

Abstract
Mammalian phagocytes carry out several essential functions, including killing and digesting infectious organisms, clearing denatured proteins, removing dead cells, and removing several types of debris from the extracellular space. Many of these functions involve phagocytosis, the engulfment of a target in a specialized endocytic process and then fusion of the new phagosome with lysosomes. Phagocytes such as macrophages can phagocytose targets that are several μm in diameter (e.g., dead cells), but in some cases they encounter much larger objects. We have studied two such examples in some detail: large deposits of lipoproteins such as those in the wall of blood vessels associated with atherosclerosis, and dead adipocytes, which are dozens of μm in diameter. We describe a process, which we call digestive exophagy, in which macrophages create a tight seal in contact with the target, acidify the sealed zone, and secrete lysosomal contents into the contact zone. By this process, hydrolysis by lysosomal enzymes occurs in a compartment that is outside the cell. We compare this process to the well characterized digestion of bone by osteoclasts, and we point out key similarities and differences.
Keywords: Lysosomes, exocytosis, phagocytosis, macrophages, exophagy
Mammalian phagocytic cells carry out essential processes to clear debris and to remove infectious agents such as bacteria and fungi1–3. Various types of debris accumulate and must be cleared, including denatured proteins and damaged or dead cells. In a typical process, macrophages, microglia or other phagocytes respond to chemoattractant gradients released by dead cells or infectious agents. They migrate toward the source of the chemoattractant and engulf the dead cell or infectious agent. The phagosome that is formed fuses with lysosomes, and the target is digested by lysosomal hydrolases. In response to infectious agents, macrophages and other immune system cells release reactive oxygen species, which aids in killing the infectious agent. This whole process has been characterized extensively and described in excellent reviews1–3.
We were interested in a particular aspect of macrophage biology: their role in formation of atherosclerotic lesions4. Early stages in the development of an atherosclerotic lesion involve the digestion of lipoproteins in the vessel wall by macrophages, including hydrolysis of cholesteryl esters in the core of the lipoproteins, leading to delivery of unesterified (free) cholesterol and a fatty acid to the macrophages. While uptake of soluble lipoproteins is well characterized4,5, as discussed below most LDL in the vessel wall is not soluble. Regardless of the mechanism of cholesterol delivery, cells closely regulate levels of free cholesterol, and excess free cholesterol is rapidly esterified and stored in cholesteryl ester rich lipid droplets. Macrophages that process large amounts of lipoproteins cannot release the cholesterol load adequately, and macrophages filled with lipid droplets are described as foam cells. This lipid loading is eventually toxic, and the foam cells die6. Other macrophages are recruited to digest the dead cells7, and they are exposed to this large load of cholesteryl esters and become foam cells themselves. Over time this can lead to an unresolved collection of dead cells, which can contribute to deforming the vessel wall, leading to a blockage of blood flow.
In studies of atherosclerosis related to an early step in this process in which macrophages clear large deposits or aggregates of lipoproteins in the wall of blood vessels, we encountered a conundrum. These aggregates were large (a few hundred nm to a few μm), but more importantly, they were tightly crosslinked to the extracellular matrix8,9. It did not seem possible that macrophages could simply take these particles up by endocytosis or phagocytosis, but we found that the hydrolysis of cholesteryl esters in the core of these aggregates required a lysosomal enzyme – lysosomal acid lipase (LIPA)10. Consideration of this biological puzzle led us to a more general question: How do phagocytic cells digest objects too large to be phagocytosed? A simple solution would be to secrete hydrolases that are active at neutral pH. The release of matrix metalloproteases by migratory cells is a well-characterized example of this mechanism. However, in our study of catabolism of aggregates of LDL, the digestion by LIPA would require a low pH. This suggested that a different type of process was occurring – one that would require a sealed compartment outside the cell that would allow sufficient acidification to activate lysosomal hydrolases.
