

Malaria parasite uses sFGL2 secreted mainly by Tregs to subvert the host innate immune response for its survival.
Abstract
Malaria parasites suppress host immune responses to facilitate their survival, but the underlying mechanism remains elusive. Here, we found that blood-stage malaria parasites predominantly induced CD4+Foxp3+CD25+ regulatory T cells to release soluble fibrinogen-like protein 2 (sFGL2), which substantially enhanced the infection. This was attributed to the capacity of sFGL2 to inhibit macrophages from releasing monocyte chemoattractant protein-1 (MCP-1) and to sequentially reduce the recruitment of natural killer/natural killer T cells to the spleen and the production of interferon-γ. sFGL2 inhibited c-Jun N-terminal kinase phosphorylation in the Toll-like receptor 2 signaling pathway of macrophages dependent on FcγRIIB receptor to release MCP-1. Notably, sFGL2 were markedly elevated in the sera of patients with malaria, and recombinant FGL2 substantially suppressed Plasmodium falciparum from inducing macrophages to release MCP-1. Therefore, we highlight a previously unrecognized immune suppression strategy of malaria parasites and uncover the fundamental mechanism of sFGL2 to suppress host innate immune responses.
INTRODUCTION
Malaria is caused by infection with parasites of the genus Plasmodium, and the blood-stage infection causes patients to exhibit clinical symptoms. The components [glycosylphosphatidylinositol (GPI) anchor and DNA] and metabolic products (hemozoin and uric acid) of the malaria blood-stage infection can be sensed by Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)–like receptor containing pyrin domain 3 on macrophages or dendritic cells (DCs) (1–3) and can sequentially orchestrate the generation of parasite-specific antibodies and the activation of CD4+ and CD8+ T cell responses to limit blood-stage infection (4–8). However, infections with the human malaria parasite always persist for several weeks or months, and immunity to malaria is slow to develop, indicating that parasites have also acquired strategies to suppress host immune responses to facilitate their survival (9).
Evidence shows that the malaria parasite can inhibit the activation of parasite-specific CD4+ T cell responses by inducing apoptosis of the DCs (10, 11), down-regulating costimulatory molecules, and/or suppressing antigen presentation of DCs (12, 13). Plasmodium parasites also induce apoptosis and anergy of the activated CD4+ T cells and B cells (14–16). However, the mechanism by which the malaria parasite dampens the host immune responses is still elusive (17).
Here, we report an immune escape mechanism of the malaria parasites that involves the induction of CD4+Foxp3+CD25+ regulatory T cells (Tregs) to release soluble fibrinogen-like protein 2 (sFGL2), a member of the fibrinogen superfamily. The released sFGL2 inhibited macrophages to release monocyte chemoattractant protein-1 (MCP-1) and sequentially reduced the recruitment of natural killer/natural killer T (NK/NKT) cells in the spleen and interferon-γ (IFN-γ) production, thereby facilitating parasite survival. In contrast to the major role played by sFGL2 to inhibit T cell activation in malignancies and autoimmune diseases (18, 19), we comprehensively describe the cellular mechanism of sFGL2 to suppress host innate immune responses.
RESULTS
sFGL2 was markedly induced by the malaria blood-stage infection and promoted parasite development
sFGL2 could suppress DC maturation and T cell proliferation (20), leading to the progress of malignancies and autoimmune diseases (18, 21), but its contribution to malaria infection has not yet been defined. Therefore, we first investigated whether sFGL2 was elevated in the sera of patients infected with Plasmodium vivax or Plasmodium falciparum, which were collected at the China-Myanmar border. Compared with the healthy donors, substantially higher levels of sFGL2 were found in both P. vivax– and P. falciparum–infected patients, and the extent of sFGL2 elevation was similar in the two infected groups (Fig. 1A). Moreover, a weak correlation was also found between parasitemia and sFGL2 levels in the sera of patients with malaria (Fig. 1B).
Fig. 1. sFGL2 was markedly induced by malaria blood-stage infection and promoted parasite development.
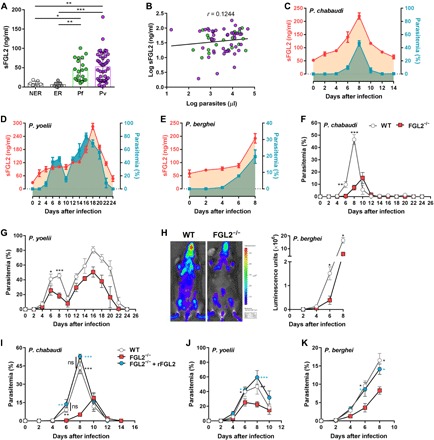
(A) sFGL2 levels in the sera of P. vivax– and P. falciparum–infected patients from the China-Myanmar border and healthy donors as measured by enzyme-linked immunosorbent assay (ELISA). NER, nonendemic healthy residents; ER, endemic healthy residents; Pf, patients with acute P. falciparum infection; Pv, patients with acute P. vivax infection. (B) Correlation between parasitemia and sFGL2 levels in patients with malaria. r, Pearson’s correlation coefficient. (C to E) The time courses of the sFGL2 levels in sera (red line) measured by ELISA and levels of parasitemia (blue line) determined by Giemsa staining in mice (n = 5) infected with P. chabaudi (C), P. yoelii (D), and P. berghei (E). (F and G) Parasitemias of wild-type (WT) and FGL2−/− mice (n = 6) infected with P. chabaudi (F) or P. yoelii (G). (H) In vivo images of parasite burden at day 6 in WT and FGL2−/− mice (n = 4) infected with P. berghei luciferase (left), and the parasite loads between the WT and FGL2−/− mice (n = 4) at the indicated times after infection (right) were compared. (I to K) The effect of recombinant FGL2 administration (+rFGL2) on the parasitemia of FGL2−/− mice (n = 5) infected with P. chabaudi (I), P. yoelii (J), and P. berghei (K). Triplicate experiments were performed. Data represent the means ± SD. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001.
Infection with the human malaria parasite was mimicked by the rodent malaria parasite Plasmodium chabaudi chabaudi AS (P. chabaudi), which can persist at subpatent levels for several months. The levels of sFGL2 in the mouse sera were also found to gradually increase after P. chabaudi infection, which were also closely correlated with the parasitemia of P. chabaudi (Fig. 1C). Similar results were obtained with two other rodent malaria parasites, Plasmodium yoelii and Plasmodium berghei (Fig. 1, D and E).
