

Abstract
Mitomycin C (MC), a potent antitumor drug, and decarbamoylmitomycin C (DMC), a derivative lacking the carbamoyl group, form highly cytotoxic DNA interstrand crosslinks. The major interstrand crosslink formed by DMC is the C1” epimer of the major crosslink formed by MC. The molecular basis for the stereochemical configuration exhibited by DMC was investigated using biomimetic synthesis. The formation of DNA-DNA crosslinks by DMC is diastereospecific and diastereodivergent: Only the 1”S-diastereomer of the initially formed monoadduct can form crosslinks at GpC sequences, and only the 1”R-diastereomer of the monoadduct can form crosslinks at CpG sequences. We also show that CpG and GpC sequences react with divergent diastereoselectivity in the first alkylation step: 1”S stereochemistry is favored at GpC sequences and 1”R stereochemistry is favored at CpG sequences. Therefore, the first alkylation step results, at each sequence, in the selective formation of the diastereomer able to generate an interstrand DNA-DNA crosslink after the “second arm” alkylation. Examination of the known DNA adduct pattern obtained after treatment of cancer cell cultures with DMC indicates that the GpC sequence is the major target for the formation of DNA-DNA crosslinks in vivo by this drug.
Keywords: antitumor agents, biomimetic synthesis, DNA crosslinking, mitomycins, oligonucleotides
Graphical Abstract
GpC sequences are crosslinked by Mitomycins in duplex DNA: Decarbamoylmitomycin C (DMC) is able to form DNA interstrand crosslinks both at GpC and CpG sequences. The crosslinks have an opposite stereochemical configuration at each sequence. The GpC sequence is the major target for DNA crosslinking in vivo by this drug, in marked contrast to the current consensus of CpG specificity for Mitomycins.
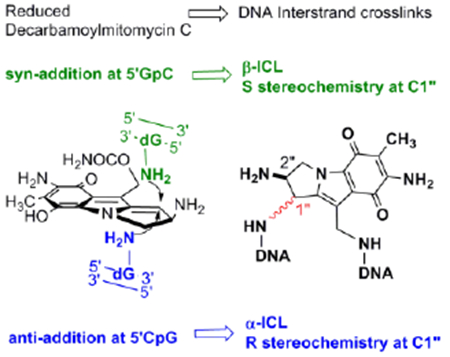
Decarbamoylmitomycin C (DMC, 1) is a derivative of the anticancer drug mitomycin C (MC, 2) lacking the O10 carbamoyl group.[1,2] MC is the prototype bioreductive drug: inert in its original structure, it is reduced in vivo to an intermediate (4b, scheme 1) that reacts with nucleophiles in two sequential steps at the C1 and C10 positions.[3] In duplex DNA, MC forms interstrand crosslinks which constitute the molecular basis for the cytotoxic effects of the drug.[4,5] The current consensus is that mitomycins target CpG sequences for crosslinking.
Scheme 1.
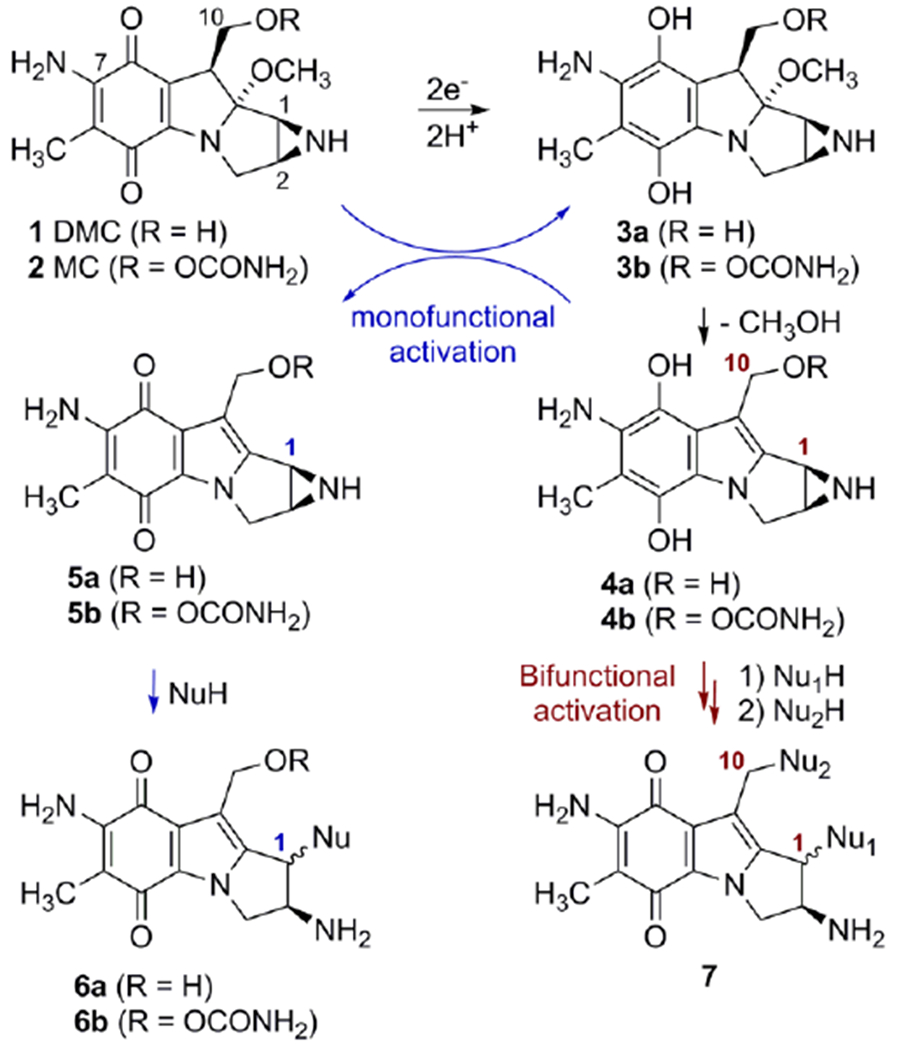
Reductive activation and formation of DNA adducts by Mitomycin C and Decarbamoylmitomycin C.
