isobutanol, H2S04, reflux, 18 h; (b) CO, DPPP, NEt3, Pd(OAc)2, H2O/DMF, 85 °C, 24 h; (c) (i) ethyl 4-methylvalerate, LiHMDS, THF, −78 °C then rt 3 h, (ii) 1 M NaOH (aq), 50 °C, 18 h, (iii) conc HCI (aq), 60 °C, 1 h; (d) CO, DPPP, NEt3, Pd(OAc)2, MeOH/DMF, 90 °C, 18 h; (e) (i) LiOH, dioxane/H20, rt, 18 h, (ii) 1 M HCI (aq); (f) 4-pentyn-2-ol, NEt3, CuBr, Pd(PPh3)4, 65 °C, 18 h; (g) H2, 10% Pd/C, EtOH, rt, 1 h; (h) DMP, DCM, rt, 2 h; (i) methyl diethylphosphonoacetate, KHMDS, THF, 0 °C 1 h then rt 4 h; Q) H2, 10% Pd/C, EtOH, rt, 1 h; (k) CH3PPh3Br, KHMDS, THF, −78 °C 1 h then rt 2 h.
To gather additional SAR information, ketone 37 was subjected to further chemical modification (Scheme 4). Using Wittig reaction chemistry, methylenation of ketone 37 afforded alkene 43 in good yield. Although the majority of alkene 43 was taken forward and hydrogenated to give ester 45, some was held back and hydrolysed using base to give unsaturated acid 44. Ester 45 was hydrolysed to acid 46 in good yield under basic conditions (Scheme 4).
Thioether, amide and ester analogues of 14b were also synthesised during the course of SAR studies (Scheme 5). The reaction between 6-bromomethyl derivative 47 and 2-methylpropan-1-thiol in the presence of sodium yielded thioether 48 which was readily converted to corresponding acid 49 by base hydrolysis (Scheme 5). Coupling of acid chloride 50 with isobutylamine under standard conditions afforded amide 51 which was hydrolysed to acid 52 using base (Scheme 5). Using identical conditions, 6-hydroxymethyl 53 and isobutyryl chloride were coupled to yield ester 54 (Scheme 5).
Scheme 5a.

aReagents and conditions: (a) 2-Methyipropan-1-thiol, Na, EtOH; (b) (i) LiOH, dioxane/H2O, rt, 18 h, (ii) 1 M HCI (aq); (c) Isobutylamine, NEt3, DCM, 0 °C then rt, 18 h; (d) Isobutyryl chloride, NEt3, DCM, 0 °C then rt, 1 h, (ii) 2 M NaHC03 (aq), 0 °C, 3 h, (iii) 1 M HCI (aq).
Further modifications to the 6-isohexyl side chain in 14b were investigated using 6-bromomethyl derivative 55 as a starting material (Scheme 6). The reaction between 55 and 2-methylpropan-1-ol in the presence of sodium yielded ether 56. Palladium catalysed carboxylation using carbon monoxide subsequently afforded acid 57 (Scheme 6). Base promoted nucleophilic substitution between 55 and ethyl 4-methylvalerate generated ester 58. Palladium catalysed carboxylation using carbon monoxide yielded di-ester 59. Base hydrolysis subsequently afforded di-acid 60 (Scheme 6).
Scheme 6a.

aReagents and conditions: (a) 2-Methylpropan-1-ol, Na, DMF, rt, 7 h; (b) CO, DPPP, NEt3, Pd(OAc)2, H2O/DMF, 85 °C, 24 h; (c) ethyl 4-methylvalerate, KHMDS, THF, −78 °C 0.5 h then rt, 18 h; (d) CO, DPPP, NEt3, Pd(OAc)2, MeOH/DMF, 80 °C, 18 h; (e) (i) LiOH, dioxane/H20, rt, 18 h, (ii) 1 M HCI (aq).
3-Hydroxy-2-naphthoic acid derivatives of 14b were also the subject of investigation (Schemes 7). The starting material required for these compounds (63) was synthesised in two steps from 7-bromo-3-hydroxy-2-naphthoic acid 69. Firstly, the 2-carboxy group was esterified using K2CO3 and methyl iodide to afford methyl ester 62. The 3-hydroxy group was in turn acetylated using acetic anhydride to afford 63 (Scheme 7). Heck coupling between 63 and 4-methylpent-1-ene and hydrogenation of the resulting alkene yielded 64. Base hydrolysis was used to deprotect a sample of this compound giving 65 (Scheme 7). The majority of 64 was deprotected at the 3-position using dimethylamine to afford phenol 66 which was in turn reacted with triflic anhydride under classical conditions to give triflate 67. Removal of the triflate group using formic acid and Pd(PPh3)4 yielded ester 68 which was subsequently hydrolysed in the presence of base to acid 69 (Scheme 7).
Scheme 7a.

aReagents and conditions: (a) Mel, K2C03, DMF, rt, 12 h; (b) Ac20, TEA, DMAP, DCM, rt, 12 h; (c) 4-methylpent-1 -ene, P(o-tolyl)3, Pd(OAc)2, NEt3, DMF, 100 °C, 18 h; (d) H2, 10% Pd/C, EtOH, rt, 1 h; (e) (i) LiOH, dioxane/H2O, rt, 18 h, (ii) 1 M HCI (aq); (f) (CH3)2NH, MeOH, 0 °C then rt 1 h; (g) Tf20, NEt3, DCM, −10 °C, 2 h; (h) HCOOH, NEt3, Pd(PPh3)4, DMF, 80 °C, 18 h.
Swapping the respective positions of the carboxy and hydroxy groups in compound 65 was desirable for investigating the SAR surrounding this compound (Scheme 8). Sonogashira coupling between triflate 70 and 4-methylpent-1-yne led to the synthesis of alkyne 71. Hydrogenation of the triple bond followed by t-BuLi promoted carboxylation afforded ester 72. Acidic deprotection of the 3-hydroxy group yielded 73 which was readily hydrolysed using base to give acid 74 (Scheme 8). Replacing the hydroxy group in 65 with a carboxy moiety was also of interest for SAR studies (Scheme 8). Heck coupling between commercially available 75 and 4-methylpent-1-ene afforded alkene 76. Subsequent hydrolysis of the nitrile groups under acidic conditions and hydrogenation of the alkene bond afforded di-acid 77 (Scheme 8).
Scheme 8a.

aReagents and conditions: (a) 4-Methylpent-1-yne, NHEt2, Cul, Pd(PPh3)2Cl2, 45 °C, 18 h; (b) H2, 10% Pd/C, rt, 18 h; (c) (i) f-BuLi, 0 °C, 1h, (ii) methyl chloroformate, −78 °C then rt 1 h; (d) 2 M HCI (aq), MeOH; (e) (i) LiOH, dioxane/H2O, rt, 18 h, (ii) 1 M HCI (aq); (0 4-Methylpent-1-ene, P(o-tolyl)3, Pd(OAc)2, NEt3, DMF, 100 °C, 18 h; (g) H2S04, AcOH/H2Q (3:1), 118 °C, 24 h; (h) H2, 10% Pd/C, EtOAc, rt, 18 h.
Heck coupling between 63 and the appropriately substituted styrene afforded a series of 7-substituted styryl derivatives (78a-j) (Scheme 9). The majority of these compounds were taken forward and de-protected under basic conditions to give 79a-j. However, to gather additional SAR information, 78a, 78b and 78d were hydrogenated to their saturated counterparts (80a-c) and then hydrolysed using base to afford phenethyl derivatives 81a-c (Scheme 9). In order to investigate it’s importance, the 3-hydroxy moiety of 79a was subjected to modification. De-acetylation of 78a using dimethylamine afforded phenol 82 which was in turn reacted with triflic anhydride under classical conditions to give triflate 83 (Scheme 9). Palladium catalysed carboxylation using carbon monoxide gave di-ester 84 which was hydrolysed using base to afford di-acid 85 in moderate yield (Scheme 9).
Scheme 9a.

aReagents and conditions: (a) Styrene, P(o-tolyl)3, NEt3, Pd(OAc)2, DMF, 100 °C, 18 h; (b) (i) NaOH (aq), THF/H20, reflux or dioxane/H20, rt, (ii) 2 M HCI (aq); (c) H2, 10% Pd/C, THF or dioxane, rt, 18 h; (d) NHMe2, 0 °C, 2 h; (e) Tf20, NEt3, DCM, −10 °C, 2 h; (0 CO, DPPP, NEt3, Pd(OAc)2, MeOH/DMF, 80 °C, 24 h; (g) (i) LiOH, dioxane/H20, rt, 18 h, (ii) 1 M HCI (aq).
The coumarin UBP608 has previously been reported as a moderately potent NAM that fully inhibited GluN2A responses with an IC50 of 18.6 ± 1.4 μM and 23-fold selectivity over GluN2D [7a]. It was thought, given styryl group addition to the naphthalene series (see 78a-j in Table 5) gave a series of interesting NMDAR NAMs, that the same strategy could be applied to create the coumarin analogue of 78a. It was hoped that by using commercially available ethyl 6-bromo-3-coumarincarboxylate (86), a range of analogues could be accessed in a similar way to the naphthalene series, through palladium catalysed cross-coupling with a variety of substituted benzene derivatives. In our hands, however, negligible yields of the desired styryl-coupled coumarin (88) were achieved under standard Heck and Suzuki conditions (Scheme 10). Instead of attempting to functionalise an existing coumarin, the next strategy was to synthesise a coumarin with the desired functional groups in place, in this case, a styryl group. A Knoevenagel condensation of Meldrum’s acid [18] with known intermediate (E)-2-hydroxy-5-styrylbenzaldehyde (87) (synthesised using a literature procedure [19]) gave the desired coumarin 88 in a moderate yield (Scheme 10).
Table 5.
SAR studies on naphthalene derivatives as NMDAR NAMs. Values are percentage response for agonist (10 μM L-glutamate/10 μM glycine) in presence of test compound (100 μM) compared to agonist alone.
 |
||||||
|---|---|---|---|---|---|---|
| NMDAR (n≥4)a | ||||||
| Compound | Formula | R | GluN2A | GluN2B | GluN2C | GluN2D |
| 7 | - | - | 28.4 ± 2.6 | 49.9 ± 9.0 | 26.1 ± 1.7 | 26.1 ± 4.6 |
| 79a | A | H | 32.7 ± 3.8 | 7.3 ± 1.6 | 21.7 ± 0.7 | 11.3 ± 3.3 |
| 85 | C | - | 55.0 ± 12.6 | 66.7 ± 14.2 | 37.1 ± 6.8 | 38.6 ± 6.1 |
| 88 | D | - | 95.1 ± 12.4 | 92.0 ± 5.6 | 114.3 ± 8.6 | 118.9 ± 7.9 |
| 81a | B | H | 71.4 ± 14.3 | 54.3 ± 9.0 | 46.8 ± 5.7 | 32.1 ± 2.9 |
| 79b | A | o-CO2H | 92.1 ± 5.9 | 84.5 ± 2.8 | 83.7 ± 3.1 | 102.6 ± 5.0 |
| 81b | B | o-CO2H | 91.8 ± 15.7 | 85.2 ± 2.4 | 94.1 ± 1.5 | 80.7 ± 1.7 |
| 79c | A | m-CO2H | 90.3 ± 4.3 | 70.0 ± 5.7 | 76.8 ± 3.7 | 61.4 ± 2.1 |
| 79d | A | p-CO2H | 89.7 ± 3.2 | 89.0 ± 6.0 | 85.2 ± 2.7 | 67.0 ± 11.0 |
| 81c | B | p-CO2H | 101.9 ± 4.3 | 95.8 ± 7.6 | 114.6 ± 6.1 | 92.6 ± 2.2 |
| 79e | A | o-OMe | 105.0 ± 12.3 | 74.0 ± 18.3 | 61.7 ± 15.6 | 71.0 ± 9.3 |
| 79f | A | m-OMe | 52.4 ± 10.2 | 24.9 ± 3.7 | 30.8 ± 4.9 | 29.9 ± 5.1 |
| 79g | A | p-OMe | 42.7 ± 20.6 | 36.0 ± 6.6 | 39.6 ± 7.2 | 39.0 ± 10.0 |
| 79h | A | o-NO2 | 80.5 ± 4.7 | 56.1 ± 7.8 | 35.1 ± 3.2 | 26.8 ± 3.4 |
| 79i | A | m-NO2 | 50.5 ± 11.0 | 28.8 ± 5.5 | 24.4 ± 5.3 | 30.7 ± 7.0 |
| 79j | A | p-NO2 | 47.4 ± 5.7 | 53.2 ± 6.7 | 43.3 ± 5.1 | 34.4 ± 2.0 |
Compounds were tested by TEVC for activity at recombinant NMDA receptors (GluN1a and the indicated GluN2 subunit) expressed in Xenopus oocytes. After obtaining a steady state NMDAR response evoked by 10 μM L-glutamate and 10 μM glycine, test compounds (100 μM) were co-applied with agonists. Values (mean ± s.e.m.) represent the % response in the presence of the test compound compared to response in the presence of agonists alone. Values >100 represent potentiation and those < 100 represent inhibition of the agonist response.
Scheme 10a.

aReagents and conditions: (a) Meldrum’s acid, piperidinium acetate, EtOH, rt 20 min, then reflux 2 h.
Biological evaluation and SAR studies
Compounds were initially evaluated at a concentration of 100 μM for their effects on agonist-induced NMDAR currents using a two-electrode voltage clamp (TEVC) electrophysiological assay [7]. Although the test concentration is high even for screening it allowed us to detect any weak NMDAR PAM effects. Compounds with significant activity were investigated more thoroughly using full concentration response curves. cRNA coding for the four GluN1/GluN2A-D diheteromeric NMDARs were individually injected into Xenopus laevis oocytes. After 2 to 5 days, NMDAR currents were induced by L-glutamate (Glu) (10 μM) and glycine (Gly) (10 μM) and after a steady-state response was obtained, the test compounds were co-applied with agonist. Data from these studies are shown in Tables 1–3 and 5. Full concentration-response curves (Figures 2 and 3) and EC50 or IC50 values across GluN2A-D were then generated for compounds with significant NMDAR potentiating or inhibitory activity identified in the initial screen (Tables 4 and 6). All compounds were soluble and showed no visible signs of precipitation at the concentrations tested in these assays. The originally described compounds (e.g. 1 and 7, Figure 1) did not display glutamate-site or glycine-site NMDAR agonist activity nor were they active in the absence of agonists [7,13]. In this study, compounds with the greatest PAM activity (14b and 46, Tables 1, 2 and 4) were similarly evaluated and found to have no NMDAR agonist activity or effect on the holding current (Saptoka et al., 2017).
Table 1.
SAR studies on naphthalene derivatives and comparsion to corresponding phenanthrene deriavtives. Values are percentage response for agonist (10 μM L-glutamate/10 μM glycine) in presence of test compound (100 μM) compared to agonist alone.
| NMDAR (n≥4)a | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Formula | R1 | R2 | R3 | R4 | GluN2A | GluN2B | GluN2C | GluN2D |
| 1 | A | I | - | - | - | 108.6 ± 4.8 | 100.9 ± 0.1 | 65.9 ± 0.1 | 47.7 ± 3.0 |
| 12a | B | Pent-1-ene-1-yl | H | H | CO2H | 51 ± 18 | 58 ± 5 | 82 ± 9 | 68 ± 6 |
| 12b | B | CH=CHCH2CH(CH3)2 | H | H | CO2H | 135 ± 11 | 152 ± 9 | 264 ± 41 | 220 ± 40 |
| 14a | B | (CH2)4CH3 | H | H | CO2H | 131 ± 5 | 102 ± 2 | 126 ± 3 | 110 ± 4 |
| 14b | B | (CH2)3CH(CH3)2 | H | H | CO2H | 171 ± 6 | 171 ± 9 | 192 ± 6 | 207 ± 15 |
| 17 | B | Cyclopropyl | H | H | CO2H | 97.0 ± 3.4 | 97.0 ± 1.6 | 97.0 ± 0.6 | 94.0 ± 3.2 |
| 18 | B | (CH2)4CH3 | H | H | CH2OH | 89 ± 3 | 84 ± 8 | 76 ± 3 | 85 ± 2 |
| 21 | B | (CH2)4CH3 | H | H | CH2CO2H | 67 ± 18 | 67 ± 10 | 60 ± 3 | 37 ± 2 |
| 22 | B | (CH2)4CH3 | Br | H | CO2H | 73 ± 11 | 52 ± 15 | 97.0 ± 4.3 | 99.0 ± 2.8 |
| 24 | B | O(CH2)3CH3 | H | H | CO2H | 55 ± 7 | 46 ± 3 | 63 ± 15 | 57 ± 3 |
| 27a | B | (CH2)4Ph | H | H | CO2H | 118 ± 13 | 120 ± 4 | 123 ± 10 | 115 ± 5 |
| 27b | B | 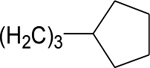 |
H | H | CO2H | 136 ± 7 | 175 ± 13 | 171 ± 13 | 161 ± 11 |
| 29 | B | 4-methylpent-1-yn-1-yl | H | H | CO2H | 104 ± 12 | 99 ± 7 | 125± 15 | 110 ± 20 |
| 74 | B | (CH2)3CH(CH3)2 | H | OH | CO2H | 178 ± 30 | 309 ± 107 | 357 ± 93 | 301 ± 113 |
| 69 | B | (CH2)3CH(CH3)2 | H | CO2H | H | 123 ± 36 | 120 ± 12 | 115 ± 22 | 80 ± 21 |
| 65 | B | (CH2)3CH(CH3)2 | H | CO2H | OH | 78 ± 23 | 113 ± 18 | 113 ± 38 | 180 ± 51 |
| 77 | B | (CH2)3CH(CH3)2 | H | CO2H | CO2H | 92.0 ± 7.9 | 88 ± 6 | 47 ± 2 | 24 ± 1 |
Compounds were tested by TEVC for activity at recombinant NMDA receptors (GluN1a and the indicated GluN2 subunit) expressed in Xenopus oocytes. After obtaining a steady state NMDAR response evoked by 10 μM L-glutamate and 10 μM glycine, test compounds (100 μM) were co-applied with agonists. Values (mean ± s.e.m.) represent the % response in the presence of the test compound compared to response in the presence of agonists alone. Values >100 represent potentiation and those < 100 represent inhibition of the agonist response.
Table 3.
SAR studies to probe the effect of heteroatom or methylene substitution of the alky chain. Values are percentage response for agonist (10 μM L-glutamate/10 μM glycine) in presence of test compound (100 μM) compared to agonist alone.
 | |||||||
|---|---|---|---|---|---|---|---|
| NMDAR (n≥4)a | |||||||
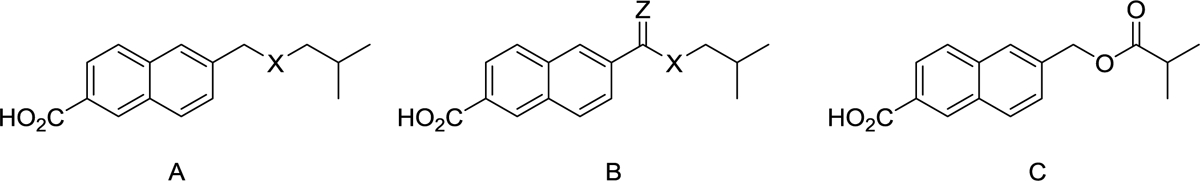
| Compound | Formula | X | Z | GluN2A | GluN2B | GluN2C | GluN2D |
| 38 | B | CH2 | O | 128 ± 10 | 82 ± 9 | 99 ± 12 | 79 ± 12 |
| 44 | B | CH2 | CH2 | 163 ±1 2 | 265 ± 29 | 349 ± 45 | 310 ± 49 |
| 49 | A | S | - | 113 ± 15 | 77 ± 11 | 105 ± 13 | 100.2 ± 7 |
| 52 | B | NH | O | 65 ± 17 | 65 ± 10 | 46 ± 14 | 99 ± 5 |
| 54 | C | - | - | 73 ± 12 | 45 ± 10 | 56 ± 12 | 42 ± 10 |
| 57 | A | O | - | 83 ± 14 | 49 ± 10 | 83 ± 9 | 72 ± 8 |
| 35 | B | O | O | 159 ± 21 | 179 ± 35 | 144 ± 17 | 169 ± 30 |
Compounds were tested by TEVC for activity at recombinant NMDA receptors (GluN1a and the indicated GluN2 subunit) expressed in Xenopus oocytes. After obtaining a steady state NMDAR response evoked by 10 μM L-glutamate and 10 μM glycine, test compounds (100 μM) were co-applied with agonists. Values (mean ± s.e.m.) represent the % response in the presence of the test compound compared to response in the presence of agonists alone. Values >100 represent potentiation and those < 100 represent inhibition of the agonist response.
Figure 2.

Potentiation of NMDAR responses by 2-naphthoic acid derivatives. Select PAMs identified in Table 1 were tested for activity at various concentrations to determine potency and efficacy. Compounds were tested on NMDA receptors containing GluN1a and the indicated GluN2 subunit expressed in Xenopus oocytes. After obtaining a steady state NMDAR response evoked by 10 μM L-glutamate and 10 μM glycine, test compounds were co-applied with agonists at various concentrations. Values (mean ± s.e.m.) represent the % potentiation of the response above the agonist-alone response.
Figure 3.

Inhibition of NMDAR responses by 2-naphthoic acid derivatives. A. 7, B. 79h, C. 79i, D. 79j. NAMs described in Table 6 were tested for activity at various concentrations to determine inhibitory potency and the percentage of maximum inhibition. Compounds were tested on NMDA receptors containing GluN1a and the indicated GluN2 subunit expressed in Xenopus oocytes. After obtaining a steady state NMDAR response evoked by 10 μM L-glutamate and 10 μM glycine, test compounds were co-applied with agonists at various concentrations. Values (mean ± s.e.m.) represent the % response in the presence of the test compound compared to response in the presence of agonists alone.
Table 4.
EC50 (μM, n≥4) values for potentiation of GluN1/GluN2 NMDAR subtypesa
| Compounds | GluN2A | GluN2B | GluN2C | GluN2D |
|---|---|---|---|---|
| 14a | 26.7 ± 5.6 (39.0 ± 5.9) |
ND | 113 ± 66 (42.7 ± 16.1) |
61 ± 33 (17.3 ± 16.2) |
| 14b | 28.0 ± 4.6 (68.6 ± 16.2) |
34.6 ± 3 (102.0 ± 17.8) |
37.2 ± 2.8 (117.2 ± 22.3) |
28.9 ± 4.1 (88.4 ±9.6) |
| 27b | 7.5 ± 2.8 (38.4 ±4.7) |
27.0 ± 6.3 (61.9 ± 10.9) |
22.4 ± 3.6 (105.3 ± 26.1) |
34.7 ± 10.3 (65.6 ± 18.4)) |
| 44 | 114.4 ± 7.2 (230.4 ± 84.6) |
116.4 ± 19.8 (416.6 ± 70.9) |
26.1 ± 4.5 (136.6 ± 11.6) |
72.3 ± 15.5 (277.2 ± 36.8) |
| 46 | 39.4 ± 27.5 (277.2 ± 36.8) |
25.0 ± 11.6 (192.3 ± 46.6) |
36.2± 5.7 (262.6 ± 33.9) |
30.6 ± 7.5 (240.3 ± 63.6) |
EC50 values (mean ± s.e.m.) for the potentiation of GluN1/GluN2 NMDAR responses responses. Values in parenthesis are the maximum potentiation expressed as a percentage (± s.e.m.) above the agonist alone response (L-glutamate, 10 μM and glycine, 10 μM). ND = not determined.
Table 6.
IC50 (μM, n≥4) values for inhbition of GluN1/GluN2 NMDAR subtypesa
| Compounds | GluN2A | GluN2B | GluN2C | GluN2D |
|---|---|---|---|---|
| 7b | 11.0 ± 5.2 (71.5 ± 6.4) |
97.8 ± 20.9 (56.8 ± 5.2) |
41.8 ± 8.4 (105.5 ± 4.4) |
49.6 ± 6.0 (98.6 ± 3.2) |
| 79a | 17.8 ± 4.6 (83.2 ± 11.4) |
7.5 ± 2.0 (93.9 ± 7.3) |
13.8 ± 3.3 (86.3 ± 4.7) |
8.6 ± 1.9 (93.6 ±4.2) |
| 79h | 6.0 ± 4.1 (30.5 ± 6.3) |
32.2 ± 16.0 (61.3 ± 18.1) |
8.2 ± 1.2 (80.1 ± 4.1) |
2.9 ± 0.4 (79.7 ± 2.8) |
| 79i | 9.7 ± 3.8 (49.7 ± 6.8) |
9.2 ± 5.3 (66.3 ± 5.2) |
7.9 ± 4.2 (74.4 ± 6.9) |
1.4 ± 0.4 (70.0 ± 4.6) |
| 79j | 5.8 ± 1.1 (54.1 ± 5.5) |
11.1 ± 2.9 (48.4± 6.0) |
6.5 ± 2.8 (55.9 ± 3.9) |
2.9 ± 0.3 (72.3 ± 2.1) |
IC50 values (mean ± s.e.m.) for the inhibition of GluN1/GluN2 NMDAR responses. Values in parenthesis are the percentage maximum inhibition (± s.e.m.) of the agonist response (L-glutamate, 10 μM and glycine, 10 μM).
Values taken from Costa et al., 2012.
Table 2.