To determine the role of sFGL2 in malaria parasite infection, we infected both wild-type (WT) and FGL2−/− mice with P. chabaudi. The parasite growth in FGL2−/− mice was markedly inhibited, as the appearance of parasites in FGL2−/− mice was delayed by 2 days, and the peak parasitemia was much lower than that in WT mice (Fig. 1F). The growth of parasites was also greatly inhibited when FGL2−/− mice were infected with two other rodent malaria parasites, P. yoelii and P. berghei (Fig. 1, G and H), indicating that FGL2 has broad effects in malaria parasite infections. Given that FGL2 can exist in two forms—membrane bound (mFGL2) and sFGL2, both of which are absent in the FGL2−/− mice—we still needed to determine which form inhibited parasite growth. mFGL2 has been reported to exhibit prothrombinase activity, resulting in fibrin deposition in affected tissues (22). However, no significant difference in fibrin deposition in the spleen and coagulation function was found between the parasite-infected WT and FGL2−/− mice (fig. S1, A and B). The administration of recombinant FGL2 (rFGL2) into FGL2−/− mice restored the growth of P. chabaudi, P. yoelii, and P. berghei to levels comparable to that in WT mice (Fig. 1, I to K). Thus, our data suggest that the elevated sFGL2 in serum promotes the development of malaria parasite infection.
sFGL2 does not affect parasite-specific antibody production or CD4+/CD8+ T cell activation
Parasite-specific antibodies and T cell responses are the predominant immune effectors against blood stages (4–7). In that context, we note that sFGL2 reportedly suppresses mainly T cell responses in malignancies and autoimmune diseases (18, 19). Therefore, we sought to determine whether sFGL2 enhances parasite growth by modulating parasite-specific antibodies and T cell responses. We found, however, that both total immunoglobulin G (IgG) and its subclasses in FGL2−/− mouse serum were comparable to those of WT mice at 7 days after infection (fig. S2A) and a similar result was obtained with germinal center B cells (B220+CD95+GL7+) (fig. S2B). No differences in either the proliferation or the IFN-γ secretion capacity of CD4+CD49dhiCD11ahi T cells, which are representative of parasite-specific CD4+ T cells (23), were detected between the WT and FGL2−/− mice at 6, 8, and 10 days after infection (fig. S3, A and B). The proliferation and IFN-γ, granzyme B, and perforin secretion of CD8aloCD11ahi T cells, which are representative of parasite-specific CD8+ T cells (24), of FGL2−/− mice were also comparable with those of WT mice (fig. S4, A to D). Thus, our data strongly suggest that parasite growth enhanced by sFGL2 is not associated with the modulation of parasite-specific antibody production or CD4+/CD8+ T cell responses.
sFGL2 promotes parasite growth through impeding the production of IFN-γ secreted by NK/NKT cells
Next, we sought to determine whether sFGL2 enhances parasite growth by manipulating the innate immune response against blood-stage infection. Given that IFN-γ plays a central role in the innate immune response against malaria blood-stage infection (25–27), we compared the serum IFN-γ levels of parasite-infected WT and FGL2−/− mice. Consistent with reduced parasitemia, the IFN-γ level in FGL2−/− mice was much higher than that in WT mice at 6 days after infection (Fig. 2A). To determine whether the elevated IFN-γ in serum contributed to the lower parasitemia in FGL2−/− mice, IFN-γ in FGL2−/− mice was depleted. The parasitemia of FGL2−/− mice returned almost to the level in WT mice after IFN-γ was depleted (Fig. 2B). Thus, we demonstrated a pivotal role of IFN-γ in the suppression of parasite growth in FGL2−/− mice.
Fig. 2. sFGL2 promotes parasite growth through impeding the production of IFN-γ secreted by NK/NKT cells.
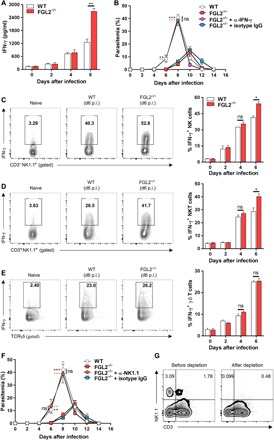
(A) The levels of IFN-γ in the sera of WT and FGL2−/− mice (n = 4) at the indicated time points after infection with P. chabaudi as measured by ELISA. (B) The parasitemia of the parasite-infected WT mice and FGL2−/− mice (n = 4) depleted with or without anti–IFN-γ monoclonal antibody (mAb). (C to E) Representative fluorescence-activated cell sorting (FACS) analyses (left) and statistical analyses (right) of IFN-γ production capacity by splenic total NK (C), NKT (D), and γδ T (E) cells from both WT and FGL2−/− mice (n = 4) at the indicated time points after infection with P. chabaudi. d6 p.i., day 6 post-infection. (F) The parasitemia of the infected WT mice (n = 5) and FGL2−/− mice (n = 5) with both NK and NKT cells depleted with or without anti-NK1.1 mAb. (G) The depletion efficacies of NK (NK1.1+ CD3−) and NKT (NK1.1+ CD3+) cells in FGL2−/− mice (n = 4) as determined by FACS. Data represent three separate experiments with at least four mice per group. Numbers represent the means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
It is well known that IFN-γ is secreted by NK, NKT, and γδ T cells at the early stage of malaria parasite infection (28, 29). Consistent with this behavior, we found that the level of IFN-γ secreted by splenic total NK cells, especially NKT cells, from FGL2−/− mice was much higher than that secreted from WT mice at 6 days after infection (Fig. 2, C and D), although no significant difference was found between WT and FGL2−/− mice in the amount of IFN-γ secreted by γδ T cells (Fig. 2E). To confirm that the enhanced NK/NKT-secreted IFN-γ contributed to the suppression of parasite growth in FGL2−/− mice, both NK and NKT cells in FGL2−/− mice were depleted using an anti-NK1.1 monoclonal antibody (mAb). The parasitemia in FGL2−/− mice was markedly increased to the level of that in WT mice (Fig. 2F), after 73% of NKT cells (NK1.1+CD3+) and 97% of NK cells (NK1.1+CD3−) were depleted (Fig. 2G). Thus, the reduced parasitemia in FGL2−/− mice was due to the enhanced NK/NKT-secreted IFN-γ.
sFGL2 reduces IFN-γ secretion by suppressing MCP-1–mediated recruitment of NK/NKT cells
We then determined how knocking out FGL2 affected the production of IFN-γ. To our surprise, knocking out FGL2 from mice did not improve the capacity of individual NK or NKT cells to secrete IFN-γ, as the mean fluorescence intensity of IFN-γ secreted by individual NK or NKT cell from FGL2−/− mice was comparable to that of WT mice (Fig. 3A). Instead, we found that the total number of splenocytes in the infected FGL2−/− mice on day 6 was much higher than that of the infected WT mice (2.12 × 108 ± 0.14 × 108 versus 1.45 × 108 ± 0.26 × 108; P < 0.05), and the numbers of both IFN-γ–secreting NK and NKT cells in the spleens of FGL2−/− mice were increased by approximately twofold compared with those in the spleens of WT mice (Fig. 3B). This finding indicates that the increased NK/NKT-secreted IFN-γ might be attributed to the recruitment of more NK/NKT cells to the spleens of FGL2−/− mice.
Fig. 3. sFGL2 reduces IFN-γ secretion by suppressing MCP-1–mediated recruitment of NK/NKT cells.