Mitomycins’ activation pathway 2→3b→4b→7, Scheme 1, is termed “bifunctional activation”, meaning it activates both C1 and C10 as electrophilic positions. An alternative “monofunctional activation” pathway of mitomycins involves the formation of 5b in an autocatalytic process, a compound that functions as a monoalkylating agent at C1 (Scheme 1).[6,7] For MC, the DNA interstrand crosslink is formed between the exocyclic amino groups of deoxyguanosine residues located on opposing DNA strands (10a, Figure 1).[8] MC also forms several monoadducts after reaction with DNA, such as 9a (Figure 1).[9]
Figure 1.
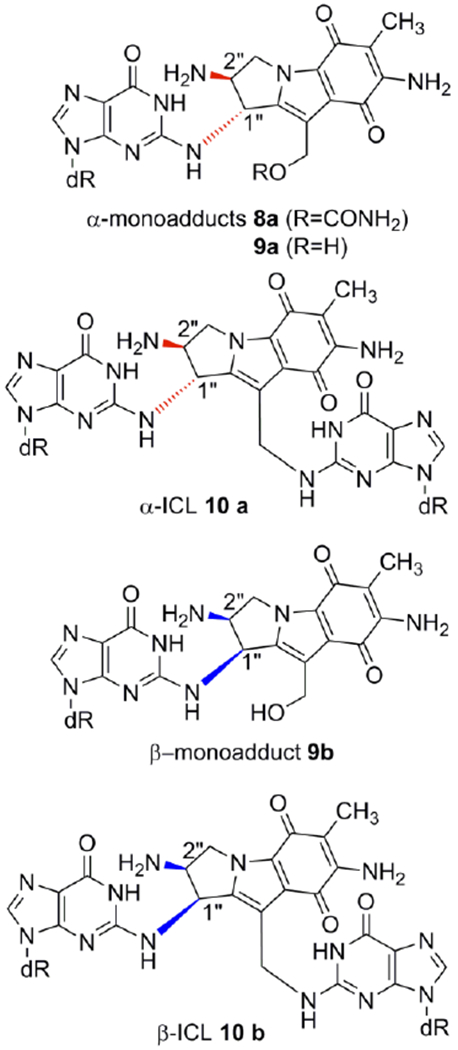
Mitomycin C and Decarbamoylmitomycin C DNA adducts. (ICL: interstrand crosslink)
DMC was originally used in several studies as a monofunctional analog of MC under the assumption that replacing the carbamoyloxy group at C10 with a poor leaving group (the hydroxyl group) would render C10 a non-alkylable position.[10,11,12] This hypothesis was, in principle, confirmed by analyzing products from cell-free reactions between DNA and reductively activated DMC: the major adduct was monoadduct 9a, and no crosslink was detected.[13]
However, in ensuing experiments, DMC showed a series of unexpected properties.[14] First, DMC displayed a higher cytotoxicity than MC in several cell lines. This was surprising because, assuming that DMC could not form DNA crosslinks, it was expected to be much less cytotoxic than MC. This paradox was later resolved by another startling finding: contrary to what was expected, DMC could indeed form DNA crosslinks.[14]
A final unexpected finding concerned the stereochemistry at C1” of DMC adducts. For MC, the stereochemical configuration at C1” of the major adducts is always R (e.g. 8a, 9a, 10a). Unpredictably, DMC major crosslink (10b) and monoadduct (9b) presented a stereochemical configuration at C1” opposed to that of MC adducts. In this communication we will refer to dG adducts with a R stereochemistry at C1”as α-adducts, while adducts with an C1”-S configuration will be denoted as β-adducts. These puzzling results could not be rationalized at the time mainly because the adduct profile observed in vivo could not be reproduced in cell-free reactions with DMC. For instance, the reaction of poly d(CpG) with DMC activated with sodium dithionite, gave exclusively α-adducts.[9,14]
In our quest to understand the molecular basis of the different adduct profiles formed by MC and DMC; we further explored the reactivity of DMC with DNA. Authentic standards 9a, 9b and 10a were synthesized as previously reported and these standards were used to identify 9a, 9b and 10a in the products of the reaction between DMC and DNA (supporting information S-5-2 and S7).[9] The stereochemistry of adducts 9a, 9b and 10a has been unambiguously determined by Circular Dichroism (CD) spectroscopy.[8,9,14.15]. Adduct 10b has been formerly isolated and characterized from cell cultures.[14] An authentic standard of 10b was synthesized in this work for the first time from reactions between Calf Thymus DNA and DMC (supporting information S-3-4 and S-5-3). The stereochemistry at C1” of 10b was established by CD spectroscopy (supporting information S-5-3). A description of the identification of adducts 9a, 9b, 10a and 10b in the digest of oligonucleotides studied and co-injection chromatograms are available in the supporting information section (S-5-2; S-5-3 and S7).
