

Abstract
Background and aims
Allograft inflammatory factor-1 (Aif1) has been characterized as a pro-inflammatory molecule expressed primarily in the monocyte/macrophage (MP) lineage and positively associated with various forms of vascular disease, including atherosclerosis. Studies of Aif1 in atherosclerosis have relied on mouse models in which Aif1 was overexpressed in either myeloid or smooth muscle cells, resulting in increased atherosclerotic plaque burden. How physiologic expression of Aif1 contributes to MP biology in atherogenesis is not known.
Methods
Effects of global Aif1 deficiency on atherosclerosis were assessed by crossing Aif1−/− and ApoE−/− mice, and provoking hyperlipidemia with high fat diet feeding. Atherosclerotic plaques were studied en face and in cross section. Bone marrow-derived MPs (BMDMs) were isolated from Aif1−/− mice for study in culture.
Results
Atherosclerotic plaques in Aif1−/−; ApoE−/− mice showed larger necrotic cores compared to those in ApoE−/− animals, without change in overall lesion burden. In vitro, lack of Aif1 reduced BMDM survival, phagocytosis, and efferocytosis. Mechanistically, Aif1 supported activation of the NF-κB pathway and expression of related target genes involved in stress response, inflammation, and apoptosis. Consistent with this in vitro BMDM phenotype, Aif1 deficiency reduced NF-κB pathway activity in vivo and increased apoptotic cell number in atherosclerotic lesions from Aif1−/−; ApoE−/− mice.
Conclusions
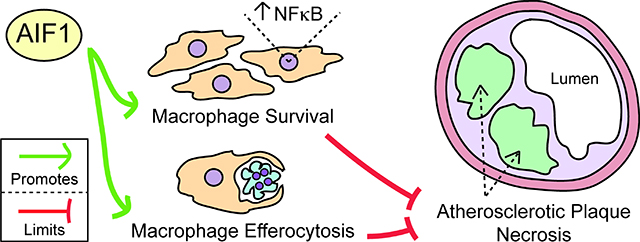
These findings characterize Aif1 as a positive regulator of the NF-κB pathway that supports MP functions such as survival and efferocytosis. In inflammatory settings such as atherosclerosis, these Aif1-dependent activities serve to clear cellular and other debris, limit necrotic core expansion, and may oppose lesion destabilization.
Keywords: Atherosclerosis, Efferocytosis, Macrophage, Necrosis, NF-κB
Graphical Abstract
INTRODUCTION
Atherosclerotic vascular disease is the leading cause of ischemic heart disease, stroke, and death worldwide1. It is a chronic inflammatory process that is initiated by endothelial dysfunction and sub-endothelial accumulation of monocytes due to lipid deposition leading to non-resolving inflammatory responses by monocyte-derived macrophages (MPs) and vascular smooth muscle cells (VSMCs) that contribute to atherosclerotic plaque formation2, 3 Classically activated MPs contribute to the inflammatory response and plaque destabilization through secretion of inflammatory molecules and matrix-degrading proteases and through their death by apoptosis or necrosis4. In early lesions, MP phagocytic and efferocytotic capacities limit plaque formation by promoting uptake of the excess of oxidized low-density lipoprotein (oxLDL) and by clearing the apoptotic cellular debris5. As lesions progress, cytokines synthesized by activated MPs promote VSMC migration and aberrant proliferation, and together with the accumulation of apoptotic lipid-loaded foam-cells and defective MP efferocytosis, these factors contribute to the expansion of large necrotic cores2, hallmarks of unstable plaques that are prone to undergo rupture6. The rupture of an advanced unstable plaque can precipitate acute, occlusive luminal thrombosis and lead directly to unstable angina, myocardial infarction, and stroke1, 7.
Allograft inflammatory factor-1 (Aif1), also known as ionized binding adapter-1 (Iba1), is a 17 kDa Ca2+ binding EF-hand protein originally identified in rat cardiac allografts undergoing chronic rejection and expressed primarily in cells of monocyte lineage8, 9. Indeed, Aif1 was initially characterized as a MP protein directly involved in phagocytosis and actin bundling10 and positively associated with inflammatory processes and various forms of vascular disease8, 11, 12. Associations of Aif1 with atherosclerosis are based largely on overexpression studies in which Aif1 was targeted to either MPs13, leading to augmented phagocytosis and lipid uptake, IL-6 and IL-12 production14 and cell proliferation and migration15; or to VSMCs16, leading to increased VSMC migration and inflammatory response. Accordingly, short-term siRNA-mediated knockdown limited MP phagocytosis, migration and proliferation10, 17 and VSMC growth18. In vivo, AIF1 has been shown to co-localize with CD68+ macrophages in human atherosclerotic arteries17, and transgenic mice overexpressing Aif1 either in MPs13 or VSMCs16 displayed increased atherosclerosis.
Here we describe how complete loss of Aif119 affects MP functions in vitro and atherosclerotic plaque composition in vivo. These loss-of-function studies depict an essential role for Aif1 in cell survival, inflammatory signaling, and efferocytosis that act in opposition to plaque necrosis, and highlight a requirement for Aif1 in activation of NF-κB signaling both in isolated MPs and in atherosclerosis lesions.
MATERIALS AND METHODS
Experimental Animals
C57BL/6 wild-type (wt) and ApoE−/− mice were purchased from Jackson Laboratory. The Aif1−/− mouse line19 was backcrossed at least 8 generations onto a C57BL/6 background. ApoE−/− single KO and Aif1−/−;ApoE−/−double KO male and female mice were used for in vivo studies of atherosclerosis. Methods for mouse genotyping by PCR for Aif1 and lacZ have been previously described19, and genotyping for ApoE was performed according to a Jackson Laboratory protocol. Mice were housed five to a cage under specific pathogen-free conditions in the Albert Einstein College of Medicine barrier facility, maintained on standard chow diet, or placed for 18 weeks on high fat diet containing 42% calories from fat (Harlan Teklad TD.88137; Envigo) at 8 weeks of age. All animal studies followed regulations of the Association for Assessment and Accreditation of Laboratory Animal Care and were approved by the Albert Einstein College of Medicine Institutional Animal Care and Use Committee.
Processing and Morphometric Analysis of Atherosclerotic Lesions
Vessels from ApoE−/− and Aif1−/−;ApoE−/− mice were embedded in optimal cutting temperature and snap frozen. Cryosections were sectioned from distal to proximal at 8 μm thickness. Lesions luminal to the internal elastic lamina were measured in Oil-Red O stained sections as the mean of six sections of the brachiocephalic artery (BCA) quantified at 200, 280, 360, 440, 520, and 600 μm proximal to the branching point of the BCA into the carotid and subclavian arteries. En face lesion area of the aorta was quantified relative to its surface area by Sudan IV staining using Photoshop. For quantification of aortic root lesion size, sections were fixed in 10% formalin, processed as previously described20 and stained with hematoxylin and eosin (H&E). Necrotic cores were quantified as previously described21, defined as the H&E-negative acellular areas in the intima. Collagen content was quantified using Masson’s trichrome staining. All plaque analyses were performed by an observer blinded to the genotype.
