

Abstract
Biological nanopores are emerging as powerful and low-cost sensors for real-time analysis of biological samples. Proteins can be incorporated inside the nanopore, and ligand binding to the protein adaptor yields changes in nanopore conductance. In order to understand the origin of these conductance changes and develop sensors for detecting metabolites, we tested the signal originating from 13 different protein adaptors. We found that the quality of the protein signal depended on both the size and charge of the protein. The engineering of a dipole within the surface of the adaptor reduced the current noise by slowing the protein dynamics within the nanopore. Further, the charge of the ligand and the induced conformational changes of the adaptor defined the conductance changes upon metabolite binding, suggesting that the protein resides in an electrokinetic minimum within the nanopore, the position of which is altered by the ligand. These results represent an important step toward understanding the dynamics of the electrophoretic trapping of proteins inside nanopores and will allow developing next-generation sensors for metabolome analysis.
Keywords: nanopore, cytolysin A, metabolite sensor, substrate-binding protein, electrochemistry
The metabolome is the entirety of all small molecules present in a biological system. These metabolites, which include vitamins, sugars, amino acids, metal ions, and other transmitter molecules,1 are involved in many essential biological processes.2 Because the metabolome is influenced by a variety of factors including gene and protein expression,3,4 as well as lifestyle factors such as diet, age, fitness, hormonal balance, and medication, the metabolome displays the actual health status of an organism5,6 and is directly related to diseases.7,8 It is also known that many diseases alter the metabolite composition before developing clinical symptoms.9 Therefore, the analysis of metabolites in blood, also called blood metabolome, is a promising tool for early stage diagnosis and continuous health-status monitoring. These specifics are crucial for cancer diagnosis and therapy10 as well as for the evaluation of the progress of dementia11 or the risk for developing cardiovascular diseases.12,13
The diagnosis of diseases by monitoring the blood metabolome requires the targeted detection of hundreds of biomarkers to compare with standard values.14 The detection of the concentration of multiple biomarkers is important because the fluctuation on individual values can be associated with multiple factors.15 Today, biomarker detection and analysis is performed utilizing either mass spectrometry- or NMR-based methods.16 These techniques are reliable, the sample preparation is kept to a minimum, and high-throughput approaches are possible.17 However, they require the use of large and complex machines and trained personnel. Therefore, such analytical techniques cannot be incorporated in home-diagnostic devices and are not amenable for real-time analysis. Real-time detection of the blood metabolome using wearable or implantable sensors would be beneficial, as it would allow early and continuous background diagnostics. Indeed, an ideal sensor for metabolite detection should be small, low-powered, selective, and eligible to communicate with silicon-based devices.
Biological nanopores are an emerging class of sensors with such characteristics. A biological nanopore is a protein that forms a water-filled channel on a voltage-clamped hydrophobic membrane. The output signal is a current of hydrated ions passing through individual nanopores. Crucially, nanopore currents can be easily digitized using small and low-cost devices that contain thousands of individually addressing elements. Most notably, portable devices containing nanopores are now commercially available for DNA sequencing.18−22 Efforts are underway toward the detection and analysis of peptides23−26 and proteins,27−35 as well as the detection of biologically relevant molecules such as amino acids36−38 or sugars39 and even whole viruses.40 The identification of metabolites is complicated by the fact that blood contains thousands of chemically similar molecules. Hence, real-time analysis of the blood metabolome will require developing nanopores that contain a sensing element that recognizes and quantifies a molecule with high specificity.
We have recently shown that folded proteins pushed by the electroosmotic flow entered inside a cytolysin A (ClyA) nanopore. Notably, changes in the nanopore current could report the switching of the protein from its open unliganded conformation to its closed liganded conformation.41 The frequency of switching from the open to the closed configurations is related to the concentration of the ligand in solution. The single-molecule nature of the nanopore approach allowed the simultaneous detection of multiple ligands.42 This is because when a protein enters inside the nanopore, the associated blocked current can be used to identify the protein adaptor. Importantly, the concentration of the ligand could be measured directly from blood or other biological samples without any sample preparation,42 indicating that this approach can be used in real-time blood analysis.
Some of the protein adaptors used in the initial experiments were substrate-binding proteins,41,42 which are associated with bacterial ATP-binding cassette importers for substrate uptake.43 Hundreds of such proteins exist that bind metabolites specifically,44 which make them ideal in developing nanopore sensors for metabolites. However, several aspects of the nanoconfinement of the protein inside ClyA are unknown, including the origin of the signal for the open and closed states, the relation between the concentration of analytes in bulk and inside the nanopore, the positioning of proteins inside the nanopore, and the relation between the signal and the size and shape of the incorporated protein. Here, we study 13 possible adaptor proteins varying in size, shape, charge, and ligand. We found that the majority of proteins can be used to determine the concentration of their cognate ligands in bulk solution. The analysis of the protein blockades revealed that inside the ClyA nanopore proteins occupy two possible binding sites depending on the charge and size of the adaptor. Conveniently, the properties of the protein adaptors can be manipulated to improve the nanopore signal.
Results and Discussion
Protein Adaptors
Thirteen different proteins were tested as adaptors in the ClyA-AS from Salmonella typhi nanopore (Figure 1). These proteins were recombinantly expressed in E. coli and purified (Figure S1) before being added to the nanopore. The protein sizes ranged from 25 to 42 kDa, and their net charge from 0 to −12, while their cognate ligands showed a variety of sizes, charges, and chemical properties (Table 1). In particular, the ligand charges ranged from +2 to −1; their size from 88 Da (putrescine, or 1,4-diaminobutane) to 1355 Da (CN-cobalamin, Figure 1b). Eleven of the 13 proteins were substrate-binding proteins of ABC transporters, which are known to specifically bind metabolites through conformational changes involving large-scale domain motions.45−47 In order to generalize our approach, as the shape- and the ligand-induced structural rearrangement of SBDs are comparable, we also tested two proteins from different families: nicotinamidase and hoefavidin. Nicotinamidase converts nicotinamide to nicotinic acid, which are part of the vitamin B3 system and are both metabolites and biomarkers.48 Hoefavidin is a dimeric avidin that binds specifically biotin,49 which is also known as vitamin B7.
Figure 1.
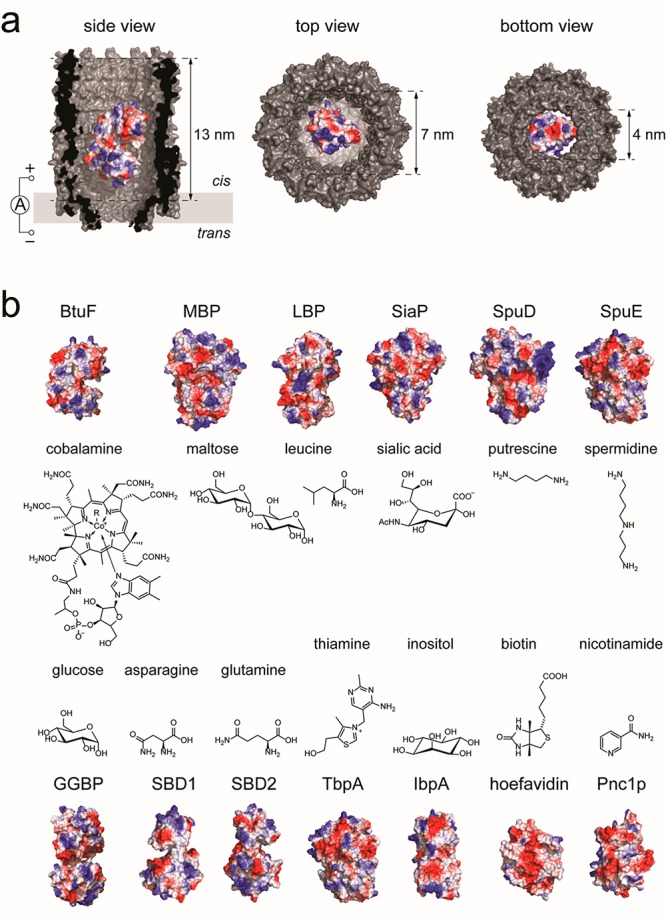
Protein adaptors for ClyA nanopores. (a) Cut-through of a surface representation of a ClyA nanopore (PDB: 2WCD) containing a single protein adaptor (BtuF, vitamin B12-binding protein, PDB: 1N2Z). (b) Surface representation of the 13 adaptor proteins studied in this work, and the chemical structure of their respective ligands. Proteins are colored according to their surface charge (Pymol). PDB files: MBP (maltose-binding protein): 1OMP; LBP (leucine-binding protein): 1USG; SiaP (sialic acid-binding protein): 4MMP; SpuD (putrescine-binding protein): 3TTM; SpuE (spermidine binding protein): 3TTL; GGBP (glucose-/galactose-binding protein): 2FVY; SBD1 (substrate-binding protein 1): 4KPT; SBD2 (substrate-binding protein 2): 4KR5; TbpA (thiamine-binding protein): 2QRY; IbpA (myo-inositol-binding protein): 4IRX; hoefavidin (dimeric avidin): 4Z27; Pnc1p (nicotinamidase): 3 V8E.
