


Abstract
Expansion of an unstable CTG repeat in the 3′UTR of the DMPK gene causes Myotonic Dystrophy type 1 (DM1). CUG-expanded DMPK transcripts (CUGexp) sequester Muscleblind-like (MBNL) alternative splicing regulators in ribonuclear inclusions (foci), leading to abnormalities in RNA processing and splicing. To alleviate the burden of CUGexp, we tested therapeutic approach utilizing antisense oligonucleotides (AONs)-mediated DMPK splice-switching and degradation of mutated pre-mRNA. Experimental design involved: (i) skipping of selected constitutive exons to induce frameshifting and decay of toxic mRNAs by an RNA surveillance mechanism, and (ii) exclusion of the alternative exon 15 (e15) carrying CUGexp from DMPK mRNA. While first strategy failed to stimulate DMPK mRNA decay, exclusion of e15 enhanced DMPK nuclear export but triggered accumulation of potentially harmful spliced out pre-mRNA fragment containing CUGexp. Neutralization of this fragment with antisense gapmers complementary to intronic sequences preceding e15 failed to diminish DM1-specific spliceopathy due to AONs’ chemistry-related toxicity. However, intronic gapmers alone reduced the level of DMPK mRNA and mitigated DM1-related cellular phenotypes including spliceopathy and nuclear foci. Thus, a combination of the correct chemistry and experimental approach should be carefully considered to design a safe AON-based therapeutic strategy for DM1.
INTRODUCTION
Myotonic Dystrophy type 1 (DM1) is an RNA-dominant disease caused by expansion of a CTG repeat in the 3′ untranslated region (3′UTR) of the DMPK gene (1). CUG-expanded DMPK mRNAs (CUGexp) form ribonuclear inclusions (foci) which interact with poly(CUG) binding proteins such as the alternative splicing regulators from the Muscleblind-like (MBNL) family (2–5). Nuclear retention of MBNL in foci perturbs the intricate balance between distinct splicing regulators, leading to abnormalities in RNA processing, metabolism and splicing. Hyperphosphorylation and stabilization of MBNLs antagonist, CUGBP Elav-like (CELF1) splicing regulator (6), further worsens alternative splicing aberrations (spliceopathy) observed in DM1 (7). DM1-related spliceopathy displays as a shift from adult-to-embryonic mRNA isoforms which, upon translation, generate proteins inept for performing proper functions in the adult organism, ultimately leading to pathological hallmarks of DM1 such as myotonia, muscle weakness and wasting (8).
Among many approaches explored to alleviate CUGexp-related toxicity, antisense oligonucleotide (AON)-based strategies were shown particularly successful in reversing the molecular hallmarks of DM1 (recently reviewed in (9)). For example, steric blocking AONs including a morpholino-based anti-CUG oligomer (10) and short locked-nucleic acid (LNA)-based oligonucleotides complementary to the repeats (11) were demonstrated to efficiently bind CUGexp in vivo and preclude MBNLs sequestration without inducing a significant degradation of the toxic RNA. Conversely, other data supports the feasibility of post-transcriptional silencing of toxic RNAs with chemically modified AONs targeting either CUGexp directly (12–16), or DMPK sequences located outside of the repeat tract (16–19) (recently reviewed in (9,20)). While the most commonly exploited mechanism for degradation of toxic transcripts in DM1 is the recruitment of diverse gapmer-based AONs compatible with RNase H endonuclease-dependent RNA cleavage (13,14,16–19), AONs promoting degradation of toxic RNAs via other unidentified yet mechanisms have been reported (12,15). Recently, an interesting approach involving the recruitment of endogenous RNA surveillance pathway was used in a DM1-unrelated study that exploited rationally designed AONs for steric inhibition of distinct exonic splicing enhancer regions (ESE) within a constitutive STAT3 exon (21). Here, ESE inhibition prompted exon skipping from mRNA and caused a frameshift error leading to premature termination codon (PTC) and further elimination of the transcript via nonsense-mediated mRNA decay (NMD), a cytoplasmic translation-dependent process preventing mis-spliced mRNAs from producing potentially toxic proteins (21,22). This strategy could be potentially used in DM1 therapy, however, reduction of DMPK mRNA may raise concerns as the exact contribution of DMPK haploinsufficiency to DM1 pathophysiology has not been established conclusively. Reports show reduction of baseline DMPK protein levels by half in DM1 patients (23), or only a slight decline in DMPK in severely affected congenital patients (24). Likewise, no adverse effects upon wild-type DMPK allele knock-down was recently demonstrated in mice (25), despite earlier reports linking genetic disruption of Dmpk with skeletal myopathy (26) and cardiac conduction defects (27).
Importantly, DMPK transcripts are subject to extensive cell-type specific alternative splicing, and imbalance in the splice isoform profile of DMPK was suggested to play pivotal role in the pathogenesis of DM1 (28,29). While splice modulating strategies, such as AON-forced exon skipping, have been extensively used as therapeutic approaches towards other neuromuscular diseases like Duchenne muscular dystrophy and spinal muscular atrophy (recently reviewed in (30,31)), DMPK splice-switching has not been tested so far as a viable therapeutic option against DM1. Interestingly, downstream of the CUG-repeat region in e15 of DMPK, is a rarely used cryptic 3′ splice acceptor site (3′SA) present only in humans, which defines a new terminal exon termed e16 (32). The use of 3′SA results in the excision of a large 5′ part portion of e15, including the CUG-repeat region, leading to translation of an alternative DMPK protein isoform with a unique C-terminal tail (32). Naturally occurring expression of these e16-splice isoforms in various skeletal muscles is estimated at 10–15% of total DMPK mRNA (32). Most importantly, e16-containing DMPK mRNAs, in contrast to CUGexp-containing transcripts, are free from nuclear retention defect and may be present in relatively large amounts in the cytosol of cells from DM1 patients (32). However, no follow-up studies have been yet conducted to fully understand the significance and contribution of this rare alternative splicing event to DM1 pathophysiology and test whether and how it could be used for therapeutic advantage against DM1.
Here, we investigated whether novel approaches utilizing AON-mediated DMPK splice-switching and post-transcriptional reduction of mutated DMPK pre-mRNA have the potential of reducing the overall burden of CUGexp-induced toxicity. For this purpose, we rationally designed two AON-based splice-switching strategies involving skipping of selected constitutive DMPK exons in order to induce frameshift error and decay of toxic mRNAs (strategy 1) and exclusion of the alternative e15 carrying CUGexp (strategy 2). We hypothesized that the resultant mutant DMPK reduction (strategy 1) or CUGexp skipping (strategy 2) could release MBNLs from toxic sequestration, induce foci dispersal, rescue the DMPK translational defect and ameliorate DM1-related spliceopathy. We tested these ideas in a well-established and physiologically relevant cell model consisting of DM1-patients’ derived fibroblasts harboring distinct lengths of CTG-repeat expansions, in which correction of spliceopathy and foci numbers as well as mutant DMPK expression can be readily measured (11,33).
MATERIALS AND METHODS
ESE analysis and AON design
AON used in this study are listed in Supplementary Table S1. Putative ESE motifs within DMPK e5, e9 and e15 were predicted using ESEfinder 3.0 (34,35). AONs targeting e5, e9 and e15 were designed based on exonic regions with highest ESE density and presence of high-score motifs (Supplementary Figure S1, Supplementary Table S2), and were synthesized as uniformly modified 2′-O-methyl oligomers with a fully phosphorothioated backbone (2′-Ome-PS) (RiboTask ApS, FutureSynthesis Sp. z o.o.) or locked nucleic acid oligomers with a fully phosphorothioated backbone (LNA-PS) (Exiqon/Qiagen), except for one AON targeting e5 which was synthesized as 2′-Ome for control experiments (RiboTask ApS). Antisense gapmers targeting DMPK intron 14 (Antisense LNATM GapmeRs, Exiqon/Qiagen) were designed using Antisense GapmeR Designer tool (Exiqon/Qiagen) and all contain LNA flanking regions (the position of LNA modifications is proprietary to Exiqon/Qiagen), central DNA gap and a fully phosphorothioated backbone. Negative control gapmer (Gap ctrl (–)) was a custom predesigned Antisense LNA™ GapmeR (Exiqon/Qiagen). Positive control gapmer (Gap ctrl (+) CAG 14) was described previously (14) and purchased from Exiqon/Qiagen.
