

Abstract
The most common treatment for patients with sickle cell disease (SCD) is the chemotherapeutic hydroxyurea, a therapy with pleiotropic effects, including increasing fetal hemoglobin (HbF) in red blood cells and reducing adhesion of white blood cells to the vascular endothelium. Hydroxyurea has been proposed to mediate these effects through a mechanism of increasing cellular cGMP levels. An alternative path to increasing cGMP levels in these cells is through the use of phosphodiesterase-9 inhibitors that selectively inhibit cGMP hydrolysis and increase cellular cGMP levels. We have developed a novel, potent and selective phosphodiesterase-9 inhibitor (IMR-687) specifically for the treatment of SCD. IMR-687 increased cGMP and HbF in erythroid K562 and UT-7 cells and increased the percentage of HbF positive erythroid cells generated in vitro using a two-phase liquid culture of CD34+ progenitors from sickle cell blood or bone marrow. Oral daily dosing of IMR-687 in the Townes transgenic mouse SCD model, increased HbF and reduced red blood cell sickling, immune cell activation and microvascular stasis. The IMR-687 reduction in red blood cell sickling and immune cell activation was greater than that seen with physiological doses of hydroxyurea. In contrast to other described phosphodiesterase-9 inhibitors, IMR-687 did not accumulate in the central nervous system, where it would inhibit phosphodiesterase-9 in neurons, or alter rodent behavior. IMR-687 was not genotoxic or myelotoxic and did not impact fertility or fetal development in rodents. These data suggest that IMR-687 may offer a safe and effective oral alternative for hydroxyurea in the treatment of SCD.
Introduction
Sickle cell disease (SCD) is a genetic disease arising from a point mutation in the HBB gene that leads to the polymerization of hemoglobin S (HbS) during deoxygenation.1–5 HbS forms long chains of polymers that deform red blood cells (RBC) into a sickle shape, which impairs RBC transit in smaller blood vessels and renders them prone to hemolysis.6,7 Increased RBC lysis and release of free HbS scavenges nitric oxide (NO) and promotes vasoconstriction, which further alters vascular biology.8–10 This process in turn promotes the activation and mobilization of white blood cells (WBC), increasing their adhesiveness to activated endothelium.11–16 These pathological manifestations in RBC and WBC in SCD ultimately result in painful vaso-occlusive crises, end-organ damage, and, in many cases, premature death.17–19
Hydroxyurea (HU) was the first approved disease modifying therapy for SCD.20–24 HU was originally developed as a chemotherapeutic agent, and is believed to mitigate disease pathology and organ damage sequelae by increasing RBC expression of fetal hemoglobin (HbF) and reducing WBC counts.8,23,25–27 HU has been proposed to stimulate soluble guanyl cyclase, resulting in the elevation of cellular cGMP levels and activation of protein kinase G, which ultimately induces HbF expression.26 HU may also indirectly affect NO biology as a result of these activities, or directly increase NO levels. Despite its activity on multiple pathways that can improve SCD pathophysiology, HU is under-used in patients with SCD and often underdosed.28,29 Use of HU is challenged by responder effects and the careful safety monitoring required due to its myelosuppressive properties, and by concerns about toxicities, including HU impact on fertility and long-term carcinogenic potential.30–35 As a result of these risks, female and male patients are advised to discontinue HU therapy when trying to conceive or during pregnancy.
The cGMP specific phosphodiesterase 9 (PDE9) enzyme degrades cGMP and therefore PDE9 inhibitors (PDE9i) increase intracellular cGMP levels recapitulating the HbF induction mechanism of HU.36–38 PDE9 is highly expressed in erythropoietic cells, and is further elevated in neutrophils and reticulocytes from patients with SCD.39 A PDE9i originally developed for the treatment of neurodegenerative diseases (BAY73-6691) has been shown to increase HbF transcripts in K562 cells.38 BAY73-6691 also reduced WBC adhesion to endothelial cells, the adhesion of patient-derived neutrophils to immobilized fibronectin, leukocyte recruitment to the microvasculature, and, in conjunction with HU, it reduced the lethality of TNF-α induced vaso-occlusion in a mouse model of SCD.38,40
We describe here a novel, potent, and selective phosphodiesterase 9A inhibitor (IMR-687) that induced cGMP and HbF in the erythroid cell line K562 and increased HbF expression in erythroid cells derived from multiple SCD patients. In murine SCD models, IMR-687 increased plasma cGMP levels and HbF expression in RBC and impacted a number of disease-relevant features of SCD, including reducing lung inflammation, RBC sickling, and occlusion of micro-vessels. Furthermore, unlike PDE9i that was developed for neurodegenerative diseases, IMR-687 did not alter cognition in mice and, unlike HU, did not induce myelosuppression. In summary, IMR-687 demonstrated disease-relevant improvements in several aspects of SCD with comparable efficacy to HU.
Methods
Phosphodiesterase enzyme inhibition
Phosphodiesterase enzyme (PDE) inhibition IC50 values were determined for IMR-687 using recombinant human PDE enzymes in a radiometric assay.41
K562 and UT-7 erythroid cells
Human erythroleukemic K562 and UT-7 cells (American Type Culture Collection) were cultured as described in the Online Supplementary Methods. Terminal cell viability was determined by use of a trypan-blue exclusion technique (Thermo Fisher Scientific, France), ATP-based assays (Cell-Titer Glo, Promega), or automated cell counts (Countess Automated Cell Counter, Life Technologies). Apoptosis was assessed by Annexin V FACS assay (Biolegend).
