

We develop a deep learning method that predicts atomic structure directly from experimental atomic force microscopy images.
Abstract
Atomic force microscopy (AFM) with molecule-functionalized tips has emerged as the primary experimental technique for probing the atomic structure of organic molecules on surfaces. Most experiments have been limited to nearly planar aromatic molecules due to difficulties with interpretation of highly distorted AFM images originating from nonplanar molecules. Here, we develop a deep learning infrastructure that matches a set of AFM images with a unique descriptor characterizing the molecular configuration, allowing us to predict the molecular structure directly. We apply this methodology to resolve several distinct adsorption configurations of 1S-camphor on Cu(111) based on low-temperature AFM measurements. This approach will open the door to applying high-resolution AFM to a large variety of systems, for which routine atomic and chemical structural resolution on the level of individual objects/molecules would be a major breakthrough.
INTRODUCTION
Scanning probe microscopy has been the engine of characterization in nanoscale systems (1). Atomic force microscopy (AFM) (2) in particular has developed into a leading technique for high-resolution studies without material restrictions (3–5). It is increasingly being used for detailed characterization in a wide variety of physical, biological, and chemical processes (6, 7). Pioneering experimental studies are now providing atomic-scale insight into, for example, friction, catalytic reactions, electron transport, and optical response. In general for AFM, the tip itself has often been the barrier to translating atomic resolution into physical understanding, with many images and processes ultimately being identified as a convolution with the tip structure (8, 9). While many partially successful efforts in tip functionalization were attempted in the last decade, the use of a CO molecule attached to a metal tip in low-temperature ultrahigh vacuum AFM (CO-AFM) measurements (5, 10) has offered a path to reliable, outstanding resolution. The use of a relatively inert tip, with respect to the molecule-substrate interaction (11), means that it can approach very close to the object of interest without excessive attractive forces resulting in unintentional lateral manipulation of the target molecule. This allows the interaction to be dominated by extremely short-ranged Pauli repulsion between atoms in the sample and at the tip apex, providing the very high resolution essential to the technique. In particular, CO-AFM now offers an unprecedented window into molecular structure on surfaces—aside from the detailed resolution of the results of molecular assembly (12, 13), it is possible to study bond order (14), charge distributions (15, 16), and the individual steps of on-surface chemical reactions (17–20).
As yet, most CO-AFM studies have been focused on planar molecular systems, where the experimental image requires almost no interpretation (5, 10, 21). Even where understanding is not immediately obvious, such as due to controversies over the nature of observed bonds (22), efficient models have been developed (13, 23–26) that explain the contrast mechanism in terms of the tip-surface interaction and CO lateral flexibility. However, the further the systems are from two-dimensional (2D) molecules containing only hydrogen and carbon, the more complex and time-consuming (if not impossible) the interpretation process becomes (18, 27–30). While recent measurements using rigid O-terminated copper tips make interpreting images of flat systems even easier (31, 32), the rigidity also means that even fewer atoms can be characterized when moving to 3D systems—the flexibility of CO allows it to sample molecular “edges” in more detail. In recent years, CO-AFM has moved toward measuring truly unknown structures (30, 33–35), where it has overcome many of the limitations of techniques such as nuclear magnetic resonance and mass spectrometry. It is clear that this trend is going to continue and potentially even accelerate, in particular for innovative studies, e.g., in life sciences or biochemistry (6, 7), demonstrated manifestly in the first CO-AFM images of DNA (36). Reliable interpretation of these data becomes a vast exploration through all possible molecules, configurations, and imaging parameters to find an agreement. This is impractical in anything beyond very simple systems, severely limiting the ultimate power of the technique.
In this work, we couple a systematic software approach with detailed experimental CO-AFM imaging to understand and predict AFM images for molecules of any size, configuration, or orientation without prior knowledge of the system being studied. We use the latest modeling approaches to efficiently synthesize 3D AFM data (37) from 134,000 isolated molecules. These were scanned from representative directions to establish physical descriptors that characterize a series of slices through the data in a given direction. For a given series of experimental images, we then apply a deep learning infrastructure (38–41) to find a descriptor match and predict the molecular structure directly. The method is validated by comparison to a systematic CO-AFM experimental study of orientations of camphor molecules on a copper surface. This automated structure discovery AFM (ASD-AFM) approach will open the door to applying high-resolution AFM to a huge variety of systems for which routine atomic and chemical structural resolution on the level of individual objects/molecules would be a major breakthrough.
RESULTS
The measured signal in CO-AFM is the shift of the resonance frequency of the cantilever (Δ𝑓), which is due to the sum of all conceivable tip-sample interactions. In CO-AFM, the Δ𝑓 signal is, to a large extent, determined by the interaction of oxygen in the CO molecule and the closest atoms of the sample directly under the tip. Nevertheless, because of the lateral flexibility of the CO, the image contrast is not related to the atomic positions in a trivial fashion. We will describe a methodology that aims to invert this imaging process and yield the atomic coordinates directly from a set of measured (or simulated) Δ𝑓 data. Briefly, this involves developing an image descriptor, i.e., a 2D representation of molecular structure, that encodes the positions of the atoms in the object molecule—this can be calculated directly if the positions are known. We train a neural network (NN) to reproduce this image descriptor directly from the Δ𝑓 data using simulated AFM images and then verify this approach using simulated images from molecules not included in the training data. Last, we will use experimental AFM images as a final test of the proposed methodology.
Inverse imaging problem
Reconstruction of molecular structures from AFM images can be seen as the search for an inverse function (Φ−1) to the imaging process , where are the positions and atomic number of nuclei, and is the value of measured frequency shift in each point of space (see Fig. 1). Analysis and understanding of the imaging process Φ are therefore crucial for obtaining (Φ−1). In particular, it is important to estimate how well conditioned the inverse operation is and to identify which information is preserved or where information is lost.
Fig. 1. Schematic illustration of the CO-tip AFM imaging process and the proposed solution for the inverse imaging problem.
