

Abstract

Phosphorylation of alcohols is a fundamentally important reaction in both life science and physical science. Product phosphate monoesters play key roles in living organisms, natural products, pharmaceuticals, and organic materials. Most of the chemical methods to date for synthesizing phosphate monoesters, however, require multistep sequences or are limited to specific types of substrates possibly due to harsh conditions. An alternative way to enable the simple production of phosphate monoesters from highly functionalized precursor alcohols is, thus, highly desired. We report herein a catalytic phosphorylation of alcohols with high functional group tolerance using tetrabutylammonium hydrogen sulfate (TBAHS) and phosphoenolpyruvic acid monopotassium salt (PEP-K) as the catalyst and phosphoryl donor, respectively. This method enables the direct introduction of a nonprotected phosphate group to the hydroxy group of a diverse menu of alcohol substrates, including functionalized small molecules, carbohydrates, and unprotected peptides. Nuclear magnetic resonance, mass spectrometric, and density functional theory analyses suggest that an unprecedented mixed anhydride species, generated from PEP-K and TBAHS, acts as an active phosphoryl donor in this reaction. This operationally simple and chemoselective catalytic phosphorylation allows for the efficient production of densely functionalized O-phosphorylated compounds, which are useful in diverse fields including biology and medicine.
Short abstract
A catalytic and chemoselective direct phosphorylation of a wide range of alcohol substrates, including functionalized small molecules, carbohydrates, and unprotected peptides, mediated by a phosphoenolpyruvic acid monopotassium salt (PEP-K)/tetrabutylammonium hydrogen sulfate (TBAHS) system is reported.
Introduction
Phosphate monoesters are fundamental motifs in various molecules and play diverse and pivotal roles in life science and physical science. Phosphate monoester motifs are widely found in biomolecules, including proteins,1 signal messengers,2 and primary3 and secondary4,5 metabolites, and in human-made functional molecules, such as pharmaceuticals6 and organic materials.7 In proteins, for example, phosphorylation of the serine, threonine, and tyrosine hydroxy groups is among the most common and important post-translational modifications.1 Such modifications may induce structural changes in the protein or lead to the formation of a new protein–protein interaction surface, triggering cellular signal cascades and constituting a fundamental regulatory mechanism of cell biology.8 The introduction of a phosphate monoester motif can dramatically alter the function of a molecule and can play a key role in mediating the bioactivity of natural products9,10 and the pharmacokinetics of pharmaceutical agents.6 Thus, a simple and robust method to enable easy access to these important motifs would be highly desirable, as the ready availability of large quantities of these functional materials would facilitate further investigation of their biological and physical functions.11−13
A straightforward way to access phosphate monoesters is via phosphorylation of alcohol starting materials. There are two major chemical approaches to the transformation of alcohols to phosphate monoesters: (1) the formation of trivalent phosphite followed by oxidation and (2) the reaction with a pentavalent phosphoryl donor (Figure 1). A standard example of the first approach is depicted in Figure 1A. Thus, an alcohol is first reacted with a trivalent phosphoramidite reagent to produce a phosphite intermediate, which is then oxidized, typically in the same reaction vessel, to afford a protected phosphate triester. Finally, the removal of the protecting groups under acidic, reductive, or basic conditions (R1 = alkyl, benzyl, or cyanoethyl, respectively) produces the desired phosphate monoester.14−16 This method requires three steps and is sometimes incompatible with substrates that are sensitive to oxidation or to the (acidic, basic, or reducing) conditions required to remove the protecting groups, although several improved methods were reported.17,18 A typical example of approach (2), outlined in Figure 1B, uses phosphoryl chloride and its derivatives as the phosphoryl donors.19−22 Although the use of these phosphoryl chloride derivatives directly provides the desired phosphate monoester after hydrolysis, the high reactivity of the phosphoryl chloride derivatives sometimes render functional groups in a substrate incompatible with the reaction conditions and complicate the reaction.
Figure 1.

Chemical approaches for phosphate monoester synthesis from alcohols. (A) Step-wise phosphorylation with phosphoramidites, followed by oxidation and deprotection. (B) Phosphorylation with phosphoryl chloride and its derivative, followed by hydrolysis. (C) Dehydrative condensation approaches developed by Ishihara and co-workers.23,24 (D) Catalytic direct phosphorylation of alcohols with phosphoenolpyruvic acid monopotassium salt (PEP-K) and tetrabutylammonium hydrogen sulfate developed in this study.
There is thus a clear need for a robust method that allows access to a diverse menu of nonprotected phosphate monoesters in a single step from alcohol precursors. Toward this end, Ishihara and co-workers have reported the synthesis of phosphate monoesters through azeotropic dehydrative condensation between phosphoric acid and alcohols promoted by N-butylimidazole and tributylamine (Figure 1C).23 In 2007, the researchers further developed a rhenium(VII) oxide-catalyzed variant that proceeded at >150 °C (Figure 1C).24 The substrate scope of those reactions, however, was mostly limited to alcohols containing simple alkyl chains, likely due to the high reaction temperature. Therefore, the development of a catalyst-promoted direct phosphorylation of alcohols with high functional group tolerance is an important goal in organic chemistry.