Fetal hemoglobin quantification
K562 cells (5×106) supernatants were assayed using an ELISA kit for HbF (Cloud Clone Corp, CEA996Hu) (see Online Supplementary Methods). Permeabolized cells were stained with PE-mouse anti-human HbF and the percentage of HbF+ cells (% HbF) and the HbF levels (MFI) determined by flow cytometry (see Online Supplementary Methods).
Sickle cell disease patient cells
Blood was collected from five adult patients with severe SCD, aged 19-33 years (median age 32 years), admitted to the Biotherapy Department of Necker Hospital for an exchange transfusion. All samples used in this study were obtained from patients who signed informed consent forms approved by the ethical committee of Necker Hospital on 11th September 2015 (study IMNIS2015-01). CD34+ cells were were cultured in the presence of 15% BIT 9500 [mixture of bovine serum albumin (BSA) + insulin + transferrin from Stem Cell Technologies], 100 U/mL penicillin-streptomycin, 2 mM L-glutamine, 10 ng/mL recombinant human (rh) IL-3 (Peprotech), 100 ng/mL rhIL-6 (Peprotech), and 100 ng/mL rhSCF (Peprotech) for seven days and then CD36+ cells, isolated and cultured in media containing 100 ng/mL rhSCF, 10 ng/mL rhIL-3 and 2 UI/mL erythropoetin (Cilag, France) supplemented with dimethyl sulfoxide (DMSO), 30 μM HU or 10 μM IMR-687 for five days, at which point the HbF+ erythroid cells (LD−/GPA+/Band3+) was determined by FACS.
Animals
Townes model
HbSS-Townes mice42 on a 129/B6 background (Jackson Laboratory, Bar Harbor, ME, USA; 10-12 weeks old, n=7 per group) were dosed daily by gavage with vehicle (polyethylene glycol in water 1:3), 50 or 25 mg/kg of HU, or 30 mg/kg of IMR-687. On day 30, mice were anesthetized and blood counts, spleen weights, and plasma bilirubin, LDH, nitrite, HbF and free Hb determined (see Online Supplementary Methods). Lung homogenate myeloperoxide (MPO) and arginase were also determined (see Online Supplementary Methods).
Hemoglobin S-Townes vaso-occlusive crisis model
HbSS-Townes mice42 (6-17 weeks old, n=3 per group) were treated with vehicle (0.08% w/v methyl cellulose), 100 mg/kg of HU, 10 or 30 mg/kg of IMR-687, or 100 mg/kg HU + 30 mg/kg IMR-687 in their drinking water. On day 7 of treatment, the mice were implanted with dorsal skin-fold chambers (DSFC). Three days later, on day 10 of treatment, mice with DSFC were anesthetized, placed on a special intravital microscopy stage, and 2023 flowing subcutaneous venules in the DSFC window were selected and mapped. Mice were then placed in a hypoxic atmosphere chamber (7% O2/ 93% N2) for 1 hour (h), after which they were returned to room air. All the selected venules were re-examined after 1 and 4 h of re-oxygenation in room air, and the number of static (no flow) venules was counted and expressed as percent stasis. After this, mice were euthanized and plasma hematocrit, bilirubin, Hb and heme were measured and WBC, RBC, sickled RBC and HbF+ RBC quantified (see Online Supplementary Methods).
Results
Phosphodiesterase enzyme selectivity
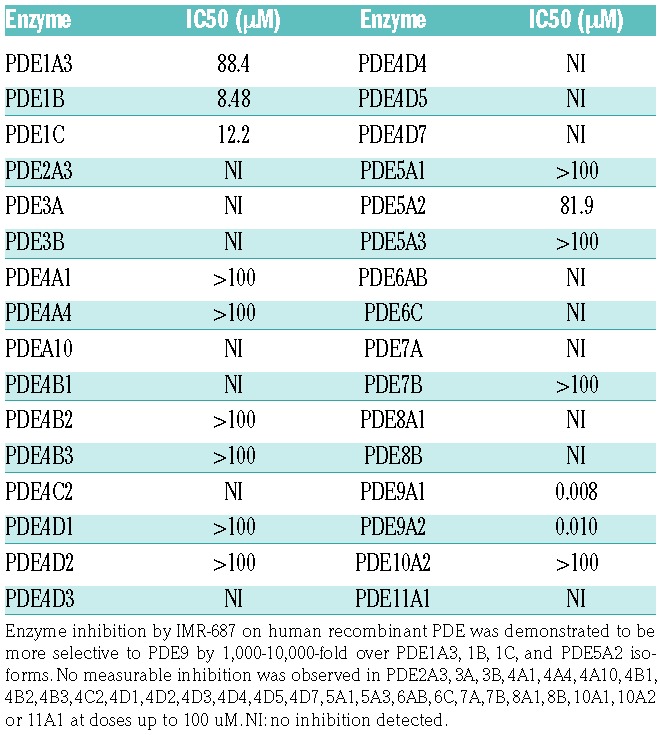
To determine the selectivity of IMR-687 for the phosphodiesterase 9A, 33 recombinant human PDE were incubated in vitro with increasing concentrations of IMR-687 and their activity determined. The IC50 of IMR-687 for PDE9A1 and PDE9A2 were 8.19 nM and 9.99 nM, respectively. IMR-687 inhibited PDE9A with more than 800-fold greater potency than PDE1A3, PDE1B, PDE1C, PDE5A2, with IC50 values of 88.4 μM, 8.48 μM, 12.2 μM, and 81.9 μM, respectively (Table 1). Significant inhibition of the other 27 PDE enzymes tested, including PDE4 and PDE10, was not observed (Table 1).