There is a very well-characterized process of this type – digestion of bone by osteoclasts, which are specialized cells of the monocyte/macrophage lineage11,12. Osteoclasts attach to bone by a process that involves αvβ3 integrin attachment followed by formation of an actin-dependent ruffled border that creates a tight seal between the bone and the cell. The resulting resorptive compartment is acidified by the vacuolar H+ pumping ATPase (V-ATPase) in conjunction with the ClC-7 Cl−/H+ antiporter13. There is polarized secretion of lysosomal hydrolases into the resorptive compartment, and the combination of low pH and acid hydrolases dissolves the bone matrix and digests the protein components of the bone. The key features of this digestion are the formation of a tightly sealed attachment zone, acidification of the resorptive compartment, and polarized secretion of lysosomal hydrolases11,12. This process has generally been considered a highly specialized activity of osteoclasts to deal with the unique challenge of bone resorption.
Description of digestive exophagy of agLDL
We considered the possibility that macrophages actually might use a similar process to degrade large, matrix-attached aggregates of lipoprotein in the wall of blood vessels. To test this idea in a cell culture model, we created large aggregates of LDL (agLDL) that are approximately 20 −50 μm across, and we exposed macrophages to the agLDL14,15. These artificial aggregates were much larger than those observed in vivo9, which were attached to extracellular matrix – preventing endocytosis or phagocytosis. It seemed that the most challenging aspect of this would be the creation of an extracellular acidified compartment since hydronium ions (H3O+) would need to be blocked from diffusion out of the compartment. To observe this, we covalently attached pH-sensitive and pH-insensitive fluorescent dyes to the agLDL, and we carried out ratiometric fluorescence imaging. We observed that at sites of contact there were regions that were acidified to pH 6 or below, and the acidification was inhibited by bafilomycin A1, confirming that it was due to the V-ATPase15. It is known that macrophages have V-ATPases on the plasma membrane16, but the maintenance of a pH gradient implies that the protons must be pumped into a relatively well-sealed compartment. In preliminary studies to characterize the compartment, we used fluorescence microscopy, and we observed that portions of the extracellular agLDL could invaginate a few μm into the cells while still attached to the bulk of the agLDL that was outside the cell14,15. (Electron microscopy studies of the compartment are described below.)
To see if there was polarized exocytosis of lysosomes, we preloaded macrophage lysosomes by an overnight incubation in biotinylated-fluorescein dextran. We then exposed the macrophages to agLDL that had been covalently coupled to streptavidin to capture any lysosomal biotinylated-fluorescein dextran that was secreted from the cells onto the agLDL. We found that lysosomal contents were deposited onto the extracellular agLDL15. This secretion was polarized, and streptavidin beads surrounding the macrophages were not labeled if they were not near to the agLDL contact area. The presence of lysosomal hydrolases in an acidified compartment suggests that there should be hydrolysis of the agLDL, and we did observe hydrolysis of cholesteryl esters in the agLDL to generate free cholesterol. This was observed by measurement of free cholesterol in the extracellular agLDL both biochemically15 and by the detection of free cholesterol in the agLDL by filipin, a fluorescent molecule that binds free cholesterol14. All of these results indicated that, indeed, macrophages can carry out an extracellular digestive process using mechanisms that are in some respects similar to digestion of bone by osteoclasts. We have called this process “digestive exophagy”.
We have carried out some characterization of the mechanisms by which macrophages carry out exophagy. As noted above, osteoclasts use F-actin in the formation of the ruffled border that seals the resorptive compartment11,12. Using phalloidin staining, we observed highly polarized formation of F-actin at the sites of contact between macrophages and agLDL14. This actin polymerization was enhanced by the release of the free cholesterol from the agLDL, and the formation of F-actin was greatly attenuated when agLDL was filled with non-hydrolysable cholesteryl ether14. This suggested that there was a positive feedback in the macrophage digestion of agLDL, with the released cholesterol inserting into the plasma membrane and activating signals that enhanced actin polymerization. We had shown previously that cholesterol delivered to the plasma membrane of macrophages would lead to actin polymerization through the activity of Rho family GTPases17.