Kinases are capable of effectively catalyzing phosphoryl-transfer reactions in highly functionalized substrates, such as proteins, carbohydrates, and other metabolites using nucleoside 5′-triphosphates (e.g., adenosine triphosphate, ATP) or phosphoenolpyruvate (PEP)25 as high-energy phosphoryl donors.26 The kinetic barrier of the phosphoryl-transfer reactions is generally high, but kinases (biocatalysts) provide enormous rate acceleration in part by efficiently activating the phosphoryl donor.26 In our effort to achieve a direct phosphorylation of alcohols, we sought to exploit those high-energy phosphates because they are stable as they are but can become reactive if properly activated. Among the phosphoryl donors, PEP has the highest energy phosphate bond (Figure 1D).27 We envisioned that, if an appropriate catalyst could be identified that is capable of activating PEP under mild conditions, then it should be possible to achieve chemoselective phosphorylation to directly access unprotected phosphate monoesters in a single step from alcohol substrates. Herein, we report a catalytic and chemoselective direct phosphorylation of a wide range of alcohol substrates, including functionalized small molecules, carbohydrates, and unprotected peptides, mediated by a phosphoenolpyruvic acid monopotassium salt (PEP-K)/tetrabutylammonium hydrogen sulfate (TBAHS) system (Figure 1D).
Results and Discussion
For our initial studies, we selected 3-phenyl-1-propanol (1) as a model substrate. Alcohol 1 was subjected to simple heating conditions in the presence of commercially available PEP-K in acetonitrile for 12 h. However, under these conditions, no phosphorylated product 2 was detected, and PEP-K was recovered (Figure 2A, eq 1). In contrast, when phosphoenolpyruvic acid (PEP-H, prepared by cation exchange resin from PEP-K; see the Supporting Information for details) was used as the phosphoryl donor under the same conditions, product 2 was obtained in 42% yield (Figure 2A, eq 2). Thus, we postulated that a Brønsted acid catalyst could protonate PEP-K and facilitate the formation of an active phosphorylating species through PEP-H. On the basis of this hypothesis, we investigated the catalytic activities of various Brønsted acids (20 mol %) in the phosphorylation of 1 using PEP-K (1.5 equiv) as a phosphoryl donor in acetonitrile at 80 °C (Figure 2B, Entries 1–8; for a complete list of reaction conditions evaluated, see Table S1). The pKa1 value of PEP-H in water is reported to be less than 2.28 Brønsted acids of weak acidity, such as tetrazole (pKa1 = 4.9) and benzoic acid derivatives (pKa1 = 2.9–4.2), did not afford any phosphorylated product 2 (Figure 2B, Entries 2–4). Stronger Brønsted acids, such as phosphoric acid (pKa1 = 2.1), 10-camphorsulfonic acid (pKa1 = 1.2), and trifluoroacetic acid (TFA, pKa1 = −0.3), gave product 2 in very low yields (2–6%, Figure 2B, Entries 5, 7, and 8). However, tetrabutylammonium hydrogen sulfate (TBAHS, pKa1 = 2.0) did catalyze the phosphorylation, affording 2 in 35% yield (Figure 2B, Entry 6). We can thus conclude that catalytic activity does not directly correlate with Brønsted acidity (i.e., pKa1 values). We next established that the hydrogen sulfate Brønsted acid is key to promoting the reaction; other quaternary ammonium salts without the hydrogen sulfate did not react to afford 2 at all (Figure 2B, Entries 9–10). A solvent screen revealed that, with DMF as solvent and an increased TBAHS catalyst loading of 30 mol %, a product yield of 80% could be obtained (Figure 2B, Entries 11–12, and Table S1). Hydrogen sulfate catalysts containing counterions other than the tetrabutylammonium ion produced less satisfactory results (Figure 2B, Entries 13–15). Finally, raising the temperature to 100 °C and using 4.5 equiv of PEP-K led to an improved yield of 2 (88%) and shorter reaction times (6 h) (Figure 2B, Entry 16).
Figure 2.

Initial trials and optimization of reaction conditions. (A) Phosphorylation of 3-phenyl-1-propanol with PEP-K (eq 1) and with PEP-H (eq 2). (B) Optimization of reaction conditions.
Having identified optimized reaction conditions, we next investigated the substrate scope of the transformation. As shown in Figure 3, a range of useful functional groups, including bromide (3: 85% yield), methoxy (4: 90% yield), cyano (5: 77% yield), alkyne (6: 81% yield), trimethylsilyl (7: 77% yield), aldehyde (8: 73% yield), and carboxylic acid (9: 71% yield), were well-tolerated, reacting to afford the corresponding phosphorylation products in good to excellent yield. Allylic (10: 78% yield), benzylic (11: 70% yield), and secondary (12: 71% yield) alcohols also underwent direct phosphorylation in good yields. Importantly, phosphorylation of alcohols bearing acid-, base-, and/or oxidation-sensitive functional groups also proved possible, as Boc carbamate (13: 72% yield), Fmoc carbamate (14: 74% yield), and trityl group (15: 49% yield; moderate yield was due to low reactivity) were all readily accommodated in this transformation. Glycosyl donors with activated anomeric leaving groups were also found to be good substrates for the direct phosphorylation. Thus, phosphorylated glycosyl fluoride (16: 72% yield) and thioglycoside (17: 78% yield) were formed from their corresponding alcohols in good yield. In particular, the reaction of a glycal substrate containing an enol ether moiety to form 18 (47% yield) is notable, because this functional group is highly sensitive to both slightly acidic and oxidative conditions. The moderate yield of 18 was due to the gradual decomposition of the substrate under the current conditions. A disaccharide with an acid-sensitive glycosidic linkage (19: 78% yield) readily underwent phosphorylation. It is also noteworthy that a sterically hindered secondary alcohol was efficiently phosphorylated using this catalytic system (20: 66% yield). Protected serine (21: 77% yield) and threonine (22: 78% yield) underwent phosphorylation in good yield, although a substrate bearing a primary amine did not react to generate the desired phosphorylation product, 23. Although 1H nuclear magnetic resonance (NMR) analysis of the reaction indicated that phosphorylation of the hydroxy group did proceed in this case, imine formation between the primary amine moiety and pyruvic acid generated after phosphorylation complicated the reaction, producing 24 as a possible byproduct.
