

Heterogeneity of treatment related myeloid neoplasms (tMN) might be partially related to inadvertent inclusion of coincidental second disease and neoplasia due to genetic predisposition. Clinical features or traditional morphology and cytogenetics are not sufficiently distinctive for possible subtypes of tMN and thus we investigated whether a therapy-related somatic molecular signature can be found. Using controls, including patients who developed myeloid neoplasms (MN) as a second cancer after surgical therapy of a primary tumor, we identified somatic mutations in TP53 and EZH2 likely related to cytotoxic therapy and radiation and compared them to those in primary cases. We further divided tMN types based on the derivation of founder mutations, into cases derived from antecedent or treatment-induced clonal hematopoiesis of indeterminate potential (CHIP) versus those to de novo driver mutations. Treatment-associated myeloid neoplasia are serious complications of cytotoxic therapies of primary cancers.1–3
Some tMN cases represent two coincidental malignancies, others may carry a germ line predisposition responsible for co-occurrence of more than one neoplastic process, or are synchronized by external or endogenous carcinogen exposures, and finally, others are truly causally linked to prior treatments. Patients who received only surgical treatment for a primary malignancy constitute the best control (second MN; sMN) to identify truly treatment-related molecular changes, distinguishing tMN from those MN without antecedent cancer (pMN). We aimed to identify mutations caused or selected by radiation (Rtx) or chemotherapy (Ctx). Furthermore, we compared mutational patterns of tMN to those found in CHIP to identify mutations created versus selected after iatrogenic exposures.
Blood and bone marrow samples were obtained from patients following informed consent in accordance with the procedures of the Cleveland Clinic Institutional Review Board and the Declaration of Helsinki. Commonly mutated genes in MN were sequenced and the data was processed using standard pipelines (Online Supplementary Table S1 and Online Supplementary Figures S1-S4). Variants were then annotated using Annovar and non-somatic lesions were excluded from further analysis. Variant allele frequencies (VAF) of mutations were adjusted to zygosity and single nucleotide polymorphism (SNP)-confirmed copy number, and analyzed by ranks or dichotomized as present or absent (Online Supplementary Table S2). Ancestral/founder and secondary/subclonal mutations were distinguished using published algorithms (Online Supplementary Materials and Online Supplementary Table S3). As follows: 1) in cases with serial samples, mutations appearing at second sampling but not the initial sampling were subclonal; 2) mutations with largest VAF were deemed ancestral (dominant); 3) mutations with VAF within 5% of the largest were co-dominant; and 4) those with >5% difference were categorized as subclonal. Co-dominant mutations were excluded from analyses to purify results. Patients with a history or Li-Fraumeni were excluded from our study cohort.
MN in patients who received prior cytotoxic treatments are heterogeneous and apart from a history of the primary cancer, only few features were distinctive to allow discrimination of these conditions on clinical grounds (Table 1). MN diagnoses were similar across pMN, sMN, and tMN (Online Supplementary Figure S5), and this held true for specific MDS, MDS/MPN, and acute myeloid leukemia (AML) subtypes (Online Supplementary Figure S6-8). However, notable differences included the absence of MPN and decreased frequency of MDS/MPN in tMN versus pMN (P<0.0001, P=0.0085, respectively). Within AML, inv(3) was 17 times more common in sMN versus pMN (P=0.033) and was never found in tMN (P=0.0509) and AML with mutated NPM1 was five times more common in pMN versus tMN (P=0.019). Using a set of unique control cohorts we investigated which somatic mutations are indeed the result of Rtx and Ctx.
Table 1.
Clinical characteristics of cohort.