Cell culture
Mouse aortic smooth muscle cells (MASMCs) were isolated from 5 week old wt and Aif1−/− mice using collagenase-elastase digestion and maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 20% fetal bovine serum (FBS), 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 2 mM L-glutamine. Bone marrow derived MPs (BMDMs) were isolated from femurs and tibias from wt and Aif1−/− mice (8–10 weeks old), flushed with cold α-modified minimal essential medium (α-MEM) and seeded in petri dishes with α-MEM supplemented with 15% FBS and 10% WEHI conditioned media for 48 hours. Non-adherent cells were collected and grown in α-MEM with 15% FBS and 1,000 U/ml colony stimulating factor (CSF)-1 to allow for macrophage differentiation. After incubation with α-MEM supplemented with 15% FBS and 10,000 U/ml colony stimulating factor CSF-1, cultures showed >99% macrophage differentiation (confirmed by FITC-F4/80). BMDMs were maintained in supplemented α-MEM containing 15% FBS, 10,000 U/ml CSF-1, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 2 mM L-glutamine. BMDMs were immortalized as previously described using a retrovirus containing SV-U-19–5 large T-antigen22. For NF-κB pathway stimulation, BMDMs were stimulated with 50 μg/ml of human medium oxidized low-density lipoprotein (oxLDL) (Kalen Biomedical; 7702026) for 15 minutes, 30 minutes, 1 hour or 6 hours.
Survival
Wt or Aif1−/− BMDMs were incubated with H2O2 for 40 minutes. Dead and live cells were distinguished using the LIVE/DEAD® system (Thermo Scientific; L3224). Percent survival was determined as the mean of (live/(live + dead) cells × 100) using ImageJ software from 15 randomly selected epifluorescent photomicrographic fields.
Phagocytosis
BMDMs were incubated at 4° or 37°C for 1 h with 10, 50, or 100 particles per cell of Alexa-488 labeled zymosan (Invitrogen; Z23373). Cells were washed several times with PBS, and treated with trypsin/EDTA at 37°C to free unbound zymosan from the cell surface. After the ingestion period, cells were washed and fixed with 2% PFA and immediately analyzed by flow cytometry. Fluorescence at 488 nm was normalized to baseline fluorescence obtained with particle incubation at 4°C. Zymosan fluorescence intensity per cell was determined for at least 50,000 cells by FACS analysis. Free zymosan particles were distinguished from cells with ingested zymosan by their distinct light scattering pattern.
In vivo efferocytosis assay
Three days before the efferocytosis assay, wt and Aif1−/− mice received an intraperitoneal injection of thioglycolate to elicit MPs. Jurkat T-cells were labeled for 2 hours with TAMRA (1 μg/mL) (Invitrogen; C1171), washed with PBS, and subjected to UV irradiation (254 nm, 20 J/cm2) for 10 min to induce apoptosis. After 3 hours of recovery, apoptotic cells were washed and injected into mice (20 × 106 cells/mouse). After 30 minutes, mice were sacrificed and peritoneal leukocytes were harvested by lavage. MPs were labeled with FITC-F4/80 for 30 min on ice. The cells were washed, fixed and analyzed by flow cytometry to quantify FITC+ MPs that had ingested TAMRA+ apoptotic T-cells.
Western blotting
Protein lysates were extracted from BMDMs using RIPA buffer (50 mM Tris-HCl pH 7.4, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 150 mM NaCl) plus protease inhibitor (Millipore; 535142–1SET) and phosphatase inhibitors (Roche; PhosSTOP Easypack 04906837001) cocktails. A BCA protein assay kit (Pierce; 23335) was used to measure protein concentrations, and equal amounts of protein were loaded (20–35 μg) in the gels. For the detection of Aif1 total protein lysates were separated by a 10–20% Tricine gel electrophoresis (Invitrogen; EC6625BOX), wet-transferred (40 V; 3.5 hours) to a 0.2 μm pore size PVDF membranes (Bio-Rad; 1620177), blocked for 1 hour at room temperature with TBST (Tris pH8.0, NaCl 150 mM, 0.1% Tween 20) plus 5% (wt/vol) nonfat milk, and incubated overnight at 4°C with anti-Aif1 (Wako; 016–20001; 1:500 dilution or Abcam; ab178847; 1:2000 dilution). Other proteins were separated by 10% polyacrylamide gel electrophoresis, semi-dry-transferred (25 V; 1 hour) to a 0.45 μm pore size PVDF membranes (Millipore; IPVH00010), blocked for 1 hour at room temperature with TBST (Tris pH8.0, NaCl 150 mM, 0.1% Tween 20) plus 5% (wt/vol) BSA, and incubated overnight at 4°C with antibodies from the NF-κB Pathway Sampler Kit (Cell Signaling; 9936T; 1:2000 dilution), anti-Mer (Novus Biologicals; AF591-SP, 1:2000 dilution), anti-MFG-E8 (R&D SYSTEMS; AF2805-SP, 1:2500 dilution) or anti-β-actin (Invitrogen; MA1–140, 1:5000 dilution) as primary antibody in blocking solution. After washing, membranes were incubated with HRP-conjugated secondary antibodies in blocking solution for 1 hour at room temperature. After washing, signals were detected by adding ECL blotting substrate (Pierce; 32106) and exposing films to membranes. Densitometric analysis was performed using ImageJ software. Protein levels were calculated relative to β-actin. Phosphorylated protein levels were normalized to the respective total protein levels.
Overexpression of Aif1 in RAW 264.7 cells
Mouse RAW 264.7 cells (1 × 106) were plated in 60 mm culture dishes and transfected with 2 μg/ml of pcDNA3.1-Aif1 or control pcDNA3.1 using TranslT-X2 (Mirus; MIR6004). After 48 hours, cells were washed, and protein lysates were collected, quantified and immunoblotted.
RNA extraction and gene expression analysis
Total RNA was isolated from unstimulated and oxLDL-stimulated (50 μg/ml for 6 hours) wt and Aif1-deficient BMDM using Trizol reagent (Invitrogen; 15596018) according to manufacturer’s instructions. RNA concentrations were calculated using NanoDrop technology (ThermoFisher Scientific; ND-1000), and RNA integrity was evaluated using RNA 6000 Nano LabChips on an Agilent 2100 Bioanalyzer (Agilent Technologies). All chips were prepared according to the manufacturer’s instructions at the Genome Facility of the Albert Einstein College of Medicine. Only samples with RNA integrity number (RIN) values greater than 9 were selected for gene expression study. Total RNA (400 ng) was reverse transcribed using the RT2 First Strand Synthesis Kit (Qiagen; 330401), and resultant cDNA was loaded onto the RT2 Profiler PCR Array (Qiagen; PAMM- 225Z). Arrays were run in the Applied Biosystems 7900HT Fast real-time PCR system following the manufacturer’s recommendations. CT values (cut-off <35) were uploaded to the Qiagen data analysis web portal (geneglobe). Samples were assigned to controls (unstimulated) and test groups (oxLDL). Gene expression changes were calculated using the ΔΔCT method23 in which ΔCT is calculated between the gene of interest and an average of housekeeping genes, followed by ΔΔCT calculations (ΔCT (Test Group) - ΔCT (Control Group)). Fold Change is then calculated using 2−(ΔΔCT) formula. Gene expression changes were represented using Volcano plots displaying statistical significance versus fold-change. We defined a threshold for fold regulation at ±1.5 and p-value at 0.05 (Student’s t-test).