Figure 3.

Substrate scope of the catalytic phosphorylation of alcohols with PEP-K and TBAHS. The general conditions are shown in the reaction scheme. Modified conditions are as follows. a: PEP-K (6.0 equiv), TBAHS (60 mol %). b: PEP-K (6.0 equiv), TBAHS (60 mol %), 3 h. c: PEP-K (10 equiv), TBAHS (60 mol %), 4.5 h. d: PEP-K (10 equiv), TBAHS (60 mol %), 3 h. e: PEP-K (50 equiv), TBAHS (200 mol %), DMF (0.0125 M), 3 h. Isolated yields are shown.
Chemo- and regioselective phosphorylation of substrates containing more than one OH group was also found to be possible (Figure 3). Thus, an aliphatic hydroxy group was selectively phosphorylated over a phenolic hydroxy group, affording 25 as the sole product in 60% yield. The primary hydroxy group in cortisone was selectively reacted over the tertiary hydroxy group to afford 26 in 88% yield. A myo-inositol derivative was regioselectively phosphorylated at the most reactive hydroxy groups to give monophosphorylated product 27 in 46% yield with 8% yield of a bisphosphorylated product.29 The regioisomer was not detected.
The high functional group tolerance of the PEP-K/TBAHS system allowed for the straightforward, single-step, chemoselective synthesis of O-phosphorylated peptides with minimal use of protecting groups (Figure 3). Serine (28: 75% yield) and threonine (29: 57% yield) residues in tripeptides underwent selective O-phosphorylation in good yield even in the presence of a tyrosine residue on the peptide sequence. The presence of carboxylic acid (aspartic acid, 30: 71% yield), indole (tryptophan, 31: 76% yield), and sulfide (methionine, 32: 81% yield) moieties on the peptide substrates did not adversely impact the reaction yields. Although the presence of an unprotected cysteine residue inhibited phosphorylation, the yield improved dramatically when the cysteine thiol group was converted to a disulfide (33, 77% yield). Peptides containing basic amino acids, such as histidine (34: 85% yield) and arginine (35: 76% yield), were also competent substrates when used as their trifluoroacetic acid salts. A peptide containing an unprotected lysine residue did not afford the desired product, likely because the O-phosphorylated product further reacted with the in situ generated pyruvic acid at the primary amine of the lysine residue, as seen in 24. Peptides containing monomethylated (36: 22% yield) and dimethylated lysines (37: 56% yield), however, did react to afford the O-phosphorylated products in moderate yield due to the slower reaction of the products with pyruvic acid. The reaction was also applicable to longer oligopeptides. Phosphorylated hexapeptide 38, a useful substrate for a sensitive serine/threonine protein phosphatase assay,30 was synthesized in 54% yield from the corresponding starting material in a single step. A 15-mer oligopeptide containing multiple acidic and basic amino acids was phosphorylated to produce 39, a potential cancer biomarker,31 in 59% yield, although more than the stoichiometric amount of TBAHS is required for the high conversion.
Next, we undertook several studies to gain a mechanistic insight into this catalytic phosphorylation reaction. Our observation that catalyst activity does not correlate with Brønsted acidity (Figure 2B) seemed to suggest a role for TBAHS beyond that of a simple Brønsted acid. Interestingly, during optimization of the reaction conditions, we found that, in the presence of 1.5 equiv of PEP-K and 1.0 equiv of TBAHS, sulfurylated compound 40 was produced as a byproduct (21% yield), together with the desired phosphorylated product 2 (68% yield) (Figure 4A). Sulfation of 1 did not proceed when PEP-K was excluded from the reaction (i.e., when TBAHS was used as the sole additive). Thus, both PEP-K and TBAHS are needed for the desired phosphorylation and the undesired sulfation to proceed under the conditions of Figure 4A. This result, coupled with a previous report indicating that an endogenous mixed anhydride, 3′-phospho-adenosine-5′-phosphosulfate (PAPS), works as an O-sulfuryl donor,32 led us to hypothesize that a new inorganic mixed anhydride, phosphosulfate (POS, 41), is generated from PEP-K and TBAHS (Figure 4B) and that this species may function as a phosphoryl or sulfuryl donor. On the basis of this hypothesis, we synthesized POS 42 (Figure S1) and assessed its reactivity with alcohol 1 in the presence of varying amounts of PEP-K (Figure 4C). As shown in Figure 4C, Entry 1, only sulfurylated compound 40 was obtained in the presence of 1.0 equiv of 42 and the absence of PEP-K; thus, POS 42 acted as a sulfuryl donor in this reaction. In striking contrast, the yield of the phosphorylated product 2 increased with the amount of PEP-K employed (Figure 4C, Entries 2–5), reaching 54% yield in the presence of 4.5 equiv of PEP-K. In the absence of POS 42, however, the reaction of 1 with 4.5 equiv of PEP-K produced only a 7% yield of 2 (Figure 4C, Entry 6). These results indicate that the hypothesized active species POS 41 is itself a sulfuryl donor and that this intermediate reacts with PEP-K to produce another true active phosphoryl donor species.