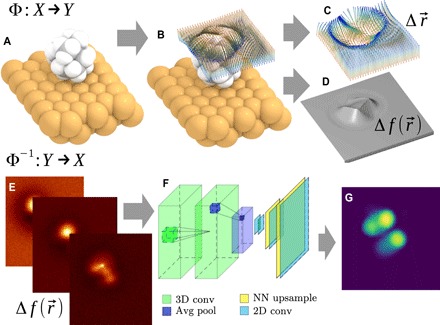
(A to D) The imaging process Φ : X→ Y of molecular geometry X (A) originates predominantly from probe particle (PP) displacement due to interactions with sample atoms (B). The resulting PP displacement is plotted in (C). The fibers show deflection of the PP as it approaches the surface, with the red-blue gradient representing the tip-sample distance (red, far; blue, close). (D) The resulting AFM frequency shift [] images Y obtained by integrating the forces felt by the relaxed PP over its path. (E to G) The inverse imaging process (i.e., reconstruction of geometry) Φ−1 : Y→ X approximated by a convolutional NN (F) transforming a 3D stack of AFM images Y (E) to a description of the molecular geometry X [represented by, e.g., van der Waals spheres (G)].
The imaging process can be decomposed into the following sequence of operations:
(1) Atoms of the sample generate various force fields in the space around them (e.g., electrostatic, van der Waals, and Pauli repulsion). Many methods ranging from empirical potentials [e.g., (42)] to ab initio calculations [e.g., (43)] were applied in the past to approximate those force fields.
(2) The tip apex (e.g., CO molecule) relaxes under the influence of those force fields as it approaches toward the sample (see Fig. 1B). This means that the force fields are sampled in distorted (relaxed) coordinates (Fig. 1C). These distortions are crucial for understanding features in AFM images. The process can be simulated by a simple mechanical model [e.g., probe particle (PP) model (21, 24)].
(3) Forces felt by the relaxed PP are integrated over its path (Fig. 1C), and this causes changes in the measured oscillation frequency (Fig. 1D). The change of frequency Δ𝑓 can be therefore calculated using a simple formula (44).
Furthermore, from previous simulations of the AFM imaging process (13, 21, 24, 25, 45), it is clear that images are extremely sensitive to even minor variations of height (z coordinate) of the topmost atoms, and conversely very insensitive to atoms >0.5 Å below this. In addition, the chemical identity of the atom cannot be easily determined from observed contrast, as it depends on the z coordinate, the chemical neighborhood, and orbital structure (e.g., nitrogen can appear both as a depression and a protrusion in carbonaceous aromatic systems). Instead, the characteristic topology of interatomic potentials (saddle ridges between nearby atoms, vertexes between those ridges, and contrast inversion) can be determined from AFM data as a fingerprint of typical chemical groups or bonding configurations. The electrostatic force has a rather small contribution to vertical force in contact but often considerably distorts the image laterally (25, 46).
Overall, the imaging process (Φ; Fig. 1, A to D) is a complex and highly nonlinear function, and its inversion (Φ−1) cannot be easily expressed by any analytic equation or practical numerical algorithm. Hence, we use an NN (Fig. 1F) as an efficient universal fitting scheme to learn an approximation to Φ−1 from example atomic structures and corresponding 3D AFM data stacks (a stack is a set of constant height images at different vertical positions; Fig. 1E). The image-like structure of input AFM data calls for the use of a deep convolutional NN (CNN) (38), optimized for machine learning (ML) of regular 3D grids.
Generation of training data
The main problem in training deep convolutional networks is providing sufficiently labeled training data (from thousands to millions of input-output pairs). High-resolution AFM experiments are time intensive, requiring several hours to acquire a single 3D data stack, which would render direct training on experimental data impractical. In addition, experimental data are a priori unlabeled (i.e., we do not know the correct interpretation), and interpretation of 3D features in AFM data is currently a difficult task, even for human experts. Hence, human labeling cannot provide us with reliable labels.
Therefore, the only feasible option is to train a model on simulated data, where correct interpretation (labels) is known a priori. For our reference simulations, the geometries of sample molecules were taken from a well-known database of 134,000 isolated small organics (47), structurally optimized with density functional theory (DFT).
Our methodology uses a new, highly efficient graphical processing unit implementation of the PP model (24, 48), which allows the generation of ~50 input-output pairs (i.e., 3D AFM data stacks and 2D image representation of structure) per second. This implementation is performance optimized, allowing rapid experimentation with new settings and CNN architectures while simultaneously generating data on the fly. This eliminates issues related to the storage of terabytes of training data otherwise needed. For each molecule, we first calculate the force field sampled on a regular 3D grid (this step takes ~0.1 s on a desktop computer), and then this force field can be rapidly interpolated to generate simulated constant-height Δ𝑓 images from 10 to 20 orientations of a given molecule (dependent on molecule symmetries), each of which takes ~0.02 s. These orientations are initially uniformly distributed over a sphere, but we then weight the final selection to orientations that expose more atoms to the tip. This avoids images where just a single atom is visible and increases the information available per stack in the training process. Here, and in general, the z coordinate is defined as the distance from the carbon in the CO-tip apex to the atom closest to the tip in a particular molecular orientation. Each scan starts at z = 8.0 Å and continues 3.0 Å toward the molecule in steps of 0.1 Å. These 30 slices of vertical force are transformed into 20 slices of frequency shift (2.0 Å of valid data) using the Giessibl formula (44), forming a stack from simulated data. Optimization of this choice of z window is possible for a given experiment, but this selection provided the best performance for the results presented here.
Image descriptors
In general, when trying to predict molecular geometries from AFM images, while it may seem most obvious to directly convert an image stack to a set of xyz coordinates, this is not an efficient descriptor in a CNN model [see expanded discussion in the Supplementary Materials (SM)]. Hence, we opt to represent the output geometry in an image-like form that is directly related to the atomic coordinates. The selection of this 2D image descriptor is critical to an efficient model and must be chosen such that it can be realistically and reliably determined from AFM data. The descriptor can be considered as the language with which we wish to analyze the problem, and the choice of language is enforced by the reference database—during the generation of the simulated image database, we also calculate 2D image descriptors for all molecules and orientations.
Then, we ask the CNN to translate the data stacks into this language. It achieves this by extracting features in a given Δ𝑓 slice as a function of their character and position. It does this simultaneously for all given Δ𝑓 slices in a data stack—features that appear in multiple slices are much more likely to be identified as important. As the deep CNN moves through its multiple layers (Fig. 1F), it filters these features according to the chosen biases and weights (manually optimized in this work, see SM), ultimately identifying a critical feature map. The CNN then begins the second half of its job, building a 2D image descriptor from this feature map. Using the reference database for that descriptor, it makes a prediction of the best match for a given feature.