Immunohistochemistry and immunofluoresence of atherosclerotic lesions
Aortic root sections from ApoE−/− and Aif1−/−;ApoE−/− mice were fixed with methanol at −20°C, followed by cold acetone at 4°C. For immunohistochemical analysis, sections were incubated with H2O2 to remove endogenous peroxidase activity and then blocked for 1 hour at room temperature with 5% normal goat serum and then incubated overnight at 4°C with anti-phospho-p65 Ser536 (Cell Signaling; 3033S, 1:25 dilution) in 3% BSA/PBS. After washing, sections were incubated with biotinylated goat anti-rabbit IgG (Vector Labs; BA-1000) for 1 hour at room temperature, washed again and then incubated with ABC reagent (Vector Labs; PK-6100) for 30 minutes. After washing, DAB substrate (Dako; K3467) was added to each section until specific signal was observed, and the reaction stopped by dipping slides in water. Sections were counterstained with hematoxylin (Vector Labs; H-3404) and coverslips mounted with toluene solution (Thermo Scientific; SP15–100). Specificity of the staining was tested by omission of the primary antibody. Images were obtained using a digital microscope (COOLSCOPE, Nikon). For immunofluoresecent analysis, sections were blocked for 1 hour at room temperature with 5% normal goat serum and then incubated overnight at 4°C with rat anti-CD68 (Bio-Rad; MCA1957,1:50 dilution), mouse anti-smooth muscle cell α-actin (SMA) (Santa Cruz; c-53015, 1:100 dilution) or rat anti-CD3 (Biolegend; 100201; 1:100 dilution). After washing, sections were incubated with goat anti-rat or anti-mouse IgG-594 (1:100 dilution) for 1 hour at room temperature, washed again, mounted with Fluoro-Gel medium containing DAPI and images acquired by Zeiss microscope.
TUNEL assay
An in situ cell death detection kit with fluorescein readout (Roche; 11684795910) was used to test apoptosis as per the manufacturer’s instructions. Briefly, aortic root sections were fixed with methanol followed by acetone, and labeled with TUNEL reaction mixture (label solution and terminal transferase), counterstained with DAPI, and analyzed by fluorescence microscopy in an Axio Observer.Z1 microscope (Zeiss). Sections incubated with label solution without terminal transferase were used as negative controls.
Statistical analysis
Statistical analysis was performed using Prism 7.0 software (GraphPad). All the in vitro experiments shown were repeated at least three independent times in triplicate. Comparisons between two groups were assessed using Student’s t-test for unpaired samples. All data are presented as mean ± standard error (SEM). Significance was accepted for values of probability p <0.05.
RESULTS
Loss of Aif1 increases necrotic core size in high fat diet induced atherosclerotic lesions, without affecting lesion burden
Forced expression of Aif1 in MPs13 or VSMCs16 has been linked to increased atherosclerotic lesion burden, but whether Aif1 expressed at endogenous levels affects atherogenesis has not been characterized. Our previous characterization of Aif1−/− mice revealed no differences in blood monocyte or T-cell subset numbers compared to wt animals19. Although Aif1 is expressed T-lymphocytes, monocytes, and macrophages8 - important cell types contributing to atherosclerotic plaque formation - its absence does not necessarily affect baseline leukocyte populations.
We tested the effect of loss of Aif1 on the development of atherosclerosis in vivo by crossing Aif1−/− and hyperlipidemic, atherosclerosis-susceptible ApoE−/− mice24. We placed 8 week old ApoE−/− single KO and Aif1−/−;ApoE−/− double KO mice on a high fat diet for 18 weeks and measured the extent of atherosclerosis by en face Sudan IV staining of aortae. Total aortic lesion area was similar in ApoE−/− and Aif1−/−;ApoE−/− mice (Fig. 1A (left panel) and B) - perhaps surprising in view of the results of previous Aif1 overexpression studies. We also performed cross-sectional analysis of lesion formation in the brachiocephalic trunk (Fig. 1A (right panel) and B) and in the aortic root (Fig. 1C (left panel) and D), and found no difference between groups. In addition, several studies have demonstrated that elevated levels of lipids and cholesterol are directly associated with the risk for atherosclerotic cardiovascular events25. We confirmed that after 18 weeks of high fat diet plasma cholesterol and triglyceride levels were similar between ApoE−/− and Aif1−/−;ApoE−/− mice (Supplementary Fig. 1B). These observations indicate that Aif1 does not affect circulating immune cell subsets or lipid levels, and is also not essential for atherosclerotic lesion formation. In addition, we observed no differences in the number of CD3+ T-cells in the plaques from ApoE−/− and Aif1−/−;ApoE−/− mice (Supplementary Fig. 1A).
Fig. 1.

Aif1 limits necrotic core expansion without affecting atherosclerotic lesion burden in vivo.
(A) Left panel, en face Sudan IV staining of thoracoabdominal aortae harvested from ApoE−/− (n=11) and Aif1−/−;ApoE−/− (n=11) mice after 18 weeks of high fat feeding (bar, 1 mm). Right panel, representative brachiocephalic artery (BCA) lesions from ApoE−/− (n=8) and Aif1−/−;ApoE−/− (n=10) mice after 18 weeks of high fat feeding stained with ORO. (B) Left panel; quantification of the en face Sudan IV staining of aortae. Right; quantification of the ORO staining of BCAs. (C) Left panel, representative sections of hematoxylin and eosin-stained aortic root sections from ApoE−/− (n=10) and Aif1−/−;ApoE−/− (n=10) mice. The boxed areas are expanded in the middle panels (*necrotic areas; bar, 200 μM). Right panel, representative sections of Masson’s trichrome stained aortic root sections from ApoE−/− (n=10) and Aif1−/−;ApoE−/− (n=10) mice. (D) Quantification of aortic root lesion size from ApoE−/− or Aif1−/−;ApoE−/− mice. (E) Quantification of necrotic (acellular) area from ApoE−/− or Aif1−/−;ApoE−/− mice. (F) Quantification of collagen content from ApoE−/− or Aif1−/−;ApoE−/− mice. Error bars indicate ± SEM (N.S. non-significant, *** p <0.0001)
We next analyzed the complexity of atherosclerotic lesions from high fat diet fed ApoE−/− and Aif1−/−;ApoE−/− mice. Quantification of plaque necrotic (acellular) areas in H&E-stained sections from aortic roots indicated that these areas were significantly larger in Aif1−/−;ApoE−/− mice (Fig. 1C (middle panel and E). Interestingly, although the differences were not statistically significant, collagen content tended to be decreased in the aortic root lesions from Aif1−/−;ApoE−/− mice (Fig. 1C (right panel and F). These observations suggest that Aif1 limits necrotic core expansion in atherosclerotic plaques and may oppose lesion destabilization.
Aif1 promotes macrophage accumulation and survival
The effects of forced expression of Aif1 on plaque formation have been attributed to altered MP13 or VSMC16 functions. How physiologic levels of Aif1 affect cellular functions has not been well studied. We first determined the baseline Aif1 expression levels in bone marrow derived MPs (BMDMs) and mouse aortic smooth muscle cells (MASMCs), and found that Aif1 transcript levels were robust in BMDMs, and markedly lower (but still detectable) in MASMCs. By Western analysis, Aif1 protein was detected only in BMDMs (Supplementary Fig. 2), which suggests that loss of Aif1 in MPs is more likely to result in cellular or atherosclerotic phenotypes.