Figure 4.

Mechanistic investigations for catalytic phosphorylation of alcohols with PEP-K and TBAHS. (A) A reaction using 1.0 equiv of TBAHS produced O-sulfate 40. (B) A postulated mechanism for the formation of POS 41 from PEP-K and TBAHS. (C) Reactivity of POS 42 in the presence of varied amounts of PEP-K. (D) 31P NMR and MS spectra of the reaction mixtures for 42 + PEP-K (above) and TBAHS + PEP-K (below), respectively. In 31P NMR, the peak at 1.5 ppm is phosphoric acid and the peak at −3.0 ppm is PEP-K.
To identify the true active phosphoryl donor species, we conducted a series of 31-phosphorus NMR (31P NMR) and mass spectrometric (MS) experiments. When we monitored the reaction between POS 42 and PEP-K (4.5 equiv) by 31P NMR in DMF at 100 °C, a new singlet peak, which is very close to but clearly different from that of 42 (−10.8 ppm, Figure S1), appeared at −10.1 ppm after 30 min (Figures 4D and S2–S5). MS analysis suggested the presence of a species with m/z of 257.0 (Figure 4D). This species corresponds to a new mixed anhydride, POSOP 43. Importantly, careful monitoring of the reaction between TBAHS and PEP-K by 31P NMR and MS revealed that POS 41 was formed first (−11.3 ppm in 31P NMR and m/z of 177.1 in MS, Figure S6A) and subsequently converted into POSOP 44 (−10.7 ppm in 31P NMR and m/z of 257.0, Figures 4D and S6B). We further confirmed the presence of POS 41 and POSOP 44 using 34S-labeled TBAHS in the MS analysis, because phosphorylation and 32S-sulfation give the same mass shift, and POS 41 and pyrophosphate could not be differentiated under the MS conditions. As a result, species with m/z corresponding to PO34S (216.8 for monopotassium salt) and PO34SOP (319.4 for potassium sodium salt) were observed when 34S-labeled TBAHS was used (Figure S7). These results suggest that the mixed anhydride species, POSOP, is formed through intermediate POS and that the POSOP species acts as an active phosphoryl donor in the PEP-K/TBAHS-promoted phosphorylation reaction.
We next investigated the origin of the differing reactivities of POS (sulfuryl donor) and POSOP (phosphoryl donor) using density functional theory (DFT) calculations combined with an automated reaction path search method called the “artificial force induced reaction” (AFIR) method (Figure 5).33−35 In the reaction between alcohol 1 and POS 45, sulfurylated product 46 was thermodynamically more stable than phosphorylated product 47 by 2.22 kcal mol–1 (Figures 5A and S8–S12). The activation barriers for the transition states (TSs) in sulfation and phosphorylation were 50.60 and 27.73 kcal mol–1, respectively. The activation barrier for sulfation markedly decreased, however, in the presence of a molecule mediating the proton transfer from ROH 1 to the phosphate group of 45. Thus, the activation barriers were 24.70, 26.43, and 26.32 kcal mol–1 in the presence of PEP (TS3), pyruvic acid (for the conditions in Figure 4A, TS4), and triethylammonium ion (for the conditions in Figure 4C, TS5), respectively. Since the activation barrier difference (3.03 kcal/mol) between sulfation in the presence of proton-transfer mediators and phosphorylation corresponds to a 60-fold difference in the reaction rate at 100 °C, sulfation was favored over phosphorylation for POS. The dramatic effects of proton-transfer mediators in reducing the activation barrier of sulfation with POS can be rationalized by considering the geometry of the TSs (Figure S15). In the TS of sulfation in the absence of a proton-transfer mediator (TS2 in Figure 5), the lengths of the formed O(ROH)–S bond and the cleaved S–O(phosphate in POS) bond were similar (2.40 and 2.55 Å, respectively; Figure S15B), indicating that this process can be categorized as an SN2 reaction. The phosphate group of POS existed proximate to ROH in TS2, because protonation of the phosphate group by ROH was a prerequisite for the elimination of the phosphate group. As a consequence, the O(ROH)–S–O(phosphate in POS) angle in TS2 (68.1°) deviated significantly from the ideal angle for an SN2 reaction (180°), leading to destabilization of TS2. In the presence of a proton-transfer mediator, however, the O(ROH)–S–O(phosphate in POS) angle was nearly ideal for an SN2 reaction (167.9–177.5° for TS3–TS5; Figure S15C–E), because the phosphate group was already protonated by the proton-transfer mediator.