We designed several physically meaningful representations of molecular structure on a grid, with specifics of AFM microscopy in mind (see discussion in the SM). In all cases, we represent the data as a single 2D image with the same lateral resolution as the input AFM data, which simplifies the computational analysis and allows for quick validation via human users. For the rest of the discussion, we use the vdW-Spheres representation—an intuitive representation of molecular structure by their van der Waals radii, commonly used in chemical visualization programs. For each molecule and orientation, we calculate the vdW-Spheres descriptor from the reference database as follows: We calculate the van der Waals radius of all atoms and then plot this in 2D using a z range starting from the position of the highest atom to 1.5 Å below it; i.e., contributions below this are ignored. The relative height of atoms in this window is represented by their brightness in the 2D image descriptor.
Geometry prediction from simulated AFM data
To benchmark the methodology, we used the trained CNN model to predict the geometry of several molecules that were not included in the training set. The internal quality of the model can be judged by how well the predicted 2D image descriptor (derived from the simulated AFM 3D image stack) matches the reference descriptor calculated directly from the molecular geometry. In the first example (Fig. 2, A to F), we picked a molecule (an isomer of C7H10O2) that has a functional group and a nonplanar geometry as representative of the types of molecule we wish to identify. The prediction qualitatively matches the reference, capturing all the key atoms except the hydrogen of the hydroxyl group, which is present in the analytically computed reference image representation. It is very difficult to identify the lower-lying atoms from the AFM images. For the molecule shown in Fig. 2 (A to F), it would not be possible for a human expert to identify the hydrogen atom of the hydroxyl group. The goal of the introduced ideal image representation, i.e., vdW-Spheres representation, is to train a CNN to extract as much as possible structural information presented in an individual AFM stack of data and store it in a compressed readable format.
Fig. 2. Examples of CNN prediction from simulated and experimental data.
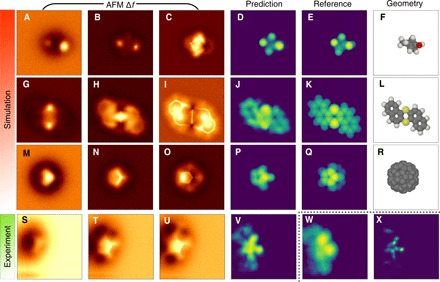
(A to F) A molecule from the validation set with formula C7H10O2. (G to L) A dibenzo[a,h]thianthrene molecule (49). (M to U) A fullerene C60 [experimental data in (S) to (U)]. (V to X) Comparison of image descriptors, vdW-Spheres, height map, and atomic disk representation (see the SM for explanation) predicted from experimental images of C60. Columns 1 to 3 show simulated AFM signal (Δf) at different heights. Column 4 shows the vdW-Spheres representation predicted by the trained CNN (naturally, the reference is not available for experiment). Column 5 shows the reference vdW-Spheres representation calculated directly from geometry. Column 6 depicts a 3D render of the molecule.
As another example, we consider a dibenzo[a,h]thianthrene molecule, which has been previously experimentally studied (Fig. 2, G to L) (49). The CNN is again able to predict most of molecular features in the vdW-Spheres representation, in particular, identifying the two dominant sulfur atoms. The remaining atoms of the aromatic system are also predicted, but they are not as well separated as in the reference. CNN-predicted properties are typically blurred, and this is somewhat dependent on the choice of 2D image descriptor (see fig. S3G).
The last example is a fullerene C60 molecule oriented with a pentagon upward. We performed a prediction of the vdW-Spheres representation based both on simulation (Fig. 2, M to R) and newly measured experimental data (Fig. 2, S to V). The pentagons are oriented slightly in an asymmetric manner with three carbon atoms up. The main features, i.e., eight top-most atoms, are reproduced rather well in the CNN prediction, while the remaining atoms remain invisible. This is true for both simulated and experimental images. In the experimental image, however, there are visible artifacts originating from dark attractive areas of C60, which are not visible in the simulated image. This is a clear indication that the simulation does not reproduce this particular experiment sufficiently well. Despite this fact, the CNN prediction is robust enough to consistently render the top-most atoms. More examples from our training set can be found in fig. S4.
To illustrate how our method can aid in the discrimination of unknown molecules and separate chemical information and physical topography, we compare three different derivatives of antraquinone with a different number of chlorine atoms in Fig. 3. In this illustrative example, the molecules are tilted so that the bottom edge is higher than the upper edge, making this a 3D problem with a peculiar image contrast over the edge that can hardly be deciphered by an expert. Although each molecule provides clearly distinct AFM images, it is rather difficult to rationalize the differences in terms of atomic structure. Any similarity between molecules in the first and second rows is hardly visible from the AFM pictures. In contrast, the predicted vdW-Spheres map shows a change in atomic radius in one or two atomic sites, while the rest of the molecular structure is preserved. While disentangling the atomic type from its z position is difficult based on the vdW-Spheres image description, the different atomic types should result in a different decay of the Δf contrast as a function of the tip-sample distance. Hence, it should be possible to differentiate atomic species. In particular, a modified CNN (shown in Fig. 3 as column type map) learned to discriminate small peripheral atoms (hydrogen, red) from larger peripheral atoms (chlorine and oxygen, green), leaving aside rather indiscriminate carbon backbone (blue). The network identified substitution of a hydrogen atom by chlorine. While showing the potential of the technique in terms of recognition, the prediction is not yet fully reliable, as can be seen from misidentified oxygen as small (red) in the second row.
Fig. 3. Discrimination of functional groups.
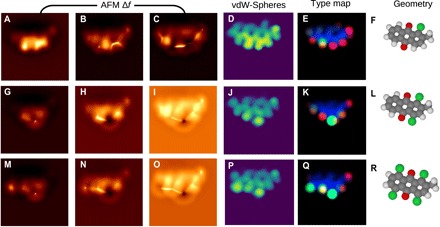
Here, we compare three hypothetical anthraquinone derivatives that have differing numbers of chlorine atoms: one chlorine (A-F), two chlorines (G-L) and four chlorines (M-R). The first three columns show simulated AFM images at far, middle, and close tip-sample distances. The fourth column shows the associated NN prediction for the vdW-Spheres representation. The fifth column shows atom-type prediction from another NN that discriminates three different types of atoms: hydrogens (red), nonhydrogen peripheral (green), and carbon backbone (blue). The final column shows the molecular geometry. Note that the molecule is tilted so that the bottom edge is higher than the upper edge.