To assess how Aif1 deficiency affects MP activities, we cultured primary BMDMs from wt and Aif1−/− mice. After 6 or more days of culture, we found a consistent decrease in the number of Aif1−/− relative to wt cells (Fig. 2A). To determine if this deficiency was due to decreased cell proliferation, we analyzed cell proliferation by measuring BrdU incorporation at various time points from wt or Aif1−/− BMDMs cultured for 10 days with CSF-1. BMDMs from Aif1−/− mice showed no proliferation defect, as the number of BrdU incorporating cells was similar to wt (Supplementary Fig. 3A). Staining of DNA content with propidium iodide also showed similar cell cycle parameters. Interestingly, more sub-G1 apoptotic cells were observed in the cell cycle profiles of Aif1−/− cells at 10 days after bone marrow retrieval (Supplementary Fig. 3B, arrow), suggesting that despite conditions optimized to support cell growth, loss of Aif1 could adversely affect MP survival. We then modeled the deleterious atherosclerotic milieu by subjecting BMDMs from wt and Aif1−/− mice to oxidative stress or oxidized (ox)-LDL overload. At all concentrations of H2O2, Aif1 deficiency caused a decrease in survival (Fig. 2B) and similarly, cell viability was reduced in Aif1−/− BMDMs after exposure to high concentrations of oxLDL (Supplementary Fig. 3C). These studies show that loss of Aif1 has little effect on MP proliferation, but increases susceptibility to oxidative stress and lipid overload.
Fig. 2.

Aif1 supports macrophage survival, phagocytosis, and efferocytosis.
(A) Cells were counted during several days after bone marrow harvest using a Coulter counter. (B) Wt and Aif1−/− BMDMs were treated with H2O2 (0, 20, 40, 75, 100 or 150 μM) for 40 minutes. Survival was assessed and quantified with a LIVE/DEAD viability kit. (C) Wt and Aif1−/− BMDMs were synchronized and incubated with the indicated numbers of Alexa 488-zymosan particles per cell (bar, 25 μM). (D) The percentage of internalized zymosan particles per cell was measured by FACS. (E) FACS quantification of the number of F4/80+ cells recruited to the site of inflammation in wt (n=5) or Aif1−/− (n=6) mice. (F) FACS quantification of F4/80+ macrophages isolated from wt (n=5) or Aif1−/− (n=6) that have ingested TAMRA-labeled apoptotic Jurkat T-cells. Error bars indicate ± SEM (* p <0.05, ** p <0.001, *** p <0.0001).
Aif1 supports macrophage phagocytosis and efferocytosis
MPs recruited into developing atherosclerotic lesions initially clear apoptotic cells efficiently via a process termed efferocytosis, and also take up modified low-density lipoprotein (LDL) — these activities temper inflammatory processes to restrict plaque progression. Accumulation of foam cells and defective efferocytosis are major drivers of lesion formation and necrotic core expansion, factors that can trigger plaque rupture and acute thrombotic cardiovascular events26–28.
In order to investigate the phagocytic ability of Aif1−/− BMDMs, we measured phagocytosis of fluorescently labeled zymosan. MPs lacking Aif1 showed a decrease in the number of ingested zymosan particles across a spectrum of zymosan concentrations (Fig. 2C and D), suggesting that loss of Aif1 reduces the ability to phagocytose these particles. Zymosan uptake occurs by phagocytosis and involves pattern recognition receptors, such as scavenger receptors, CD36 and others28, 29. These evolutionarily conserved receptors also bind oxLDL27. Since Aif1-deficient BMDM showed impaired zymosan phagocytosis, we also determined if Aif1 plays a role in oxLDL uptake. When wt or Aif1−/− BMDMs were incubated with oxLDL (50 μg/ml) for 18 hours, Aif1−/− BMDMs ingested less oxLDL than wt cells (Supplementary Fig. S3D). Accordingly we checked the expression changes of genes involved in lipid uptake (Sr-a1, CD36) and cholesterol efflux (Abca1, Abcg1) after oxLDL stimulation. The gene expression study showed significantly decreased scavenger receptor A (Sr-a1) levels in Aif1−/− MPs after stimulation (Supplementary Fig. 3E). These findings demonstrate that loss of Aif1 impairs lipid uptake and phagocytosis in MPs.
Because loss of Aif1 impaired phagocytosis of zymosan particles and resulted in decreased oxLDL uptake, we next performed an in vivo efferocytosis assay by injecting apoptotic Jurkat T-cells into the peritoneal cavities of Aif1−/− and wt mice with thioglycolate induced peritonitis30. As shown in Fig. 2E, Aif1 deficiency decreased the percentage of F4/80+ MPs among cells elicited from the peritoneum, suggesting that loss of Aif1 impaired MP recruitment to the site of inflammation. Furthermore, compared to wt, Aif1−/− MPs ingested fewer apoptotic cells (Fig. 2F), suggesting that Aif1 is also required for efficient efferocytosis. In order to elucidate this pro-efferocytotic mechanism, we assessed the effect of Aif1 deficiency on the expression of Mer Tyrosine Kinase (MerTK) and Lactadherin (MFG-E8) levels, two of the most well known proteins involved in the process of efferocytosis31. There were no differences in baseline MerTK levels. In contrast, we observed reduced baseline MFG-E8 levels in the Aif1−/− MPs (Supplementary Fig. 3F), suggesting that Aif1 might be directly involved in the regulation of efferocytosis.
In summary, the findings described above show that Aif1 limits necrotic core expansion by supporting MP functions directly involved in atherogenesis, including survival under oxidative stress or lipid overload, phagocytosis, and efferocytosis.
Aif1 supports NF-κB pathway activation in macrophages
The NF-κB pathway serves as a pivotal regulator of inflammatory responses by promoting the survival and activation of immune cells, including MPs32. Interestingly, in humans, the AIF1 gene maps to the major histocompatibility complex class III region on chromosome 6p21.3, a region densely clustered with genes involved in the inflammatory response, including NF-κB9. In vitro studies using epithelial human breast cancer33 and mouse adipocyte-like34 cell lines have shown an induction in NF-κB activity associated with Aif1. In addition, VSMCs overexpressing Aif1 showed increased activation of the NF-κB pathway16. Whether Aif1 contributes to MP NF-κB activity, specifically in the context of atherosclerosis, is not known.
Given our findings linking Aif1 to a pro-survival MP phenotype (Fig. 2B), we asked if Aif1 affected NF-κB signaling pathway in these cells. We stimulated wt and Aif1−/− BMDMs with oxLDL, and at different time points, measured levels of total and phosphorylated (active) forms of the upstream IKKβ and IKKα kinases, the inhibitory complex IkBα, and downstream transcription factor RelA/p65 - NF-κB. Stimulation by oxLDL significantly increased phosphorylated p65 Ser536 levels, relative to total p65 levels, in wt but not Aif1−/− MPs (Fig. 3A, Fig. 3B). Interestingly, loss of Aif1 did not induce any changes in the level of the phosphorylated IKKα/β and phosphorylated IkBα, relative to total IKKα/IKKβ and total IkBα levels, respectively (Supplementary Fig. 4A–C).
Fig. 3.

Aif1 promotes NF-κB signaling activity.