Figure 5.

DFT calculation to rationalize the contrasting reaction pattern between POS (41; sulfuryl donor) and POSOP (44; phosphoryl donor). Sulfation (left) and phosphorylation (right) reactions of alcohol 1 with POS (A) and POSOP (B) are shown. The references of relative Gibbs free energies ΔG (in kcal mol–1) are 1 + 45 + proton-transfer mediators (A) and 1 + 48 (B). Detailed Gibbs free energy profiles are shown in Figures S8–S14.
In contrast, in the reaction between POSOP 48 and 1, the phosphorylation pathway was favored over sulfation, both thermodynamically and kinetically (Figures 5B and S13–S14). The activation barrier of phosphorylation (TS6) was only 16.58 kcal mol–1, which was much smaller than that of sulfation by 48 (30.45 kcal mol–1, TS7). The activation barrier of TS6 was also significantly smaller than that of sulfation by POS 45 in the presence of a proton-transfer mediator (24.70–26.43 kcal mol–1, TS3–TS5). This energy difference between TS6 (phosphorylation by POSOP) and TS3–TS5 (sulfation by POS) corresponds to about a 60 000-fold kinetic difference at 100 °C. Thus, the phosphorylation pathway through TS6 should be predominant compared to the sulfation pathway through TS3–TS5 even if POS and POSOP coexist in the reaction mixture (Figure 6).
Figure 6.
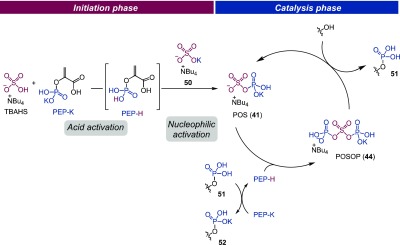
Postulated mechanism of the catalytic phosphorylation of alcohols with PEP-K and TBAHS.
The reactivity difference between POS 41 and POSOP 44 can be explained as follows. As noted above, O-sulfation of alcohols by POS can be categorized as an SN2 reaction. Protonation of the phosphate leaving group in POS by a proton-transfer mediator facilitated the sulfation pathway by enabling the TS geometry favorable for the SN2 reaction (Figure 5A). In contrast, O-phosphorylation by POSOP can be viewed as an SN1 reaction: the P–O bond in POSOP was cleaved prior to the formation of the O(ROH)–P(phosphate) bond in the reaction coordinate (Figures S13 and S16A). The exceptionally high ability of POS to act as a leaving group can rationalize this switching of the reaction mechanism. Indeed, the bond order of P–O in POSOP was markedly smaller (0.48) than that of other bonds (0.79, 0.56, and 0.67 for S–O in POSOP, P–O in POS, and S–O in POS, respectively; see Table S2). Moreover, POSOP was a better electrophile than POS: the HOMO–LUMO gap between an alcohol nucleophile and POSOP was smaller than that of POS (Figure S17). Taken together, we may conclude that alcohol O-phosphorylation by POSOP is the most favorable pathway among many possible pathways in the developed PEP-K/TBAHS system.
On the basis of these mechanistic investigations, a postulated mechanism for the catalytic phosphorylation of alcohols with the PEP-K/TBAHS system is shown in Figure 6. First, TBAHS catalyst works as a Brønsted acid to protonate PEP-K, generating PEP-H and sulfate SO42– (50). PEP-H then reacts with 50 to afford POS intermediate 41 with the release of pyruvic acid (initiation phase). Although this POS intermediate itself acts as a sulfuryl donor, in the presence of excess PEP-K, the sulfate anionic oxygen atom of POS 41 selectively reacts with a second PEP-H to produce the active phosphoryl donor, POSOP 44. Nucleophilic addition of the hydroxy group of the substrate alcohol to POSOP 44 occurs at the phosphorus atom to produce phosphate monoester 51 with a concomitant regeneration of POS 41. Finally, 51 protonates PEP-K to afford product phosphate 52 and PEP-H, the latter of which reacts with POS 41 to enter another catalytic cycle (catalysis phase).
Two major features of this method are its high functional group tolerance and single-step introduction of an unprotected phosphate group to various alcohols. Although temperature-sensitive substrates are incompatible with the reaction conditions (i.e., 100 °C), these characteristics should prove advantageous for the late-stage functionalization of bioactive molecules, such as pharmaceuticals.36 Many bioactive compounds contain hydroxy groups, and phosphorylation of these groups can have a dramatic impact on biological activity and pharmacokinetics.6,9,10 Therefore, the application of this method to bioactive compounds or their advanced synthetic intermediates is expected to deliver valuable compound libraries with new bioactivities.