Geometry prediction from experimental AFM data
The true validation of our ML approach is to make predictions directly from experimental AFM images. Ultimately, this would be done from images of an unknown system, but as a benchmark for our first iteration of the method, we apply it to find molecular configurations of a known molecule. Here, we selected 1S-camphor as the target molecule due to its 3D geometry and potential for adopting multiple distinct adsorption geometries on a Cu(111) surface. Combined scanning tunneling microscopy (STM) and AFM imaging allowed us to distinguish eight characteristic adsorption geometries with reproducible data in each case. Further analysis reduced this to a set of five distinct configurations clean enough for good comparison, and we acquired a set of constant-height Δ𝑓 images in each case (see the SM for details). Even highly trained experts were not able to decipher the molecular structure from these images, and they provided an excellent challenge and example for the CNN model. The 3D experimental image stack (Fig. 4, A to C) is fed into the CNN model, and a 2D image descriptor (vdW-Spheres) is predicted on the basis of this data (Fig. 4D). This experimental descriptor is then compared via cross-correlation to a set of descriptors calculated directly from atomic coordinates taken from a set of uniformly distributed molecular rotations (Fig. 4E). The best fit gives us a prediction of the molecular configuration corresponding to the original descriptor from experimental data (Fig. 4F). Qualitatively, the match between experimental and simulated descriptors is very good, reproducing the performance seen with purely simulated data (Fig. 2). To explore the plausibility of the predicted geometries, we now reverse the inverse imaging process and consider the predicted simulated images for the best fit descriptor (Fig. 4, G to I). In all cases, the simulated images qualitatively capture the main features seen in the experimental images. In cases 1 to 4, agreement is generally good at all heights, but the simulated image tends to be somewhat sharper than the experiments at close approach. For case 5, the core of the simulated image is representative of experiments, but some of the extended features are clearly absent. Furthermore, note that experimental image 5A in Fig. 4 shows no atomic features (the interactions are purely attractive), whereas the simulated image 5G clearly does (showing the onset of repulsive short-range interactions). This is because the CNN was consciously trained only on data containing atomic-like features, as those are critical for identification, and not the kind of large tip-sample distance used in 5A.
Fig. 4. Identification of the 1S-camphor adsorption configurations on Cu(111) with ASD-AFM.
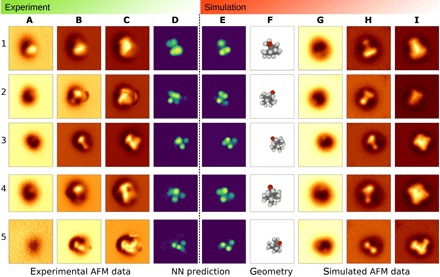
1 to 5 refer to distinct molecular configurations with experiments in columns (A) to (D) and simulations in columns (E) to (I). Selected experimental AFM images (out of 10 slices used for input): at (A), far; (B), middle; (C), close tip-sample distances; and NN prediction (D) for the vdW-Spheres representation. The vdW-Spheres representation shown in (E) corresponds to the full molecular configuration (F) resulting from the best match to the experiment. The corresponding simulated AFM images are given in (G) to (I) (far, middle, and close).
DISCUSSION
The aim of this work was to establish a reliable and rapid method for solving a problem that expert humans cannot—the interpretation of high-resolution AFM images of complex 3D molecules. We have demonstrated that our ML method based on a CNN architecture can solve this problem with trivial computational effort. In its current form, the model can, e.g., identify adsorption configurations accurately. On a complex system, this allows us to markedly reduce the number of possible molecular solutions from a set of experimental images.
However, we believe that this is only the first step in a developing analysis field, and it is clear that several further problems need to be tackled if we wish to increase prediction accuracy even further. Simple improvements include introducing a bigger variety of atoms into the training set (with a very large initial computational cost) and creating an integral model that can predict multiple 2D image representations simultaneously, improving model robustness for features recognition. In the medium term, while our current approach using the PP method (i.e., reusing a precalculated force-field grid for scans from multiple directions) is highly efficient, it prevents a simple implementation of more sophisticated nonspherical electrostatics (e.g., quadrupoles) that have been shown to be important for CO tip simulations in certain systems (45, 50). While we consider this limitation of the underlying simulation model a secondary issue in the development of a reliable ML architecture, we have already begun exploring efficient solvers for more sophisticated models based on the electron density from DFT (26). A more pressing concern for accuracy in simulated images is the role of surface- and tip-induced molecular displacements. For the latter, this has generally been ignored in previous simulations of CO-tip AFM experiments, and fixed geometries are considered throughout. In this work, we considered how molecular tilting and functional group rotations affected the predicted images (see section S3). It is clear that these can change the predicted simulated images, particularly at close approach, and finding a systematic way to include these in the matching process could notably improve accuracy. We also considered the possible changes of molecular configurations when adsorbed on the surface (see section S2), but any errors seen were not in the predictions of the CNN model, and improvements would require advances beyond the standard methods used to obtain accurate adsorption structures—a separate research field.
Last, the nature of the AFM measurement itself causes a particular difficulty in the uniqueness of the molecular solutions. For certain configurations, common in small nonplanar molecules, AFM data may provide information only about a very limited number of atoms, and this may lead to several molecular solutions being almost equivalent in the quality of best fit to experiments (see section S3). In systems where this is a problem, considering several experimental configurations of the same molecule, as done here, makes identification notably easier. More generally, we are looking at including multiple channels of information for a single configuration by using an image descriptor incorporating tip-dependent electrostatic information available via other tip terminations (25, 46, 51). This could also be extended to incorporate simultaneous fitting to the Kelvin probe force microscopy data (52–55), further improving the uniqueness of predictions.
Despite these challenges, the approach is immediately applicable to a wide variety of complex molecular systems where conventional interpretation approaches either have failed or cannot even be attempted. Hence, it promises the availability of atomic and chemical structural resolution in systems where it offers the prospect of major impact.