Out of 266 tMN cases, 145 were sequenced (49 Ctx, 44 Rtx and 52 combination), while 65 of 109 sMN and all 683 pMN cases were sequenced (Online Supplementary Table S4-5).Complex karyotypes were two- to three-fold more common in tMN versus pMN and sMN (both P<0.001). Chromosome 5 and 7 aberrations were also more prevalent in tMN; 27% of tMN patients had - 7/del(7q) compared to 12% in pMN and 8% in sMN (both P<0.001). Normal cytogenetics were found in 30% of tMN cases (46% and 44% in pMN and sMN; P<0.001, P=0.0117; Table 1). The most frequently mutated genes in pMN, sMN, and tMN were TET2, DNMT3A, ASXL1 and SRSF2 (Figure 1A). Mutations in SF3B1 and JAK2 were less frequent in tMN, while those in KIT (P=0.016), WT1 (P=0.08), and EZH2 (P=0.0083) were more common. Notably, TP53 mutations were two to three times more likely in tMN than pMN and sMN (P=0.002, P=0.158; Figure 1B). Separate subset analysis revealed that ETV6 and EZH2 mutations associated with Rtx (P=0.046, P=0.004) and TP53 mutations with Ctx (P=0.617) and combination (P=0.0105; Figure 1E-G). Our analysis also noted the mutations more frequent in sMN including IDH1, which was 12 times more common in sMN versus tMN (P=0.015) and three times more common versus pMN (P=0.041; Figure 1F, Online Supplementary Table S6). Other mutations include: SRSF2, PHF6, and CUX1. These may be more related to the predisposition to develop a myeloid neoplasm or may be suppressed by cytotoxic therapy. When we compared tMDS versus tMDS/MPN versus tAML, FLT3 mutations were only observed in tAML (P=0.04), while CBL, EZH2, NRAS, and TET2 associated with overlap syndromes versus tMDS (P=0.0043, P=0.0156, P=0.0006, P=0.025, respectively). All other genes tested were not significantly different.
Figure 1.
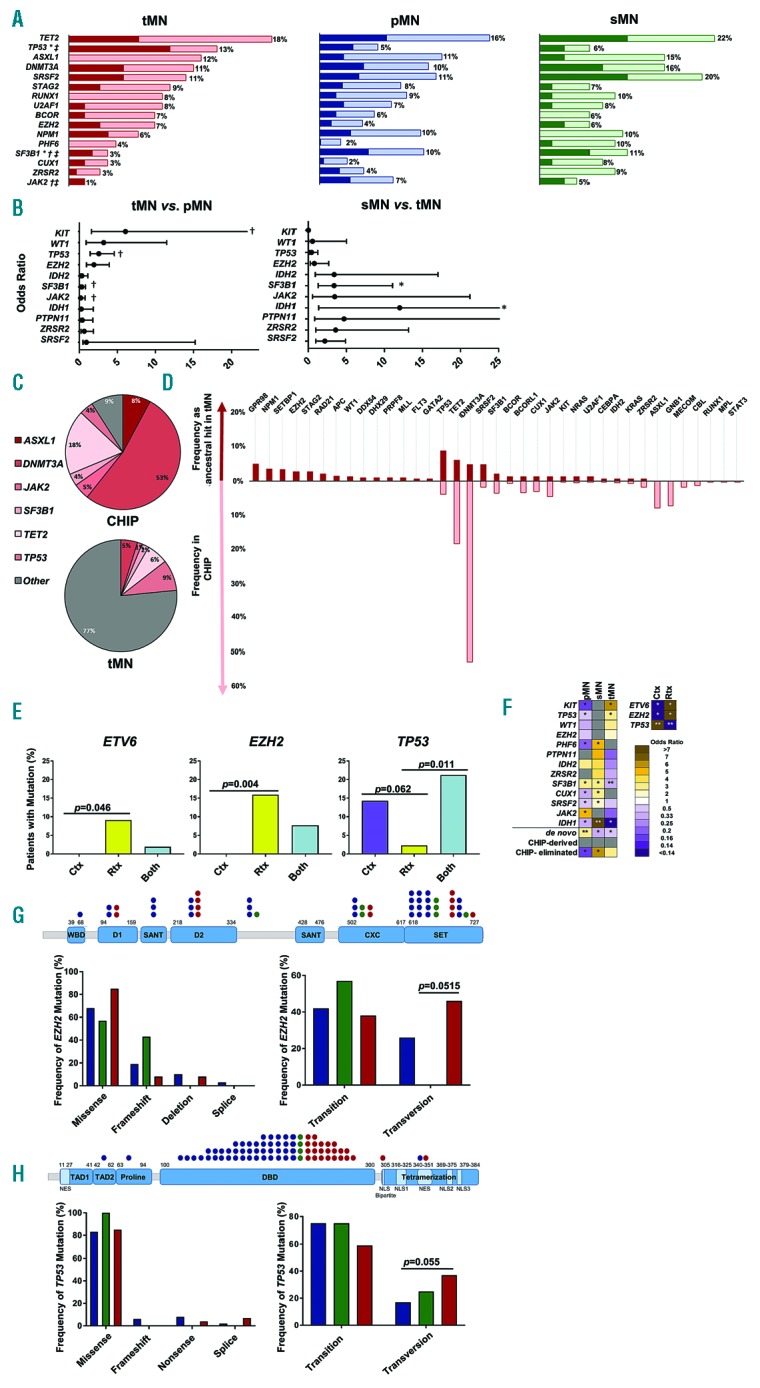
Mutational Landscape of therapy-related myeloid neoplasms. (A) Frequencies of the top 10 most common mutations observed. Dark red, blue, and green indicate ancestral events while light red, blue, and green represent secondary, subclonal events. *Denotes statistical significance between tMN and sMN; †denotes statistical significance between tMN and pMN; ‡denotes statistical significance between tMN and pMN+sMN. (B) Forest plot of odds ratios for mutations in tMN versus pMN and sMN versus tMN; †denotes statistical significance between tMN and pMN; *denotes statistical significance between tMN and sMN. (C) Relationship of common mutations found in CHIP versus ancestral mutations in tMN. (D) Frequencies of mutations occurring as ancestral events in tMN (maroon) versus frequencies found in CHIP based on meta-analysis (pink). (E) Effect of treatment modality on mutational acquisition. Frequencies