Using Calf Thymus DNA as substrate, we searched for reaction conditions that maximized the bifunctional route (4→7, Scheme 1) to better mimic cellular systems since, in cells, the activation occurs purely by the bifunctional pathway.[9] We developed reactions that produced an adduct profile that closely resembles the adduct profile observed in cell cultures treated with DMC.[16] These reaction conditions involve the use of slightly acidic pH and addition of a fast reducing agent (sodium dithionite) in excess.[16] We found that at more acidic pH (pH 4.5), the adduct profile presented a high proportion of products from N7 guanine monoalkylation in addition to the expected N2 adducts. This is in agreement with previous observations on the reaction of acid activated mitomcyins with DNA.[7,17] At higher pH (pH 7.4), as anticipated, the ratio crosslinks /monoadducts was lower than at pH 5.8.[7,16] At pH 5.8, the adduct profile from DMC treated Calf Thymus DNA was very similar to the adduct profile observed in cells and this pH was chosen for further reactions.[16] A key finding was to discover that the temperature plays a determinant role in the reaction outcome: At 0°C, only monoadducts are formed, but higher temperatures started to promote the formation of crosslinks (supporting information, S6). The reactions performed at 37 °C gave the maximum yield of crosslink adducts, while also increasing the β/α adduct ratio, and this temperature was used in succeeding crosslinking reactions.[16]
These biomimetic reaction conditions were applied to short oligonucleotides containing either CpG or GpC sequences, the two possible targets for interstrand crosslinks. The selected oligonucleotides had to guarantee duplex formation at 37 °C, and include several target sequences to maximize the yield of adducts. With these criteria in mind, we designed the following oligonucleotides:
-
11: d(TGCTTGCTTGCTTGCTTGCTTGCT.
AGCAAGCAAGCAAGCAAGCAAGCA)
-
12: d(TGCATGCATGCAAGCTAGCTAGCT.
AGCTAGCTAGCTTGCATGCATGCA)
-
13: d(TCGTTCGTTCGTTCGTTCGTTGCT.
ACGAACGAACGAACGAACGAACGA)
-
14: d(TCGATCGATCGAACGTACGTACGT.
ACGTACGTACGTTCGATCGATCGA)
Two of them, 11 and 12, include GpC sequences (11 has six d(TGCT·AGCA) sequences and 12 has three d(TGCA·TGCA) and three d(AGCT·AGCT)). The other two, 13 and 14, provide the CpG target sequences (13 contains six d(TCGT·ACGA) sequences and 14 has three d(TCGA·TCGA) and three d(ACGT·ACGT)). All oligonucleotides were alkylated with DMC using our optimized bifunctional activation conditions. The adduct profile was determined by HPLC after purification and enzymatic digestion (Figure 2).
Figure 2.