MATERIALS AND METHODS
ML model architecture
The architecture of our CNN is similar to the encoder-decoder–type networks that have been used in, for example, image segmentation (56). At the input side, it comprises three layers of 3D convolutional filters (3 × 3 × 3) interleaved by average pooling (2 × 2 × 2), which reduces the size of the input image by a factor of 8 in the 𝑥, 𝑦 dimensions. This information bottleneck is motivated by the fact that input AFM images are mostly rather smooth and carry a limited amount of information (i.e., just position and size of a few atoms). Downsampling also helps to facilitate long-range correlations in the image using only local and cheap 3 × 3 × 3 filters. This should help to recognize larger features such as atoms and bonds spanning over tens of pixels. The data are collapsed in the z direction from 3D to 2D by the action of the pooling layers while gradually being expanded to several independent channels (2× channels by each layer). Therefore, the features obtained after this operation should encode varying z dependence of the frequency shift. The signal is further processed by three layers of purely convolutional filters operating independently on each of the 64 channels of the 2D image. In the last part of the CNN architecture, the image is expanded back to original resolution (8× in each dimension) by three bilayers of 2D convolution interleaved by NN-upsample operations. The final convolution is followed by a rectified linear unit [ReLU; (57)] activation, which basically cuts the negative part of activations from the convolution layer, leaving “unchanged” positive values. Other convolutions are followed by LeakyReLU activations with a factor of 0.1 on the negative side, so as not to completely block learning when values are under 0 (they are leaked through). The model is implemented in Keras (58) running a TensorFlow (59) backend. Optimization of kernel sizes in the convolutional layer has not been systematically tested, but for our image recognition network, small kernel sizes with additional layers have been quite effective.
The structure was motivated by the idea that the central part—i.e., the 8× downsampled representation with 64 channels—will learn to represent AFM images in terms of abstract and physically meaningful features (e.g., slope of frequency shift curve, blobs representing atoms, and characteristic sharp-line features between nearby atoms). Various physical properties, such as height maps or positions of atoms in the second upsampling stage, can then be identified from this internal abstract representation.
To make the model more robust to experimental artifacts and limitations, we added 5% white noise (representing electronic noise in the measurements) and random rectangular cutouts (60) (representing sudden jumps in the measurements) to the simulation data. Note that this also aids in avoiding problems in relation to the ill-posed nature of the force-frequency shift conversion (61, 62).
Molecular database
The original structures of the molecules in the database were optimized with DFT at the B3LYP/6-31G level (63). Using the quantum chemistry software Psi4 (64, 65), we performed single-point coupled-cluster calculations (singles and doubles, cc-pvdz basis) for all the 134,000 molecules, thus obtaining charge densities and Mulliken populations necessary to operate the PP simulator.
Experimental methods
Polished Cu(111) and Au(111) single crystals (MaTecK, Germany) were prepared by repeated Ne + sputtering (0.75 keV, 15 mA, 20 min) and annealing (850 to 900 K, 5 min) cycles. Surface cleanliness and structure were verified by STM. Sample temperatures during annealing were measured with a pyrometer (Sensortherm Metis MI16). 1S-camphor (Sigma-Aldrich; purity, >98.5%) was introduced into the vacuum system via a leak valve and deposited onto the Cu(111) surface at a low temperature (T = 20 K) to increase the number of distinct adsorption configurations and to achieve individual molecules rather than clusters on the surface. Fullerene C60 (Sigma-Aldrich; purity, >99.9%) was sublimed onto an Au(111) substrate held at ~200 K.
The STM and CO-AFM images were taken with a CreaTec LT-STM/AFM (low-temperature scanning tunneling microscope and atomic force microscope) with a commercial qPlus sensor with a Pt/Ir tip, operating at approximately T = 5 K in ultrahigh vacuum at a pressure of 1 × 10−10 mbar. The quartz cantilever (qPlus sensor) had a resonance frequency of 𝑓0 = 29939 Hz, a quality factor 𝑄 = 101099, and was operating with an oscillation amplitude of 𝐴 = 50 pm. Tip conditioning was performed by repeatedly bringing the tip into contact with the copper surface and applying bias pulses until the necessary STM resolution was achieved. The tip apex was functionalized with a CO molecule (66) before AFM measurements. The STM images were recorded in constant-current mode, while the AFM operated in constant-height mode. Raw data were used as input for the ML infrastructure. To minimize experimental artifacts that would cause problems with interpretation, we have implemented the following measures: checking the background Δ𝑓 before CO pickup (smaller value indicates sharper overall tip), scanning another CO to ensure the symmetry of the CO tip after tip passivation and prior to further AFM imaging, and confirming that the excitation (dissipation) signal remains flat/featureless during the AFM measurements.
Supplementary Material
Acknowledgments
Funding: Computing resources from the Aalto Science-IT project and CSC, Helsinki are gratefully acknowledged. This research made use of the Aalto Nanomicroscopy Center (Aalto NMC) facilities and was supported by the European Research Council (ERC 2017 AdG no. 788185 “Artificial Designer Materials”), and the Academy of Finland (project numbers 311012, 314862, and 314882; Centres of Excellence Program project no. 284621; and Academy Professor funding numbers 318995 and 320555). A.S.F. has been supported by the World Premier International Research Center Initiative (WPI), MEXT, Japan. Author contributions: F.S., P.L., and A.S.F. conceived the research. P.H., N.O., F.U., O.K., and F.F.C. developed the software and ran the simulations. B.A. performed the experiments. All authors were involved in the results analysis and contributed to the manuscript. Competing interests: The authors declare that they have no competing financial interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/9/eaay6913/DC1
Section S1. Image representations of output molecular structure
Section S2. Matching experiment to relaxed on-surface simulated configurations
Section S3. Effect of small perturbations on AFM imaging and matching
Section S4. Neural network architecture
Section S5. PP simulations
Fig. S1. Different 2D image representations of the output geometry X for simulated AFM images of a C7H10O2 molecule from the training set.
Fig. S2. Different 2D image representations of the output geometry X for simulated AFM images of a C60 molecule.
Fig. S3. Different 2D image representations of the output geometry X for simulated AFM images of dibenzo[a,h]thianthrene molecule (71).
Fig. S4. Molecules from the validation data set together with the vdW-Spheres representation predicted by the CNN.
Fig. S5. Matching between simulated relaxed configurations of 1S-camphor and experiment.
Fig. S6. Effect of tilt of molecules on simulated AFM images 1 to 5.
Fig. S7. Adjustment of simulated configuration by –CH3 group rotations.
Fig. S8. Matching experimental configuration 2 of 1S-camphor with the closest simulated configurations.