Table 1.
Phosphodiesterase enzyme (PDE) selectivity of IMR-687.
cGMP and fetal hemoglobin induction in erythroid cells
To determine if IMR-687 would increase cGMP levels in an erythroid cell line, actively growing K562 cells were cultured in media containing increasing concentrations of IMR-687 or HU. cGMP levels were assessed using a non-radioactive cGMP enzyme immunoassay (ENZO Life Sciences, France) with the acetylation protocol and protein levels were quantified by the BCA assay (Pierce, France). IMR-687 incubated for 6 h induced cGMP in a dosedependent manner at a dose that was well tolerated (Figure 1A).
Figure 1.

IMR-687 recapitulates the cGMP and fetal hemoglobin (HbF) induction mechanism of hydroxyurea (HU) in erythroid cells. (A) Phosphodiesterase 9A inhibitor (IMR-687) (triangles) and HU (squares) treatment for 6 hours (h) in erythroid K562 cells increases cGMP in a dose-dependent manner. (B) IMR-687 (triangles) and HU (squares) treatment in erythroid K562 cells for 72 h also induced HbF in a dose-dependent manner, evaluated by an ELISA assay using an antibody against human HbF. (C) HU (red symbols) demonstrates greater cytotoxicity than IMR-687 (black symbols) as assessed by cell counts at the end of a 72-h culture with each drug. *P<0.05; **P<0.01; ***P<0.001. Data are presented as means±Standard Errors.
Almeida et al. reported that exposure to the PDE9i BAY73-6691 and the sGC activator BAY 41-2271, increased HbF mRNA expression in K562 cells.38 To confirm this finding with IMR-687, actively growing K562 cells were exposed to increasing concentrations of IMR-687 or HU, and HbF expression was assessed by ELISA after 72 h. IMR-687 dose-dependently induced more HbF than either BAY73-6691 or BAY 41-2271 and was 4.6 times more potent at 10 μM than a dose of HU that demonstrated cytotoxicity (Figure 1B). Induction of HbF by IMR-687 was observed with the GM-CSF dependent erythroid line UT-7 (data not shown). While HU produced more HbF at higher concentrations, the induction was accompanied by cytotoxicity which was not observed with IMR-687 (Figure 1C).
Improved sickle cell disease phenotypes in vivo in murine model of sickle cell disease
We next tested the impact of IMR-687 and HU on F-cells, RBC sickling and markers of hemolysis in HbSS-Townes mice. After 30 days of treatment at 30 mg/kg/day of IMR-687, we observed a greater than 3-fold increase in the percent of HbF+ F-cells (8.4% in vehicle treated and 27.3% in IMR-687 treated; P<0.001) (Figure 2A) and a corresponding 2-fold decrease in sickled RBC (56.3% in vehicle treated and 24.4% in IMR-687 treated; P<0.0001) (Figure 2B). We saw a similar induction of HbF and reduction in sickled RBC with mice treated with HU doses of 100 mg/kg/day (29.3% F cells and 28.8% sickled RBC). This dose, which resulted in mortality in mice, was higher than the dose employed in patients. At HU doses that were tolerated in mice, the induction of HbF was modest and not significant compared to vehicle control (25 and 50 mg/mL/day increased F-cells to 13% and 18% compared to 8.4% for vehicle). There was a minimal decrease in the percent of sickled RBC with 25-50 mg/kg/day of HU compared to vehicle control (percentage of sickled RBC was decreased to 52% and 49%, respectively, compared to 56% for vehicle) (Figure 2A and B).
Figure 2.

Treatment of phosphodiesterase 9A inhibitor (IMR-687) in sickle mice for 30 days results in fetal hemoglobin (HbF) induction, reduced hemolysis and reduced reticulocytosis. Townes-HbSS mice were dosed orally for 30 days with IMR-687 at 30 mg/kg or hydroxyurea (HU) at 25, 50 or 100 mg/kg. Treatment with IMR-687, or HU at the highest dose resulted in an increase in HbF (A) in Ter-119+ red blood cells (RBC), reduction in the percentage of RBC with a sickle shape observed on blood smear (B), and a reduction in hemolysis as indicated by reduced plasma free hemoglobin (C), plasma bilirubin levels (D), and lactate dehydrogenase (LDH) levels (E), and indirectly with an increase in plasma nitrate levels (F) and reduction in lung arginase levels (G). Commensurate with these changes there was a reduction in evidence of reticulocytosis including reduced reticulocyte counts (H), increased mature RBC counts (I), increased hematocrit (J) and hemoglobin (K). Statistical significance was calculated for each agent and dose compared to a vehicle-treated control (n=7). *P<0.05; **P<0.01; ***P<0.001; ns: not significant (P>0.05). Data are presented as means±Standard Errors.