(A) Wt and Aif1−/− BMDMs were stimulated with oxLDL (50 μg/ml) for 15, 30, or 60 minutes. Lysates were immunoblotted for total p65 and phosphorylated p65 Ser536 and Ser276. (B) Quantification of phosphorylated p65 Ser536 protein levels, relative to total p65 levels. (C) Quantification of phosphorylated p65 Ser276 protein levels, relative to total p65 levels. (D) RAW 264.7 cells were transfected with 2 μg/ml of pcDNA3.1-Aif1 or control pcDNA3.1. 48 hours after transfection lysates were immunoblotted for total p65 and phosphorylated p65 Ser536 and Ser276. (E) Quantification of phosphorylated p65 Ser536 and Ser276 protein levels, relative to total p65 levels. Dashed grey line represents distinct gels. Error bars indicate ± SEM (* p <0.05, ** p <0.001).
Studies of p65 phosphorylation have focused mainly on two phosphorylation sites within the subunit: Ser536 in the C-terminal transactivation domain and Ser276 within the N-terminal Rel homology domain35, 36 Ser536 is targeted by several kinases, including IKKβ, and numerous effects have been ascribed to this modification, such as the regulation of p65 nuclear localization and interaction with transcriptional co-activators37. The p65 Ser276 site is phosphorylated by the catalytic subunit of PKA (PKAc) after accumulation of cAMP, facilitating the binding to transcriptional co-activators and to DNA38. Our studies found that after oxLDL stimulation, the level of phosphorylated p65 Ser276 levels was not different in wt vs. Aif1−/− BMDMs (Fig. 3A and C). Together, these results suggest that Aif1 is specifically required for phosphorylation of RelA/p65 at Ser536.
Interestingly, Aif1 overexpression in the murine MP RAW 264.7 cell line also increased levels of p65 Ser536 phosphorylation (Fig. 3D and E). Phosphorylated IKKα/β and phosphorylated IkBα levels were also not affected using this approach (Supplementary Fig. 4D and E). Similarly, the overexpression study did not show any differences on phosphorylated p65 Ser276 levels, relative to total p65 levels, between control and Aif1 overexpressing RAW 264.7 cells (Fig. 3D and E). These observations characterize Aif1 as a positive regulator of the NF-κB pathway that is specifically required in MPs for phosphorylation of p65 at Ser536.
Aif1 promotes NF-κB pathway-regulated inflammatory, anti-apoptotic, and stress response gene expression in macrophages
NF-κB is a pleiotropic transcription factor that triggers the expression of various target genes directly involved in the production of inflammatory cytokines, chemokines, and adhesion molecules, and in the regulation of cell survival, proliferation, apoptosis, morphogenesis, and differentiation32. In view of the observation that Aif1 promotes NF-κB activation in MPs, we looked for differences in NF-κB target gene expression between wt and Aif1-deficient MPs stimulated with oxLDL.
The NF-κB signaling target gene array showed 14 upregulated (fold change > 1.5) and 7 downregulated genes (fold change > −1.5) (Fig. 4A and Supplementary Table 1) in wt BMDMs after oxLDL stimulation. Among these genes, changes in Cxcl10, Gadd45b, Ifnb1, Il1a, Map2k6, Nqo1, Nr4a2, Ptgs2 and Tnfrsf1b expression levels were statistically significant (p <0.05) (Fig. 4B and Supplementary Table 1). On the other hand, in oxLDL-stimulated Aif1-deficient BMDMs, 7 NF-κB signaling target genes were upregulated and 15 genes were downregulated (Fig. 4C and Supplementary Table 1). Among these genes, changes in Bcl2l1, Ccl12, Ccnd1, Cd83, Cxcl10, Gadd45b, Il1b, Map2k6, Nqo1, Pdgfb, Plau and Stat1 levels were statistically significant (p <0.05) (Fig. 4D and Supplementary Table 1). These findings are consistent with the observation that Aif1 promotes NF-κB pathway activation and expression of its target genes, while loss of Aif1 impairs MP responsiveness.
Fig. 4.

Aif1 promotes expression of NF-κB signaling target genes in macrophages.
(A) Volcano plot displaying statistical significance (p <0.05) versus fold-change (± 1.5) on the y- and x-axes, respectively. Each dot is a single gene. Red dots highlight upregulated genes and green dots highlight downregulated genes in wt BMDMs after 6 hours of oxLDL stimulation (50 μg/ml). (B) Statistically significant gene expression changes in wt BMDMs after 6 hours of oxLDL stimulation. (C) Volcano plot displaying statistical significance (p <0.05) versus fold-change (± 1.5) on the y- and x-axes, respectively. Each dot is a single gene. Red dots highlight upregulated genes and green dots highlight downregulated genes in Aif1−/− BMDMs after 6 hours of oxLDL stimulation (50 μg/ml). (D) Statistically significant gene expression changes in Aif1−/− BMDMs after 6 hours of oxLDL stimulation. (E) Comparison of gene expression changes of the genes identified in B an D between wt and Aif1−/− BMDMs. (F) Additional observation of genes that show differential tendencies between wt and Aif1−/− BMDPs after oxLDL stimulation. Error bars indicate ± SEM (* p <0.05, ** p <0.001, *** p <0.0001, **** p <0.00001).
We then focused on genes found to be differentially regulated in either wt or Aif1−/− MPs after oxLDL stimulation, and compared these changes in expression between the two types of cells. This analysis revealed significantly different levels of Bcl2l1, Ccnd1, Ifn1b, Il1a, Plau, Ptgs2 and Tnfrsf1b between the wt and Aif1−/− MPs after oxLDL stimulation. Notably, anti-apoptotic (Bcl2l1), pro-inflammatory (Il1a, Tnfrsf1b), pro-survival (Ccnd1) and stress-related (Plau, Ptgs2) genes were downregulated in the Aifi−/− cells compared to wt (Fig. 4E). In contrast, the anti-inflammatory cytokine Ifnb1 was upregulated in the Aif1-deficient cells.
In addition, several functionally relevant target genes upregulated in wt MPs were not induced or decreased in Aif1-deficient MPs. These genes could be grouped according to their involvement in anti-apoptotic processes (Akt, Csf2, Myd88, Rela, Relb), stress-response (Mmp9, Birc2, Adm, Xiap) and cytokine and chemokine expression (Ccl22, Il6, IL15, Ccr5). In contrast, anti-inflammatory Il4 and Csf3 mRNA levels tended to be upregulated in the Aif1−/− BMDMs (Fig. 4F and Supplementary Table 1).
Accordingly, compared to the wt cells, Aif1-deficient MPs showed decreased IL-6 and TNF-α pro-inflammatory cytokine production after oxLDL stimulation (Supplementary Fig. 5).
Taken together, these studies show that upon MP stimulation with oxLDL Aif1 promotes the expression of pro-inflammatory, anti-apoptotic, and stress response-related genes that are regulated through the NF-κB pathway.
Aif1 induces NF-κB activity and limits apoptosis in atherosclerotic plaques in vivo
A number of processes may trigger cellular death and necrosis in atherosclerotic lesions, including the detrimental effect of NF-κB suppression2, 39. Furthermore, NF-κB pathway activity has been detected in human and mouse atherosclerotic tissues40. To determine if NF-κB activation in vivo requires Aif1 expression, we analyzed atherosclerotic lesions from ApoE−/− and Aif1−/−;ApoE−/− mice after 18 weeks on high fat diet. Immunohistochemical analysis showed that in comparison to single KO samples, double KO aortic roots had markedly decreased staining for phosphorylated p65 Ser536 levels (Fig. 5A and B). This suggests that Aif1 is required both for NF-κB pathway activation in isolated MPs and for in vivo NF-κB activation in atherosclerotic plaques.
Fig. 5.