Cell culture and transfection of AONs
The following cell lines were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research: GM07492 - human primary fibroblasts from unaffected individual (non-DM1; control); GM04033 and GM03989 - from DM1 probands expressing mutant DMPK transcript containing 1,000 and 2,000 CUG repeats, respectively (DM1-1000CUG and DM1-2000CUG, respectively). Cells were grown in a high glucose EMEM (Lonza) supplemented with 20% fetal bovine serum, 1% antibiotic-antimycotic (Gibco) and 1% non-essential amino acids solution (Sigma), in a 37°C humidified incubator containing 5% CO2. Indicated AON amounts were transfected at ∼80% cell confluency using Lipofectamine™ 2000 Transfection Reagent (Invitrogen) per manufacturer's instructions. For combined treatment with splice-switchers and gapmers, both types of AONs were delivered subsequently using lipofectamine, with a 12 h lag time in between the two transfections. All molecular analyses were performed at indicated time-points post transfection (48–96 h) and mock samples refer to lipofectamine treated cells.
RT-PCR splicing assays and qPCR gene expression analyses
RNA was isolated at indicated time-points following AON treatment using TRI reagent (Sigma-Aldrich) per the manufacturer's recommended protocol. cDNA was synthesized using GoScript™ Reverse Transcriptase (Promega) with Random Primers (Promega) per manufacturer's protocol. Exon skipping and alternative splicing assays were analyzed by standard RT-PCR reactions using GoTaq Flexi DNA Polymerase kit (Promega). The PCR products were separated in a 0.5–2% agarose gel (depending on the amplicon size) stained with 0.5 μg/ml ethidium bromide. PCR images were captured using G:Box EF2 (Syngene) and analyzed using GeneTools image analysis software (Syngene). Efficiency of AON-mediated exon skipping (Δexon) and the strength of splicing changes in the alternative exons of DM1 biomarker transcripts (PSI), respectively, were calculated according to the formula: Δexon or PSI = (b*100)/(a + b), where Δexon is the % exon skipping, PSI the % alternative exon inclusion (which in the case of analyzed exons represents adult-to-embryonic splicing shift characteristic for DM1), a is the intensity of the PCR product with exon inclusion and b is the intensity of the PCR product with exon exclusion. Results of splicing assays were plotted on bar graphs. For standard RT-PCR gene expression analyses results were related to GAPDH expression. Real-time quantitative PCR analyses (qPCR) were performed in a 7900 HT Fast Real-Time PCR System (Applied Biosystems) using Power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufactures’ instructions. Quantitative PCR reactions were performed using at least three independent cDNAs per sample type, each in three technical replicates. Raw Ct data were analyzed in Microsoft Excel using 2−ΔΔCt method. Results represent a relative expression of the analyzed target gene compared to an indicated reference gene, and normalized to averaged control/calibrator samples. All PCR primers are listed in Supplementary Table S3.
Immunoblotting
Protein isolation and western blotting were performed as described previously (36), using the following primary antibodies: polyclonal rabbit anti-human DMPK antibody recognizing an epitope within the N-terminal region between aa11–39 (LS-C167452, LifeSpan BioSciences, Inc.), rabbit anti-human MBNL1 (a kind gift from C. Thornton; University of Rochester Medical Center), mouse anti-CUGBP1, clone 3B1 (Merck-Millipore) and mouse anti-human GAPDH antibody (sc-47724; Santa Cruz). DMPK antibody was validated using a DMPK blocking peptide (LS-E4669, LifeSpan BioSciences, Inc.).
Luciferase constructs and luciferase assay
Luciferase vectors pmirGLO-DT960 and pmirGLO-DMPKs were constructed by subcloning a NheI digested fragment of DT960 (37) or DMPKs (38) plasmids (a gift from Thomas Cooper) spanning human DMPK exons 11–15 with either 960 interrupted CTG repeats or no repeats at all, respectively, into an MCS within the 3′UTR of firefly luciferase in a pmirGLO vector backbone. Positive clones and correct insert orientation were verified by restriction digestion and sequencing. For luciferase assay, simian Cos7 cells were plated at a high confluency in 96-well plates and transfected with 25–50 ng luciferase plasmids using X-tremeGENE™ HP DNA Transfection Reagent (Roche) per manufacturer's instructions. Indicated amounts of splice-switching AONs or gapmers were transfected 12 h later using Lipofectamine™ 2000 Transfection Reagent (Invitrogen) per manufacturer's instructions. Luciferase assay was performed 48 h post last transfection using Dual-Glo® Luciferase Assay System (Promega) per manufacturer's protocol. Mock refers to transfection reagent treated samples.
Fluorescent in situ hybridization
The RNA fluorescent in situ hybridization (FISH) was performed using DNA/LNA probe (CAG)6-CA, labeled at the 5′-end with Cy-3 as previously described (11). Microscopic slides were mounted using VECTASHIELD Antifade Mounting Medium with DAPI (Vector laboratories) and sealed with a clear nail polish. The quantitation of RNA foci was performed using standard fluorescence microscope and 60× oil immersion objective, by visual inspection of fluorescent spots present in cell nuclei. Foci were counted in at least 200 nuclei per experimental sample and each experiment was repeated at least twice to verify reproducibility of the results. Mock (lipofectamine-treated) samples were used for reference.
Statistical analyses
Group data are expressed as mean ± standard deviation (error bars). Statistical significance was determined by a two-tailed Student's t-test (Microsoft Excel) by comparing average results from two to three independent experiments to results obtained for mock or ctrl-AON (scrambled sequence) treated samples (*P < 0.05; **P < 0.01 and ***P < 0.001). All experiments involved two to three independent biological replicates per sample type and were repeated at least twice to verify reproducibility of the results.
RESULTS
Experimental strategies and designing of AONs
In splice-switching strategy 1 (Figure 1A), we intended to shift the open reading frame (ORF) of DMPK transcripts by AON-induced skipping of internal constitutive exons, and subsequently induce a PTC in DMPK mRNA, thus increasing the likelihood of transcript decay via RNA surveillance mechanism NMD. For this purpose, we designed steric-blocking splice-switching antisense RNA oligomers fully modified with 2′-O-methyl (2′-Ome) sugar moieties and phosphorothioate (PS) linkages (2′-Ome-PS) that target putative ESEs within specific internal constitutive DMPK exons (Supplementary Figure S1, Supplementary Tables S1 and S2). Target exons e5 (AONs 1–5), encoding part of the kinase domain of the protein, and e9 (AON-6), encoding part of the α-helical coiled-coil domain of the protein, were chosen based on a prediction that exclusion of either one of them from mature DMPK mRNA would shift the ORF and generate a PTC in e7 or e10, respectively, in agreement with the 55-nt NMD rule (39) and hypothetically leading to degradation of e5- and e9-deficient and frameshifted mRNAs.
Figure 1.

Experimental strategies for AON-mediated DMPK splice-switching. (A, B) Schematic outline of human DMPK pre-mRNA (not to scale) and two distinct AON-based strategies for testing therapeutic potential of DMPK splice-switching. (C, D) Representative RT-PCR analyses of AON-induced e5 (C) and e15 (D) skipping in control fibroblasts (non-DM1) transfected with indicated amounts of 2′-Ome-PS AONs targeting respective exons. Two lanes (C) or three lanes (D) represent distinct samples, while %Δe5 and %Δe15 indicate percentages of e5 or e15 spliced out, respectively. Schematics of distinct PCR amplicons and primer locations are indicated. Gel images shown in C and D were cropped to show only selected samples. In D, note the appearance of several e16-containing (Δe15/ΔCUG) splice isoforms differing in the alternative inclusion of e13 and e14. Chromatograms presented in panel D show sequencing results across indicated splice-junctions (vertical red dashed line) of three distinct e16-containing DMPK splice products.