of mutations found in Ctx, chemotherapy; Rtx, radiation; Both, combination of chemotherapy and radiation. (F) Heat map of events occurring in tMN, pMN, and sMN, expressed as odds ratios. Events include mutations, relationship to CHIP, and associations of treatment modality with mutations. Gray squares indicate opposing or no relationships between groups. Events marked with * are significant in one direction, while those marked with **are significant in two directions. (G) Effect of therapy on EZH2. WBD, WD-40 binding domain; D1: domain 1; SANT contains Switching-defecting protein 3 (Swi3), adaptor 2 (Ada2), nuclear receptor co-repressor (N-CoR), and transcription factor TFIIIB; D2: domain 2; CXC: cysteine-rich domain; and SET: the catalytic domain; MCSS: motif connecting SANT1L and SANT2L. Red, tMN; blue, pMN; green, sMN. (H) Effect of therapy on TP53. TAD1: transactivation 1 domain; TAD2: transactivation 2 domain; Proline: proline-rich domain; DBD: DNA-binding domain; Tetramerization: tetramerization domain; NES: nuclear export signal; NLS: nuclear localization signal. Red: tMN; blue: pMN; green: sMn. Only values of P<0.05 are considered significant.
Most TP53 mutations occurred in the DNA binding domain, however those in tMN were enriched in residues involved in nuclear trafficking, which correlated with poorer overall survival (Figure 1H; Online Supplementary Figure S9B). The majority EZH2 mutations were found in the SET domain, however those in tMN were enriched in domain two mutations (P=0.064), with no impact on survival (Figure 1G, Online Supplementary Figure S9A). In both cases, the distribution of alterations was similar in pMN (blue), sMN (green), and tMN (red); missense mutations were predominantly transitions, and included aging-associated C>T, with elevated transversions in tMN (Figure 1G-H). However, this had no effect on survival versus transitions (Online Supplementary Figure S9).
Mechanisms of transversion formation include 8-oxoguanine creation and subsequent mispairing with A instead of C and association with DNA-adduct forming carcinogens such as alkylating agents and topoisomerase II inhibitors.4–6 However, JAK2 V617F associated with background processes in pMN rather than therapies in tMN, suggesting that other transversion mechanisms may be involved in tMN. Although a totally unique signature of therapy was not found, the type of treatment used influenced the molecular signature of MN. While certain genes were commonly mutated regardless of preceding malignancy or therapy, notable mutational differences were also present in tMN. The site of mutation, as well as the mutation type, may be pertinent to tMN pathogenesis.