Aif1 promotes NF-κB pathway and limits apoptosis in atherosclerotic plaques in vivo.
(A) Immunohistochemical analysis for phosphorylated p65 Ser536 was performed in atherosclerotic lesions (aortic roots) from Apoe−/− (n=4) and Aif1−/−;ApoE−/− (n=5) mice maintained on high fat diet for 18 weeks. (B) Quantification of phosphorylated p65 Ser536 levels by immunohistochemistry. (C) Representative epifluorescent micrographs showing TUNEL staining of aortic roots from Apoe−/− (n=8) and Aif1−/−;ApoE−/− (n=8) mice (bar, 50 μM) (D) Quantification of TUNEL positive cells on lesions from ApoE−/− or Aif1−/−;ApoE−/− mice. (E) Representative immunofluorescence analysis of CD68+, left, and SMA+, right, cells in aortic roots from Apoe−/− (n= 3) and Aif1−/−;ApoE−/− (n=3) mice maintained on high fat diet for 18 weeks. (F) Quantification of CD68+ cells on lesions from ApoE−/− or Aif1−/−;ApoE−/− mice. (G) Quantification of SMA+ cells on lesions from ApoE−/− or Aif1−/−;ApoE−/− mice. Error bars indicate ± SEM (* p <0.05, *** p <0.0001).
Apoptotic cell death has been observed in atherosclerotic lesions and is thought to contribute to plaque instability. Interestingly, our data demonstrate that Aif1 is required for MP resistance to oxidative stress and lipid overload in vitro. To determine if loss of Aif1 increased cell death in vivo, we performed in situ TUNEL staining of atherosclerotic lesions from ApoE−/− and Aif1−/−;ApoE−/− mice. Consistent with in vitro findings, aortic root lesions from Aif1−/−;ApoE−/− mice showed over twice as many TUNEL-positive nuclei compared to ApoE−/− mice (Fig. 5C and D). Accordingly, the number of CD68+ cells (Fig. 5E (left panel) and F) and SMA+ cells (Fig. 5G (right panel) and H) were reduced in the aortic roots from Aif1−/−;ApoE−/− mice.
Together, these studies of Aif1−/− BMDMs revealed decreased survival after oxidative stress or lipid overload and impaired phagocytic capabilities and apoptotic cell uptake. These findings in MPs lacking Aif1 show decreased phosphorylated p65 S536 levels and lower expression of genes involved in stress response, inflammatory, and anti-apoptotic processes - including many genes that are regulated at least partly through the induction of NF-κB pathway. Functional deficits in both survival and efferocytosis likely contribute to the atherosclerotic phenotype of Aif1−/−;ApoE−/− mice, in which loss of Aif1 and impaired NF-κB activity increase apoptosis, impair clearance of cell corpses, and permit expansion of the necrotic core.
DISCUSSION
The present study provides the first analysis of complete loss of Aif1 function in atherosclerotic lesions in vivo and in MP biology in vitro. Our results suggest that the physiological importance of Aif1 in the context of atherogenesis depends on its contributions to MP functions such as survival under stress and effective efferocytosis, both of which oppose the accumulation of dead cells and expansion of necrotic areas within plaques. Moreover, this study shows that Aif1 effects are mediated at least in part through the activation of the NF-κB pathway and its respective target genes.
Although Aif1 is expressed most strongly in cells of monocyte lineage, its expression has also been described in activated VSMCs and T-cells41,42. Moreover, previous studies have reported pro-atherogenic effects, focusing on lesion size, of Aif1 using overexpression strategies in MPs13 and VSMCs16. Such gain of function studies do not necessarily reveal how this protein contributes to plaque formation when expressed at endogenous levels. We have addressed this question through a loss of function strategy by crossing global Aif1−/−19 and ApoE−/− mice to assess the development of atherosclerosis in vivo. Surprisingly, Aif1−/−;ApoE−/− mice placed on high fat diet did now show differences in plaque size compared to control ApoE−/− mice. However, analysis of plaque composition revealed larger necrotic cores and a trend toward decreased collagen content in mice lacking Aif1. These observations point to Aif1 as a molecule that supports more fibrotic and less unstable plaques. In addition, we identified Aif1 protein expression in MPs, but not in VSMCs, which implicates loss of Aif1 in MPs - and the resulting cellular functional deficits - as the main factor contributing to the in vivo phenotypes we observed. The lack of plaque expansion could be due in part to decreased collagen accumulation, consistent with the trend that we observed. In addition, decreased recruitment of CD68+ and SMA+ cells into the lesion, perhaps as a result of lower cytokine and chemokine production by Aif1−/− macrophages, could also counterbalance the effect of impaired clearance of dead cells.
A multitude of studies over the last 30 years have led to the recognition that MPs are major drivers of atherosclerosis and its complications. Monocyte-derived MPs are recruited to the lesion site, accumulate, ingest lipids and cellular debris, and produce a diverse repertoire of inflammatory mediators that exacerbate disease2, 5. In addition, it is known that decreased efferocytosis results in larger necrotic cores in animal models of atherosclerosis induced by prolonged high fat feeding43. Our assessment of how loss of Aif1 affects a repertoire of cellular activities showed that Aif1 supports important MP functions directly involved in plaque progression and rupture: survival, lipid uptake, phagocytosis, and efferocytosis2, 44. Interestingly, our data showing reduced Sr-a1 expression in Aif1−/− macrophages upon oxLDL stimulation, suggests a possible role of Aif1 in regulating target gene expression. Moreover, baseline levels of MFG-E8, a bridging molecule between phosphatidylserines exposed on the surface of apoptotic cells and integrins on efferocytotic macrophages43, were reduced in Aif1−/− cells. This observation suggests that loss of Aif1 not only affects macrophage survival but also limits the recognition signals of the dying macrophages to be cleared by efferocytosis. Considering the reported role of Aif1 in membrane ruffling and actin bundling45, we hypothesize that its effects on receptor-mediated lipid and corpse ingestion might be associated with the regulation of the mechanical properties of the cytoskeleton and actin remodeling steps that facilitate corpse engulfment.
We sought to understand how Aif1 regulates MP survival and stress response. Previous in vitro studies have linked the pro-inflammatory and pro-survival NF-κB pathway to Aif1 functions in breast cancer cells, adipocytes, and VSMCs16, 33, 34. In addition, active NF-κB pathway has been detected in human and mouse atherosclerotic plaques46, 47. Therefore, we wanted to assess if Aif1 regulates the NF-κB pathway in MPs. Our in vitro studies, using both loss and gain of function approaches, demonstrated a relation between Aif1 and the phosphorylation of p65 at Ser536, but not Ser276.
The activated NF-κB pathway orchestrates the regulation of several genes involved in pro- and anti-inflammatory processes, apoptosis and stress response. To determine if the activation of the pathway by Aif1 also leads to a specific gene expression pattern, we evaluated a panel of known NF-κB target genes. Overall, out of 84 genes tested, the dominant effect of Aif1 deficiency on NF-κB target gene expression pattern was loss of responsiveness or down-regulation compared to wt BMDMs. We observed reduced expression of the anti-apoptotic gene Bcl2l1, which has been associated with increased apoptosis and reduced MP functional competence48, 49; reduced Ccnd1 and Plau expression, which are associated with decreased MP motility, proliferation and plaque growth50–52; reduced expression of Ptgs2 (Cox2), which has been associated with both atheroprotective and anti-thrombotic effects53, 54; and reduced expression of pro-inflammatory cytokines that have been classically linked to atherosclerosis, such as Il1a, IL-6, and TNF-α55,56. Although these negative effects of Aif1 loss of function are consistent, in the absence of effective NFkB silencing in MPs, which we were not able to achieve for technical reasons, these links between Aif1 and NF-κB target gene expression remain correlative, and this is a limitation to our study.