An important issue is the structure of the acidified compartments, which we have called “lysosomal synapses”. We initially tried to observe this with conventional thin section transmission electron microscopy, but the compartments were much too large to capture a significant fraction of a lysosomal synapse in a single thin section. We then tried EM tomography on sections that were 220–250 nm in thickness18, but even this did not provide enough depth to capture an entire lysosomal synapse. We then used focused ion beam-scanning electron microscopy (FIB-SEM) in which a thin slice of the sample is milled off between each round of scanning. With about a thousand sequential 40 nm slices, we were able to capture most of a cell, and we were able to see the full scale of a lysosomal synapse between a macrophage and agLDL18. This is best viewed from the movies that are in the supplementary material of that paper. FIB-SEM data are shown in Figures 1 and 2, and these illustrate the deep penetration of the lysosomal synapse into the cell. By confocal microscopy, we also found that there were often F-actin rings around the dead end projections of the lysosomal synapse, which would help with sealing in these regions18.
Figure 1. FIB-SEM microscopy of agLDL.
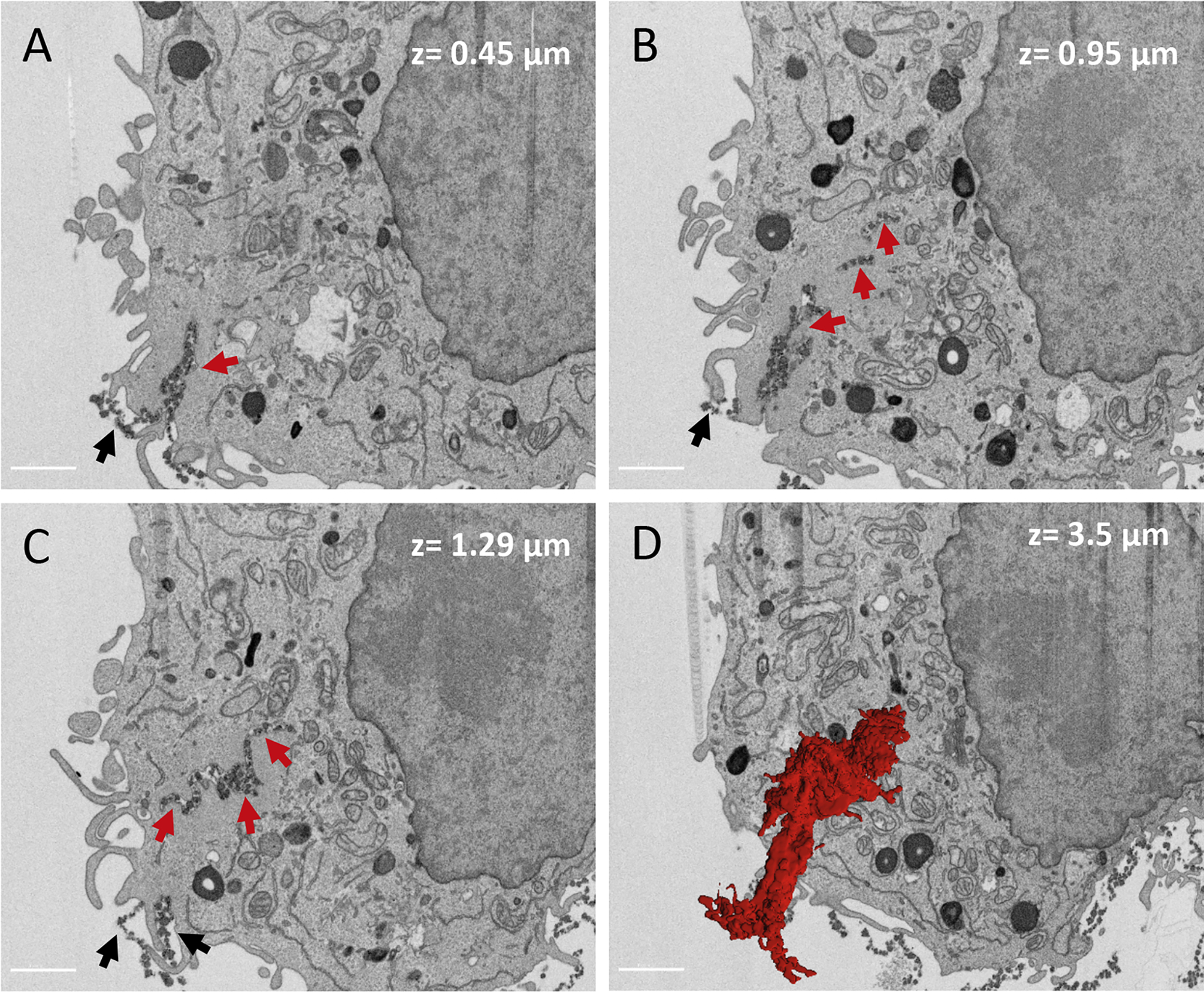
J774 macrophages were incubated with colloidal-gold-labeled agLDL for 1 h, followed by fixation and staining for EM, as described previously18. (A-C) SEM images at different depths (0.45 μm, 0.95 μm, and 1.29 μm) show agLDL being internalized, as indicated by the red arrows. Black arrows indicate agLDL that has not been internalized. (D) 3D reconstruction of agLDL allows visualization of deeply internalized agLDL that it is continuously connected to the extracellular space, as shown in red. Segmentation of agLDL was made manually and was carried out on 320 slices with a total z depth of 3.5 μm. Segmentation and 3D rendering were made using Imaris (Bitplane). Size bars 2 μm. Images were obtained with a Zeiss Crossbeam 540 FIB-SEM in a backscatter mode with a z step size of 10 nm.
Figure 2. 3D reconstruction of agLDL interacting with a J774 macrophages from a FIB-SEM image series.