Table 1. Adaptor Proteins Used in This Worka.
| protein |
ligand |
V |
IRES |
KD |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| protein | PDB | ligand | size [kDa] | charge | size [Da] | charge | [mV] | σB/σO | open [%] | closed [%] | bulk [nM] | pore [nM] |
| SBD1 | 4kpt | asparagine | 25.5 | –2 | 132.1 | 0 | –70 | 1.45 ± 0.2 | 66.2 ± 0.4 | 65.4 ± 0.1 | 20046 | 470 ± 341 |
| Pnc1p | 3v8e | nicotinamide | 26.1 | –9 | 122.1 | 0 | –60 | 13.7 ± 2.2 | 55.8 ± 0.3 | n.d. | n/a | n.d. |
| Pnc1p_dipole | nicotinamide | 25.0 | –6 | 122.1 | 0 | –60 | 1.40 ± 0.2 | n.d. | n.d. | n/a | n.d. | |
| SBD2 | 4kr5 | glutamine | 27.8 | –5 | 146.1 | 0 | –70 | 0.87 ± 0.2 | 64.0 ± 0.3 | 62.9 ± 0.3 | 90046 | 830 ± 1041 |
| BtuF | 1n2z | CN-cobalamine | 28.0 | 0 | 1355.4 | 0 | –55 | 15.7 ± 8.5 | 68.0 ± 0.4 | 60.6 ± 0.5 | 14.850 | 26 ± 8 |
| hoefavidin | 4z27 | biotin | 30.8 | –12 | 244.3 | 0 | –90 | n.d. | n.d. | n.d. | 35849 | n.d. |
| IbpA | 4irx | myo-inositol | 31.8 | –2 | 180.2 | 0 | –60 | 7.52 ± 4.1 | 64.5 ± 0.5 | n.d. | 76051 | n.d. |
| GGBP | 2fvy | glucose | 34.3 | –6 | 180.2 | 0 | –90 | 0.84 ± 0.0 | 68.3 ± 0.6 | 66.2 ± 0.7 | 20052 | 42 ± 3 |
| SiaP | 4mmp | sialic acid | 35.2 | –5 | 309.3 | –1 | –50 | 2.93 ± 0.9 | 58.1 ± 1.2 | 55.9 ± 1.4 | 19.753 | 154 ± 18 |
| TbpA | 2qry | thiamine | 36.5 | –1 | 265.4 | +1 | –35 | 4.84 ± 3.4 | 49.8 ± 0.2 | 47.6 ± 0.1 | 3354 | 40 ± 10 |
| LBP | 1usg | leucine | 38.0 | –9 | 131.2 | 0 | –60 | 3.24 ± 2.6 | 65.8 ± 0.3 | 60.2 ± 0.3 | 40055 | 86 ± 19 |
| SpuE | 3ttl | spermidine | 38.8 | –8 | 145.3 | +2 | –70 | n.d. | n.d. | n.d. | 1456 | n.d. |
| SpuD | 3ttm | putrescine | 39.2 | –3 | 88.2 | +2 | –80 | 0.74 ± 0.1 | 46.2 ± 0.7 | 44.0 ± 0.7 | 356 | 2.3 ± 0.5 |
| MBP | 1omp | maltose | 41.9 | –9 | 342.3 | 0 | –70 | 1.50 ± 0.3 | 59.3 ± 0.7 | 56.7 ± 0.8 | 120057 | 1780 ± 240 |
V represents the electric potential applied to the trans side of the nanopore. IRES is the residual current after protein capture and was determined for the open and closed levels depicted in Figure 3. KD is the apparent binding constant, and σB/σO is the signal-to-noise ratio of the protein adaptor. All experiments were performed in triplicates; the error represents the standard deviation.
Protein Signal
In a typical experiment, a negative potential is applied to the trans side of the nanopore, whereas proteins are added to the cis side (Figure 2a,b). The entry of the protein in the pore is observed by the specific reduction of the open pore current IO to the blocked pore current IB. In a 150 mM NaCl solution, pH 7.5, each protein adaptor showed an individual current signal as measured by the residual current IRES = 100 × IB/IO, the average residence time (τ), and the current noise (Figure 2). The latter is quantified using σB/σO, which is the standard deviation of the Gaussian distribution of the protein blockade (σB) divided by the standard deviation of the Gaussian distribution of the open pore (σO).
Figure 2.

Protein signals in the ClyA-AS nanopore. (a) Typical trace showing the main measurands of an adaptor protein in the pore: dwell time (τprotein) and residual current (IRES = IB/IO × 100%). In a typical experiment, thousands of dwell times are collected and exponential fits are used to determine the average τprotein. IB and IO were determined by Gaussian fitting to all-point histograms of protein blockades. (b) Representation of a protein (BtuF) electrophoretically captured in the pore. The gray arrows indicate the flux of ions across the nanopore. (c) Example of the three different signals and corresponding full-point histograms of a 1 s trace. σB/σO represents the ratio of the standard deviation of the Gaussian fitting of the open pore current and the protein block current as measured from full-point histograms. (d) Protein signal optimization. On the left: Surface representation of Pnc1p and Pnc1p_dipole (D82K, D83K, K216E, Δ6xHis) with the arrow showing the direction of the protein dipole. The current trace shows a typical protein blockade. The histograms were calculated from a representative 10 s of protein block. All measurements were performed in 15 mM Tris and 150 mM NaCl, pH 7.5, under negative bias (trans) and sampling at 10 kHz with a 2 kHz Bessel filter. For figure preparation, all traces were additionally filtered with a 500 Hz Gaussian filter.
Three classes of signals were recorded (Figure 2c). GGBP, SBD1, SBD2, MBP, and SpuD (see the legend in Figure 1 for the full name of the proteins) showed a σB/σO of ∼1, or even <1 (Table 1), suggesting that such proteins are lodged in a well-defined minimum within the nanopore where Brownian motions are largely suppressed. Within this group, sometimes alternative blockades with higher noise levels occurred (e.g., SpuD and MBP; see also Figures S2 and S7 for longer traces), which can be excluded from analysis. LBP, IbpA, TbpA, and SiaP showed a more dynamic but reproducible current signal with a σB/σO between 3 and 8, while Pnc1p and BtuF exhibited a σB/σO above 10. Multiple levels were observed within a blockade, suggesting that these proteins are either intrinsically dynamic or moving within the lumen of ClyA on a time scale similar to the sampling rate (20 μs). SpuE and hoefavidin represented a third kind of signal, in which each blockade showed a different signal (IRES ranging from 44% to 57% for SpuE) and varying noise levels (Figure 2c). Since both proteins have a large net negative charge (net charge −12, hoefavidin) or are relatively large (39 kDa, net charge −8, SpuE), the unfavorable signal might originate from a combination of electrostatic and steric interactions with the nanopore inner surface.
In order to understand the origin of the highly dynamic protein blockades, we further investigated Pnc1p. Pnc1p has a net charge of −9 that is evenly spread across the surface of the protein, as shown by its net dipole perpendicular to the protein main axis (Figure 2d; see also Figure S15 for dipole of all tested proteins). If the dynamic current levels (σB/σO Pnc1p = 10) are reflecting the tumbling of the protein within the nanopore, then the introduction of a dipole on the protein surface should reduce the current noise. We introduced a negative charge at the C-terminus (K216E) and two positive charges (D82K, D83K) at the opposite end of the folded structure, and we deleted the C-terminal 6xHis-tag. The σB/σO decreased from 10 to 1 (Figure 2d), and the signal appeared uniform and clear, suggesting correct folding. The noise reduction indicated that the highly dynamic signal was due to the fast tumbling of the protein inside the nanopore.