Fig. S9. Illustration of the layers of the CNN model.
Fig. S10. The mean squared loss for height maps, vdW-Spheres, and atomic disks.
Table S1. Losses on the training and test sets for the trained models.
Table S2. Model architecture.
Table S3. Lennard-Jones parameters in PP simulation and rigid body relaxation of surface.
REFERENCES AND NOTES
- 1.Loos J., The art of SPM: Scanning probe microscopy in materials science. Adv. Mater. 17, 1821–1833 (2005). [Google Scholar]
- 2.Binnig G., Quate C., Gerber C., Atomic force microscope. Phys. Rev. Lett. 56, 930–933 (1986). [DOI] [PubMed] [Google Scholar]
- 3.Giessibl F. J., Advances in atomic force microscopy. Rev. Mod. Phys. 75, 949–983 (2003). [Google Scholar]
- 4.S. Morita, F. J. Giessibl, E. Meyer, R. Wiesendanger, Noncontact Atomic Force Microscopy (Springer International Publishing, Cham, 2015). [Google Scholar]
- 5.Pavliček N., Gross L., Generation, manipulation and characterization of molecules by atomic force microscopy. Nat. Rev. Chem. 1, 0005 (2017). [Google Scholar]
- 6.Mueller D. J., Dufrene Y. F., Atomic force microscopy as a multifunctional molecular toolbox in nanobiotechnology. Nat. Nanotechnol. 3, 261–269 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Dufrene Y. F., Ando T., Garcia R., Alsteens D., Martínez-Martín D., Engel A., Gerber C., Müller D. J., Imaging modes of atomic force microscopy for application in molecular and cell biology. Nat. Nanotechnol. 12, 295–307 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Hofer W., Foster A., Shluger A., Theories of scanning probe microscopes at the atomic scale. Rev. Mod. Phys. 75, 1287–1331 (2003). [Google Scholar]
- 9.Barth C., Foster A. S., Henry C. R., Shluger A. L., Recent trends in surface characterization and chemistry with high-resolution scanning force methods. Adv. Mater. 23, 477–501 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Gross L., Mohn F., Moll N., Liljeroth P., Meyer G., The chemical structure of a molecule resolved by atomic force microscopy. Science 325, 1110–1114 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Sweetman A., Jarvis S. P., Rahe P., Champness N. R., Kantorovich L., Moriarty P., Intramolecular bonds resolved on a semiconductor surface. Phys. Rev. B 90, 165425 (2014). [Google Scholar]
- 12.Gross L., Recent advances in submolecular resolution with scanning probe microscopy. Nat. Chem. 3, 273–278 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Hämäläinen S. K., van der Heijden N., van der Lit J., den Hartog S., Liljeroth P., Swart I., Intermolecular contrast in atomic force microscopy images without intermolecular bonds. Phys. Rev. Lett. 113, 186102 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Gross L., Mohn F., Moll N., Schuler B., Criado A., Guitian E., Peña D., Gourdon A., Meyer G., Bond-order discrimination by atomic force microscopy. Science 337, 1326–1329 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Gross L., Mohn F., Liljeroth P., Repp J., Giessibl F. J., Meyer G., Measuring the charge state of an adatom with noncontact atomic force microscopy. Science 324, 1428–1431 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Mohn F., Gross L., Moll N., Meyer G., Imaging the charge distribution within a single molecule. Nat. Nanotechnol. 7, 227–231 (2012). [DOI] [PubMed] [Google Scholar]
- 17.de Oteyza D. G., Gorman P., Chen Y. C., Wickenburg S., Riss A., Mowbray D. J., Etkin G., Pedramrazi Z., Tsai H. Z., Rubio A., Crommie M. F., Fischer F. R., Direct imaging of covalent bond structure in single-molecule chemical reactions. Science 340, 1434–1437 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Kawai S., Haapasilta V., Lindner B. D., Tahara K., Spijker P., Buitendijk J. A., Pawlak R., Meier T., Tobe Y., Foster A. S., Meyer E., Thermal control of sequential on-surface transformation of a hydrocarbon molecule on a copper surface. Nat. Commun. 7, 12711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawai S., Takahashi K., Ito S., Pawlak R., Meier T., Spijker P., Canova F. F., Tracey J., Nozaki K., Foster A. S., Meyer E., Competing annulene and radialene structures in a single anti-aromatic molecule studied by high-resolution atomic force microscopy. ACS Nano 11, 8122–8130 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Schulz F., Jacobse P. H., Canova F. F., van der Lit J., Gao D. Z., van den Hoogenband A., Han P., Gebbink R. J. M. K., Moret M.-E., Joensuu P. M., Swart I., Liljeroth P., Precursor geometry determines the growth mechanism in graphene nanoribbons. J. Phys. Chem. C 121, 2896–2904 (2017). [Google Scholar]
- 21.Jelínek P., High resolution SPM imaging of organic molecules with functionalized tips. J. Phys. Condens. Matter 29, 343002 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Jarvis S. P., Rashid M. A., Sweetman A., Leaf J., Taylor S., Moriarty P., Dunn J., Intermolecular artifacts in probe microscope images of of C60 assemblies. Phys. Rev. B 92, 241405 (2015). [Google Scholar]
- 23.Boneschanscher M. P., Hämäläinen S. K., Liljeroth P., Swart I., Sample corrugation affects the apparent bond lengths in atomic force microscopy. ACS Nano 8, 3006–3014 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Hapala P., Kichin G., Wagner C., Tautz F. S., Temirov R., Jelínek P., Mechanism of high-resolution STM/AFM imaging with functionalized tips. Phys. Rev. B 90, 085421 (2014). [DOI] [PubMed] [Google Scholar]
- 25.van der Lit J., Di Cicco F., Hapala P., Jelínek P., Swart I., Submolecular resolution imaging of molecules by atomic force microscopy: The influence of the electrostatic force. Phys. Rev. Lett. 116, 096102 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Ellner M., Pou P., Pérez R., Molecular identification, bond order discrimination, and apparent intermolecular features in atomic force microscopy studied with a charge density based method. ACS Nano 13, 786–795 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Gross L., Mohn F., Moll N., Meyer G., Ebel R., Abdel-Mageed W. M., Jaspars M., Organic structure determination using atomic-resolution scanning probe microscopy. Nat. Chem. 2, 821–825 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Albrecht F., Pavliček N., Herranz-Lancho C., Ruben M., Repp J., Characterization of a surface reaction by means of atomic force microscopy. J. Am. Chem. Soc. 137, 7424–4728 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Albrecht F., Bischoff F., Auwärter W., Barth J. V., Repp J., Direct identification and determination of conformational response in adsorbed individual nonplanar molecular species using noncontact atomic force microscopy. Nano Lett. 16, 7703–7709 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Schuler B., Meyer G., Peña D., Mullins O. C., Gross L., Unraveling the molecular structures of asphaltenes by atomic force microscopy. J. Am. Chem. Soc. 137, 9870–9876 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Mönig H., Hermoso D. R., Arado O. D., Todorović M., Timmer A., Schueer S., Langewisch G., Pérez R., Fuchs H., Submolecular imaging by noncontact atomic force microscopy with an oxygen atom rigidly connected to a metallic probe. ACS Nano 10, 1201–1209 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Mönig H., Copper-oxide tip functionalization for submolecular atomic force microscopy. Chem. Commun. 