J774 macrophages were incubated with colloidal gold-labeled agLDL for 1 h, fixed and processed for FIB-SEM. Images were acquired every 40 nm depth. Images were segmented and outlined in each image slice and compiled to generate a 3D reconstruction. Red, agLDL. Grey, macrophage plasma membrane. Green, macrophage plasma membrane in contact with agLDL. Reprinted from reference18.
From these studies we learned that there were similarities but also important differences between osteoclast digestion of bone and macrophage digestion of agLDL. Both depend on F-actin to form a sealed extracellular compartment and acidify it by the action of V-ATPase. Both also involve polarized secretion of lysosomal hydrolases and digestion topologically outside the cell. However, bone is stiff, and the resorptive compartment on bone is formed on a relatively flat substrate. In contrast, the agLDL becomes invaginated deeply into the macrophages. We also noted that the lysosomal synapses of macrophages are very dynamic with significant movement and changes in pH in some regions within 10 seconds15.
It should be noted that other investigators have examined mechanisms for uptake and digestion of lipoproteins in the vessel wall by macrophages19,20. One model has proposed that pinocytosis plays a key role. It is not clear how the agLDL that is tightly linked to the extracellular matrix could be pinocytosed directly. However, we have observed that fragments of agLDL are released during exophagy, and these fragments are endocytosed by the engaged macrophage and could be engulfed by nearby macrophages.
We have observed exophagy of agLDL in cultured macrophage-like cells (J774 and RAW 264.7), in human monocyte-derived macrophages, and in murine bone marrow-derived macrophages. We also observed exophagy of agLDL by human and murine monocyte-derived dendritic cells21. The observation in dendritic cells is significant because it has been suggested that these cells might be involved in the earliest stages of atherosclerotic plaque development22. In addition to our cell culture studies, we have observed lysosomal synapse activity in murine models of atherosclerosis23,24. Additionally, pathologists have reported the presence of filipin-labeled extracellular lipid deposits in human atherosclerotic lesions25, which are likely similar to our observation that agLDL in contact with macrophages become enriched in free cholesterol due to hydrolysis of the cholesteryl esters.