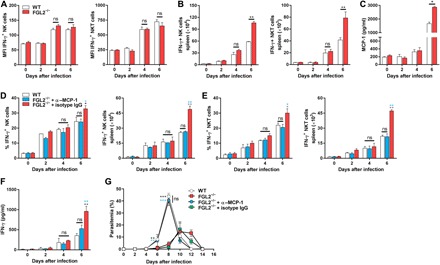
(A) The level of IFN-γ secreted by a single NK (left) or NKT (right) cell from P. chabaudi–infected WT and FGL2−/− mice (n = 4) was analyzed by FACS at the indicated time points. (B) The numbers of NK (left) and NKT (right) cells in the spleens of P. chabaudi–infected WT and FGL2−/− mice (n = 4) were statistically analyzed at the indicated time after infection. (C) The level of MCP-1 in the serum of P. chabaudi–infected WT and FGL2−/− mice (n = 4) was analyzed by ELISA at the indicated time points. (D and E) The percentages (left) and numbers (right) of IFN-γ–secreting NK cells (D) or NKT cells (E) in the spleens of both P. chabaudi–infected WT and FGL2−/− mice (n = 4) were analyzed by flow cytometry, at the indicated time points after MCP-1 was depleted. (F) The IFN-γ levels in the sera of parasite-infected WT and FGL2−/− mice (n = 5) were measured by ELISA after MCP-1 was depleted. (G) The parasitemia of the parasite-infected WT mice and FGL2−/− mice (n = 5) with or without MCP-1 depletion was determined. Each experiment was repeated three times with at least four mice per group. Data represent the means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
We then considered what other factors might be involved in recruiting NK and NKT cells. Although the predominant role of MCP-1 is to recruit monocytes and macrophages (30), a recent study reported that MCP-1 could also recruit NK cells (31), leading us to investigate whether the increased NK and NKT cells in the spleens of FGL2−/− mice were recruited by MCP-1. We observed that the MCP-1 levels in the sera of FGL2−/− mice were approximately twofold that of WT mice at 6 days after infection (Fig. 3C). Depletion of MCP-1 sharply reduced both the number of IFN-γ–secreting NK/NKT cells and serum IFN-γ levels to the levels of those of WT mice (Fig. 3, D to F). The parasitemia of FGL2−/− mice also returned to a level comparable to that of WT mice after MCP-1 was depleted (Fig. 3G), indicating the essential role of MCP-1–mediated recruitment of NK/NKT cells in the suppression of parasite growth in FGL2−/− mice.
sFGL2 inhibits parasite-stimulated macrophages from secreting MCP-1 through blocking c-Jun N-terminal kinase activation
Next, we sought to explore the cellular source of MCP-1. Because MCP-1 is produced mainly by macrophages (30), we compared the capacities of macrophages from infected WT and FGL2−/− mice to generate MCP-1. Consistent with the observation that FGL2−/− mice harbored a higher level of MCP-1 in serum than did WT mice during early infection, a much higher level of MCP-1 was generated by macrophages from the spleens of the FGL2−/− mice on day 6 after infection than by macrophages from the spleens of WT mice (Fig. 4A). Depletion of macrophages in FGL2−/− mice with clodronate liposomes greatly reduced both serum MCP-1 and IFN-γ amount at day 6 after infection to the level lower than those of the WT mice (Fig. 4B), and the parasitemia in the macrophage-depleted FGL2−/− mice was much higher than that of the infected WT mice on day 8 after infection, resulting in the death of 100% of the mice (Fig. 4C). Together, these results further strengthen the notion that the reduced parasitemia and higher level of serum IFN-γ in FGL2−/− mice were attributed to the enhanced capacity of macrophage to release MCP-1.
Fig. 4. sFGL2 inhibits parasite-stimulated macrophages from secreting MCP-1 through blocking JNK activation.
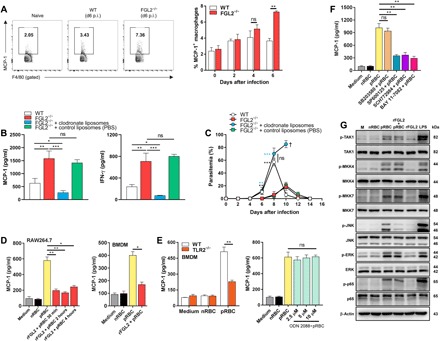
(A) Representative FACS (left) and statistical (right) analyses of the production of MCP-1 by macrophages from the spleens of both WT and FGL2−/− mice (n = 4) at the indicated time points. (B) The levels of MCP-1 (left) and IFN-γ (right) in the serum of the parasite-infected WT mice and FGL2−/− mice (n = 5) with or without macrophages depleted by clodronate liposomes at day 6 after infection. (C) The parasitemia of both parasite-infected WT mice and FGL2−/− mice (n = 5) with or without macrophage depletion by clodronate liposomes. (D) MCP-1 levels in the supernatants of pRBC lysate–stimulated RAW264.7 macrophages (left) and bone marrow–derived macrophages (BMDMs) (right) treated with or without rFGL2 for the indicated times as detected by ELISA. (E) MCP-1 levels released by WT and TLR2−/− BMDMs incubated with the pRBC lysate preparation as measured by ELISA (left), and the MCP-1 levels produced by the pRBC lysate–stimulated macrophages pretreated with or without the TLR9 antagonist ODN2088 as determined by ELISA (right). (F) MCP-1 levels in the supernatants of pRBC lysate–stimulated macrophages pretreated with or without nuclear factor κB (NF-κB) inhibitor BAY 11-7082, p38 inhibitor SB203580, c-Jun N-terminal kinase (JNK) inhibitor SP600125, and extracellular signal–regulated kinase 1/2 (ERK1/2) inhibitor SCH772984. (G) Western blots of the phosphorylation of TAK1, MKK4, MKK7, JNK, ERK1/2, and p65 in pRBC lysate–stimulated RAW264.7 cells pretreated with or without rFGL2. nRBC, normal red blood cell. Two to four independent experiments were performed. Data represent the means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001. The symbol “†” indicates the time point when all mice died.
We then investigated whether sFGL2 can directly suppress the ability of the parasite to induce MCP-1 production in macrophages. Pretreatment of bone marrow–derived macrophages (BMDMs) with rFGL2 markedly reduced the MCP-1 released by parasitized red blood cell (pRBC) lysate–stimulated BMDMs to a baseline level, and a similar result was obtained with the RAW264.7 macrophage cell line (Fig. 4D). Together, these results suggest that blood-stage malaria parasites could use sFGL2 to inhibit macrophages from secreting MCP-1.
It is still unclear how parasite infection induces macrophages to release MCP-1. Key triggers for stimulating macrophages to release inflammatory cytokines are TLR9 and TLR2, the predominant pattern recognition receptors that bind hemozoin and GPI, respectively (1, 2). Therefore, we next tested whether the induction of macrophages to release MCP-1 by pRBC lysate was TLR dependent. We found that the capacity of pRBC lysates to trigger macrophages to release MCP-1 was greatly reduced in BMDMs from TLR2−/− mice, but not in macrophages treated with the TLR9 antagonist ODN2088 (Fig. 4E), indicating that macrophages induced to release MCP-1 by the parasite were mainly TLR2 but not TLR9 dependent. Although both nuclear factor κB (NF-κB) and mitogen-activated protein kinase [including p38, extracellular signal–regulated kinase 1/2 (ERK1/2), and c-Jun N-terminal kinase (JNK)] could be activated by the malaria parasite–triggered TLR2 signaling pathway, only NF-κB, JNK, and ERK1/2 inhibitor, but not p38 inhibitor, could sharply block macrophages from releasing MCP-1 (Fig. 4F).
Next, we sought to establish the intracellular target in macrophages that sFGL2 blocked. We observed that the addition of rFGL2 had no significant effect on the activation of upstream adaptors of TGF beta-activated kinase 1 (TAK1) and mitogen-activated protein kinase kinase 4/7 (MKK4/7) and only the phosphorylation of JNK, but not ERK1/2 and p65, in parasite-stimulated macrophages was greatly suppressed by rFGL2 (Fig. 4G). From this, we conclude that sFGL2 suppressed the parasite-stimulated macrophages from releasing MCP-1 through the depression of JNK phosphorylation in the TLR2 signaling pathway.
sFGL2 attenuates parasite-stimulated macrophages from releasing MCP-1 dependent on FcγRII B receptors
sFGL2 reportedly binds specifically to FcγRII and FcγRIII receptors that are expressed on the surfaces of antigen-presenting cells (APCs), including B cells, macrophages, and DCs (32). We tested whether sFGL2 inhibited pRBC lysate–stimulated macrophages from releasing MCP-1 through binding to FcγRII and FcγRIII receptors. Although rFGL2 notably inhibited pRBC lysates from inducing either BMDMs or RAW264.7 cells to release MCP-1, pretreatment with anti-FcγRIII/FcγRII abolished the inhibitory effect of rFGL2 (Fig. 5A), strongly suggesting that sFGL2 suppressed the pRBC lysate–stimulated macrophages from releasing MCP-1 through binding to FcγRIII/FcγRII.