54, 9874–9888 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Fatayer S., Coppola A. I., Schulz F., Walker B. D., Broek T. A., Meyer G., Druffel E. R. M., McCarthy M., Gross L., Direct visualization of individual aromatic compound structures in low molecular weight marine dissolved organic carbon. Geophys. Res. Lett. 45, 5590–5598 (2018). [Google Scholar]
- 34.Fatayer S., Poddar N. B., Quiroga S., Schulz F., Schuler B., Kalpathy S. V., Meyer G., Pérez D., Guitián E., Peña D., Wornat M. J., Gross L., Atomic force microscopy identifying fuel pyrolysis products and directing the synthesis of analytical standards. J. Am. Chem. Soc. 140, 8156–8161 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Schulz F., Commodo M., Kaiser K., Falco G. D., Minutolo P., Meyer G., D’Anna A., Gross L., Insights into incipient soot formation by atomic force microscopy. Proc. Combust. Inst. 37, 885–892 (2019). [Google Scholar]
- 36.Pawlak R., Vilhena J. G., Hinaut A., Meier T., Glatzel T., Baratoff A., Gnecco E., Pérez R., Meyer E., Conformations and cryo-force spectroscopy of spray-deposited single-strand DNA on gold. Nat. Commun. 10, 685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albers B. J., Schwendemann T. C., Baykara M. Z., Pilet N., Liebmann M., Altman E. I., Schwarz U. D., Three-dimensional imaging of short-range chemical forces with picometre resolution. Nature 4, 307–310 (2009). [DOI] [PubMed] [Google Scholar]
- 38.LeCun Y., Bengio Y., Hinton G., Deep learning. Nature 521, 436–444 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Kalinin S. V., Sumpter B. G., Archibald R. K., Big–deep–smart data in imaging for guiding materials design. Nat. Mater. 14, 973–980 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Kalinin S. V., Strelcov E., Belianinov A., Somnath S., Vasudevan R. K., Lingerfelt E. J., Archibald R. K., Chen C., Proksch R., Laanait N., Jesse S., Big, deep, and smart data in scanning probe microscopy. ACS Nano 10, 9068–9086 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Ziatdinov M., Maksov A., Kalinin S. V., Learning surface molecular structures via machine vision. npj Comput. Mater. 3, 31 (2017). [Google Scholar]
- 42.Jorgensen W. L., Tirado-Rives J., The opls [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110, 1657–1666 (1988). [DOI] [PubMed] [Google Scholar]
- 43.Moll N., Gross L., Mohn F., Curioni A., Meyer G., The mechanisms underlying the enhanced resolution of atomic force microscopy with functionalized tips. New J. Phys. 12, 125020 (2010). [Google Scholar]
- 44.Giessibl F. J., A direct method to calculate tip–sample forces from frequency shifts in frequency-modulation atomic force microscopy. Appl. Phys. Lett. 78, 123–125 (2001). [Google Scholar]
- 45.Peng J., Guo J., Hapala P., Cao D., Ma R., Cheng B., Xu L., Ondráček M., Jelínek P., Wang E., Jiang Y., Weakly perturbative imaging of interfacial water with submolecular resolution by atomic force microscopy. Nat. Commun. 9, 122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hapala P., Švec M., Stetsovych O., van der Heijden N. J., Ondráček M., van der Lit J., Mutombo P., Swart I., Jelínek P., Mapping the electrostatic force field of single molecules from high-resolution scanning probe images. Nat. Commun. 7, 11560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramakrishnan R., Dral P. O., Rupp M., von Lilienfeld O. A., Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, 201422 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hapala P., Temirov R., Tautz F. S., Jelínek P., Origin of high-resolution iets-stm images of organic molecules with functionalized tips. Phys. Rev. Lett. 113, 226101 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Pavliček N., Fleury B., Neu M., Niedenführ J., Herranz-Lancho C., Ruben M., Repp J., Atomic force microscopy reveals bistable configurations of dibenzo[a,h]thianthrene and their interconversion pathway. Phys. Rev. Lett. 108, 086101 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Ellner M., Pavliček N., Pou P., Schuler B., Moll N., Meyer G., Gross L., Peréz R., The electric field of co tips and its relevance for atomic force microscopy. Nano Lett. 16, 1974–1980 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Gross L., Schuler B., Mohn F., Moll N., Pavliček N., Steurer W., Scivetti I., Kotsis K., Persson M., Meyer G., Investigating atomic contrast in atomic force microscopy and kelvin probe force microscopy on ionic systems using functionalized tips. Phys. Rev. B 90, 155455 (2014). [Google Scholar]
- 52.Melitz W., Shen J., Kummel A. C., Lee S., Kelvin probe force microscopy and its application. Surf. Sci. Rep. 66, 1–27 (2011). [Google Scholar]
- 53.S. Sadewasser, T. Glatzel, Kelvin Probe Force Microscopy: Measuring and Compensating Electrostatic Forces (Springer International Publishing, Cham, 2012). [Google Scholar]
- 54.S. Sadewasser, T. Glatzel, Kelvin Probe Force Microscopy: From Single Charge Detection to Device Characterization (Springer International Publishing, Cham, 2018). [Google Scholar]
- 55.Schulz F., Ritala J., Krejčí O., Seitsonen A. P., Foster A. S., Liljeroth P., Elemental identification by combining atomic force microscopy and kelvin probe force microscopy. ACS Nano 12, 5274–5283 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Badrinarayanan V., Kendall A., Cipolla R., Segnet: A deep convolutional encoder-decoder architecture for image segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 39, 2481–2495 (2017). [DOI] [PubMed] [Google Scholar]
- 57.X. Glorot, A. Bordes, Y. Bengio, Proceedings of the Fourteenth International Conference on Artificial Intelligence and Statistics (PMLR, 2011), vol. 15, pp. 315–323. [Google Scholar]
- 58.F. Chollet, et al., Keras, (2015); https://keras.io.