Regulation of exophagy of agLDL
In contrast to studies of bone resorption by osteoclasts, which has been studied by many laboratories over several decades11,12, we are just starting to characterize the signaling processes that regulate exophagy of agLDL, and again there are some similarities and some differences with osteoclasts. Both cell types use a Vav guanine nucleotide exchanger as part of the regulation cascade for actin polymerization11,12,23, but for osteoclasts Rac seems to be the main Rho-GTPase involved while Cdc42 plays a key role in regulating F-actin formation during exophagy. As described earlier, cholesterol elevation in the macrophage plasma membrane provides a positive signal for increased actin polymerization associated with exophagy of agLDL, and this signaling also involves Rho-GTPases Rac or Cdc4217. In contrast another hydrolysis product provides negative feedback for exophagy of agLDL. Acid sphingomyelinase hydrolyses sphingomyelin in the outer monolayer surrounding agLDL, producing ceramide. During exophagy, macrophages take up ceramide from agLDL, and increased ceramide in the cells impairs actin polymerization through a process involving RhoA and Rho kinase24. This effect tends to slow down catabolism of agLDL. (We are not aware that products of bone resorption feed back on the activity of osteoclasts.)
We are carrying out more systematic analyses of signaling pathways involved in exophagy of agLDL, and a preliminary description of these studies has been provided26. Some of these signaling pathways are outlined in Figure 3. These studies are far from complete as seen in a comparison with the level of detail describing osteoclast regulation. Perhaps most interesting is the role of TLR4 signaling in exophagy of agLDL. Macrophages lacking TLR4 have greatly reduced actin polymerization and lysosome secretion in response to agLDL26.
Figure 3. Schematic of a TLR4-dependent macrophage signaling cascade regulating degradative exophagy of agLDL.

AgLDL induced TLR4/CD14 signaling activates MyD88, PI3K and AKT and leads to formation of the compartment. Activation of signaling through AKT1/2 stimulates lysosome exocytosis, which delivers lysosomal acid lipase (LAL) to the compartment. V-ATPase pumps protons into the compartment and establishes a low pH. This enables LAL activity in the compartment, which stimulates agLDL degradation and generation of free cholesterol (FC). This FC inserts into the plasma membrane and further stimulates signaling. The activity of PI3K and AKT2 lead to Vav and Cdc42 activation that support longer term stabilization of the compartment.
Consequences of exophagy vs. phagocytic or pinocytic uptake of lipoproteins
It is reasonable to wonder if the mode of cholesterol uptake from agLDL actually matters. Whether by a conventional endocytic/phagocytic process or by exophagy, cholesterol is delivered to the macrophage, esterified, and stored in lipid droplets. One important difference is that delivery of cholesterol to the plasma membrane activates signaling pathways that increase actin polymerization and may have other effects17. TLR4 signaling in response to agLDL would activate the innate immune signaling, and chronic inflammation is a key aspect of atherosclerosis26. A potentially important consequence of extracellular hydrolysis of cholesteryl esters is the presence of very high levels of free cholesterol in the extracellular agLDL during catabolism. This extracellular free cholesterol could lead to the formation of cholesterol monohydrate crystals in the extracellular space. Such cholesterol crystals are an important feature of atherosclerotic plaques, and there is evidence that they might play a role in plaque rupture27.
Exophagy of dead adipocytes
There is normal turnover of adipocytes, and this is greatly increased under some metabolic disorder conditions, such as, obesity and type 2 diabetes. The expansion of white adipose tissue (WAT) under these circumstances is followed by the accumulation of immune cells, predominantly by adipose tissue macrophages (ATMs), contributing to a low-grade chronic inflammation that ultimately leads to hypertrophy and death of WAT28. An adipocyte is as much as 500 times the volume of a macrophage, so the typical phagocytic clearance of dead cells cannot be used to remove dead adipocytes. Macrophages encircle dead adipocytes in white adipose tissue forming a crown-like structure29. The macrophages surrounding the dead adipocyte accumulate lipid from the adipocyte and become foam cells loaded with triacylglycerides (TAG). We examined how the macrophages digest the TAG in the adipocyte, take up the released fatty acids, and then re-esterify them to form TAG lipid droplets in the macrophages. We used many of the methods that we had developed to study exophagy of agLDL30.
Using electron microscopy, and especially FIB-SEM, we found that individual macrophages formed extensive areas of very close contact with dead adipocytes. These contact areas did not involve invagination into the macrophage as had been seen with agLDL. The adipocyte surface remained nearly flat with openings of a few hundred nm in depth and up to several μm wide surrounded by tight apposition of the two cells. By linking pH-sensitive and insensitive fluorescent dyes to the surface of the adipocytes, we could observe acidification of these contact regions to pH values below 6. Again, the acidification was inhibited by bafilomycin A1, indicating that it was due to V-ATPase activity. We preloaded macrophage lysosomes with biotin-fluorescein-dextran, and we coupled streptavidin to the surface of the adipocytes. Within 90 minutes, we could observe extensive deposition of lysosomal contents into lysosomal synapses between the macrophages and the dead adipocytes.