Current Modulation by Ligand Binding
In order to use adaptor proteins as sensors, the addition of ligands to the cis side of the nanopore should induce a change in the current signal. Nine of the tested proteins showed a change in the electrical signal when the ligands were added in the solution. For some proteins (SBD1, GGBP, SpuD, and TbpA, signal type 1a, Figures 3, S2, S3, S8, and S9), a single well-defined additional level corresponding to the shift from the open to the closed conformation of the protein was observed. Other proteins (LBP, SiaP, MBP, and BtuF, signal type 2, Figures 3 and S4–S7) showed often a noisy signal already in the unliganded form; thus the shift toward the closed conformation of the protein was more difficult to assign due to the high noise. Probably, intrinsic motions and unspecific interactions of the proteins with the nanopore are in this group more pronounced. SBD2 was a special case (classified type 1b), as the current blockade showed two well-defined current levels, and the addition of the ligand introduced a third well-defined level. Previous work demonstrated that the two levels of the apo protein correspond to the different orientation of the protein in the open configuration inside the nanopore, while the third level corresponds to the closed protein conformation.41
Figure 3.

Current modulation of adaptor proteins upon ligand binding. In each box the left trace shows the current blockade of the apo-protein; the right the blockade in the presence of the cognate ligand added to the cis side. The ligand concentration for each protein was SBD1, 470 nM asparagine; GGBP, 500 nM glucose; SBD2, 2.4 μM glutamine; SpuD, 64 nM putrescine; BtuF, 500 nM CN-cobalamin; MBP, 2 μM maltose; LBP, 2 μM leucine; SiaP, 400 nM sialic acid; Tbpa, 2 μM thiamine; SpuE, 1 mM spermidine; IbpA, 40 μM myo-inositol; hoefavidin, 10 μM biotin; Pnc1p_wt, 2 mM nicotinamide; Pnc1p_dipole, 100 μM nicotinamide. The blue line represents the unbound state; the red line the bound state. All measurements were performed in 15 mM Tris and 150 mM NaCl, pH 7.5, under a negative bias (trans). Traces were sampled at 10 kHz and filtered with a 2 kHz Bessel filter. For figure preparation, all traces were additionally filtered with a 100 Hz Gaussian filter.
In all blockades the closed state showed less current compared to the open state. Since a more condensed structure is expected to block less current than a less condensed structure, this finding suggests that the signal is most likely associated with a change in the position of the protein within the pore. Most likely, as the structure of the protein becomes more compacted, the protein penetrates deeper inside the nanopore, resulting in more current being blocked (i.e., less overall current). Interestingly, we also noticed that all the apo protein showed conformational transitions to the closed state also in the absence of their cognate ligand. This could reflect spontaneous opening and closing of the protein. Indeed, intrinsic conformational changes were observed for GGBP58 and MBP59 as well as SBD1 and SBD2,46 in NMR and single-molecule fluorescence studies, respectively. However, this could also result from the binding of contaminant to the protein adaptors.
Hoefavidin, a dimeric avidin that binds to biotin, did not manifest ligand-induced signal changes. Previous work using a tetrameric avidin (60 kDa) revealed that the binding of biotin induced a conductance change in the E. coli ClyA nanopore.60 Possibly, therefore, a tight fit between the protein and the pore is required to observe the rather small conformational change that follows the binding of biotin to avidin. Pnc1p and Pnc1p-dipole also showed no change in signal upon binding to nicotinamide, as well as two SBD proteins (Figure 3). These proteins have a comparable size and charge to other proteins that display a ligand-induced signal (Table 1). Possibly, these proteins bind inside the nanopore in a configuration that prevents the ligands from reaching the active site or the conformational change upon ligand binding is too small to cause significant current alteration.
Affinity of Adaptor Proteins for Their Cognate Ligands
The ability of the nanopore sensor to identify analytes was tested by applying increasing concentration of ligands to the cis side of the nanopore and measuring the open and closed protein levels. In GGBP, SBD1, SBD2, and SpuD the association kon and dissociation koff rate constants could be measured by sampling the dwell time of the open (τon) and closed (τoff) levels (Figure S2b,c). Typically, more than 500 events per condition were measured. kon were then determined as 1/(τoncligand) and koff as 1/τoff. For this class of adaptors, the apparent binding constant KD was measured by increasing the ligand concentration and observing the change in the binding frequency (Figure 4a). Conveniently, KD can be also estimated at a single substrate concentration as koff/kon.
Figure 4.

Dissociation constants and binding rates. (a) Representative example of rate constant determination. Binding and unbinding frequencies were determined from event dwell times (see also Figure S2bc). (b) Representative example of a ligand binding curve. The percentage of protein in the closed state was determined by the analysis of full-point histograms followed by ratio calculation of the peak height of the bound and unbound protein state over concentration (see also Figure S5bc). (c) Difference between the KD determined in the nanopore (KD pore) and the KD reported in the literature (KD bulk); see also Table 1. (d) Change in the percentage of SiaP in the closed state with increasing negative potential at a fixed sialic acid concentration of 150 nM. (e) Binding and unbinding rates for three different proteins with increasing negative potential at a fixed ligand concentration (SBD2: 830 nM glutamine; GGBP: 50 nM glucose; SpuD: 8 nM putrescine). Kon and koff were determined and then normalized to the highest value of every measurement. All experiments were performed in triplicates. The error bars represent the standard error of the mean.
For the other proteins, because their binding rates could not be easily determined, the KD was measured from full-point histograms of the whole trace (typically 2–3 min per concentration) based on the IRES (see Experimental Section). Within the histogram, the peaks representing the open and ligand-bound (closed) protein were easily identified (Figure S2d,e). The KD was then measured by plotting the fraction of the protein in the closed configuration (see Experimental Section) as a dependency of the ligand concentration and fitting on a binding curve (Figure 4b). BtuF displayed a noisy signal that prevented histogram analysis. However, KD could still be determined by analyzing the fast-current blockades (see SI, Experimental Section, and Figure S6b,c).
The apparent KD values measured by the nanopore were for six proteins comparable to the values measured in bulk (Table 1, Figure 4c). GGBP and LBP exhibited ∼4-fold higher affinity then determined in bulk measurements. A notable exception was represented by the binding of sialic acid to SiaP, which showed an 8-fold higher KD than in the bulk. A 100-fold decreased affinity compared to bulk values was also observed for the binding of NADPH to DHFR61,62 and for the binding of oxoglutarate to AlkB.63 A possible explanation is that the diffusion of negatively charged ligands across the nanopore is retarded by the negative bias applied to the trans chamber. To test for this effect, we measured the voltage dependency of the kon and koff rates for SiaP (net charge of sialic acid −1), SBD2 and GGBP, which bind to glutamine and glucose, respectively (both neutral), and SpuD, which binds to putrescine (net charge +2) (Figure 4d,e). The signal of SiaP is complex and prevented measuring association and dissociation rate constants. Instead, we determined the fraction of the protein in the closed conformation as a function of the bias. We found that the fraction of SiaP in the closed state decreased with increasing potential (Figure 4d), which is compatible with an electrophoretic reduced diffusion of the negative ligand across the nanopore. According to this interpretation, when measuring neutral ligands (asparagine and glucose), the dissociation and association rate constants remained largely unaffected by the increased potential (Figure 4e). Unexpectedly, however, the binding frequency of positively charged spermidine to SpuD decreased four times by increasing the applied bias from −50 mV to −90 mV, while the dissociation rate remained constant. This is surprising, because the diffusion of putrescine toward the negative trans side is expected to be facilitated by the applied potential. A possible explanation is that SpuD, which is bigger than the other proteins tested (Table 1), sits tightly inside the nanopore. The increased electroosmotic flow may push the protein deeper inside the nanopore, thus reducing the accessibility of the ligand for its binding site. Noticeably, however, at −80 mV the KD of SpuD is similar to the value measured in bulk (Figure 4c), suggesting that the effect of the potential does not significantly compromise the integrity of the protein inside the nanopore.