The significant reduction in the RBC sickling by IMR-687 produced a corresponding decrease in markers of hemolysis. This was seen in a reduction of free plasma Hb (Figure 2C) where IMR-687 reduced plasma free Hb levels over 55%. HU treatment also reduced free Hb levels in a dose-dependent fashion with the highest dose, 100 mg/kg, reducing levels by approximately 55%.
Consistent with the reduction in hemolysis and reduction in free Hb, plasma bilirubin levels and LDH activity, markers of hemolysis46 were significantly increased in vehicle treated SS mice compared to AA mice and reduced over 2-fold in IMR-687 treated mice (4.7 mg/dL, P<0.01 and 102 AU, P<0.05) (Figure 2D and E). The impact of the 100 mg/kg HU treatment was less pronounced, reducing bilirubin levels to 5 mg/dL (P<0.01) and LDH levels to 121 AU (not significant). HU dosed at 25 and 50 mg/kg did not produce a significant reduction in either marker of hemolysis.
Red cell lysis results in the release of Hb which consumes the plasma pool of NO and increases the vasculopathy associated with SCD7 nitrite generated in the plasma from an excess of NO produced by endothelial NO synthase (eNOS), can be converted back to NO as levels drop, acting as a biochemical reserve for NO.47 In HbSS-Townes mice, plasma nitrate levels are 41% lower than those in control AA mice (0.56 mg/mL vs. 0.95 mg/mL) (Figure 2F). Hemolysis results in the release of Hb and heme, which acts as a scavenger of NO. Treatment of SS mice with 30 mg/kg of IMR-687 increased plasma nitrite levels almost 2.5-fold to 1.39 mg/mL (P<0.05). HU in a dose-dependent manner increased nitrite levels as well, with a peak of 1.23 mg/mL in the 100 mg/kg dose group; however, these changes were not significant and were modest at therapeutic doses of HU. The difference in plasma nitrite levels in IMR-687 and 100 mg/kg HU treated mice were not significantly different.
Hemolysis also results in the release of arginase which reduces NO bioavailability and is correlated with SCD mortality.10 IMR-687 reduced lung arginase 25% (P<0.0001) (Figure 2G) compared to vehicle controls. This effect was less pronounced in the mice treated with 100 mg/kg of HU.
Reticulocytosis reflects the bone marrow’s response to anemia due to hemolysis. IMR-687 treated mice demonstrated significant changes in all measures of reticulocytosis including a 36% reduction in reticulocyte counts (Figure 2H), a 27% increase in mature RBC (Figure 2I), a 10% increase in hematocrit (Figure 2J), and a 1.5g/dL increase in Hb (Figure 2K). HU at a dose of 100 mg/kg produced smaller changes in reticulocyte counts, RBC, hematocrit and Hb; the changes in Hb were not significant. At HU doses of 25 and 50 mg/kg, only the change in reticulocyte counts was significant.
Townes mice have elevated circulating WBC counts, the major component of which are neutrophils. WBC were 36% lower in IMR-687 (24.1×109/L vs. 38.2×109/L; P<0.05) and 21% lower in 100 mg/kg HU treatment groups (30.4×109/L vs. 38.2×109/L) (Figure 3A). While the reduction in WBC with HU treatment can result from the myelotoxicity of HU, the IMR-687 reduction in peripheral WBC was not due to myelotoxicity as demonstrated in long-term toxicology studies conducted in normal rats (data not shown) and dogs (Figure 3B) treated with IMR-687 for up to 6 and 9 months, respectively. In these studies, super-physiological doses of IMR-687 did not result in any reduction in peripheral WBC counts. Furthermore, a histological examination of bone marrow smears from IMR-687-treated rats and dogs did not demonstrate any myelotoxicity (data not shown). This reduction in WBC counts with IMR-687 treatment likely reflects reduced WBC activation or mobilization in this sickle cell model.
Figure 3.

Treatment of phosphodiesterase 9A inhibitor (IMR-687) in sickle mice for 30 days results in reduced immune cell activity. White blood cell (WBC) counts are elevated in Townes-HbSS mice above normal controls (A). Townes-HbSS mice were dosed orally for 30 days with IMR-687 at 30 mg/kg or hydroxyurea (HU) at 100 mg/kg. Treatment with IMR-687 or HU reduced circulating WBC counts (A) (n=3). NS: not significant; *P>0.05. Data are presented as means±Standard Errors. This decrease in WBC counts is not seen in normal mice, rats or dogs dosed with IMR-687, including long-term 9-month toxicology studies in dogs dosed orally daily with 10, 25, or 50 mg/kg of IMR-687 (B). Along with the reduction in circulating WBC levels in IMR-687-treated Townes mice, there was a significant reduction in lung myeloperoxidase activity (C) (n=7). **P<0.01; ***P<0.001; ns: not significant (P>0.05). Data are presented as means±Standard Errors. MPO: myeloperoxide.