In splice-switching strategy 2 (Figure 1B), we targeted AONs to putative ESEs within DMPK’s last coding alternative e15 (AONs 8–10) harboring 3′UTR with the CUG-repeat expansion region (Supplementary Figure S1, Supplementary Table S1–2). Based on previous reports (32), we predicted that AON-mediated inhibition of e15 inclusion could redirect the splicing from exons preceding e15 to a cryptic alternative 3′SA located downstream of the CUG-repeat region. This would result in DMPK mRNAs lacking the entire 5′ part of e15 including a 3′UTR fragment harboring pathogenic CUG expansion (referred to as e15/CUG), but containing novel terminal e16 devoid of the repeat region (referred to as Δe15/ΔCUG). We hypothesized that these two approaches could reduce DMPK mRNA (strategy 1), mitigate its nuclear retention and translational defect due to CUGexp (strategy 2), and reduce nuclear foci leading to MBNLs release and spliceopathy correction (both strategies).
Splice-switching AONs effectively induce DMPK exon skipping
We first tested the ability of distinct AONs to induce constitutive exon skipping in primary fibroblasts derived from control (unaffected; non-DM1) as well as DM1-affected patients harboring distinct lengths of CTG-repeat expansion (Figure 1C). RT-PCR analyses with primers flanking targeted exons revealed efficient skipping of e5 and e9 in both control and DM1 cells dosed with increasing AON amounts, while no skipping was induced by control AONs with scrambled sequences (Figure 1C, Supplementary Figure S2A–E, S3A). AONs targeting e5 within the middle region (AON-4) and at the 3′ exon/intron boundary (AON-5) induced the most efficient e5 exclusion (Supplementary Figure S2A). For e9, only one AON targeting ESE region proximal to 3′ exon/intron boundary was tested (Supplementary Figure S2D and E). The rate of e5 and e9 skipping was higher in unaffected vs DM1 cells and ranged from ∼30% upon 25 nM AONs to ∼40% (in the case of e9) or ∼60% (in the case of e5) upon 125 nM AONs (Figure 1C, Supplementary Figure S2B–E). Importantly, neither further increment of AONs dose up to 250 nM (upon solo treatment) nor combined treatment with both e5- and e9-directed AONs enhanced the exon skipping rates (Supplementary Figure S2B–D, F). Lower exon skipping upon high AON amounts resulted most likely from increased cell mortality, as judged by microscopic evaluation of cells. Importantly, locked nucleic acid oligomers with a fully phosphorothioated backbone (LNA-PS), which were equivalents of the most efficient 2′-Ome-PS AONs (LNA-PS: +AON-4 and +AON-6 vs 2′-Ome-PS: AON-4 and AON-6, as listed in Supplementary Table S1), did not compromise cell viability as judged by microscopic observations. In agreement, cell death and cytotoxicity were elevated in control fibroblasts treated with 2′-Ome-PS but not equivalent 2′-Ome or LNA-PS AONs targeting the same core sequence within e5 (Supplementary Figure S3C). However, LNA-PS equivalents induced much weaker constitutive exon skipping compared to 2′-Ome-PS AONs (Supplementary Figure S3A). Noteworthy, majority of oligomers tested in our study have high GC content (Supplementary Table S1) and may be prone to self-pairing, especially when modified with high-affinity modifications such as LNA, which could in part explain the poorer activity of LNA-modified AONs in our exon skipping experiments. Therefore, AONs with the 2′-Ome-PS chemical modification were used in subsequent experiments at doses inducing efficient skipping, but no detrimental effects on cell viability (50–75 nM).
We next tested the ability of splice-switching AONs to exclude CUG-expansions from mature DMPK mRNA by skipping of the alternative e15 (strategy 2, Figure 1B). Two out of five tested 2′-Ome-PS AONs, namely AON-8 and AON-10 targeting the 5′-most one-third and 3′-most one-third of e15, respectively, were able to induce a significant splice-shift in fibroblast (Figure 1D, Supplementary Figures S1, S2G, Supplementary Table S1). Using PCR primers located within e11 and cryptic e16, we detected several DMPK splice isoforms devoid of the entire 5′ part of e15 including the CUG-repeat tract, but containing novel terminal e16 (Δe15/ΔCUG) (Figure 1D). These isoforms included previously described e14-e16 splice isoform (32) as well as two isoforms in which either e13 or e12 were directly joined to e16. The identity of these three distinct splice isoforms was confirmed by sequencing of the PCR products across the splice junctions (Figure 1D). The overall efficiency of AON-induced e15 vs e16 splice-switching was similar (∼80%) in control and DM1-affected cells (Figure 1D, Supplementary Figure S2G), however, like with e5 and e9, LNA-PS AONs prompted weaker splice-switch compared to their 2′-Ome-PS equivalents (Supplementary Figure S3B).
AON-mediated skipping of constitutive exons fails to reduce DMPK mRNA levels
Having confirmed AONs efficiency, we next examined how strategy 1 affected the overall expression of DMPK mRNA and protein. Quantitation of total DMPK mRNA in control and DM1 fibroblast demonstrated lack of significant transcript decay, despite very efficient e5 and e9 skipping (Figure 1C, Supplementary Figure S2C, Figure 2A and Supplementary Figure S4A, B). As expected, treatment with e5 or e9 directed AONs reduced expression of DMPK splice isoforms containing e5 or e9, respectively (Supplementary Figure S4A), however, total DMPK mRNA level remained unaffected regardless of the analyzed RT-PCR amplicon (3′UTR vs internal mRNA fragment spanning e9–e10 or e5–e7) (Supplementary Figure S4A). Although, a trend in the overall DMPK protein level reduction was observed upon treatment with AONs targeting e5 and e9, it was not statistically significant (Figure 2B) and no truncated protein isoforms were detected (Mw of truncated proteins: ∼24-25 kDa w/o e5, ∼43-45 kDa w/o e9) (whole-gel scan in Supplementary Figure S4C). Summarizing, despite efficient skipping of constitutive DMPK exons, experimental strategy 1 failed to induce DMPK mRNA decay as initially predicted.
Figure 2.
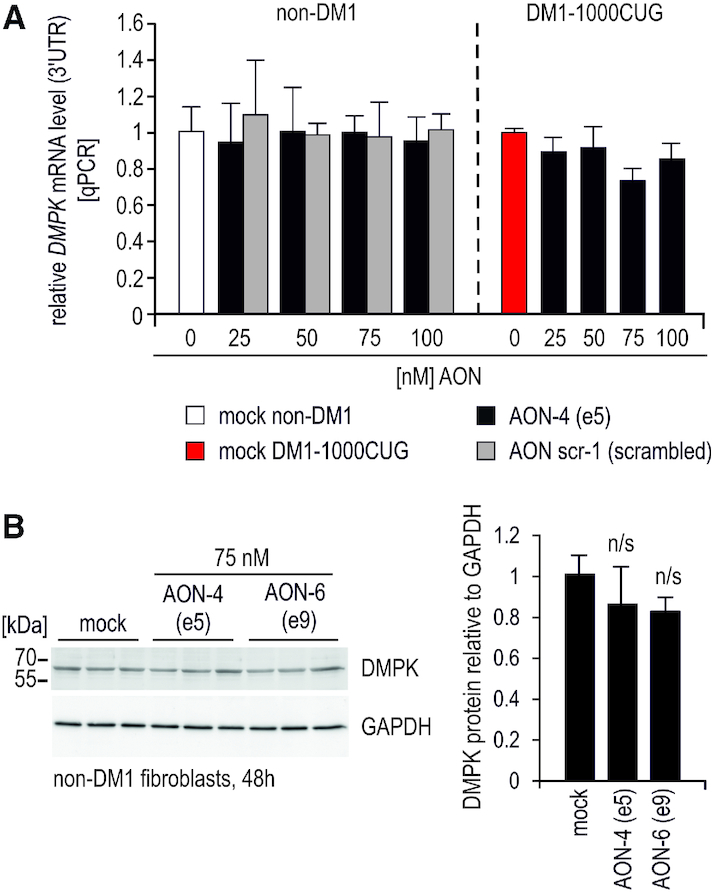
AON-mediated skipping of constitutive exons fails to reduce DMPK RNA. (A) Quantitation of total DMPK mRNA by qPCR (real-time PCR) expression analyses of the 3′UTR fragment demonstrates lack of significant DMPK transcript reduction upon e5 skipping in control and DM1-affected fibroblasts. DMPK expression is shown relative to GAPDH, increasing doses of AON targeting e5 (AON-4), control scrambled sequence AON (scr-1) are indicated. (B) Representative results of western blot analyses showing DMPK protein level in control fibroblasts dosed with 75 nM of indicated AONs. Three lanes indicate individual samples. Quantitation is shown to the right. Whole-membrane scan of DMPK western blot is shown in Supplementary Figure S4C.