Bone marrow-derived macrophages from TLR2−/− mice showed about a 70% reduction in lysosome secretion upon contact with dead adipocytes31. NOX2 deficient (Gp91−/−) mice also had greatly reduced lysosome exocytosis, and this correlated with reduced lipid uptake by the macrophages31. It is interesting that TLR2 knockout reduced lysosome secretion to dead adipocytes but had no effect on lysosome secretion to agLDL. The converse was observed for TLR4 knockout, which reduced exophagy of agLDL but not dead adipocytes. In both cases, knockout of Myd88 greatly reduced lysosome secretion and catabolism of the targets. When Nox2 deficient mice were placed on a high fat diet for 16 weeks, there was a large increase in crown-like structures, which is consistent with an inability to clear dead adipocytes31. Altogether, exophagy of dead adipocytes and agLDL share key features (tightly sealed zones, acidification, and lysosome secretion), but they differ in the shape of the compartment and the cell surface Toll-like receptor that is used for signaling.
Other potential targets of exophagy
One can now speculate on other potential targets of exophagy based on their large size but observed clearance by phagocytes. A recent paper showed that Kupffer cells, the resident macrophages of the liver, form crown-like structures and use a similar process to digest remnant lipid droplets and dead hepatocytes in a model of steatosis32. Alzheimer’s amyloid deposits seem like a candidate for this type of clearance – in this case by microglia, which are the main phagocytic cells in the central nervous system. It also seems possible that dead neurons could trigger a similar process. The neuronal processes can extend for great lengths, and a single microglial cell could not clear them by phagocytosis. Using exophagy, these neuronal processes could be broken into pieces that could then be phagocytosed and digested. Debris-filled extracellular pockets have been observed in the brains of wild type and Alzheimer’s disease mice33, and it will be interesting to see if these are examples of exophagy. There are many other biological systems that may use digestive exophagy.
Relationship to other processes
The closest analogy to exophagy by macrophages is clearly digestion of bone by osteoclasts, and we have pointed out some similarities and differences between these processes. Cell biologists have used “frustrated phagocytosis” to characterize early steps of phagocytosis34,35. Macrophages are plated onto an opsonized surface (e.g., a glass coverslip with attached immune complexes), and signaling through surface receptors is activated leading to an attempt to phagocytose the object. The cells generally become spread as far as possible and then the process stalls. Examination of these cells reveals many similarities to initial steps of phagocytosis. This includes enhanced actin polymerization around the circumference of the “phagosome”. Lysosome secretion has also been observed36. We have been unable to detect significant acidification, probably because the gaps around the phagocytic ring are too wide to block diffusion effectively.
Summary
It is well known that osteoclasts create an extracellular resorptive compartment to digest bone as part of a normal cycle to maintain healthy bone structure. The digestion relies on low pH and lysosomal hydrolases, which require tight sealing to prevent diffusion out if the compartment. This has generally been considered a unique specialization because of the special properties of bone. However, in considering how phagocytes digest other large objects, we wondered if macrophages might be able to create similar compartments to digest large objects such as aggregates of LDL that are tightly crosslinked to the extracellular matrix in atherosclerotic lesions. As summarized in this review, we have found that macrophages do use a similar strategy to digest this agLDL in an extracellular hydrolytic compartment, a lysosomal synapse. While the overall process is similar (tight seal, low pH, lysosomal enzymes), many of the details are different. These differences include the morphology of the digestive compartments, the precise nature of the seal, and apparently some of the signaling and regulation. Realizing that this was not a unique specialization, we also examined the digestion of dead adipocytes by macrophages, and we found that the basic principles were similar to digestive exophagy of agLDL. However, there were also differences in the morphology of the lysosomal synapse and the signaling and regulation. Recently, others have found evidence for digestive exophagy by Kupffer cells associated with steatosis and perhaps in microglia. It now seems likely that as we learn how to study these processes, they will be found more frequently for a variety of purposes and with adaptations for each particular example of digestive exophagy.