Not only were peripheral WBC counts increased in Townes mice, but soluble WBC-derived factors were elevated, including lung-associated myeloperoxidase (MPO), which is released by activated neutrophils, reduces plasma NO, and contributes to vascular damage.48 MPO levels were elevated over 5-fold in HbSS-Townes mice compared to control mice (3.1 mU/L vs. 0.57 mU/L in control mice; P<0.0001) (Figure 3C). MPO levels were reduced 2.3-fold in IMR-687-treated mice and 2.1-fold in 100 mg/kg HU-treated mice (1.3 mU/L and 1.5 mU/L, respectively; P<0.0001). Lower doses of HU also reduced MPO levels in the lungs.
Reduced vaso-occlusion in hemoglobin S-Townes mice
Occlusion of vessels by sickled RBC and adhesive WBC in SCD leads to multi-organ pathology. To assess the impact of IMR-687 on vessel occlusion, HbSS-Townes mice were exposed to 1 h of hypoxia (7% O2/ 93% N2) and the percentage of static venules (no blood flow) was quantified after return to normoxic conditions using a DSFC and intravital microscopy. After vehicle-treated mice were returned to normoxia, microvascular stasis was 33% and 16% after 1 h (Figure 4A) and 4 h (Figure 4B), respectively. Treatment with IMR 687 for ten days decreased stasis to 12% (P<0.01 vs. vehicle) and 7% (P<0.01) at 30 mg/kg/day and 20% (P<0.05) and 14% (ns) at 10 mg/kg/day after 1 h and 4 h in normoxia. Treatment of SS mice with HU at 100 mg/kg/day for ten days decreased microvascular stasis to 13% (P<0.05) and 8% (P<0.05) after 1 h and 4 h, respectively. When mice were given the combination of IMR 687 (30 mg/kg/day) and HU (100 mg/kg/day), stasis was 7% (P<0.01) and 4% (P<0.01) at 1 h and 4 h, respectively, suggesting a potential synergistic effect of the two agents.
Figure 4.

Treatment with phosphodiesterase 9A inhibitor (IMR-687) reduces vessel-occlusion in the Townes-HbSS sickle cell disease model. Townes-HbSS mice were dosed orally for 30 days with IMR-687 at 30 or 10 mg/kg or hydroxyurea (HU) at 100 mg/kg or 30 mg/kg IMR-687 in combination with 100 mg/kg HU. After ten days of treatment, animals were exposed to hypoxic conditions for quantification of microvessel occlusion via dorsal skin-fold chambers implanted on day 7 of treatment. On day 10 of treatment, 20-23 flowing venules in the chamber window were selected and mapped. Mice were then exposed to 1 h of hypoxia (7% O2) and then returned to room air. The same venules were re-examined at 1 h (A) and 4 h (B) post hypoxia for blood flow, and static (no flow) venules were counted and the data expressed as percent stasis. Data are presented as means±Standard Deviations. Statistical significance was calculated for each agent and dose compared to a vehicle-treated control. *P<0.05; **P<0.01; ns: not significant (P>0.05). Data are presented as means±Standard Errors.
Fetal hemoglobin induction in sickle cell disease patient erythroblasts
Erythroblasts were generated in vitro using two-phase liquid culture from CD34+ progenitors from nine SCD blood or bone marrow (SCD patients undergoing hip replacement for osteonecrosis) donors. These cells were treated with IMR-687 to determine if the drug could increase HbF expression in patient-derived erythrocytes.
F-cells were determined by their expression of HbF in the LiveDead-GPA+Band3+ population (Figure 5) by FACS. The mean for the DMSO control group (n=9) was 13.3% HbF positive. IMR-687 increased the percentage of F-cells to 21.9% (P<0.01, n=9). HU increased the percentage of F-cells to 22.2% (P<0.01, n=7, due to cytotoxicity induced by HU in 2 cultures). HU had a greater impact on the intensity of HbF staining in blood-derived CD34+ cells, increasing the MFI of the cells to 9744±2805 compared to 6073±1217 in control cells (P=0.041, n=7, due to cytotoxicity in 2 cultures), while IMR-687 significantly increased the MFI to 7813±1374 (P<0.01, n=9). This difference may be due in part to the greater cytotoxic stress of culturing the cells in 30 μM HU, evidenced by the loss of 2 of the 9 HU cultures.
Figure 5.

IMR-687 increases F-cells in patient-derived sickle cell disease (SCD) CD36+ cells. CD36+ cells derived from CD34+ adult SCD peripheral blood cells were cultured as described in the Methods section. Increase in the percentage of F-cells for each treatment is shown. Statistical significance was calculated for each agent compared to a vehicle-treated control (n=9). **P<0.01; ns: not significant (P>0.05). Errors are presented as Standard Error.