Skipping of the alternative e15 results in accumulation of excised pre-mRNA fragment containing CUG-expansions
We next examined how e15 exclusion (strategy 2) affected the levels of total DMPK mRNA as well as spliced-out pre-mRNA fragment consisting of intron 14 (i14) and the entire 5′ part of e15 including CUG-repeat region (referred to as i14/e15/CUG; Figure 1B). While the overall level of DMPK transcript was unaltered by AON-8-mediated e15/CUG skipping in DM1 fibroblasts, control experiments with gapmer AONs complementary to i14 (Gap i14-1 and Gap i14-2) reduced DMPK transcript by half, even upon prior e15 splice-switching (Figure 3A, Supplementary Figure S5B and C). Intriguingly, RNA reduction was not accompanied by DMPK protein downregulation even after long incubation with gapmers up to 84 h (Figure 3B) and 96 h (Supplementary Figure S5D), which might suggest high protein stability. Because potential therapeutic effect of AONs could be masked by excised pre-mRNA fragment containing CUG expansion (depending on the half-life after splicing), we examined its level upon AON-8-mediated e15 splice-switching. Interestingly, significant accumulation of i14/e15/CUG was evident in DM1 fibroblasts treated with AON-8, but not after dosing the cells with scrambled control AON (Figure 3C). Importantly, intronic gapmers efficiently neutralized this effect as i14/e15/CUG pre-mRNA accumulation in AON-8 treated cells was almost completely abrogated by sequential treatment with either of the i14 gapmers (Figure 3C; compare green vs checked bars). Our results also indicated that in DM1 cells intronic gapmers can effectively degrade pre-mRNA DMPK transcripts as well as the spliced-out fragment containing expanded CUG-repeats (Figure 3A and C, compare red vs violet and green vs checked bars).
Figure 3.

AON-mediated skipping of e15 triggers accumulation of spliced out pre-mRNA fragment containing CUG-expansions and mitigates nuclear retention of DMPK. (A) RT-PCR quantitation of total DMPK mRNA in DM1-1000CUG fibroblasts 72 h post transfection with indicated AONs (PCR amplicon within 3′UTR). Mock, scrambled sequence AON (scr-1) and gapmer AONs were used as controls and representative RT-PCR image is shown below bar graph. (B) Representative immunoblots showing no changes in DMPK protein expression in DM1 fibroblasts 48 h (upper gel) and 84 h (bottom gel) post AON dosing. (C) qPCR quantitation of i14/e15/CUG spliced-out pre-mRNA fragment vs total DMPK pre-mRNA in DM1-1000CUG fibroblasts 72 h after dosing the cells with e15 splice-switching AON-8 either alone or in a combination with i14-1 and i14-2 gapmer AONs targeting distinct regions of i14. Scrambled-sequence AON scr-1 and gapmer AONs were used as controls. i14/e15/CUG pre-mRNA was amplified with primers located within i14 and e15 (upstream of CUG-repeat region), primers for DMPK pre-mRNA amplification were located within i9 and e10. (D) Outline and results of the firefly luciferase reporter assay modeling the DM1 translational defect. Luciferase reporter constructs (schematic) with a 3′UTR harboring genomic DMPK fragment spanning e11–e15 along with entire 3′UTR carrying either expanded (pmirGLO-DT960) or no repeats (pmirGLO-DMPKs) were transfected into Cos7 cells and dosed with indicated AONs. Mock and scr-1 were used as controls. Horizontal dashed lines indicate luciferase expression level in control and mutant mock treated samples.
Skipping of e15/CUG improves nuclear export of DMPK transcripts
Because CUG-expansions are known to compromise nuclear export of mutant DMPK mRNAs, we next tested how e15/CUG skipping affected nuclear retention of DMPK transcripts. For this purpose, we designed two luciferase reporters harboring in their 3′UTR a genomic DMPK fragment spanning e11–e15 and the entire 3′UTR sequence with either expanded (mutant) or no CTG repeats (control) (Figure 3D). Luciferase assay in Cos7 cells transiently transfected with these reporters confirmed that the presence of CUGexp in the 3′UTR of firefly luciferase reduced nucleocytoplasmic transport of the mutant transcript, as expected, and thereby suppressed luciferase translation compared to control reporters with no repeats, as shown in control experiments (mock- and scrambled AON-treated cells) (Figure 3D). Exclusion of e15 from mature luciferase transcript via AON-8 and AON-10 significantly improved luciferase production and unexpectedly, this was evident in both mutant and control reporters (Figure 3D). This result suggests that the sole presence of CUG-repeat flanking region has an inhibitory role in DMPK nuclear export and translation, whereas CUG-repeated sequence might have an additive effect (Figure 3D; compare green and red bars in AON-treated samples vs mock samples). Importantly, gapmers alone almost entirely switched off luciferase translation, confirming high efficiency of intron-directed gapmers in reducing DMPK regardless of the presence of CUG-expansion (Figure 3D). Taken together, these data strongly support therapeutic potential of AON-mediated e15/CUG exclusion in mitigating DM1-related DMPK mRNA nuclear retention and translational defect.
DM1 spliceopathy can be reversed by gapmer-mediated degradation of DMPK pre-mRNA, but not by AON-induced DMPK splice-switching
To further explore therapeutic potential of splice-switching AONs targeting e15, we examined how AON-8, either alone or combined with i14 gapmers, affects the alternative splicing of known DM1 biomarker transcripts in DM1-patients’ derived fibroblasts. These biomarkers included MBNL1-regulated alternative exons of MBNL1 – e1 (36,40) and e5 (40–46), MBNL2 – e7 and NCOR2 (nuclear receptor co-repressor 2) – e47a (47). Expectedly, i14 gapmer-mediated DMPK pre-mRNA reduction mitigated DM1 related spliceopathy of all four tested biomarkers in DM1–1000CUG fibroblasts (Figure 4A-B). This therapeutic effect was modest yet evident in the case of MBNL1 e5, MBNL2 e7 and NCOR2 e45a, but very significant in the case of MBNL1 e1, whose exon inclusion shifted back to the levels observed in control fibroblasts (Figure 4A-B). Unexpectedly, however, dosing DM1 fibroblasts with e15-directed AON-8 as well as control scrambled sequence-AON scr-1 consistently aggravated the already pre-existing spliceopathy in all of the tested transcripts (Figure 4B, compare red versus green and gray bars), while exposure of cells to AON-8 prior to i14 gapmers transfection completely prevented the therapeutic effect of gapmers (Figure 4, checked bars). Interestingly, DM1-spliceopathy was consistently enhanced in DM1 fibroblasts dosed with AONs targeting e5/e9 and additional control AON scr-2 (Supplementary Figure S6). Moreover, DM1-like splicing aberrations were evident in control cells regardless of the AON-targeted sequence, and failed to diminish over time up to 72 h post AON treatment (Supplementary Figures S6 and S7). These data suggested that aggravation of DM1-like alternative splicing defects was unlikely caused by DMPK pre-mRNA splicing manipulation or by oligomer sequence alone, but rather due to AON chemistry‐dependent toxicity. In support of this hypothesis, no alternative splicing abnormalities were detected in analyzed exons upon e5 or e15 skipping induced with LNA-PS-modified AONs targeting the same core sequence as their 2′-Ome-PS equivalents (Supplementary Figure S7, Supplementary Table S1).