Two Protein Residence Sites inside the Nanopore
It is generally accepted that in nanopore experiments the current blockade arises from the excluded volume of the analyte inside the nanopore.64,65 Hence, for a protein inside ClyA, the residual current is expected to depend on the size of the protein. However, the charge of the protein and its relative position within the nanopore are also likely to play a role. Two main forces drive proteins in and out the nanopore. Under a negative applied potential, the electrophoretic force moves negatively charged proteins toward the cis side, hence resisting their entry inside the pore. By contrast, the electroosmotic flow (EOF) moves the protein toward the trans side,66,67 facilitating the entry of proteins inside the nanopore independently of the charge of the protein. The balance between these forces is, therefore, likely to define the IRES and dwell time of proteins inside ClyA. We found that for all proteins the dwell time inside ClyA increased by about 10-fold every 10 mV (Figure S16), indicating that despite the charge of the protein, the EOF is the dominant force in driving the proteins inside the nanopore.68,69 For five proteins (SBD1, SBD2, BtuF, IbpA, and GGBP) we observed a decrease in dwell time with the potential, suggesting that above a certain potential a protein can translocate through the nanopore.62,70
The voltage dependency of the proteins’ IRES showed two behaviors (Figure 5a). Noteworthy, SpuE and hoefavidin are not included in the analysis because of the too large variation in their signal. The IRES of Pnc1p, GGBP, LBP, and MBP remained constant with the applied bias, while the IRES of the other proteins decreased when increasing the potential. The former proteins have a relatively large negative net charge (−9, −9, −9, and −6, respectively, Table 1), suggesting that the trans to cis electrophoretic force almost perfectly opposes the cis to trans electroosmotic flow. By contrast, the reduced IRES with the bias for the latter proteins most likely represent a deeper penetration of the protein toward the narrower trans constriction of the nanopore as the EOF is increased. Interestingly, the blockades of the highly negatively charged proteins (LBP, MBP, and GGBP, but not Pnc1p) were associated with higher current compared to the blockade of the other group of proteins (Figure 5b). It has been previously shown that human thrombin (37 kDa, pI 8.8) occupies two residence sites (R1 and R2) within the lumen of the ClyA nanopore depending on the applied potential,71 where R1 reflects a site located closer to the cis entrance of the nanopore and R2 a more sterically constrained site deeper in the pore.72 Therefore, a possible explanation is that the large and/or highly negatively charged proteins occupy the more superficial R1 residence site, while smaller and less negatively charged proteins occupy the deeper R2 residence site.
Figure 5.

Characterization of the protein binding behavior in the nanopore. In the absence of ligands, a negative potential was applied to the trans side of the pore. All plots show the mean of at least three independent experiments. The threshold for classification as “constant IRES” was set for linear fits with a slope ≤0.03. (a) Dependency of residual current (IRES) on applied potential. IRES is constant (blue, red) or variable (black) over potential. (b) IRES obtained at −80 mV plotted against the protein size in kDa. The mutants TbpA Y27A, and Pnc1p_dipole were used.
Conclusions
The ability to sense metabolites in real-time using low-cost sensing devices is of great interest in personalized health-care monitoring.5,6 We recently showed that nanopores with internalized protein adaptors can be used to quantify glucose and asparagine directly from biological samples.42 Such adaptors belonged to the SBD protein family, which comprises more than 120 proteins that are capable of recognizing a variety of molecules including sugars, amino acids such as Glu, Ile, Val, Met, Pro, Arg, Cys, His, and GABA, metals such as tungsten, iron, and molybdenum, and vitamins such as riboflavin.44,73 In this work we investigated 13 different protein adaptors, which bind to a wide range of metabolites (Figure 1). All proteins were tested under the same condition of pH (7.5) and ionic strength (150 mM NaCl), so that multiple protein adaptors could be used simultaneously under physiological conditions.
Ligand binding was detected in nine of the 11 SBD proteins examined within the ClyA nanopore, suggesting that the majority of SBDs are suitable adaptor proteins for metabolite sensing. Incidentally, the adaptors were either negatively charged or neutral, while their size comprised between 25 kDa (SBD1) and 42 kDa (MBP). Although a large negative charge reduced the residence time of the protein inside the nanopore, all proteins entered the nanopore and remained trapped for several seconds. The analysis of the current signal revealed that inside the nanopore proteins most likely occupy two residence sites where they are in a relatively tight interaction with the nanopore lumen. Noisy signals were occasionally observed, and they most likely originated from the tumbling of the protein inside the nanopore with a frequency comparable to the sampling frequency (10 kHz) or the intrinsic motions of the protein itself. Nonetheless, the introduction of a dipole within the surface of the protein allowed orienting the protein with the electric field lines inside the nanopore and drastically reduced the current noise.
The affinity binding constant measured for the majority of the internalized proteins that bound to noncharged ligands compared reasonably well to bulk values, suggesting that the proteins remain folded inside the nanopore and that the concentration of ligands across the nanopore is comparable with bulk concentrations. It also suggests that the electrostatic interactions between the proteins and the interior of the nanopore, the relatively strong transmembrane electric fields, and the confinement of solvent inside the nanopore do not fundamentally change the function of proteins, which is rather surprising, as protein folding is known to be affected by electric fields in solid-state nanopores.64,78 Nonetheless, the small deviation in affinity of the adaptors can be compensated for sensor application by the usage of standard curves. The association rates measured for charged ligands, however, depended on the applied potential, indicating that the diffusion of charged molecules is affected by the electric field drop inside the nanopore. This finding might be useful for a sensor as well, because the applied potential can be used to tune the affinity of the nanopore sensor for charged analytes.
Investigations with solid-state nanopores revealed that, despite the fact that the dipole and shape of the protein might be displayed by ionic currents,74−77 the protein signal is originated almost exclusively by the excluded volume of the protein.64,78,79 By contrast, by using biological nanopores such as ClyA, which has a size similar to the size of the protein adaptor, we found that the protein signal is also influenced by the position of the protein within the nanopore, which in turn is determined by a complex relation between the protein size, shape, surface charge distribution, and residence site occupancy. This is advantageous since the binding of ligands to the protein is likely to change the position of the protein within the nanopore, which in turn is reflected by a change in the signal that is used to detect the metabolite.
Experimental Section
Materials
All chemicals were purchased from Sigma-Aldrich, enzymes from Thermo Scientific, and DNA from Integrated DNA Technologies (IDT).
Cloning
The genes encoding for the proteins used in this work, which were designed to include NdeI and XhoI restriction sites, were first digested and then ligated to either pT7SC, pET22b, or pET101TOPO vectors. The resulting plasmids were transformed into E. coli cells to amplify the DNA. The identity of the plasmid was checked by sequencing. We used three different plasmids in this study, all controlled by a T7 promoter. pT7SC: ClyA, GGBP; pET101TOPO: TbpA, SiaP, LBP, SpuD, IbpA; pET22b: SpuE, BtuF, Pnc1p, hoefavidin, MBP. The point mutations D82K,D83K,K216E introduced into Pnc1p and the removal of the His-Tag on the C-terminus were done by a mega-primer PCR80 followed by restriction with NdeI and XhoI and ligation to the pET22b vector.
Expression and Purification of ClyA Nanopores
ClyA nanopores were expressed and purified as previously reported.42 In brief, the protein was expressed in E. coli BL21(DE3) cells in 2-YT medium. When the optical density at 600 nm reached 0.6, 0.5 mM IPTG was added, the temperature was set to 25 °C, and the cells were grown for 20 h. Cells were then harvested by centrifugation (8000g, 5 min) and lysed by three freeze–thaw cycles and resuspension in lysis buffer containing 10 μg/mL lysozyme, 0.2 U/mL DNase, and 5 mM MgCl2. After centrifugation at 7500 rpm for 30 min, the supernatant resulted from a 100 mL culture was loaded on 100 μL of Ni-NTA resin for purification utilizing the His-tag at the C-terminus of the protein. Proteins were eluted with 300 mM imidazole in Tris buffer (15 mM Tris, 150 mM NaCl, pH 7.5), and the protein was oligomerized with 0.2% β-dodecylmaltoside (DDM) for 30 min at 37 °C. Oligomers were separated from monomers using native polyacrylamide gel electrophoresis. The band corresponding to the 12-mer of ClyA was cut out, and the protein was extracted by adding 0.2% DDM and 10 mM EDTA in Tris buffer.
Expression and Purification of Adapter Proteins
Purified SBD1 and SBD2 were kindly provided by Bert Poolman, University of Groningen, and prepared as described before.41 Purified MBP was kindly provided by Giorgos Gouridis, KU Leuven.