Phosphodiesterase 9 inhibitor IMR-687 demonstrated low central nervous system accumulation and did not alter behavior
Many PDE9i were originally developed for neurological diseases.49–55 In contrast, IMR-687 is a novel PDE9i selected specifically for low CNS exposure to reduce the potential impact of neuronal PDE9 inhibition on cognitive development and function. C57Bl/6J mice were dosed with IMR-687 at 10 mg/kg/day for five days or a CNS-active PDE9i, PF-04447943, originally developed for the treatment of neurological disorders. Plasma concentrations of the two PDE9i were very similar, while the brain exposure levels of IMR-687 were 5-fold lower than those seen with PF-04447943 (Figure 6A). Comparing the brain/plasma exposure profiles of the two drugs confirmed a very low concentration of IMR-687 in the CNS (7% brain/plasma ratio) compared to the PF-04447943, (41% brain/plasma ratio). Not unexpectedly, given its low brain exposure, IMR-687 showed no effect on locomotor activity or behavioral responses in toxicology studies (data not shown) nor in a classical fear conditioning mouse model of learning and memory (Figure 6B) (see Online Supplementary Methods). In contrast, the brain penetrant PF-04447943 significantly increased conditioned fear responses in mice at a similar dose. Besides confirming the lack of CNS activity of IMR-687, this finding suggests that brain-penetrant PDE9i treatment could trigger cognitive modulation of unknown consequences with chronic therapy.
Figure 6.

A brain-penetrant phosphodiesterase-9 inhibitor (PDE9i), but not IMR-687, increases fear responses in a model of learning and memory. (A) Fear conditioning responses are increased and persistent in mice treated with a brain-penetrant PDE9i compared to vehicle-treated or IMR-687-treated mice. (B) Drug exposure of the brain-penetrant PDE9i is 5-fold greater than that of IMR-687. Errors are presented as Standard Error. ns: not significant.
Discussion
Previous groups have described that reticulocytes and neutrophils from SCD patients express elevated levels of PDE9 and that exposure to a PDE9 inhibitor reduced the adhesive properties and extravagation of neutrophils in sickle cell models.38,39 They also reported the ability of this PDE9i to increase HbF mRNA levels in K562 cells. We describe a novel non-brain penetrant PDE9i, IMR-687, and its ability to increase HbF protein expression in human cell lines, patient-derived cells, and mouse models of SCD, and reduce many of the associated disease pathologies, including reduced RBC sickling and hemolysis, and normalization of WBC counts. Normalization of hemolysis is one of the major key improvements in SCD pathophysiology, having the potential to impact hemolytic-related complications. This is the first demonstration of the reduction in hemolysis by a PDE9i. IMR-687 treatment was also efficacious in a model of vasoocclusive crisis, preventing in vivo microvascular occlusion following a transient hypoxic insult. These effects were similar to the benefits seen with a high dose of HU, associated with mortality in the mouse model that was associated with some lethality in mice and cellular toxicity in vitro.
Hydroxyurea has been associated with activity in multiple pathways beyond cGMP, including cAMP, c-Jun kinases, epigenetic modification, and regulation of miRNA.56 It is, therefore, intriguing that many of the beneficial RBC and WBC effects of HU therapy in models of SCD are recapitulated by inhibitors to a PDE9 enzyme at daily doses that were safe and well tolerated. This suggests that an optimized dose of IMR-687 may be useful as a single agent therapy for SCD. That said, IMR-687 may also have a role in combination with low-dose HU in refractory patients. This may open the way for a new group of patients to see the full benefits of HU. Data in the Townes mouse model suggested that IMR-687 and HU together had an additive effect in reducing vasoocclusion. This effect did not seem to be mediated by an additive effect on induction of HbF or reduction in RBC sickling. It may have been through an additive effect in NO modulation; this remains hypothetical, although not unexpected, given the robust reduction in hemolysis seen with IMR-687 which would reduce the release of heme, an NO scavenger. Clinically, IMR-687 is being tested in adult SCD patients both as a solo therapy and in those taking HU.
IMR-687 was purposefully developed for SCD, selected not only for its potency and selectivity, but also its low brain exposure to avoid concerns about modulating cognitive function, especially in children with SCD. The data presented in this report indicate that, in the context of SCD models, IMR-687 has many of the beneficial in vitro and in vivo properties of HU without its attendant toxicities. Furthermore, many of the positive changes associated with HU are sufficiently recapitulated by selective targeting of the PDE9 pathway, which acts through increases in cGM, culminates in increased HbF and ameliorates RBC pathology. This offers significant advantages over drugs that increase cGMP systemically, impacting cells that are not necessarily suitable targets, and mediating side effects such as hypotension. The clinical development of a safe, well-tolerated, orally available drug like IMR-687, with low CNS exposure, acting through the PDE9 pathway, may offer an improved single treatment option for patients living with SCD. In the light of these findings, clinical studies are underway to determine if IMR-687 might offer a safe, well-tolerated and efficacious alternative to HU therapy for SCD patients.