Figure 4.
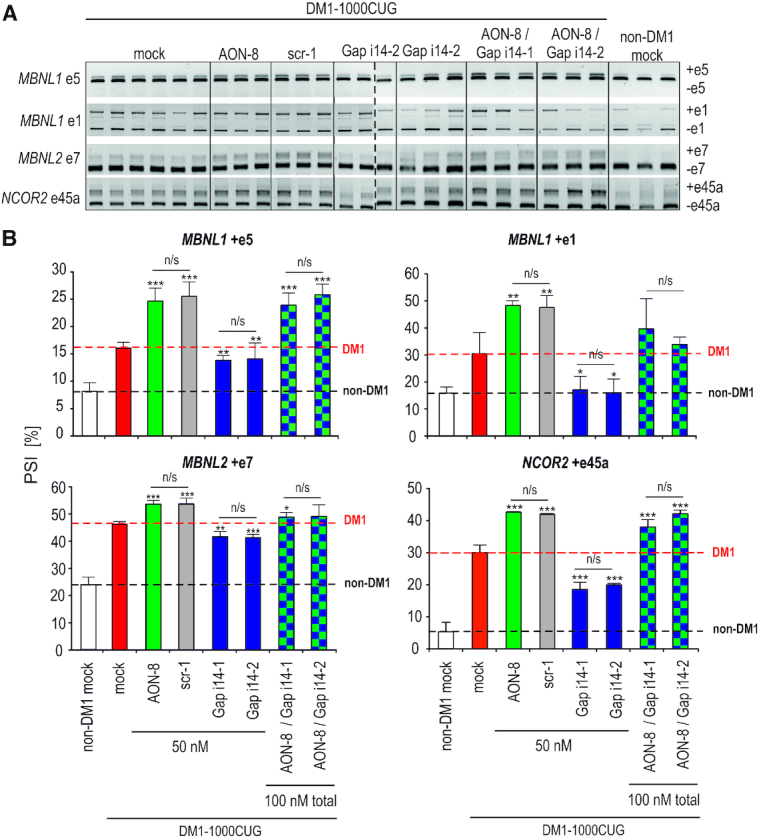
DM1 spliceopathy can be reversed by gapmer-mediated degradation of DMPK pre-mRNA, but not by AON-induced skipping of e15/CUG. (A) Representative RT-PCR products showing splicing changes in indicated alternative exons of MBNL1, MBNL2 and NCOR2 in DM1-1000CUG fibroblasts, 72 h post transfection with indicated reagents. Each lane represents individual biological sample and vertical dashed line marks boundaries of two electrophoretic separations. (B) Quantitation of splicing changes based on three distinct RT-PCR analyses of indicated alternative exons in DM1-1000CUG fibroblasts transfected with indicated amounts of e15 splice-switching AON-8 (green), scrambled control AON scr-1 (gray), gapmers Gap i14-1 and Gap i14-2 targeting distinct sequences within DMPK i14 (violet) or combination of gapmers and AON-8 (checked). Mock samples from unaffected (white) and DM1-affected (red) fibroblasts are shown for reference. Black horizontal dashed line marks the percentage of alternative exon inclusion in non-DM1 mock controls, while red dashed line – in untreated DM1-1000CUG mock controls. Statistical significance relative to DM1 mock samples as well as between indicated experimental samples is indicated. PSI refers to % exon spliced in (DM1-like shift).
To explore the basis for the aberrant splicing observed upon 2′-Ome-PS AONs, we analyzed the expression of two antagonistic splicing regulators implicated in DM1 pathogenesis, namely MBNL1 and CELF1 (Figure 5). Intriguingly, the level of both proteins was significantly upregulated in control fibroblasts dosed with AON targeting e5 (Figure 5A). Although the ∼40% CELF1 protein increase was paralleled by ∼40% elevated mRNA content (Figure 5A, C), the MBNL1 mRNA level was unaltered (data not shown). We observed, however, a ∼40% higher inclusion of e1 into MBNL1 mRNA, which could alone explain the observed ∼40% rise in MBNL1 protein (Figure 5C). This is in agreement with our recent observations demonstrating autoregulation of MBNL1 expression via e1 repression (36,40). Similar results were observed in a DM1 cell model (Figure 5B, Figure 4B). However, while CELF1 protein reached even higher levels than in non-DM1 cells, MBNL1 content was only slightly elevated (Figure 5B). The latter could be explained by higher basal levels of e1 in non-treated DM1 cells (Figure 4B, compare white vs red bars in MBNL1 e1 splicing quantification) and the resulting lower impact of AON addition on MBNL1 splicing (compare e1 splicing in Figure 4B, red vs green bar, and Figure 5C) and protein level (Figure 5B). Taken together, we conclude that DM1-like splicing aberrations upon 2′-Ome-PS AONs are likely triggered by toxicity related to their chemical modification, and not by their specific sequence, and these events correlate with altered expression and/or activity of two antagonistic splicing regulators implicated in DM1, MBNL1 and CELF1.
Figure 5.
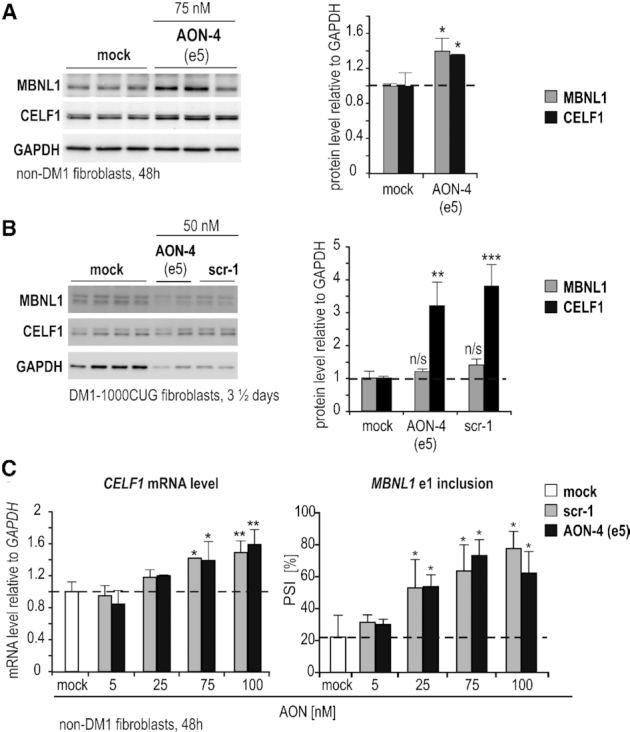
2′-Ome-PS-induced splicing abnormalities correlate with aberrant expression and splicing of MBNL1 and CELF1. (A, B) Representative immunoblot analyses of MBNL1 and CELF1 protein level relative to GAPDH in DM1-unaffected (A) or DM1-1000CUG (B) fibroblasts transfected with indicated amounts of distinct 2′-Ome-PS AONs targeting DMPK e5. Mock (lipofectamine) and scrambled AON (scr-1) were used as controls. Each lane represents an individual sample. Quantitation is shown to the right. Horizontal dashed line marks the protein level in untreated controls. (C) RT-PCR quantitation of CELF1 total mRNA and the alternative splicing of MBNL1 e1 in non-DM1 fibroblasts transfected with indicated amounts of 2′-Ome-PS AONs. AON scr-1 was used as a control. Horizontal dashed lines mark the percentage of CELF1 level or MBNL1 e1 inclusion in mock samples, respectively. Statistical significance relative to mock is indicated.