SAR studies to probe the effect of substitution of the alkyl chain at the 6-position of the naphthalene ring. Values are percentage response for agonist (10 μM L-glutamate/10 μM glycine) in presence of test compound (100 μM) compared to agonist alone.
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| NMDAR (n≥4)a | ||||||||
| Compound | Formula | R1 | R2 | R3 | GluN2A | GluN2B | GluN2C | GluN2D |
| 33 | A | H | H | CO2H | 110 ± 6 | 85 ± 6 | 98 ± 2 | 107 ± 12 |
| 31 | B | - | - | - | 107 ± 4 | 97 ± 4 | 105 ± 0.5 | 97 ± 1 |
| 42 | A | H | H | CH2CO2H | 60 ± 10 | 64 ± 8 | 96 ± 10 | 71 ± 13 |
| 46 | A | CH3 | H | CH3 | 281 ± 42 | 225 ± 25 | 337 ± 55 | 317 ± 37 |
| 60 | A | H | CO2H | CH3 | 104 ± 6 | 78 ± 6 | 118 ± 10 | 85 ± 5 |
Compounds were tested by TEVC for activity at recombinant NMDA receptors (GluN1a and the indicated GluN2 subunit) expressed in Xenopus oocytes. After obtaining a steady state NMDAR response evoked by 10 μM L-glutamate and 10 μM glycine, test compounds (100 μM) were co-applied with agonists. Values (mean ± s.e.m.) represent the % response in the presence of the test compound compared to response in the presence of agonists alone. Values >100 represent potentiation and those < 100 represent inhibition of the agonist response.
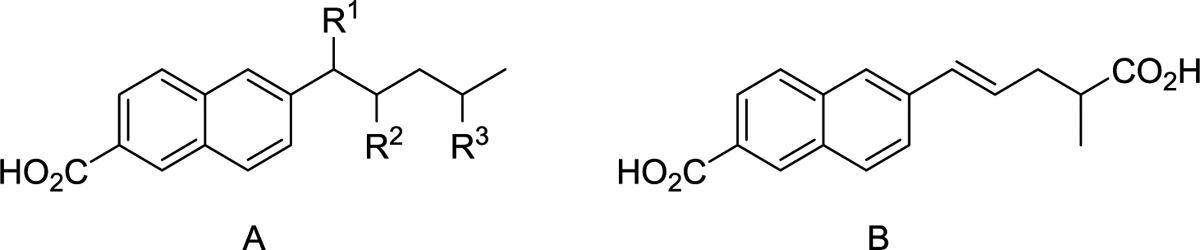
SAR studies to identify novel NMDAR positive allosteric modualtors.
SAR studies were focused on identifying novel NMDAR PAMs and investigating their structural requirements. The following structural changes were investigated: 1. Effect of removing the unsubstituted aromatic ring from the phenanthrene based compounds such as UBP512 (1) to form naphthalene derivatives, 2. Effect of changing the 6-substituent on the naphthyl ring, 3. Effect of changing the position of the carboxylic acid group attached to the naphthalene ring, 4. Effect of adding substituents to the naphthyl ring system, 5. Effect of adding substituents to the alkyl chain attached to the 6-position of the naphthalene ring, 6. Effect of adding heteroatoms to the alkyl chain attached to the 6-position of the naphthalene ring.
Effect of removing unsubstitued aromatic ring from phenanthrene lead compounds.
Previous studies have shown that 9-substituted phenanthrene-3-carboxylic acids such as UBP512 (1, Figure 1) and 9-alkyl derivatives have potentiating effects on NMDARs [7]. The 9-iodo derivative (1) weakly potentiated the GluN2A response, had no effect on GluN2B and inhibited GluN2C/GluN2D [7] (Table 1).
Removal of the unsubstituted aromatic ring from 1 (Figure 1) to afford 6-iodo-2-naphthoic acid eliminated NMDAR PAM activity on GluN2A (at 100 μM it inhibited the agonist response on GluN2A by 26 ± 2% and had 9–12% inhibitory activity on GluN2B-D). 2-Naphthoic acid analogues with short chain alkyl substituents at the 6-position (e.g. ethyl or n-propyl) were found to have no NMDAR PAM activity. The 6-ethyl derivative (100 μM) instead inhibited GluN2A-D with percentage values in the range of 17–52%, while the 6-n-propyl derivative (100 μM) had percentage inhibition values on GluN2A-D in the range of 0–14%). A similar outcome was observed for the 6-cyclopropyl derivative (17, Table 1). In contrast, the corresponding 9-ethyl, 9-n-propyl and 9-cyclopropyl phenanthrene-3-carboxylic acid derivatives showed weak to moderate NMDAR PAM activity [7]. Like the parent phenanthrenes [7], the 6-n-pentyl (14a) and 6-isohexyl (14b, UBP684) derivatives of 2-naphthoic acid showed potentiating effects on NMDARs. The 6-n-pentyl derivative (14a) showed potentiating activity on NMDARs containing GluN2A, GluN2C and GluN2D (Tables 1 and 4) but not GluN2B. When tested at a concentration of 100 μM, the 6-isohexyl naphthyl derivative (14b) showed potentiation in excess of 170% across GluN2A-D. This was higher than that of the 6-n-pentyl derivative (14a) (Table 1), suggesting that 14b was better at increasing the efficacy of L-glutamate/and or glycine. The pan-potentiator 14b (EC50 values ranging from 28.0–37.2 μM across GluN2A-D) had similar potency to that of 14a on GluN2A but was a more potent PAM on GluN2B-D (Tables 1 and 4, Figure 2). These data suggest that the unsubstituted aromatic ring of the phenanthrene series is not needed for NMDAR PAM activity when long chain alkyl substituents are present at the 6-position of the naphthalene ring.
Effect of changes to the 6-alkyl substituent on the naphthalene ring.
Conformational restriction of the isohexyl side chain in 14b by incorporation of a trans double bond gave 12b, which displayed an increase in potentiation of agonist response on GluN2C and GluN2D compared to 14b (Table 1). However, a similar conformational restriction of the n-pentyl side chain of 14a to give 12a resulted in weak inhibitory activity across the GluN2 subunits (Table 1). Incorporation of a triple bond into the isohexyl side chain to give 29, led to the loss of the potentiating effect on GluN2A and GluN2B and a much reduced potentiating effect on GluN2C and GluN2D compared to 14b (Table 1). Although restricting conformational freedom by incorporating a double or triple bond was detrimental for PAM activity on GluN2A and GluN2B, adding a trans double bond into the side chain of 14b can be used to increase selectivity for GluN2C/GluN2D versus GluN2A/GluN2B.
Adding a 4-phenylbut-1-yl substituent to the 6-position of the naphthalene ring (Figure 2, Table 1) to give 27a reduced potentiation compared to 14b. Adding a 3-cyclopentylprop-1-yl substituent to the 6-position of the naphthalene ring to give 27b (Tables 1 and 4) led to a similar level of potentiating activity on GluN2A-D to that of 14b, with the former being more potent on GluN2A (Table 4). Thus, incorporating the two methyl groups at the end of the isohexyl chain of 14b into a cyclopentyl ring enhances PAM potency and selectivity for GluN2A.
Effect of changing the nature and position of the carboxylic acid group attached to the naphthalene ring.
Reduction of the carboxylic acid group in 14a to give the corresponding hydroxymethyl derivative 18 replaced potentiating activity with weak inhibitory activity at GluN2A-D (Table 1). A similar observation was made when a CH2 linker was introduced between the naphthalene ring and the carboxylic acid group in 14a. The resultant acetic acid derivative, 21, had weak to moderate inhibitory activity at GluN2A-D (Table 1). Taken together, the activities of 18 and 21 suggest that the carboxylic acid group is necessary for potentiating activity and that this group must be directly attached to the naphthalene ring. Furthermore, moving the carboxylic acid of 14b to an adjacent position on the naphthalene ring to give 7-isohexyl-2-naphthoic acid (69) resulted in weak inhibitory activity at GluN2D and a weaker potentiating effect on GluN2A-C compared to the parent compound (Table 1). Thus, switching the position of the carboxylic acid group relative to the alkyl substituent is detrimental for NMDAR PAM activity, especially for GluN2D.
Effect of adding substituents to the naphthalene ring.
Adding a bromo substituent to the 5-position of the naphthalene ring of 14a to give 22 (Table 1) resulted in weak inhibitory activity on GluN2A and GluN2B and no activity on GluN2C and GluN2D, suggesting the bromo group is in an area of excluded volume in the NMDAR PAM binding site or that this addition prevents the PAM’s allosteric action. The 2,3-dicarboxy analogue (77) of 14b resulted in little or no activity on GluN2A and GluN2B but weak to moderate inhibitory activity on GluN2C and GluN2D (Table 1).
Adding a hydroxyl group to the 3-position of the naphthalene ring of 14b to give 74 produced a pan-potentiator with similar, or possibly enhanced activity on GluN2B-D compared to 14b (Table 1). Thus, it may be possible to reduce the hydrophobicity and increase water solubility of the NMDAR PAMs described here by adding a hydroxyl group to the 3-position of the 2-naphthoic acid derivatives. Switching the placement of the hydroxyl and carboxyl groups on the naphthalene ring of 74 to give 65 resulted in weak inhibitory activity on GluN2A and weaker potentiating activity on GluN2B-D compared to that observed for 14b and 74 (Table 1). However, unlike the 7-isohexyl derivative 69, which showed weak inhibitory activity on GluN2D, 65 potentiated GluN2D activity (Table 1), suggesting that the 3-hydroxyl group is required for this activity. Interestingly, replacing the isohexyl group of 65 with a bromo [13], phenyl (7, Figure 1) [13], styryl (79a, Table 5) or phenethyl (81a, Table 5) group leads to moderate NMDAR NAM activity on GluN2A-D.
Effect of adding substituents to the 6-alkyl chain.
Substitution at the 1-position of the isohexyl side chain of 14b with a methyl group to give 46 resulted in a pan-potentiator with similar potency on GluN2A-D (EC50 values ranged from 25.0 to 39.4 μM) to that of 14b (Tables 2 and 4, Figure 2). However, the maximum potentiating effect of 46 across GluN2A-D was greater than that observed for 14b (Figure 2). Conformational restriction of 46 by incorporating a double bond to give 44 (Table 3) produced a pan-potentiator with similar potency to its parent compound on GluN2C but weaker potency on the other GluN2 subunits (EC50 values ranged from 26.1 to 116.4 μM across GluN2A-D, Table 4). Interestingly, 44 showed different degrees of maximal potentiation across GluN2A-D and the value for GluN2B was higher than for any other compound that was tested (Figure 2E, Table 4). Adding a carboxylic acid at the 4-position of the n-pent-1-yl side chain of 14a to give 33 resulted in little or no activity at GluN2B or GluN2C and very weak potentiating activity on GluN2A and GluN2D (Table 2). Conformational restriction of 33 by including a trans double bond to give 31 (Table 2) resulted in little or no activity across GluN2A-D. Furthermore, adding a CH2CO2H group to the 4-position of the n-pent-1-yl side chain of 14a to give 42 removed potentiating activity and instead weak inhibition was observed on GluN2A, GluN2B and GluN2D (Table 2). Adding a carboxylic acid at the 2-position of the isohexyl side chain of 14b to give 60 resulted in selective weak potentiation of GluN2C with little or no activity on GluN2A and weak inhibition of GluN2B and GluN2D (Table 2). Thus, it appears that polar substituents on the alkyl chain cannot be accommodated, while methylene substitution (44) at the 1-position of the alkyl chain of 14b reduces PAM potency on GluN2A and GluN2B but increases agonist efficacy to a greater degree than 14b.
Effect of adding heteroatoms to the 6-alkyl chain.
A SAR study was undertaken to investigate the effect of adding heteroatoms to the isohexyl side chain of 14b, with the aim of reducing hydrophobicity. Adding a ketone group to the isohexyl chain of 14b to produce 38 (Table 3) led to a loss of potentiating activity on GluN2B-D and a weak potentiating effect on GluN2A. Replacing the CH2 group adjacent to the naphthalene ring of 14a with an oxygen atom to give 24 (Table 1) resulted in inhibitory activity across GluN2A-D. Similarly, replacing the second CH2 group in the isohexyl chain of 14b with either a sulphur atom (49) or an oxygen atom (57) resulted in either loss of potentiating activity or weak inhibitory effects (Table 3). Incorporating either an amide or ester group into the isohexyl chain of 14b to give 52 and 54 respectively, led to either inhibitory effects or loss of activity on GluN2A-D. However, incorporating an ester group at a different position in the isohexyl chain of 14b to give 35 resulted in a pan-potentiator with similar levels of activity across GluN2A-D to that observed with 14b (Table 3). The potentiating activity of the ester 35 was unexpected given that the ketone 38 and the ether derivative 57 lacked potentiating activity. A possible explanation is that both oxygen atoms of the ester group are required for potentiating activity.
SAR studies on 2-naphthoic acid derivatives as NMDAR negative allosteric modulators
In the preceding SAR studies, NMDAR NAM activity was observed for some 2-naphthoic acid derivatives. For example, the 6-pentyl compound (21, Table 1) and 6-isohexyl derivatives with heteroatoms in the side chain (24 Table 1, 52, 54 and 57 Table 3) showed 50% or greater inhibition on at least one of the GluN1/GluN2 subtypes when tested at a concentration of 100 μM. These data show that inserting heteroatoms into the 6-alkyl chain such as an O atom (24 and 57), a carboxamide (52) or an ester (54) converts PAMs into NMDAR NAMs. This suggests that the PAM and NAM binding sites for 6-substituted 2-naphthoic acid derivatives differ in that the NAM binding site appears to be much more polar in the area where the 6-substituent binds. Chain extension of the carboxylic acid in 14a by insertion of a CH2 group leads to preferential GluN2D NAM activity for compound 21, while adding a carboxy group to the 3-position of 14b leads to preferential GluN2C and GluN2D NAM activity for compound 77 (Table 1). Thus, while acidic group chain extension and 2,3-dicarboxy substitution is acceptable for NMDAR NAMs it is detrimental to PAM activity.
The 7-styryl substituted 2-naphthoic acid 79a (Tables 5 and 6) showed greater NMDAR NAM activity than any of the 6-substituted 2-naphthoic acid derivatives. This 2,7 relationship between the carboxy group and styryl side chain favours NAM activity but the 2,7 relationship of the 7-alkyl derivatives 65 and 69 was detrimental to PAM activity (Table 1). Compound 79a is structurally similar to the previously reported NMDAR NAM 7, (Figure 1) [13], which has a phenyl substituent at the position occupied by the styryl group. Compared to 7, (Figure 1), the parent styryl-substituted compound, 79a was found to have similar activity on GluN2A, ~10-fold greater inhibitory activity on GluN2B and 3–5 fold greater inhibitory on GluN2C and GluN2D (Table 6). In addition, 79a showed 83–94% maximal inhibition across GluN2A-D, whereas 7 showed only 57–72% maximal inhibition of GluN2A and GluN2B (Table 6) [13].
Given that 79a showed interesting NMDAR NAM activity, a SAR study was undertaken with the aim of improving NAM potency and GluN2 subunit selectivity. When the hydroxyl group of 79a was replaced by a carboxyl group to give compound 85 (formula C, Table 5) inhibitory activity was reduced. Similarly, replacement of the naphthalene ring of 79a with a coumarin ring to give compound 88 (formula D, Table 5) dramatically reduced inhibitory activity and indeed led to potentiating activity on GluN2C and GluN2D. This suggests that the hydroxyl group of 79a is necessary for optimal NAM activity. Next, the effect of saturating the double bond linking the phenyl ring of 79a to the naphthalene ring was studied. The saturated analogue, compound 81a (Table 5) had weaker inhibitory activity than the parent compound 79a, suggesting a degree of conformational restriction is necessary for optimal NMDAR NAM activity.
Having established that the core structure was necessary for NMDAR NAM activity of 79a, a study was undertaken to investigate the effect of substitutions on the phenyl ring. Carboxyl group substitution at the ortho, meta or para positions (compounds 79b, 79c and 79d, Table 5) led to much reduced inhibitory activity compared to the parent compound. Saturation of the double bond in the ortho and para carboxy derivatives to give compounds 81b and 81c, respectively, did not improve upon the already weak inhibitory activity of the parent compounds. Interestingly, ortho methoxy substitution (compound 79e, Table 5) led to weak potentiation of GluN2A responses and weak inhibition of GluN2B-D. Either meta or para methoxy substitution to give compounds 79f and 79g (Table 5), respectively, led to improved inhibitory activity compared to the ortho derivative. Ortho nitro substitution (79h, Table 5) improved inhibitory activity compared to the corresponding methoxy derivative, especially on GluN2C and GluN2D. Meta and para nitro substitution (79i and 79j, Table 5) had a similar effect on inhibitory activity to that observed with the corresponding methoxy substituted compounds.
Full concentration response curves were obtained for the inhibitory activity of 79h, 79i and 79j across GluN1/GluN2A-D (Figure 3B–D). Surprisingly, unlike the parent compound 79a (Figure 3A), all three compounds failed to produce 100% maximal inhibition of GluN2A-D responses. The ortho nitro derivative 79h only maximally inhibited GluN2A by 30% and GluN2B-D by 60–80% (Table 6). Similar findings were observed with the meta (79i) and para (79j) nitro derivatives in terms of their maximal inhibition. Regarding inhibitory activity, IC50 values for 79h were 3-fold lower on GluN2A and GluN2D and 4-fold higher on GluN2B compared to 79a (Table 6). A similar trend was observed for 79i and 79j, with the former having the most potent inhibitory activity on GluN2D of all the analogues tested (IC50 value 1.4 μM). Compared to the previously reported 7-phenyl derivative UBP617 (7, Table 6) (Costa et al., 2012), the meta-nitrostyryl derivative 79i had similar activity on GluN2A and 10-fold, 5-fold and 35-fold higher inhibitory activity on GluN2B, GluN2C and GluN2D, respectively (Table 6). Thus, meta-nitrostyryl substitution produced a NAM that was more potent than the parent compound 79a and was selective for inhibiting NMDARs containing GluN2D. In addition, unlike 79a, 79i was a partial NAM on GluN2D.
Testing 79h at two different agonist concentrations (10/10 μM Glu/Gly and 300/300 μM Glu/Gly) showed that the degree of inhibition of the agonist response was not reduced at higher agonist concentrations (Figure 4). This suggests that 79h is not competing with either Glu or Gly for their binding sites on the GluN1 or GluN2 LBD, respectively.
Figure 4.

79h does not compete with L-glutamate or glycine at NMDARs. 30 μM 79h did not display reduced inhibition of GluN1/GluN2C (blue) and GluN1/GluN2D (gray) NMDAR responses evoked by high concentrations of agonists (300 μM glutamate / 300 μM glycine; dark blue/gray) than by low agonist concentrations (10 μM glutamate / 10 μM glycine; light blue/gray).
Discussion
Structure-activity relationship studies on 2-naphthoic acid derivatives based on the lead phenanthrene UBP512 (1, Table 1) [7] have led to the discovery of novel series of both NMDAR PAMs and NAMs. We have shown that 6-alkyl substitued 2-naphthoic acid derivatives such as 14b (UBP684) can act as pan potentiators of agonist-induced currents through NMDARs (i.e. they potentiate regardless of the type of GluN2 subunit incorporated into the NMDAR). The NMDAR PAM 27b (Table 1) showed a preference for the GluN2A subunit and further modification of this compound may lead to GluN2A selective NMDAR potentiators. In contrast to 6-alkyl derivatives such as 14b, the 7-styryl derivative 79a and analogues thereof were found to be NMDAR NAMs. NAM activity and selectivity for GluN2D could be enhanced by 3-nitro substitution to produce 79i (Table 6), which unlike the parent compound 79a, was a partial NAM.
Our SAR studies revealed different structural requirements for NMDAR NAMs and PAMs based on 2-naphthoic acid. Whilst 6-alkyl substituents were required for optimal PAM activity, NAM activity was predominantly observed when heteroatoms were added to the 6-alkyl chain. In addition, with regards to relative positioning of the carboxy and alkyl or alkenyl groups, a 2,6-substitution pattern favours PAM activity (e.g. 14a and 14b, Table 1), whereas a 2,7-substitution pattern was either detrimental for PAM activity (e.g. 65 and 69) or produced moderately potent NAM activity (e.g. 79a, Table 5). These differences in the structural requirements suggest that either there are distinct binding sites for PAMs and NAMs or that they contact different areas within the same binding site on the NMDAR complex.
Experiments with structurally related analogues of the NMDAR PAM 14b (Table 1) and the NAM 79a showed that they are not binding to the known sites for glutamate antagonists, glycine site antagonists, channel blockers or at the N-terminal domain site where NMDAR NAMs such as ifenprodil bind [7,13]. The NMDAR PAM 14b (UBP684) has been shown to slow deactivation upon glutamate removal and increases open channel probability by stabilizing the NMDAR LBDs in an active conformation [20,21]. It has been proposed that 14b may be binding at the LBD dimer interface [6d,20,21], as has been observed for the GluN2A selective PAM GNE-6901 (4, Figure 1) [10a] or at the LBD/transmembrane domain linker region as proposed for neurosteroids that act as NMDAR PAMs [22]. A feasible binding mode for 14b was observed when it was docked into the GluN1/GluN2A LBD dimer interface using the Induced Fit module in Maestro (Schrodinger LLC) [21]. The 6-isohexyl side chain of 14b interacts with a number of hydrophobic residues in the dimer interface in agreement with the current SAR study, which showed that a 6-alkyl substituent is preferred for NMDAR PAM activity. However, structural studies are required to definitively show that 14b is binding to the LBD dimer interface.
It has been reported that the GluN2A selective NAM MPX-004 (9, Figure 1) binds at a similar site to the GluN2A PAM GNE-6901 at the dimer interface of the LBDs of GluN1/GluN2A. The major diffrences between these two binding sites is the conformation of Y535 in GluN1 and the position of V783 in GluN2A, which is displaced upon MPX-004 but not PAM binding [10a,17]. It is possible that the 2-naphthoic acid based NAMs and PAMs bind at these sites. However, the inhibitory activity of NMDAR NAMs structurally related to 79h were not dependent on glycine concentration [13] in the same way as MPX-004 [17]. In the case of 79h it did not show dependence on glycine concentration for its inhibitory activity (Figure 4) and so it is unlikely that 79h and derivatives thereof are binding to the NMDAR LBD dimer interface or the glycine binding site on the GluN1 LBD. Further studies are needed to definitively identify the binding sites on the NMDAR for the NAMs based on 2-naphthoic acid. This will enable computer modelling studies to gain an understanding of the binding modes of the NMDAR NAMs we have identified.
The NMDAR PAMs described herein are useful tools to study synaptic function and represent possible leads for the development of drugs for disorders that involve NMDAR hypofunction, such as schizophrenia. Since NMDARs are involved in mechanisms that have been proposed to underlie learning and memory processes in the hippocampus, NMDAR potentiators may have application in the treatment of disorders that involve a loss of cognitive function, such as Alzheimer’s disease.
A new class of partial NMDAR NAMs that show some selectivity for GluN2D has been identified. NMDAR inhibitors have been proposed as treatments for disorders which arise from NMDAR overactivation such as neuropathic pain, epilepsy, depression and for neurodegenerative disorders such as ischaemia. However, inhibition of NMDARs involved in normal CNS activity causes adverse effects such as psychotomimetic effects, ataxia and cognitive dysfunction. The use of partial NAMs, together with improved subtype-selectivity, may be a way of counteracting overactivation of NMDARs in CNS disorders whilst allowing essential physiological signalling, thus avoiding adverse CNS effects.
Experimental Methods
Chemistry experimental
General Procedures
Melting points were determined using an Electrothermal IA9100 capillary apparatus and are uncorrected.1H-NMR spectra were measured on either a Jeol JNM-LA300 spectrometer at 300.53 MHz, a Jeol JNM-ECP400 spectrometer at 400.18 MHz, a Varian 400MR spectrometer at 399.77 MHz, or a Varian 500 spectrometer at 500 MHz.13C-NMR spectra were recorded on either a Jeol JNM-LA300 spectrometer at 75.57 MHz, a Jeol JNM-ECP400 spectrometer at 100.63 MHz, a Varian 400MR spectrometer at 100.52 MHz or a Varian 500 spectrometer at 125 MHz. Chemical shifts (δ) are reported in parts per million (ppm) with tetramethylsilane in CDCl3 or DMSO-d6 used as internal standards. Mass spectrometry was performed in the mass spectroscopy laboratories of the School of Chemistry, University of Bristol, UK. Elemental analyses were performed in the microanalytical laboratories of the School of Chemistry, University of Bristol, UK. The purity of all compounds sent for biological testing was determined by combustion analysis, which confirmed that there were ≥ 95% pure. Thin layer chromatography was performed on Merck silica gel 60 F254 plastic sheets. Flash chromatography was performed on Merck silica gel 60 (220–440 mesh) from Fisher. All anhydrous reactions were conducted under argon. All anhydrous solvents were obtained from Sigma-Aldrich, UK or Acros Organics, UK. 47 [23] (methyl 6-(bromomethyl)-2-naphthoate), 50 [24] (methyl 6-(chlorocarbonyl)-2-naphthoate), 53 [25] (6-(hydroxymethyl)-2-naphthoic acid), 55 [26] (2-bromo-6-(bromomethyl)naphthalene), 61 [27] (7-bromo-3-hydroxy-2-naphthoic acid) and 70 [28,29] (6-(methoxymethoxy)naphthalen-2-yl trifluoromethane-sulfonate) were synthesized according to literature procedures.