Fig. 5. sFGL2 attenuates parasite-stimulated macrophages from releasing MCP-1 dependent on FcγRIIB receptors.
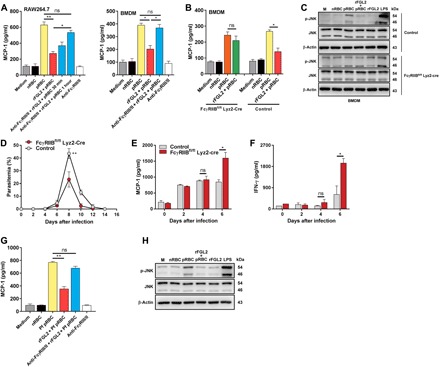
(A) RAW264.7 cells (left) and BMDMs (right) were pretreated with anti-FcγRIII/FcγRII for the indicated time and then incubated with pRBC lysate and rFGL2, and MCP-1 levels in the supernatants were measured by ELISA. (B) BMDMs isolated from control and FcγRIIBfl/fl Lyz2-Cre mice were incubated with pRBC lysate and rFGL2, and MCP-1 levels in the supernatants were detected by ELISA. (C) Western blots of JNK activation in pRBC lysate–stimulated control and FcγRIIB-deficient BMDMs preincubated with or without rFGL2. (D) The parasitemia levels of control and FcγRIIBfl/fl Lyz2-Cre mice (n = 5) infected with P. chabaudi were determined by Giemsa staining. (E) The MCP-1 levels in the sera of control and FcγRIIBfl/fl Lyz2-Cre mice (n = 4) after infection with P. chabaudi were detected by ELISA. (F) The IFN-γ levels in the serum of the infected control and FcγRIIBfl/fl Lyz2-Cre mice (n = 4) were determined by ELISA. (G) MCP-1 levels in the supernatants of P. falciparum lysate–stimulated human THP-1–derived macrophages treated with or without rFGL2 and anti-FcγRIII/FcγRII 2 hours before parasite stimulation were detected by ELISA. (H) Western blots of JNK activation in P. falciparum lysate–stimulated macrophages pretreated with or without rFGL2. Each experiment was repeated three times. Data represent the means ± SD. *P < 0.05, **P < 0.01.
Given that the maturation of DCs is suppressed by sFGL2 through binding to FcγRIIB but not FcγRIII receptors (32), we investigated whether suppression of the activation of pRBC lysate–stimulated macrophages by sFGL2 was FcγRIIB receptor dependent. To this end, we constructed FcγRIIBfl/fl Lyz2-Cre mice, in which the FcγRIIB gene of macrophages was conditionally knocked out. We found that rFGL2 completely lost its capacity to suppress the action of pRBC lysate upon inducing FcγRIIB-deficient BMDMs to release MCP-1 and activate JNK, although rFGL2 could greatly inhibit the parasite lysate to induce WT BMDMs to release MCP-1 and phosphorylate JNK (Fig. 5, B and C). Moreover, the parasitemia in FcγRIIBfl/fl Lyz2-Cre mice infected with P. chabaudi was greatly reduced compared with that in control mice (Fig. 5D), and the levels of both MCP-1 and IFN-γ were markedly increased in FcγRIIBfl/fl Lyz2-Cre mice compared with those in control mice (Fig. 5, E and F). Thus, we demonstrated that sFGL2 suppressing macrophages from releasing MCP-1 is dependent on FcγRIIB receptors.
Next, the effect of sFGL2 on MCP-1 released by human THP-1–derived macrophages stimulated with P. falciparum lysate was investigated. We found that the addition of rFGL2 greatly inhibited P. falciparum lysate from inducing human THP-1–derived macrophages to produce MCP-1 and anti-FcγRIII/FcγRII abolished the inhibitory effect of rFGL2 (Fig. 5G). Moreover, rFGL2 also greatly suppressed the phosphorylation of JNK in human THP-1–derived macrophages stimulated by P. falciparum lysate (Fig. 5H). Together, these results suggest that sFGL2 may also play a key role in human malaria.
sFGL2 is secreted mainly by CD4+Foxp3+CD25+ Tregs and promotes the growth of blood-stage malaria parasites
Given that Tregs can inducibly secrete sFGL2 (33), we tested whether Tregs were the main T cell source of sFGL2 in malaria parasite infection. We found that the abundance of Tregs was greatly up-regulated at 2 to 8 days in the spleens of mice infected with P. chabaudi (Fig. 6A) and sFGL2 transcripts were detected mainly in CD4+CD25+ Tregs and much less in either CD4− or CD4+CD25− T cells (Fig. 6B). Moreover, sFGL2 levels in the sera of infected mice were greatly reduced after Tregs were depleted with anti-CD25 mAb at −1, 1, 3, 5, and 7 days after infection but returned to increased levels when anti-CD25 mAb was withdrawn (Fig. 6, C and D), indicating that sFGL2 in the serum of parasite-infected mice was predominantly secreted by Tregs.
Fig. 6. sFGL2 is secreted mainly by CD4+Foxp3+CD25+ Tregs and promotes the growth of malaria blood stage.
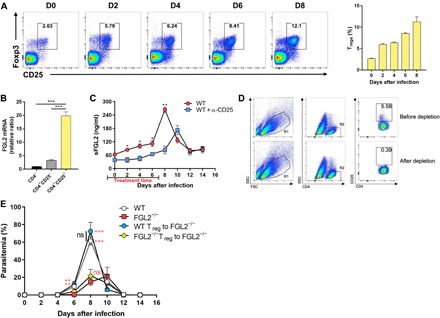
(A) The expansion of CD4+Foxp3+CD25+ Tregs in mice (n = 4) infected with P. chabaudi was analyzed at the indicated time points by flow cytometry (left), and a statistical analysis was also performed (right). (B) The transcript levels of FGL2 in CD4−, CD4+CD25+, and CD4+CD25− T cells were determined by real-time polymerase chain reaction (PCR). (C) ELISA of the level of sFGL2 in the serum of P. chabaudi–infected mice (n = 4) after Tregs were depleted with anti-CD25 mAb at −1, 1, 3, 5, and 7 days after infection. (D) The depletion of Tregs was determined by FACS. (E) The effects of the adoptive transfer of WT or FGL2−/− mice–derived Tregs on the parasitemia of FGL2−/− mice (n = 4) infected with P. chabaudi. Each experiment was performed three times, with at least four mice per group. Data represent the means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
The adoptive transfer of Tregs from WT mice, but not FGL2−/− mice, into FGL2−/− mice before infection notably increased the parasitemia of the infected FGL2−/− mice to a level comparable to that of the infected WT mice (Fig. 6E). These data strongly suggest that sFGL2 secreted by Tregs could promote the progression of malaria blood-stage infection.