His6MBP was expressed and purified as previously.47 Briefly, cells harboring the plasmid expressing His6MBP was introduced to E. coli BL21(DE3) cells and grown until an optical density (OD600) of 0.5 was reached, and protein expression was induced by 0.25 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). The soluble material (50 000 g, 30 min, 4 °C) was purified on a Ni2+-sepharose resin (equilibrated: 50 mM Tris-HCl, pH 8, 1 M KCl, 10% glycerol,10 mM imidazole; washed: 50 mM Tris-HCl, pH 8, 50 mM KCl, 10% glycerol, 10 mM imidazole and subsequently with 50 mM Tris-HCl, pH 8, 50 mM KCl, 10% glycerol, 30 mM imidazole; eluted: 50 mM Tris-HCl, pH 8, 50 mM KCl, 10% glycerol, 300 mM imidazole), concentrated (Amicon, Merck-Millipore), dialyzed (50 mM Tris-HCl, pH 8, 50 mM KCl, 50% glycerol), aliquoted, and stored at −20 °C.
For all other proteins, plasmids containing the desired gene were transformed in E. coli BL21(DE3), and a starter culture in 2-YT medium was grown at 37 °C overnight. The starter was transferred the next day in 100 mL of TB medium (or 2-YT medium for GGBP and TbpA) to an optical density of 0.15 and grown at 37 °C until OD ≥ 0.6. Expression was induced by addition of 0.5 mM IPTG, and cells were grown at 25 °C overnight. The next day, cells were harvested by centrifugation (8000g, 5 min). All periplasmic substrate binding proteins (BtuF, GGBP, IbpA, SiaP, TbpA, LBP) were prepared as follows: the pellet containing overexpressed proteins was resuspended in 50 mL of ice-cold sucrose buffer (20% sucrose, 50 mM Tris, 1 mM EDTA, pH 7.5) per 100 mL of culture and incubated with gentle shaking and cooling for 30 min. The suspension was centrifuged at 7500 rpm for 20 min, and the pellet was resuspended in 30 mL of ice-cold water, shaken, and centrifuged as before. The supernatant was taken for Ni-NTA purification. Hoefavidin and Pnc1p were prepared by resuspending the pellet containing overexpressed proteins in 10 mL of lysis buffer (1 mM MgCl2, 0.2 U/mL Dnase I, 10 μg/mL lysozyme in protein buffer (150 mM NaCl, 15 mM Tris), pH 7.5), incubated 30 min at room temperature followed by 2 × 30 sweeps of sonication and 20 min of centrifugation at 7500 rpm.
All proteins except for hoefavidin were purified using Ni-NTA affinity chromatography. Typically the cell lysate containing the overexpressed protein was loaded on ca. 500 μL in protein buffer equilibrated Ni-NTA beads. The collected flow-through was loaded again to achieve maximum loading. The beads were washed with 20 mL of washing buffer (10 mM imidazole in protein buffer, pH 7.5). The protein was eluted with 5 mL of elution buffer (300 mM imidazole in protein buffer) in two steps and concentrated with Amicon centrifugal filters to a final volume of 500 μL. Hoefavidin was purified by affinity chromatography using 2-iminobiotin agarose beads (Sigma-Aldrich). The cell lysate was loaded twice on the beads using wash buffer (1 M NaCl, 50 mM Na2CO3, pH 11) followed by washing with 20 mL of the same wash buffer. Elution was obtained with 5 mL of elution buffer (50 mM NH4CH3CO2, pH 4). The protein was concentrated with Amicon centrifugal filters. Buffer was changed to protein buffer (150 mM NaCl, 15 mM Tris, pH 7.5) by diluting 1:100 in protein buffer and concentrating again by using the Amicon filters. Protein concentrations were determined by absorption measurement at 280 nm. Protein identity was confirmed by SDS-PAGE analysis. Proteins were stored with 20% glycerol at −80 °C.
Electrical Recordings in Planar Lipid Bilayers
The measuring setup consists of a vertical chamber containing two 500 μL compartments separated by a 20 μm PTFE film with a central aperture of ∼100 μm diameter. A lipid bilayer was formed on the aperture by adding a drop of hexadecane (2.5% (v/v) in pentane) on each side of the PTFE film directly above the aperture. After this, the compartments were filled with the recording buffer, and two drops of 1,2-diphytanoyl-sn-glycero-3-phosphocholine was added on each side. An electric potential was applied to the trans side utilizing Ag/AgCl electrodes. By lowering and raising the buffer level in one compartment across the aperture, a lipid bilayer was formed within the aperture. After letting the bilayer stabilize for 5 min, a pipet tip was dipped into the ClyA-AS solution and dipped afterward into the buffer of the cis compartment. The formation of single pores in the lipid bilayer was monitored by applying a −35 mV bias, resulting in a current of −60 pA. Adaptor proteins and ligands were added to the cis compartment. All experiments were done in triplicates, which means that three different single pores were used.
Electrophysiological Data Recording and Processing
Electrophysiological data were recorded under a negative potential, which was varied between −20 and −150 mV by using an Axopatch 200B patch clamp amplifier (Axon Instruments) connected to a DigiData 1440 A/D converter (Axon Instruments). The data were sampled with a frequency of 10 kHz, and a low-pass Bessel filter of 2 kHz was applied. After data recording, the traces were additionally filtered with a Gaussian low-pass filter with a 100 Hz cutoff. Data recording was executed using Clampex 10.7 software (Molecular Devices), and analysis was realized with Clampfit 10.7 software (Molecular Devices).
Analysis of Current Traces
Open pore (IO) and protein block currents (IB) were determined from Gaussian fits to all-point histograms using a bin width of 0.1 pA. IRES are then calculated as 100 × (IB/IO). Standard deviations of the mean values of Gaussian fits to all-point histograms of protein blockades (σ) were calculated from 1 s traces using Clampfit (Molecular Devices). The dwell times for proteins inside the nanopore were measured applying a single-channel search function in Clampfit (Molecular Devices). About 100 events at IB were collected, binned, and fitted to a single exponential to determine the average dwell time (τ). Single-channel search and dwell time analysis were also used to determine the association and dissociation rate constants for the ligands to their cognate protein adaptor. For that, levels referring to the open and closed protein state were predefined and picked by the program. Events with a duration of less than 10 ms were ignored. We collected at least 500 events per level per concentration. Association and dissociation rates were then calculated as 1/τon and 1/τoff, respectively. kon = 1/(τoncligand) and koff = 1/τoff. KD were calculated for individual ligand concentrations and used to measure the KD as koff/kon.
The KD for BtuF was measured by performing single-channel searches (Clampfit, Molecular Devices) on individual current blockades. Once the open and closed protein levels were identified, an analysis was performed without excluding short events. We picked at least 1000 events. The relative time the protein spent on the closed configuration was then estimated by counting events on the closed and open levels. Although this analysis did not reveal the association and dissociation rate constant, we could plot the fraction of the overall number of events in the closed conformation versus the concentration of the ligand to produce binding curves that could be fitted to a binding isotherm (Prism5 software, GraphPad, one-site total fit) to determine KD (Figure S6c).
KD can also be determined by plotting all-point histograms of full traces (2–3 min length) based on the IRES using a bin width of 0.1 pA. For that, IO was determined as described above. All amplitude values within the histogram were divided by IO to finally plot the IRESversus the amount of data points. The resulting peaks corresponding to the open (no ligand bound) and the closed (ligand bound) protein conformation were then identified by changing the ligand concentration in the cis chamber. Typically one peak decreased (protein’s open form) and one peak increased (protein’s closed form). We identified the IRES of the peak maxima of the open and the closed state and determined the counts at this particular IRES for the open state (NO) and the closed state (NC) at different ligand concentrations (added to the cis chamber). Parts of the histogram that did not change with the concentration of the ligands were excluded from histogram analysis (e.g., for SpuD, Figure S2). The amount of closed protein in the sample was calculated by NC/(NC + NO) and was plotted against ligand concentration to fit a binding isotherm and to calculate the respective KD. Full-point histograms corresponding to GGBP, SBD1, and SBD2 blockades showed well-defined peaks corresponding to the open and closed states. Hence, for these proteins a Gaussian fit was performed, and the area under the curve was used to calculate the fraction of the protein in the closed conformation.
Acknowledgments
We thank B. Poolman for providing us the purified SBD1 and SBD2 protein and F. Lucas and C. Wloka to provide the KD data for the thiamine binding protein. S.Z. was funded by an ERC consolidator grant number 726151.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b09434.