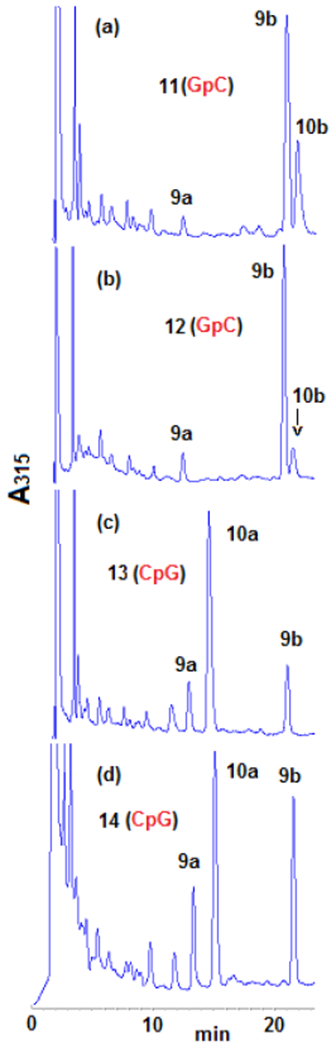
HPLC chromatograms (315 nm) of oligonucleotides digests (a) 11; (b) 12; (c) 13; (d) 14. Oligonucleotides were treated with DMC using optimized bifunctional activation conditions at 37°C. Enzymatic digestion: Oligonucleotides (1 A260 unit) were incubated with 1 unit of nuclease P1 at 37°C for 2 hours in 0.8 mL of 0.02 M ammonium acetate, pH 5.5. The pH was adjusted to 8.2 by addition of 0.2 M NaOH. MgCl2 (0.1 M solution, 20μL) was added followed by Snake Venom Diesterase (2 units) and Alkaline Phosphatase (2 units). Incubation continued at 37°C for 2.5 h. Digestion mixtures were analysed by HPLC on a Kromasil C-18 reverse phase column. The elution system was: 6-18% acetonitrile in 0.03 M potassium phosphate, pH 5.4, in 60 min, 1 mL/min flow rate. The temperature of the column was set at 45°C. More information is provided in the supporting information section (S-5-1 and S-4-2).
These experiments provided a clear picture of DMC DNA crosslinking: Alkylation of oligonucleotides containing GpC sequences resulted in the formation of β-crosslink 10b, β-monoadduct 9b and a small amount of 9a. However, α-crosslink 10a was not detected. In contrast, at CpG steps, α-crosslink 10a was the major adduct, together with epimeric monoadducts 9a and 9b but β-crosslink 10b was not detected (Figure 2 and Table 1). These results demonstrate that DMC can crosslink DNA both at GpC and CpG sequences. This is the first time that a mitomycin derivative is shown to significantly target GpC sequences for crosslinking. Previous research has unambiguously shown that MC crosslinks DNA at CpG sequences, but that GpC sequences are not a target for DNA crosslinking.[18,19,20] The CpG sequence-specificity has also been observed for related mitomycinoid alkaloids.[21]
Table 1.
Percent yield of monoadducts and crosslink adducts formed with DMC at 37°C. ND: non detected.
| Oligonucleotide | 9a | 9b | R 9b/9a | 10a | 10b | Crosslinking efficiency | Total dG adducts |
|---|---|---|---|---|---|---|---|
| 11 (GpC) | 0.03a | 0.34 (±0.02) | 11.4b | ND | 0.22 (±0.03) | 39% | 0.57(±0.02) |
| 12 (GpC) | 0.14 (±0.007) | 0.55 (±0.005) | 7.1 (±0.8) | ND | 0.12 (±0.02) | 18% | 0.82(±0.005) |
| 13 (CpG) | 0.09 (±0.02) | 0.11 (±0.007) | 1.3 (±0.02) | 0.40 (±0.01) | ND | 82% | 0.60(±0.02) |
| 14 (CpG) | 0.17 (±0.01) | 0.22 (±0.04) | 1.63 (±0.2) | 0.41 (±0.01) | ND | 71% | 0.82(±0.01) |
9a could only be detected in 1 experiment out of three repeats.
The ratio 9b/9a could only be determined in one case out of three repeats.
The results of these experiments also indicate that the formation of crosslinks by DMC occurs by a diastereospecific and diastereodivergent reaction. At GpC sequences only β-crosslink 10b is formed. Even though α-monoadduct 9a is present, it did not evolve to form α-crosslink 10a (Figures 2a, 2b). In contrast, at CpG steps α-crosslink 10a is formed specifically and 9b did not generate 10 b (Figures 2c, 2d). The diastereospecificity of the crosslinking reaction was further demonstrated by dissecting it into its two alkylation steps. We performed the reaction of DMC with oligonucleotides 11, 12, 13 and 14 using the optimal conditions for monoalkylation. Figure 3, (top), and Table 2 show that, as expected, monoadducts 9a and 9b were the only adducts formed at 0°C with the four oligonucleotides and, importantly, that they are generated to a significant extent both at CpG and GpC steps (Figure 3, top). The monoalkylated duplex oligonucleotides were then reduced at 37°C to trigger the second arm alkylation. Analysis of the products confirmed the formation of β-crosslink 10b exclusively at GpC steps (oligonucleotides 11 and 12, Figures 3b and 3d) and the formation of α-crosslink 10a solely at CpG steps (oligonucleotides 13 and 14, Figures 3f and 3h). This experiment clearly proves that α-monoadduct 9a, when formed at GpC sequences, is not competent to evolve toward the formation of a crosslink. Analogously, β-monoadduct 9b is not able to form crosslinks at CpG sequences (Scheme 2). The diastereospecificity observed in this reaction can be attributed to opposed orientations of the monoadduct in the minor groove of DNA, with each orientation determined by the monoadduct stereochemistry at C1”, as proposed before for MC.[19]
Figure 3.