DISCUSSION
In this study, we found that sFGL2 deficiency, which did not affect parasite-specific antibody production or CD4+ and CD8+ T cell responses, promoted macrophages to secrete MCP-1, resulting in enhanced NK/NKT cell recruitment to the spleen and IFN-γ production. Thus, in contrast to the major role of sFGL2 to inhibit host adaptive immune responses (18, 19), we revealed a previously unidentified role of sFGL2 to manipulate host innate immune responses. Furthermore, we found that sFGL2 was mainly secreted by Tregs (CD4+Foxp3+CD25+ Tregs) expanded after malaria parasite infection, and adoptive transfer assays showed that sFGL2 released by Tregs markedly promoted the growth of Plasmodium blood stages. In contrast, the elimination of Tregs greatly enhances CD4+ T cell responses against P. yoelii (34) or prevents the development of parasite-specific T helper 1 (TH1) cells involved in the induction of cerebral malaria (35). Therefore, our data strongly suggest that Tregs can also manipulate host innate immune responses against malaria parasite through releasing sFGL2.
To our surprise, we did not observe a significant effect of Treg depletion on the growth of P. chabaudi, which is consistent with a previous study (36). However, a remarkable protective effect of FGL2 knockout on P. chabaudi was found in this study. This discrepancy might be interpreted in such a way that a small proportion of sFGL2 spontaneously secreted by CD4+ and CD8+ T cells (37) would also be defective in the FGL2−/− mice and their deficiency contributed to its protective effect against P. chabaudi. In contrast, the depletion of Treg not only reduces the secretion of sFGL2 but also might affect other unknown functions of Tregs. It has also been pointed out that the Tregs function was dynamically modulated during the course of malaria parasite infection and the depletion of Tregs at different temporal windows has different effects on parasites (38).
We found that sFGL2 inhibits macrophages from releasing MCP-1 and demonstrated that this inhibition is an FcγRIIB-dependent action. This finding not only supports previous reports regarding the resistance to malaria parasite infection in populations with a dysfunctional polymorphism of the FcγRIIB-encoding gene FCGR2B or a defect in FcγRIIB (39, 40) but also suggests an underlying mechanism to explain the resistance of FcγRIIB defects or dysfunctional polymorphisms to malaria parasite infection. Either FCGR2B dysfunctional polymorphism or FcγRIIB defect was also associated with systemic lupus erythematosus (39, 41), an autoimmunity disease with TH17/Treg imbalance (42). Thus, the defects of sFGL2 receptor, FcγRIIB, might be linked with autoimmunity disease/Treg dysfunction and the reduced malaria risk.
Notably, several lines of evidence indicate that our findings are clinically relevant: (i) sFGL2 was markedly elevated in the sera of patients with malaria; (ii) the addition of rFGL2 greatly inhibited P. falciparum–stimulated human macrophages from releasing MCP-1; and (iii) we found a weak correlation between sFGL2 and parasitemia in patients with malaria. Together, these observations suggest a potential role for sFGL2 as a prognostic marker for predicting the severity of disease in patients with malaria. Furthermore, blocking sFGL2 could also become a novel immune intervention to limit the malaria parasite at early stages, helping the ensuing adaptive immune responses to control the parasite.
MATERIALS AND METHODS
Mice and experimental malaria model
C57BL/6J mice were purchased from the Animal Institute of the Academy of Medical Science (Beijing, China). TLR2−/− C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). FGL2 knockout mice (FGL2−/−) and mice carrying the lysozyme-M Cre (Lyz2-Cre) recombinase transgene in a C57BL/6J genetic background were gifts from S. Smiley (Trudeau Institute, NY, USA) and Z. Zhang (Army Medical University, Chongqing, China), respectively. FcγRIIBfl/fl C57BL/6J mice provided by S. Izui (University of Geneva, Switzerland) were subsequently crossed with Lyz2-Cre mice to generate FcγRIIBfl/fl Lyz2-Cre mice and with FcγRIIBfl/fl mice used as controls. All mice were housed under specific pathogen–free conditions and were generally used at 6 to 8 weeks of age. P. chabaudi, P. yoelii BY265 (P. yoelii), P. berghei ANKA (P. berghei), and a transgenic P. berghei ANKA parasite expressing luciferase under the control of the ef1a promoter (P. berghei A-luc) were maintained in our laboratory. Mice were inoculated intraperitoneally with 1 × 105 pRBCs. Animal care and all experiments were performed in accordance with approved protocols by the Institutional Animal Care and Use Committee of the Third Military Medical University.
Patients with malaria and healthy donors
Sera from healthy volunteers were obtained from Shenyang of China and the China-Myanmar border. Sera from P. vivax– and P. falciparum–infected patients were collected from clinics located at the China-Myanmar border. Infection with P. vivax or P. falciparum was confirmed by microscopic examination, and parasite density was estimated as the number of parasites per microliter. Detailed information about patients with malaria is included in table S1. Serum was collected from healthy volunteers and P. vivax– and P. falciparum–infected patients after individuals signed a written informed consent form. The experiments in this study were approved by the Institutional Review Board (no. PRAMS0034319) of the Pennsylvania State University.
Detection of parasitemia and parasite burden
Parasitemia was determined by examination of Giemsa-stained thin blood smears every other day after infection. Parasite burden in mice infected with P. berghei–luc was assessed by an IVIS Lumina In Vivo Imaging System (PerkinElmer) after intraperitoneal injection of d-luciferin (100 mg/kg).
Cell culture and stimulation
The RAW264.7 cell line was cultured in Dulbecco’s modified Eagle’s medium (HyClone) with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (Life Technologies). The human THP-1 cell line was cultured in RPMI 1640 (HyClone) with 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin and 0.05 mM β-Mer (Invitrogen). BMDMs were prepared as previously described (43). Briefly, bone marrow cells were collected from the femurs and tibias of female C57BL/6J mice and FcγRIIB fl/fl Lyz2-Cre mice at 8 weeks of age. Then, the cells were maintained in macrophage colony-stimulating factor (PeproTech) differentiation medium for 7 days and allowed to differentiate into mature macrophages. Both cell lines and BMDMs were tested for mycoplasma contamination using the Mycoplasma PCR (polymerase chain reaction) Detection Kit (Sigma-Aldrich) according to the manufacturer’s instructions. P. chabaudi and P. falciparum pRBC lysates were prepared by three freeze-thaw cycles of parasite-infected RBCs isolated from P. chabaudi–infected mice or P. falciparum NF54 schizonts synchronically cultured in vitro. For MCP-1 enzyme-linked immunosorbent assay (ELISA) and Western blot experiments, both cell lines and BMDMs were treated with rFGL2 proteins (R&D Systems) and with or without anti-FcγRIII/FcγRII (15 μg/ml) (Bio X Cell) before pRBC lysate stimulation. For cell signaling experiments, the RAW264.7 cell line was pretreated with NF-κB inhibitor (10 μM/liter) (BAY 11-7082, Selleck), p38 inhibitor (20 μM/liter) (SB203580, Cell Signaling Technology), JNK inhibitor (50 μM/liter) (SP600125, Cell Signaling Technology), or ERK1/2 inhibitor (10 μM/liter) (SCH772984, Cell Signaling Technology), followed by stimulation with pRBC lysate.