Figures S1–S16: SDS-PAGE of purified adaptor proteins, exemplary traces for all tested proteins including description of dissociation constant determination, dipole orientation of all tested proteins, voltage dependency of the protein dwell time. (PDF)
Author Contributions
S.Z. and G.M. designed the study and wrote the paper. S.Z. performed all experiments unless otherwise stated, analyzed data, and wrote the paper. N.H. cloned TbpA, SiaP, LBP, SpuD, and IbpA and expressed and purified TbpA. In addition, NH expressed, purified, and oligomerized the ClyA-AS nanopore. N.G. cloned, expressed, and purified GGBP and performed electrophysiological measurements of GGBP. G.G. cloned, expressed, and purified MBP. G.M. supervised the project. All authors verified the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Wishart D. S.; Tzur D.; Knox C.; Eisner R.; Guo A. C.; Young N.; Cheng D.; Jewell K.; Arndt D.; Sawhney S.; Fung C.; Nikolai L.; Lewis M.; Coutouly M.-A.; Forsythe I.; Tang P.; Shrivastava S.; Jeroncic K.; Stothard P.; Amegbey G.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–526. 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W. B.; Broadhurst D.; Begley P.; Zelena E.; Francis-McIntyre S.; Anderson N.; Brown M.; Knowles J. D.; Halsall A.; Haselden J. N.; Nicholls A. W.; Wilson I. D.; Kell D. B.; Goodacre R. Procedures for Large-Scale Metabolic Profiling of Serum and Plasma Using Gas Chromatography and Liquid Chromatography Coupled to Mass Spectrometry. Nat. Protoc. 2011, 6, 1060–1083. 10.1038/nprot.2011.335. [DOI] [PubMed] [Google Scholar]
- Fiehn O.; Kind T.. Metabolite Profiling in Blood Plasma. In Metabolomics; Weckwerth W., Ed.; Walker J. M., Series Ed.; Humana Press: Totowa, NJ, 2007; Vol. 358, pp 3–17. [DOI] [PubMed] [Google Scholar]
- Raamsdonk L. M.; Teusink B.; Broadhurst D.; Zhang N.; Hayes A.; Walsh M. C.; Berden J. A.; Brindle K. M.; Kell D. B.; Rowland J. J.; Westerhoff H. V.; van Dam K.; Oliver S. G. A Functional Genomics Strategy That Uses Metabolome Data to Reveal the Phenotype of Silent Mutations. Nat. Biotechnol. 2001, 19, 45–50. 10.1038/83496. [DOI] [PubMed] [Google Scholar]
- Nicholson J. K.; Holmes E.; Wilson I. D. Gut Microorganisms, Mammalian Metabolism and Personalized Health Care. Nat. Rev. Microbiol. 2005, 3, 431–438. 10.1038/nrmicro1152. [DOI] [PubMed] [Google Scholar]
- Beger R. D. A Review of Applications of Metabolomics in Cancer. Metabolites 2013, 3, 552–574. 10.3390/metabo3030552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson J. K.; Everett J. R.; Lindon J. C. Longitudinal Pharmacometabonomics for Predicting Patient Responses to Therapy: Drug Metabolism, Toxicity and Efficacy. Expert Opin. Drug Metab. Toxicol. 2012, 8, 135–139. 10.1517/17425255.2012.646987. [DOI] [PubMed] [Google Scholar]
- van der Greef J.; Hankemeier T.; McBurney R. N. Metabolomics-Based Systems Biology and Personalized Medicine: Moving towards n = 1 Clinical Trials?. Pharmacogenomics 2006, 7, 1087–1094. 10.2217/14622416.7.7.1087. [DOI] [PubMed] [Google Scholar]
- Zhang A.; Sun H.; Wang X. Serum Metabolomics as a Novel Diagnostic Approach for Disease: A Systematic Review. Anal. Bioanal. Chem. 2012, 404, 1239–1245. 10.1007/s00216-012-6117-1. [DOI] [PubMed] [Google Scholar]
- Kaddurah-Daouk R.; Kristal B. S.; Weinshilboum R. M. Metabolomics: A Global Biochemical Approach to Drug Response and Disease. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 653–683. 10.1146/annurev.pharmtox.48.113006.094715. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Zhu Z.; Shi J.; An Y.; Zhang K.; Wang Y.; Li S.; Jin L.; Ye W.; Cui M.; Chen X. Metabolomics in the Development and Progression of Dementia: A Systematic Review. Front. Neurosci. 2019, 13, 343. 10.3389/fnins.2019.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap I. K. S.; Brown I. J.; Chan Q.; Wijeyesekera A.; Garcia-Perez I.; Bictash M.; Loo R. L.; Chadeau-Hyam M.; Ebbels T.; De Iorio M.; Maibaum E.; Zhao L.; Kesteloot H.; Daviglus M. L.; Stamler J.; Nicholson J. K.; Elliott P.; Holmes E. Metabolome-Wide Association Study Identifies Multiple Biomarkers That Discriminate North and South Chinese Populations at Differing Risks of Cardiovascular Disease: Intermap Study. J. Proteome Res. 2010, 9, 6647–6654. 10.1021/pr100798r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. J.; Larson M. G.; Vasan R. S.; Cheng S.; Rhee E. P.; McCabe E.; Lewis G. D.; Fox C. S.; Jacques P. F.; Fernandez C.; O’Donnel C. J.; Carr S. A.; Mootha V. K.; Flores J. C.; Souza A.; Melander O.; Clish C. B.; Gerszten R. E. Metabolite Profiles and the Risk of Developing Diabetes. Nat. Med. 2011, 17, 448–453. 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesenfeld D. B.; Habermann N.; Owen R. W.; Scalbert A.; Ulrich C. M. Review of Mass Spectrometry–Based Metabolomics in Cancer Research. Cancer Epidemiol., Biomarkers Prev. 2013, 22, 2182–2201. 10.1158/1055-9965.EPI-13-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanaukee; Pönniö; Jääskeläinen Occurrence of Sialic Acids in Healthy Humans and Different Disorders. Eur. J. Clin. Invest. 1999, 29, 413–425. 10.1046/j.1365-2362.1999.00485.x. [DOI] [PubMed] [Google Scholar]
- Fuhrer T.; Zamboni N. High-Throughput Discovery Metabolomics. Curr. Opin. Biotechnol. 2015, 31, 73–78. 10.1016/j.copbio.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Manzi M.; Riquelme G.; Zabalegui N.; Monge M. E. Improving Diagnosis of Genitourinary Cancers: Biomarker Discovery Strategies through Mass Spectrometry-Based Metabolomics. J. Pharm. Biomed. Anal. 2020, 178, 112905. 10.1016/j.jpba.2019.112905. [DOI] [PubMed] [Google Scholar]
- Kasianowicz J. J.; Brandin E.; Branton D.; Deamer D. W. Characterization of Individual Polynucleotide Molecules Using a Membrane Channel. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13770–13773. 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler T. Z.; Pavlenok M.; Derrington I. M.; Niederweis M.; Gundlach J. H. Single-Molecule DNA Detection with an Engineered MspA Protein Nanopore. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 20647–20652. 10.1073/pnas.0807514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomidis S. K.; Hooyberghs J.; Maglia G.; Carlon E. DNA Capture into the ClyA Nanopore: Diffusion-Limited versus Reaction-Limited Processes. J. Phys.: Condens. Matter 2018, 30, 304001. 10.1088/1361-648X/aacc01. [DOI] [PubMed] [Google Scholar]
- Moon J.; Kim N.; Kim T.-J.; Jun J.-S.; Lee H. S.; Shin H.-R.; Lee S.-T.; Jung K.-H.; Park K.-I.; Jung K.-Y.; Kim M.; Lee S. K.; Chu K. Rapid Diagnosis of Bacterial Meningitis by Nanopore 16S Amplicon Sequencing: A Pilot Study. Int. J. Med. Microbiol. 2019, 309, 151338. 10.1016/j.ijmm.2019.151338. [DOI] [PubMed] [Google Scholar]