Synopsis:
Phagocytic cells have developed mechanisms, which we call digestive exophagy, to digest very large objects outside the cell using lysosomal enzymes. To do this, they create a tightly sealed extracellular compartment (a lysosomal synapse) that is attached to the target. Lysosomes are secreted into the lysosomal synapse, which is acidified by the proton pumping V-ATPase. Known targets of digestive exophagy include aggregates of LDL deposited in atherosclerotic lesions and dead adipocytes.
Acknowledgement
We thank Leona Cohen-Gould at the Electron Microscopy & Histological Core and Optical Microscopy Core at Weill Cornell Medical College for helping with FIB-SEM sample preparation. We also thank Vincent Cavaliere, Ruth Redman, and Pal Pedersen at Carl Zeiss Microscopy LLC, Thornwood, NY for their generous help and assistance in generating the FIB-SEM data, shown in Figure 1. The work described here has been supported by NIH grant R01-HL093324.
Footnotes
The authors declare they have no conflicts of interest.
References
- 1.Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. 2012;7:61–98. [DOI] [PubMed] [Google Scholar]
- 2.Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev. 2014;262(1):193–215. [DOI] [PubMed] [Google Scholar]
- 3.Gordon S. Phagocytosis: An Immunobiologic Process. Immunity. 2016;44(3):463–475. [DOI] [PubMed] [Google Scholar]
- 4.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438(7068):612–621. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Apoptosis Tabas I. and plaque destabilization in atherosclerosis: the role of macrophage apoptosis induced by cholesterol. Cell Death Differ. 2004;11 Suppl 1:S12–16. [DOI] [PubMed] [Google Scholar]
- 7.Yurdagul A Jr., Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front Cardiovasc Med. 2017;4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832–1844. [DOI] [PubMed] [Google Scholar]
- 9.Tamminen M, Mottino G, Qiao JH, Breslow JL, Frank JS. Ultrastructure of early lipid accumulation in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19(4):847–853. [DOI] [PubMed] [Google Scholar]
- 10.Buton X, Mamdouh Z, Ghosh R, et al. Unique cellular events occurring during the initial interaction of macrophages with matrix-retained or methylated aggregated low density lipoprotein (LDL). Prolonged cell-surface contact during which ldl-cholesteryl ester hydrolysis exceeds ldl protein degradation. J Biol Chem. 1999;274(45):32112–32121. [DOI] [PubMed] [Google Scholar]
- 11.Feng X, Teitelbaum SL. Osteoclasts: New Insights. Bone Res. 2013;1(1):11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soysa NS, Alles N. Osteoclast function and bone-resorbing activity: An overview. Biochem Biophys Res Commun. 2016;476(3):115–120. [DOI] [PubMed] [Google Scholar]
- 13.Marcoline FV, Ishida Y, Mindell JA, Nayak S, Grabe M. A mathematical model of osteoclast acidification during bone resorption. Bone. 2016;93:167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grosheva I, Haka AS, Qin C, Pierini LM, Maxfield FR. Aggregated LDL in contact with macrophages induces local increases in free cholesterol levels that regulate local actin polymerization. Arterioscler Thromb Vasc Biol. 2009;29(10):1615–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haka AS, Grosheva I, Chiang E, et al. Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Mol Biol Cell. 2009;20(23):4932–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brisseau GF, Grinstein S, Hackam DJ, et al. Interleukin-1 increases vacuolar-type H+-ATPase activity in murine peritoneal macrophages. J Biol Chem. 1996;271(4):2005–2011. [DOI] [PubMed] [Google Scholar]
- 17.Qin C, Nagao T, Grosheva I, Maxfield FR, Pierini LM. Elevated plasma membrane cholesterol content alters macrophage signaling and function. Arterioscler Thromb Vasc Biol. 2006;26(2):372–378. [DOI] [PubMed] [Google Scholar]