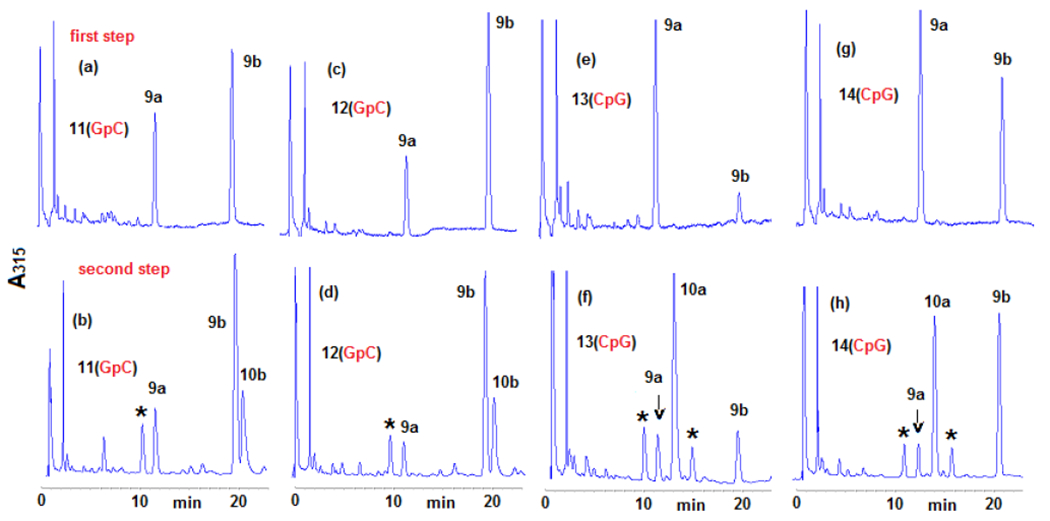
HPLC chromatograms of the enzymatic digestion of oligonucleotides 11, 12, 13 and 14 after two-step crosslinking reactions (315 nm). Oligonucleotides were reacted with DMC using optimized bifunctional activation conditions at 0° C (first step), purified, then crosslinked at 37°C with sodium dithionite (second step). Peaks labelled with an asterisk indicate bisulfite adducts. (a) 11, first alkylation step, 0°C; (b) 11, second alkylation step, 37°C; (c) 12, first alkylation step 0°C; (d) 12, second alkylation step, 37°C; (e) 13, first alkylation step, 0°C; (f) 13, second alkylation step, 37°C; (g) 14, first alkylation step, 0°C; (h) 14, second alkylation step, 37°C. Digestion and analysis were performed as described in Figure 2. More information is provided in the supporting information section (S-5-1 and S-4-2)
Table 2.
Percent yield of monoadducts and crosslink adducts formed in the two-step crosslinking reactions with DMC. ND: non detected. NA: non available.
| Oligonucleotide | Alkylation step | 9a | 9b | R 9b/9a | 10a | 10b | Crosslinking efficiency |
|---|---|---|---|---|---|---|---|
| 11 (GpC) | first | 0.54 (±0.04) | 0.98 (±0.04) | 1.8 (±0.07) | ND | ND | NA |
| second | 0.31 (±0.2) | 1.20 (±0.6) | 4.2 (±0.6) | ND | 0.60 (±0.4) | 33% | |
| 12 (GpC) | first | 0.50 (±0.02) | 1.52 (±0.07) | 3.10 (±0.04) | ND | ND | NA |
| second | 0.12 (±0.02) | 0.80 (±0.02) | 6.90 (±1) | ND | 0.40 (±0.09) | 33% | |
| 13 (CpG) | first | 0.92 (±0.2) | 0.19 (±0.04) | 0.21 (±0.06) | ND | ND | NA |
| second | 0.11 (±0.02) | 0.13 (±0.01) | 1.13 (±0.3) | 0.50 (±0.2) | ND | 82% | |
| 14 (CpG) | first | 1.1 (±0.07) | 0.88 (±0.08) | 0.8 (±0.07) | ND | ND | NA |
| second | 0.14 (±0.001) | 0.70 (±0.03) | 4.90 (±0.2) | 0.76 (±0.01) | ND | 84% |
Scheme 2.