- Branton D.; Deamer D. W.; Marziali A.; Bayley H.; Benner S. A.; Butler T.; Di Ventra M.; Garaj S.; Hibbs A.; Huang X.; Jovanovich S. B.; Krstic P. S.; Lindsay S.; Ling X. S.; Mastrangelo C. H.; Meller A.; Oliver J. S.; Pershin Y. V.; Ramsey J. M.; Riehn R.; et al. The Potential and Challenges of Nanopore Sequencing. Nat. Biotechnol. 2008, 26, 1146–1153. 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G.; Willems K.; Soskine M.; Wloka C.; Maglia G. Electro-Osmotic Capture and Ionic Discrimination of Peptide and Protein Biomarkers with FraC Nanopores. Nat. Commun. 2017, 8, 935. 10.1038/s41467-017-01006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G.; Voet A.; Maglia G. FraC Nanopores with Adjustable Diameter Identify the Mass of Opposite-Charge Peptides with 44 Da Resolution. Nat. Commun. 2019, 10, 835. 10.1038/s41467-019-08761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Huang J.; Holden M. A.; Chen M. Peptide-Mediated Membrane Transport of Macromolecular Cargo Driven by Membrane Asymmetry. Anal. Chem. 2017, 89, 12369–12374. 10.1021/acs.analchem.7b03421. [DOI] [PubMed] [Google Scholar]
- Larimi M. G.; Mayse L. A.; Movileanu L. Interactions of a Polypeptide with a Protein Nanopore Under Crowding Conditions. ACS Nano 2019, 13, 4469–4477. 10.1021/acsnano.9b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahie M. A.; Yang B.; Pham B.; Chen M. Tuning the Selectivity and Sensitivity of an OmpG Nanopore Sensor by Adjusting Ligand Tether Length. ACS Sens. 2016, 1, 614–622. 10.1021/acssensors.6b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastoriza-Gallego M.; Rabah L.; Gibrat G.; Thiebot B.; van der Goot F. G.; Auvray L.; Betton J.-M.; Pelta J. Dynamics of Unfolded Protein Transport through an Aerolysin Pore. J. Am. Chem. Soc. 2011, 133, 2923–2931. 10.1021/ja1073245. [DOI] [PubMed] [Google Scholar]
- Thakur A. K.; Movileanu L. Real-Time Measurement of Protein–Protein Interactions at Single-Molecule Resolution Using a Biological Nanopore. Nat. Biotechnol. 2019, 37, 96–101. 10.1038/nbt.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell N. A. W.; Keyser U. F. Specific Protein Detection Using Designed DNA Carriers and Nanopores. J. Am. Chem. Soc. 2015, 137, 2035–2041. 10.1021/ja512521w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell N. A. W.; Keyser U. F. Digitally Encoded DNA Nanostructures for Multiplexed, Single-Molecule Protein Sensing with Nanopores. Nat. Nanotechnol. 2016, 11, 645–651. 10.1038/nnano.2016.50. [DOI] [PubMed] [Google Scholar]
- Si W.; Aksimentiev A. Nanopore Sensing of Protein Folding. ACS Nano 2017, 11, 7091–7100. 10.1021/acsnano.7b02718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Lee K.; Chen J.; Chen M. A ClyA Nanopore Tweezer for Analysis of Functional States of Protein-Ligand Interactions. bioRxiv 2019, 727503.(accessed Jan 31, 2020). [Google Scholar]
- Ohayon S.; Girsault A.; Nasser M.; Shen-Orr S.; Meller A. Simulation of Single-Protein Nanopore Sensing Shows Feasibility for Whole-Proteome Identification. PLoS Comput. Biol. 2019, 15, e1007067 10.1371/journal.pcbi.1007067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varongchayakul N.; Song J.; Meller A.; Grinstaff M. W. Single-Molecule Protein Sensing in a Nanopore: A Tutorial. Chem. Soc. Rev. 2018, 47, 8512–8524. 10.1039/C8CS00106E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Niu A.; Jian F.; Wang Y.; Yao F.; Wei Y.; Tian L.; Kang X. Metal–Organic Complex-Functionalized Protein Nanopore Sensor for Aromatic Amino Acids Chiral Recognition. Analyst 2017, 142, 1048–1053. 10.1039/C7AN00097A. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Bayley H. Continuous Stochastic Detection of Amino Acid Enantiomers with a Protein Nanopore. Angew. Chem., Int. Ed. 2012, 51, 9606–9609. 10.1002/anie.201205687. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Brain K. L.; Bayley H. Real-Time Stochastic Detection of Multiple Neurotransmitters with a Protein Nanopore. ACS Nano 2012, 6, 5304–5308. 10.1021/nn301125y. [DOI] [PubMed] [Google Scholar]
- Ramsay W. J.; Bayley H. Single-Molecule Determination of the Isomers of d-Glucose and d-Fructose That Bind to Boronic Acids. Angew. Chem., Int. Ed. 2018, 57, 2841–2845. 10.1002/anie.201712740. [DOI] [PubMed] [Google Scholar]
- Miyagawa T.; Hongo S.; Nakamura N.; Horiguchi Y.; Miyahara Y.; Shibata H.. A Novel Diagnostic System for Infectious Diseases Using Solid-State Nanopore Devices. In 2018 40th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC); 2018; pp 2833–2836. [DOI] [PubMed]
- Van Meervelt V.; Soskine M.; Singh S.; Schuurman-Wolters G. K.; Wijma H. J.; Poolman B.; Maglia G. Real-Time Conformational Changes and Controlled Orientation of Native Proteins Inside a Protein Nanoreactor. J. Am. Chem. Soc. 2017, 139, 18640–18646. 10.1021/jacs.7b10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galenkamp N. S.; Soskine M.; Hermans J.; Wloka C.; Maglia G. Direct Electrical Quantification of Glucose and Asparagine from Bodily Fluids Using Nanopores. Nat. Commun. 2018, 9, 4085. 10.1038/s41467-018-06534-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K. J.; Song S.; Mason K.; Pinkett H. W. Selective Substrate Uptake: The Role of ATP-Binding Cassette (ABC) Importers in Pathogenesis. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 868–877. 10.1016/j.bbamem.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berntsson R. P.-A.; Smits S. H. J.; Schmitt L.; Slotboom D.-J.; Poolman B. A Structural Classification of Substrate-Binding Proteins. FEBS Lett. 2010, 584, 2606–2617. 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- Husada F.; Gouridis G.; Vietrov R.; Schuurman-Wolters G. K.; Ploetz E.; de Boer M.; Poolman B.; Cordes T. Watching Conformational Dynamics of ABC Transporters with Single-Molecule Tools. Biochem. Soc. Trans. 2015, 43, 1041–1047. 10.1042/BST20150140. [DOI] [PubMed] [Google Scholar]
- Gouridis G.; Schuurman-Wolters G. K.; Ploetz E.; Husada F.; Vietrov R.; de Boer M.; Cordes T.; Poolman B. Conformational Dynamics in Substrate-Binding Domains Influences Transport in the ABC Importer GlnPQ. Nat. Struct. Mol. Biol. 2015, 22, 57–64. 10.1038/nsmb.2929. [DOI] [PubMed] [Google Scholar]
- de Boer M.; Gouridis G.; Vietrov R.; Begg S. L.; Schuurman-Wolters G. K.; Husada F.; Eleftheriadis N.; Poolman B.; McDevitt C. A.; Cordes T. Conformational and Dynamic Plasticity in Substrate-Binding Proteins Underlies Selective Transport in ABC Importers. eLife 2019, 8, e44652 10.7554/eLife.44652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiner D. R.; Hegde S. S.; Blanchard J. S. Kinetics and Inhibition of Nicotinamidase from Mycobacterium Tuberculosis. Biochemistry 2010, 49, 9613–9619. 10.1021/bi1011157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham O.; Meir A.; Fish A.; Bayer E. A.; Livnah O. Hoefavidin: A Dimeric Bacterial Avidin with a C-Terminal Binding Tail. J. Struct. Biol. 2015, 191, 139–148. 10.1016/j.jsb.2015.06.020. [DOI] [PubMed] [Google Scholar]
- Cadieux N.; Bradbeer C.; Reeger-Schneider E.; Koster W.; Mohanty A. K.; Wiener M. C.; Kadner R. J. Identification of the Periplasmic Cobalamin-Binding Protein BtuF of Escherichia Coli. J. Bacteriol. 2002, 184, 706–717. 10.1128/JB.184.3.706-717.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrou J.; Crosson S. Myo-Inositol and d-Ribose Ligand Discrimination in an ABC Periplasmic Binding Protein. J. Bacteriol. 2013, 195, 2379–2388. 10.1128/JB.00116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. M.; Olson J. S.; Pflugrath J. W.; Quiocho F. A. Rates of Ligand Binding to Periplasmic Proteins Involved in Bacterial Transport and Chemotaxis. J. Biol. Chem. 1983, 258, 13665–13672. [PubMed] [Google Scholar]