Our observation that Aif1 regulates the MP NF-κB pathway led us to determine if activation of the pathway in vivo was Aif1-dependent. Compared to those from ApoE−/− mice, atherosclerotic lesions from Aif1−/−;ApoE−/− mice placed on high fat diet revealed decreased phosphorylation of p65 at Ser536. MP-induced plaque necrosis has been linked to the detrimental effect of NF-κB suppression39 - consistent with this observation, mice lacking Aif1 showed increased necrosis in atherosclerotic lesions. Moreover, mice lacking Aif1 showed increased apoptosis and reduced CD68+ macrophage and SMA+ cell content in the plaque, which could reflect loss of marker gene expression and/or decreased cell recruitment into the lesion, perhaps as a result of lower cytokine and chemokine production by Aif1−/− macrophages. Thus, our results provide the first observations that link Aif1 expression, activation of NF-κB signaling, effective efferocytosis, and reduced apoptosis and necrosis. While loss of Aif1 also decreased NF-κB-dependent inflammatory gene expression -and so might be expected to decrease plaque size - our findings suggest that Aif1-dependent MP survival and clearance functions remain critical determinants of plaque development and size.
In summary, atherosclerosis remains a major unresolved health problem and accounts for most of the health care expenditure in Western-style societies. Accurate understanding of plaque pathobiology is important for the development of new therapeutic approaches to stabilize plaques and decrease the incidence of acute coronary syndromes and strokes that result from plaque rupture. At present, there are no therapeutics that support MP clearance functions for plaque-stabilizing activity. This study provides insight into the significance of endogenous levels of Aif1 in a specific inflammatory disease process, such as atherosclerosis, and serves also to underscore the importance of MP survival and efferocytosis as limiting processes in the expansion of atherosclerotic lesion necrotic cores.
Supplementary Material
HIGHLIGHTS.
Aif1 limits necrotic core expansion in atherosclerotic lesions.
Aif1 supports macrophage survival and efferocytotic activities.
Aif1 promotes NF-κB pathway activity in macrophages.
Aif1 induces NF-κB-dependent stress response, inflammatory, and anti-apoptotic gene expression in macrophages.
Aif1 promotes NF-κB pathway and limits apoptosis in atherosclerotic lesions in vivo.
ACKNOWLEDGMENTS
We wish to thank Richard Stanley and Violeta Chitu (Albert Einstein College of Medicine) for providing CSF-1 and for helping with macrophage cultures, Laura Santambrogio (Albert Einstein College of Medicine) for technical advice with phagocytosis assays, George Kuriakose (Columbia University Medical Center) for technical advice with atherosclerotic lesion harvesting and analysis, Ira Tabas (Columbia University Medical Center) for helpful discussions and Sergio Espeso-Gil (Icahn School of Medicine at Mount Sinai) for assistance with figure editing.
FINANCIAL SUPPORT
This work was supported by National Institutes of Health (NIH) grants R01HL067944 and R01HL128066 to N.E.S.S. C.L. was supported by the Summer Undergraduate Research Program (SURP, 2017) of the Albert Einstein College of Medicine, Graduate Division.
Footnotes
CONFLICT OF INTEREST
The authors declared they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu jH , Alger HM, Wong SS, Muntner P, American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 2.Moore KJ and Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett MR, Sinha S and Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. [DOI] [PubMed] [Google Scholar]
- 5.Moore KJ, Sheedy FJ and Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorp E and Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol. 2009;86:1089–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Virmani R, Burke AP, Kolodgie FD and Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J Interventional Cardiol. 2002;15:439–46. [DOI] [PubMed] [Google Scholar]
- 8.Utans U, Arceci RJ, Yamashita Y and Russell ME. Cloning and characterization of allograft inflammatory factor-1: a novel macrophage factor identified in rat cardiac allografts with chronic rejection. J Clin Invest. 1995;95:2954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iris FJ, Bougueleret L, Prieur S, Caterina D, Primas G, Perrot V, Jurka J, Rodriguez-Tome P, Claverie JM, Dausset J and et al. Dense Alu clustering and a potential new member of the NF kappa B family within a 90 kilobase HLA class III segment. Nature Genet. 1993;3:137–45. [DOI] [PubMed] [Google Scholar]
- 10.Ohsawa K, Imai Y, Kanazawa H, Sasaki Y and Kohsaka S. Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J Cell Sci. 2000;113 ( Pt 17):3073–84. [DOI] [PubMed] [Google Scholar]
- 11.Postler E, Rimner A, Beschorner R, Schluesener HJ and Meyermann R. Allograft-inflammatory-factor-1 is upregulated in microglial cells in human cerebral infarctions. J Neuroimmunol. 2000;104:85–91. [DOI] [PubMed] [Google Scholar]
- 12.Sommerville LJ, Kelemen SE and Autieri MV. Increased smooth muscle cell activation and neointima formation in response to injury in AIF-1 transgenic mice. Arterioscl Thromb Vasc Biol. 2008;28:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mishima T, Iwabuchi K, Fujii S, Tanaka SY, Ogura H, Watano-Miyata K, Ishimori N, Andoh Y, Nakai Y, Iwabuchi C, Ato M, Kitabatake A, Tsutsui H and Onoe K. Allograft inflammatory factor-1 augments macrophage phagocytotic activity and accelerates the progression of atherosclerosis in ApoE−/− mice. Int J Molec Med. 2008;21:181–7. [PubMed] [Google Scholar]
- 14.Watano K, Iwabuchi K, Fujii S, Ishimori N, Mitsuhashi S, Ato M, Kitabatake A and Onoe K. Allograft inflammatory factor-1 augments production of interleukin-6, −10 and −12 by a mouse macrophage line. Immunology. 2001;104:307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang ZF, Ho DW, Lau CK, Lam CT, Lum CT, Poon RT and Fan ST. Allograft inflammatory factor-1 (AIF-1) is crucial for the survival and pro-inflammatory activity of macrophages. Int Immunol. 2005;17:1391–7. [DOI] [PubMed] [Google Scholar]
- 16.Sommerville LJ, Kelemen SE, Ellison SP, England RN and Autieri MV. Increased atherosclerosis and vascular smooth muscle cell activation in AIF-1 transgenic mice fed a high-fat diet. Atherosclerosis. 2012;220:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian Y, Kelemen SE and Autieri MV. Inhibition of AIF-1 expression by constitutive siRNA expression reduces macrophage migration, proliferation, and signal transduction initiated by atherogenic stimuli. Am J Physiol Cell Physiol. 2006;290:C1083–91. [DOI] [PubMed] [Google Scholar]
- 18.Sommerville LJ, Xing C, Kelemen SE, Eguchi S and Autieri MV. Inhibition of allograft inflammatory factor-1 expression reduces development of neointimal hyperplasia and p38 kinase activity. Cardiovasc Res. 2009;81:206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casimiro I, Chinnasamy P and Sibinga NE. Genetic inactivation of the allograft inflammatory factor-1 locus. Genesis. 2013;51:734–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paigen B, Morrow A, Holmes PA, Mitchell D and Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–40. [DOI] [PubMed] [Google Scholar]