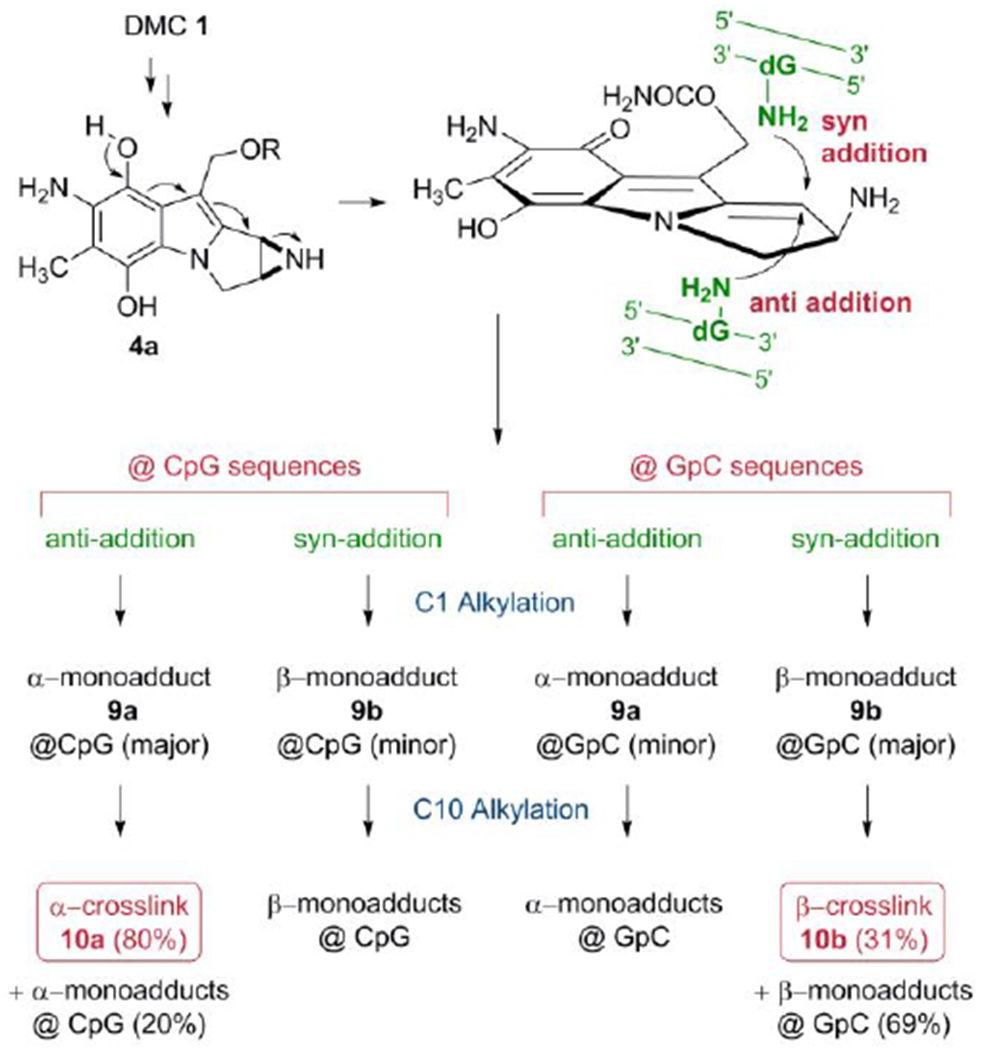
Formation of DNA-DNA crosslinks by DMC.
The results obtained in the first reaction of the two-step crosslinking experiment showed that the monoalkylation reaction of DMC is diastereoselective and diastereodivergent at both sequences: 9b is the major adduct formed at GpC sequences by a factor of 2 to 3-fold (Figures 3a and 3c), while 9a is the major adduct formed at CpG steps by 5-fold for oligonucleotide 13 (Figure 3b) and 1.25-fold for oligonucleotide 14 (Figure 2d). The monoalkylation reaction, therefore, displays a “smart” diastereoselectivity. With this term, we emphasize that the first alkylation results, at each sequence, in the selective formation of the diastereomer able to generate a crosslink. If that is the case, then DMC would constitute an extraordinary example of a chemically intelligent semisynthetic drug.
Our results also show that crosslink formation is significantly more efficient at CpG steps than at GpC steps. On average, the conversion of monoadduct to crosslink occurs with 31% efficiency at GpC steps and with 80% efficiency at CpG steps (Tables 1 and 2). This is probably the consequence of the different spatial configurations adopted by the reacting dG residues at CpG and GpC (figure 4). The second alkylation step is likely to be governed by kinetic factors: the C10 electrophilic position in the short-lived activated monoadduct must react faster with the dG amino group in the opposite strand than it does with solvent or other nucleophiles. At CpG sequences the distance between the two reacting amino group is on average 3.3 Å, while at GpC sequences it is 4.1 Å (both measured from PDB file 1D65).[22] The distance between C1” and C10” in the monoadduct formed by MC is 3.4 Å (as measured from PDB file 199D),[23] a perfect match for CpG crosslinking, but significantly shorter than the distance between the two nucleophiles at GpC sequences. A consequence of these structural differences is that the second alkylation step occurs with high efficiency at CpG sequences, while at GpC steps only a small population of conformers (about one third) adopts a viable conformation for crosslinking, and, therefore, most of the conformers react with water or other nucleophiles, such as bisulfite.[24]
Figure 4.