- 18.Singh RK, Barbosa-Lorenzi VC, Lund FW, Grosheva I, Maxfield FR, Haka AS. Degradation of aggregated LDL occurs in complex extracellular sub-compartments of the lysosomal synapse. J Cell Sci. 2016;129(5):1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruth HS. Sequestration of aggregated low-density lipoproteins by macrophages. Curr Opin Lipidol. 2002;13(5):483–488. [DOI] [PubMed] [Google Scholar]
- 20.Kruth HS, Jones NL, Huang W, et al. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem. 2005;280(3):2352–2360. [DOI] [PubMed] [Google Scholar]
- 21.Haka AS, Singh RK, Grosheva I, et al. Monocyte-Derived Dendritic Cells Upregulate Extracellular Catabolism of Aggregated Low-Density Lipoprotein on Maturation, Leading to Foam Cell Formation. Arterioscler Thromb Vasc Biol. 2015;35(10):2092–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106(2):383–390. [DOI] [PubMed] [Google Scholar]
- 23.Singh RK, Haka AS, Bhardwaj P, Zha X, Maxfield FR. Dynamic Actin Reorganization and Vav/Cdc42-Dependent Actin Polymerization Promote Macrophage Aggregated LDL (Low-Density Lipoprotein) Uptake and Catabolism. Arterioscler Thromb Vasc Biol. 2019;39(2):137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh RK, Haka AS, Brumfield A, et al. Ceramide activation of RhoA/Rho kinase impairs actin polymerization during aggregated LDL catabolism. J Lipid Res. 2017;58(10):1977–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarig S, Utian WH, Sheean LA, Gorodeski GI. Distribution of unesterified cholesterol-containing particles in human atherosclerotic lesions. Am J Pathol. 1995;146(1):139–147. [PMC free article] [PubMed] [Google Scholar]
- 26.Singh RK, Haka AS, Asmal A, et al. TLR4 (Toll-Like Receptor 4)-Dependent Signaling Drives Extracellular Catabolism of LDL (Low-Density Lipoprotein) Aggregates. Arterioscler Thromb Vasc Biol. 2019:ATVBAHA119313200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grebe A, Latz E. Cholesterol crystals and inflammation. Curr Rheumatol Rep. 2013;15(3):313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russo L, Lumeng CN. Properties and functions of adipose tissue macrophages in obesity. Immunology. 2018;155(4):407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46(11):2347–2355. [DOI] [PubMed] [Google Scholar]
- 30.Haka AS, Barbosa-Lorenzi VC, Lee HJ, et al. Exocytosis of macrophage lysosomes leads to digestion of apoptotic adipocytes and foam cell formation. J Lipid Res. 2016;57(6):980–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coats BR, Schoenfelt KQ, Barbosa-Lorenzi VC, et al. Metabolically Activated Adipose Tissue Macrophages Perform Detrimental and Beneficial Functions during Diet-Induced Obesity. Cell Rep. 2017;20(13):3149–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ioannou GN, Subramanian S, Chait A, et al. Cholesterol crystallization within hepatocyte lipid droplets and its role in murine NASH. Journal of Lipid Research. 2017;58(6):1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El Hajj H, Savage JC, Bisht K, et al. Ultrastructural evidence of microglial heterogeneity in Alzheimer’s disease amyloid pathology. Journal of Neuroinflammation. 2019;16(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heiple JM, Wright SD, Allen NS, Silverstein SC. Macrophages form circular zones of very close apposition to IgG-coated surfaces. Cell Motil Cytoskeleton. 1990;15(4):260–270. [DOI] [PubMed] [Google Scholar]
- 35.Masters TA, Pontes B, Viasnoff V, Li Y, Gauthier NC. Plasma membrane tension orchestrates membrane trafficking, cytoskeletal remodeling, and biochemical signaling during phagocytosis. Proc Natl Acad Sci U S A. 2013;110(29):11875–11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Labrousse AM, Meunier E, Record J, et al. Frustrated phagocytosis on micro-patterned immune complexes to characterize lysosome movements in live macrophages. Front Immunol. 2011;2:51. [DOI] [PMC free article] [PubMed] [Google Scholar]