(E)-Methyl 6-(pent-1-en-1-yl)-2-naphthoate (11a).
A flask was charged with 10 (3.00 g, 11.3 mmol), palladium acetate (25 mg, 1 mol%) and tri-o-tolylphosphine (138 mg, 4 mol%). The flask was then briefly evacuated and backfilled with argon three times. Degassed anhydrous DMF (100 mL) was added followed by pent-1-ene (1.55 mL, 14.1 mmol) and triethylamine (1.97 mL, 14.1 mmol). The resultant mixture was heated at 100 °C overnight. After being allowed to cool to room temperature the reaction mixture was filtered through a celite pad to remove any precipitated Pd(0) and then poured into a stirred solution of EtOAc (100 mL), water (100 mL) and aqueous 1 M HCl (10 mL). The organic layer was subsequently isolated and the aqueous phase further extracted with EtOAc (2 × 50 mL). The organic extracts were pooled, washed with water (5 × 100 mL), brine (100 mL) and dried over MgSO4. Concentration in vacuo afforded a light brown solid which was purified by flash chromatography (2 → 5% EtOAc in hexane) to afford 11a as a clear oil (2.62 g, 91%);1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H), 8.02 (dd, J = 8.4 & 1.6 Hz, 1H), 7.86 (d, J = 8.8 Hz, 1H, ArH), 7.81 (d, J = 8.4 Hz, 1H, ArH), 7.70 (s, 1H, ArH), 7.63 (dd, J = 8.8 & 2.0 Hz, 1H, ArH), 6.56 (d, J = 16.0 Hz, 1H, ArCH=CH-), 6.42 (dt, J = 16.0 & 6.8 Hz, 1H, ArCH=CH-), 3.97 (s, 3H, -CO2CH3), 2.32–2.23 (m, 2H, -CH2CH2CH3), 1.55 (sex, J = 7.2 Hz, 2H, -CH2CH2CH3), 0.99 (t, J = 7.2 Hz, 3H, -CH2CH2CH3);13C NMR (100 MHz, CDCl3) δ 167.3, 137.8, 135.9, 133.1, 131.6, 130.7, 129.7, 129.4, 127.9, 126.8, 125.6, 125.0, 124.4, 52.2, 35.3, 22.5, 13.8; HRMS-ESI calcd for C17H18O2 [M + H]+ 255.1385; found 255.1392.
(E)-Methyl 6-(4-methylpent-1-en-1-yl)-2-naphthoate (11b).
Method identical to that described for 11a. 10 (4.00 g, 15.1 mmol), palladium acetate (34 mg, 1 mol%), tri-o-tolylphosphine (184 mg, 4 mol%), 4-methylpent-1-ene (2.39 mL, 18.9 mmol) and triethylamine (2.63 mL, 18.9 mmol) afforded a light brown solid which was purified by flash chromatography (2 → 5% EtOAc in hexane) to afford 11b as a as a clear oil (3.61 g, 89%);1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H, ArH), 8.02 (dd, J = 8.8 & 2.0 Hz, 1H, ArH), 7.87 (d, J = 8.8 Hz, 1H, ArH), 7.81 (d, J = 8.4 Hz, 1H, ArH), 7.70 (s, 1H, ArH), 7.64 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 6.54 (d, J = 15.6 Hz, 1H, ArCH=CH-), 6.42 (dt, J = 15.6 & 7.2 Hz, 1H, ArCH=CH-), 3.97 (s, 3H, -CO2CH3), 2.20–2.15 (m, 2H, -CH2CH(CH3)2), 1.84–1.70 (m, 1H, -CH2CH(CH3)2), 0.98 (d, J = 6.8 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 167.3, 137.8, 135.9, 132.0, 131.6, 130.7, 130.6, 129.4, 128.0, 126.8, 125.6, 125.0, 124.4, 52.2, 42.6, 28.6, 22.4; HRMS-ESI calcd for C18H20O2 [M + H]+ 269.1536; found 269.1540.
(E)-6-(Pent-1-en-1-yl)-2-naphthoic acid (12a).
To a stirring solution of 11a (1.29 g, 5.1 mmol) in a THF/water mix (3:1, 100 mL) was added LiOH (486 mg, 20.3 mmol) dissolved in water (10 mL). The resultant mixture was stirred at room temperature overnight. In the morning, TLC indicated incomplete hydrolysis so the mixture was heated at 65 °C until all the ester had been consumed. After 2 h the mixture was allowed to cool to room temperature before the THF was removed in vacuo. The resultant aqueous solution was topped up with water (40 mL) and then acidified to pH 1 using aqueous 2 M HCl. The solid which precipitated out of solution was filtered off, washed copiously with water and then dried over P2O5 overnight. Recrystallisation from toluene (twice) afforded 12a as a white solid (507 mg, 42%); mp: 157–160°;1H NMR (500 MHz, DMSO-d6) δ 13.00 (br s, 1H, -CO2H), 8.53 (s, 1H, ArH), 8.02 (d, J = 8.5 Hz, 1H, ArH), 7.94 (dd, J = 8.5 & 1.5 Hz, 1H, ArH), 7.92 (d, J = 8.5 Hz, 1H, ArH), 7.86 (s, 1H, ArH), 7.74 (dd, J = 8.5 & 1.5 Hz, 1H, ArH), 6.59 (d, J = 16.0 Hz, 1H, ArCH=CH-), 6.53 (dt, J = 16.0 & 6.5 Hz, 1H, ArCH=CH-), 2.23 (q, J = 7.0 Hz, 2H, -CH2CH2CH3), 1.50 (sex, J = 7.5 Hz, 2H, -CH2CH2CH3), 0.94 (d, J = 7.5 Hz, 3H, -CH2CH2CH3);13C NMR (100 MHz, DMSO-d6) δ 167.3, 137.1, 135.3, 132.7, 131.2, 130.1, 129.4, 129.4, 127.9, 127.5, 125.4, 124.6, 124.3, 34.6, 21.8, 13.5; HRMS-ESI calcd for C16H16O2 [M - H]− 239.1078; found 239.1081; Anal. (C16H16O2) C, H.
(E)-6-(4-Methylpent-1-en-1-yl)-2-naphthoic acid (12b).
To a stirring solution of 11b (889 mg, 3.31 mmol) in a dioxane/water mix (3:1, 80 mL) was added aqueous 1 M NaOH (13.2 mL, 13.2 mmol). The resultant mixture was stirred at 80 °C until TLC indicated complete hydrolysis of the ester. The mixture was then allowed to cool to room temperature before the dioxane was removed in vacuo. The resultant aqueous solution was topped up with water (40 mL), extracted with diethyl ether (25 mL), and then acidified to pH 1 using aqueous 2 M HCl. The solid which precipitated out of solution was filtered off, washed copiously with water and then dried over P2O5 overnight. Recrystallisation from toluene afforded 12b as a white solid (376 mg, 45%); mp: 178–182°;1H NMR (400 MHz, DMSO-d6) δ 13.01 (br s, 1H, -CO2H), 8.54 (s, 1H, ArH), 8.03 (d, J = 8.8 Hz, 1H, ArH), 7.95 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.92 (d, J = 8.8 Hz, 1H, ArH), 7.87 (s, 1H, ArH), 7.76 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 6.58 (d, J = 16.0 Hz, 1H, ArCH=CH-), 6.52 (dt, J = 16.0 & 6.4 Hz, 1H, ArCH=CH-), 2.14 (t, J = 6.4 Hz, 2H, -CH2CH(CH3)2), 1.75 (sep, J = 6.8 Hz, 1H, -CH2CH(CH3)2), 0.94 (d, J = 6.8 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 167.3, 137.1, 135.3, 131.7,131.2, 130.4, 130.1, 129.4, 127.9, 127.5, 125.5, 124.7, 124.4, 41.9, 27.9, 22.2; HRMS-ESI calcd for C17H18O2 [M - H]− 253.1234; found 253.1235; Anal. (C17H18O2·0.34C7H8) C, H.
Methyl 6-n-pent-1-yl-2-naphthoate (13a).
A solution of 11a (1.30 g, 5.1 mmol) in EtOAc (100 mL) was hydrogenated under 3 bar of hydrogen in the presence of 10 wt % palladium on activated carbon (50 mg) for 18 h. The reaction mixture was then filtered through a celite pad before being concentrated in vacuo to afford 13a as a clear oil (1.28 g, 98%);1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H, ArH), 8.03 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.87 (d, J = 8.4 Hz, 1H, ArH), 7.81 (d, J = 8.4 Hz, 1H, ArH), 6.64 (s, 1H, ArH), 7.40 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 3.98 (s, 3H, -CO2CH3), 2.79 (t, J = 7.6 Hz, 2H, ArCH2-), 1.76–1.67 (m, 2H, -CH2CH2 CH2-), 1.38–1.31 (m, 4H, -CH2CH2CH2-), 0.90 (t, J = 7.2 Hz, 3H, -CH3);13C NMR (100 MHz, CDCl3) δ 167.4, 143.4, 135.8, 130.9, 130.8, 129.2, 128.3, 127.6, 126.5, 126.2, 125.2, 52.1, 36.2, 31.5, 30.9, 22.5, 14.0; HRMS-CI calcd for C17H20O2 [M + H]+ 257.1542; found 257.1550.
Methyl 6-(4-methylpent-1-yl)-2-naphthoate (13b).
Method identical to that described for 13a. 11b (2.70 g, 10.1 mmol) and 10 wt % palladium on activated carbon (100 mg) afforded 13b as a clear oil (2.68 g, 98%);1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H, ArH,), 8.03 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.87 (d, J = 8.8 Hz, 1H, ArH), 7.81 (d, J = 8.8 Hz, 1H, ArH), 6.55 (s, 1H, ArH), 7.40 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 3.98 (s, 3H, -CO2CH3), 2.77 (t, J = 7.6 Hz, 2H, ArCH2-), 1.76–1.67 (m, 2H, -CH2CH2-), 1.64–1.55 (m, 1H, -CH(CH3)2), 1.32–1.23 (m, 2H, -CH2CH2-), 0.89 (d, J = 6.8 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 167.4, 143.4, 135.8, 130.9, 130.8, 129.2, 128.3, 127.6, 126.6, 126.2, 125.3, 52.1, 38.6, 36.5, 29.1, 27.9, 22.6; HRMS-CI calcd for C18H22O2 [M + H]+ 271.1698; found 271.1703.
6-n-Pent-1-yl-2-naphthoic acid (14a).
To a stirring solution of 13a (1.14 g, 4.5 mmol) in a THF/water mix (3:1, 80 mL) was added aqueous 1 M NaOH (22.5 mL, 22.5 mmol). The resultant mixture was stirred at 65 °C until TLC indicated complete hydrolysis of the ester. The mixture was then allowed to cool to room temperature before the THF was removed in vacuo. The resultant aqueous solution was topped up with water (40 mL) and then acidified to pH 1 using aqueous 2 M HCl. The solid which precipitated out of solution was filtered off, washed copiously with water and then dried over P2O5 overnight to afford 13a as an off-white solid (913 mg, 84%); mp: 128–132°;1H NMR (500 MHz, DMSO-d6) δ 13.00 (br s, 1H, -CO2H), 8.54 (s, 1H, ArH), 8.01 (d, J = 9.0 Hz, 1H, ArH), 7.94 (dd, J = 9.0 & 1.5 Hz, 1H, ArH), 7.91 (d, J = 8.5 Hz, 1H, ArH), 7.76 (s, 1H, ArH), 7.46 (dd, J = 8.5 & 1.5 Hz, 1H, ArH), 2.76 (t, J = 7.5 Hz, 2H, ArCH2-), 1.66 (pent, J = 7.5 Hz, 2H, -CH2CH2CH2-), 1.37–1.25 (m, 4H, -CH2CH2CH2-), 0.86 (t, J = 7.0 Hz, 3H, -CH3);13C NMR (100 MHz, DMSO-d6) δ 167.5, 142.8, 135.1, 130.6, 130.2, 129.1, 128.2, 127.6, 127.2, 125.9, 125.1, 35.3, 30.9, 30.3, 21.9, 13.9; HRMS-ESI calcd for C16H18O2 [M - H]− 241.1234; found 241.1235; Anal. (C16H18O2·0.2H2O) C, H.
6-(4-Methylpent-1-yl)-2-naphthoic acid (14b).
To a stirring solution of 13b (2.66 g, 9.84 mmol) in a dioxane/water mix (3:1, 80 mL) was added NaOH (1.58 g, 39.5 mmol) dissolved in water (20 mL). The resultant mixture was stirred at 80 °C until TLC indicated complete hydrolysis of the ester. The mixture was then allowed to cool to room temperature before the dioxane was removed in vacuo. The resultant aqueous solution was topped up with water (40 mL) and then acidified to pH 1 using aqueous 2 M HCl. The solid which precipitated out of solution was filtered off, washed copiously with water and then dried over P2O5 overnight. Recrystallisation from acetonitrile afforded 14b as a fluffy white solid (1.67 g, 66%); mp: 149–153°;1H NMR (400 MHz, DMSO-d6) δ 13.05 (br s, 1H, -CO2H), 8.55 (s, 1H, ArH), 8.02 (d, J = 8.4 Hz, 1H, ArH), 7.95 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 7.91 (d, J = 8.4 Hz, 1H, ArH), 7.76 (s, 1H, ArH), 7.47 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 2.74 (t, J = 7.6 Hz, 2H, ArCH2-), 1.71–1.61 (m, 2H, -CH2CH2-), 1.56 (sep, J = 6.8 Hz, 1H, -CH(CH3)2), 1.24–1.17 (m, 2H, CH2CH2-), 0.85 (d, J = 6.8 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 167.4, 142.7, 135.1, 130.5, 130.2, 129.1, 128.2, 127.5, 127.2, 125.9, 125.1, 38.0, 35.5, 28.4, 27.2, 22.4; HRMS-ESI calcd for C17H20O2 [M - H]− 255.1391; found 255.1396; Anal. (C17H20O2·0.13H2O) C, H.
Methyl 6-vinyl-2-naphthoate (15).
A flask containing 10 (2.00 g, 7.6 mmol) was evacuated and backfilled with argon three times. Anhydrous toluene (50 mL) was cannulated into the flask and the resultant solution de-gassed with argon for approx. 30 mins. Pd(PPh3)4 (266 mg, 3 mol%) was then added and the mixture de-gassed for a further 10 mins before vinyl(tri-n-butyl)tin (2.55 mL, 8.7 mmol) was added. The resultant mixture was refluxed for 4 h before being allowed to cool to room temperature and filtered through celite to remove any precipitated Pd(0). The filtrate was then poured into a stirring mixture of EtOAc and saturated aqueous NH4Cl (50 mL each). The organic layer was isolated and washed with aqueous 1M KF (2 × 50 mL) to remove any tin by-products. The white solid (Bu3SnF) which precipitated from solution after the first wash was removed via filtration through celite. The organic layer was then isolated, washed with water (50 mL), brine (50 mL), dried over MgSO4 and concentrated in vacuo to yield a light peach coloured residue. Purification by flash chromatography (5% EtOAc in hexane) afforded 15 as a viscous clear oil which partially solidified on standing to a white solid (1.01 g, 63%);1H NMR (300 MHz, CDCl3) δ 8.56 (s, 1H, ArH), 8.05 (d, J = 8.7 & 1.8 Hz, 1H, ArH), 7.90 (d, J = 8.7 Hz, 1H, ArH), 7.85 (d, J = 8.7 Hz, 1H, ArH), 7.78 (s, 1H, ArH), 7.69 (dd, J = 8.7 & 1.8 Hz, 1H, ArH), 6.89 (dd, J = 17.4 & 10.8 Hz, 1H, ArCH=CH2), 5.93 (d, J = 17.4 Hz, 1H, ArCH=CH2), 5.41 (d, J = 10.8 Hz, 1H, ArCH=CH2), 3.98 (s, 3H, -CO2CH3);13C NMR (100 MHz, CDCl3) δ 167.2, 137.3, 136.5, 135.8, 132.2, 130.7, 129.6, 128.2, 127.3, 126.0, 125.7, 124.0, 115.6, 52.2; HRMS-ESI calcd for C14H12O2 [M + H]+ 213.0910; found 213.0915.
Methyl 6-cyclopropyl-2-naphthoic acid (17).
Diiodomethane (0.91 mL, 11.32 mmol) was dissolved in anhydrous DCM (30 mL) and ZnEt2 (1.0 M solution in hexane, 5.66 mL, 5.66 mmol) added to this solution at 0 °C followed by a solution of 15 in DCM (600 mg, 2.83 mmol in 10 mL). The reaction mixture was then stirred vigorously overnight before being quenched with aqueous saturated NH4Cl. The mixture was diluted with diethyl ether (50 mL) and the organic layer isolated, washed with water (25 mL), brine (25 mL), dried over MgSO4 and concentrated in vacuo. The resultant residue was purified by flash chromatography (5% EtOAc in hexane) to afford the ester (16) as a viscous clear oil (465 mg, 73%) which was taken forward and hydrolysed without further characterisation. To a mixture of the 16 (465 mg, 2.05 mmol) in a THF/water mix (3:1, 40 mL) was added aqueous 1 M NaOH (8.2 mL, 8.2 mmol). The resultant mixture was stirred at 65 °C until TLC indicated complete hydrolysis of the ester. The mixture was then allowed to cool to room temperature before the THF was removed in vacuo. The resultant aqueous solution was topped up with water (40 mL), extracted with diethyl ether (25 mL) and then acidified to pH 1 using aqueous 2 M HCl. The solid which precipitated out of solution was filtered off, washed copiously with water and then dried over P2O5 overnight to yield a white solid. Recrystallisation from toluene afforded 17 as a white solid (179 mg, 41%); mp: 125–129°;1H NMR (400 MHz, DMSO-d6) δ 12.96 (br s, 1H, -CO2H), 8.52 (s, 1H, ArH), 7.98 (d, J = 8.4 Hz, 1H, ArH), 7.92 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.87 (d, J = 8.4 Hz, 1H, ArH), 7.68 (s, 1H, ArH), 7.31 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 2.16–2.08 (m, 1H, ArCH(CH2)2), 1.09–1.03 (m, 2H, ArCH(CH2)2), 0.87–0.81 (m, 2H, ArCH(CH2)2);13C NMR (100 MHz, DMSO-d6) δ 167.9, 144.8, 135.6, 130.9, 130.7, 129.7, 127.8, 127.4, 125.8, 125.6, 123.5, 16.0, 10.4; HRMS-ESI calcd for C14H12O2 [M - H]− 211.0765; found 211.0770; Anal. (C14H12O2) C, H.
2-Hydroxymethyl-6-n-pent-1-yl-naphthalene (18).
A flask was charged with 14a (4.50 g, 18.6 mmol) before being briefly evacuated and backfilled with argon. Anhydrous THF (100 mL) was cannulated into the flask and the resultant solution cooled to 0 °C. A 1 M LiAlH4 solution in THF (27.9 mL, 27.9 mmol) was then added dropwise with care. After complete addition the solution was allowed to warm to room temperature before being refluxed for 4 h. After being allowed to cool to room temperature the solution was further cooled to 0 °C and excess LiAlH4 destroyed via the dropwise addition of saturated aqueous NH4Cl. The THF was then removed in vacuo and the resultant residue partitioned between EtOAc and water (100 mL each). The organic layer was isolated, dried over MgSO4 and concentrated in vacuo to yield a cream coloured solid which was purified by flash chromatography (20 → 30% EtOAc in hexane) to afford 18 as a white solid (3.20 g, 75%); mp: 72–76°;1H NMR (400 MHz, CDCl3) δ 7.80–7.73 (m, 3H, ArH), 7.61 (s, 1H, ArH), 7.45 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 7.35 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 4.84 (s, 2H, -CH2OH), 2.77 (t, J = 7.6 Hz, 2H, ArCH2-), 1.77–1.67 (m, 2H, -CH2CH2CH2-), 1.39–1.32 (m, 4H, CH2CH2CH2-), 0.91 (t, J = 7.2 Hz, 3H, -CH3);13C NMR (100 MHz, CDCl3) δ 140.6, 137.4, 133.1, 131.8, 127.9, 127.8, 127.7, 126.1, 125.3, 125.2, 65.6, 36.1, 31.5, 31.0, 22.6, 14.0; HRMS-CI calcd for C16H20O [M]+ 228.1514; found 228.1516; Anal. (C16H20O) C, H.
2-Bromomethyl-6-n-pent-1-yl-naphthalene (19).
A flask containing 18 (3.00 g, 13.1 mmol) was briefly evacuated and backfilled with argon. Anhydrous DCM (100 mL) was then cannulated into the flask and the resultant solution cooled to 0 °C. PBr3 (4.92 mL, 52.4 mmol) was added dropwise and after complete addition the solution was allowed to warm to room temperature and stirred until TLC showed complete conversion. After 1 h the solution was cooled to 0 °C and excess PBr3 destroyed via the dropwise addition of saturated aqueous NaHCO3. The DCM was removed in vacuo to afford an oily residue which was diluted with diethyl ether (100 mL). The organic layer was isolated, dried over MgSO4 and concentrated in vacuo to yield an amber oil which solidified on standing. Dissolving the crude product in hexane and passing it through a short silica plug (10 cm) afforded 19 as an oil which partially solidified on standing to a light brown solid (2.98 g, 78%);1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1H, ArH), 7.76 (d, J = 8.4 Hz, 1H, ArH), 7.73 (d, J = 8.4 Hz, 1H, ArH), 7.59 (s, 1H, ArH), 7.47 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 7.35 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 4.67 (s, 2H, -CH2Br), 2.76 (t, J = 7.6 Hz, 2H, ArCH2-), 1.74–1.66 (m, 2H, -CH2CH2CH2-), 1.38–1.31 (m, 4H, -CH2CH2CH2-), 0.90 (t, J = 7.2 Hz, 3H, -CH3);13C NMR (100 MHz, CDCl3) δ 141.4, 134.2, 133.3, 131.6, 128.3, 128.1, 127.8, 127.6, 126.7, 126.2, 36.1, 34.3, 31.5, 31.0, 22.6, 14.0; HRMS-CI calcd for C16H19Br [M + H]+ 291.0748; found 291.0746.
(6-n-Pent-1-ylnapthalen-2-yl)acetonitrile (20).
19 (1.90 g, 6.5 mmol) was dissolved in anhydrous DCM (25 mL) and stirred vigorously with a solution of sodium cyanide (480 mg, 9.8 mmol) and tetra-n-butylammonium bromide (316 mg, 0.98 mmol) in water (25 mL). After 48 hours TLC indicated complete conversion. The organic layer was subsequently isolated and the aqueous phase extracted with DCM (2 × 20 mL). The organic layers were combined, dried over MgSO4 and concentrated in vacuo to afford a golden brown oil. Purification by flash chromatography (10% EtOAc in hexane) yielded 20 as a yellow oil (1.20 g, 78%);1H NMR (400 MHz, CDCl3) δ 7.81–7.77 (m, 2H, ArH), 7.75 (d, J = 8.4 Hz, 1H, ArH), 7.61 (s, 1H, ArH), 7.38 (dd, J = 8.4 & 2.0, 1H, ArH), 7.35 (dd, J = 8.0 & 2.0 Hz, 1H, ArH), 3.90 (s, 2H, -CH2CN), 2.77 (t, J = 7.6 Hz, 2H, ArCH2-), 1.71 (pent, J = 7.6 Hz, 2H, -CH2CH2CH2-), 1.40–1.30 (m, 4H, -CH2CH2CH2-), 0.90 (t, J = 7.2 Hz, 3H, -CH3);13C NMR (100 MHz, CDCl3) δ 141.3, 132.9, 131.8, 128.6, 128.4, 127.5, 126.6, 126.2, 126.2, 125.4, 118.0, 36.1, 31.5, 31.0, 23.8, 22.6, 14.0; HRMS-ESI cald for C17H19N [M + Na]+ 260.1410; found 260.1420.
(6-n-Pent-1-ylnaphthalen-2-yl)acetic acid (21).
A stirring mixture of 20 (1.17 g, 4.9 mmol) in glacial acetic acid (20 mL), conc H2SO4 (10 mL) and water (10 mL) was heated under reflux for 18 h. The mixture was then allowed to cool to room temperature before being diluted with water (100 mL). The aqueous mixture was extracted with EtOAc (100 mL then 2 × 50 mL) and the organic layers pooled, washed with water (3 × 100 mL), brine (100 mL), dried over Na2SO4 and concentrated in vacuo to afford a light brown solid. The crude product was dissolved in diethyl ether (100 mL) and the resultant organic solution extracted with aqueous 2 M NaOH (30 mL). The sodium salt which formed had poor solubility so additional water (approximately 150 mL) was added in order to get it fully into solution. The aqueous phase was isolated and the organic solution further extracted with aqueous 2 M NaOH (2 × 30 mL). The alkaline phases were combined and acidified to pH 1 using aqueous 2 M HCl. The aqueous solution was then extracted with diethyl ether (2 × 100 mL) and the organic layers pooled, washed with water (4 × 100 mL), brine (100 mL), dried over MgSO4 and concentrated in vacuo to afford 21 as a light brown solid (994 mg, 79%); mp: 120–124°;1H NMR (400 MHz, DMSO-d6) δ 12.36 (br s, 1H, -CH2CO2H), 7.79 (d, J = 6.0 Hz, 1H, ArH), 7.77 (d, J = 6.0 Hz, 1H, ArH), 7.71 (s, 1H, ArH), 7.65 (s, 1H, ArH), 7.38 (dd, J = 6.0 & 2.0 Hz, 1H, ArH), 7.36 (dd, J = 6.0 & 2.0 Hz, 1H, ArH), 3.71 (s, 2H, -CH2CO2H), 2.73 (d, J = 7.2 Hz, 2H, ArCH2-), 1.66 (pent, J = 7.2 Hz, 2H, -CH2CH2CH2-), 1.39–1.25 (m, 4H, -CH2CH2CH2-), 0.86 (t, J = 7.2 Hz, 3H, -CH3);13C NMR (100 MHz, DMSO-d6) δ 172.7, 139.6, 131.9, 131.7, 131.4, 127.8, 127.4, 127.3, 127.2, 127.1, 125.7, 40.7, 35.1, 30.8, 30.4, 21.9, 13.8; HRMS-ESI cald for C17H20O2 [M + Na]+ 279.1356; found 279.1360; Anal. (C17H20O2) C, H.