- 59.M. Abadi, A. Agarwal, P. Barham, E. Brevdo, Z. Chen, C. Citro, G. S. Corrado, A. Davis, J. Dean, M. Devin, S. Ghemawat, I. Goodfellow, A. Harp, G. Irving, M. Isard, Y. Jia, R. Jozefowicz, L. Kaiser, M. Kudlur, J. Levenberg, D. Mané, R. Monga, S. Moore, D. Murray, C. Olah, M. Schuster, J. Shlens, B. Steiner, I. Sutskever, K. Talwar, P. Tucker, V. Vanhoucke, V. Vasudevan, F. Viégas, O. Vinyals, P. Warden, M. Wattenberg, M. Wicke, Y. Yu, X. Zheng, TensorFlow: Large-scale machine learning on heterogeneous systems (2015); tensorflow.org.
- 60.T. Devries, G. W. Taylor, Improved regularization of convolutional neural networks with cutout. arXiv:1708.04552 (2017).
- 61.Dagdeviren O. E., Zhou C., Altman E. I., Schwarz U. D., Quantifying tip-sample interactions in vacuum using cantilever-based sensors: An analysis. Phys. Rev. Appl. 9, 044040 (2018). [Google Scholar]
- 62.Sader J. E., Hughes B. D., Huber F., Giessibl F. J., Interatomic force laws that evade dynamic measurement. Nat. Nanotechnol. 13, 1088–1091 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Becke A. D., Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993). [Google Scholar]
- 64.Parrish R. M., Burns L. A., Smith D. G. A., Simmonett A. C., DePrince III A. E., Hohenstein E. G., Bozkaya U., Sokolov A. Y., Di Remigio R., Richard R. M., Gonthier J. F., James A. M., McAlexander H. R., Kumar A., Saitow M., Wang X., Pritchard B. P., Verma P., Schaefer H. F., Patkowski K., King R. A., Valeev E. F., Evangelista F. A., Turney J. M., Crawford T. D., Sherrill C. D., Psi4 1.1: An open-source electronic structure program emphasizing automation, advanced libraries, and interoperability. J. Chem. Theory Comput. 13, 3185–3197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.T. D. Crawford, H. F. Schaefer III, An Introduction to Coupled Cluster Theory for Computational Chemists (John Wiley & Sons, Ltd, 2007), pp. 33–136. [Google Scholar]
- 66.Bartels L., Meyer G., Rieder K.-H., Controlled vertical manipulation of single co molecules with the scanning tunneling microscope: A route to chemical contrast. Appl. Phys. Lett. 71, 213–215 (1997). [Google Scholar]
- 67.Behler J., Parrinello M., Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Bartók A. P., Kondor R., Csányi G., On representing chemical environments. Phys. Rev. B 87, 184115 (2013). [Google Scholar]
- 69.Kuzminykh D., Polykovskiy D., Kadurin A., Zhebrak A., Baskov I., Nikolenko S., Shayakhmetov R., Zhavoronkov A., 3d molecular representations based on the wave transform for convolutional neural networks. Mol. Pharm. 15, 4378–4385 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Connolly M., Solvent-accessible surfaces of proteins and nucleic acids. Science 221, 709–713 (1983). [DOI] [PubMed] [Google Scholar]
- 71.P. Hapala, Simple simulation engine : Rigidmol, (2019); available at https://github.com/ProkopHapala/SimpleSimulationEngine/blob/master/cpp/libs/Molecular/RigidMol.cpp.
- 72.Hutter J., Iannuzzi M., Schiffmann F., VandeVondele J., cp2k: Atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev. 4, 15–25 (2014). [Google Scholar]
- 73.A. Odena, V. Dumoulin, C. Olah, Deconvolution and checkerboard artifacts (Distill, 2016).
- 74.W. Shi, J. Caballero, L. Theis, F. Huszar, A. Aitken, C. Ledig, Z. Wang, Is the deconvolution layer the same as a convolutional layer? arXiv:1609.07009 (2016).
- 75.A. Aitken, C. Ledig, L. Theis, J. Caballero, Z. Wang, W. Shi, Checkerboard artifact free sub-pixel convolution: A note on sub-pixel convolution, resize convolution and convolution resize. arXiv:1707.02937 (2017).
- 76.D. Kingma, J. Ba, Adam: A method for stochastic optimization. arXiv:1412.6980 (2014).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/9/eaay6913/DC1
Section S1. Image representations of output molecular structure
Section S2. Matching experiment to relaxed on-surface simulated configurations
Section S3. Effect of small perturbations on AFM imaging and matching
Section S4. Neural network architecture
Section S5. PP simulations
Fig. S1. Different 2D image representations of the output geometry X for simulated AFM images of a C7H10O2 molecule from the training set.
Fig. S2. Different 2D image representations of the output geometry X for simulated AFM images of a C60 molecule.
Fig. S3. Different 2D image representations of the output geometry X for simulated AFM images of dibenzo[a,h]thianthrene molecule (71).
Fig. S4. Molecules from the validation data set together with the vdW-Spheres representation predicted by the CNN.
Fig. S5. Matching between simulated relaxed configurations of 1S-camphor and experiment.
Fig. S6. Effect of tilt of molecules on simulated AFM images 1 to 5.
Fig. S7. Adjustment of simulated configuration by –CH3 group rotations.
Fig. S8. Matching experimental configuration 2 of 1S-camphor with the closest simulated configurations.
Fig. S9. Illustration of the layers of the CNN model.
Fig. S10. The mean squared loss for height maps, vdW-Spheres, and atomic disks.
Table S1. Losses on the training and test sets for the trained models.
Table S2. Model architecture.
Table S3. Lennard-Jones parameters in PP simulation and rigid body relaxation of surface.