Enzyme-linked immunosorbent assay
The levels of sFGL2 in serum from healthy volunteers, P. vivax–infected patients, and P. falciparum–infected patients and in rodent malaria parasite (P. chabaudi, P. berghei, or P. yoelii)–infected mice were detected by a human or mouse FGL2 ELISA Kit (BioLegend) according to the manufacturer’s instructions. The concentrations of IFN-γ and MCP-1 in the sera of P. chabaudi–infected mice were determined using a corresponding ELISA kit (Dakewei). For the in vitro stimulation assay, the culture supernatant was collected, and MCP-1 was detected using a mouse or human MCP-1 ELISA kit (Dakewei). The malaria-specific total IgG, IgG1, and IgG2a levels in the sera of P. chabaudi–infected mice were measured as previously described. Briefly, mouse blood was lysed with saponin (Sigma-Aldrich) and sonicated in phosphate-buffered saline (PBS). Nunc MaxiSorp immunoplates (Nalge Nunc) were coated with parasite antigen at a concentration of 10 μg/ml overnight at 4°C and then incubated with serial dilutions of sera from WT and FGL2−/− mice. Biotin-conjugated anti-mouse IgG1 and IgG2a (BioLegend) were added to the plates to detect IgG1 and IgG2a. After a washing step with wash buffer, the plates were incubated with horseradish peroxidase (HRP)–conjugated anti-mouse IgG or HRP-conjugated streptavidin (BioLegend), and 3,3′, 5,5′-tetramethylbenzidine was added (BioLegend). The absorbance at a wavelength of 450 nm was read using a spectrophotometer.
In vivo depletion and rescue assays
For the depletion assay, 200 μg of the corresponding isotype control IgG, anti–IFN-γ mAb (clone XMG1.2, Bio X Cell), or anti–MCP-1 mAb (clone 2H5, Bio X Cell) was injected intraperitoneally into P. chabaudi–infected mice at −1, 0, 1, 3, and 5 days after infection. Depletion of NK/NKT cells was accomplished by intraperitoneal injection of 200 μg of anti-NK1.1 mAb (clone PK136, Bio X Cell) at −1, 0, 1, 3, and 5 days after infection. Depletion of Tregs was accomplished by intraperitoneal injection of 400 μg of anti-CD25 mAb (clone PC61.5, Bio X Cell) at −1, 0, 1, 3, 5, and 7 days after infection. Macrophage depletion in FGL2−/− mice was accomplished by intravenous injection of 200 μl of clodronate liposomes (Liposoma BV) on days −1, 2, and 5 of malaria parasite infection and with the injection of control liposomes (PBS) as a control. For rescue experiments, FGL2−/− mice received 25 μg of rFGL2 proteins (R&D Systems) by intravenous injection from the first day of infection until 5 days after infection.
Flow cytometry analysis
The following antibodies were used for flow cytometry analysis: anti-CD3 (clone 17A2, BioLegend), anti-CD8a (clone 53-6.7, BioLegend), anti-CD4 (clone GK1.5, BioLegend), anti-CD11a (clone M17/4, BioLegend), anti-CD49d (clone R1-2, BioLegend), anti-B220 (clone RA3-6B2, BioLegend), anti-CD95 (clone SA367H8, BioLegend), anti-GL7 (clone GL7, BioLegend), anti-NK1.1 (clone PK136, BioLegend), anti-F4/80 (clone BM8, BioLegend), anti-CD25 (clone PC61, eBioscience), anti-TCR γ/δ (clone GL3, BioLegend), anti-FoxP3 (clone MF-14, BioLegend), anti–MCP-1 (clone 2H5, BioLegend), anti–IFN-γ (clone XMG1.2, BioLegend), anti–granzyme B (clone NGZB, BioLegend), and anti-perforin (clone eBioOMAK-D, BioLegend). For surface staining, cells were first incubated with anti-CD16/CD32 antibodies (BioLegend) to block Fc receptors and were then incubated with the antibodies of interest for 45 min. For intracellular cytokine staining, cells were stimulated for 5 hours in the presence of phorbol 12-myristate 13-acetate (50 ng/ml), ionomycin (1 μg/ml), and GolgiStop (BD Biosciences), then stained with antibodies against surface proteins, and lastly fixed and permeabilized using a BD Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s protocol and stained for intracellular cytokines. All data were collected on a BD FACS Canto II cytometer (BD Biosciences) and analyzed with FlowJo v10 software (Tree Star).
5-Bromo-2′-deoxyuridine incorporation
Mice received 2 mg of 5-bromo-2′-deoxyuridine (BrdU) (Sigma-Aldrich) by intraperitoneal injection and BrdU (0.8 mg/ml) was added in their drinking water for a total of 4 days. Splenocytes were isolated on the indicated days, and parasite-specific CD4+ T cells and CD8+ T cells were intracellularly stained for BrdU incorporation using an APC BrdU Flow Kit (BD Biosciences).
Western blot analysis
Cells were lysed by radioimmunoprecipitation assay buffer (Beyotime) supplemented with protease inhibitor cocktail (Sigma-Aldrich), and proteins were then separated by SDS–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Millipore). The membranes were blocked with 5% (w/v) reagent-grade nonfat milk (Cell Signaling Technology), followed by incubation with antibodies against TAK1 (no. 4505, Cell Signaling Technology), phospho-TAK1 (Thr187) (no. 4536, Cell Signaling Technology), p65 (no. 8242, Cell Signaling Technology), phospho-p65 (Ser536) (no. 3033, Cell Signaling Technology), ERK1/2 (no. 4695, Cell Signaling Technology), phospho-ERK1/2 (Thr202/Tyr204) (no. 4370, Cell Signaling Technology), MKK4 (no. 9152, Cell Signaling Technology), phospho-MKK4 (Ser257/Thr261) (no. 9156, Cell Signaling Technology), MKK7 (no. 4172, Cell Signaling Technology), phospho-MKK7 (Ser271/Thr275) (no. 4171, Cell Signaling Technology), JNK (no. 9252, Cell Signaling Technology), and phospho-JNK (Thr183/Tyr185) (no. 9251, Cell Signaling Technology). The protein bands were visualized using the Western BLoT Hyper HRP Substrate (TAKARA) and exposed using a Chemiluminescence Imaging System (Fusion Solo S, Vilber, France).
Adoptive transfer analysis
Total splenocytes were obtained from WT or FGL2−/− mice 4 days after infection with P. chabaudi, and CD4+CD25− T cells and CD4+CD25+ T cells were enriched to a purity >95% using a mouse CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. Naive FGL2−/− recipients were injected intravenously with 5 × 105 CD4+CD25+ T cells from WT or FGL2−/− mice. One day after adoptive transfer, FGL2−/− recipient and control mice were challenged with P. chabaudi as described above.
Quantitative real-time PCR analysis
CD4− T cells and CD4+CD25− T cells and CD4+CD25+ T cells were isolated from P. chabaudi–infected mice using a mouse CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. Then, FGL2 mRNA expression levels were determined by quantitative PCR using an SYBR Premix Ex Taq II kit (TAKARA) on a CFX Connect Real-Time System (Bio-Rad). Expression levels were normalized to endogenous expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and data are shown relative to a control sample after GAPDH normalization.
Immunofluorescence
Snap-frozen spleens were surgically removed from P. chabaudi–infected WT or FGL2−/− mice, cryosectioned at 5-mm thickness, and mounted on glass slides. Sections were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature (RT), washed three times with PBS, and permeabilized with 0.1% saponin in PBS for 10 min at RT. Then, fixed sections were incubated overnight at 4°C with fibrinogen polyclonal rabbit antibody (1:100; Dako), diluted in blocking solution, and incubated for 1 hour at RT with a fluorescent secondary antibody, anti-rabbit IgG Alexa Fluor 488 (1:100; Invitrogen). The sections were lastly mounted in PBS/glycerol and photographed on a fluorescence microscope.