5-Bromo-6-n-pent-1-yl-2-naphthoic acid (22).
To a stirring solution of 14a (1.00 g, 4.1 mmol) in glacial acetic acid (65 mL) at 50 °C was added dropwise a solution of bromine (0.21 mL, 4.1 mmol) in glacial acetic acid (5 mL). The resultant mixture was stirred at 50 °C until TLC showed complete consumption of the starting material. The mixture was then allowed to cool to room temperature before being diluted with water (100 mL), causing precipitation of a solid. Excess bromine was destroyed via the dropwise addition of a saturated aqueous Na2SO3 solution. Filtration of the suspension afforded an off-white solid which was washed copiously with water and then dried over P2O5 overnight. Re-crystallisation from glacial acetic acid (three times) afforded 22 as a white solid (293 mg, 22%); mp: 193–196°;1H NMR (400 MHz, DMSO-d6) δ 13.18 (br s, 1H, -CO2H), 8.61 (d, J = 1.6 Hz, 1H, ArH), 8.27 (d, J = 8.8 Hz, 1H, ArH), 8.12–8.08 (m, 2H, ArH), 7.58 (d, J = 8.4 Hz, 1H, ArH), 2.95 (t, J = 7.2 Hz, 2H, ArCH2-), 1.65 (pent, J = 7.2 Hz, 2H, -CH2CH2CH2-), 1.40–1.29 (m, 4H, -CH2CH2CH2-), 0.88 (t, J = 7.2 Hz, 3H, -CH3);13C NMR (100 MHz, DMSO-d6) δ 167.5, 143.1, 134.1, 132.5, 131.2, 129.7, 129.6, 128.6, 127.4, 127.4, 122.8, 37.1, 31.5, 29.7, 22.4, 14.3; HRMS-ESI calcd for C16H17O2Br [M + Na]+ 343.0304; found 343.0312; Anal. (C16H17O2Br) C, H.
6-Butoxy-2-naphthoic acid (24).
Method adapted from the literature.30 To a stirring solution of 23 (1.00 g, 5.3 mmol) in an EtOH/water mix (3:1, 40 mL) was added 1-bromobutane (0.86 mL, 8.0 mmol). The resultant mixture was refluxed for 18 h and then allowed to cool to room temperature before a 10% aqueous NaOH solution (10 mL) was added. The mixture was then refluxed for a further 2 h before being allowed to cool to room temperature. After being diluted with water (150 mL) the solution was acidified to pH 1 using aqueous 2 M HCl. The solid which precipitated out of solution was filtered off, washed copiously with water and then dried over P2O5 overnight to afford 24 as a white solid (1.10 g, 85%); mp: 188–192° (lit30: 198°);1H NMR (400 MHz, DMSO-d6) δ 12.93 (br s, 1H, -CO2H), 8.51 (s, 1H, ArH), 8.00 (d, J = 8.8 Hz, 1H, ArH), 7.92 (dd, J = 8.8 & 2.0 Hz, 1H, ArH), 7.86 (d, J = 8.8 Hz, 1H, ArH), 7.39 (d, J = 2.4 Hz, 1H, ArH), 7.23 (dd, J = 8.8 & 2.4 Hz, 1H, ArH), 4.12 (t, J = 6.4 Hz, 2H, ArOCH2-), 1.81–1.73 (m, 2H, -CH2CH2CH3), 1.48 (sex, J = 7.2 Hz, 2H, -CH2CH2CH3), 0.96 (t, J = 7.2 Hz, 3H, -CH2CH2CH3);13C NMR (100 MHz, DMSO-d6) δ 167.5, 158.4, 136.7, 130.8, 130.3, 127.3, 126.8, 125.6, 125.5, 119.6, 106.5, 67.4, 30.6, 18.7, 13.6.
(E)-Methyl 6-(4-phenylbut-1-en-1-yl)-2-naphthoate (25a).
Method identical to that described for 11a. 10 (1.50 g, 5.7 mmol), palladium acetate (13 mg, 1 mol%), tri-o-tolylphosphine (70 mg, 4 mol%), 4-phenylbut-1-ene (1.06 mL, 7.1 mmol) and triethylamine (0.99 mL, 7.1 mmol) afforded 25a as a light brown solid (1.75 g, 98%) which was utilised immediately in the next step.
(E)-Methyl 6-(3-cyclopentylprop-1-en-1-yl)naphthalene-2-carboxylate (25b).
Method identical to that described for 11a. 10 (1.50 g, 5.7 mmol), palladium acetate (13 mg, 1 mol%), tri-o-tolylphosphine (70 mg, 4 mol%), allyl cyclopentane (0.99 mL, 7.1 mmol) and triethylamine (0.99 mL, 7.1 mmol) afforded 25b as a light brown solid (1.60 g, 96%) which was utilised immediately in the next step.
Methyl 6-(4-phenylbut-1-yl)-2-naphthoate (26a).
Method identical to that described for 13a. 25a (1.75 g, 5.5 mmol) and 10 wt % palladium on activated carbon (100 mg) yielded a viscous golden coloured oil which was purified by flash chromatography (5% EtOAc in hexane) to afford 26a as a viscous clear oil (1.29 g, 73%);1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H, ArH), 8.03 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.87 (d, J = 8.4 Hz, 1H, ArH), 7.80 (d, J = 8.4 Hz, 1H, ArH), 7.63 (s, 1H, ArH), 7.38 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 7.31–7.25 (m, 2H, ArH), 7.21–7.15 (m, 3H, ArH), 3.98 (s, 3H, -CO2CH3), 2.83 (t, J = 7.2 Hz, 2H, ArCH2-), 2.67 (m, 2H, -CH2CH2CH2Ph), 1.82–1.67 (m, 4H, -CH2CH2CH2Ph);13C NMR (100 MHz, CDCl3) δ 167.4, 142.9, 142.4, 135.8, 131.0, 130.8, 129.2, 128.4, 128.3, 128.2, 127.6, 126.6, 126.2, 125.7, 125.3, 52.1, 36.1, 35.8, 31.1, 30.7; HRMS-CI calcd for C22H22O2 [M + H]+ 319.1698; found 319.1705; Anal. (C22H22O2) C, H.
Methyl 6-(3-cyclopentylprop-1-yl)naphthalene-2-carboxylate (26b).
Method identical to that described for 13a. 25b (1.60 g, 5.4 mmol) and 10 wt % palladium on activated carbon (100 mg) yielded a golden coloured oil which was purified by flash chromatography (1 → 5% EtOAc in hexane) to afford 26b as a viscous clear oil (1.10 g, 68%);1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H, ArH), 8.03 (dd, J = 8.8 & 2.0 Hz, 1H, ArH), 7.87 (d, J = 8.4 Hz, 1H, ArH), 7.81 (d, J = 8.8 Hz, 1H, ArH), 7.44 (s, 1H, ArH), 7.40 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 3.98 (s, 3H, -CO2CH3), 2.79 (t, J = 7.6 Hz, 2H, ArCH2-), 1.84–1.68 (m, 5H, -CH2CH2cPe), 1.65–1.45 (m, 4H, CH2CH2cPe), 1.43–1.34 (m, 2H, CH2CH2cPe), 1.13–1.02 (m, 2H, CH2CH2cPe);13C NMR (100 MHz, CDCl3) δ 167.4, 143.4, 135.8, 130.9, 130.8, 129.2, 128.3, 127.6, 126.5, 126.2, 125.2, 52.1, 40.1, 36.5, 35.9, 32.7, 30.4, 25.2; HRMS-ESI calcd for C20H24O2 [M + Na]+ 319.1669; found 319.1674.
6-(4-Phenylbut-1-yl)naphthalene-2-carboxylic acid (27a).
Method identical to that described for 14a. 26a (1.29 g, 4.1 mmol) and aqueous 1 M NaOH (12.3 mL, 12.3 mmol) afforded 27a as a white solid (1.17 g, 95%); mp: 151–155°;1H NMR (400 MHz, DMSO-d6) δ 12.91 (br s, 1H, -CO2H), 8.53 (s, 1H, ArH), 8.00 (d, J = 8.4 Hz, 1H, ArH), 7.94 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.89 (d, J = 8.4 Hz, 1H, ArH), 7.74 (s, 1H, ArH), 7.44 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.28–7.22 (m, 2H, ArH), 7.19–7.12 (m, 3H, ArH), 2.79 (d, J = 7.2 Hz, 2H, ArCH2CH2-), 2.61 (d, J = 7.2 Hz, 2H, ArCH2CH2-), 1.73–1.56 (m, 4H, -CH2CH2Ph);13C NMR (100 MHz, DMSO-d6) δ 167.6, 142.4, 142.1, 135.0, 130.6, 130.0, 129.1, 128.2, 128.2, 128.1, 127.4, 125.9, 125.6, 125.3, 35.1, 34.9, 30.6, 30.2; HRMS-ESI calcd for C21H20O2 [M - H]− 303.1385; found 303.1393; Anal. (C21H20O2) C, H.
6-(3-Cyclopentylprop-1-yl)-2-naphthoic acid (27b).
Method identical to that described for 14a. 26b (1.10 g, 3.7 mmol) and aqueous 1 M NaOH (11.1 mL, 11.1 mmol) yielded a white solid which was recrystallised from acetonitrile to afford 27b as an off-white solid (476 mg, 45%); mp: 147–150°;1H NMR (400 MHz, DMSO-d6) δ 13.00 (br s, 1H, -CO2H), 8.53 (s, 1H, ArH), 8.01 (d, J = 8.4 Hz, 1H, ArH), 7.94 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.91 (d, J = 8.4 Hz, 1H, ArH), 7.75 (s, 1H, ArH), 7.46 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 2.75 (t, J = 7.6 Hz, 2H, ArCH2-), 1.81–1.61 (m, 5H, -CH2CH2cPe), 1.57–1.41 (m, 4H, CH2CH2cPe), 1.36–1.27 (m, 2H, CH2CH2cPe), 1.08–0.97 (m, 2H, CH2CH2cPe);13C NMR (100 MHz, DMSO-d6) δ 167.4, 142.8, 135.1, 130.5, 130.2, 129.1, 128.2, 127.5, 127.2, 125.9, 125.1, 39.4, 35.6, 35.2, 32.1, 29.8, 24.6; HRMS-ESI calcd for C19H22O2 [M - H]− 281.1547; found 281.1552; Anal. (C19H22O2) C, H.
Methyl 6-(4-methylpent-1-yn-1-yl)-2-naphthoate (28).
A stirring solution of 10 (1.32g, 5.0 mmol), PdCl2(Ph3)2 (211 mg, 0.3 mmol) and CuI (57 mg, 0.3 mmol) in anhydrous THF (15 mL) was briefly evacuated and backfilled with argon. An identical procedure was conducted on a solution of 4-methylpent-1-yne (0.71 mL, 6.0 mmol) and diethylamine (1.0 mL, 10 mmol) in anhydrous THF (5 mL). The alkyne solution was then added to the bromide solution and the resultant mixture stirred at 50° overnight. The reaction was then quenched with saturated NH4Cl (10 mL) and extracted with diethyl ether (2 × 40 mL). The organic extracts were combined, dried over MgSO4 and concentrated in vacuo. The resultant residue was purified by flash chromatography (10% EtOAc in hexane) to afford 28 as a clear oil (1.25 g, 94%);1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H, ArH), 8.01 (d, J = 8.2 Hz, 1H, ArH), 7.90 (s, 1H, ArH), 7.82 (d, J = 8.2 Hz, 1H, ArH), 7.76 (d, J = 8.2 Hz, 1H, ArH), 7.49 (d, J = 8.2 Hz, 1H, ArH), 3.95 (s, 3H, -CO2CH3), 2.34 (d, J = 6.4 Hz, 2H, -CH2CH(CH3)2), 1.99–1.89 (m, 1H, -CH2CH(CH3)2), 1.07 (d, J = 6.4 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 167.0, 135.1, 131.4, 130.7, 129.6, 129.1, 127.7, 125.8, 124.1, 91.5, 81.6, 52.2, 28.7, 28.2, 22.1; HRMS-ESI calcd for C18H19O2 [M + H]+ 267.1386; found 267.1380.
6-(4-Methylpent-1-yn-1-yl)-2-naphthoic acid (29).
To a stirring solution of 28 (1.25 g, 4.7 mmol) in dioxane (10 mL) was added LiOH (562 mg, 23.5 mmol) dissolved in a few mL of water. The resultant mixture was stirred at room temperature until TLC indicated complete hydrolysis. The dioxane was then removed in vacuo and the resultant residue dissolved in water (40 mL), extracted with diethyl ether (10 mL), and acidified to pH 1 using aqueous 2 M HCl. The solid which precipitated from solution was filtered off, washed copiously with water and then dried over P2O5 overnight to afford 29 as white solid (1.05 g, 88%); mp: 125–129°;1H NMR (300 MHz, DMSO-d6) δ 8.54 (s, 1H, ArH), 8.06–8.03 (m, 2H, ArH), 7.95–7.94 (m, 2H, ArH), 7.49 (dd, J = 7.6 & 1.5 Hz, 1H, ArH), 2.36 (d, J = 6.5 Hz, 2H, -CH2CH(CH3)2), 1.92–1.79 (m, 1H, -CH2CH(CH3)2), 1.01 (d, J = 6.6 Hz, 6H, -CH2CH(CH3)2);13C NMR (75 MHz, DMSO-d6) δ 167.8, 135.1, 131.7, 131.0, 130.8, 130.1, 129.6, 129.1, 128.4, 126.4, 123.5, 92.0, 82.1, 28.3, 28.2, 22.4; HRMS-ESI calcd for C17H15O2 [M-H]− 251.1074; found 251.1077; Anal. (C17H16O2) C, H.
(RS)-(E)-Ethyl 6-(4-ethylcarbonylpent-1-en-1-yl)-2-naphthoate (30).
Method identical to that described for 11a. 10 (3.00 g, 11.3 mmol), palladium acetate (25 mg, 1 mol%), tri-o-tolylphosphine (138 mg, 4 mol%), ethyl 2-methyl-4-pentenoate (2.28 mL, 14.1 mmol) and triethylamine (1.97 mL, 14.1 mmol) yielded a dark orange oil which was purified by flash chromatography (5 → 20% EtOAc in hexane) to afford 30 as a viscous light yellow oil (3.35 g, 91%);1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H, ArH), 8.03 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 7.86 (d, J = 8.4 Hz, 1H, ArH), 7.81 (d, J = 8.8 Hz, 1H, ArH), 7.70 (s, 1H, ArH), 7.61 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 6.60 (d, J = 15.6 Hz, 1H, -CH=CHCH2-), 6.35 (dt, J = 15.6 & 7.2Hz, 1H, -CH=CHCH2-), 4.15 (q, J = 7.2 Hz, 2H, -CH(CH3)CO2CH2CH3), 3.97 (s, 3H, -CO2CH3), 2.69–2.58 (m, 2H, -CH=CHCH2-), 2.47–2.37 (m, 1H, -CH(CH3)CO2CH2CH3), 1.28–1.22 (m, 6H, -CH(CH3)CO2CH2CH3);13C NMR (100 MHz, CDCl3) δ 175.9, 167.2, 137.2, 135.8, 131.8, 131.8, 130.7, 129.5, 129.3, 128.0, 127.0, 125.6, 125.3, 124.4, 60.4, 52.2, 39.6, 37.2, 16.8, 14.3; HRMS-ESI calcd for C20H22O4 [M + Na]+ 349.1410; found 349.1423.
(RS)-(E)-6-(4-Carboxypent-1-en-1-yl)-2-naphthoic acid (31).
Method identical to that described for 14b with the exception that the reaction mixture was stirred at room temperature until TLC indicated complete hydrolysis of the ester. 30 (1.41 g, 4.3 mmol) and NaOH (1.04 g, 25.9 mmol) afforded 31 as an off-white solid (1.19 g, 96%); mp: 226–230° (dec);1H NMR (400 MHz, DMSO-d6) δ 12.61 (br s, 2H, -CO2H), 8.53 (s, 1H, ArH), 8.03 (d, J = 8.4 Hz, 1H, ArH), 7.96–7.93 (m, 2H, ArH), 7.87 (s, 1H, ArH), 7.73 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 6.63 (d, J = 16.0 Hz, 1H, -CH=CHCH2-), 6.47 (dt, J = 16.0 & 6.8 Hz, 1H, -CH=CHCH2-), 2.59–2.51 (m, 2H, -CH=CHCH2-), 2.39–2.29 (m, 1H, -CH(CH3)CO2H), 1.13 (d, J = 6.8 Hz, 3H, -CH(CH3)CO2H);13C NMR (100 MHz, DMSO-d6) δ 176.7, 167.3, 136.8, 135.2, 131.3, 131.1, 130.1, 129.9, 129.5, 128.0, 127.6, 125.5, 124.9, 124.3, 38.7, 36.6, 16.5; HRMS-ESI calcd for C17H16O4 [M - H]− 283.0976; found 283.0982; Anal. (C17H16O4) C, H.
(RS)-Ethyl 6-(4-ethoxycarbonylpent-1-yl)-2-naphthoate (32).
Method identical to that described for 13a. 30 (1.91 g, 5.9 mmol) and 10 wt % palladium on activated carbon (100 mg) afforded 32 as a viscous light yellow oil (1.90 g, 99%);1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H, ArH), 8.03 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.87 (d, J = 8.4 Hz, 1H, ArH), 7.80 (d, J = 8.4 Hz, 1H, ArH), 7.64 (s, 1H, ArH), 7.38 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 4.12 (q, J = 7.2 Hz, 2H, -CH(CH3)CO2CH2CH3), 3.97 (s, 3H, -CO2CH3), 2.80 (t, J = 7.2 Hz, 2H, ArCH2-), 2.47 (sex, J = 7.2 Hz, 1H, -CH2CH2-), 1.81–1.68 (m, 3H, -CH2CH2-), 1.54–1.46 (m, 1H, -CH(CH3)CO2CH2CH3), 1.24 (t, J = 7.2 Hz, 3H, -CH(CH3)CO2CH2CH3), 1.15 (d, J = 7.2 Hz, 3H, -CH(CH3)CO2CH2CH3);13C NMR (100 MHz, CDCl3) δ 176.6, 167.3, 142.6, 135.8, 131.0, 130.8, 129.3, 128.1, 127.6, 126.7, 126.3, 125.3, 60.2, 52.2, 39.4, 36.0, 33.4, 28.8, 17.1, 14.3; HRMS-ESI calcd for C20H24O4 [M + Na]+ 351.1567; found 351.1558.
(RS)-6-(4-Carboxypent-1-yl)-2-naphthoic acid (33).
Method identical to that described for 14b with the exception that the reaction mixture was stirred at room temperature until TLC indicated complete hydrolysis of the ester. 32 (1.90 g, 5.8 mmol) and NaOH (1.39 g, 34.8 mmol) afforded 33 as a white solid (1.60 g, 95%); mp: 195–199° (dec);1H NMR (400 MHz, DMSO-d6) δ 12.51 (br s, 2H, CO2H), 8.55 (s, 1H, ArH), 8.02 (d, J = 8.4 Hz, 1H, ArH), 7.94 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.91 (d, J = 8.8 Hz, 1H, ArH), 7.76 (s, 1H, ArH), 7.46 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 2.77 (t, J = 6.8 Hz, 2H, ArCH2-), 2.36 (sex, J = 6.8 Hz, 1H, -CH2CH2-), 1.72–1.55 (m, 3H, -CH2CH2-), 1.44–1.34 (m, 1H, -CH(CH3)CO2H), 1.04 (d, J = 6.8 Hz, 3H, -CH(CH3)CO2H);13C NMR (100 MHz, DMSO-d6) δ 177.3, 167.4, 142.4, 135.1, 130.6, 130.2, 129.2, 128.1, 127.6, 127.2, 125.9, 125.2, 38.5, 35.2, 32.8, 28.3, 16.9; HRMS-ESI calcd for C17H18O4 [M - H]− 285.1132; found 285.1125; Anal. (C17H18O4) C, H.
6-(Isobutoxycarbonyl)-2-naphthoic acid (35).
A stirring mixture of 10 (2.65g, 10.0 mmol), 2-methylpropan-1-ol (20 mL) and concentrated H2SO4 (0.5 mL) was heated at 70° overnight. After cooling to room temperature, the reaction was diluted with diethyl ether (50 mL) and the solution washed with water (2 × 20 mL) and dried over MgSO4. Concentration in vacuo yielded a tacky solid which was re-crystallised from EtOAc to afford the crude bromo (34) as an off-white solid (1.24 g, 40%) which was taken forward without further purification or characterisation.
A flask was charged with 34 (920 mg, 3.0 mmol), Pd(OAc)2 (67 mg, 0.3 mmol) and DPPP (161 mg, 0.39 mmol). The flask was then evacuated and backfilled with carbon monoxide three times. A solution of triethylamine (2.09 mL, 15 mmol) and water (5.4 mL, 0.3 mol) in anhydrous DMF (20 mL) was then added and the resultant mixture stirred at 85° for 24 h. After cooling to room temperature the reaction was diluted with water (25 mL) and EtOAc (50 mL). The organic layer was isolated and washed with aqueous 1 M NaOH (3 × 15 mL). The aqueous extracts were combined and acidified to pH 2 with aqueous 2 M HCl. The solid which precipitated from solution was filtered off, washed copiously with water and then dried over P2O5 overnight to afford 35 as a white solid (678 mg, 83%); mp: 210–212°;1H NMR (400 MHz, DMSO-d6) δ 8.53 (br s, 2H), 8.01 (dd, J = 8.0 & 1.6 Hz, 1H), 8.00 (dd, J = 8.4 & 1.6 Hz, 1H), 7.96 (d, J = 8.8 Hz, 2H), 4.07 (d, J = 6.4 Hz, 2H, -CH2CH(CH3)2), 2.10–2.00 (m, 1H, -CH2CH(CH3)2), 0.98 (d, J = 6.8 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 167.8, 166.0, 134.5, 134.4, 130.6, 130.5, 130.3, 129.6, 129.6, 129.5, 126.3, 125.8, 71.2, 27.9, 19.3; MS (ESI) 273 (MH+), 295 (MNa+); Anal. (C16H16O4) C, H.
1-(6-Bromonaphthalen-2-yl)-4-methylpentan-1-one (36).
To a stirring solution of ethyl 4-methylvalerate (6.64 mL, 40.0 mmol) in anhydrous THF (20 mL) at −78° was added LiHMDS (1.0 M in THF, 40.0 mL, 40.0 mmol). The resultant mixture was stirred at −78° for 30 min before a solution of 10 (5.30g, 20.0 mmol) in anhydrous THF (100 mL) was added. After complete addition, the reaction was stirred at room temperature for 3 h. The THF was then removed in vacuo and hexane (50 mL) added to the remaining residue. The solid which precipitated from solution was filtered off, re-dissolved in aqueous 1 M NaOH (40 mL) and stirred at 50° overnight. After cooling to room temperature, the pH was adjusted to 1 using conc aqueous HCl and the mixture heated at 60° for 1 h. After cooling to room temperature, the reaction was extracted with diethyl ether (3 × 30 mL). The organic extracts were combined, washed with water (25 mL), brine (25 mL), dried over MgSO4 and concentrated in vacuo. The resultant residue was purified by flash chromatography (10% EtOAc in hexane) to afford 36 as a light brown oil (4.57 g, 75%);1H NMR (400 Mz, CDCl3) δ 8.39 (s, 1H, ArH), 8.03–8.00 (m, 2H, ArH), 7.79 (d, J = 8.8 Hz, 1H, ArH), 7.76 (d, J = 8.8 Hz, 1H, ArH), 7.58 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 2.36 (t, J = 7.6 Hz, 2H, ArCOCH2-), 1.54–1.39 (m, 3H, -CH2CH(CH3)2), 0.85 (d, J = 6.4 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 Mz, CDCl3) δ 211.7, 136.3, 134.7, 131.0, 131.0, 130.2, 129.9, 129.3, 127.4, 125.1, 122.6, 40.7, 32.7, 27.7, 22.3.
Methyl 6-(4-methylpentanoyl)-2-naphthoate (37).