Our analysis of mutations typical for CHIP versus de novo founder mutations reveals a relationship between CHIP and cancer treatment (Figure 1C-D).7–10 The role of CHIP in aging is recapitulated in our analysis; patients without antecedent malignancy were younger than those with a history of cancer and had more de novo mutations, while those with prior cancers were older and had fewer de novo hits. Patients with CHIP-derived tMN were 6 years older than those with de novo tMN (P=0.019). This held true for the age of primary malignancy diagnosis, where CHIP-derived cases were 10 years older than de novo tMN (P=0.017; Online Supplementary Table S7).
Given CHIP’s prevalence, the types of mutations present in CHIP, and the distribution of these mutations in tMN, it is unlikely that tMN are all derived from preexisting CHIP. Interestingly, the proportion of CHIP-derived cases was similar in pMN, sMN, and tMN (Online Supplementary Figure S10-12) and within tMN disease subtypes of tMDS, tMDS/MPN, and tAML. This suggests that some ancestral CHIP mutations were eliminated by the dominant clone of the disease and that evolution to MN is selective, i.e. not all CHIP mutations are leukemogenic drivers; e.g. when mutations in GNB1, MPL, and STAT3 are present in the disease they are usually subclonal. In CHIP, 23% of such hits occur concomitant with mutations in TET2, DNMT3A, SF3B1, BCOR, and PTPN11 (Online Supplementary Figure S13).
Limitations of our analysis include the lack of serial samples from primary cancer diagnosis. Several groups reported on therapy-related CHIP, particularly on TP53 mutations at time of primary malignancy, prior to initiation of therapy.11–15 CHIP may precede Ctx/Rtx, which could accelerate the malignant progression of preexisting clones. Also observed, however, are CHIP-derived TP53 mutations, which disappeared after therapy and some de novo TP53 hits acquired after post-cytotoxic therapy CHIP.12,14 We estimate that approximately half of TP53-mutated tMN are CHIP-derived and our analysis suggests that therapies may accelerate progression of CHIP to tMN, but also may result in either in post-therapy CHIP or de novo non-CHIP hits. The situation for TP53 is further complicated by the presence of biallelic inactivation. We observed a two-fold increase in TP53 double mutants in tMN versus pMN and sMN (P=0.198) and have correlated the presence of two mutations versus one with poorer overall survival (Online Supplementary Figure S9B). Thus, if a preexisting heterozygous hit exists, either as germ line (Li-Fraumeni) or CHIP, systemic Ctx may increase the frequency of secondary deletions or biallelic mutations and generate selection pressure for pre-existing TP53 clones in blood or marrow. The presence of CHIP, primary malignancy, and therapeutic modality affect molecular lesions observed in tMN and further work is warranted to elucidate the role of a germ line predisposition.
Footnotes
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17(9):513–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shih AH, Rapaport F, Chung SS, et al. Mutation profiling of therapy-related myeloid neoplasms using next-generation sequencing demonstrates distinct profiles from de novo disease. Blood. 2014; 124(21):4611–4611. [Google Scholar]
- 3.Nishiyama T, Ishikawa Y, Kawashima N, et al. Mutation analysis of therapy-related myeloid neoplasms. Cancer Genet. 2018;222-223: 38–45. [DOI] [PubMed] [Google Scholar]
- 4.Ohno M, Sakumi K, Fukumura R, et al. 8-oxoguanine causes spontaneous de novo germline mutations in mice. Sci Rep. 2014;4:4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawley PD, Phillips DH. DNA adducts from chemotherapeutic agents. Mutat Res. 1996;355(1-2):13–40. [DOI] [PubMed] [Google Scholar]
- 7.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017; 377(2):111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zink F, Stacey SN, Norddahl GL, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130(6):742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coombs CC, Zehir A, Devlin SM, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017; 21(3):374–382.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong TN, Miller CA, Jotte MRM, et al. Cellular stressors contribute to the expansion of hematopoietic clones of varying leukemic potential. Nat Commun. 2018;9(1):455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi K, Wang F, Kantarjian H, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. 2017;18(1):100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coombs CC, Gillis NK, Tan X, et al. Identification of clonal hematopoiesis mutations in solid tumor patients undergoing unpaired next-generation sequencing assays. Clin Cancer Res. 2018; 24(23):5918–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]