Reduction of DMPK pre-mRNA diminishes CUGexp foci number
Finally, we tested whether DMPK splicing manipulation and pre-mRNA reduction affect CUGexp foci formation. FISH analysis comparing nuclear foci in DM1 fibroblasts transfected with distinct splice-switching AONs revealed no differences in cumulative foci numbers or foci distribution upon skipping of constitutive e5 or e9 (Figure 6A). This result was not unexpected considering lack of significant DMPK mRNA reduction upon e5 or e9 exclusion. Similarly, skipping of e15/CUG failed to reduce nuclear foci numbers most likely due to accumulation of the excised pre-mRNA fragment containing expanded repeats (Figure 6B, black dotted line). However, additional treatment with intronic gapmer significantly reduced foci numbers compared to AON-only treated cells (Figure 6B, black line). This is most likely due to gapmer-mediated reduction of overall DMPK pre-mRNA, as demonstrated in Figure 3A and Supplementary Figure S6. The most significant reduction in total foci numbers was evident upon solo gapmer treatment (Figure 6B, violet line), however, analyses of foci number distribution in distinct nuclei indicated that both gapmer alone as well as AON/gapmer-treated cells contained less nuclei with 3–5 foci, and more nuclei with single focus or no foci at all, compared to mock, scramble or AON-only treated cells (data not shown), further corroborating therapeutic potential of gapmers directed at intronic DMPK sequences.
Figure 6.

Targeting DMPK with intronic gapmers, but not splice-switching AONs, diminishes CUGexp foci numbers in DM1 fibroblasts. Representative FISH analyses of CUGexp foci in DM1-1000CUG fibroblasts upon AON-induced skipping of DMPK e5 or e9 (A), and upon skipping of e15/CUG either alone or in a combination with Gap i14-1 treatment (B). Scrambled sequence AONs (scr-1 and scr-2) were used as controls. Statistical significance is indicated in the legend.
DISCUSSION
In this work, we investigated whether antisense oligomer-mediated DMPK pre-mRNA splice-switching can ameliorate the CUGexp-related molecular phenotypes of DM1, including nuclear foci formation, spliceopathy and DMPK translational defect. Using well characterized and physiologically relevant cell model consisting of DM1-patients’ derived fibroblasts, we tested two novel experimental strategies exploiting AON splice-switchers to induce either DMPK mRNA frame-shifting followed by decay (strategy 1), or CUG-repeat region skipping (strategy 2). Importantly, DMPK splicing approach has not been tested before in therapeutic context despite extensive literature data on DMPK splice isoforms and their suggested role in DM1 pathogenesis.
Both splice-switching strategies, although fairly efficient in control as well as in DM1-fibroblasts, were unsuccessful in considerably improving molecular hallmarks of DM1. While constitutive exon skipping failed to stimulate DMPK mRNA decay at all, exclusion of the alternative e15 carrying CUGexp was able to rescue only the nuclear export and translation of DMPK in an artificial luciferase reporter system, but triggered persistent accumulation of potentially harmful spliced out pre-mRNA fragment containing i14, e15 and CUG expansion region. This pre-mRNA fragment could be ‘neutralized’ by i14-directed gapmers and most importantly, intronic gapmers alone were able to reverse major molecular hallmarks of DM1 like ribonuclear foci accumulation, spliceopathy and DMPK translational defect. Notably, while majority of AON-based approaches for DM1 reported to-date targeted antisense oligomers either to the repeated CUG-sequence or to exonic and 3′UTR sequences, none tested the potential of intron-targeted AONs for DMPK transcript reduction. Noteworthy, antisense gapmers have been previously tested in animal models of DM1 where they have shown beneficial effects in DMPK mRNA reduction, muscle histology and reversal of aberrant splicing without detectable toxicity (17–19).
We observed that gapmer AONs were generally more effective compared to splice-switchers. This difference might be determined by AONs’ mode of action (RNase H1 recruitment vs steric blocking of splice machinery and competing with splicing factors like SRSF proteins, respectively) but also their chemistries (2′-OmePS versus LNA-PS/DNA-PS), length, cellular uptake efficiency as well as intracellular routing (nucleus or cytoplasm). Also, while design of splice-switchers is constrained by ESE motif location within targeted exons, numerous designs are possible for gapmers depending on optimal sequence or secondary structure to ensure target accessibility and high potency. Our data also revealed that the exclusion of alternative e15 was more potent compared to constitutive e5 and e9 and in general, exon skipping was more effective in control than DM1-affected fibroblasts. These differences might stem from distinct location of targeted exons within the pre-mRNA (internal constitutive exons vs 3′ proximal alternative exon) and their accessibility to AONs, as well as distinct subcellular localizations and accessibility of non-expanded vs mutated DMPK transcripts (i.e. RNA foci), respectively. Nonetheless, despite fairly efficient splice-switching in DM1 fibroblasts, no effect on DMPK mRNA reduction and no truncated DMPK proteins were observed with strategy 1. In agreement with the 55-nt rule of the NMD (22,48), skipping of e5 and e9 in our approach was predicted to induce PTCs more than 100 nt upstream of the 3′-most exon-exon junction (within e7 and e10, respectively) therefore it could hypothetically induce NMD of misprocessed DMPK mRNAs. Currently, we cannot explain why these misprocessed mRNAs failed to be degraded. One possibility is that crucial factors of the NMD RNA surveillance pathway are inefficiently expressed in our experimental cells, or that upstream ORFs present within 5′UTR of DMPK transcript could possibly initiate translation from a different site and avoid NMD pathway. Alternatively, translation re-initiation from an in-frame downstream AUG codon might have bypassed NMD and accounted for the lack of DMPK reduction. The latter scenario, which frequently generates N-terminally truncated proteins, has been reported as a likely cause for bypassing NMD of other transcripts with nonsense mutations, for example HBB mRNA (human β-globin) or DMD (Duchenne Muscular Dystrophy) (49,50). DMPK antibody used in this study recognizes N-terminal part of the protein, therefore we are unable to conclude at the moment, whether such N-terminally truncated protein fragments were generated by AON-induced e5 and e9 skipping.
Our results demonstrated that AON-induced skipping of DMPK’s e15 triggers accumulation of potentially harmful spliced-out i14/e15/CUG pre-mRNA fragment which could be prevented by additional treatment with i14 directed gapmers. The exact determinants of the i14/e15/CUG stability in this context are currently unknown and remain to be further investigated. Notably, full functionality in vivo of the cryptic 3′SA and its ability to generate naturally occurring e16 DMPK splice isoforms devoid of CUG-expansion places DM1 mutation formally within the last intron of the gene (32). This closely resembles the situation observed in myotonic dystrophy type 2 (DM2) where massive intronic CCTG expansion within the first intron of CNBP (CCHC-type zinc finger nucleic acid binding protein) triggers similar yet milder disease course. Interestingly, significant retention of CNBP intron 1 has been reported in DM2 tissues and cells (51). In light of this finding, it would be interesting to test whether gapmers directed at intron 1 of CNBP could also neutralize the RNA containing retained intron and hence alleviate the molecular phenotype of DM2. However, similarly as in the case of DMPK reduction, decrease of CNBP transcripts may raise concerns due to controversial influence of the repeat expansions on RNA and protein expression (52–55). In our experiments, a significant reduction of DMPK transcripts in DM1 fibroblasts transfected with i14 gapmers was evident, yet molecular hallmarks of DM1 like spliceopathy and foci accumulation were effectively alleviated with no apparent detrimental effects on cells’ viability. Importantly, we observed no reduction of DMPK protein level even after long incubation with gapmers up to 96h, which might suggest high stability of the protein in this context. In addition, our experiments with luciferase reporters modeling CUGexp-induced DMPK translational defect demonstrated that AON-mediated exclusion of e15/CUG is a viable option for boosting nucleocytoplasmic transport and DMPK biosynthesis. The improvement was evident not only in the case of e15 splice-switching in mutant luciferase reporter carrying long repeats, but unexpectedly, also in the case of control reporter harboring no repeats. These results imply that the sole presence of CUG-neighboring/flanking sequences might have inhibitory role in DMPK mRNA nuclear export and translation and suggest their functional role in DMPK transcript metabolism and cellular trafficking, whereas CUG-repeated sequence might have an additive effect in this context. Interestingly, sequences within the 3′UTR of DMPK, including the CUG-repeated sequence, have been recently reported as binding sites for several distinct microRNAs (miRNAs) that may contribute to regulation of DMPK expression (56).