A flask was charged with Pd(OAc)2 (225 mg, 1.0 mmol) and DPPP (536 mg, 1.3 mmol) before being briefly evacuated and backfilled with carbon monoxide three times. A degassed solution of 36 (3.05g, 10.0 mmol) and triethylamine (3.1 mL, 22.0 mmol) in a mixture of anhydrous MeOH (9 mL) and DMF (27 mL) was added and the resultant mixture heated at 90° for 18 h. After cooling to room temperature, the reaction mixture was extracted with EtOAc (3 × 30 mL). The organic extracts were combined, washed with aqueous 1 M HCl (25 mL), water (25 mL), brine (25 mL) dried over MgSO4 and concentrated in vacuo. The resultant residue was purified via flash chromatography (10% EtOAc in hexane) to afford 37 as a colourless oil (2.07 g, 73%);1H NMR (400 Mz, CDCl3) δ 8.62 (s, 1H, ArH), 8.48 (s, 1H, ArH), 8.12 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 8.07 (dd, J = 1.6 Hz, 1H, ArH), 8.02–7.99 (m, 2H, ArH), 3.99 (s, 3H, -CO2CH3), 3.10 (t, J = 7.6 Hz, 2H, ArCOCH2-), 1.71–1.67 (m, 3H, -CH2CH(CH3)2), 0.97 (d, J = 6.4 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 Mz, CDCl3) δ 200.4, 166.8, 136.2, 134.7, 134.5, 130.6, 129.8, 129.7, 129.5, 129.0, 126.0, 124.7, 52.4, 36.8, 30.9, 27.9, 22.5; MS (ESI) 285 (MH+), 307 (MNa+); HRMS (ESI) for C18H21O3 required 285.1485; found 285.1481.
6-(4-Methylpentanoyl)-2-naphthoic acid (38).
Method as that described for 29. 37 (1.50 g, 5.3 mmol) and LiOH (634 mg, 26.5 mmol) afforded 38 as an off-white solid (1.17 g, 82%); mp: 202–204°;1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H, ArH), 8.63 (s, 1H, ArH), 8.20 (d, J = 7.6 Hz, 1H, ArH), 8.19 (d, J = 8.4 Hz, 1H, ArH), 8.04 (d, J = 7.6 Hz, 1H, ArH), 8.02 (d, J = 8.4 Hz, 1H, ArH), 3.16 (t, J = 7.6 Hz, 2H, ArCOCH2-), 1.66–1.53 (m, 3H, -CH2CH(CH3)2), 0.92 (d, J = 6.4 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 200.7, 167.7, 136.1, 134.7, 134.6, 130.4, 130.4, 130.3, 130.2, 129.7, 126.5, 124.7, 36.6, 33.2, 27.7, 22.9; MS (ESI) 270 (M−); Anal. (C17H18O3) C, H.
Methyl 6-(4-hydroxypent-1-yn-1-yl)-2-naphthoate (39).
A flask was charged with 10 (1.33 g, 5.0 mmol), Pd(PPh3)4 (116 mg, 0.1 mmol) and CuBr (43 mg, 0.30 mmol). The flask was then briefly evacuated and backfilled with argon three times. A degassed solution of 4-pentyn-2-ol (0.57 mL, 6.0 mmol) in triethylamine (15 mL) was then added and the reaction heated at 65° overnight. The mixture was then concentrated in vacuo and the residue re-dissolved in diethyl ether (30 mL) and aqueous 1 M HCl (10 mL). The organic layer was isolated, washed with water (10 mL), brine (10 mL), dried over MgSO4 and concentrated in vacuo. The resultant residue was purified via flash chromatography (30% EtOAc in hexane) to afford 39 as colourless oil (1.12 g, 84%);1H NMR (400 Mz, CDCl3) δ 8.55 (s, 1H, ArH), 8.06 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 7.95 (s, 1H, ArH), 7.86 (d, J = 8.4 Hz, 1H, ArH), 7.80 (d, J = 8.4 Hz, 1H, ArH), 7.51 (dd, J = 8.4 & 2.0 Hz, 1H, ArH), 4.14–4.06 (m, 1H, -CH2CH(OH)CH3), 3.96 (s, 3H, -CO2CH3), 2.69 (dd, J = 16.8 & 5.6 Hz, 1H, -CH2CH(OH)CH3), 2.61 (dd, J = 16.9 & 6.4 Hz, 1H, -CH2CH(OH)CH3), 1.36 (d, J = 6.4 Hz, 3H, -CH2CH(OH)CH3);13C NMR (100Mz, CDCl3) δ 167.0, 135.0, 131.6, 131.0, 130.7, 129.4, 129.3, 127.8, 127.9, 125.9, 123.2, 88.1, 83.0, 66.6, 52.3, 30.1, 22.5.
Methyl 6-(4-oxopent-1-yl)-2-naphthoate (40).
A solution of 39 (1.34g, 5.0 mmol) in ethanol (100 mL) was hydrogenated under 3 bar pressure of hydrogen in the presence of 10 wt % palladium on carbon (100 mg) for 18 h. The reaction mixture was then filtered through a celite pad before being concentrated in vacuo to afford the crude alcohol which was taken up in anhydrous DCM (20 mL) and treated with Dess-Martin periodinane (2.12 g, 5 mmol). The resultant mixture was stirred at room temperature for 2 h and then extracted with diethyl ether (30 mL). The organic layer was isolated, washed with saturated aqueous NaHCO3 (10 mL), water (10 mL), brine (10 mL), dried over Na2SO4 and concentrated in vacuo. The resultant residue was purified via flash chromatography (10% EtOAc in hexane) to give 40 as a viscous oil (1.24 g, 92% over two steps);1H NMR (400 Mz, CDCl3) δ 8.53 (s, 1H, ArH), 8.00 (d, J = 8.4 Hz, 1H, ArH), 7.83 (d, J = 8.4 Hz, 1H, ArH), 7.76 (d, J = 8.4 Hz, 1H, ArH), 7.59 (s, 1H, ArH), 7.33 (d, J = 8.4 Hz, 1H, ArH), 3.93 (s, 3H, -CO2CH3), 2.75 (t, J = 7.6 Hz, 2H, ArCH2-), 2.43 (t, J = 7.6 Hz, 2H, -CH2CH2COCH3), 2.08 (s, 3H, -CH2CH2COCH3), 1.99–1.92 (m, 2H, -CH2CH2COCH3);13C NMR (100Mz, CDCl3) δ 208.4, 167.2, 142.0, 135.7, 131.0, 130.8, 129.4, 128.0, 127.6, 126.7, 126.4, 125.4, 52.1, 42.6, 35.2, 29.9, 24.8.
(RS)-Methyl 6-(6-methoxy-4-methyl-6-oxohex-1-yl)-2-naphthoate (41).
To a stirring solution of methyl diethylphosphonoacetate (841 mg, 4.0 mmol) in anhydrous THF (20 mL) at 0° was added dropwise KHMDS (0.5 M in toluene, 8.0 mL, 4.0 mmol). The resultant mixture was stirred at room temperature for 1 h before a solution of 40 (830 mg, 3.1 mmol) dissolved in anhydrous THF (5 mL) was added dropwise. The reaction was then stirred for 4 h at room temperature before being quenched by the addition of aqueous 1 M HCl (10 mL). The reaction was extracted with diethyl ether (2 × 20 mL) and the organic layers combined, washed with water (10 mL), brine (10 mL), dried over Na2SO4 and concentrated in vacuo to afford the crude alkene which was subsequently dissolved in ethanol (20 mL) and hydrogenated under 3 bar of hydrogen in the presence of 10 wt % palladium on carbon (50 mg) for 18 h. The reaction mixture was then filtered through a celite pad before being concentrated in vacuo to afford 41 as a colourless oil (763 mg, 75%);1H NMR (400 Mz, CDCl3) δ 8.56 (s, 1H, ArH), 8.03 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.85 (d, J = 8.4 Hz, 1H, ArH), 7.79 (d, J = 8.8 Hz, 1H, ArH), 7.62 (s, 1H, ArH), 7.37 (d, J = 8.4 Hz, 1H, ArH), 3.96 (s, 3H, -CO2CH3), 3.63 (s, 3H, -CO2CH3), 2.79–2.74 (m, 2H, -CH2CH2CH2CH-), 2.29 (dd, J = 14.8 & 6.4 Hz, 1H, -CH(CH3)CH2CO2CH3), 2.12 (dd, J = 14.8 & 7.6 Hz, 1H, -CH(CH3)CH2CO2CH3), 2.05–1.96 (m, 1H, -CH2CH2CH2CH-), 1.81–1.63 (m, 2H, -CH2CH2CH2CH-), 1.45–1.36 (m, 1H, -CH2CH2CH2-), 1.31–1.22 (m, 1H, -CH2CH2CH2CH-), 0.94 (d, J = 6.8 Hz, 3H, -CH(CH3)CH2CO2CH3);13C NMR (100Mz, CDCl3) δ 173.6, 167.3, 142.9, 135.7, 131.0, 130.8, 129.2, 128.1, 127.6, 126.6, 126.2, 125.3, 52.3, 51.1, 41.5, 36.2, 36.2, 30.2, 28.5, 19.7; HRMS-ESI calcd for C20H24O4 [M + H]+ 329.1675; found 329.1677.
(RS)-6-(5-Carboxy-4-methylpent-1-yl)-2-naphthoic acid (42).
Method as that described for 29. 41 (763 mg, 2.3 mmol) and LiOH (275 mg, 11.5 mmol) afforded 42 as a white solid (551 mg, 79%); mp: 146–148°;1H NMR (400 MHz, DMSO-d6) δ 8.51 (s, 1H, ArH), 7.98 (d, J = 8.4 Hz, 1H, ArH), 7.93 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.87 (d, J = 8.8 Hz, 1H, ArH), 7.74 (s, 1H, ArH), 7.46 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 2.73 (t, J = 7.2 Hz, 2H, ArCH2-), 2.19 (dd, J = 14.8 & 6.0 Hz, 1H, -CH(CH3)CH2CO2H), 1.99 (dd, J = 14.8 & 7.6 Hz, 1H, -CH(CH3)CH2CO2H), 1.90–1.82 (m, 1H, -CH2CH2CH-), 1.71–1.58 (m, 2H, -CH2CH2CH-), 1.39–1.29 (m, 1H, -CH2CH2CH-), 1.24–1.15 (m, 1H, -CH2CH2CH-), 0.87 (d, J = 6.8 Hz, 3H, -CH(CH3)CH2CO2H);13C NMR (100 MHz, DMSO-d6) δ 174.4, 168.1, 142.9, 135.5, 131.1, 130.53, 129.6, 128.6, 128.5, 127.9, 126.4, 125.8, 41.8, 36.2, 36.0, 30.0, 28.6, 20.0; MS (ESI) 300 (M+), 256; Anal. (C18H20O4) C, H.
Methyl 6-(5-methylhex-1-en-2-yl)-2-naphthoate (43).
KHMDS (0.5 M in toluene, 4.4 mL, 2.2 mmol) was added dropwise to a stirring −78° solution of methyl triphenylphosphonium bromide (643 mg, 1.8 mmol) in anhydrous THF (15 mL). The resultant mixture was stirred at room temperature for 1 h before a solution of 37 (427 mg, 1.5 mmol) dissolved in anhydrous THF (5 mL) was added dropwise. After complete addition, the reaction mixture was stirred for 2 h at room temperature before being quenched with aqueous 1 M HCl (2 mL). The reaction mixture was extracted with diethyl ether (3 × 20 mL) and the organic extracts combined, washed with water (25 mL), brine (25 mL), dried over Na2SO4 and concentrated in vacuo. The resultant residue was purified via flash chromatography (10% EtOAc in hexane) to afford 43 as a colourless oil (380 mg, 90%);1H NMR (400 Mz, CDCl3) δ 8.58 (s, 1H, ArH), 8.06 (d, J = 8.8 Hz, 1H, ArH), 7.90 (d, J = 8.4 Hz, 1H, ArH), 7.87 (d, J = 8.8 Hz, 1H, ArH), 7.86 (s, 1H, ArH), 7.64 (d, J = 8.4 Hz, 1H, ArH), 5.44 (s, 1H, =CH2), 5.22 (s, 1H, =CH2), 3.98 (s, 3H, -CO2CH3), 2.62 (t, J = 8.0 Hz, 2H, -CH2CH2CH(CH3)2), 1.67–1.59 (m, 1H, -CH2CH2CH(CH3)2), 1.43–1.37 (m, 2H, -CH2CH2CH(CH3)2), 0.93 (d, J = 6.8 Hz, 6H, -CH2CH2CH(CH3)2);13C NMR (100 Mz, CDCl3) δ 167.3, 148.5, 141.2, 135.6, 131.8, 130.6, 129.2, 128.3, 125.5, 125.5, 125.3, 124.4, 113.5, 52.2, 37.6, 33.1, 27.8, 22.5; HRMS-ESI calcd for C19H22O2 [M + H]+ required 283.1693; found 283.1683.
6-(5-Methylhex-1-en-2-yl)-2-naphthoic acid (44).
Method as that described for 29. 43 (185 mg, 0.66 mmol) and LiOH (79 mg, 3.30 mmol) afforded 44 as a white solid (147 mg, 83%); mp: 178–180°;1H NMR (400 MHz, DMSO-d6) δ 8.55 (s, 1H, ArH), 8.05 (d, J = 8.8 Hz, 1H, ArH), 8.01–7.99 (m, 2H, ArH), 7.95 (d, J = 8.8 Hz, 1H, ArH), 7.70 (d, J = 8.4 Hz, 1H, ArH), 5.49 (s, 1H, =CH2), 5.22 (s, 1H, =CH2), 2.60 (t, J = 8.0 Hz, 2H, -CH2CH2CH(CH3)2), 1.62–1.52 (m, 1H, -CH2CH2CH(CH3)2), 1.33–1.28 (m, 2H, -CH2CH2CH(CH3)2), 0.86 (d, J = 6.8 Hz, 6H, -CH2CH2CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 167.9, 148.2, 140.6, 135.5, 131.9, 130.5, 129.7, 128.9, 128.5, 125.9, 125.7, 124.6, 114.2, 37.6, 27.6, 22.8; MS (ESI) 268 (M−), 228; Anal. (C18H20O2) C, H.
(RS)-Methyl 6-(5-methylhexan-2-yl)-2-naphthoate (45).
A solution of 43 (175 mg, 0.62 mmol) in ethanol (50 mL) was hydrogenated under 3 bar of hydrogen in the presence of 10 wt % palladium on activated carbon (50 mg) for 18 h. The reaction mixture was then filtered through a celite pad before being concentrated in vacuo to afford 45 as a colourless oil (170 mg, 96%);1H NMR (400 Mz, CDCl3) δ 8.59 (s, 1H, ArH), 8.05 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.89 (d, J = 8.4 Hz, 1H, ArH), 7.84 (d, J = 8.4 Hz, 1H, ArH), 7.64 (s, 1H, ArH), 7.42 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 3.98 (s, 3H, -CO2CH3), 2.88–2.79 (m, 1H, -CHCH2CH2CH-), 1.75–1.60 (m, 2H, -CHCH2CH2CH-), 1.57–1.47 (m, 1H, -CHCH2CH2CH-), 1.33 (d, J = 6.8 Hz, 3H, ArCH(CH3)-), 1.24–1.15 (m, 1H, -CHCH2CH2CH-), 1.10–1.01 (m, 1H, -CHCH2CH2CH-), 0.85 (d, J = 6.8 Hz, 6H, -CH(CH3)2);13C NMR (100Mz, CDCl3) δ 167.4, 148.3, 135.8, 131.2, 130.8, 129.3, 127.2, 126.7, 126.6, 125.2, 125.0, 52.1, 40.5, 37.0, 35.9, 28.1, 22.6, 22.5, 22.2; HRMS-ESI calcd for C19H24O2 [M + H]+ 285.1776; found 285.1768.
(RS)-6-(5-Methylhexan-2-yl)-2-naphthoic acid (46).
Method as that described for 29. 45 (170 mg, 0.60 mmol) and LiOH (72 mg, 3.0 mmol) afforded 46 as a white solid (143 mg, 88%); mp: 151–153°;1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H, ArH), 8.01 (d, J = 8.4 Hz, 1H, ArH), 7.94–7.90 (m, 2H, ArH), 7.74 (s, 1H, ArH), 7.48 (d, J = 8.4 Hz, 1H, ArH), 2.85–2.79 (m, 1H, -CHCH2CH2CH-), 1.65–1.57 (m, 2H, -CHCH2CH2CH-), 1.49–1.42 (m, 1H, -CHCH2CH2CH-), 1.26 (d, J = 6.8 Hz, 3H, ArCH(CH3)-), 1.16–1.07 (m, 1H, -CHCH2CH2CH-), 0.99–0.90 (m, 1H, -CHCH2CH2CH-), 0.78 (d, J = 6.4 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 168.0, 148.2, 135.6, 131.3, 130.9, 129.8, 128.2, 127.7, 127.0, 125.6, 125.2, 37.0, 35.7, 28.0, 23.0, 22.9, 22.6; MS (ESI) 270 (M−); Anal. (C18H22O2) C, H.
Methyl 6-((Isobutylthio)methyl)-2-naphthoate (48).
To a solution of sodium (58 mg, 2.5 mmol) dissolved in anhydrous ethanol (20 mL) was added isobutyl mercaptan (0.76 mL, 7.0 mmol). The resultant mixture was stirred for 10 mins at room temperature before 47 (650 mg, 2.3 mmol) was added. After complete addition the reaction was stirred at room temperature for 30 mins before being diluted with diethyl ether (100 mL). The organic solution was washed with aqueous 1 M HCl (10 mL), water (20 mL), brine (10 mL) and dried over MgSO4. Concentration in vacuo afforded a viscous oil which was purified by flash chromatography (5% EtOAc in hexane) to give the 48 as a colourless oil (623 mg, 94%);1H NMR (300 MHz, CDCl3) δ 8.57 (s, 1H, ArH), 8.05 (dd, J = 8.4 & 1.5 Hz, 1H, ArH), 7.90 (d, J = 8.4 Hz, 1H, ArH), 7.81 (d, J = 8.4 Hz, 1H, ArH), 7.71 (s, 1H, ArH), 7.54 (d, J = 8.4 & 1.8 Hz, 1H, ArH), 3.97 (s, 3H, -CO2CH3), 3.84 (s, 2H, ArCH2-), 2.30 (d, J = 4.8 Hz, 2H, -CH2CH(CH3)2), 1.85–1.71 (m, 1H, -CH2CH(CH3)2), 0.94 (d, J = 6.6 Hz, 6H, -CH2CH(CH3)2);13C NMR (75 MHz, CDCl3) δ 167.3, 139.0, 135.5, 131.6, 130.9, 129.8, 128.1, 127.9, 127.3, 127.1, 125.7, 52.3, 40.6, 37.1, 28.3, 22.1.
6-((Isobutylthio)methyl)-2-naphthoic acid (49).
Method identical to that described for 29. 48 (550 mg, 1.9 mmol) and LiOH (227 mg, 9.5 mmol) afforded 49 as an off-white solid (454 mg, 87%); mp: 170–172°;1H NMR (400 MHz, DMSO-d6) δ 8.54 (s, 1H, ArH), 8.04 (d, J = 8.4 Hz, 1H, ArH), 7.97–7.90 (m, 2H, ArH), 7.83 (s, 1H, ArH), 7.55 (d, J = 8.4 Hz, 1H, ArH), 3.87 (s, 2H, ArCH2-), 2.27 (d, J = 6.8 Hz, 2H, -CH2CH(CH3)2), 1.75–1.65 (m, 1H, -CH2CH(CH3)2), 0.86 (d, J = 6.8 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 167.8, 139.6, 135.2, 131.5, 130.7, 129.9, 128.6, 128.3, 128.3, 127.2, 125.9, 40.2, 36.3, 28.1, 22.2; MS (ESI) 274 (M−), 230; Anal. (C16H18O2S) C, H, S.
Methyl 6-(isobutylcarbamoyl)-2-naphthoate (51).
50 (500 mg, 2.0 mmol) was added to a stirring mixture of isobutylamine (0.22 mL, 2.2 mmol) and triethylamine (0.84 mL, 6.0 mmol) in anhydrous DCM (20 mL) at 0°. After complete addition the reaction mixture was stirred at room temperature overnight. The reaction was then concentrated in vacuo and the resultant residue was taken up in EtOAc (20 mL) and washed sequentially with water (10 mL), aqueous 1 M HCl (5 mL), water (10 mL), aqueous 1 M NaOH (5 mL), water (10 mL), brine (10 mL) and dried over MgSO4. Concentration in vacuo afforded a tacky residue which was purified by flash chromatography (10% EtOAc in hexane) to afford 51 as a clear oil (540 mg, 94%);1H NMR (400 MHz, CDCl3) δ 8.60 (s, 1H, ArH), 8.28 (s, 1H, ArH), 8.08 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.98 (d, J = 8.8 Hz, 1H, ArH), 7.93 (d, J = 8.8 Hz, 1H, ArH), 1H, 7.86 (dd, J = 8.8 & 2.0 Hz, 1H, ArH), 3.98 (s, 3H, -CO2CH3), 3.34 (t, J = 6.0 Hz, 2H, -CH2CH(CH3)2), 1.99–1.89 (m, 1H, -CH2CH(CH3)2), 1.00 (d, J = 6.8 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 167.2, 166.9, 134.8, 134.4, 133.7, 130.7, 129.9, 129.1, 128.9, 126.8, 126.1, 124.4, 52.4, 47.5, 28.7, 20.2; HRMS-EI calcd for C17H19NO3 [M]+ 285.1367; found: 285.1365.
6-(Isobutylcarbamoyl)-2-naphthoic acid (52).
Method identical to that described for 29. 51 (540 mg, 1.9 mmol) and LiOH (227 mg, 9.5 mmol) afforded 52 as an off-white solid (439 mg, 85%); mp: >250°;1H NMR (400 MHz, DMSO-d6) δ 8.61 (s, 1H, ArH), 8.46 (s, 1H, ArH), 8.14 (d, J = 8.4 Hz, 1H, ArH), 8.09 (d, J = 8.8 Hz, 1H, ArH), 7.98 (dd, J = 8.8 & 2.0 Hz, 1H, ArH), 7.97 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 3.11 (t, J = 6.8 Hz, 2H, -CH2CH(CH3)2), 1.90–1.80 (m, 1H, -CH2CH(CH3)2), 0.88 (d, J = 6.8 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 167.7, 166.5, 134.7, 134.6, 133.6, 130.6, 129.8, 129.8, 129.6, 127.5, 126.2, 125.47, 47.3, 28.6, 20.7; MS (ESI) 270 (M−), 226; Anal. (C16H17NO3) C, H, N.
6-((Isobutyryloxy)methyl)-2-naphthoic acid (54).
To a stirring solution of 53 (405 mg, 2.0 mmol) and triethylamine (0.70 mL, 5.0 mmol) in anhydrous DCM (10 mL) at 0° was added isobutyryl chloride (0.52 mL, 5.0 mmol) dissolved in 3 mL of anhydrous DCM. After complete addition the reaction mixture was stirred at room temperature for 1 h. The DCM was then removed in vacuo and aqueous 2 M NaHCO3 (40 mL) added to the resultant residue. The mixture was stirred vigorously at 0° for 3 h and then extracted with diethyl ether (15 mL). The aqueous phase was isolated and then acidified with aqueous 1 M HCl. The solid which precipitated from solution was filtered off, washed copiously with water and then dried over P2O5 overnight to afford 54 as a white solid 457 mg, 84%); mp: 152–154°;1H NMR (400 MHz, CDCl3) δ 8.60 (s, 1H, ArH), 8.05 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.88 (d, J = 8.8 Hz, 1H, ArH), 7.80 (d, J = 8.8 Hz, 1H, ArH), 7.78 (s, 1H, ArH), 7.45 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 5.23 (s, 2H, ArCH2-), 2.13–2.06 (m, 1H -CH(CH3)2), 0.91 (d, J = 6.4 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 172.8, 171.1, 136.5, 135.6, 132.0, 131.6, 129.8, 128.2, 127.0, 126.7, 126.4, 125.8, 65.7, 25.7, 20.8; MS (ESI): 273 (MH+), 295 (MNa+); Anal. (C16H16O4) C, H.
2-Bromo-6-(isobutoxymethyl)naphthalene (56).
To a stirring suspension of 60% NaH (1.2 g, 30.0 mmol) in anhydrous DMF (150 mL) was added 2-methylpropan-1-ol (5.5 mL, 60.0 mmol). The mixture was stirred at room temperature for 4 h before 55 (3.0 g, 10.0 mmol) was added. After complete addition, the reaction was stirred at room temperature for 3 h before being quenched with water (50 mL) and extracted with hexane (100 mL). The organic layer was separated, dried over Na2SO4 and concentrated in vacuo. The resultant residue was purified by flash chromatography (2% EtOAc in hexane) to afford 56 as a colourless oil (2.7 g, 92%);1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 2.0 Hz, 1H, ArH), 7.75 (s, 1H, ArH), 7.73 (d, J = 8.8 Hz, 1H, ArH), 7.69 (d, J = 8.8 Hz, 1H, ArH), 7.54 (dd, J = 8.8 & 2.0 Hz, 1H, ArH), 7.49 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 4.65 (s, 2H, ArCH2-), 3.29 (d, J = 7.8 Hz, 2H, -CH2CH(CH3)2), 2.00–1.90 (m, 1H, -CH2CH(CH3)2), 0.97 (d, J = 6.4 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 137.0, 133.9, 131.7, 129.7, 129.5, 129.4, 127.1, 126.7, 126.0, 119.6, 72.8, 28.56, 22.7, 19.4.
6-(Isobutoxymethyl)-2-naphthoic acid (57).