Acknowledgments
PDE selectivity assays were performed by SB Drug Discovery (Glasgow, UK). We thank Dr. Michael Dussiot for his assistance on Amnis Imaging Flow Cytometer experiment and data analysis.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/105/3/623
References
- 1.Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature.1956;178(4537):792–794. [DOI] [PubMed] [Google Scholar]
- 2.Bertles JF, Rabinowitz R, Dobler J. Hemoglobin interaction: modification of solid phase composition in the sickling phenomenon. Science.1970;169(3943):375–377. [DOI] [PubMed] [Google Scholar]
- 3.Ross PD, Hofrichter J, Eaton WA. Thermodynamics of gelation of sickle cell deoxyhemoglobin. J Mol Biol.1977;115(2):111–134. [DOI] [PubMed] [Google Scholar]
- 4.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med.1994;330(23):1639–1644. [DOI] [PubMed] [Google Scholar]
- 5.Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet.2017;390(10091):311–323. [DOI] [PubMed] [Google Scholar]
- 6.Berger SA, King WS. The flow of sickle-cell blood in the capillaries. Biophys J.1980; 29(1):119–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev.2007;21(1):37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gladwin MT, Schechter AN, Ognibene FP, et al. Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation.2003;107(2):271–278. [DOI] [PubMed] [Google Scholar]
- 9.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med.2002;8(12):1383–1389. [DOI] [PubMed] [Google Scholar]
- 10.Morris CR, Kato GJ, Poljakovic M, et al. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA.2005;294(1):81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vasoocclusion. Blood.2000;96(7):2451–2459. [PubMed] [Google Scholar]
- 12.Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N Engl J Med.1980;302(18):992–995. [DOI] [PubMed] [Google Scholar]
- 13.Hebbel RP, Yamada O, Moldow CF, Jacob HS, White JG, Eaton JW. Abnormal adherence of sickle erythrocytes to cultured vascular endothelium: possible mechanism for microvascular occlusion in sickle cell disease. J Clin Invest.1980;65(1):154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A.2002;99(5):3047–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zennadi R, Chien A, Xu K, Batchvarova M, Telen MJ. Sickle red cells induce adhesion of lymphocytes and monocytes to endothelium. Blood.2008;112(8):3474–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol. 2002;9(2):101–106. [DOI] [PubMed] [Google Scholar]
- 17.Nath KA, Hebbel RP. Sickle cell disease: renal manifestations and mechanisms. Nat Rev Nephrol.2015;11(3):161–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thornburg CD, Dixon N, Burgett S, et al. A pilot study of hydroxyurea to prevent chronic organ damage in young children with sickle cell anemia. Pediatr Blood Cancer.2009;52(5):609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassim AA, DeBaun MR. Sickle cell disease, vasculopathy, and therapeutics. Annu Rev Med.2013;64:451–466. [DOI] [PubMed] [Google Scholar]
- 20.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med.1995;332(20):1317–1322. [DOI] [PubMed] [Google Scholar]
- 21.Heeney MM, Ware RE. Hydroxyurea for children with sickle cell disease. Pediatr Clin North Am.2008;55(2):483–501, x. [DOI] [PubMed] [Google Scholar]
- 22.Maier-Redelsperger M, de Montalembert M, Flahault A, et al. Fetal hemoglobin and F-cell responses to long-term hydroxyurea treatment in young sickle cell patients. The French Study Group on Sickle Cell Disease. Blood.1998;91(12):4472–4479. [PubMed] [Google Scholar]
- 23.Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest.1984; 74(2):652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet.2011;377(9778):1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erard F, Dean A, Schechter AN. Inhibitors of cell division reversibly modify hemoglobin concentration in human erythroleukemia K562 cells. Blood.1981; 58(6):1236–1239. [PubMed] [Google Scholar]
- 26.Cokic VP, Smith RD, Beleslin-Cokic BB, et al. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J Clin Invest.2003;111(2):231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Odievre MH, Bony V, Benkerrou M, et al. Modulation of erythroid adhesion receptor expression by hydroxyurea in children with sickle cell disease. Haematologica.2008;93(4):502–510. [DOI] [PubMed] [Google Scholar]
- 28.Stettler N, McKiernan CM, Melin CQ, Adejoro OO, Walczak NB. Proportion of adults with sickle cell anemia and pain crises receiving hydroxyurea. JAMA.2015; 313(16):1671–1672. [DOI] [PubMed] [Google Scholar]
- 29.Adams-Graves P, Bronte-Jordan L. Recent treatment guidelines for managing adult patients with sickle cell disease: challenges in access to care, social issues, and adherence. Expert Rev Hematol.2016;9(6):541–552. [DOI] [PubMed] [Google Scholar]
- 30.Sheehan VA, Luo Z, Flanagan JM, et al. Genetic modifiers of sickle cell anemia in the BABY HUG cohort: influence on laboratory and clinical phenotypes. Am J Hematol.2013;88(7):571–576. [DOI] [PubMed] [Google Scholar]
- 31.Steinberg MH. Determinants of fetal hemoglobin response to hydroxyurea. Semin Hematol.1997;34(3 Suppl 3):8–14. [PubMed] [Google Scholar]
- 32.Dover GJ, Humphries RK, Moore JG, et al. Hydroxyurea induction of hemoglobin F production in sickle cell disease: relationship between cytotoxicity and F cell production. Blood.1986;67(3):735–738. [PubMed] [Google Scholar]