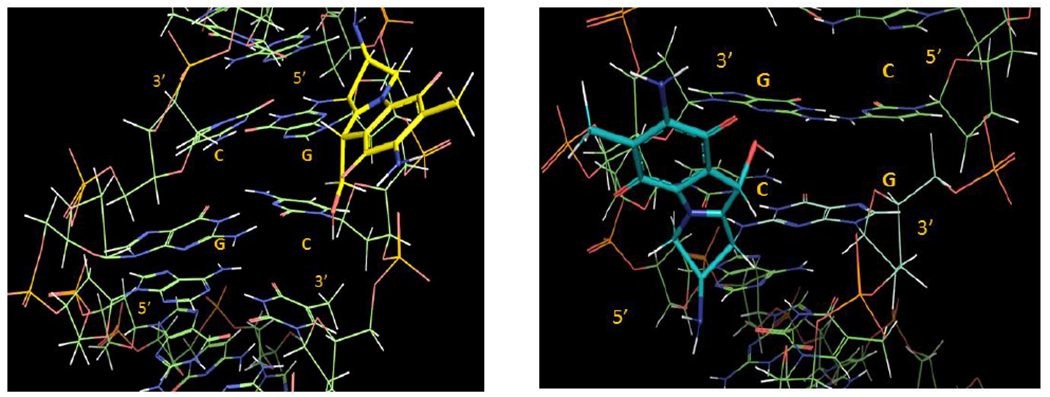
3D representations of adduct 9b, left (9b is in yellow and in a 5’-GpC sequence context) and 9a, right (9a is in blue and in a 5’-CpG sequence context). The DNA segments under view are centred around the mitomycin complexation site and involve the d(AG[9b]CT).d(AGCT) (left) and d(ACG[9a]GT).d(ACGT) (right) segments. For details about the molecular modelling protocol see supporting information section S8.
Why was it not discovered earlier that DMC targets GpC sequences for crosslinking? We propose that the answer to this question lies in the competition between the monofunctional and bifunctional activation pathways (Scheme 1). Until now, research on the alkylation of DNA by MC or DMC has mostly used sequential additions of sub-stoichiometric amounts of dithionite or slow enzymatic reductions of DNA by MC or DMC.[20] Under such conditions, the monofunctional pathway is promoted (Scheme 1), and the first alkylation step is effected by aziridinomitosene 5a (from DMC) or 5b (from MC). The monoalkylation of DNA by 5b is selective for CpG sequences and, more importantly, produces mostly α-monoadducts.[7,25] Consequently, crosslinking reactions that involve monoalkylation by 5b in the first step show specificity for CpG sequences.[18,19,20] A similar scenario has probably occurred in previous research with DMC: The alkylation of DNA by reductively activated DMC resulted in the selective (and, in some cases, exclusive) formation of α-adducts,[14] an outcome that can be attributed to alkylation by decarbamoyl aziridinomitosene 5a. The development of reaction conditions promoting the bifunctional activation pathway permitted us to discover a novel target sequence for mitomycin drugs.
The results presented here also show that the formation of each diastereomeric crosslink adduct can be used as a proxy for the targeted sequence: If the α-crosslink 10a is formed, it means that DMC has targeted a CpG sequence whereas if the β-crosslink 10b is formed, it means that DMC has targeted a GpC sequence. Previous cell-culture experiments have established that β-crosslink 10b is the major crosslink formed in EMT6 cells, normal and FA-A human fibroblasts and MCF-7 human breast cancer cells upon DMC treatment.[9,26] For example, in EMT6 cells, 10b is formed with a 10-fold higher frequency than 10a. Therefore, the adduct profiles from cell cultures imply that GpC sequences are the major target for DNA crosslinking by DMC in mammalian cells. In contrast, MC generates the α-isomer 10a as the major crosslink adduct in the same cells, implying a CpG target. The two drugs, therefore, target different sequences for crosslinking and this may constitute the molecular basis for the contrasting biological responses triggered in cells by MC and DMC.[26–28]
As a final remark, it must be noted that adduct profiles obtained from MC-treated cell cultures showed that the β-crosslink 10b, which is a marker of GpC targeting, accounts for about one third of the total crosslinks.[9] Therefore, a corollary of the discoveries presented is that MC targets GpC sequences significantly for crosslinking in cells. We hypothesize that in vitro crosslinking reactions of DNA with MC using purely bifunctional conditions will also show a significant targeting of GpC sequences, in marked contrast to the current consensus of CpG specificity.
Experimental Section
Experimental details are available in supporting information.