A flask was charged with 56 (1.8 g, 6.14 mmol), Pd(OAc)2 (138 mg, 0.61 mmol) and DPPP (327 mg, 0.79 mmol). The flask was then briefly evacuated and backfilled with carbon monoxide three times. A solution of triethylamine (4.28 mL, 30.7 mmol) and water (11.0 mL, 0.61 mmol) in anhydrous DMF (30 mL) was added and the resultant mixture heated at 80° for 24 h. After cooling to room temperature the reaction was diluted with water (30 mL) and EtOAc (100 mL). The organic layer was isolated and washed with aqueous 1 M NaOH (3 × 20 mL). The aqueous extracts were combined and acidified to pH 2 with aqueous 2 M HCl. The solid which precipitated from solution was filtered off, washed copiously with water and then dried over P2O5 overnight to afford 57 as an off-white solid (1.14 g, 72%); mp: 160–162°;1H NMR (300 MHz, DMSO-d6) δ 8.55 (s, 1H, ArH), 8.05 (d, J = 8.4 Hz, 1H, ArH), 7.97–7.91 (m, 2H, ArH), 7.87 (s, 1H, ArH), 7.51 (dd, J = 8.4 & 1.5 Hz, 1H, ArH), 4.61 (s, 2H, ArCH2-), 3.21 (d, J = 6.6 Hz, 2H, -CH2CH(CH3)2), 1.91–1.76 (m, 1H, -CH2CH(CH3)2), 0.85 (d, J = 6.9 Hz, 6H, -CH2CH(CH3)2);13C NMR (75 MHz, DMSO-d6) δ 167.8, 139.6, 135.3, 132.0, 130.7, 129.8, 128.5, 128.4, 126.8, 125.9, 125.8, 77.0, 72.3, 28.5, 19.8; MS (ESI): 259 (HM+); Anal. (C16H18O3) C, H.
(RS)-Methyl 6-(2-(ethoxycarbonyl)-4-methylpent-1-yl)-2-naphthoate (59).
KHMDS (0.5 M in toluene, 12.0 mL, 6.0 mmol) was added dropwise to a stirred −78° solution of ethyl 4-methylvalerate (1.00 mL, 6.0 mmol) in anhydrous THF (10 mL). The mixture was stirred at −78° for 30 min before 55 (900 mg, 3.0 mmol) in THF (10 mL) was added. After complete addition, the mixture was allowed to warm to room temperature and stirred overnight. The reaction was then quenched with water (20 mL) and extracted with diethyl ether (2 × 30 mL). The organic extracts were combined, washed with aqueous 2 M HCl (10 mL), water (10 mL), brine (10 mL), dried over MgSO4 and concentrated in vacuo. The resultant residue was dissolved in ethyl acetate and passed through a short silica plug (10 cm). After concentration in vacuo the crude bromide (58) was dissolved in a mixture of anhydrous methanol (3 mL) and DMF (9 mL). Triethylamine (0.92 mL, 6.6 mmol) was added and the solution subsequently de-gassed. A flask containing Pd(OAc)2 (67 mg, 0.3 mmol) and DPPP (136 mg, 0.33 mmol) was briefly evacuated and backfilled with carbon monoxide three times. The de-gassed bromide solution was then added and the resultant mixture heated at 80° for 24 h. The reaction was then quenched with water (20 mL) and extracted with ethyl acetate (30 mL). The organic phase was washed with aqueous 2 M HCl (10 mL), water (10 mL), brine (10 mL), dried over MgSO4 and concentrated in vacuo. The resultant residue was purified by flash chromatography (10% EtOAc in hexane) to afford 59 as a colourless oil (690 mg, 67% over two steps);1H NMR (400 MHz, CDCl3) δ 8.56 (s, 1H, ArH), 8.03 (d, J = 8.6 Hz, 1H, ArH), 7.86 (d, J = 8.6 Hz, 1H, ArH), 7.80 (d, J = 8.6 Hz, 1H, ArH), 7.64 (s, 1H, ArH), 7.37 (d, J = 8.6 Hz, 1H, ArH), 4.01–3.98 (m, 2H, -CH(CO2CH2CH3)), 3.97 (s, 3H, -CO2CH3), 3.07 (dd, J = 13.5 & 8.8 Hz, 1H, ArCH2-), 2.98–2.80 (m, 2H, ArCH2CH-), 1.74–1.67 (m, 1H, -CH2CH(CH3)2), 1.65–1.55 (m, 1H, -CH2CH(CH3)2), 1.36–1.30 (m, 1H, -CH2CH(CH3)2), 1.05 (t, J = 7.1 Hz, 3H, -CH(CO2CH2CH3)), 0.91–0.89 (m, 6H, -CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 175.6, 167.3, 139.8, 135.6, 131.2, 130.8, 129.3, 128.2, 127.7, 127.1, 126.9, 125.3, 60.1, 52.2, 45.6, 41.6, 39.3, 26.1, 23.0, 22.0, 14.1; HRMS-ESI calcd for C21H26O4 [M + H]+ 243.1831; found 243.1832.
(RS)-6-(2-Carboxy-4-methylpent-1-yl)-2-naphthoic acid (60).
Method identical to that described for 29. 59 (650 mg, 1.90 mmol) and LiOH (227 mg, 9.50 mmol) afforded 60 as an off-white solid (405 mg, 71%); mp: 197–199°;1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H, ArH), 8.00 (d, J = 8.4 Hz, 1H, ArH), 7.93 (d, J = 8.4 Hz, 1H, ArH), 7.90 (d, J = 8.4 Hz, 1H, ArH), 7.75 (s, 1H, ArH), 7.45 (d, J = 8.4 Hz, 1H, ArH), 2.95 (dd, J = 13.6 & 9.2 Hz, 1H, ArCH2-), 2.87 (dd, J = 13.6 & 5.6 Hz, 1H, ArCH2-), 2.76–2.68 (m, 1H, -CHCH2CH-), 1.58–1.53 (m, 2H, -CHCH2CH-), 1.28–1.21 (m, 1H, -CHCH2CH-), 0.84 (d, J = 6.4 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 176.8, 167.9, 140.4, 135.4, 131.3, 130.7, 129.6, 128.8, 128.1, 128.0, 127.3, 125.7, 45.5, 41.7, 39.1, 26.2, 23.5, 22.3; MS (ESI) 300 (M−); Anal. (C18H20O4) C, H.
Methyl 7-bromo-3-hydroxy-2-naphthoate (62).
61 (500 mg, 1.87 mmol) was added to a suspension of K2CO3 (129 mg, 0.94 mmol) in anhydrous DMF (18 mL). The reaction was stirred for 1 h, and then methyl iodide (174 μL, 2.81 mmol) was added. The reaction was stirred for 12 h at room temperature, and then concentrated under reduced pressure. The solid was taken up in a 1:1 mixture of ethyl acetate and water (50 ml each). The aqueous layer was removed and the organic layer was washed with water (2 × 10 mL) and then brine (10 mL). After drying over MgSO4, the solvent was removed under reduced pressure, and the product obtained by flash chromatography (0 → 25% EtOAc in hexane) to give 62 as white crystals (525 mg, 99%); mp: 153–155°;1H NMR (500 MHz, CDCl3) δ 10.46 (s, 1H, -OH), 8.39 (d, J = 0.7 Hz, 1H, ArH), 7.95 (dd, J = 1.4 & 0.7 Hz, 1H, ArH), 7.57–7.54 (m, 2H, ArH), 7.29 (s, 1H, ArH), 4.03 (s, 3H, -CO2CH3);13C (125 MHz, CDCl3) δ 170.0, 156.7, 136.2, 132.3, 131.3, 130.9, 128.0, 127.9, 117.4, 115.0, 111.9, 52.7; HRMS-CI calcd for C12H10O3Br [M + H]+ 279.9727; found 279.9735.
Methyl 3-acetoxy-7-bromo-2-naphthoate (63).
To a solution of 62 (184 mg, 0.65 mmol) in CHCl3 (6.5 mL), was added acetic anhydride (100 μL, 0.98 mmol), followed by pyridine (80 μL, 0.98 mmol), and then a catalytic amount of DMAP. The reaction was stirred for 12 h, and then diluted with DCM (50 mL). The organic layer was washed with water (2 × 10 mL), and then brine (10 mL). After drying over MgSO4, the solvent was removed under reduced pressure, to give 63 as an amber oil (168 mg, 80%) which was used without further purification;1H NMR (500 MHz, CDCl3) δ 8.49 (s, 1H, ArH), 8.08 (d, J = 1.4 Hz, 1H, ArH), 7.68 (d, J = 8.8 Hz, 1H, ArH), 7.65 (dd, J = 8.8 & 1.4 Hz, 1H, ArH), 7.50 (s, 1H, ArH), 3.93 (s, 3H, -CO2CH3), 2.40 (s, 3H, -OAc);13C (125 MHz, CDCl3) δ 170.1, 164.6, 147.0, 133.1, 132.6, 132.2, 131.6, 130.9, 128.8, 122.9, 121.2, 120.5, 52.4, 21.0; HRMS-CI calcd for C14H11O4Br [M + H]+ 321.9841; found 321.9838.
Methyl 3-acetoxy-7-(4-methylpent-1-yl)-2-naphthoate (64).
A flask was charged with 63 (7.95g, 24.6 mmol), palladium acetate (55 mg, 1 mol%), and tri-o-tolylphosphine (30 mg, 4 mol%). The flask was then briefly evacuated and backfilled with argon three times. A degassed mixture of 4-methylpent-1-ene (4.67 mL, 36.9 mmol) and triethylamine (8.57 mL, 61.5 mmol) in anhydrous DMF (80 mL) was added and the resultant mixture heated at 100° overnight. After cooling to room temperature the mixture was diluted with diethyl ether (200 mL). The organic layer was isolated, washed with water (100 mL), aqueous 2M HCl (20 mL), brine (20 mL), dried over Na2SO4 and concentrated in vacuo to afford the crude alkene which was taken forward to the next step without further characterisation or purification.
A solution of the crude alkene in ethanol (40 mL) was hydrogenated under 3 bar of hydrogen in the presence of 10 wt % palladium on activated carbon (100 mg) for 18 h. The reaction mixture was then filtered through a celite pad before being concentrated in vacuo. The resultant residue was purified by flash chromatography (10% ethyl acetate in hexane) to afford 64 as a colourless oil (7.0 g, 87% over two steps);1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H, ArH), 7.72 (d, J = 8.4 Hz, 1H, ArH), 7.68 (s, 1H, ArH), 7.48 (s, 1H, ArH), 7.43 (d, J = 8.4 Hz, 1H, ArH), 3.92 (s, 3H, -CO2CH3), 2.74 (t, J = 6.6 Hz, 2H, ArCH2-), 2.39 (s, 3H, -OAc), 1.72–1.64 (m, 2H, -CH2CH2CH-), 1.60–1.54 (m, 1H, -CH2CH2CH-), 1.29–1.20 (m, 2H, -CH2CH2CH-), 0.87 (d, J = 6.8 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 170.5, 165.2, 146.3, 141.4, 134.2, 133.4, 131.0, 130.7, 127.5, 127.2, 121.6, 120.9, 52.3, 38.6, 36.3, 29.1, 28.0, 22.7, 21.2; HRMS-ESI calcd for C20H24O4 [M + Na]+ 351.1572; found 351.1567.
3-Hydroxy-7-(4-methylpent-1-yl)-2-naphthoic acid (65).
Method identical to that described for 29. 64 (3.50 g, 10.7 mmol) and LiOH (1.28 g, 53.5 mmol) afforded 65 as a yellow solid (2.62 g, 90%); mp: 150–152°;1H NMR (300 MHz, DMSO-d6) δ 8.41 (s, 1H, ArH), 7.68 (br s, 1H, ArH), 7.65 (d, J = 8.4 Hz, 1H, ArH), 7.36 (dd, J = 8.4 & 1.5 Hz, 1H, ArH), 7.23 (s, 1H, ArH), 2.61 (t, J = 7.5 Hz, 2H, ArCH2-), 1.63–1.44 (m, 3H, -CH2CH2CH-), 1.18–1.11 (m, 2H, -CH2CH2CH-), 0.80 (d, J = 0.6 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 167.9, 135.2, 131.8, 131.0, 130.8, 130.1, 129.7, 129.2, 128.4, 126.4, 123.5, 92.0, 82.1, 28.3, 28.1, 22.4; HRMS-ESI calcd for C17H20O3 [M-H]− 271.1340; found 271.1335. Anal. (C17H20O3) C, H.
Methyl 3-hydroxy-7-(4-methylpent-1-yl)-2-naphthoate (66).
To a stirring solution of 64 (3.28g, 10.0 mmol) in methanol (50 mL) at 0° was added dimethylamine (2.0 M in methanol, 10.0 mL, 20.0 mmol). After 1 h the reaction mixture was diluted with diethyl ether (100 mL) and aqueous 1 M HCl (30 mL). The organic phase was separated and washed with water (25 mL), brine (25 mL), dried over Na2SO4 and concentrated in vacuo to afford 66 (1.45 g, 51%) as a tacky solid which was utilised without further purification;1H NMR (400 MHz, CDCl3) δ 10.36 (s, 1H, -OH), 8.43 (s, 1H, ArH), 7.61 (d, J = 8.8 Hz, 1H, ArH), 7.56 (s, 1H, ArH), 7.27 (d, J = 8.8 Hz, 1H, ArH), 7.26 (s, 1H, ArH), 4.02 (s, 3H, -CO2CH3), 2.70 (t, J = 7.6 Hz, 2H, ArCH2-), 1.72–1.64 (m, 2H, -CH2CH2CH-), 1.62–1.54 (m, 1H, -CH2CH2CH-), 1.28–1.23 (m, 2H, -CH2CH2CH-), 0.89 (d, J = 7.6 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 170.4, 155.8, 138.4, 136.4, 131.8, 131.0, 127.3, 127.2, 126.2, 114.0, 111.4, 52.5, 38.6, 36.0, 29.0, 27.9, 22.6.
Methyl 7-(4-methylpent-1-yl)-3-(((trifluoromethyl)sulfonyl)oxy)-2-naphthoate (67).
To a stirring solution of 66 (1.45 g, 5.1 mmol) and triethylamine (1.42 mL, 10.2 mmol) in anhydrous DCM (10 mL) at −5° was added Tf2O (1.1 mL, 6.5 mmol). The resultant mixture was stirred at −5° for 1.5 h and then quenched by the addition of aqueous 2 M HCl (5 mL). The organic phase was separated, dried over Na2SO4 and concentrated in vacuo. The resultant residue was dissolved in ethyl acetate and passed through a short silica plug (10 cm) to give 67 (1.92g, 90%) which was utilised without further purification or characterisation.
Methyl 7-(4-methylpent-1-yl)-2-naphthoate (68).
A flask was charged with 67 (418 mg, 1.0 mmol) and Pd(PPh3)4 (12 mg, 1 mol%) before being evacuated and backfilled with argon three times. A degassed mixture of formic acid (0.11 mL, 3.0 mmol) and triethylamine (0.42 mL, 3.0 mmol) in anhydrous DMF (10 mL) was then added and the resultant mixture heated at 80° overnight. After cooling to room temperature, the reaction was diluted with water (40 mL) and extracted with diethyl ether (3 × 20 mL). The organic extracts were combined, washed with aqueous 1 M HCl (20 mL), water (20 mL), brine (25 mL), and dried over Na2SO4. Concentration in vacuo yielded a viscous oil which was purified by flash chromatography (5% ethyl acetate in hexane) to afford 68 as a colourless oil (232 mg, 86%);1H NMR (400 MHz, CDCl3) δ 8.55 (s, 1H, ArH), 7.99 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.83 (d, J = 8.8 Hz, 1H, ArH), 7.79 (d, J = 8.4 Hz, 1H, ArH), 7.71 (s, 1H, ArH), 7.44 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 3.97 (s, 3H, -CO2CH3), 2.76 (t, J = 7.6 Hz, 2H, ArCH2-), 1.75–1.66 (m, 2H, -CH2CH2CH-), 1.64–1.54 (m, 1H, -CH2CH2CH-), 1.29–1.23 (m, 2H, -CH2CH2CH-), 0.88 (d, J = 6.8 Hz, 6H, -CH(CH3)2);13C NMR (125 MHz, CDCl3) δ 167.4, 141.4, 134.0, 132.7, 130.6, 129.9, 127.8, 127.7, 127.6, 127.3, 124.39, 52.2, 38.6, 36.2, 29.1, 27.9, 22.6; HRMS-ESI calcd for C18H22O2 [M+] 270.1620; found 270.1618.
7-(4-Methylpent-1-yl)-2-naphthoic acid (69).
Method identical to that described for 29. 68 (232 mg, 0.86 mmol) and LiOH (103 mg g, 4.3 mmol) afforded 69 as a white solid (203 mg, 92%); mp: 140–142°;1H NMR (400 MHz, DMSO-d6) δ 8.48 (s, 1H, ArH), 7.94–7.85 (m, 4H, ArH), 7.50 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 2.72 (t, J = 8.0 Hz, 2H, ArCH2-), 1.64–1.61 (m, 2H, -CH2CH2CH-), 1.58–1.50 (m, 1H, -CH2CH2CH-), 1.22–1.17 (m, 2H, -CH2CH2CH-), 0.83 (d, J = 6.4 Hz, 6H, -CH(CH3)2);13C NMR (125 MHz, DMSO-d6) δ 167.9, 141.5, 133.9, 132.8, 130.4, 130.1, 128.7, 128.2, 128.0, 127.8, 124.8, 38.4, 35.9, 29.0, 27.8, 22.9; MS (ESI): 257 (MH+), 279 (MNa+); Anal. (C17H20O2) C, H.
2-(Methoxymethoxy)-7-(4-methylpent-1-yn-1-yl)naphthalene (71).
A flask was charged with CuI (248 mg, 1.3 mmol) and Pd(PPh3)2Cl2 (913 mg, 1.3 mmol) before being briefly evacuated and backfilled with argon three times. A degassed solution of 70 (10.4 g, 30.9 mmol), 4-methylpent-1-yne (4.36 mL, 37.1 mmol) and diethylamine (6.39 mL, 61.8 mmol) in anhydrous THF (100 mL) was added and the resultant mixture heated at 45° for 18 h. The reaction was then extracted with diethyl ether (3 × 75 mL) and the organic layer isolated and washed with saturated aqueous NH4Cl (40 mL), water (50 mL) and brine (50 mL). After drying over Na2SO4, concentration in vacuo yielded a viscous oil which was purified by flash chromatography (5% EtOAc in hexane) to afford 71 as a colourless oil (7.97 g, 96%);1H NMR (300 MHz, CDCl3) δ 7.81 (s, 1H, ArH), 7.70 (d, J = 9.3 Hz, 1H, ArH), 7.66 (d, J = 8.7 Hz, 1H, ArH), 7.34 (dd, J = 9.3 & 1.2 Hz, 1H, ArH), 7.32 (s, 1H, ArH), 7.18 (dd, J = 8.7 & 2.4 Hz, 1H, ArH), 5.28 (s, 2H, ArOCH2OCH3), 3.51 (s, 3H, ArOCH2OCH3), 2.34 (d, J = 8.8 Hz, 2H, -CH2CH(CH3)2), 1.94 (m, 1H, -CH2CH(CH3)2), 1.07 (d, J = 8.8 Hz, 6H, -CH2CH(CH3)2);13C NMR (75 MHz, CDCl3) δ 155.5, 134.2, 130.3, 129.3, 128.6, 127.6, 127.1, 122.1, 119.3, 109.7, 94.6, 89.9, 82.0, 56.2, 28.8, 28.4, 22.2.
Methyl 3-(methoxymethoxy)-6-(4-methylpent-1-yl)-2-naphthoate (72).
A solution of 71 (7.97 g, 29.7 mml) in ethanol (100 mL) was hydrogenated under 3 bar pressure of hydrogen in the presence of 10 wt % palladium on carbon (200 mg) for 18 h. The reaction mixture was then filtered through a celite pad before being concentrated in vacuo. The resultant residue was purified by flash chromatography (5% EtOAc in hexane) to afford the saturated alkyl as a viscous colourless oil which was taken forward without further characterisation.
t-Butyl lithium (1.9 M in pentene, 20.3 mL, 38.6 mmol) was added dropwise to a stirring 0° solution of the hydrogenation product (8.08 g, 29.7 mmol) in a mixture of diethyl ether and hexane (1:1, 200 mL). After complete addition, the mixture was stirred at 0° for 1 h before being cooled to −78°. Methyl chloroformate (3.44 mL, 44.6 mmol) was added dropwise and the resultant mixture stirred at room temperature for 1 hour before being quenched with methanol (10 mL). The organic mixture was washed with water (50 mL), brine (50 mL), dried over Na2SO4 and concentrated in vacuo. The resultant residue was purified by flash chromatography (DCM/hexane from 3:5 to 4:5 to 6:5) to afford 72 as a colourless oil (5.49 g, 56% over two steps);1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H, ArH), 7.73 (d, J = 8.4 Hz, 1H, ArH), 7.52 (s, 1H, ArH), 7.44 (s, 1H, ArH), 7.24 (d, J = 8.4 Hz, 1H, ArH), 5.36 (s, 2H, ArOCH2OCH3), 3.95 (s, 3H, -CO2CH3), 3.57 (s, 3H, ArOCH2OCH3), 2.73 (t, J = 7.6 Hz, 2H, ArCH2-), 1.73–1.65 (m, 2H, -CH2CH2CH-), 1.62–1.53 (m, 1H, -CH2CH2CH-), 1.28–1.19 (m, 2H, -CH2CH2CH-), 0.89 (d, J = 6.8 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 166.7, 153.2, 143.4, 136.2, 132.4, 128.5, 126.7, 126.6, 125.3, 121.3, 111.2, 95.2, 56.3, 52.1, 38.6, 36.5, 29.0, 27.9, 22.6; HRMS-EI calcd for C20H26O4 [M+] 330.1834; found 330.1831.
Methyl 3-hydroxy-6-(4-methylpent-1-yl)-2-naphthoate (73).
To a stirring solution of 72 (5.28 g, 16.0 mmol) in methanol (40 mL) was added aqueous 2 M HCl (5 mL). The mixture was stirred at room temperature for 4 h before being extracted with diethyl ether (2 × 50 mL). The organic extracts were combined, washed with saturated aqueous NaHCO3 (6 ml), water (20 mL), brine (20 mL), dried over Na2SO4 and concentrated in vacuo. The resultant residue was purified via flash chromatography (20% EtOAc in hexane) to give 73 as a viscous oil (4.48 g, 98%);1H NMR (400 MHz, CDCl3) δ 10.45 (s, 1H, -OH), 8.42 (s, 1H, ArH), 7.70 (d, J = 8.4 Hz, 1H, ArH), 7.44 (s, 1H, ArH), 7.24 (s, 1H, ArH), 7.18 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 4.00 (s, 3H, -CO2CH3), 2.71 (t, J = 8.0 Hz, 2H, ArCH2-), 1.74–1.66 (m, 2H, -CH2CH2CH-), 1.64–1.55 (m, 1H, -CH2CH2CH-), 1.30–1.25 (m, 2H, -CH2CH2CH-), 0.91 (d, J = 6.8 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 170.4, 156.5, 144.3, 138.3, 132.1, 129.1, 125.7, 124.6, 113.3, 111.1, 52.4, 38.7, 36.6, 28.9, 27.9, 22.6; HRMS-EI for C18H22O3 [M+] 286.1566; found 286.1569.
3-Hydroxy-6-(4-methylpent-1-yl)-2-naphthoic acid (74).
Method as that described for 29. 73 (3.50 g, 12.2 mmol) and LiOH (1.46 g, 61.0 mmol) afforded 74 as a pale yellow solid (2.36 g, 71%); mp: 168–170°;1H NMR (400 MHz, DMSO-d6) δ 8.46 (s, 1H, ArH), 7.85 (d, J = 8.4 Hz, 1H, ArH), 7.51 (s, 1H, ArH), 7.21 (s, 1H, ArH), 7.19 (d, J = 8.4 Hz, 1H, ArH), 2.65 (t, J = 7.6 Hz, 2H, ArCH2-), 1.65–1.47 (m, 3H, -CH2CH2CH-), 1.20–1.14 (m, 2H, -CH2CH2CH-), 0.82 (d, J = 6.4 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, DMSO-d6) δ 172.1, 156.7, 144.1, 138.0, 132.7, 129.6, 125.9, 125.7, 124.6, 114.7, 110.7, 38.5, 36.2, 28.8, 27.8, 22.9; MS (ESI) 271 (M−), 227; Anal. (C17H20O3) C, H.
(E)-(6-(4-Methylpent-1-en-1-yl)naphthalene-2,3-dicarbonitrile (76).