Another attractive potential application of this method is the synthesis of radioactive O-phosphorylated peptides containing a 32P-nucleus at a serine/threonine residue. Such peptides are useful for sensitive assays of protein phosphatase activity30 and anticancer radiotherapy.37 It is not currently realistic to access radioactive O-phosphorylated peptides via chemical synthesis. All available methods are time- and labor-intensive, requiring the preparation of protected serine/threonine building blocks containing 32P-labeled protected phosphate groups, followed by the merger of these amino acid building blocks via solid-phase peptide synthesis, deprotection, cleavage from the solid phase, and finally HPLC purification. Therefore, the synthesis of radioactive O-phosphorylated peptides has thus far relied on kinase-mediated methods using a γ-32P-adenosine 5′-triphosphpate (ATP) phosphoryl donor. The efficiency of enzymatic methods, however, depends on the nature of the peptide sequence.38 This restriction significantly narrows the range of available 32P-labeled peptide sequences. 32P-Labeled PEP is routinely synthesized from readily available γ-32P-ATP39 with pyruvate kinase40 or phosphoenolpyruvate carboxykinase.41 Using 32P-PEP as a phosphoryl donor, the application of our phosphorylation method to preassembled peptides would directly produce 32P-labeled peptides without significant sequence biases.
Conclusions
We have developed the first catalytic and chemoselective phosphorylation of alcohols using PEP-K as a phosphoryl donor and TBAHS as a catalyst. This method displays high functional group tolerance and thus enables efficient access to a variety of phosphate monoesters, including functionalized small molecules, carbohydrates, and unprotected peptides. Mechanistic studies suggest that TBAHS plays a significant role as both a Brønsted acid and a nucleophilic activator. This dual activation mode generates an unprecedented mixed anhydride species, POSOP 44, as an active phosphoryl donor, enabling the functional group-tolerant O-phosphorylation reactions. Since functionalized phosphate monoesters are prevalent as important chemical entities ranging from biologically active compounds to functional materials in diverse fields, this operationally simple and efficient synthetic method will broadly contribute to both life science and physical science.
Acknowledgments
We thank Christopher W. Adamson for providing useful comments on the manuscript. We acknowledge the computer resources provided by the Academic Center for Computing and Media Studies (ACCMS) at Kyoto University. This research was supported by JSPS KAKENHI Grant nos. JP17H06442 (M.K.), JP17H06445 (M.H.) (Hybrid Catalysis), and JP17K15420 (K.Y.). This manuscript is dedicated to Prof. Laura L. Kiessling for her 60th birthday.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.9b01272.
Materials and methods, Cartesian coordinates of the critical points, spectral data, synthesis of O-benzyl phosphosulfate and 31P NMR change, 31P NMR and MS spectra, MS analyses of POS and POSOS, Gibbs free energy profiles, geometries of TSs for phosphorylation and sulfation by POS and POSOP, frontier orbitals and their energy levels, list of reaction conditions for optimization of the phosphorylation of 3-phenyl-1-propanol (1), bond orders/distances of POS 45 and POSOP 48 (PDF)
Author Contributions
K.D., S.A.K., K.Y., and M.K. conceived and designed the study. K.D., K.F, and K.Y. performed all the synthetic experiments. M.P. and M.H. performed the DFT calculations. K.D, K.F., M.H., S.A.K., K.Y., and M.K. analyzed the data. K.D., M.H., K.Y., and M.K. cowrote the paper with comments from all the authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Olsen J. V.; Blagoev B.; Gnad F.; Macek B.; Kumar C.; Mortensen P.; Mann M. Global, in Vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Berridge M. J.; Irvine R. F. Inositol phosphates and cell signaling. Nature 1989, 341, 197–205. 10.1038/341197a0. [DOI] [PubMed] [Google Scholar]
- Hers H. G.; Hue L. Glucogenesis and related aspects of glycolysis. Annu. Rev. Biochem. 1983, 52, 617–653. 10.1146/annurev.bi.52.070183.003153. [DOI] [PubMed] [Google Scholar]
- Kato Y.; Fusetani N.; Matsunaga S.; Hashimoto K.; Fujita S.; Furuya T. Bioactive marine metabolites. Part 16. Calyculin A. A novel antitumor metabolite from the marine sponge Discodermia calyx. J. Am. Chem. Soc. 1986, 108, 2780–2781. 10.1021/ja00270a061. [DOI] [Google Scholar]
- Stampwala S. S.; Bunge R. H.; Hurley T. R.; Willmer N. E.; Brankiewicz A. J.; Steinman C. E.; Smitka T. A.; French J. C. Novel anti-tumor agents CI-920, PD 113,270 and PD 113,271 0.2. Isolation and characterization. J. Antibiot. 1983, 36, 1601–1605. 10.7164/antibiotics.36.1601. [DOI] [PubMed] [Google Scholar]