Statistical analysis
All analyses were performed using Prism 7.0 (GraphPad Software, La Jolla, CA). Data are shown as the means ± SD unless otherwise stated. Statistically significant differences between two groups were determined by unpaired Student’s t tests, and P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank K. Wang for critical comments on the statistical analysis of our data. Funding: This work was supported by the State Key Program of the National Natural Science Foundation of China (no. 81830067), the NIH (no. U19AI089672), the National Key R&D Program of China (no. 2018YFA0507300), and the National Natural Science Foundation of China (no. 81802033). Author contributions: Conceptualization: W.X. Methodology: Y.F., T.L., Y.T., and Y.D. Investigation: Y.F., T.L., Y.D., F.Z., X.L., and Q.W. Writing (original draft): W.X. and Y.F. Writing (review and editing): W.X. and L.C. Funding acquisition: W.X., L.C., and Y.D. Resources: B.G., Y.C., L.C., and Q.Z. Supervision: W.X. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/9/eaay9269/DC1
Fig. S1. FGL2 deficiency has no significant effect on both fibrin deposition in the spleen and coagulation function of the parasite-infected mice.
Fig. S2. sFGL2 has no effect on parasite-specific antibody production.
Fig. S3. sFGL2 has no effect on parasite-specific CD4+ T cell activation.
Fig. S4. sFGL2 has no effect on parasite-specific CD8+ T cell activation.
Fig. S5. Full blots of the effect of rFGL2 administration on both mitogen-activated protein kinase and NF-κB activity in P. chabaudi lysate–stimulated macrophages by rFGL2.
Fig. S6. Full blots of the FcγRIIB-dependent inhibition of JNK in P. chabaudi lysate–activated macrophages by sFGL2.
Fig. S7. Full blots of the inhibition of JNK in P. falciparum lysate–activated macrophages by rFGL2.
Table S1. Detailed information of patients with malaria.
REFERENCES AND NOTES
- 1.Krishnegowda G., Hajjar A. M., Zhu J., Douglass E. J., Uematsu S., Akira S., Woods A. S., Gowda D. C., Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: Cell signaling receptors, glycosylphosphatidylinositol (GPI) structural requirement, and regulation of GPI activity. J. Biol. Chem. 280, 8606–8616 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coban C., Ishii K. J., Kawai T., Hemmi H., Sato S., Uematsu S., Yamamoto M., Takeuchi O., Itagaki S., Kumar N., Horii T., Akira S., Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J. Exp. Med. 201, 19–25 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffith J. W., Sun T., McIntosh M. T., Bucala R., Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J. Immunol. 183, 5208–5220 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen S., McGregor I. A., Carrington S., Gamma-globulin and acquired immunity to human malaria. Nature 192, 733–737 (1961). [DOI] [PubMed] [Google Scholar]
- 5.Meding S. J., Langhorne J., CD4+ T cells and B cells are necessary for the transfer of protective immunity to Plasmodium chabaudi chabaudi. Eur. J. Immunol. 21, 1433–1438 (1991). [DOI] [PubMed] [Google Scholar]
- 6.Stephens R., Albano F. R., Quin S., Pascal B. J., Harrison V., Stockinger B., Kioussis D., Weltzien H.-U., Langhorne J., Malaria-specific transgenic CD4+ T cells protect immunodeficient mice from lethal infection and demonstrate requirement for a protective threshold of antibody production for parasite clearance. Blood 106, 1676–1684 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Imai T., Ishida H., Suzue K., Taniguchi T., Okada H., Shimokawa C., Hisaeda H., Cytotoxic activities of CD8+ T cells collaborate with macrophages to protect against blood-stage murine malaria. eLife 4, e04232 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Junqueira C., Barbosa C. R. R., Costa P. A. C., Teixeira-Carvalho A., Castro G., Sen Santara S., Barbosa R. P., Dotiwala F., Pereira D. B., Antonelli L. R., Lieberman J., Gazzinelli R. T., Cytotoxic CD8+ T cells recognize and kill Plasmodium vivax-infected reticulocytes. Nat. Med. 24, 1330–1336 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stanisic D. I., Barry A. E., Good M. F., Escaping the immune system: How the malaria parasite makes vaccine development a challenge. Trends Parasitol. 29, 612–622 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Woodberry T., Minigo G., Piera K. A., Amante F. H., Pinzon-Charry A., Good M. F., Lopez J. A., Engwerda C. R., McCarthy J. S., Anstey N. M., Low-level Plasmodium falciparum blood-stage infection causes dendritic cell apoptosis and dysfunction in healthy volunteers. J. Infect. Dis. 206, 333–340 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Pinzon-Charry A., Woodberry T., Kienzle V., McPhun V., Minigo G., Lampah D. A., Kenangalem E., Engwerda C., López J. A., Anstey N. M., Good M. F., Apoptosis and dysfunction of blood dendritic cells in patients with falciparum and vivax malaria. J. Exp. Med. 210, 1635–1646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urban B. C., Ferguson D. J. P., Pain A., Willcox N., Plebanski M., Austyn J. M., Roberts D. J., Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature 400, 73–77 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Wilson N. S., Behrens G. M. N., Lundie R. J., Smith C. M., Waithman J., Young L., Forehan S. P., Mount A., Steptoe R. J., Shortman K. D., de Koning-Ward T. F., Belz G. T., Carbone F. R., Crabb B. S., Heath W. R., Villadangos J. A., Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat. Immunol. 7, 165–172 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Horne-Debets J. M., Faleiro R., Karunarathne D. S., Liu X. Q., Lineburg K. E., Poh C. M., Grotenbreg G. M., Hill G. R., MacDonald K. P. A., Good M. F., Renia L., Ahmed R., Sharpe A. H., Wykes M. N., PD-1 dependent exhaustion of CD8+ T cells drives chronic malaria. Cell Rep. 5, 1204–1213 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Illingworth J., Butler N. S., Roetynck S., Mwacharo J., Pierce S. K., Bejon P., Crompton P. D., Marsh K., Ndungu F. M., Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J. Immunol. 190, 1038–1047 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirunpetcharat C., Good M. F., Deletion of Plasmodium berghei-specific CD4+ T cells adoptively transferred into recipient mice after challenge with homologous parasite. Proc. Natl. Acad. Sci. U.S.A. 95, 1715–1720 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierce S. K., Miller L. H., World Malaria Day 2009: What malaria knows about the immune system that immunologists still do not. J. Immunol. 182, 5171–5177 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shalev I., Liu H., Koscik C., Bartczak A., Javadi M., Wong K. M., Maknojia A., He W., Liu M. F., Diao J., Winter E., Manuel J., McCarthy D., Cattral M., Gommerman J., Clark D. A., Phillips M. J., Gorczynski R. R., Zhang L., Downey G., Grant D., Cybulsky M. I., Levy G., Targeted deletion of fgl2 leads to impaired regulatory T cell activity and development of autoimmune glomerulonephritis. J. Immunol. 180, 249–260 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Yan J., Kong L.-Y., Hu J., Gabrusiewicz K., Dibra D., Xia X., Heimberger A. B., Li S., FGL2 as a multimodality regulator of tumor-mediated immune suppression and therapeutic target in gliomas. J. Natl. Cancer Inst. 107, djv137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan C. W. Y., Kay L. S., Khadaroo R. G., Chan M. W. C., Lakatoo S., Young K. J., Zhang L., Gorczynski R. M., Cattral M., Rotstein O., Levy G. A., Soluble fibrinogen-like protein 2/fibroleukin exhibits immunosuppressive properties: Suppressing T cell proliferation and inhibiting maturation of bone marrow-derived dendritic cells. J. Immunol. 