A flask was charged with 75 (1.00 g, 3.89 mmol), palladium acetate (8.7 mg, 1 mol%) and tri-o-tolylphosphine (47.4 mg, 4 mol%). The flask was then briefly evacuated and backfilled with argon three times. Degassed anhydrous DMF (20 mL) was added followed by 4-methylpent-1-ene (0.62 mL, 4.86 mmol) and triethylamine (0.68 mL, 4.86 mmol). The resultant mixture was heated at 100 °C overnight. After being allowed to cool to room temperature the reaction mixture was filtered through a celite pad to remove any precipitated Pd(0) and then poured into a stirred solution of EtOAc (100 mL), water (100 mL) and aqueous 1 M HCl (10 mL). The organic layer was subsequently isolated and the aqueous phase further extracted with EtOAc (2 × 50 mL). The organic extracts were pooled, washed with water (2 × 100 mL), brine (100 mL) and dried over MgSO4. Concentration in vacuo afforded a clay coloured solid which was purified by flash chromatography (10% EtOAc in hexane) to afford 76 as a light yellow oil (773 mg, 77%);1H NMR (400 MHz, CDCl3) δ 8.29–8.25 (m, 2H, ArH), 7.90–7.86 (m, 2H, ArH), 7.77 (s, 1H, ArH), 6.59–6.47 (m, 2H, -CH=CH-), 2.20 (t, J = 6.4 Hz, 2H, -CH2CH(CH3)2), 1.81 (sep, J = 6.4 Hz, 1H, -CH2CH(CH3)2), 0.98 (d, J = 6.4 Hz, 6H, -CH2CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 140.5, 135.5, 135.5, 135.0, 133.8, 132.2, 129.4, 128.7, 128.6, 125.3, 116.1, 116.0, 110.4, 109.1, 42.6, 28.5, 22.4; HRMS-ESI calcd for C18H16N2 [M + Na]+ 283.1206; found 283.1214.
6-(4-Methylpent-1-yl)naphthalene-2,3-dicarboxylic acid (77).
To a stirring suspension of 76 (400 mg, 1.54 mmol) in a 3:1 glacial acetic acid/water mix (40 mL) was added concentrated sulphuric acid (5 mL). The resultant mixture was heated at reflux until TLC indicated complete hydrolysis. After 24 h the now solution was allowed to cool to room temperature before being poured into a stirring mix of EtOAc and water (75 mL each). The organic layer was isolated and the aqueous phase further extracted with EtOAc (2 × 25 mL). The organics were pooled, washed with water (2 × 25 mL), brine (2 × 25 mL), dried over MgSO4 and concentrated in vacuo to afford an orange residue. The crude product was then suspended in water (50 mL) and basified using aqueous 1 M NaOH. The resultant aqueous solution was extracted with diethyl ether (25 mL) and then acidified with aqueous 1 M HCl before being extracted with EtOAc (3 × 30 mL). The organic extracts were combined, washed with water (25 mL), brine (25 mL), dried over MgSO4 and concentrated in to afford the di-acid as a viscous orange oil which used without further purification or characterisation. A solution of the crude alkene (427 mg, 1.43 mmol) in EtOAc (100 mL) was hydrogenated under 3 bar of hydrogen in the presence of 10 wt % palladium on activated carbon (50 mg) for 18 h. The reaction mixture was then filtered through a celite pad before being concentrated in vacuo to afford 77 as a glassy yellow solid (275 mg, 60%); mp: >250°;1H NMR (400 MHz, CDCl3) δ 7.98 (s, 1H, ArH), 7.93–7.89 (m, 2H, ArH), 7.79 (s, 1H, ArH), 7.52 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 2.80 (t, J = 7.6 Hz, 2H, ArCH2-), 1.73 (quin, J = 7.6 Hz, 2H, -CH2CH2CH(CH3)2), 1.58 (sep, J = 6.8 Hz, 1H, -CH2CH2CH(CH3)2), 1.26–1.19 (m, 2H, -CH2CH2CH(CH3)2), 0.85 (d, J = 6.8 Hz, 6H, -CH(CH3)2);13C NMR (100 MHz, CDCl3) δ 167.8, 167.7, 141.0, 133.9, 132.5, 131.1, 129.1, 129.1, 128.3, 127.1, 126.9, 38.9, 35.9, 28.3, 27.8, 22.8; HRMS-ESI calcd for C18H20O4 [M - H]− 299.1289; found 299.1283; Anal. (C18H20O4) C, H.
Methyl (E)-3-Acetoxy-7-styryl-2-naphthoate (78a).
Method identical to that described for 11a. 63 (1.00 g, 3.1 mmol), palladium acetate (7 mg, 1 mol%), tri-o-tolylphosphine (38 mg, 4 mol%), styrene (0.45 mL, 3.9 mmol), triethylamine (0.54 mL, 3.9 mmol) and anhydrous DMF (25 mL) yielded an orange/yellow solid which was re-crystallized from toluene to afford 78a (843 mg, 79 %) as an off-white solid; mp: 195–197 °C;1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 7.93 (s, 1H), 7.86 (dd, J = 8.8 & 2.0 Hz, 1H, ArH), 7.79 (d, J = 9.0 Hz, 1H, ArH), 7.59–7.55 (m, 2H, ArH), 7.51 (s, 1H, ArH), 7.42–7.37 (m, 2H, ArH & -CH=CH-), 7.33–7.27 (m, 1H, -CH=CH-), 7.25 (s, 2H, ArH), 3.94 (s, 3H, -CO2CH3), 2.41 (s, 3H, -OAc);13C NMR (125 MHz, CDCl3) δ 170.3, 165.0, 146.8, 137.0, 135.7, 135.2, 133.7, 131.0, 130.0, 128.8, 128.0, 127.9, 127.6, 127.3, 126.7, 126.6, 122.2, 121.0, 52.3, 21.1; MS (ESI+) m/z 369 (M + Na, 100 %); HRMS-ESI calcd for C22H18O4 [M + Na]+ 369.1097; found 369.1107; Anal (C22H18O4.0.3C7H8) C, H.
(E)-3-Hydroxy-7-styryl-2-naphthoic acid (79a).
To a stirring suspension of 78a (250 mg, 0.72 mmol) in a dioxane/water mix (2:1, 60 mL) was added aqueous 1 M NaOH (2.88 mL, 2.88 mL). The resultant mixture was stirred at room temperature until TLC indicated complete de-protection. After 18 h, the dioxane was removed in vacuo causing precipitation of an orange solid. The aqueous mixture was topped up with water and acidified to pH 1 using aqueous 2 M HCl. The precipitate was filtered off, washed copiously with water and then dried over P2O5 overnight to afford 79a as an orange solid (199 mg, 95 %); mp: >250 °C;1H NMR (400 MHz, DMSO-d6) δ 8.33 (s, 1H, ArH), 7.92 (s, 1H, ArH), 7.78 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.67–7.60 (m, 3H, ArH), 7.41–7.33 (m, 3H, ArH & -CH=CH-), 7.31–7.23 (m, 2H, ArH & -CH=CH-), 7.05 (s, 1H, ArH);13C NMR (100 MHz, DMSO-d6) δ 170.8, 159.3, 137.3, 136.0, 131.0, 130.8, 128.6, 128.6, 127.7, 127.2, 126.9, 126.2, 126.2, 125.9, 124.6, 121.5, 109.3; MS (ESI−) m/z 289 (M - H, 100 %), 245 (25); HRMS-ESI calcd for C19H14O3 [M - H]+ 289.0870; found 289.0874; Anal. (C19H14O3.0.75H2O) C, H.
(E)-7-(2-carboxystyryl)-3-hydroxy-2-naphthoic acid (79b).
Method identical to that described for 11a. 63 (600 mg, 1.86 mmol), palladium acetate (4.2 mg, 1 mol%), tri-o-tolylphosphine (22.6 mg, 4 mol%), methyl 2-vinylbenzoate (378 mg, 2.33 mmol), triethylamine (0.33 mL, 2.33 mmol) and anhydrous DMF (25 mL) yielded a viscous orange oil. Purification by flash chromatography (2 % EtOAc in hexane) afforded the protected styryl intermediate 78b as a white solid (752 mg) which was taken forward without further characterisation.
To a stirring suspension of 78b (752 mg, 1.86 mmol), in a THF/water mix (3:1, 80 mL) was added aqueous 1 M NaOH (11.16 mL, 11.16 mmol). The resultant mixture was heated at reflux until TLC indicated complete de-protection. After 4 h the reaction was allowed to cool to room temperature and the THF removed in vacuo. The resultant aqueous solution was topped up with water and acidified to pH 1 using aqueous 2 M HCl. The solid which precipitated from solution was filtered off, washed copiously with water and dried over P2O5 overnight to afford 79b (187 mg, 30%) as a yellow solid; mp: >250 °C;1H NMR (DMSO-d6, 500 MHz) δ 8.52 (s, 1H, ArH), 8.05 (s, 1H, ArH), 7.99 (d, J = 16.5 Hz, 1H, -CH=CH-), 7.87 (m, 2H, ArH), 7.81 (m, 2H, ArH), 7.60 (m, 1H, ArH), 7.40 (m, 1H, ArH), 7.33 (s, 1H, ArH), 7.28 (d, J = 16.5 Hz, 1H, -CH=CH-);13C NMR (DMSO-d6, 125 MHz) δ 171.4, 168.6, 156.5, 137.9, 136.9, 132.8, 132.5, 131.9, 130.4, 130.3, 129.7, 128.1, 127.4, 127.0, 126.8, 126.6, 126.6, 126.5, 115.9, 111.1; MS (ESI−) m/z 333 (M – H, 100 %); HRMS-ESI calcd for C20H13O5, 333.0768; found, 333.0766; Anal. (C20H14O5.0.89H2O) C, H.
(E)-7-(3-carboxystyryl)-3-hydroxy-2-naphthoic acid (79c).
Method identical to that described for 11a. 63 (500 mg, 1.55 mmol), palladium acetate (3.5 mg, 1 mol%), tri-o-tolylphosphine (18.9 mg, 4 mol%), methyl 3-vinylbenzoate (315 mg, 1.94 mmol) [32] (Erdelyi et al, 2008), triethylamine (0.27 mL, 1.94 mmol) and anhydrous DMF (25 mL) yielded the crude protected styryl intermediate 78c (627 mg) as a viscous orange oil which was taken forward without further purification or characterization.
De-protection was carried out in an identical fashion to 79b. 78c (627 mg, 1.55 mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (9.30 mL, 9.30 mmol) afforded 79c as a yellow solid (278 mg, 53 %); mp: >250 °C;1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1H, ArH), 8.14 (s, 1H, ArH), 8.06 (s, 1H, ArH), 7.94–7.72 (m, 4H, ArH & -CH=CH-), 7.49 (t, J = 7.6 Hz, 1H, ArH), 7.41 (s, 2H, ArH), 7.28 (s, 1H, ArH);13C NMR (100 MHz, DMSO-d6) δ 171.4, 167.1, 156.4, 137.5, 136.9, 132.5, 132.4, 131.3, 130.4, 129.2, 129.1, 128.3, 128.1, 127.4, 127.2, 126.8, 126.7, 126.5, 115.7, 111.1; MS (ESI−) m/z 333 (M - H, 100 %); HRMS-ESI calcd for C20H13O5, 333.0768; found, 333.0772; Anal. (C20H14O5.H2O) C, H.
Methyl (E)-3-Acetoxy-7-(4-(methoxycarbonyl)styryl)-2-naphthoate (78d).
Method identical to that described for 11a. 63 (1.00 g, 3.10 mmol), palladium acetate (7 mg, 1 mol%), tri-o-tolylphosphine (38 mg, 4 mol%), methyl 4-vinylbenzoate (629 mg, 3.88 mmol), triethylamine (0.54 mL, 3.88 mmol) and anhydrous DMF (25 mL) yielded a yielded a yellow solid which was re-crystallized from toluene to afford 78d (891 mg, 71 %) as a pale yellow solid; mp: 185–189 °C;1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H, ArH), 8.06 (d, J = 8.4 Hz, 2H, ArH), 7.95 (s, 1H, ArH), 7.86 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.81 (d, J = 8.8 Hz, 1H, ArH), 7.62 (d, J = 8.4 Hz, 2H, ArH), 7.52 (s, 1H, ArH), 7.36, (d, J = 16.4 Hz, 1H, -CH=CH-), 7.26 (d, J = 16.4 Hz, 1H, -CH=CH-), 3.94 (s, 3H, -CO2CH3), 3.94 (s, 3H, -CO2CH3), 2.41 (s, 3H, -OAc);13C NMR (100 MHz, CDCl3) δ 170.2, 166.8, 164.9, 147.1, 141.4, 135.4, 135.1, 133.8, 130.9, 130.4, 130.1, 129.2, 128.8, 127.9, 127.7, 126.5, 126.4, 122.4, 121.1, 52.4, 52.1, 21.0; MS (ESI+) m/z 427 (M + Na, 100 %); HRMS-ESI calcd for C24H20O6 [M + Na]+ 427.1152; found 427.1143; Anal. (C24H20O6) C, H.
(E)-7-(4-Carboxystyryl)-3-hydroxy-2-naphthoic acid (79d).
Method identical to that described for 79b. 78d (250 mg, 0.62 mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (3.72 mL, 3.72 mmol) afforded 79d (141 mg, 68%) as a yellow/orange solid; mp: >250 °C;1H NMR (400 MHz, DMSO-d6) δ 12.91 (br s, 1H, -CO2H), 11.25 (br s, 1H), 8.52 (s, 1H, ArH), 8.10 (s, 1H, ArH), 7.98–7.92 (m, 3H, ArH), 7.80 (d, J = 8.4 Hz, 1H, ArH), 7.74 (d, J = 8.4 Hz, 2H, ArH), 7.51 (d, J = 16.4 Hz, 1H, -CH=CH-), 7.42 (d, J = 16.4 Hz, 1H, -CH=CH-), 7.34 (s, 1H, ArH);13C NMR (100 MHz, DMSO-d6) δ 171.3, 167.0, 156.5, 141.4, 136.9, 132.4, 132.2, 130.6, 129.7, 129.3, 128.3, 127.2, 126.7, 126.5, 126.5, 126.3, 115.8, 111.0; MS (ESI−) m/z 333 (M - H, 96 %), 289 (75), 245 (100); HRMS-ESI calcd for C20H14O5 [M - H]+ 333.0768; found 333.0767; Anal. (C20H14O5.0.4H2O) C, H.
(E)-3-hydroxy-7-(2-methoxystyryl)-2-naphthoic acid (79e).
Method identical to that described for 11a. 63 (1.00 g, 3.10 mmol), palladium acetate (7 mg, 1 mol%), tri-o-tolylphosphine (38 mg, 4 mol%), 2-vinylanisole (521 mg, 3.88 mmol), triethylamine (0.54 mL, 3.88 mmol) and anhydrous DMF (25 mL) yielded the crude protected styryl intermediate 78e (1.17 g) as a viscous yellow oil which was taken forward without further purification or purification.
De-protection was carried out in an identical fashion to 79b. 78e (1.17 g, 3.10mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (12.4 mL, 12.4 mmol) afforded 79e (100 mg, 10 %) as a yellow solid; mp: >235 °C (dec);1H NMR (DMSO-d6, 400 MHz) δ 8.52 (s, 1H, ArH), 8.03 (br.s, 1H, ArH), 7.86 (dd, J = 9.0 & 1.5 Hz, 1H, ArH), 7.77 (d, J = 9.0 Hz, 1H, ArH), 7.69 (dd, J = 8.0 & 1.5 Hz, 1H, ArH), 7.51 (d, J = 16.5 Hz, 1H, -CH=CH-), 7.34 (d, J = 16.5 Hz, 1H, -CH=CH-), 7.32 (br s, 1H, ArH), 7.28 (m, 1H, ArH), 7.05 (d, J = 8.0 Hz, 1H, ArH), 6.99 (pt, J = 7.0 Hz, 1H, ArH), 3.88 (s, 3H, -OMe);13C NMR (100 MHz, DMSO-d6) δ 171.5, 156.5, 156.3, 136.7, 133.1, 132.4, 128.9, 128.6, 127.5, 126.9, 126.7, 126.5, 126.3, 125.5, 122.9, 120.7, 115.7, 111.5, 111.0, 55.5; MS (ESI−) m/z 319 (100 %); Anal. (C20H16O4) C, H.
(E)-3-Hydroxy-7-(3-methoxystyryl)-2-naphthoic acid (79f).
Method identical to that described for 11a. 63 (500 mg, 1.55 mmol), palladium acetate (3.5 mg, 1 mol%), tri-o-tolylphosphine (19 mg, 4 mol%), 3-vinylanisole (260 mg, 1.94 mmol), triethylamine (0.27 mL, 1.94 mmol) and anhydrous DMF (25 mL) yielded the crude protected styryl intermediate 78f (583 mg) as a viscous orange oil which was taken forward without further purification or characterisation.
De-protection was carried out in an identical fashion to 79b. 78f (583 g, 1.55 mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (6.20 mL, 6.20 mmol) afforded 79f as a yellow solid (391 mg, 79%); mp: > 250 °C;1H NMR (400 MHz, DMSO-d6) δ 8.30 (br s, 1H, ArH), 7.89 (br s, 1H, ArH), 7.74 (dd, J = 8.8 & 1.6 Hz, 1H, ArH), 7.61 (d, J = 8.8 Hz, 1H, ArH), 7.36 (d, J = 16.4 Hz, 1H, -CH=CH-), 7.29 (m, 1H, ArH), 7.23 (d, J = 16.4 Hz, 1H, -CH=CH-), 7.19 (m, 2H, ArH), 6.99 (br s, 1H, ArH), 6.83 (m, 1H, ArH), 3.81 (s, 3H, -OMe);13C NMR (100 MHz, DMSO-d6) δ 170.8, 160.1, 159.6, 138.9, 136.0, 130.7, 130.6, 129.7, 129.2, 127.9, 126.7, 126.2, 125.9, 124.3, 122.9, 118.9, 113.2, 111.2, 109.1, 55.1; MS (ESI−) m/z 319 (M – Na, 100 %); Anal. (C20H15NaO4) C, H.
(E)-3-hydroxy-7-(4-methoxystyryl)-2-naphthoate (79g).
Method identical to that described for 11a. 63 (500 mg, 1.55 mmol), palladium acetate (3.5 mg, 1 mol%), tri-o-tolylphosphine (19 mg, 4 mol%), 4-vinylanisole (260 mg, 1.94 mmol), triethylamine (0.27 mL, 1.94 mmol) and anhydrous DMF (25 mL) yielded the crude protected styryl intermediate 78g (583 mg) as a viscous yellow/orange oil which was taken forward without further purification or characterisation.
De-protection was carried out in an identical fashion to 79b. 78g (583 g, 1.55 mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (6.20 mL, 6.20 mmol) afforded 79g as a yellow solid (140 mg, 28%); mp: > 250 °C;1H NMR (400 MHz, DMSO-d6,) δ 8.43 (s, 1H, ArH), 7.94 (s, 1H, ArH), 7.83 (d, J = 8.5 Hz, 1H, ArH), 7.71 (d, J = 8.5 Hz, 1H, ArH), 7.56 (d, J = 8.5 Hz, 2H, ArH), 7.27 (d, J = 16.0 Hz, 1H, -CH=CH-), 7.21 (s, 1H, ArH), 7.19 (d, J = 16.0 Hz, 1H, -CH=CH-), 6.96 (d, J = 8.5 Hz, 2H, ArH), 3.78 (s, 3H, -OMe);13C NMR (100 MHz, DMSO-d6) δ 171.3, 158.9, 157.4, 136.3, 132.4, 131.7, 129.8, 127.7, 127.5, 127.1, 126.7, 126.2, 126.1, 125.9, 114.2, 110.4, 55.1; MS (ESI−) m/z 319 (100 %); Anal. (C20H15NaO4) C, H.
(E)-3-Hydroxy-7-(2-nitrostyryl)-2-naphthoic acid (79h).
Method identical to that described for 11a. 63 (737 mg, 2.28 mmol), palladium acetate (5 mg, 1 mol%), tri-o-tolylphosphine (28 mg, 4 mol%), 2-nitrostyrene (425 mg, 2.85 mmol), triethylamine (0.40 mL, 2.85 mmol) and anhydrous DMF (25 mL) yielded the crude protected styryl intermediate 78h (892 mg) as a viscous dark orange oil which was taken forward without further purification or characterisation.
De-protection was carried out in an identical fashion to 79b. 78h (892 mg, 2.28 mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (9.12 mL, 9.12 mmol) afforded 79h as a yellow solid (343 mg, 45%); mp: > 250 °C;1H NMR (500 MHz, DMSO-d6) δ 8.55 (br s, 1H, ArH), 8.13 (br.s, 1H, ArH), 8.01 (m, 2H, ArH), 7.88 (dd, J = 9.0 & 1.5 Hz, 1H, ArH), 7.81 (d, J = 9.0 Hz, 1H, ArH), 7.76 (m, 1H, ArH), 7.56 (m, 2H, ArH & -CH=CH-), 7.44 (d, J = 16.5 Hz, 1H, -CH=CH-), 7.34 (br s, 1H, ArH);13C NMR (125 MHz, DMSO-d6) δ 171.4, 156.7, 147.9, 137.1, 133.5, 133.1, 132.7, 132.0, 131.8, 128.8, 128.5, 128.0, 126.7, 126.7, 126.7, 124.5, 122.6, 116.0, 111.1; MS (ESI−) m/z 334 (M – H, 100 %); HRMS-ESI calcd for C19H12NO5, 414.0948; found, 414.0941; Anal. (C19H13NO5.0.1H2O) C, H, N.
(E)-3-Hydroxy-7-(3-nitrostyryl)-2-naphthoic acid (79i).
Method identical to that described for 11a. 63 (1.00 g, 3.10 mmol), palladium acetate (7 mg, 1 mol%), tri-o-tolylphosphine (38 mg, 4 mol%), 3-nitrostyrene (521 mg, 3.88 mmol), triethylamine (0.54 mL, 3.88 mmol) and anhydrous DMF (25 mL) yielded the crude protected styryl intermediate 78i (1.21 g) as a viscous yellow oil which was taken forward without further purification or purification.
De-protection was carried out in an identical fashion to 79b. 78i (1.21 g, 3.10 mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (12.4 mL, 12.4 mmol) afforded 79i as a brown solid (120 mg, 12 %); mp: > 250 °C;1H NMR (400 MHz, DMSO-d6) δ 8.44 (pt, J = 1.5 Hz, 1H, ArH), 8.34 (s, 1H, ArH), 8.09 (dd, J = 8.0 & 2.0 Hz, 2H, ArH), 7.98 (br s, 1H, ArH), 7.80 (dd, J = 9.0 & 1.5 Hz, 1H, ArH), 7.67 (m, 2H, ArH), 7.58 (d, J = 16.5 Hz, 1H, -CH=CH-), 7.44 (d, J = 16.5 Hz, 1H, -CH=CH-), 7.04 (br s, 1H, ArH);13C NMR (100 MHz, DMSO-d6) δ 170.8, 160.0, 148.4, 139.5, 136.4, 132.3, 131.8, 131.0, 130.4, 130.2, 128.7, 126.2, 126.1, 124.7, 124.5, 122.2, 121.6, 120.5, 109.4; MS (ESI−) m/z 334 (100 %); Anal. (C19H13NO5.0.38H2O) C, H, N.
(E)-3-Hydroxy-7-(4-nitrostyryl)-2-naphthoic acid (79j).
Method identical to that described for 11a. 63 (500 mg, 1.55 mmol), palladium acetate (3.5 mg, 1 mol%), tri-o-tolylphosphine (19 mg, 4 mol%), 4-nitrostyrene (289 mg, 1.94 mmol), triethylamine (0.27 mL, 1.94 mmol) and anhydrous DMF (25 mL) yielded the crude protected styryl intermediate 78j (607 mg) as a viscous yellow/orange oil which was taken forward without further purification or characterisation.
De-protection was carried out in an identical fashion to 79b. 78j (607 mg, 1.55 mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (6.20 mL, 6.20 mmol) afforded 79j as a yellow solid (154 mg, 30%); mp: >250 °C;1H NMR (500 MHz, DMSO-d6) δ 8.53 (s, 1H, ArH), 8.25 (d, J = 8.5 Hz, 2H, ArH), 8.13 (br s, 1H, ArH), 7.96 (d, J = 8.5 Hz, 1H, ArH), 7.89 (d, J = 8.5 Hz, 2H, ArH), 7.82 (d, J = 8.5 Hz, 1H, ArH), 7.64 (d, J = 16.0 Hz, 1H, -CH=CH-), 7.51 (d, J = 16.0 Hz, 1H, -CH=CH-), 7.35 (s, 1H, ArH);13C NMR (125 MHz, DMSO-d6) δ 171.4, 156.8, 146.1, 144.1, 137.2, 133.0, 132.7, 132.0, 129.1, 127.2, 126.7, 126.6, 126.2, 124.1, 115.9, 111.2; MS (ESI−) m/z 334 (M – H, 100 %); HRMS-ESI calcd for C19H12NO5, 334.0721; found, 334.0727; Anal. (C19H13NO5) C, H, N.
3-Hydroxy-7-phenethyl-2-naphthoic acid (81a).
78a (450 mg, 1.30 mmol) was dissolved in dioxane (100 mL) and the resulting solution hydrogenated under 3 bar of hydrogen in the presence of 10 wt % palladium on activated carbon (50 mg) for 18 h. The reaction mixture was then filtered through a Celite pad before being concentrated in vacuo to yield a clear oil which solidified on standing. Purification by flash chromatography (10 → 20% EtOAc in hexane) afforded the protected phenethyl intermediate 80a (300 mg, 66%) as a white solid which was utilised in the next step without further characterisation.