Supplementary Material
Acknowledgements
This research was supported by a grant from the National Institute of Health (5SC3GM105460-03) to E.C. We want to thank Dr G. Proni for her help.
Footnotes
Dedication: This paper is dedicated to the late Professor Maria Tomasz for her contribution to Mitomycin C chemistry
References
- [1].Hata T, Hoshi T, Kanamori K, Matsumae A, Sano Y, Shima T, Sugawara R, J. Antibiot 1956, 9, 141–146. [PubMed] [Google Scholar]
- [2].Bradner WT, Cancer Treat. Rev 2001, 27, 35–50. [DOI] [PubMed] [Google Scholar]
- [3].Paz MM, Pritsos CA, “The Molecular Toxicology of Mitomycin C” in Adv. Mol. Toxicol Vol. 6 (Ed.: Fishbein JC) Elsevier, 2012. pp. 244–286. [Google Scholar]
- [4].Iyer VN, Szybalski W, Proc. Natl. Acad. Sci. U. S. A 1963, 50, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Iyer VN, Szybalski W, Science 1964, 145, 55–58 [DOI] [PubMed] [Google Scholar]
- [6].Tomasz M, Chawla AK, Lipman R, Biochemistry 1988, 27, 3182–3187 [DOI] [PubMed] [Google Scholar]
- [7].Suresh Kumar G, Lipman R, Cummings J, Tomasz M, Biochemistry 1997, 36, 14128–14136. [DOI] [PubMed] [Google Scholar]
- [8].Tomasz M, Lipman R, Chowdary D, Pawlak J, Verdine GL, Nakanishi K, Science 1987, 235, 1204–1208. [DOI] [PubMed] [Google Scholar]
- [9].Paz MM, Ladwa S, Champeil E, Liu Y, Rockwell S, Boamah EK, Bargonetti J, Callahan J, Roach J, Tomasz M, Chem. Res. Toxicol 2008, 21, 2370–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Carrano AV, Thompson LH, Stetka DG, Minkler JL, Mazrimas JA, Fong S, Mutat. Res, 1979, 63, 175–188. [DOI] [PubMed] [Google Scholar]
- [11].Rockwell S, Kim SY, Oncol. Res 1995, 7, 39–47. [PubMed] [Google Scholar]
- [12].Hoban PR, Walton MI, Robson CN, Godden J, Stratford IJ, Workman P, Harris AL, Hickson ID, Cancer. Res 1990, 50, 4692–4797. [PubMed] [Google Scholar]
- [13].Tomasz M, Lipman R, McGuiness BF, Nakanishi K, J. Am. Chem. Soc 1988, 110, 5892–5896. [Google Scholar]
- [14].Palom Y, Suresh Kumar G, Tang LQ, Paz MM, Musser SM, Rockwell S, Tomasz M, Chem. Res. Toxicol 2002, 15, 1398–1406. [DOI] [PubMed] [Google Scholar]
- [15].Tomasz M, Jung M, Verdine GL, Nakanishi K, J. Am. Chem. Soc 1984, 106, 7367–7370. [Google Scholar]
- [16].Manuscript in preparation.
- [17].Tomasz M, Lipman R, Lee MS, Verdine GL, Nakanishi K, Biochemistry, 1987, 26, 2010–2017. [DOI] [PubMed] [Google Scholar]
- [18].Teng SP, Woodson SA, Crothers DM, Biochemistry 1989, 28, 3901–3907. [DOI] [PubMed] [Google Scholar]
- [19].Millard JT, Weidner MF, Raucher S, Hopkins PB, J. Am. Chem. Soc 1990, 112, 3637–3641. [Google Scholar]
- [20].Borowy-Borowski H, Lipman R, Tomasz M, Biochemistry 1990, 29, 2999–3006. [DOI] [PubMed] [Google Scholar]
- [21].Bass PD, Gubler DA, Judd TC, Williams RM, Chem Rev. 2013, 113, 6816–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Edwards KJ, Brown DG, Spink N, Skelly JV, Neidle S, J. Mol Biol 1992, 226, 1161–1173. [DOI] [PubMed] [Google Scholar]
- [23].Sastry M, Fiala R, Lipman R, Tomasz M, Patel DJ, J. Mol. Biol 1995, 247, 338–359. [DOI] [PubMed] [Google Scholar]
- [24].McGuiness BF, Lipman R, Nakanishi K, Tomasz M, J. Org. Chem 1991, 56, 4826–4829. [Google Scholar]
- [25].Li V-S, Kohn H, J. Am. Chem. Soc 1991, 113, 275–283. [Google Scholar]
- [26].Bargonetti J, Champeil E, Tomasz M, J. Nucleic Acids 2010, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cheng S-Y, Seo J, Huang BT, Napolitano T, Champeil E, Int. J. Oncol 2016, 49, 1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xiao G, Kue P, Bhosle R, Bargonetti J, Cell Cycle 2015, 14, 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.