170, 4036–4044 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Latha K., Yan J., Yang Y., Gressot L. V., Kong L.-Y., Manyam G., Ezhilarasan R., Wang Q., Sulman E. P., Eric Davis R., Huang S., Fuller G. N., Rao A., Heimberger A. B., Li S., Rao G., The role of fibrinogen-like protein 2 on immunosuppression and malignant progression in glioma. J. Natl. Cancer Inst. 111, 292–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding J. W., Ning Q., Liu M. F., Lai A., Leibowitz J., Peltekian K. M., Cole E. H., Fung L. S., Holloway C., Marsden P. A., Yeger H., Phillips M. J., Levy G. A., Fulminant hepatic failure in murine hepatitis virus strain 3 infection: Tissue-specific expression of a novel fgl2 prothrombinase. J. Virol. 71, 9223–9230 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butler N. S., Moebius J., Pewe L. L., Traore B., Doumbo O. K., Tygrett L. T., Waldschmidt T. J., Crompton P. D., Harty J. T., Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat. Immunol. 13, 188–195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rai D., Pham N.-L. L., Harty J. T., Badovinac V. P., Tracking the total CD8 T cell response to infection reveals substantial discordance in magnitude and kinetics between inbred and outbred hosts. J. Immunol. 183, 7672–7681 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Favre N., Ryffel B., Bordmann G., Rudin W., The course of Plasmodium chabaudi chabaudi infections in interferon-gamma receptor deficient mice. Parasite Immunol. 19, 375–383 (1997). [DOI] [PubMed] [Google Scholar]
- 26.van der Heyde H. C., Pepper B., Batchelder J., Cigel F., Weidanz W. P., The time course of selected malarial infections in cytokine-deficient mice. Exp. Parasitol. 85, 206–213 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Su Z., Stevenson M. M., Central role of endogenous gamma interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect. Immun. 68, 4399–4406 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ing R., Stevenson M. M., Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect. Immun. 77, 770–782 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teirlinck A. C., McCall M. B. B., Roestenberg M., Scholzen A., Woestenenk R., de Mast Q., van der Ven A. J. A. M., Hermsen C. C., Luty A. J. F., Sauerwein R. W., Longevity and composition of cellular immune responses following experimental Plasmodium falciparum malaria infection in humans. PLOS Pathog. 7, e1002389 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deshmane S. L., Kremlev S., Amini S., Sawaya B. E., Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 29, 313–326 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrison B. E., Park S. J., Mooney J. M., Mehrad B., Chemokine-mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. J. Clin. Invest. 112, 1862–1870 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu H., Shalev I., Manuel J., He W., Leung E., Crookshank J., Liu M. F., Diao J., Cattral M., Clark D. A., Isenman D. E., Gorczynski R. M., Grant D. R., Zhang L., Phillips M. J., Cybulsky M. I., Levy G. A., The FGL2-FcγRIIB pathway: A novel mechanism leading to immunosuppression. Eur. J. Immunol. 38, 3114–3126 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Shalev I., Wong K. M., Foerster K., Zhu Y., Chan C., Maknojia A., Zhang J., Ma X. Z., Yang X. C., Gao J. F., Liu H., Selzner N., Clark D. A., Adeyi O., Phillips M. J., Gorczynski R. R., Grant D., McGilvray I., Levy G., The novel CD4+CD25+ regulatory T cell effector molecule fibrinogen-like protein 2 contributes to the outcome of murine fulminant viral hepatitis. Hepatology 49, 387–397 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Hisaeda H., Maekawa Y., Iwakawa D., Okada H., Himeno K., Kishihara K., Tsukumo S. I., Yasutomo K., Escape of malaria parasites from host immunity requires CD4+CD25+ regulatory T cells. Nat. Med. 10, 29–30 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Nie C. Q., Bernard N. J., Schofield L., Hansen D. S., CD4+ CD25+ regulatory T cells suppress CD4+ T-cell function and inhibit the development of Plasmodium berghei-specific TH1 responses involved in cerebral malaria pathogenesis. Infect. Immun. 75, 2275–2282 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cambos M., Bélanger B., Jacques A., Roulet A., Scorza T., Natural regulatory (CD4+CD25+FOXP+) T cells control the production of pro-inflammatory cytokines during Plasmodium chabaudi adami infection and do not contribute to immune evasion. Int. J. Parasitol. 38, 229–238 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Marazzi S., Blum S., Hartmann R., Gundersen D., Schreyer M., Argraves S., von Fliedner V., Pytela R., Rüegg C., Characterization of human fibroleukin, a fibrinogen-like protein secreted by T lymphocytes. J. Immunol. 161, 138–147 (1998). [PubMed] [Google Scholar]
- 38.Kurup S. P., Obeng-Adjei N., Anthony S. M., Traore B., Doumbo O. K., Butler N. S., Crompton P. D., Harty J. T., Regulatory T cells impede acute and long-term immunity to blood-stage malaria through CTLA-4. Nat. Med. 23, 1220–1225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clatworthy M. R., Willcocks L., Urban B., Langhorne J., Williams T. N., Peshu N., Watkins N. A., Floto R. A., Smith K. G. C., Systemic lupus erythematosus-associated defects in the inhibitory receptor FcγRIIb reduce susceptibility to malaria. Proc. Natl. Acad. Sci. U.S.A. 104, 7169–7174 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willcocks L. C., Carr E. J., Niederer H. A., Rayner T. F., Williams T. N., Yang W., Scott J. A. G., Urban B. C., Peshu N., Vyse T. J., Lau Y. L., Lyons P. A., Smith K. G. C., A defunctioning polymorphism in FCGR2B is associated with protection against malaria but susceptibility to systemic lupus erythematosus. Proc. Natl. Acad. Sci. U.S.A. 107, 7881–7885 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waisberg M., Tarasenko T., Vickers B. K., Scott B. L., Willcocks L. C., Molina-Cruz A., Pierce M. A., Huang C. Y., Torres-Velez F. J., Smith K. G. C., Barillas-Mury C., Miller L. H., Pierce S. K., Bolland S., Genetic susceptibility to systemic lupus erythematosus protects against cerebral malaria in mice. Proc. Natl. Acad. Sci. U.S.A. 108, 1122–1127 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Talaat R. M., Mohamed S. F., Bassyouni I. H., Raouf A. A., Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: Correlation with disease activity. Cytokine 72, 146–153 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Warren M. K., Vogel S. N., Bone marrow-derived macrophages: Development and regulation of differentiation markers by colony-stimulating factor and interferons. J. Immunol. 134, 982–989 (1985). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/9/eaay9269/DC1
Fig. S1. FGL2 deficiency has no significant effect on both fibrin deposition in the spleen and coagulation function of the parasite-infected mice.
Fig. S2. sFGL2 has no effect on parasite-specific antibody production.
Fig. S3. sFGL2 has no effect on parasite-specific CD4+ T cell activation.
Fig. S4. sFGL2 has no effect on parasite-specific CD8+ T cell activation.
Fig. S5. Full blots of the effect of rFGL2 administration on both mitogen-activated protein kinase and NF-κB activity in P. chabaudi lysate–stimulated macrophages by rFGL2.
Fig. S6. Full blots of the FcγRIIB-dependent inhibition of JNK in P. chabaudi lysate–activated macrophages by sFGL2.
Fig. S7. Full blots of the inhibition of JNK in P. falciparum lysate–activated macrophages by rFGL2.
Table S1. Detailed information of patients with malaria.