- 21.Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR and Tabas IA. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest. 2009;119:886–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jat PS and Sharp PA. Large T antigens of simian virus 40 and polyomavirus efficiently establish primary fibroblasts. J Virol. 1986;59:746–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 24.Zhang SH, Reddick RL, Piedrahita JA and Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–71. [DOI] [PubMed] [Google Scholar]
- 25.Linton MF, Yancey PG, Davies SS, Jerome WGJ, Linton EF and Vickers KC. The Role of Lipids and Lipoproteins in Atherosclerosis. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F and Vinik A, eds. Endotext South Dartmouth (MA); 2000. [Google Scholar]
- 26.Yurdagul A Jr., Doran AC, Cai B, Fredman G and Tabas IA. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front Cardiovasc Med. 2017;4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boullier A, Bird DA, Chang MK, Dennis EA, Friedman P, Gillotre-Taylor K, Horkko S, Palinski W, Quehenberger O, Shaw P, Steinberg D, Terpstra V and Witztum JL. Scavenger receptors, oxidized LDL, and atherosclerosis. Annals NY Acad Sci. 2001;947:214–22; discussion 222–3. [DOI] [PubMed] [Google Scholar]
- 28.Kume N, Moriwaki H, Kataoka H, Minami M, Murase T, Sawamura T, Masaki T and Kita T. Inducible expression of LOX-1, a novel receptor for oxidized LDL, in macrophages and vascular smooth muscle cells. Annals NY Acad Sci. 2000;902:323–7. [DOI] [PubMed] [Google Scholar]
- 29.Matsuura E, Kobayashi K, Tabuchi M and Lopez LR. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Progress Lipid Res. 2006;45:466–86. [DOI] [PubMed] [Google Scholar]
- 30.Gautier EL, Ivanov S, Lesnik P and Randolph GJ. Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood. 2013;122:2714–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linton MF, Babaev VR, Huang J, Linton EF, Tao H and Yancey PG. Macrophage Apoptosis and Efferocytosis in the Pathogenesis of Atherosclerosis. Circ J. 2016;80:2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu T, Zhang L, Joo D and Sun SC. NF-kappaB signaling in inflammation. Signal Transduction Targeted Ther. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu S, Tan WY, Chen QR, Chen XP, Fu K, Zhao YY and Chen ZW. Daintain/AIF-1 promotes breast cancer proliferation via activation of the NF-kappaB/cyclin D1 pathway and facilitates tumor growth. Cancer Sci. 2008;99:952–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren J, Lin Y, Tang J, Yue H and Zhao Y. Allograft Inflammatory Factor-1 Mediates Macrophage-Induced Impairment of Insulin Signaling in Adipocytes. Cell Physiol Biochem. 2018;47:403–413. [DOI] [PubMed] [Google Scholar]
- 35.Christian F, Smith EL and Carmody RJ. The Regulation of NF-kappaB Subunits by Phosphorylation. Cells. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oeckinghaus A and Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor Perspectives Biol. 2009;1:a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–30. [DOI] [PubMed] [Google Scholar]
- 38.Zhong H, Voll RE and Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–71. [DOI] [PubMed] [Google Scholar]
- 39.Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, Clausen BE, Forster I, Kockx MM, Rajewsky K, Kraal G, Hofker MH and de Winther MP. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003; 112:1176–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Winther MP, Kanters E, Kraal G and Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:904–14. [DOI] [PubMed] [Google Scholar]
- 41.Autieri MV, Carbone C and Mu A. Expression of allograft inflammatory factor-1 is a marker of activated human vascular smooth muscle cells and arterial injury. Arterioscler Thromb Vasc Biol. 2000;20:1737–44. [DOI] [PubMed] [Google Scholar]
- 42.Kelemen SE and Autieri MV. Expression of allograft inflammatory factor-1 in T lymphocytes: a role in T-lymphocyte activation and proliferative arteriopathies. Am J Pathol. 2005;167:619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ait-Oufella H, Kinugawa K, Zoll J, Simon T, Boddaert J, Heeneman S, Blanc-Brude O, Barateau V, Potteaux S, Merval R, Esposito B, Teissier E, Daemen MJ, Leseche G, Boulanger C, Tedgui A and Mallat Z. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation. 2007;115:2168–77. [DOI] [PubMed] [Google Scholar]
- 44.Kojima Y, Weissman IL and Leeper NJ. The Role of Efferocytosis in Atherosclerosis. Circulation. 2017;135:476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohsawa K, Imai Y, Sasaki Y and Kohsaka S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J Neurochem. 2004;88:844–56. [DOI] [PubMed] [Google Scholar]
- 46.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA and Neumeier D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. J Clin Invest. 1996;97:1715–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye X, Jiang X, Guo W, Clark K and Gao Z. Overexpression of NF-kappaB p65 in macrophages ameliorates atherosclerosis in apoE-knockout mice. Am J Physiol Endocrinol Metab. 2013;305:E1375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silbiger VN, Luchessi AD, Hirata RD, Lima-Neto LG, Cavichioli D, Carracedo A, Brion M, Dopazo J, Garcia-Garcia F, dos Santos ES, Ramos RF, Sampaio MF, Armaganijan D, Sousa AG and Hirata MH. Novel genes detected by transcriptional profiling from whole-blood cells in patients with early onset of acute coronary syndrome. Clin Chim Acta. 2013;421:184–90. [DOI] [PubMed] [Google Scholar]
- 49.Sevilla L, Zaldumbide A, Carlotti F, Dayem MA, Pognonec P and Boulukos KE. Bcl-XL expression correlates with primary macrophage differentiation, activation of functional competence, and survival and results from synergistic transcriptional activation by Ets2 and PU.1. J Biol Chem. 2001;276:17800–7. [DOI] [PubMed] [Google Scholar]
- 50.Neumeister P, Pixley FJ, Xiong Y, Xie H, Wu K, Ashton A, Cammer M, Chan A, Symons M, Stanley ER and Pestell RG. Cyclin D1 governs adhesion and motility of macrophages. Mol Biol Cell. 2003;14:2005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Novak ML, Bryer SC, Cheng M, Nguyen MH, Conley KL, Cunningham AK, Xue B, Sisson TH, You JS, Hornberger TA and Koh TJ. Macrophage-specific expression of urokinase-type plasminogen activator promotes skeletal muscle regeneration. J Immunol. 2011;187:1448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farris SD, Hu JH, Krishnan R, Emery I, Chu T, Du L, Kremen M, Dichek HL, Gold E, Ramsey SA and Dichek DA. Mechanisms of urokinase plasminogen activator (uPA)-mediated atherosclerosis: role of the uPA receptor and S100A8/A9 proteins. J Biol Chem. 2011;286:22665–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kirkby NS, Lundberg MH, Wright WR, Warner TD, Paul-Clark MJ and Mitchell JA. COX-2 protects against atherosclerosis independently of local vascular prostacyclin: identification of COX-2 associated pathways implicate Rgl1 and lymphocyte networks. PLoS One. 2014;9:e98165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grosser T, Fries S and FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rader DJ. IL-1 and atherosclerosis: a murine twist to an evolving human story. J Clin Invest. 2012;122:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tousoulis D, Oikonomou E, Economou EK, Crea F and Kaski JC. Inflammatory cytokines in atherosclerosis: current therapeutic approaches. Eur Heart J. 2016;37:1723–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.