Intriguingly, we observed that 2′-Ome-PS AONs, but not their equivalents with otherwise modified backbone chemistry or gapmer-AONs alone, aggravated already pre-existing spliceopathy in DM1 cells and stimulated DM1-like splicing shifts in DM1-unaffected cells. These unexpected changes, likely due to AON chemistry‐dependent toxicity, were accompanied by increase in MBNL1 and CELF1 proteins, two antagonistic alternative splicing regulators implicated in DM1. While CELF1 upregulation was paralleled by mRNA increase, the increase in MBNL1 protein level may be explained by significantly increased DM1-like inclusion of e1 in MBNL1 mRNA, which we detected in control non-DM1 fibroblasts dosed with 2′-Ome-PS AONs. Our recent data demonstrated that MBNL1 is capable of switching off its own expression by binding to e1 and prompting its exclusion from its own pre-mRNA, and that functional depletion of MBNL1 such as in DM1, due to sequestration by CUGexp, hinders MBNL1 splicing activity resulting in e1 inclusion and increased production of a fully functional MBNL1 (36,40). Our current work indicates that delivery of 2′-Ome-PS AONs, regardless of their sequence, hinders functionality of MBNL1 and this releases the block of e1 skipping from MBNL1 mRNA to increase the protein content and prevent further aggravation of the phenotype by AONs toxicity. How exactly 2′-Ome-PS chemistry affects MBNL1 is unclear at the moment, however, experimental data from other groups indicates that upon exposure to a variety of environmental stress conditions, both MBNL1 and CELF1 can actively shuttle between the nucleus and cytoplasmic stress granules (57,58). Whether increased stress response is the case upon 2′-Ome-PS exposure, and how it might potentially affect MBNL1 and CELF1 splicing activities, however, awaits further investigation.
In conclusion, although neither of our novel DMPK splice-switching strategies was as successful as initially predicted, our data showed that molecular hallmarks of DM1 can be reversed by pre-mRNA reduction of DMPK. This has important implications for designing and testing novel therapeutic strategies using intron-directed gapmers not only in DM1, but also in DM2 associated with intronic repeat expansions. Targeting introns can be an ‘early prevention step’ i.e. down-modulating mutated pre-mRNA likely before it accumulates in ribonuclear foci and hinders splicing regulators. As a closing remark, it is important to point that a combination of both, the correct experimental approach and non-toxic AON chemistry, should be carefully considered to design safe therapeutics.
Supplementary Material
ACKNOWLEDGEMENTS
E.S.K., K.S. and P.K. designed the study; E.S.K., P.K., J.D. and P.C. performed experiments; E.S.K., P.K. and K.S. analysed the data; E.S.K. prepared figures and wrote the manuscript, with contributions from co-authors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Centre [2014/15/B/NZ5/00142 to E.S.K., 2018/30/E/NZ5/00065 to P.K.]; Foundation for Polish Science-TEAM program co-financed by the European Union within the European Regional Development Fund (to K.S.); Ministry of Science and Higher Education of the Republic of Poland, from the quality promoting subsidy, under the Leading National Research Centre (KNOW) program for the years 2014–2019. Funding for open access charge: National Science Centre [2014/15/B/NZ5/00142 to E.S.K.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Brook J.D., McCurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.P., Hudson T. et al.. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992; 69:385. [DOI] [PubMed] [Google Scholar]
- 2. Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., Swanson M.S.. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000; 19:4439–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mankodi A., Urbinati C.R., Yuan Q.P., Moxley R.T., Sansone V., Krym M., Henderson D., Schalling M., Swanson M.S., Thornton C.A.. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001; 10:2165–2170. [DOI] [PubMed] [Google Scholar]
- 4. Fardaei M., Rogers M.T., Thorpe H.M., Larkin K., Hamshere M.G., Harper P.S., Brook J.D.. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002; 11:805–814. [DOI] [PubMed] [Google Scholar]
- 5. Jiang H., Mankodi A., Swanson M.S., Moxley R.T., Thornton C.A.. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004; 13:3079–3088. [DOI] [PubMed] [Google Scholar]
- 6. Kuyumcu-Martinez N.M., Wang G.-S.S., Cooper T.A.. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell. 2007; 28:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang E.T., Ward A.J., Cherone J.M., Giudice J., Wang T.T., Treacy D.J., Lambert N.J., Freese P., Saxena T., Cooper T.A. et al.. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015; 25:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Osborne R.J., Thornton C.A.. RNA-dominant diseases. Hum. Mol. Genet. 2006; 15:R162–R169. [DOI] [PubMed] [Google Scholar]
- 9. Konieczny P., Stepniak-Konieczna E., Sobczak K.. Jurga S, Erdmann VA, Barciszewski J. Modified Nucleic Acids in Biology and Medicine. 2016; Cham: Springer International Publishing; 243–271. [Google Scholar]
- 10. Wheeler T.M., Sobczak K., Lueck J.D., Osborne R.J., Lin X., Dirksen R.T., Thornton C.A.. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science (New York, N.Y.). 2009; 325:336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wojtkowiak-Szlachcic A., Taylor K., Stepniak-Konieczna E., Sznajder L.J., Mykowska A., Sroka J., Thornton C.A., Sobczak K.. Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Res. 2015; 43:3318–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mulders S.A., van den Broek W.J., Wheeler T.M., Croes H.J., van Kuik-Romeijn P., de Kimpe S.J., Furling D., Platenburg G.J., Gourdon G., Thornton C.A. et al.. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. PNAS. 2009; 106:13915–13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakamori M., Gourdon G., Thornton C.A.. Stabilization of expanded (CTG)•(CAG) repeats by antisense oligonucleotides. Mol. Ther. 2011; 19:2222–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee J., Bennett C., Cooper T.. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. PNAS. 2012; 109:4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalez-Barriga A., Mulders S.A., van de Giessen J., Hooijer J.D., Bijl S., van Kessel I.D., van Beers J., van Deutekom J.C., Fransen J.A., Wieringa B. et al.. Design and analysis of effects of triplet repeat oligonucleotides in cell models for myotonic dystrophy. Mol. Ther. Nucleic Acids. 2013; 2:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Manning K.S., Rao A.N., Castro M., Cooper T.A.. BNA(NC) gapmers revert splicing and reduce RNA foci with low toxicity in myotonic dystrophy cells. ACS Chem. Biol. 2017; 12:2503–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wheeler T.M., Leger A.J., Pandey S.K., MacLeod A.R., Nakamori M., Cheng S.H., Wentworth B.M., Bennett C.F., Thornton C.A.. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012; 488:111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pandey S.K., Wheeler T.M., Justice S.L., Kim A., Younis H.S., Gattis D., Jauvin D., Puymirat J., Swayze E.E., Freier S.M. et al.. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J. Pharmacol. Exp. Ther. 2015; 355:310–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jauvin D., Chretien J., Pandey S.K., Martineau L., Revillod L., Bassez G., Lachon A., MacLeod A.R., Gourdon G., Wheeler T.M. et al.. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Ther. Nucleic Acids. 2017; 7:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thornton C.A., Wang E., Carrell E.M.. Myotonic dystrophy: approach to therapy. Curr. Opin. Genet. Dev. 2017; 44:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ward A.J., Norrbom M., Chun S., Bennett C.F., Rigo F.. Nonsense-mediated decay as a terminating mechanism for antisense oligonucleotides. Nucleic Acids Res. 2014; 42:5871–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Popp M.W., Maquat L.E.. Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell. 2016; 165:1319–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Davis B.M., McCurrach M.E., Taneja K.L., Singer R.H., Housman D.E.. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. PNAS. 1997; 94:7388–7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maquat L.E. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004; 5:89–99. [DOI] [PubMed] [Google Scholar]