- Wire M. B.; Shelton M. J.; Studenberg S. Fosamprenavir. Clin. Pharmacokinet. 2006, 45, 137–168. 10.2165/00003088-200645020-00002. [DOI] [PubMed] [Google Scholar]
- Yao C.; Wei C.; Huang Z.; Lu Y.; El-Toni A. M.; Ju D.; Zhang X.; Wang W.; Zhang F. Phosphorylated peptide functionalization of lanthanide upconversion nanoparticles for tuning nanomaterial–cell interactions. ACS Appl. Mater. Interfaces 2016, 8, 6935–6943. 10.1021/acsami.6b01085. [DOI] [PubMed] [Google Scholar]
- Johnson L. N.; Lewis R. J. Structural basis for control by phosphorylation. Chem. Rev. 2001, 101, 2209–2242. 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- Strader C. R.; Pearce C. J.; Oberlies N. H. Fingolimod (FTY720): A recently approved multiple sclerosis drug based on a fungal secondary metabolite. J. Nat. Prod. 2011, 74, 900–907. 10.1021/np2000528. [DOI] [PubMed] [Google Scholar]
- Swingle M. R.; Amable L.; Lawhorn B. G.; Buck S. B.; Burke C. P.; Ratti P.; Fischer K. L.; Boger D. L.; Honkanen R. E. Structure-activity relationship studies of fostriecin, cytostatin, and key analogs, with PP1, PP2A, PP5, and (β12−β13)-chimeras (PP1/PP2A and PP5/PP2A), provide further insight into the inhibitory actions of fostriecin family inhibitors. J. Pharmacol. Exp. Ther. 2009, 331, 45. 10.1124/jpet.109.155630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamady S.; Taylor S. D. Synthesis of Nucleoside Tetraphosphates and Dinucleoside Pentaphosphates via Activation of Cyclic Trimetaphosphate. Org. Lett. 2013, 15, 2612–2615. 10.1021/ol4007822. [DOI] [PubMed] [Google Scholar]
- Shepard S. M.; Cummins C. C. Functionalization of Intact Trimetaphosphate: A Triphosphorylating Reagent for C, N, and O Nucleophiles. J. Am. Chem. Soc. 2019, 141, 1852–1856. 10.1021/jacs.8b12204. [DOI] [PubMed] [Google Scholar]
- Shepard S. M.; Windsor I. W.; Raines R. T.; Cummins C. C. Nucleoside Tetra- and Pentaphosphates Prepared Using a Tetraphosphorylation Reagent Are Potent Inhibitors of Ribonuclease A. J. Am. Chem. Soc. 2019, 141, 18400–18404. 10.1021/jacs.9b09760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perich J. W.; Johns R. B. A new, convenient and efficient general procedure for the conversion of alcohols into their dibenzyl phosphorotriesters using N,N-diethyl dibenzyl phosphoramidite. Tetrahedron Lett. 1987, 28, 101–102. 10.1016/S0040-4039(00)95660-0. [DOI] [Google Scholar]
- Perich J. W.; Johns R. B. Di-t-butyl N, N-diethylphosphoramidite and dibenzyl N, N-diethylphosphoramidite. Highly reactive reagents for the ‘phosphite-triester’ phosphorylation of serine-containing peptides. Tetrahedron Lett. 1988, 29, 2369–2372. 10.1016/S0040-4039(00)86062-1. [DOI] [Google Scholar]
- Jordan P. A.; Kayser-Bricker K. J.; Miller S. J. Asymmetric phosphorylation through catalytic P(III) phosphoramidite transfer: Enantioselective synthesis ofD-inositol-6-phosphate. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20620–20624. 10.1073/pnas.1001111107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoharan M.; Lu Y.; Casper M. D.; Just G. Allyl Group as a Protecting Group for Internucleotide Phosphate and Thiophosphate Linkages in Oligonucleotide Synthesis: Facile Oxidation and Deprotection Conditions. Org. Lett. 2000, 2, 243–246. 10.1021/ol9910518. [DOI] [PubMed] [Google Scholar]
- Wei X. Coupling activators for the oligonucleotide synthesis via phosphoramidite approach. Tetrahedron 2013, 69, 3615–3637. 10.1016/j.tet.2013.03.001. [DOI] [Google Scholar]
- Bonnet H. Ueber die einwirkung des königswassers auf alkohol. Justus Liebigs Ann. Chem. 1857, 104, 337–338. 10.1002/jlac.18571040310. [DOI] [Google Scholar]
- Yoshikawa M.; Kato T.; Takenishi T. Studies of Phosphorylation. III. Selective Phosphorylation of Unprotected nucleosides. Bull. Chem. Soc. Jpn. 1969, 42, 3505–3508. 10.1246/bcsj.42.3505. [DOI] [Google Scholar]
- Sowa T.; Ouchi S. The Facile Synthesis of 5′-Nucleotides by the Selective Phosphorylation of a Primary Hydroxyl Group of Nucleosides with Phosphoryl Chloride. Bull. Chem. Soc. Jpn. 1975, 48, 2084–2090. 10.1246/bcsj.48.2084. [DOI] [Google Scholar]
- Sculimbrene B. R.; Miller S. J. Discovery of a catalytic asymmetric phosphorylation through selection of a minimal kinase mimic: a concise total synthesis of D-myo-inositol-1-phosphate. J. Am. Chem. Soc. 2001, 123, 10125–6. 10.1021/ja016779+. [DOI] [PubMed] [Google Scholar]
- Sakakura A.; Katsukawa M.; Ishihara K. Selective synthesis of phosphate monoesters by dehydrative condensation of phosphoric acid and alcohols promoted by nucleophilic bases. Org. Lett. 2005, 7, 1999–2002. 10.1021/ol0504796. [DOI] [PubMed] [Google Scholar]