De-protection was carried out in an identical fashion to 79b. 80a (300 mg, 0.86 mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (3.44 mL, 3.44 mmol) afforded 81a (238 mg, 95%) as a pale yellow/green solid; mp: >250 °C;1H NMR (400 MHz, DMSO-d6) δ 8.37 (s, 1H, ArH), 7.70 (s, 1H, ArH), 7.65 (d, J = 8.4 Hz, 1H, ArH), 7.41 (dd, J = 8.4 & 1.6 Hz, 1H, ArH), 7.30–7.22 (m, 4H, ArH), 7.20–7.14 (m, 2H, ArH), 3.03–2.91 (m, 4H, -CH2CH2-);13C NMR (100 MHz, DMSO-d6) δ 171.3, 156.4, 141.4, 136.2, 135.5, 131.2, 129.9, 128.3, 128.1, 127.2, 126.5, 125.7, 125.6, 117.2, 110.0, 36.8, 36.7; MS (ESI−) m/z 291 (M - H, 100 %); HRMS-ESI calcd for C19H16O3 [M - H]+ 291.1027; found 291.1038; Anal. (C19H16O3.0.1H2O) C, H.
3-Hydroxy-7-(2-carboxyphenethyl)-2-naphthoic acid (81b).
Method identical to that described for 80a with the exception that EtOAc (100 mL) was used instead of dioxane.78b (336 mg, 0.83 mmol) and 10 wt % palladium on activated carbon (50 mg) yielded the protected phenethyl intermediate 80b as a tacky white solid (337 mg) which was taken forward without further purification or characterization.
De-protection was carried out in an identical fashion to 79b. 80b (337 mg, 0.83 mmol), THF/water (3:1, 80 mL), and aqueous 1 M NaOH (4.98 mL, 4.98 mmol) yielded a light brown solid that1H-NMR indicated was a mixture of the desired product and the methyl ester. As base hydrolysis was not sufficient to bring the reaction to completion, the crude product was suspended in a mixture of glacial acetic acid and water (25 mL each), and 6 M HCl added (50 mL). The resulting suspension was heated at reflux for 18 hours before being allowed to cool to room temperature. The solid which precipitated from solution was filtered off, washed with plenty of water and then dried over P2O5 overnight to yield a yellow/mustard coloured solid. Re-crystallisation from methanol (H2O was added to precipitate the product) afforded 81b as a pale yellow solid (191 mg, 68 %); mp: >250 °C;1H NMR (400 MHz, DMSO-d6) δ 12.93 (br s, 1H, -CO2H), 10.97 (br s, 1H, -OH), 8.43 (s, 1H, ArH), 7.83 (dd, J = 8.0 & 1.2 Hz, 1H, ArH), 7.73 (s, 1H, ArH), 7.71 (d, J = 8.8 Hz, 1H, ArH), 7.49–7.42 (m, 2H, ArH), 7.34–7.29 (m, 2H, ArH), 7.28 (s, 1H, ArH), 3.30–3.24 (m, 2H, -CH2CH2-), 2.92–2.90 (m, 2H, -CH2CH2-);13C NMR (100 MHz, DMSO-d6) δ 171.5, 168.7, 155.5, 142.5, 137.1, 135.8, 131.7, 131.6, 130.9, 130.5, 130.3, 127.2, 126.6, 126.1, 125.9, 115.1, 110.6, 37.2, 35.6; MS (ESI+) m/z 335 (M - H, 100 %); HRMS-ESI calcd for C20H16O5 [M - H]+ 335.0925; found 335.0928.
7-(4-Carboxyphenethyl)-3-hydroxy-2-naphthoic acid (81c).
Method identical to that described for 80a with the exception that THF (100 mL) was used instead of dioxane. 78d (500 mg, 1.24 mmol) and 10 wt % palladium on activated carbon (50 mg) yielded the protected phenethyl intermediate 80c (504 mg) as a white solid which was taken forward without further characterisation or purification.
De-protection was carried out in an identical fashion to 79b. 80c (504 mg, 1.24 mmol), THF/water (3:1, 80 mL) and aqueous 1 M NaOH (1.24 mL, 1.24 mmol) afforded 81c (388 mg, 93 %) as a yellow solid; mp: >250 °C;1H NMR (400 MHz, DMSO-d6) δ 12.5 (br s, 1H, -CO2H), 8.36 (s, 1H, ArH), 7.85 (d, J = 8.4 Hz, 2H, ArH), 7.70 (s, 1H, ArH), 7.63 (d, J = 8.4 Hz, 1H, ArH), 7.42–7.34 (m, 3H, ArH), 7.16 (s, 1H, ArH), 3.02 (s, 4H, -CH2CH2-);13C NMR (100 MHz, DMSO-d6) δ 171.3, 167.2, 156.7, 146.8, 135.8, 135.4, 131.1, 129.7, 129.3, 128.6, 128.4, 127.3, 126.5, 125.6, 109.8, 36.6, 36.3; MS (ESI−) m/z 335 (M - H, 100 %); HRMS-ESI calcd for C20H16O5 [M - H]+ 335.0925; found 335.0928; Anal. (C20H16O5.0.25H2O) C, H.
Dimethyl 6-Styrylnaphthalene-2,3-dicarboxylate (84).
Methylamine (2.0 M in methanol, 3.2 mL, 6.4 mmol) was added dropwise to a stirring 0° solution of 78a (1.10 g, 3.2 mmol) in toluene (20 mL). The resultant mixture was stirred at 0° for 2 hours. The solid which precipitated from solution was filtered off, washed with methanol (20 mL) and dried under vacuum to afford the crude phenol (82) as a pale yellow solid which was taken forward without further purification or characterisation.
To a solution of 82 (974 mg, 3.2 mmol) in anhydrous DCM (20 mL) at −10° was added triethylamine (0.89 mL, 6.4 mmol) and Tf2O (0.7 mL, 4.2 mmol). The resultant mixture was stirred at room temperature for 2 h before aqueous 1 M HCl (10 mL) was added. The organic layer was separated, washed with water (10 mL), brine (10 mL), dried over MgSO4 and concentrated in vacuo. The resultant residue was passed through a short silica plug to afford the crude triflate (83) which was taken forward without further purification or characterisation.
A flask was charged with Pd(OAc)2 (72 mg, 0.32 mmol) and DPPP (172 mg, 0.42 mmol) before being briefly evacuated and backfilled with carbon monoxide. A degassed mixture of 83 (1.39 g, 3.2 mmol) and triethylamine (0.89 mL, 6.4 mmol) in a mixture of anhydrous DMF and methanol (3:1, 12 mL) was added. The resultant mixture was heated at 80° for 24 h. After being allowed to cool to room temperature, the reaction was diluted with aqueous 1 M HCl (10 mL) and then extracted with ethyl acetate (2 × 25 mL). The organic phase was isolated, washed with water (15 mL), brine (15 mL), dried over MgSO4 and concentrated in vacuo. The resultant residue was purified via flash chromatography (20% EtOAc in hexane) to afford a white solid which was further purified by re-crystallisation from toluene to give 84 (459 mg, 41% over 3 steps);1H NMR (400 MHz, CDCl3) 8.24 (s, 1H, ArH), 8.21 (s, 1H, ArH), 7.91–7.86 (m, 3H, ArH), 7.59–7.57 (m, 2H, ArH), 7.42–7.38 (m, 2H, ArH & -CH=CH-), 7.33–7.28 (m, 3H, ArH & -CH=CH-), 3.97 (s, 3H, -CO2CH3), 3.96 (s, 3H, -CO2CH3);13C NMR (100 MHz, CDCl3) 168.3, 168.0, 137.7, 137.8, 133.9, 132.8, 130.9, 130.0, 129.9, 129.2, 129.0, 128.8, 128.2, 128.0, 127.7, 126.8, 126.7, 126.2, 52.7, 52.7; MS (ESI) 347 (MH+), 369 (MNa+), 315; HRMS (ESI) for C22H19O4 required 347.1278, obtained 347.1283.
(E)-6-Styrylnaphthalene-2,3-dicarboxylic acid (85).
Method identical to that described for 29. 84 (415 mg, 1.2 mmol) and LiOH (144 mg, 6.0 mmol) afforded 85 as an off-white solid (329 mg, 86%); mp: >250 °C;1H NMR (400 MHz, DMSO-d6) 8.28 (s, 1H, ArH), 8.24 (s, 1H, ArH), 8.16 (s, 1H, ArH), 8.08 (d, J = 8.4 Hz, 1H, ArH), 8.01 (d, J = 8.4 Hz, 1H, ArH), 7.67–7.64 (m, 2H, ArH), 7.46–7.38 (m, 4H, ArH & -CH=CH-), 7.31–7.28 (m, 1H, -CH=CH-);13C NMR (100 MHz, DMSO-d6) 169.2, 169.0, 137.6, 137.2, 133.7, 132.7, 131.1, 130.9, 130.0, 129.6, 129.4, 129.4, 129.3, 128.5, 128.3, 127.2, 127.1, 126.5; MS (ESI) 318 (M−), 374; Anal. (C20H14O4) C, H.
(E)-2-oxo-6-styryl-2H-chromene-3-carboxylic acid (88).
(E)-2-hydroxy-5-styrylbenzaldehyde [19] (64 mg, 0.29 mmol), Meldrum’s acid (41 mg, 1.34 mmol) and piperidinium acetate (1mg, 0.01 mmol) were mixed in ethanol (5 mL) and stirred at rt for 20 min before heating at reflux for 2 h. The mixture was then cooled to 0 °C for 1 h and the resulting precipitate was filtered, washed with ethanol and dried under vacuum to give 88 as a white solid (50 mg, 60%); mp: >250 °C;1H NMR (500 MHz, DMSO-d6) δ 13.28 (br s, 1H, -CO2H), 8.71 (s, 1H, ArH), 8.09 (d, J = 2.0 Hz, 1H, ArH), 7.99 (dd, J = 8.5 & 2.0 Hz, 1H, ArH), 7.62 (br d, J = 7.0 Hz, 2H, ArH), 7.46 (d, J = 8.5 Hz, 1H, ArH), 7.40 (m, 2H, ArH & -CH=CH-), 7.30 (m, 3H, ArH & -CH=CH-);13C NMR (125 MHz, DMSO-d6) δ 164.0, 156.6, 153.8, 148.2, 136.7, 133.9, 129.5, 128.8, 128.0, 127.5, 126.6, 126.5, 118.7, 118.2, 116.6; MS (ESI−) m/z 291 (100 %); Anal. C18H12O4: C, H.
Biology Experimental
Expression and electrophysiological analysis of NMDAR activity.
cDNA encoding the rat NMDAR subunits was generously provided by Dr. Shigetada Nakanishi, Kyoto, Japan (GluN1a), Dr. Peter Seeburg, Heidelburg, Germany (GluN2A, GluN2C, and GluN2D) and Drs. Dolan Pritchett and David Lynch, Philadelphia, USA (GluN2B). Plasmids were linearized with Not I (GluN1a, GluN2C, and GluN2D), EcoR I (GluN2A) or Sal I (GluN2B) and transcribed in vitro with T7 (GluN1a, GluN2A, GluN2C, and GluN2D) or SP6 (GluN2B) RNA polymerase using mMessage mMachine transcription kits (Ambion, Austin, TX, USA).
Oocytes from mature female Xenopus laevis (Xenopus One, Ann Arbor, MI, USA) were removed and isolated.31 GluN1a and GluN2 RNAs were mixed in a molar ratio of 1:1–3 and microinjected (50 nl, 15–30 ng total) into the oocyte cytoplasm. Oocytes were incubated in ND-96 solution at 17°C prior to electrophysiological assay (1–5 days). Electrophysiological responses were measured using a standard two-electrode voltage clamp (TEVC) using a Warner Instruments (Hamden, CT, U.S.A.) model OC-725B Oocyte Clamp amplifier and a Digidata 1440 data acquisition system with pClamp 10 software (Molecular Devices, Sunnyvale, CA, USA). The recording buffer contained 116 mM NaCl, 2 mM KCl, 0.3 mM BaCl2 and 5 mM HEPES, pH 7.4. Response magnitude was determined by the steady plateau response elicited by bath application of 10 μM L-glutamate plus 10 μM glycine at a holding potential of −60 mV. Dose-response results were fit using GraphPad Prism (ISI Software, San Diego, CA, U.S.A.) (Costa et al., 2010).
Acknowledgements
Research reported in this publication was supported by the National Institute of Mental Health of the National Institutes of Health under Award Number R01MH060252, the MRC (Grants G0601509, G0601812) and the BBSRC (grant BB/L001977/1).
Abbreviations
- AMPAR
(S)-2-amino-3-hydroxy-5-methyl-4-isoxazolepropanoic acid receptor
- CNS
central nervous system
- GFP
green fluorescent protein
- HEK
human embryonic kidney
- HEPES
4-(2-hydroxethyl)-1-piperazineethanesulfonic acid)
- LBD
ligand binding domain
- LTP
long-term potentiation
- NAM
negative allosteric modulator
- NMDAR
N-methyl-D-aspartic acid receptor
- PAM
positive allosteric modulator
- STP
short-term potentiation
- TEVC
two electrode voltage clamp
Footnotes
Supporting Information Available.
Proton and carbon NMR spectra for final compounds. Elemental analyses for intermediates and final compounds.
References
- (1).(a) Watkins JC; Jane DE The glutamate story. Br. J. Pharmacol 2006, 147, S100–S108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kew JNC; Kemp JA Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology 2005, 179, 4–29. [DOI] [PubMed] [Google Scholar]; (c) Traynelis SF; Wollmuth LP; McBain CJ; Menniti FS; Vance KM; Ogden KK; Hansen KB; Yuan H; Myers SJ; Dingledine R Glutamate receptor ion channels: structure, regulation, and function. Pharmacological Reviews 2010, 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Collingridge GL; Olsen RW; Peters J; Spedding M A nomenclature for ligand-gated ion channels. Neuropharmacology 2009, 56, 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Monaghan DT; Jane DE (2008) Pharmacology of the NMDA receptor In Biology of the NMDA receptor. Ed. Antonius VanDongen, Taylor and Francis, London, UK: pp 257–282. [Google Scholar]
- (4).Collingridge GL Synaptic plasticity. The role of NMDA receptors in learning and memory. Nature 1987, 330, 604–605. [DOI] [PubMed] [Google Scholar]
- (5).Kalia LV; Kalia SK; Salter MW NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 2008, 7, 742–755. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hizue M; Pang CH, Yokoyama M Involvement of N-methyl-D-aspartate-type glutamate receptor epsilon I and epsilon 4 subunits in tonic inflammatory pain and neuropathic pain. Neuroreport 2005, 16, 1667–1670. [DOI] [PubMed] [Google Scholar]; (c) Sanacora G; Zarate CA; Krystal JH; Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat. Rev. Drug. Discov 2008, 7, 426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Sapkota K; Mao Z; Synowicki P; Lieber D; Liu M,; Ikezu T; Gautam V; Monaghan DT GluN2D N-methyl-D-aspartate receptor subunit contribution to the stimulation of brain activity and gamma oscillations by ketamine: implications for schizophrenia. J. Pharmacol. Exp. Ther 2016, 356, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Monaghan DT; Irvine MW; Costa BM; Fang G; Jane DE Pharmacological modulation of NMDA receptor activity and the advent of negative and positive allosteric modulators. Neurochemistry International 2012, 61, 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Collingridge GL; Volianskis A; Bannister N; France G; Hanna L; Mercier M; Tidball P; Fang G; Irvine MW; Costa BM; Monaghan DT; Bortolotto ZA; Molnar E; Lodge D; Jane DE (2013) The NMDA receptor as a target for cognitive enhancement. Neuropharmacology 2013, 64, 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Burnell ES; Irvine M; Fang G; Sapkota K; David E Jane DE, Monaghan DT Positive and negative allosteric modulators of N‑methyl‑D‑aspartate (NMDA) receptors: Structure-activity relationships and mechanisms of action. J. Med. Chem (In Press) DOI: 10.1021/acs.jmedchem.7b01640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Costa BM; Irvine MW; Fang G, Eaves RJ; Mayo-Martin MB; Skifter DA; Monaghan DT; Jane DE A novel family of negative and positive allosteric modulators of NMDA receptors. J. Pharmacol. Exp. Ther 2010, 335, 614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Irvine MW; Fang G; Eaves R; Mayo-Martin MB; Burnell ES, Costa BM; Culley GR; Volianskis A; Collingridge GL; Monaghan DT; Jane DE Synthesis of a series of novel 3,9-disubstituted phenanthrenes as analogues of known N-methyl-D-aspartate receptor allosteric modulators. Synthesis 2015, 47, 1593–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Mullasseril P; Hansen KB; Vance KM; Ogden KK; Yuan H; Kurtkaya NL; Santangelo R; Orr AG; Le P; Vellano KM; Liotta DC; Traynelis SF A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat. Commun 2010, 1, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Santangelo Freel RM; Ogden KK; Strong KL; Khatri A; Chepiga KM; Jensen HS; Traynelis SF; Liotta DC Synthesis and structure activity relationship of tetrahydroisoquinoline-based potentiators of GluN2C and GluN2D containing N-methyl-Daspartate receptors. J. Med. Chem 2013, 56, 5351–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Khatri A; Burger PB; Swanger SA; Hansen KB; Zimmerman S; Karakas E; Liotta DC; Furukawa H; Snyder JP; Traynelis SF Structural determinants and mechanism of action of a GluN2C-selective NMDA receptor positive allosteric modulator. Mol. Pharmacol 2014, 86, 548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zimmerman SS; Khatri A; Garnier-Amblard EC; Mullasseril P; Kurtkaya NL; Gyoneva S; Hansen KB; Traynelis SF; Liotta DC Design, synthesis, and structure-activity relationship of a novel series of GluN2C-selective potentiators. J. Med. Chem 2014, 57, 2334–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Hackos DH; Lupardus PJ; Grand T; Chen Y; Wang TM; Reynen P; Gustafson A; Wallweber HJA; Volgraf M; Sellers BD; Schwarz J; Paoletti P; Sheng M; Zhou Q; Hanson JE Positive allosteric modulators of GluN2A-containing NMDARs with distinct modes of action and impacts on circuit function. Neuron 2016, 89, 1–17. [DOI] [PubMed] [Google Scholar]; (b) Volgraf M; Sellers BD; Jiang Y; Wu G; Ly CQ; Villemure E; Pastor RM; Yuen P; Lu A; Luo X; Liu M; Zhang S; Sun L; Fu Y; Lupardus PJ; Wallweber HJA; Liederer BM; Deshmukh G; Plise E; Tay S; Reynen P; Herrington J; Gustafson A; Liu Y; Dirksen A; Dietz MGA; Liu Y; Wang T-M; Hanson JE; Hackos D; Scearce-Levie K; Schwarz JB Discovery of GluN2A-selective NMDA receptor positive allosteric modulators (PAMs): Tuning deactivation kinetics via structure-based design. J. Med. Chem 2016, 59, 2760–2779. [DOI] [PubMed] [Google Scholar]
- (11).(a) Ceccon M; Rumbaugh G; Vicini S Distinct effect of pregnenolone sulfate on NMDA receptor subtypes. Neuropharmacology 2001, 40, 491–500; [DOI] [PubMed] [Google Scholar]; (b) Paul SP; Doherty JJ; Robichaud AJ; Belfort GM; Chow BY; Hammond RS; Crawford DC; Linsenbardt AJ; Shu H-J; Izumi Y; Mennerick SJ; Zorumski CF The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J. Neurosci 2013, 33, 17290–17300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Petrovic M; Sedlacek M; Cais O; Horak M; Chodounska H; Vyklicky L Jr. Pregnenolone sulfate modulation of N-methyl-D-aspartate receptors is phosphorylation dependent. Neuroscience 2009, 160, 616–628. [DOI] [PubMed] [Google Scholar]
- (13).Costa BM; Irvine MW; Fang G; Eaves RJ; Mayo-Martin MB; Laube B; Jane DE; Monaghan DT Structure-activity relationships for allosteric NMDA receptor inhibitors based on 2-naphthoic acid. Neuropharmacology 2012,. 62, 1730–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mosley CA, Acker TM, Hansen KB, Mullasseril P, Andersen KT, Le P, Vellano KM, Bräuner-Osborne H, Liotta DC, Traynelis SF, 2010. Quinazolin-4-one derivatives: A novel class of noncompetitive NR2C/D subunit-selective N-methyl-D-aspartate receptor antagonists. J. Med. Chem 53, 5476–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Katzman BM, Perszyk RE, Yuan H, Tahirovic YA, Sotimehin AE, Traynelis SF, Liotta DC, 2015. A novel class of negative allosteric modulators of NMDA receptor function. Bioorganic & Medicinal Chemistry Letters 25, 5583–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, Negri M, Andreetta F, Senar-Sancho SA, Guiral LG, Cardullo F Identification and characterisation of novel NMDA receptor antagonists selective for NR2A-over NR2B-containing receptors. The Journal of Pharmacology and Experimental Therapeutics 2010, 335, 636–644. [DOI] [PubMed] [Google Scholar]
- (17).Yi F, Mou T-C, Dorsett KN, Volkman RA, Menniti FS, Sprang SR, Hansen KB, 2016. Structural basis for negative allosteric modulation of GluN2A-containing NMDA receptors. Neuron 91, 1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Song A, Wang X, Lam KS A convenient synthesis of coumarin-3-carboxylic acids via Knoevenagel condensation of Meldrum’s acid with ortho-hydroxyaryl aldehydes or ketones (2003) Tetrahedron Letters, 44, 1755–1758. [Google Scholar]
- (19).Weng F, Wand C Xu, B. Direct C-H bond arylation of 2-hydroxybenzaldehydes with arylboronic acids via ligand-free palladium catalysis (2010) Tetrahedron Letters, 51, 2593–2596. [Google Scholar]
- (20).Sapkota K; Irvine MW; Fang G; Burnell ES; Bannister N; Volianskis A; Culley GR; Dravid SM; Collingridge GL; Jane DE; Monaghan DT Mechanism and properties of positive allosteric modulation of N-methyl-D -aspartate receptors by 6-alkyl 2-naphthoic acid derivatives. Neuropharmacology 2017, 125, 64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chopra DA; Sapkota K; Irvine MW; Fang G; Jane DE; Monaghan DT; Dravid SM A single-channel mechanism for pharmacological potentiation of GluN1/GluN2A NMDA receptors. Sci. Rep 2017, 7, 6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kostakis E, Jang MK, Russek SJ, Gibbs TT, Farb DH, 2011. A steroid modulatory domain in NR2A collaborates with NR1 exon-5 to control NMDAR modulation by pregnenolone sulfate and protons. J. Neurochem 119, 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Filosa R; Peduto A; Aparoy P; Schaible AM; Luderer S; Krauth V; Petronzi C; Massa A; de Rosa M; Reddanna P; Werz O Discovery and biological evaluation of novel 1,4-benzoquinone and related resorcinol derivatives that inhibit 5-lipoxygenase. Eur. J. Med. Chem 2013, 67, 269–279. [DOI] [PubMed] [Google Scholar]
- (24).Wendt MD; Rockway TW; Geyer A; McClellan W; Weitzberg M; Zhao X; Mantei R; Nienaber VL; Stewart K; Klinghofer V; Giranda VL; Identification of novel binding interactions in the development of potent, selective 2-naphthamide inhibitors of urokinase. Synthesis, structural analysis, and SAR of N-phenyl amide 6-substitution. J. Med. Chem 2004, 47, 303–324. [DOI] [PubMed] [Google Scholar]
- (25).De Groot FMH; Loos WJ; Koekkoek R; van Berkom LWA; Busscher GF; Seelen AE; Albrecht C; de Bruijn P; Scheeren HW; Elongated multiple electronic cascade and cyclization spacer systems in activatable anticancer prodrugs for enhanced drug release. J. Org. Chem 2001, 66, 8815–8830. [DOI] [PubMed] [Google Scholar]
- (26).Zhang X; Xiaoqiang Y; Feng X; Hesong L; He R; Yamamoto Y; Bao M; Regioselective control by a catalyst switch in palladium-catalyzed benzylallylation of arylethylidene malononitriles. J. Organomet. Chem 2013, 745.-, 177–185. [Google Scholar]
- (27).Murphy RA; Kung HF; Kung M -P.; Billings, J.; Synthesis and characterization of iodobenzamide analogues: Potential D-2 Dopamine Receptor Imaging Agents. J. Med. Chem 1990, 33, 171–178. [DOI] [PubMed] [Google Scholar]
- (28).Frigoli M; Moustrou C; Samat A; Guglielmetti R; Synthesis of new thiophene-substituted 3,3-diphenyl-3H-naphtho[2,1-b]pyrans by cross-coupling reactions, precursors of photomodulated materials. Eur. J. Org. Chem 2003, 2799–2812. [Google Scholar]
- (29).Ortar G; Cascio MG; De Petrocellis L; Morera E; Rossi F; Schiano-Moriello A; Nalli M; de Novellis V; Woodward DF; Maione S; Di Marzo V; J. Med. Chem 2007, 50, 6554–6569. [DOI] [PubMed] [Google Scholar]
- (30).Gray GW; Jones B The preparation of 4- and 5-n-alkoxy-1-naphthoic and 6- and 7-n-alkoxy-2-naphthoic acids. J. Chem. Soc 1954, 678–683. [Google Scholar]
- (31).Buller AL; Larson HC; Schneider BE; Beaton JA; Morrisett RA; Monaghan DT The molecular basis of NMDA receptor subtypes: native receptor diversity is predicted by subunit composition. J. Neurosci 1994, 14, 5471–5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Erdelyi M; Varedian M; Skold C; Niklasson IB; Nurbo J; Persson A; Bergquist J; Gogoll A Chemistry and folding of photomodulable peptides – stilbene and thioaurone-type candidates for conformational switches. Org. Biomol. Chem 2008, 6, 4356–4373. [DOI] [PubMed] [Google Scholar]