- 25. Carrell S.T., Carrell E.M., Auerbach D., Pandey S.K., Bennett C.F., Dirksen R.T., Thornton C.A.. Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet. 2016; 25:4328–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reddy S., Smith D.B.J., Rich M.M., Leferovich J.M., Reilly P., Davis B.M., Tran K., Rayburn H., Bronson R., Cros D. et al.. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996; 13:325–335. [DOI] [PubMed] [Google Scholar]
- 27. Berul C.I., Maguire C.T., Aronovitz M.J., Greenwood J., Miller C., Gehrmann J., Housman D., Mendelsohn M.E., Reddy S.. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Invest. 1999; 103:R1–R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Groenen P.J.T.A., Wansink D.G., Coerwinkel M., van den Broek W., Jansen G., Wieringa B.. Constitutive and regulated modes of splicing produce six major myotonic dystrophy protein kinase (DMPK) isoforms with distinct properties. Hum. Mol. Genet. 2000; 9:605–616. [DOI] [PubMed] [Google Scholar]
- 29. Wansink D.G., van Herpen R.E., Coerwinkel-Driessen M.M., Groenen P.J., Hemmings B.A., Wieringa B.. Alternative splicing controls myotonic dystrophy protein kinase structure, enzymatic activity, and subcellular localization. Mol. Cell. Biol. 2003; 23:5489–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aartsma-Rus A., Straub V., Hemmings R., Haas M., Schlosser-Weber G., Stoyanova-Beninska V., Mercuri E., Muntoni F., Sepodes B., Vroom E. et al.. Development of exon skipping therapies for duchenne muscular dystrophy: a critical review and a perspective on the outstanding issues. Nucleic Acid Ther. 2017; 27:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meijboom K.E., Wood M.J.A., McClorey G.. Splice-Switching therapy for spinal muscular atrophy. Genes-Basel. 2017; 8:E161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tiscornia G., Mahadevan M.S.. Myotonic dystrophy: the role of the CUG triplet repeats in splicing of a novel DMPK exon and altered cytoplasmic DMPK mRNA isoform ratios. Mol. Cell. 2000; 5:959–967. [DOI] [PubMed] [Google Scholar]
- 33. Childs-Disney J.L., Stepniak-Konieczna E., Tran T., Yildirim I., Park H., Chen C.Z., Hoskins J., Southall N., Marugan J.J., Patnaik S. et al.. Induction and reversal of myotonic dystrophy type 1 pre-mRNA splicing defects by small molecules. Nat. Commun. 2013; 4:2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cartegni L., Wang J., Zhu Z., Zhang M.Q., Krainer A.R.. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003; 31:3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smith P.J., Zhang C., Wang J., Chew S.L., Zhang M.Q., Krainer A.R.. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum. Mol. Genet. 2006; 15:2490–2508. [DOI] [PubMed] [Google Scholar]
- 36. Konieczny P., Stepniak-Konieczna E., Taylor K., Sznajder L.J., Sobczak K.. Autoregulation of MBNL1 function by exon 1 exclusion from MBNL1 transcript. Nucleic Acids Res. 2017; 45:1760–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ho T.H., Charlet-B N., Poulos M.G., Singh G., Swanson M.S., Cooper T.A.. Muscleblind proteins regulate alternative splicing. EMBO J. 2004; 23:3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Philips A.V., Timchenko L.T., Cooper T.A.. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998; 280:737–741. [DOI] [PubMed] [Google Scholar]
- 39. Popp M.W., Maquat L.E.. The dharma of nonsense-mediated mRNA decay in mammalian cells. Mol. Cells. 2014; 37:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Konieczny P., Stepniak-Konieczna E., Sobczak K.. MBNL expression in autoregulatory feedback loops. RNA Biol. 2018; 15:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin X., Miller J.W., Mankodi A., Kanadia R.N., Yuan Y., Moxley R.T., Swanson M.S., Thornton C.A.. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006; 15:2087–2097. [DOI] [PubMed] [Google Scholar]
- 42. Terenzi F., Ladd A.N.. Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA Biol. 2010; 7:43–55. [DOI] [PubMed] [Google Scholar]
- 43. Fernandez-Costa J.M., Artero R.. A conserved motif controls nuclear localization of Drosophila Muscleblind. Mol. Cells. 2010; 30:65–70. [DOI] [PubMed] [Google Scholar]
- 44. Tran H., Gourrier N., Lemercier-Neuillet C., Dhaenens C.M., Vautrin A., Fernandez-Gomez F.J., Arandel L., Carpentier C., Obriot H., Eddarkaoui S. et al.. Analysis of exonic regions involved in nuclear localization, splicing activity, and dimerization of Muscleblind-like-1 isoforms. J. Biol. Chem. 2011; 286:16435–16446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kino Y., Washizu C., Kurosawa M., Oma Y., Hattori N., Ishiura S., Nukina N.. Nuclear localization of MBNL1: splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum. Mol. Genet. 2015; 24:740–756. [DOI] [PubMed] [Google Scholar]
- 46. Sznajder L.J., Michalak M., Taylor K., Cywoniuk P., Kabza M., Wojtkowiak-Szlachcic A., Matloka M., Konieczny P., Sobczak K.. Mechanistic determinants of MBNL activity. Nucleic Acids Res. 2016; 44:10326–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nakamori M., Sobczak K., Puwanant A., Welle S., Eichinger K., Pandya S., Dekdebrun J., Heatwole C.R., McDermott M.P., Chen T. et al.. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013; 74:862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nagy E., Maquat L.E.. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 1998; 23:198–199. [DOI] [PubMed] [Google Scholar]
- 49. Neu-Yilik G., Amthor B., Gehring N.H., Bahri S., Paidassi H., Hentze M.W., Kulozik A.E.. Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA. 2011; 17:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gurvich O.L., Maiti B., Weiss R.B., Aggarwal G., Howard M.T., Flanigan K.M.. DMD exon 1 truncating point mutations: amelioration of phenotype by alternative translation initiation in exon 6. Hum. Mutat. 2009; 30:633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sznajder L.J., Thomas J.D., Carrell E.M., Reid T., McFarland K.N., Cleary J.D., Oliveira R., Nutter C.A., Bhatt K., Sobczak K. et al.. Intron retention induced by microsatellite expansions as a disease biomarker. PNAS. 2018; 115:4234–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Margolis J.M., Schoser B.G., Moseley M.L., Day J.W., Ranum L.P.. DM2 intronic expansions: evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum. Mol. Genet. 2006; 15:1808–1815. [DOI] [PubMed] [Google Scholar]
- 53. Pelletier R., Hamel F., Beaulieu D., Patry L., Haineault C., Tarnopolsky M., Schoser B., Puymirat J.. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol. Dis. 2009; 36:181–190. [DOI] [PubMed] [Google Scholar]
- 54. Huichalaf C., Schoser B., Schneider-Gold C., Jin B., Sarkar P., Timchenko L.. Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J. Neurosci. 2009; 29:9042–9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Raheem O., Olufemi S.E., Bachinski L.L., Vihola A., Sirito M., Holmlund-Hampf J., Haapasalo H., Li Y.P., Udd B., Krahe R.. Mutant (CCTG)n expansion causes abnormal expression of zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2. Am. J. Pathol. 2010; 177:3025–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koscianska E., Witkos T.M., Kozlowska E., Wojciechowska M., Krzyzosiak W.J.. Cooperation meets competition in microRNA-mediated DMPK transcript regulation. Nucleic Acids Res. 2015; 43:9500–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Onishi H., Kino Y., Morita T., Futai E., Sasagawa N., Ishiura S.. MBNL1 associates with YB-1 in cytoplasmic stress granules. J. Neurosci. Res. 2008; 86:1994–2002. [DOI] [PubMed] [Google Scholar]
- 58. Fujimura K., Kano F., Murata M.. Dual localization of the RNA binding protein CUGBP-1 to stress granule and perinucleolar compartment. Exp. Cell Res. 2008; 314:543–553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.