- 33.Lanzkron S, Haywood C, Jr., Hassell KL, Rand C. Provider barriers to hydroxyurea use in adults with sickle cell disease: a survey of the Sickle Cell Disease Adult Provider Network. J Natl Med Assoc.2008; 100(8):968–973. [PubMed] [Google Scholar]
- 34.Meyappan JD, Lampl M, Hsu LL. Parents’ assessment of risk in sickle cell disease treatment with hydroxyurea. J Pediatr Hematol Oncol.2005;27(12):644–650. [DOI] [PubMed] [Google Scholar]
- 35.Berthaut I, Guignedoux G, Kirsch-Noir F, et al. Influence of sickle cell disease and treatment with hydroxyurea on sperm parameters and fertility of human males. Haematologica.2008;93(7):988–993. [DOI] [PubMed] [Google Scholar]
- 36.Soderling SH, Bayuga SJ, Beavo JA. Identification and characterization of a novel family of cyclic nucleotide phosphodiesterases. J Biol Chem.1998; 273(25): 15553–15558. [DOI] [PubMed] [Google Scholar]
- 37.Soderling SH, Bayuga SJ, Beavo JA. Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc Natl Acad Sci U S A.1998;95(15):8991–8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Almeida CB, Scheiermann C, Jang JE, et al. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood.2012;120(14):2879–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Almeida CB, Traina F, Lanaro C, et al. High expression of the cGMP-specific phosphodiesterase, PDE9A, in sickle cell disease (SCD) and the effects of its inhibition in erythroid cells and SCD neutrophils. Br J Haematol.2008;142(5):836–844. [DOI] [PubMed] [Google Scholar]
- 40.Miguel LI, Almeida CB, Traina F, et al. Inhibition of phosphodiesterase 9A reduces cytokine-stimulated in vitro adhesion of neutrophils from sickle cell anemia individuals. Inflamm Res.2011;60(7):633–642. [DOI] [PubMed] [Google Scholar]
- 41.Thompson WJ, Appleman MM. Multiple cyclic nucleotide phosphodiesterase activities from rat brain. Biochemistry.1971; 10(2):311–316. [PubMed] [Google Scholar]
- 42.Ryan TM, Townes TM, Reilly MP, et al. Human sickle hemoglobin in transgenic mice. Science.1990;247(4942):566–568. [DOI] [PubMed] [Google Scholar]
- 43.EC L. Peripheral Blood Smear. In: Walker HKHW, Hurst JW, ed. Clinical Methods: The History, Physical, and Laboratory Examinations. Boston: Butterworths; 1990. [PubMed] [Google Scholar]
- 44.Kleihauer E, Braun H, Betke K. [Demonstration of fetal hemoglobin in erythrocytes of a blood smear]. Klin Wochenschr.1957;35(12):637–638. [DOI] [PubMed] [Google Scholar]
- 45.Fairbanks VF, Ziesmer SC, O’Brien PC. Methods for measuring plasma hemoglobin in micromolar concentration compared. Clin Chem.1992;38(1):132–140. [PubMed] [Google Scholar]
- 46.Kato GJ, McGowan V, Machado RF, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg ulceration, pulmonary hypertension, and death in patients with sickle cell disease. Blood.2006;107(6):2279–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dezfulian C, Raat N, Shiva S, Gladwin MT. Role of the anion nitrite in ischemia-reperfusion cytoprotection and therapeutics. Cardiovasc Res.2007;75(2):327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H, Xu H, Weihrauch D, et al. Inhibition of myeloperoxidase decreases vascular oxidative stress and increases vasodilatation in sickle cell disease mice. J Lipid Res.2013;54(11):3009–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.da Silva FH, Pereira MN, Franco-Penteado CF, De Nucci G, Antunes E, Claudino MA. Phosphodiesterase-9 (PDE9) inhibition with BAY 73-6691 increases corpus cavernosum relaxations mediated by nitric oxide-cyclic GMP pathway in mice. Int J Impot Res.2013;25(2):69–73. [DOI] [PubMed] [Google Scholar]
- 50.Hutson PH, Finger EN, Magliaro BC, et al. The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943 (6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-py ran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one) enhances synaptic plasticity and cognitive function in rodents. Neuropharmacology. 2011;61(4):665–676. [DOI] [PubMed] [Google Scholar]
- 51.Vardigan JD, Converso A, Hutson PH, Uslaner JM. The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943 attenuates a scopolamine-induced deficit in a novel rodent attention task. J Neurogenet.2011;25(4):120–126. [DOI] [PubMed] [Google Scholar]
- 52.van der Staay FJ, Rutten K, Barfacker L, et al. The novel selective PDE9 inhibitor BAY 73-6691 improves learning and memory in rodents. Neuropharmacology.2008;55(5): 908–918. [DOI] [PubMed] [Google Scholar]
- 53.Prickaerts J, Heckman PRA, Blokland A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin Investig Drugs.2017;26(9):1033–1048. [DOI] [PubMed] [Google Scholar]
- 54.Heckman PR, Wouters C, Prickaerts J. Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer’s disease: a translational overview. Curr Pharm Des.2015;21(3):317–331. [DOI] [PubMed] [Google Scholar]
- 55.Saavedra A, Giralt A, Arumi H, Alberch J, Perez-Navarro E. Regulation of hippocampal cGMP levels as a candidate to treat cognitive deficits in Huntington’s disease. PLoS One.2013;8(9):e73664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pule GD, Mowla S, Novitzky N, Wiysonge CS, Wonkam A. A systematic review of known mechanisms of hydroxyurea-induced fetal hemoglobin for treatment of sickle cell disease. Expert Rev Hematol.2015;8(5):669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]