- Sakakura A.; Katsukawa M.; Ishihara K. The oxorhenium(VII)-catalyzed direct condensation of phosphoric acid with an alcohol. Angew. Chem., Int. Ed. 2007, 46, 1423–1426. 10.1002/anie.200604333. [DOI] [PubMed] [Google Scholar]
- Deutscher J.; Ake F. M. D.; Derkaoui M.; Zebre A. C.; Cao T. N.; Bouraoui H.; Kentache T.; Mokhtari A.; Milohanic E.; Joyet P. The Bacterial Phosphoenolpyruvate:Carbohydrate Phosphotransferase System: Regulation by Protein Phosphorylation and Phosphorylation-Dependent Protein-Protein Interactions. Microbiol. Mol. Biol. Rev. 2014, 78, 231–256. 10.1128/MMBR.00001-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassila J. K.; Zalatan J. G.; Herschlag D. Biological Phosphoryl-Transfer Reactions: Understanding Mechanism and Catalysis. Annu. Rev. Biochem. 2011, 80, 669–702. 10.1146/annurev-biochem-060409-092741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodwell V. W.; Bender D.; Botham K. M.; Kennelly P. J.; Weil P. A.. Harper’s Illustrated Biochemistry, 31st ed., McGraw-Hill Education: 2018. [Google Scholar]
- Wold F.; Ballou C. E. Studies on the enzyme enolase: I. equilibrium studies. J. Biol. Chem. 1957, 227, 301–312. [PubMed] [Google Scholar]
- Sculimbrene B. R.; Morgan A. J.; Miller S. J. Nonenzymatic peptide-based catalytic asymmetric phosphorylation of inositol derivatives. Chem. Commun. 2003, 1781–1785. 10.1039/b304015c. [DOI] [PubMed] [Google Scholar]
- Pinna L. A.; Donella-Deana A. Phosphorylated synthetic peptides as tools for studying protein phosphatases. Biochim. Biophys. Acta, Mol. Cell Res. 1994, 1222, 415–431. 10.1016/0167-4889(94)90050-7. [DOI] [PubMed] [Google Scholar]
- Zhai G.; Wu X.; Luo Q.; Wu K.; Zhao Y.; Liu J.; Xiong S.; Feng Y.-Q.; Yang L.; Wang F. Evaluation of serum phosphopeptides as potential cancer biomarkers by mass spectrometric absolute quantification. Talanta 2014, 125, 411–417. 10.1016/j.talanta.2014.03.025. [DOI] [PubMed] [Google Scholar]
- Mueller J. W.; Shafqat N. Adenosine-5-phosphosulfate - a multifaceted modulator of bifunctional 3-phospho-adenosine-5-phosphosulfate synthases and related enzymes. FEBS J. 2013, 280, 3050–3057. 10.1111/febs.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S.; Harabuchi Y.; Takagi M.; Saita K.; Suzuki K.; Ichino T.; Sumiya Y.; Sugiyama K.; Ono Y. Implementation and performance of the artificial force induced reaction method in the GRRM17 program. J. Comput. Chem. 2018, 39, 233–251. 10.1002/jcc.25106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S.; Harabuchi Y.; Takagi M.; Taketsugu T.; Morokuma K. Artificial force induced reaction (AFIR) method for exploring quantum chemical potential energy surfaces. Chem. Rec. 2016, 16, 2232–2248. 10.1002/tcr.201600043. [DOI] [PubMed] [Google Scholar]
- Maeda S.; Ohno K.; Morokuma K. Systematic exploration of the mechanism of chemical reactions: the global reaction route mapping (GRRM) strategy using the ADDF and AFIR methods. Phys. Chem. Chem. Phys. 2013, 15, 3683–3701. 10.1039/c3cp44063j. [DOI] [PubMed] [Google Scholar]
- Dominguez-Huerta A.; Dai X. J.; Zhou F.; Querard P.; Qiu Z. H.; Ung S.; Liu W. B.; Li J. B.; Li C. J. Exploration of new reaction tools for late-stage functionalization of complex chemicals. Can. J. Chem. 2019, 97, 67–85. 10.1139/cjc-2018-0357. [DOI] [Google Scholar]
- Abraham J. M.; Cheng Y.; Hamilton J. P.; Paun B.; Jin Z.; Agarwal R.; Kan T.; David S.; Olaru A.; Yang J.; Ito T.; Selaru F. M.; Mori Y.; Meltzer S. J. Generation of small 32P-labeled peptides as a potential approach to colorectal cancer therapy. PLoS One 2008, 3, e2508. 10.1371/journal.pone.0002508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donelladeana A.; Krinks M. H.; Ruzzene M.; Klee C.; Pinna L. A. Dephosphorylation of phosphopeptides by calcineurin (protein phosphatase 2B). Eur. J. Biochem. 1994, 219, 109–117. 10.1111/j.1432-1033.1994.tb19920.x. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Wang Y.; Wang T.; Hawke D. H.; Zheng Y.; Li X.; Zhou Q.; Majumder S.; Bi E.; Liu D. X.; Huang S.; Lu Z. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat. Commun. 2014, 5, 5566. 10.1038/ncomms6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roossien F. F.; Brink J.; Robillard G. T. A simple procedure for the synthesis of [32P]phosphoenolpyruvate via the pyruvate kinase exchange reaction at equilibrium. Biochim. Biophys. Acta, Gen. Subj. 1983, 760, 185–187. 10.1016/0304-4165(83)90141-1. [DOI] [PubMed] [Google Scholar]
- Mattoo R. L.; Waygood E. B. An enzymatic method for [32P]phosphoenolpyruvate synthesis. Anal. Biochem. 1983, 128, 245–249. 10.1016/0003-2697(83)90372-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.