- Gangi Setty T.; Cho C.; Govindappa S.; Apicella M. A.; Ramaswamy S. Bacterial Periplasmic Sialic Acid-Binding Proteins Exhibit a Conserved Binding Site. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 1801–1811. 10.1107/S139900471400830X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes J. W.; Chatterjee D.; Soriano E. V.; Ealick S. E.; Begley T. P. Construction of a Thiamin Sensor from the Periplasmic Thiamin Binding Protein. Chem. Commun. 2011, 47, 2273–2275. 10.1039/C0CC04228E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson U.; Salopek-Sondi B.; Luck L. A.; Mowbray S. L. X-Ray Structures of the Leucine-Binding Protein Illustrate Conformational Changes and the Basis of Ligand Specificity. J. Biol. Chem. 2004, 279, 8747–8752. 10.1074/jbc.M311890200. [DOI] [PubMed] [Google Scholar]
- Wu D.; Lim S. C.; Dong Y.; Wu J.; Tao F.; Zhou L.; Zhang L.-H.; Song H. Structural Basis of Substrate Binding Specificity Revealed by the Crystal Structures of Polyamine Receptors SpuD and SpuE from Pseudomonas Aeruginosa. J. Mol. Biol. 2012, 416, 697–712. 10.1016/j.jmb.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Telmer P. G.; Shilton B. H. Insights into the Conformational Equilibria of Maltose-Binding Protein by Analysis of High Affinity Mutants. J. Biol. Chem. 2003, 278, 34555–34567. 10.1074/jbc.M301004200. [DOI] [PubMed] [Google Scholar]
- Unione L.; Ortega G.; Mallagaray A.; Corzana F.; Pérez-Castells J.; Canales A.; Jiménez-Barbero J.; Millet O. Unraveling the Conformational Landscape of Ligand Binding to Glucose/Galactose-Binding Protein by Paramagnetic NMR and MD Simulations. ACS Chem. Biol. 2016, 11, 2149–2157. 10.1021/acschembio.6b00148. [DOI] [PubMed] [Google Scholar]
- Millet O.; Hudson R. P.; Kay L. E. The Energetic Cost of Domain Reorientation in Maltose-Binding Protein as Studied by NMR and Fluorescence Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 12700–12705. 10.1073/pnas.2134311100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B.; Stokes C.; Fahie M.; Chen M.; Golovchenko J. A.; Hau L. V. Protein Motion and Configurations in a Form-Fitting Nanopore: Avidin in ClyA. Biophys. J. 2018, 115, 801–808. 10.1016/j.bpj.2018.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penner M. H.; Frieden C. Kinetic Analysis of the Mechanism of Escherichia Coli Dihydrofolate Reductase. J. Biol. Chem. 1987, 262, 15908–15914. [PubMed] [Google Scholar]
- Willems K.; Ruić D.; Biesemans A.; Galenkamp N. S.; Van Dorpe P.; Maglia G. Engineering and Modeling the Electrophoretic Trapping of a Single Protein Inside a Nanopore. ACS Nano 2019, 13, 9980–9992. 10.1021/acsnano.8b09137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; Maglia G. Single-Molecule Analyte Recognition with ClyA Nanopores Equipped with Internal Protein Adaptors. J. Am. Chem. Soc. 2015, 137, 5793–5797. 10.1021/jacs.5b01520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talaga D. S.; Li J. Single-Molecule Protein Unfolding in Solid State Nanopores. J. Am. Chem. Soc. 2009, 131, 9287–9297. 10.1021/ja901088b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Fologea D.; Rollings R.; Ledden B. Characterization of Protein Unfolding with Solid-State Nanopores. Protein Pept. Lett. 2014, 21, 256–265. 10.2174/09298665113209990077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch R. B.; Han J.; Renaud P. Transport Phenomena in Nanofluidics. Rev. Mod. Phys. 2008, 80, 839–883. 10.1103/RevModPhys.80.839. [DOI] [Google Scholar]
- Bhamidimarri S. P.; Prajapati J. D.; van den Berg B.; Winterhalter M.; Kleinekathöfer U. Role of Electroosmosis in the Permeation of Neutral Molecules: CymA and Cyclodextrin as an Example. Biophys. J. 2016, 110, 600–611. 10.1016/j.bpj.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firnkes M.; Pedone D.; Knezevic J.; Döblinger M.; Rant U. Electrically Facilitated Translocations of Proteins through Silicon Nitride Nanopores: Conjoint and Competitive Action of Diffusion, Electrophoresis, and Electroosmosis. Nano Lett. 2010, 10, 2162–2167. 10.1021/nl100861c. [DOI] [PubMed] [Google Scholar]
- Gu L.-Q.; Cheley S.; Bayley H. Electroosmotic Enhancement of the Binding of a Neutral Molecule to a Transmembrane Pore. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 15498–15503. 10.1073/pnas.2531778100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesemans A.; Soskine M.; Maglia G. A Protein Rotaxane Controls the Translocation of Proteins across a ClyA Nanopore. Nano Lett. 2015, 15, 6076–6081. 10.1021/acs.nanolett.5b02309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; Moeyaert B.; Cheley S.; Bayley H.; Maglia G. An Engineered ClyA Nanopore Detects Folded Target Proteins by Selective External Association and Pore Entry. Nano Lett. 2012, 12, 4895–4900. 10.1021/nl3024438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; De Maeyer M.; Maglia G. Tuning the Size and Properties of ClyA Nanopores Assisted by Directed Evolution. J. Am. Chem. Soc. 2013, 135, 13456–13463. 10.1021/ja4053398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheepers G. H.; Lycklama A Nijeholt J. A.; Poolman B. An Updated Structural Classification of Substrate-Binding Proteins. FEBS Lett. 2016, 590, 4393–4401. 10.1002/1873-3468.12445. [DOI] [PubMed] [Google Scholar]
- Houghtaling J.; Ying C.; Eggenberger O. M.; Fennouri A.; Nandivada S.; Acharjee M.; Li J.; Hall A. R.; Mayer M. Estimation of Shape, Volume, and Dipole Moment of Individual Proteins Freely Transiting a Synthetic Nanopore. ACS Nano 2019, 13, 5231–5242. 10.1021/acsnano.8b09555. [DOI] [PubMed] [Google Scholar]
- Yusko E. C.; Bruhn B. R.; Eggenberger O. M.; Houghtaling J.; Rollings R. C.; Walsh N. C.; Nandivada S.; Pindrus M.; Hall A. R.; Sept D.; Li J.; Kalonia D. S.; Mayer M. Real-Time Shape Approximation and Fingerprinting of Single Proteins Using a Nanopore. Nat. Nanotechnol. 2017, 12, 360–367. 10.1038/nnano.2016.267. [DOI] [PubMed] [Google Scholar]
- Waduge P.; Hu R.; Bandarkar P.; Yamazaki H.; Cressiot B.; Zhao Q.; Whitford P. C.; Wanunu M. Nanopore-Based Measurements of Protein Size, Fluctuations, and Conformational Changes. ACS Nano 2017, 11, 5706–5716. 10.1021/acsnano.7b01212. [DOI] [PubMed] [Google Scholar]
- Hu R.; Rodrigues J. V.; Waduge P.; Yamazaki H.; Cressiot B.; Chishti Y.; Makowski L.; Yu D.; Shakhnovich E.; Zhao Q.; Wanunu M. Differential Enzyme Flexibility Probed Using Solid-State Nanopores. ACS Nano 2018, 12, 4494–4502. 10.1021/acsnano.8b00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman K. J.; Jürgens M.; Prabhu A.; Ahn C. W.; Jemth P.; Edel J. B.; Kim M. J. Chemical, Thermal, and Electric Field Induced Unfolding of Single Protein Molecules Studied Using Nanopores. Anal. Chem. 2011, 83, 5137–5144. 10.1021/ac2001725. [DOI] [PubMed] [Google Scholar]
- Wilson J.; Sarthak K.; Si W.; Gao L.; Aksimentiev A. Rapid and Accurate Determination of Nanopore Ionic Current Using a Steric Exclusion Model. ACS Sens. 2019, 4, 634–644. 10.1021/acssensors.8b01375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammann M.; Laufs J.; Schell J.; Gronenborn B. Rapid Insertional Mutagenesis of DNA by Polymerase Chain Reaction (PCR). Nucleic Acids Res. 1989, 17, 5404. 10.1093/nar/17.13.5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.